
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An orthogonal approach for the precise synthesis of phenylpropanoid sucrose esters†
Li Lin
Ong
ab,
Pooi Wen Kathy
Wong
ab,
Surhabi
Deva Raj
a,
Duc Thinh
Khong
a,
Parthasarathi
Panda
a,
Mardi
Santoso
 c and
Zaher M. A.
Judeh
*a
c and
Zaher M. A.
Judeh
*a
aSchool of Chemical and Biomedical Engineering, Nanyang Technological University, Singapore, 62 Nanyang Drive, N1.2-B1-14, Singapore 637459. E-mail: zaher@ntu.edu.sg; Fax: +65-6794-7553; Tel: +65-6790-6738
bInstitute of Health Technologies, Interdisciplinary Graduate School, Nanyang Technological University, Singapore, 61 Nanyang Drive, ABN-02b-07, Singapore 637335
cDepartment of Chemistry, Faculty of Science, Institut Teknologi Sepuluh Nopember, Sukolilo, Surabaya 60111, Indonesia
First published on 25th April 2022
Abstract
Phenylpropanoid sucrose esters (PSEs) are plant-derived metabolites that exist widely in medicinal plants and possess important bioactivities. Their precise synthesis is challenging due to the distinct and diverse substitution patterns at the sugar framework, and it is scarcely reported. Orthogonal protection/deprotection strategies for disaccharides are more complex and less developed than those for monosaccharides. We disclose a precise synthesis of PSEs starting from 2,1′:4,6-di-O-diisopropylidene sucrose 7via an orthogonal protection/deprotection and selective cinnamoylation strategy. We demonstrate the strategy for the synthesis of several PSEs cinnamoylated at the O-3 and O-4′ positions of diisopropylidene sucrose 7. The strategy is enabled by a carefully selected and synergistic set of protecting groups and deprotecting agents under the optimized conditions. It potentially gives access to the ∼150 reported PSEs and opens the door for the custom synthesis of unnatural PSEs for industrial applications. The reported work also presents a viable strategy for the general orthogonal protection/deprotection of disaccharides for the precise synthesis of other classes of phenylpropanoid esters and related compounds.
Introduction
Phenylpropanoid sucrose esters (PSEs) are secondary metabolites widely distributed in various medicinal plants.1 More than 150 PSEs have been isolated and characterized during the past decades.1 They possess important biological activities1 including antiproliferation,2–7 antioxidation,8–15 anti-inflammation,16–18 and α-glucosidase inhibition activities.12,19,20 The core sucrose unit of PSEs is selectively decorated with one or more (substituted) cinnamoyl moieties (e.g. cinnamoyl, coumaroyl, feruloyl, sinapoyl, etc.) via ester linkages, especially at O-3, O-3′, O-4′, and O-6′ (Fig. 1).1 The variation in the type, number, and position of the cinnamoyl moieties causes structural (simple and mixed) and biological diversities among the PSEs. Examples of simple and mixed PSEs include sibiricose A51,26–29 sibiricose A62,28–30 reiniose A 3,29,30 heterosmilaside 49 and glomeratose B 531 (Fig. 1). The challenging synthesis of PSEs and their low natural abundance in the plant species hinder their exploitation as new lead drug candidates as well as food and cosmetic additives for industrial applications.1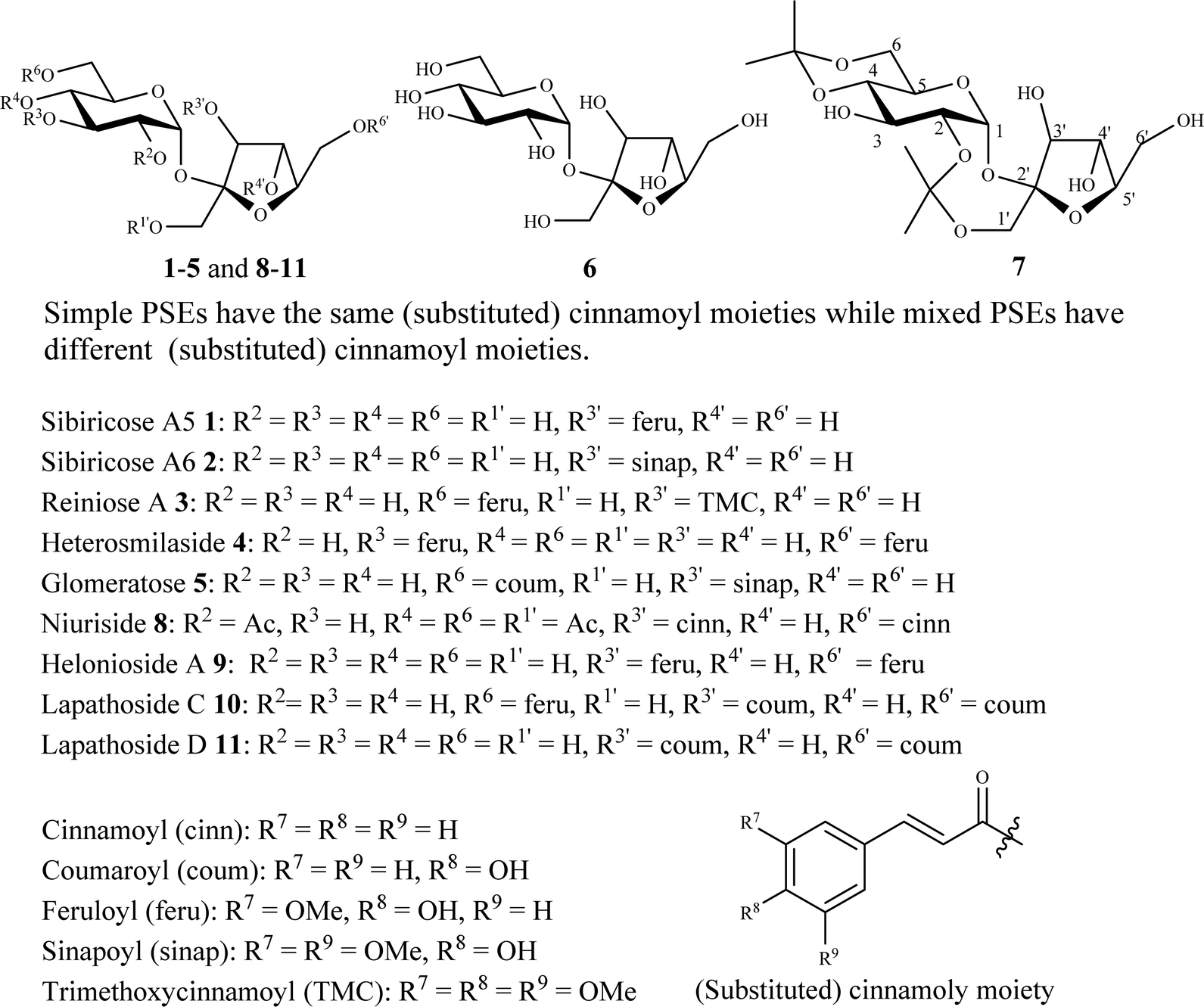 | ||
| Fig. 1 Structures of PSEs 1–5 and 8–11, sucrose 6, and 2,1′:4,6-di-O-diisopropylidene sucrose 7. | ||
Although their structures look simple (Fig. 1), only four reports dealing with their synthesis have appeared including two from our laboratory.3,6,21,22 Direct cinnamoylation of unprotected sucrose 6 is inconceivable due to the poor chemoselection and regioselection between its eight free hydroxyl groups.23–25 Therefore, we3,6 and others22 have used the partially protected 2,1′:4,6-di-O-diisopropylidene sucrose 7 as the starting material for direct acylation with cinnamoyl chlorides to synthesize niruriside 8, helonioside A 9, lapathoside C 10, lapathoside D 11, and several other analogues (Fig. 1). However, this approach suffers from several limitations including the following:3,6,21,22 (i) direct acylation of diisopropylidene sucrose 7 gave mixtures of differently acylated products which compromised the yield and complicated the purification significantly; (ii) the products and yields of the direct acylation were unpredictable and significantly