
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of new chiral N-heterocyclic diselenides and their application in the alkoxyselenylation reaction†
Karolina
Kamińska
 * and
Elżbieta
Wojaczyńska
*
* and
Elżbieta
Wojaczyńska
*
Faculty of Chemistry, Wrocław University of Science and Technology, Wybrzeże Wyspiańskiego 27, 50 370 Wrocław, Poland. E-mail: karolina.kaminska@pwr.edu.pl; elzbieta.wojaczynska@pwr.edu.pl
First published on 21st June 2022
Abstract
This paper describes the synthesis of chiral diselenides based on 2-azabicycloalkane or the pyrrolidine skeleton using in situ generated Na2Se2 as an efficient selenylating reagent. All the diselenides were obtained in moderate yields under mild conditions. The well-defined enantio- and diastereomerically pure compounds were tested in the asymmetric alkoxyselenylation of alkenes yielding products with diastereoisomeric excess up to >99%.
Introduction
Significant interest in organoselenium compounds is associated with their biological activity. Selenium is a key microelement responsible for the antioxidant protection of the living cells, due to the action of glutathione peroxidase, a Se-containing enzyme.1,2 The most important synthetic Gpx mimic, ebselen (2-phenyl-1,2-benzoselenazol-3(2H)-one, Fig. 1),3,4 has been investigated for the treatment of various conditions, including diabetes,5 Alzheimer's disease,6 bipolar disorder,7 ischemic stroke,8 hearing loss,9 and cancer.10 Recently, its derivatives have been considered as efficient inhibitors of a protease of the SARS-CoV-2 virus.11,12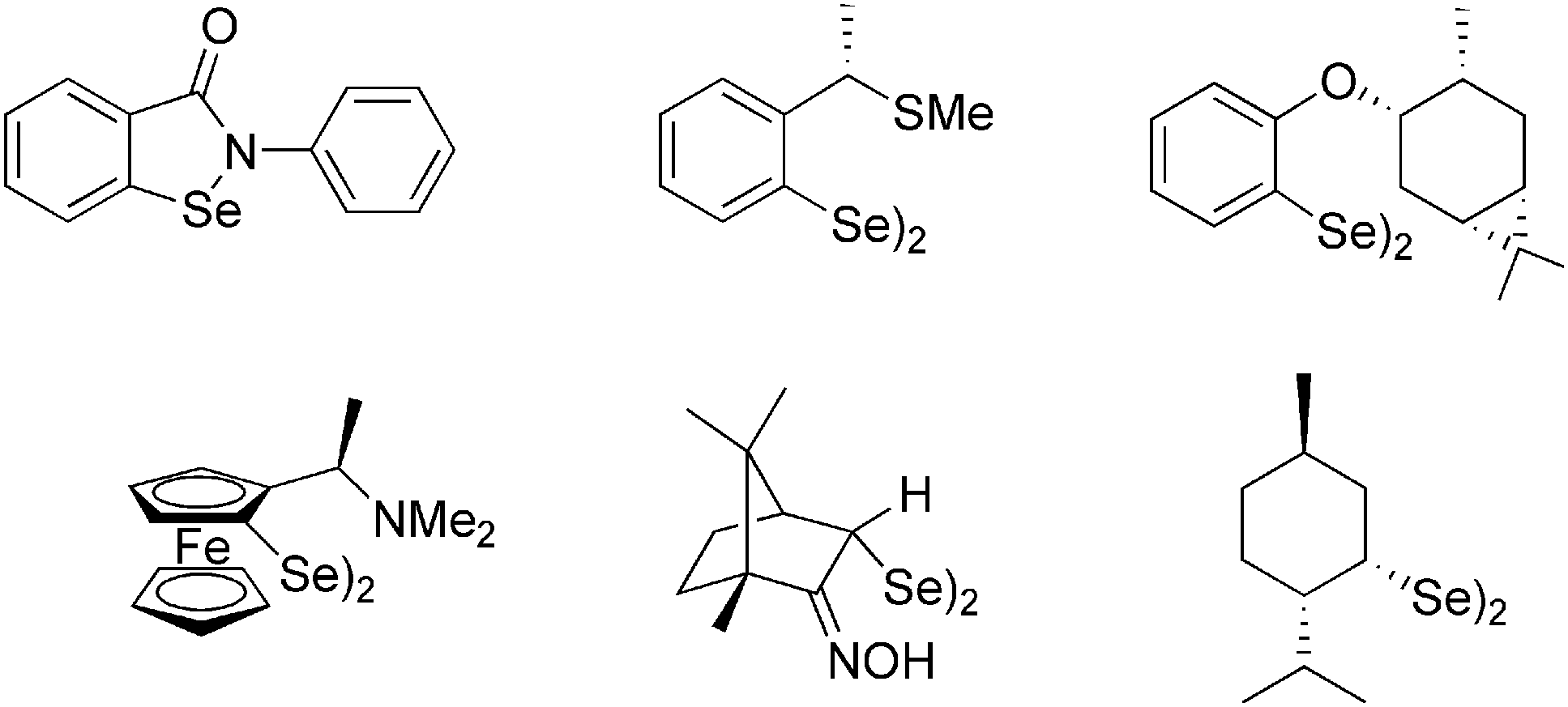 | ||
| Fig. 1 Ebselen and examples of chiral diselenides. | ||
Organoselenium compounds have also been recognized as valuable reagents widely used in organic synthesis, e.g., for introduction of various functional groups under mild conditions.13 Among various known derivatives containing Se at an oxidation state from −2 to +6, diselenides RSeSeR, analogues of peroxides and disulfides, have gained considerable attention due to their relatively high stability and possible transformations into electrophilic (RSe+), nucleophilic (RSe−), and