
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Acid–base properties of an antivirally active acyclic nucleoside phosphonate: (S)-9-[3-hydroxy-2-(phosphonomethoxy)propyl]adenine (HPMPA)
Claudia A.
Blindauer
 *ab,
Antonín
Holý†
c,
Astrid
Sigel
a,
Bert P.
Operschall
a,
Rolf
Griesser
a and
Helmut
Sigel
*a
*ab,
Antonín
Holý†
c,
Astrid
Sigel
a,
Bert P.
Operschall
a,
Rolf
Griesser
a and
Helmut
Sigel
*a
aDepartment of Chemistry, Inorganic Chemistry, University of Basel, Spitalstrasse 51, CH-4056 Basel, Switzerland. E-mail: helmut.sigel@unibas.ch
bDepartment of Chemistry, University of Warwick, Coventry CV4 7AL, UK. E-mail: c.blindauer@warwick.ac.uk
cInstitute of Organic Chemistry and Biochemistry, Centre of Novel Antivirals and Antineoplastics, Academy of Sciences, 16610 Prague, Czech Republic
First published on 16th March 2022
Abstract
HPMPA is an acyclic nucleoside phosphonate analogue of AMP which displays antiviral properties. Therefore, its acid–base behavior as well as that of related compounds like PMEA, 9-[2-(phosphonomethoxy)ethyl]adenine, are for many reasons (e.g., binding to enzymes, coordination of metal ions) of general interest. HPMPA can accept two protons at the phosphonate and two more at the adenine residue, but not all acidity constants are accessible by potentiometric pH titrations. Therefore, we measured the chemical shifts of the nine non-exchangeable HPMPA protons by 1H NMR in D2O in dependence on pD in the range from 1 to 12. The corresponding results allowed identifying the protonation sites and, transferred to aqueous solution, they gave also the acidity constants. The most basic site is the phosphonate group followed by N1 of adenine. The pKa values increase from ca. −0.27 [–N7(H)+] via 1.27 [–PO(OH)2] and 4.23 [–N1(H)+] to 6.86 [–PO(OH)−]. In the fully protonated species charge repulsion exists between N1(H)+ and N7(H)+; therefore, the affinity of N7 for H+ is not correctly reflected by the measured acidity constant (ca. −0.27). Needed is the intrinsic micro acidity constant which reflects the H+ affinity of N7 under conditions where N1 is unprotonated; we abbreviate this species as +H·N7(HPMPA)N1. The corresponding microconstant is estimated to be pkN7–N1H·N7–N1 3.5; the minor species +H·N7(HPMPA)N1 occurs with an estimated formation degree between about 5 to 20%. The basicity of the adenine nitrogens decreases in the order N1 > N7 > N3.
3.5; the minor species +H·N7(HPMPA)N1 occurs with an estimated formation degree between about 5 to 20%. The basicity of the adenine nitrogens decreases in the order N1 > N7 > N3.
1. Introduction
Nucleoside phosphates (NPs) and their metal ion complexes play key roles in many metabolic transformations.1–4 It is thus not surprising that attempts to exploit nucleoside/nucleotide derivatives as drugs have a long history. For example, the antiviral activity of benzimidazole (=1,3-dideazapurine) derivatives was first described5,6 in 1947 and 1-β-D-ribofuranosylbenzimidazoles have been tested against the influenza B virus,7,8 as well as against several strains of polio virus and herpes simplex virus.9,10 Typically, for antiviral activity, these compounds are required to be present in phosphorylated form; however, their phosphorylated derivatives suffer from a serious obstacle, namely that due to the wide occurrence of non-specific dephosphorylation enzymes11 phosphorus–oxygen ester bonds are easily cleaved, rendering these anti-metabolites inefficient.11 This problem is circumvented by using phosphonate derivatives, as the phosphorus–carbon bond cannot be hydrolysed.11–14With the foregoing facts in mind, it is not surprising that acyclic nucleoside phosphonates (ANPs) have been tested for several illnesses and effects such as Schistosomiasis,15 Malaria16 and Sleeping Sickness,17 and have also been shown to activate macrophages.18 However, more importantly for the present context, the ANPs constitute a class of highly successful antiviral compounds.11,19,20 (S)-9-[3-Hydroxy-2-(phosphonomethoxy)propyl]adenine (HPMPA),21 which is active against a range of DNA viruses,21 was the first to be evaluated in 1986, yet it was itself never commercialised as a drug. However, it led to the three structurally related successful drug molecules19,20 (S)-9-[3-hydroxy-2-(phosphonomethoxy)propyl]cytosine (HPMPC; Cidofovir), 9-[2-(phosphonomethoxy)ethyl]adenine (PMEA; Adefovir; Fig. 1, vide infra), and (R)-9-[2-(phosphonomethoxy)propyl]adenine (PMPA; Tenofovir).22 HPMPC differs from HPMPA by the replacement of the adenine by the cytosine residue; PMEA lacks the hydroxymethyl group compared to HPMPA, and PMPA lacks the 3′-hydroxyl group present in HPMPA.