depended on the type of the acylating reagent (e.g. cinnamoyl chloride, coumaroyl chloride, feruloyl chloride, etc.) and the reaction conditions (moles of the acylating reagent, solvent, temperature, time and concentration); (iii) the preparation of mixed PSEs (Fig. 1) decorated with different types of cinnamoyl moieties using this approach was significantly challenging and practically tedious. Therefore, it is advantageous to develop a systematic and precise route for the efficient synthesis of natural and unnatural PSEs to overcome the above challenges. This is specifically important to study the structure–activity relationships (SARs) of these compounds to develop lead drug candidates.32
Our earlier investigation of PSEs as potential antidiabetic lead candidates proved that the type, number, and position of the (substituted) cinnamoyl moieties greatly impact the α-glucosidase and α-amylase inhibition activities.32 In particular, the moieties at O-3, O-3′, O-4′, and O-6′ play a critical role in the inhibition. To further broaden the SAR studies to identify efficient antidiabetic lead PSE candidates, we specifically needed to synthesize PSEs precisely decorated with diverse (substituted) cinnamoyl moieties at the O-3, O-3′, O-4′ and/or O-6′ positions of diisopropylidene sucrose 7. Direct acylation methods failed to provide such compounds efficiently.2,6
Orthogonal protection/deprotection of monosaccharides and disaccharides is not unreported.33–39 A few approaches exist for monosaccharides34–36 and far less focus has been placed on disaccharides due to a higher complexity.37–39 Therefore, further development is needed in the case of disaccharides to address their complex chemo- and regio-selection during the reactions. Existing approaches are not ideal for our purpose because of several incompatibilities of the protecting groups and deprotecting conditions with the vulnerable structure of the PSEs as they possess a sucrose core esterified with cinnamoyl moieties (see below).
Herein, we develop an orthogonal strategy for the precise synthesis of PSEs starting from sucrose 6. The strategy is demonstrated by the synthesis of several complex PSEs selectively cinnamoylated at either C3-OH or C4′-OH. The strategy is applicable to the orthogonal protection of disaccharides in general for the preparation of complex disaccharide-based natural products.
Results and discussion
We envisioned that orthogonal protection of diisopropylidene sucrose 7 would give 12 which upon orthogonal deprotection would afford 13 (Fig. 2). Selective cinnamoylation of 13, followed by removal of its protecting groups, would then give a wide range of simple and mixed PSEs 14. This strategy should precisely give any natural or unnatural PSE with substituents at O-3, O-3′, O-4′, and O-6′.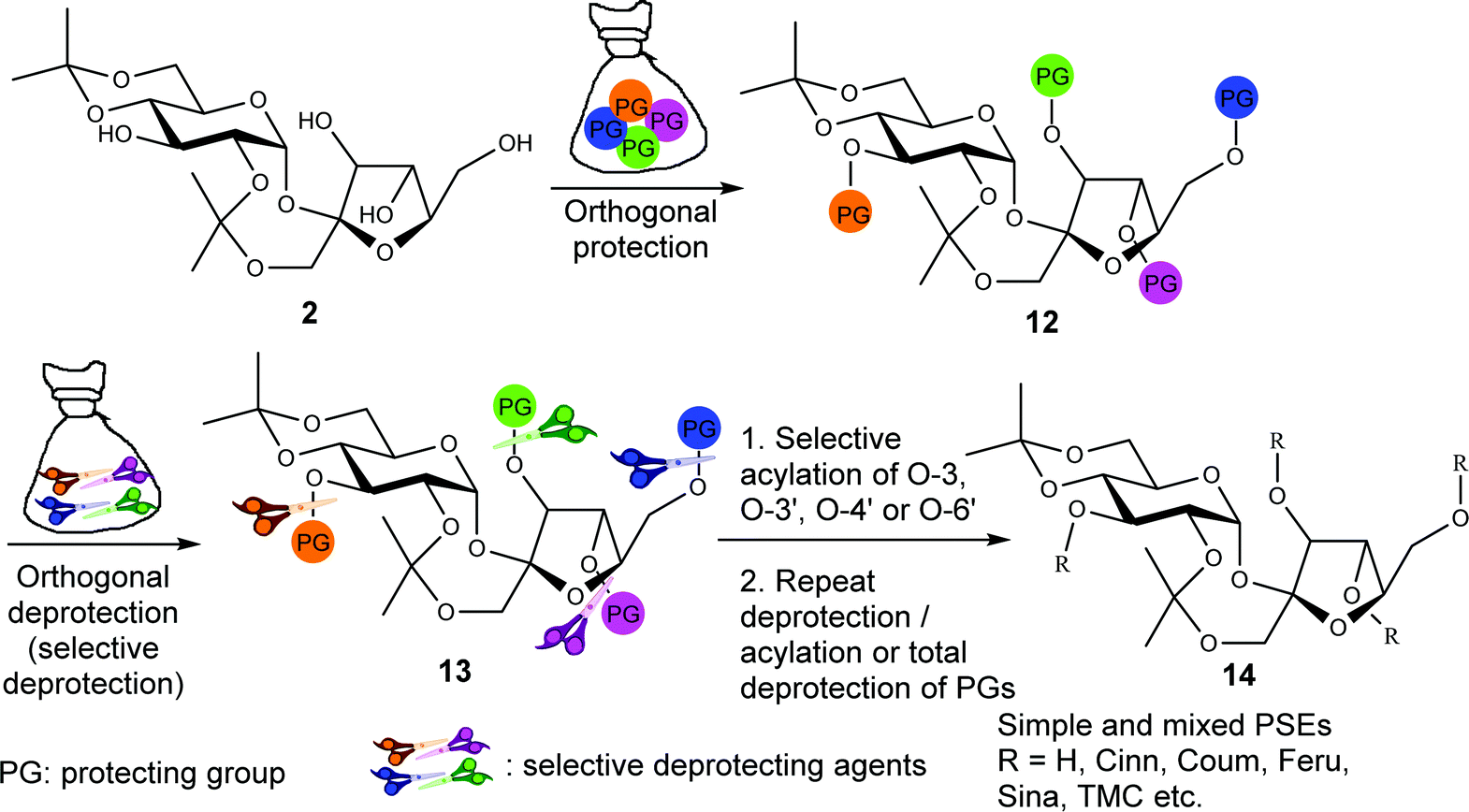 | ||
| Fig. 2 Orthogonal protection/deprotection/cinnamoylation strategy for the synthesis of simple and mixed PSEs from diisopropylidene sucrose 7. | ||
To synthesize simple and mixed PSEs, the above strategy must (i) achieve orthogonal chemoselective and regioselective protection of C4-OH, C6-OH, C3′-OH, C4′-OH and C6′-OH (Fig. 1, 7 → 12), (ii) achieve orthogonal chemoselective and regioselective deprotection of any desired protecting group in any order without affecting the integrity of the other groups (Fig. 1, 12 → 13), (iii) preserve the integrity of the isopropylidene rings during protection, deprotection, and purification (Fig. 1, 7 → 14), (iv) preserve the integrity of the cinnamoyl moieties and avoid transesterification (Fig. 1, 13 → 14), and (v) preserve the integrity of the core sucrose unit. The cinnamoyl and diisopropylidene moieties of 13 are incompatible with many common protecting/deprotecting conditions. The susceptibility of the cinnamoyl moieties to hydrogenation, acid/base hydrolysis, and transesterification prohibit employing common protecting groups such as benzoyl, pivaloyl, acetyl, or benzyl groups.33 Additionally, the diisopropylidene moieties (Fig. 2) are cleaved under acidic conditions and such conditions must be avoided/controlled. Therefore, the choice of the protecting groups and their sequence of introduction and removal as well as the reagents and reaction conditions are critical in this strategy.