radical reagents (RSe˙).14,15 Chiral, enantiomerically pure diselenides are particularly attractive in asymmetric reactions in which they are converted into electrophiles used for selenylations or selenocyclizations with alkenes or alkynes. Such additions in the presence of various nucleophiles lead to the formation of new carbon–nitrogen, carbon–oxygen, and carbon–carbon bonds, often accompanied by the creation of a new stereogenic center.16–19 The presence of a selenium moiety in the addition product opens the route for further modifications, including oxidative or reductive cleavage, which was utilized in the synthesis of natural products, like samin, a furofuran lignan,20 or salsolidine, a chiral tetraisoquinoline derivative.21 Dialkyl diselenides are conveniently prepared through the reaction of tosylates or alkyl halides with the Se2− ions generated in situ from selenium and a reducing agent in the presence of a base.22–26 On the other hand, diaryl derivatives are typically obtained by the oxidation of selenol or selenolate formed by the treatment of aryllithium or arylmagnesium halide with elementary Se.27,28 A route involving the solvolysis of arylselenocyanates prepared from the corresponding anilines has been recently described.29 Only a small part of a variety of chiral, non-racemic diselenides applied as auxiliaries, ligands or catalysts in asymmetric synthesis (Fig. 1) bear a selenium atom directly attached to a stereogenic center.30–34 On the other hand, most derivatives contain a heteroatom in the vicinity of Se which enhances selectivity in asymmetric transformations and is crucial for their biological activity.14,35
In our previous paper, we reported on the synthesis and configuration assignment of an enantiopure diselenide based on a bicyclic, N-heterocyclic backbone 1 (Fig. 2).36 The compound was prepared from the corresponding halides via a stereoselective transformation. Selenium atoms were attached directly to stereocenters whose configuration was established by the analysis of the 2D NMR spectra and confirmed by X-ray crystallography. A preferential formation of the unique stereoisomer