Our previous studies showed the reactivity order of the four hydroxyl groups towards acylation as C6′-OH > C3′-OH > C4′-OH > C3-OH.3,6 C6′-OH is a primary OH and is most reactive. Secondary C3-OH is the least reactive due to the steric hindrance caused by the diisopropylidene rings of 7. We envisioned that introducing a bulky protecting group at O-6′ should cause a steric hindrance to C4′-OH and reduce its reactivity in comparison to C3′-OH. Therefore, the ideal sequence of introducing the protecting groups should follow the above reactivity order employing a bulky protecting group at C6′-OH.40–46 After numerous preliminary experiments using several protecting groups and conditions, we focused on tert-butyldimethylsilyl (TBS), carboxybenzyl (Cbz), p-nitrobenzoyl (PNB), and levulinoyl (Lev) as the ideal protecting groups.
At the start, selective silylation of the C6′-OH of diisopropylidene sucrose 7 using TBSCl efficiently gave 6′-O-TBS 15 in 95% yield (Scheme 1). The TBS was selected to provide enough bulkiness to favor the reactivity of C3′-OH over C4′-OH in subsequent steps. Next, optimized acylation of the C3′-OH of 6′-O-TBS 15 using CbzCl in the presence of tetramethylethylenediamine (TMEDA) efficiently gave 3′-O-Cbz 16 in 75% yield along with 3′,4′-O,O-diCbz 17 in 20% yield. The reaction was sluggish when using Et3N, N,N-diisopropylethylamine (DIEA), and 1,4-diazabicyclo[2.2.2]octane (DABCO) bases. In comparison, acylation of the same C3′-OH with PNBCl or LevOH was less promising as it gave a mixture of products presumed to be 3-O-, 3′-O- and 3,3′-O,O-PNB/Lev substituted 6′-O-TBS 15 along with the starting material (TLC). The acylation of the C4′-OH of 3′-O-Cbz 16 using PNBCl in the presence of DMAP/Et3N at room temperature gave three products: 4′-O-PNB 18 in 51% yield, 3-O-PNB 19 in 10% yield and 3,4′-O,O-diPNB 20 in 36% yield. However, at an optimum temperature of 4 °C, the same reaction gave a 70% yield of the desired 4′-O-PNB 18 along with a 10% yield of 3-O-PNB 19 and a 19% yield of 3,4′-O-PNB 20 (Scheme 1). Finally, Steglich esterification of the C3-OH of 4′-O-PNB 18 using LevOH in the presence of DCC/DMAP in CH2Cl2 gave the desired key compound 21 in 85% yield (Scheme 1). Based on these results, we concluded that the success of the orthogonal protection of diisopropylidene 7 depended on the choice of the protecting group and the reaction conditions employed. It is noted that any of these compounds 15–21 are intermediate structures that can be used to synthesize PSEs depending on the substitution pattern of the PSEs.
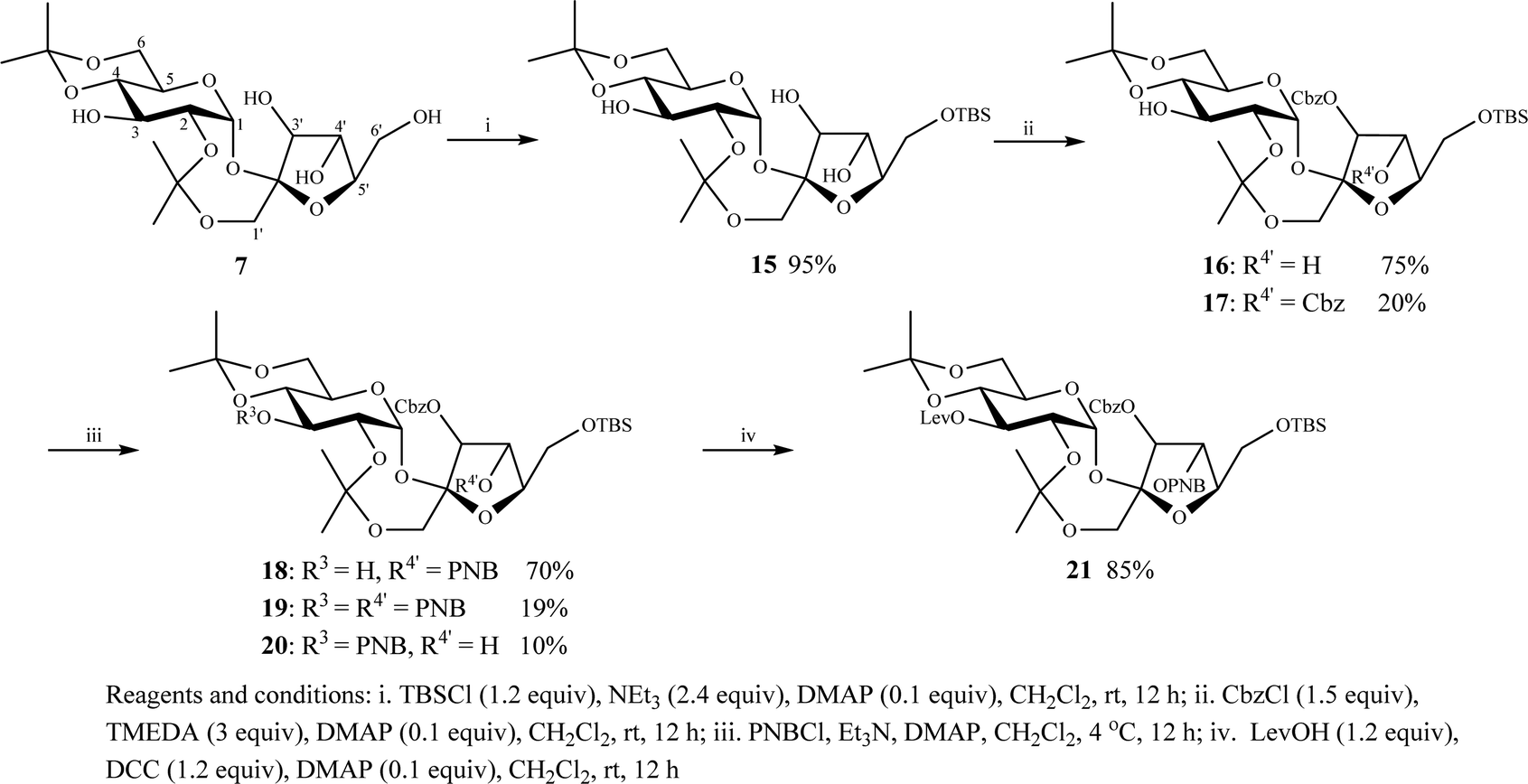 | ||
| Scheme 1 Synthesis of key compound 21via orthogonal protection of diisopropylidene sucrose 7. | ||
The orthogonal deprotection of the TBS, CBZ, PNB, and Lev groups of compound 21 was then attempted (Scheme 2). An important requirement is to achieve selective deprotection of these groups in any order to realize the planned cinnamoylation step at any desired position (Fig. 2). Extensive studies were performed before an ideal set of deprotection conditions were found. Initially, TBS removal using tetra-n-butylammonium fluoride (TBAF) in THF-buffer (K2HPO4, pH 7) gave product 22 in 60% yield along with side products, attributed to transesterification and/or removal of other protecting groups. The buffered medium was used to minimize/prevent possible migration of the secondary O-3′-Cbz or O-4′-PNB groups to the primary O-6′ position.40 Fortunately, Et3N·3HF cleanly removed TBS at room temperature to give product 22 in 86% yield (Scheme 2). Removal of the Cbz by classical hydrogenolysis is not possible in this protocol to avoid the potential reduction of the double bonds of the cinnamoyl moieties which will be introduced later (Fig. 2). Attempts to remove the Cbz of 21via thermal treatment at 100 °C in water or 1,4-dioxane were unsuccessful since 21 was insoluble in water and it decomposed in 1,4-dioxane. However, Pd(OAc)2/Et3SiH removed Cbz successfully to give 23 in 68% yield. Next, under optimized conditions, Mg(OMe)2 in MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :THF (9:1) removed PNB from 21 at 4 °C within one hour and gave 24 in 70% yield. This reaction also gave 25 as a side product in 15% yield. However, under these conditions, compound 26 was not detected. The reaction was monitored closely using TLC and quenched with 1N HCl once the starting material was consumed completely to avoid further removal of Cbz. This kinetically controlled reaction using the weakly basic Mg(OMe)2 selectively removed the PNB in the presence of Cbz and Lev groups.39 The use of Mg(OMe)2 is advantageous since concurrent removal of both PNB and Cbz can also be achieved in one step by just extending the reaction time or increasing the moles of Mg(OMe)2. Finally, treatment of 21 with NaBH4 in 1,4-dioxane cleanly removed the Lev group to give 18 in 72% yield as a single product representing another approach to obtain compound 18 in a more selective fashion (see Scheme 1). The orthogonal deprotection protocol in Scheme 2 allows selective removal of any protecting group for subsequent introduction of the cinnamoyl moieties at any position to potentially obtain any desired simple and mixed PSEs.