was explained by the proposed mechanism involving the formation of aziridinium cations, supported by DFT calculations. Here, as a continuation of these studies, we decided to synthesise a series of diselenides with a diversified structure (bicyclic systems of various size, monocyclic systems, and diverse nature of the substituent at the amine group) and tested them as chiral electrophiles in the stereoselective methoxyselenylation of unsaturated compounds.
 | ||
| Fig. 2 Enantiopure diselenide 1.36 | ||
Results and discussion
We commenced our research with the preparation of a diselenide based on the 2-azabicyclo[3.2.1]octane skeleton with an increased steric hindrance on the tertiary amine. In the first stage, a novel cycloadduct 2 was obtained via the aza-Diels–Alder reaction between cyclopentadiene and an imine generated in situ from (1R)-(+)-1-(1-naphthyl) ethylamine and ethyl glyoxylate. Chloride 5 was prepared by transforming the exo-2 cycloadduct into the alcohol 4 and a subsequent nucleophilic substitution using thionyl chloride accompanied by ring expansion.37 Compound 5 was the appropriate starting material in the synthesis of the target diselenide 6 (25%, Scheme 1). The reaction proceeded without an inversion of the configuration as indicated by the analysis of 2D NMR spectra.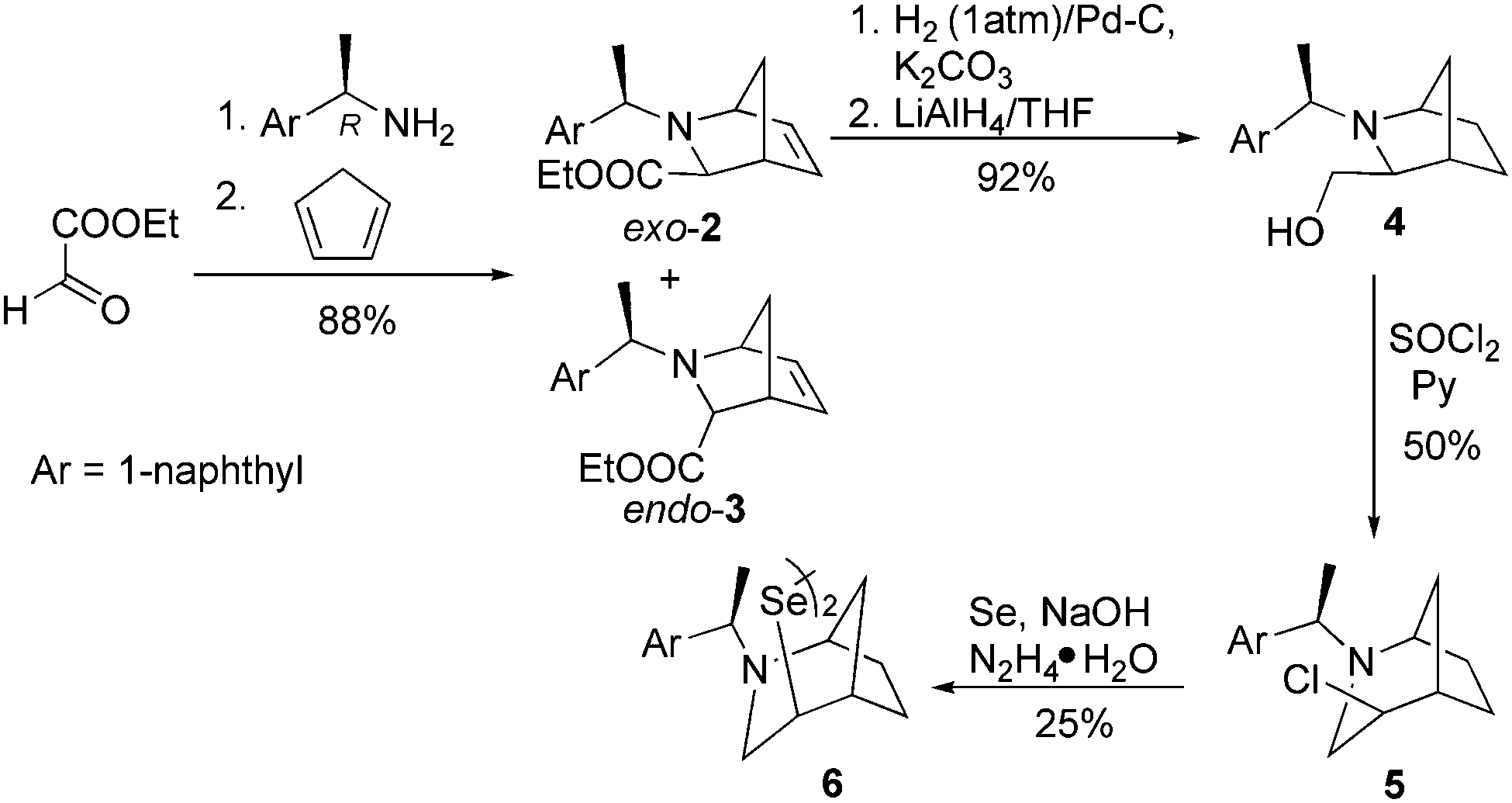 | ||
| Scheme 1 The synthesis of bridged azepane-based diselenide with naphthyl substituent 6. | ||
In order to obtain a diselenide containing a six-membered bicyclic 2-azabicyclo[2.2.1]heptane skeleton, alcohol 7 was used as a substrate.38 The presence of an electron-withdrawing substituent, a tert-butoxycarbonyl group, on the amine moiety reduces the nucleophilicity of the nitrogen atom, which allows the preparation of mesylate 8 (via a reaction with MsCl and Et3N) without enlarging the bicyclic system (Scheme 2). The resulting mesylate 8 was used in the synthesis of diselenide 9 using an optimized methodology. The reaction proceeded with a moderate yield (57%). In the final step, the protecting group was quantitatively removed using a standard procedure with trifluoroacetic acid.