:THF (9:1) removed PNB from 21 at 4 °C within one hour and gave 24 in 70% yield. This reaction also gave 25 as a side product in 15% yield. However, under these conditions, compound 26 was not detected. The reaction was monitored closely using TLC and quenched with 1N HCl once the starting material was consumed completely to avoid further removal of Cbz. This kinetically controlled reaction using the weakly basic Mg(OMe)2 selectively removed the PNB in the presence of Cbz and Lev groups.39 The use of Mg(OMe)2 is advantageous since concurrent removal of both PNB and Cbz can also be achieved in one step by just extending the reaction time or increasing the moles of Mg(OMe)2. Finally, treatment of 21 with NaBH4 in 1,4-dioxane cleanly removed the Lev group to give 18 in 72% yield as a single product representing another approach to obtain compound 18 in a more selective fashion (see Scheme 1). The orthogonal deprotection protocol in Scheme 2 allows selective removal of any protecting group for subsequent introduction of the cinnamoyl moieties at any position to potentially obtain any desired simple and mixed PSEs.
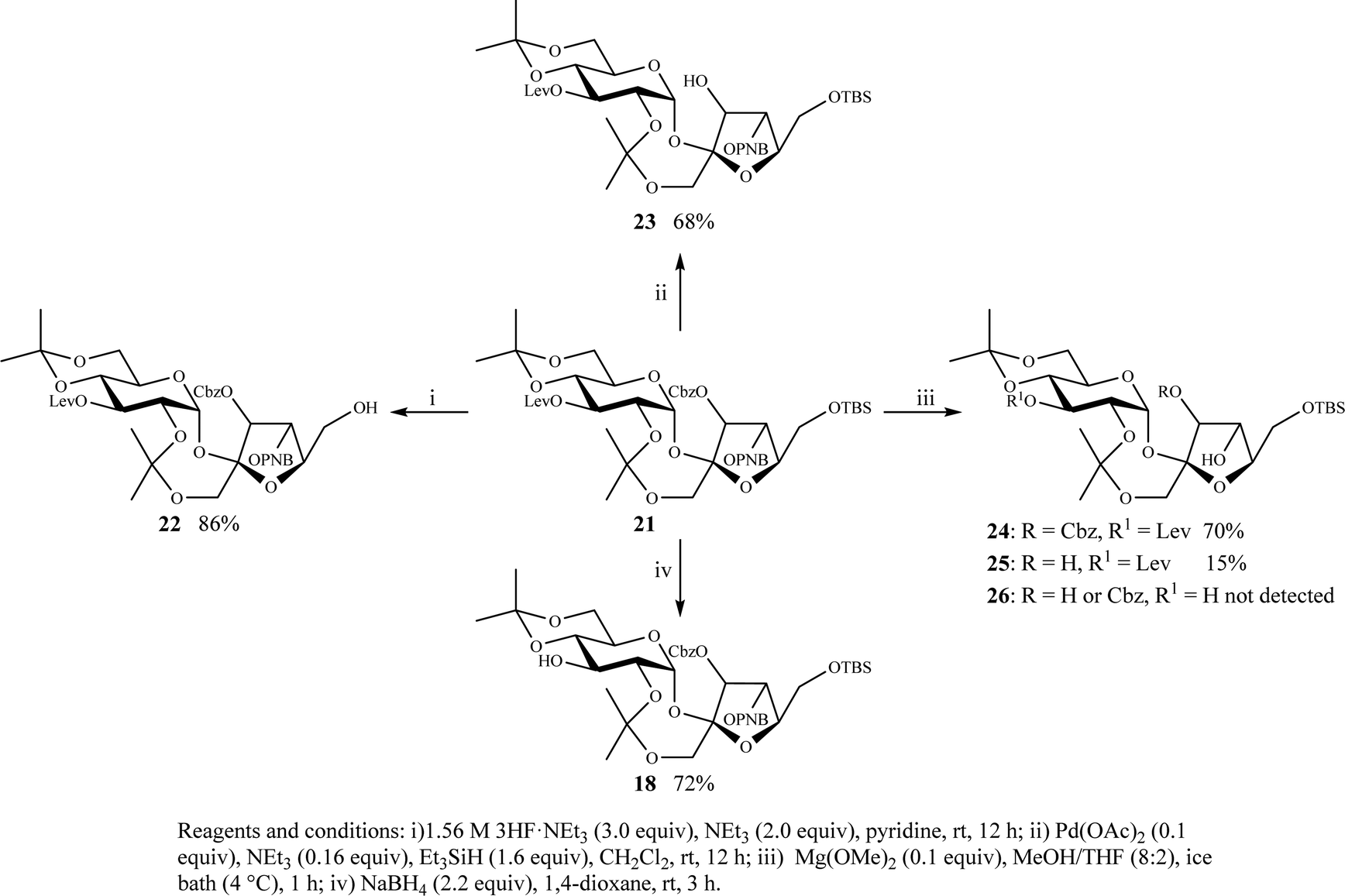 | ||
| Scheme 2 Selective removal of the TBS, Cbz, PNB, and Lev protecting moieties from 21. | ||
To demonstrate the synthetic practicality of the above strategy, compound 18 was used to synthesize PSEs 27–33 cinnamoylated at the most challenging and least reactive C3-OH (Scheme 3). Many natural PSEs are substituted at this position and their synthesis poses major challenges.1 Compound 18 can be obtained from compound 16 (Scheme 1) or compound 21 (Scheme 2) as discussed above. Steglich esterification between 18 and (substituted) cinnamic acids 34–40 gave the corresponding cinnamoylated products 41–47 in 76–89% yields. Concurrent removal of the Cbz and PNB groups using Mg(OMe)2 over 12 h followed by TBS removal using HF·NEt3 in one-pot over a two-step process gave the desired 3-cinnamoylated PSEs 27–33 in 62–81% yields (Scheme 3).
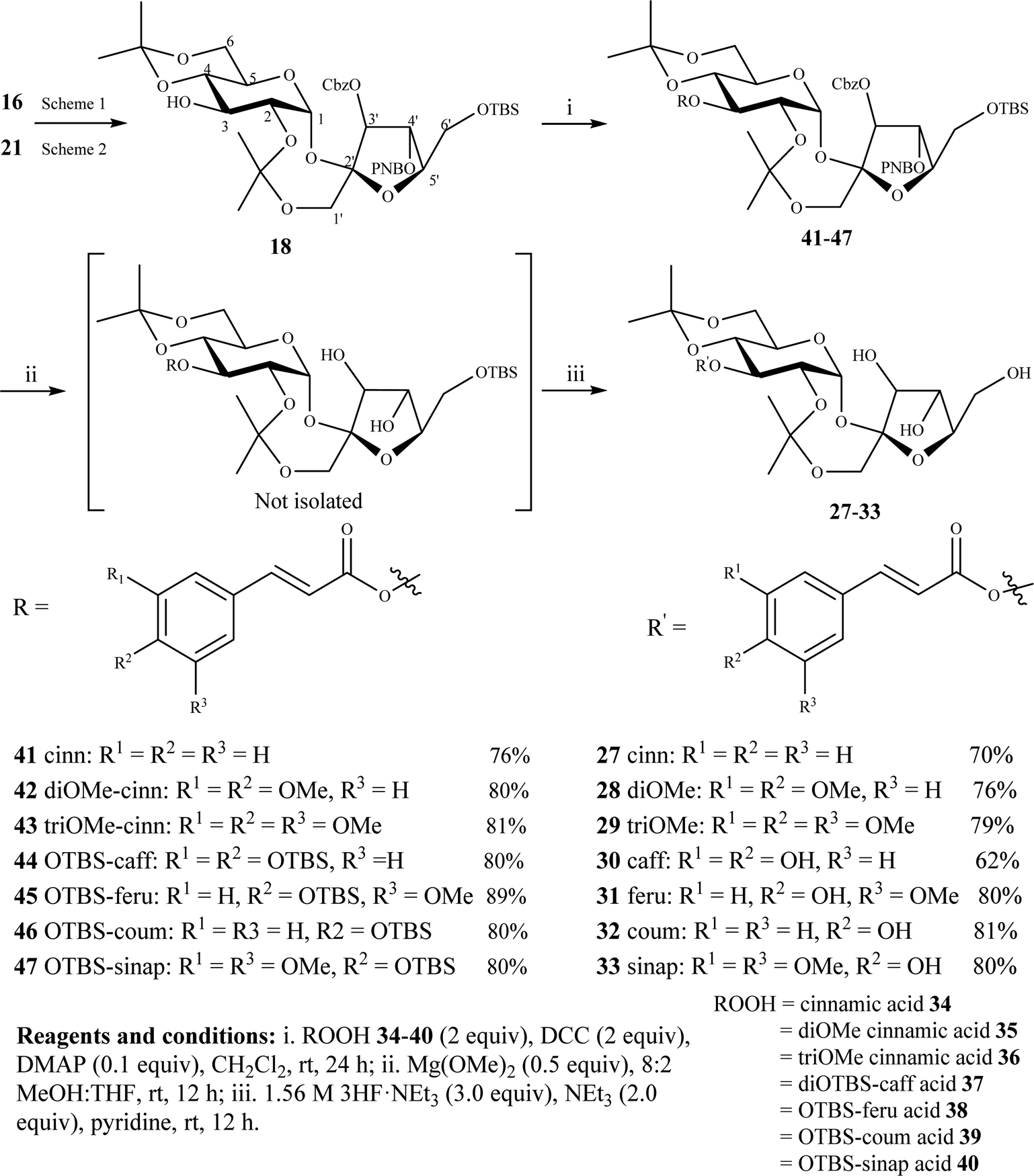 | ||
| Scheme 3 Synthesis of PSEs 27–33 substituted with cinnamoyl moieties at C3-OH. | ||
To further demonstrate the robustness of the strategy, cinnamoylation of the C4′-OH of 3′-O-Cbz 16 was also attempted to synthesize PSEs 48–54 (Scheme 4). The esterification of 16 with cinnamic acids 34–40 proceeded selectively at C4′-OH due to its higher nucleophilicity in comparison with C3-OH and gave the corresponding compounds 55–61 in 81–87% yields. Step-wise removal of the Cbz using Pd(OAc)2/Et3SiH and then TBS using 3HF·NEt3 gave PSEs 48–54 in 82–88% yields.
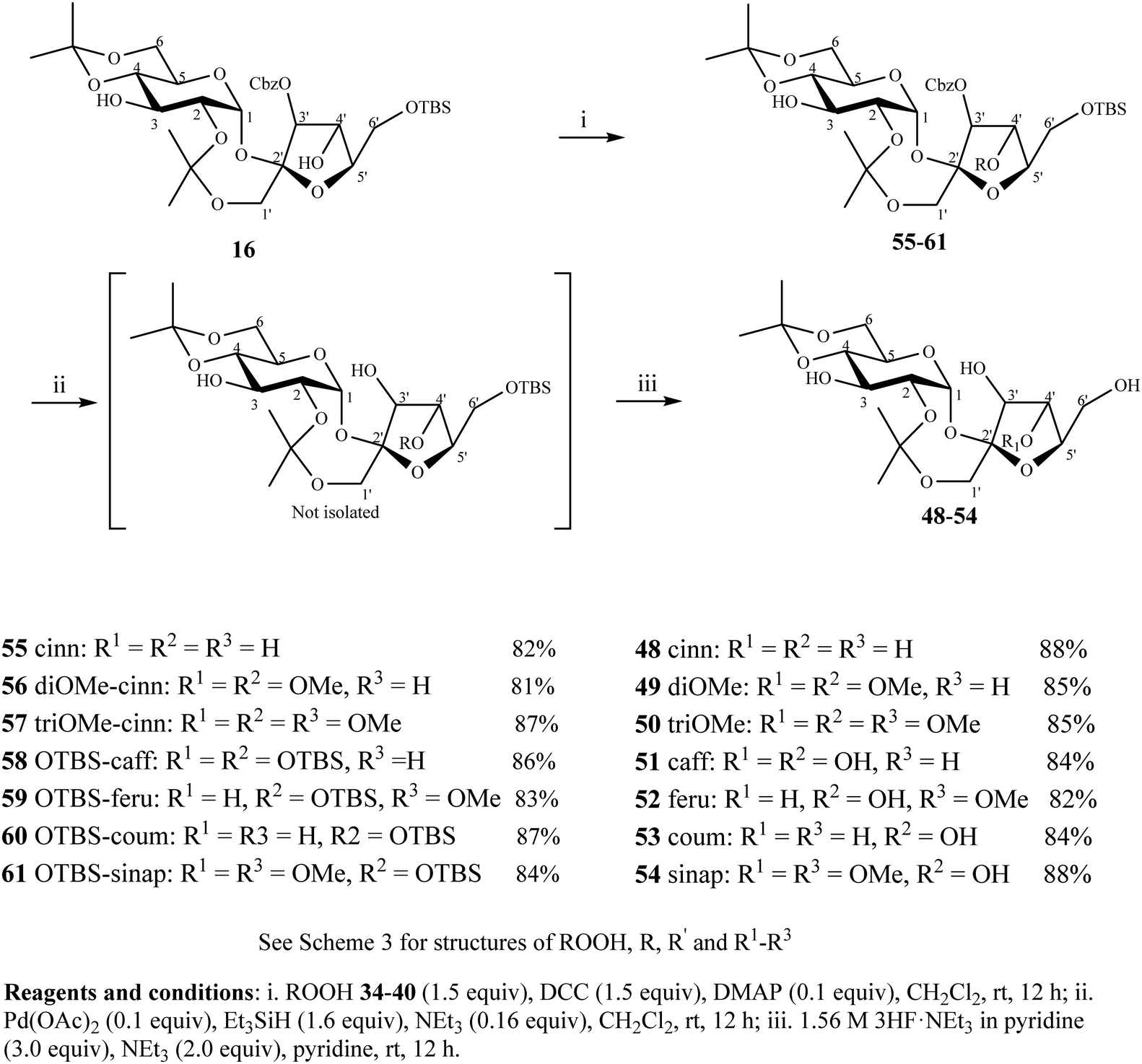 | ||
| Scheme 4 Synthesis of PSEs 48–50 substituted with cinnamoyl moieties at C4′-OH. | ||
In this work, the diisopropylidene rings of compounds 27–33 and 48–54 were not cleaved since our preliminary structure–activity relationship (SAR) studies on several PSEs as alpha glucosidase inhibitors revealed their positive role in increasing the % inhibition of α-glucosidase and decreasing the % inhibition of α-amylase enzymes.32 However, we previously established a convenient process to selectively remove the diisopropylidene rings easily without affecting the (substituted) cinnamoyl moieties using 60% aq. AcOH at 80 °C within 20–30 minutes.3,6 In future work, compounds 27–33 and 48–50 will be tested as alpha glucosidase inhibitors for the treatment of diabetes as part of an extensive study in our lab.32
Conclusion
We developed an efficient strategy for the precise synthesis of PSEs starting from 2,1′:4,6-di-O-diisopropylidene 7. With this strategy, synthesis of natural and unnatural PSEs is now achievable since cinnamoylation is attainable at any desired OH group of diisopropylidene sucrose 7. In broader terms, this strategy solves a complex problem of orthogonal protection/deprotection of disaccharides which is far more complex than monosaccharides and opens the door for the synthesis of a wide range of not only PSEs but also phenylpropanoid esters and related compounds in general. We are currently using this strategy to prepare simple and mixed PSEs as alpha glucosidase inhibitor antidiabetic lead compounds.Experimental section
Materials and methods
All commercial reagents and solvents used in this work were obtained from Sigma-Aldrich, Acros, and Merck and were used as received unless stated. Routine 1H NMR spectra were recorded using a Bruker Avance DPX 300 spectrometer (300 MHz). 1H NMR multiplicities were designated as singlet (s), doublet (d), doublet of doublet (dd), triplet (t), quartet (q), and multiplet (m). 13C NMR spectra were measured using a Bruker Avance DPX 300 spectrometer (75.47 MHz). HRMS spectra were recorded on a Finnigan MAT95XL-T spectrometer in the ESI positive mode. Flash chromatography and column chromatography were carried out using Merck silica gel 60 230–400 mesh. Analytical TLC was performed using Merck 60 F254 precoated silica gel plates (0.2 mm thickness). The products on the TLC plates were visualized under UV light (254 nm) or by using a solution of 5% H2SO4 in EtOH (v/v).General procedure for the acylation of compound 18: synthesis of compounds 41–47
To a stirred solution of compound 18 (500 mg, 0.610 mmol) and DMAP (7.5 mg, 0.0610 mmol) in CH2Cl2 (10 mL) was added the respective (substituted) cinnamic acid (0.610 mmol) and DCC (252 mg, 1.22 mmol) at room temperature. After 24 hours (TLC), CH2Cl2 was removed under vacuum and the residue was triturated with cold diethyl ether (20 mL) and filtered. Diethyl ether was then removed under vacuum, and the crude product was purified by column chromatography using 8:1 hexane/EtOAc as the eluent. This procedure was used to synthesize compounds 41–47.
General procedure for acylation of 3′-O-Cbz 16: synthesis of compounds 55–61
The (substituted) cinnamic acid (1.12 mmol) and DCC (231 mg, 1.12 mmol) were added to a stirred solution of 3′-O-Cbz 16 (500, 0.746 mmol) and DMAP (9.2 mg, 0.0746 mmol) in CH2Cl2 (10 mL) at room temperature. After 12 hours (TLC), CH2Cl2 was removed under vacuum and the crude residue was triturated in cold diethyl ether (20 mL) and filtered. Diethyl ether was then removed under vacuum and the residue was purified by column chromatography using 4:1 hexane/EtOAc as the eluent. This procedure was used to synthesize compounds 55–61.