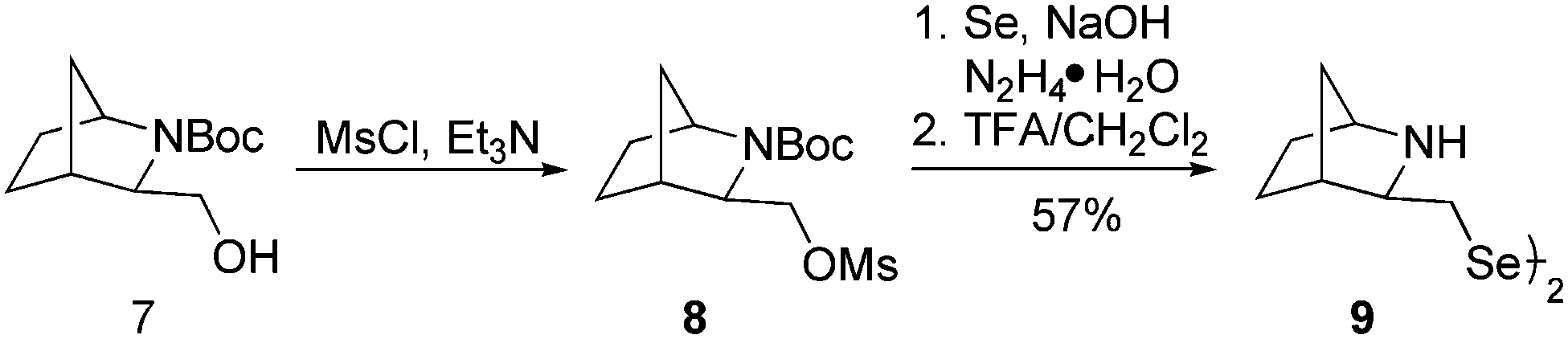 | ||
| Scheme 2 Synthetic route for 2-azanorbornane-based diselenide 9. | ||
The series of chiral diselenides was further extended by the replacement of a bicyclic system with a single heterocycle. N-Boc-L-prolinol 10 and (S)-(+)-1-benzyl-3-pyrolidinol 13 were used for the synthesis of chiral diselenides containing a pyrrolidine ring. Both alcohols 10 and 13 were converted into the corresponding mesylates, 11 and 14, respectively, using mesyl chloride and triethylamine. The obtained compounds were then transformed into the appropriate diselenides (12 and (R,R)-16) following the previously used procedure (Scheme 3). The synthesis of diselenide (R,R)-16 proceeded with inversion, giving the product with the opposite configuration to that of the starting alcohol (S)-13. The enantiomeric diselenide (S,S)-16 was obtained by converting the same alcohol (S)-13 into chloride (R)-15 under SN2 reaction conditions, which led to a change in the configuration of the intermediate. Re-inversion during conversion to diselenide resulted in the (S,S)-16 enantiomer. Measurement of the optical rotation of both diselenides (R,R)-16 and (S,S)-16 left no doubts as to the stereochemical course of the reaction according to the SN2 mechanism.
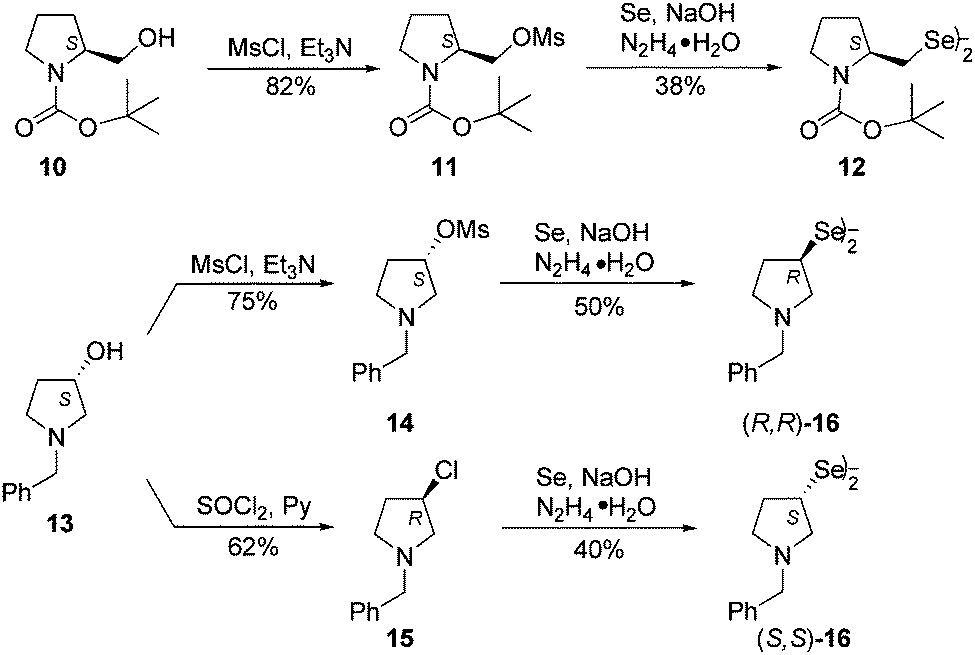 | ||
| Scheme 3 The synthesis of chiral pyrrolidine based diselenides 12 and 16. | ||
This way, a series of chiral diselenides were obtained, in which the Se2 bridge connected two mono- or bicyclic subunits containing a nitrogen atom. In some of them, selenium atoms are directly attached to stereogenic centers. It could be assumed that these factors should influence the activity of these compounds in the stereoselective alkoxyselenylation reaction of unsaturated compounds, which was examined further in this research. Structures of all compounds were confirmed by spectroscopic techniques, including 2D NMR measurements.
Optically pure diselenide derivatives were tested as chiral electrophiles in the alkoxyselenylation of alkenes (Scheme 4).23 In this asymmetric transformation, the presence of a nitrogen atom in the bi- or monocyclic system is crucial for stereoselectivity. The diselenides used differ in the ring size (bicyclic derivatives: 2-azanorbornyl and bridged azepane vs. pyrrolidine based derivatives), the nature of the substituents at the nitrogen atom, the size of the steric hindrance and the proximity of the stereogenic carbon atom to the selenium atom.