Author contributions
Z. J. supervised the work and revised the manuscript; L. L., W. P. W. K., and S. D. R. synthesized the compounds and wrote the initial draft of the manuscript; D. D. K., P. P. and M. S. revised the manuscript and analysed the NMR data.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank the Interdisciplinary Graduate School and the School of Chemical and Biomedical Engineering, Nanyang Technological University, Singapore for financial support (CoE, Startup Grant).References
- P. Panda, M. Appalashetti and Z. M. A. Judeh, Phenylpropanoid sucrose esters: Plant-derived natural products as potential leads for new therapeutics, Curr. Med. Chem., 2011, 18(21), 3234–3251 CrossRef CAS PubMed.
- W. Zhao, X. X. Huang, L.-H. Yu, Q.-B. Liu, L.-Z. Li, Q. Sun and S.-J. Song, Tomensides A–D, new antiproliferative phenylpropanoid sucrose esters from Prunus tomentosa leaves, Bioorg. Med. Chem. Lett., 2014, 24(11), 2459–2462 CrossRef CAS PubMed.
- P. Panda, M. Appalashetti, M. Natarajan, C.-P. Mary, S. S. Venkatraman and Z. M. A. Judeh, Synthesis and antiproliferative activity of helonioside A, 3′,4′,6′-tri-O-feruloylsucrose, lapathoside C and their analogs, Eur. J. Med. Chem., 2012, 58, 418–430 CrossRef CAS PubMed.
- L. Lepore, N. Malafronte, F. B. Condero, M. J. Gualtieri, S. Abdo, F. D. Piaz and N. De Tommasi, Isolation and structural characterization of glycosides from an anti-angiogenic extract of Monnina obtusifolia H.B.K, Fitoterapia, 2011, 82(2), 178–183 CrossRef CAS PubMed.
- M. Takasaki, T. Konoshima, S. Kuroki, H. Tokuda and H. Nishino, Cancer chemopreventive activity of phenylpropanoid esters of sucrose, vanicoside B and lapathoside A, from Polygonum lapathifolium, Cancer Lett., 2001, 173(2), 133–138 CrossRef CAS PubMed.
- P. Panda, M. Appalashetti, M. Natarajan, M. B. Chan-Park, S. S. Venkatraman and Z. M. A. Judeh, Synthesis and antitumor activity of lapathoside D and its analogs, Eur. J. Med. Chem., 2012, 53, 1–12 CrossRef CAS PubMed.
- Y.-H. Kuo, Y.-W. Hsu, C.-C. Liaw, J. K. Lee, H.-C. Huang and L.-M. Y. Kuo, Cytotoxic phenylpropanoid glycosides from the stems of smilax china, J. Nat. Prod., 2005, 68(10), 1475–1478 CrossRef CAS PubMed.
- P. V. Kiem, N. X. Nhiem, N. X. Cuong, T. Q. Hoa, H. T. Huong, L. M. Huong, C. V. Minh and Y. H. Kim, New phenylpropanoid esters of sucrose from Polygonum hydropiper and their antioxidant activity, Arch. Pharmacal Res., 2008, 31(11), 1477–1482 CrossRef CAS PubMed.
- N. X. Nhiem, P. Kiem, C. Minh, N. K. Ban, N. X. Cuong, B. H. Tai and Y. H. Kim, Phenylpropanoid glycosides from Heterosmilax erythrantha and their antioxidant activity, Arch. Pharmacal Res., 2009, 32(10), 1373–1377 CrossRef PubMed.
- N. Fabre, P. Urizzi, J. P. Souchard, A. Fréchard, C. Claparols, I. Fourasté and C. Moulis, An antioxidant sinapic acid ester isolated from Iberis amara, Fitoterapia, 2000, 71(4), 425–428 CrossRef CAS PubMed.
- M. Wang, Y. Shao, J. Li, N. Zhu, M. Rangarajan, E. J. LaVoie and C.-T. Ho, Antioxidative phenolic glycosides from sage (Salvia officinalis), J. Nat. Prod., 1999, 62(3), 454–456 CrossRef CAS PubMed.
- P. Fan, L. Terrier, A.-E. Hay, A. Marston and K. Hostettmann, Antioxidant and enzyme inhibition activities and chemical profiles of Polygonum sachalinensis F.Schmidt ex Maxim (Polygonaceae), Fitoterapia, 2010, 81(2), 124–131 CrossRef CAS PubMed.
- L. Zhang, C.-C. Liao, H.-C. Huang, Y.-C. Shen, L.-M. Yang and Y.-H. Kuo, Antioxidant phenylpropanoid glycosides from Smilax bracteata, Phytochemistry, 2008, 69(6), 1398–1404 CrossRef CAS PubMed.
- J. Hu, X. Shi and J. Chen, Four new antioxidant phenylpropanoid glycosides from Microlepia pilosissima, Arch. Pharmacal Res., 2012, 35(12), 2127–2133 CrossRef CAS PubMed.
- T. Hosoya, Y. S. Yun and A. Kunugi, Antioxidant phenylpropanoid glycosides from the leaves of Wasabia japonica, Phytochemistry, 2008, 69(3), 827–832 CrossRef CAS PubMed.
- C.-L. Chang, L.-J. Zhang, R. Y. Chen, L.-M. Y. Kuo, J.-P. Huang, H.-C. Huang, K.-H. Lee, Y.-C. Wu and Y.-H. Kuo, Antioxidant and anti-inflammatory phenylpropanoid derivatives from calamus quiquesetinervius, J. Nat. Prod., 2010, 73(9), 1482–1488 CrossRef CAS PubMed.
- N. Wang, X. Yao, R. Ishii and S. Kitanaka, Bioactive sucrose esters from Bidens parviflora, Phytochemistry, 2003, 62(5), 741–746 CrossRef CAS PubMed.
- Y. Sook Yun, M. Satake, S. Katsuki and A. Kunugi, Phenylpropanoid derivatives from edible canna, Canna edulis, Phytochemistry, 2004, 65(14), 2167–2171 CrossRef CAS PubMed.
- B. Shao, H. Guo, Y. Cui, M. Ye, J. Han and D. Guo, Steroidal saponins from Smilax china and their anti-inflammatory activities, Phytochemistry, 2007, 68(5), 623–630 CrossRef CAS PubMed.
- X.-Y. Chen, R.-F. Wang and B. Liu, An Update on oligosaccharides and their esters from traditional chinese medicines: Chemical structures and biological activities, J. Evidence-Based Complementary Altern. Med., 2015, 2015, 512675 Search PubMed.
- Y. Wu, Q. Shi, H. Lei, X. Liu and L. Luan, Studies on the total synthesis of tenuifoliside B, Tetrahedron, 2014, 70(24), 3757–3761 CrossRef CAS.
- Y. Kawai, H. Kumagai, H. Kurihara, K. Yamazaki, R. Sawano and N. Inoue, β-Glucosidase inhibitory activities of phenylpropanoid glycosides, vanicoside A and B from Polygonum sachalinense rhizome, Fitoterapia, 2006, 77(6), 456–459 CrossRef CAS PubMed.
- H. I. Duynstee, H. Ovaa, G. A. van der Marel and J. H. van Boom, Synthesis of niruriside a HIV REV/RRE binding inhibitor, Recl. Trav. Chim. Pays-Bas, 1996, 115(6), 339–340 CrossRef CAS.
- Y. Queneau, J. Fitremann and S. Trombotto, The chemistry of unprotected sucrose: The selectivity issue, C. R. Chim, 2004, 7(2), 177–188 CrossRef CAS.
- S. Thévenet, A. Wernicke, S. Belniak, G. Descotes, A. Bouchu and Y. Queneau, Esterification of unprotected sucrose with acid chlorides in aqueous medium: Kinetic reactivity versus acyl- or alkyloxycarbonyl-group migrations, Carbohydr. Res., 1999, 318(1–4), 52–66 CrossRef.
- Y. Queneau, S. Jarosz, B. Lewandowski and J. Fitremann, in Sucrose chemistry and applications of sucrochemicals. Advances in Carbohydrate Chemistry and Biochemistry, ed. H. Derek, Academic Press, 2007, vol. 61, pp. 217–292 Search PubMed.
- M. I. Choudhary, A. Begum, A. Abbaskhan, R. Shafiq-ur and R. Atta-ur, Cinnamate derivatives of fructo-oligosaccharides from Lindelofia stylosa, Carbohydr. Res., 2006, 341(14), 2398–2405 CrossRef CAS PubMed.
- M. Ono, C. Takamura, F. Sugita, C. Masuoka, H. Yoshimitsu, T. Ikeda and T. Nohara, Two new steroid glycosides and a new sesquiterpenoid glycoside from the underground parts of trillium kamtschaticum, Chem. Pharm. Bull., 2007, 55(4), 551–556 CrossRef CAS PubMed.
- W. Kobayashi, T. Miyase, S. Suzuki, H. Noguchi and X.-M. Chen, Oligosaccharide esters from the roots of polygala arillata, J. Nat. Prod., 2000, 63(8), 1066–1069 CrossRef CAS PubMed.
- H. Saitoh, T. Miyase, A. Ueno and A.-J. Reinioses, Oligosaccharide multi-esters from the roots of Polygala reinii Fr. et Sav, Chem. Pharm. Bull., 1994, 42, 1879–1885 CrossRef CAS PubMed.
- W. Kobayashi, T. Miyase, S. Suzuki, H. Noguchi and X.-M. Chen, Tetrasaccharide multi-esters and xanthone glycosides from the roots of Polygala wattersii, J. Nat. Prod., 2000, 63, 1121–1126 CrossRef CAS PubMed.
- S. Devaraj, Y. M. Yip, P. Panda, L. L. Ong, P. W. K. Wong, D. Zhang, Y. Ali and Z. Judeh, Feruloyl sucrose esters: Potent and selective inhibitors of α-glucosidase and α-amylase, Curr. Med. Chem., 2021, 68(9), 1606–1621 CrossRef PubMed.
- K. Ágoston, H. Streicher and P. Fügedi, Orthogonal protecting group strategies in carbohydrate chemistry, Tetrahedron: Asymmetry, 2016, 27(16), 707–728 CrossRef.
- C.-H. Wong, X.-S. Ye and Z. Zhang, Assembly of oligosaccharide libraries with a designed building block and an efficient orthogonal protection–deprotection strategy, J. Am. Chem. Soc., 1998, 120(28), 7137–7138 CrossRef CAS.
- K. Ágoston, G. M. Watt and P. Fügedi, A new set of orthogonal protecting groups on a monosaccharide scaffold, Tetrahedron Lett., 2015, 56(35), 5010–5012 CrossRef.
- W. Muramatsu, K. Mishiro, Y. Ueda, T. Furuta and T. Kawabata, Perfectly Regioselective and Sequential Protection of Glucopyranosides, Eur. J. Org. Chem., 2010,(5), 827–831 CrossRef CAS.
- M. M. L. Zulueta, S.-Y. Lin, Y.-T. Lin, C.-J. Huang, C.-C. Wang, C.-C. Ku, Z. Shi, C.-L. Chyan, D. Irene, L.-H. Lim, T.-I. Tsai, Y.-P. Hu, S. D. Arco, C.-H. Wong and S.-C. Hung, α-Glycosylation by D-glucosamine-derived donors: Synthesis of heparosan and heparin analogues that interact with mycobacterial heparin-binding hemagglutinin, J. Am. Chem. Soc., 2012, 134(21), 8988–8995 CrossRef CAS PubMed.
- S. B. Dulaney, Y. Xu, P. Wang, G. Tiruchinapally, Z. Wang, J. Kathawa, M. H. El-Dakdouki, B. Yang, J. Liu and X. Huang, Divergent synthesis of heparan sulfate oligosaccharides, J. Org. Chem., 2015, 80(24), 12265–12279 CrossRef CAS PubMed.
- T. J. Boltje, C. Li and G.-J. Boons, Versatile set of orthogonal protecting groups for the preparation of highly branched oligosaccharides, Org. Lett., 2010, 12(20), 4636–4639 CrossRef CAS PubMed.
- S. K. Mulani, J.-H. Guh and K.-K. T. Mong, A general synthetic strategy and the anti-proliferation properties on prostate cancer cell lines for natural phenylethanoid glycosides, Org. Biomol. Chem., 2014, 12(18), 2926–2937 RSC.
- X. Yao-Chang, A. Bizuneh and C. Walker, Selective deprotection of alkyl esters using magnesium methoxide, Tetrahedron Lett., 1996, 37(4), 455–458 CrossRef.
- A. M. DiLauro, W. Seo and S. T. Phillips, Use of catalytic fluoride under neutral conditions for cleaving silicon–oxygen bonds, J. Org. Chem., 2011, 76(18), 7352–7358 CrossRef CAS PubMed.
- R. S. Coleman and J. A. Shah, Chemoselective cleavage of benzyl ethers, esters, and carbamates in the presence of other easily reducible groups, Synthesis, 1999,(Suppl 1), 1399–1400 CrossRef CAS.
- M. C. Pirrung, S. W. Shuey, D. C. Lever and L. Fallon, A convenient procedure for the deprotection of silylated nucleosides and nucleotides using triethylamine trihydrofluoride, Bioorg. Med. Chem. Lett., 1994, 4(11), 1345–1346 CrossRef CAS.
- C. Zinelaabidine, O. Souad, J. Zoubir, B. Malika and A. Nour-Eddine, A Simple and efficient green method for the deprotection of N-Boc in various structurally diverse amines under water-mediated catalyst-free conditions, Int. J. Chem., 2012, 4(3), 73–79 CAS.
- P. G. M. Wuts and T. W. Greene, Protection for the hydroxyl group, including 1,2- and 1,3-diols, in Greene's Protective Groups in Organic Synthesis, John Wiley & Sons, Inc., 2006, pp. 16–366 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of the 1H NMR, 13C NMR, and COSY spectra of the synthesized compounds. See DOI: https://doi.org/10.1039/d2nj00881e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2022 |