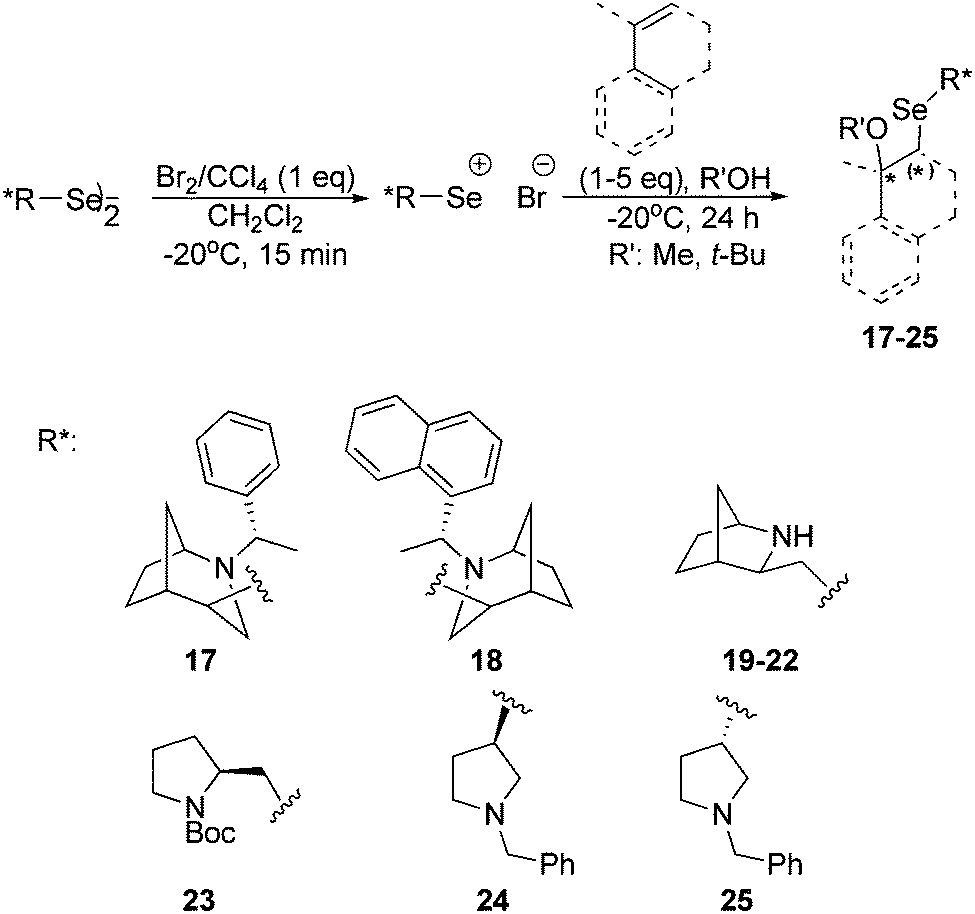 | ||
| Scheme 4 Asymmetric reaction of styrene alkoxyselenylation using different chiral electrophiles generated from diselenides 1, 6, 9, 12, (R,R)-16 and (S,S)-16. | ||
All diselenides were treated with a solution of bromine in carbon tetrachloride (Br2/CCl4) to obtain electrophilic reagents. The obtained chiral electrophiles were then reacted with an excess of styrene in the presence of methanol as the nucleophile at −20 °C (Scheme 4).23 The transformation could be easily followed as the reaction mixture gradually turned from dark brown to yellow. As a result, a variety of products were obtained in the form of diastereomeric mixtures. The diastereomeric excess was determined on the basis of the analysis of 1H NMR spectra.
The highest diastereomeric excess (de > 99%) was observed using diselenide 9 as an electrophile. Apart from the short distance between atoms of selenium and nitrogen, which creates a possibility for their interactions, this derivative is unique with the presence of a rigid azanorbornane system and a secondary amine group in the bicyclic unit. Moreover, bicyclic systems are characterized by the presence of additional stereogenic centers that may contribute to a higher asymmetric induction. Interestingly, monocyclic electrophiles led preferentially to a diastereomer with a different configuration of a new stereogenic center as compared to the bicyclic electrophiles. The presence of a stereogenic carbon atom directly connected to a selenium atom (diselenides 17, 18, 24 and 25) is not a prerequisite for efficient stereocontrol in the asymmetric reaction. In hope to improve the diastereoselectivity, we lowered the temperature from −20 °C to −78 °C, but it only decreased the chemical yield, while maintaining a similar diastereomeric excess.
Though the diastereomeric excess observed for compound 9 was promising, the conversion was not satisfactory (only 11% yield, Table 1, entry 3). Interestingly, when we repeated the reaction at room temperature instead of −20 °C, a quantitative yield was achieved, and, to our delight, the diastereoselectivity did not drop down, but even slightly increased (Table 1, entry 4).
| Entry | Alkene | Nucleophile | Diselenide | de [%] | Yield [%] |
|---|---|---|---|---|---|
| a Reaction performed at room temperature. b Diastereoisomeric excess was determined using 1H NMR measurements. | |||||
| 1 |
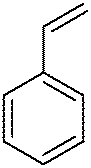
|
MeOH (5 eq.) | 1 | 26 | 72 |
| 2 | MeOH (5 eq.) | 6 | 22 | 91 | |
| 3 | MeOH (5 eq.) | 9 | >95 | 11 | |
| 4 | MeOH (5 eq.)a | 9 | >99 | 100 | |
| 5 | MeOH (1 eq.)a | 9 | >99 | 100 | |
| 6 | t-BuOH (5 eq.)a | 9 | 90 | 66 | |
| 7 | MeOH (5 eq.) | 12 | <30 | 97 | |
| 8 | MeOH (5 eq.) | (R,R)-16 | <10 | 17 | |
| 9 | MeOH (5 eq.) | (S,S)-16 | <5 | 69 | |
| 10 |
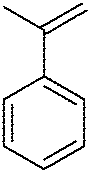
|
MeOH (5 eq.)a | 9 | 23 | 52 |
| 11 |
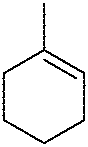
|
MeOH (5 eq.)a | 9 | nd | <10 |
In order to further investigate the utility of chiral diselenide 9 in the alkoxyselenylation reaction, we performed additional transformations using α-methylstyrene and 1-methylcyclohexene as alkenes and MeOH/t-BuOH (Table 1, entries 6, 10, and 11). The alkoxyselenylation products 19–22 were isolated by column chromatography and their diastereomeric excess was estimated from 1H NMR spectra of the crude reaction mixture. The only exception was the reaction of the electrophile obtained from 9 with 1-methylcyclohexene 22 (Table 1, entry 11); due to the low yield and a complex stereochemistry (two new stereogenic centers are formed) the diastereomeric excess was not determined. We can conclude that the steric hindrance provided by the alkene significantly worsens the reaction outcome. On the other hand, the tolerance for the large substituent of the alcohol reactant is much higher. Still, the best diastereomeric excess and yield were obtained for reaction with styrene and methanol. As the final check, we performed this reaction with diselenide 9 at room temperature, using a stoichiometric amount of alkene, and the result was identical to that with five equivalents (Table 1, entry 5). This experiment shows that an excess of alkene is not necessary to carry out the reaction in an efficient and stereoselective manner.
Experimental section
Commercial grade reagents were purchased from Merck and used without further purifications. IR spectra were recorded using a 2000 FTIR spectrophotometer. 1H NMR and 13C NMR spectra were recorded using 500 MHz and 400 MHz spectrometers using the solvent residual peak (chloroform-d, δ (1H) = 7.28 ppm, δ(13C) = 77.0 ppm) as a reference. The reported J values are those observed from the splitting patterns in the spectrum and may not reflect the true coupling constant values. High resolution mass spectra were recorded (Waters LCT Premier XE TOF spectrometer, Waters Corporation, Milford, MA, USA) utilizing the electrospray ionization mode. An automatic polarimeter model AA-5 (Optical Activity Ltd, Ramsey, UK) was used to determine the optical rotation ([α]D20 values are expressed in 10−1 deg cm2 g−1). Chromatographic separations were performed using silica gel 60 (70–230 mesh). Thin layer chromatography was carried out using silica gel 60 precoated plates. All alkoxyselenylation transformations were performed under an inert atmosphere. Diastereomeric excesses were determined by the integration of the 1H NMR signals of the methoxy groups in the crude reaction mixture.Preparation of starting compounds
The bicyclic aza-Diels–Alder cycloadducts and the respective alcohols 4 and 7 were prepared as described previously.36,38 In the case of compounds 2 and 3, imine obtained in situ via the reaction of glyoxylate and (R)-(+)-1-(1-naphthyl)ethylamine was used as an azadienophile.General procedure for the preparation of chlorides36
Alcohol and pyridine (1.0 equiv.) were dissolved in dry dichloromethane and cooled in an ice bath. Thionyl chloride (2.0 equiv.) was then slowly added. The reaction mixture was stirred overnight at room temperature. The crude reaction mixture was rendered alkaline with 20% aqueous NaOH and extracted with dichloromethane. The organic phases were collected, dried over Na2SO4 and evaporated. After evaporation of the solvent, the residue was chromatographed on a silica column using n-hexane/ethyl acetate (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v), yielding a fraction containing pure chloride.
:1 v/v), yielding a fraction containing pure chloride.
General procedure for the preparation of mesylate
Alcohol and triethylamine (1.3 equiv.) were dissolved in dry dichloromethane and cooled in an ice bath. Mesyl chloride (1.1 equiv.) was then slowly added. The reaction mixture was stirred overnight at room temperature. The crude reaction mixture was extracted with dichloromethane. The organic phases were collected, dried over Na2SO4 and evaporated. After evaporation of the solvent, the residue was chromatographed on a silica column using n-hexane/ethyl acetate (v/v), yielding a fraction containing mesylate. Its spectral characteristics were identical to those of the reported parameters.General procedure for the synthesis of diselenides36
Hydrazine monohydrate (0.11 mL; 2.2 mmol) was slowly added to a suspension of selenium (0.17 g; 2.2 mmol) and sodium hydroxide (0.12 g; 3.0 mmol) in DMF. The mixture was stirred at 100 °C under argon for 3 h. Next, chloride or mesylate (2.0 mmol) was added portionwise and the mixture was stirred for 24 h. The resulting crude reaction mixture was dissolved in petroleum ether and washed several times with water and brine. The organic phase was dried over sodium sulfate, filtered, and evaporated under a reduced pressure. The reaction product was chromatographed on a silica column using n-hexane/ethyl acetate (9:1 v/v), yielding a fraction containing a pure diselenide.
General procedure for the asymmetric methoxyselenylation of styrene23
The diselenide (100 mg) was dissolved in dry dichloromethane (3 mL) under argon. A 1 M solution of bromine (1 eq.) in tetrachloromethane was added dropwise at −20 °C (or at room temperature). After 15 min, a 0.70 M alcoholic solution of alkene (1–5 eq.) was added and stirred at the same temperature for 24 h. The reaction was quenched with aqueous NaHCO3 solution, diluted with dichloromethane, washed with water and brine, dried, filtered, and concentrated in vacuo. The crude product was purified by chromatography on silica gel. Diastereoisomeric excesses were determined using 1H NMR measurements (available in the ESI† file).Conclusions
Five new enantiopure diselenides were synthesised using stereoselective methodologies. All obtained compounds and previously described diselenide 1 were tested as chirality inducers in the stereoselective alkoxyselenylation reactions. The desired products were obtained in very good yields and high diastereoselectivity when diselenide 9 was used as an electrophile. The performed research indicated that a rigid azanorbornane bicyclic system with a secondary amine group is preferred in the electrophile structure leading to an excellent stereochemical outcome.Author contributions
Karolina Kamińska: conceptualization, investigation, methodology, and writing – original draft. Elżbieta Wojaczyńska: conceptualization, supervision, and writing – review & draft.Conflicts of interest
There are no conflicts to declare.Notes and references
- R. Brigelius-Flohé and M. Maiorano, Biochim. Biophys. Acta, 2013, 1830, 3289 CrossRef PubMed.
- E. Lubos, J. Loscalzo and D. E. Handy, Antioxid. Redox Signaling, 2011, 15, 1957 CrossRef CAS PubMed.
- C. Santi, C. Scimmi and L. Sancineto, Molecules, 2021, 26, 4230 CrossRef CAS PubMed.
- Q. Lu, Y. Cai, C. Xiang, T. Wu, Y. Zhao, J. Wang, H. Wang and L. Zou, Expert Rev. Mol. Med., 2021, 23, e12 CrossRef CAS.
- J. Mahadevan, S. Parazzoli, E. Oseid, A. V. Hertzel, D. A. Bernlohr, S. N. Vallerie, C. Liu, M. Lopez, J. S. Harmon and R. P. Robertson, Diabetes, 2013, 62, 3582 CrossRef CAS PubMed.
- F. Martini, S. G. Rosa, I. P. Klann, B. C. W. Fulco, F. B. Carvalho, F. L. Rahmeier, M. C. Fernandes and C. W. Nogueira, J. Psychiatr. Res., 2019, 109, 107 CrossRef PubMed.
- A. L. Sharpley, C. Williams, A. A. Holder, B. R. Godlewska, N. Singh, M. Shanyinde, O. MacDonald and P. J. Cowen, Psychopharmacology, 2020, 237, 3773 CrossRef CAS PubMed.
- T. Yamaguchi, K. Sano, K. Takakura, I. Saito, Y. Shinohara, T. Asano and H. Yasuhara, Stroke, 1998, 29, 12 CrossRef CAS PubMed.
- E. D. Lynch and J. Kill, Drug Discovery Today, 2005, 10, 1291 CrossRef CAS PubMed.
- Q. Feng, X. Li, W. Sun, Y. Li, Y. Yuan, B. Guan and S. Zhang, Cancer Manage. Res., 2020, 12, 6921 CrossRef CAS PubMed.
- E. Weglarz-Tomczak, J. M. Tomczak, M. Talma, M. Burda-Grabowska, M. Giurg and S. Brul, Sci. Rep., 2021, 11, 3640 CrossRef CAS PubMed.
- K. Amporndanai, X. Meng, W. Shang, Z. Jin, M. Rogers, Y. Zhao, Z. Rao, Z.-J. Liu, H. Yang, L. Zhang, P. M. O’Neill and S. S. Hasnain, Nat. Commun., 2021, 12, 3061 CrossRef CAS PubMed.
- M. Tiecco, Electrophilic selenium, Selenocyclizations. in Topics in Current Chemistry: Organoselenium Chemistry: Modern Developments in Organic Synthesis, ed. T. Wirth, Springer-Verlag: Heidelberg, 2000, pp. 7–54 Search PubMed.
- A. Ivanova and P. Arsenyan, Coord. Chem. Rev., 2018, 370, 55 CrossRef CAS.
- A. D. Sonawane, R. A. Sonawane, M. Ninomya and M. Koketsu, Dalton Trans., 2021, 50, 12764 RSC.
- C. Santi and S. Santoro, Electrophilic Selenium Reagents. in Organoselenium Chemistry: Synthesis and Reactions, ed. T. Wirth, Wiley-VCH, 2012, pp. 1–51 Search PubMed.
- J. Ścianowski and Z. Rafiński, Electrophilic Selenium Reagents: Addition Reactions to Double Bonds and Selenocyclizations. in Organoselenium Chemistry: Between Synthesis and Biochemistry, ed. C. Santi, Bentham, 2014, pp. 8–60 Search PubMed.
- L. Sancineto, M. Palomba, L. Bagnoli, F. Marini and C. Santi, Curr. Org. Chem., 2015, 20, 122 CrossRef.
- C. Santi, C. Tomassini and L. Sancineto, Chimia, 2017, 71, 592 CrossRef CAS PubMed.
- T. Wirth, K. J. Kulicke and G. Fragale, J. Org. Chem., 1996, 61, 2686 CrossRef CAS PubMed.
- T. Wirth and G. Fragale, Synthesis, 1998, 162 CrossRef CAS.
- J. Ścianowski, Tetrahedron Lett., 2005, 46, 3331 CrossRef.
- J. Ścianowski, Z. Rafiński and A. Wojtczak, Eur. J. Org. Chem., 2006, 3216 CrossRef.
- Z. Rafiński and J. Ścianowski, Tetrahedron: Asymmetry, 2008, 19, 1237 CrossRef.
- Z. Rafiński, J. Ścianowski and A. Wojtczak, Lett. Org. Chem., 2009, 6, 321 CrossRef.
- N. R. Panguluri, V. Panduranga, G. Prabhu, T. M. Vishwanatha and V. V. Sureshbabu, RSC Adv., 2015, 5, 51807 RSC.
- I. Kuwajima, M. Shimizu and H. Urabe, J. Org. Chem., 1982, 47, 837 CrossRef CAS.
- H. J. Reich, M. L. Cohen and P. S. Clark, Org. Synth., 1979, 59, 141 CrossRef.
- F. Krätzschmar, S. Ortgies, R. Y. N. Willing and A. Breder, Catalysts, 2019, 9, 153 CrossRef.
- T. G. Back, B. P. Dyck and M. J. Parvez, J. Org. Chem., 1995, 60, 703 CrossRef CAS.
- M. J. Potrzebowski, M. Michalska, J. Błaszczyk, M. W. Wieczorek, W. Ciesielski, S. Kaźmierski and J. Pluskowski, J. Org. Chem., 1995, 60, 3139 CrossRef CAS.
- S. Czernecki and D. Randriamandimby, J. Carbohydr. Chem., 1996, 15, 183 CrossRef CAS.
- M. Tiecco, L. Testaferri, C. Santi, C. Tomassini, S. Santoro, F. Marini, L. Bagnoli, A. Temperini and F. Costantino, Eur. J. Org. Chem., 2006, 4867 CrossRef CAS.
- J. Ścianowski, J. Szumera, P. Kleman and A. Pacuła, Tetrahedron: Asymmetry, 2016, 27, 238 CrossRef.
- M. Bortoli, A. Madabeni, P. A. Nogara, F. B. Omage, G. Ribaudo, D. Zeppliil, J. B. T. Rocha and L. Orian, Catalysts, 2020, 11, 114 CrossRef.
- K. Kamińska, E. Wojaczyńska, C. Santi, L. Sancineto, M. F. Pensa, A. Kochel, R. Wieczorek, J. Wojaczyński and G. Slupski, Tetrahedron: Asymmetry, 2017, 10, 1367 CrossRef.
- E. Wojaczyńska, I. Turowska-Tyrk and J. Skarżewski, Tetrahedron, 2012, 38, 7848–7854 CrossRef.
- D. Iwan, K. Kamińska, E. Wojaczyńska, M. Psurski, J. Wietrzyk and M. Daszkiewicz, Materials, 2020, 13, 5010 CrossRef CAS PubMed.
- H. Jin, S. E. Lazer-With, T. A. T. Martin, E. M. Bacon, J. J. Cotteli, Z. R. Cai, H.-J. Pyun, P. A. Morganelli and M. Ji, WO2014/100323 A1, 2014.
- R. Yang, L. Qi, Y. Liu, Y. Ding, M. S. Y. Kwek and C.-F. Liu, Tetrahedron Lett., 2013, 54, 3777–3780 CrossRef CAS.
- C. Thomas, F. Orecher and P. Gmeiner, Synthesis, 1998, 1491 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of NMR spectra and spectral characterization of alkoxyselenylation products. See DOI: https://doi.org/10.1039/d2nj01434c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2022 |