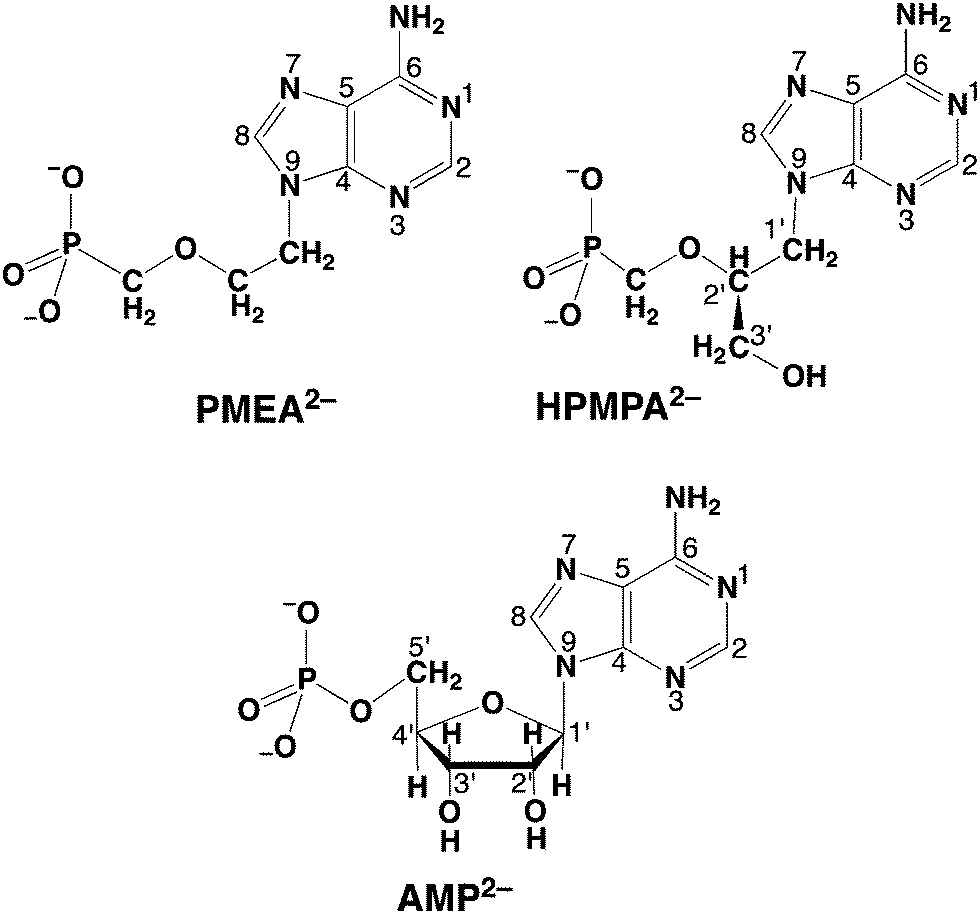 | ||
| Fig. 1 Chemical structures of acyclic nucleoside phosphonates, that is, of the dianions of (S)-9-[3-hydroxy-2-(phosphonomethoxy)propyl]adenine (HPMPA2−) and 9-[2-(phosphonomethoxy)ethyl]adenine (PMEA2−). It is assumed that the preferred orientation of HPMPA2− corresponds to the one observed for PMEA2− in solution36 and in the solid state,37 which resembles the anti conformation of the parent nucleotide, adenosine 5′-monophosphate (AMP2−), which is also shown in its dominating anti conformation.38–41 These compounds are abbreviated as NP2− = nucleoside phosph(on)ate derivative meaning further that P represents the phosph(on)ate group and N the nitrogen sites of the adenine moiety. | ||
Clearly, understanding the solution chemistry of ANPs, that is, their acid–base and metal ion-binding properties (e.g., ref. 23–29), helps to reveal their antiviral mode of action (e.g. ref. 29–34) because it is “Chemistry” that ultimately governs drug action. Evidently, the knowledge of the acid–base properties of liganding sites is important because (a) their metal ion affinities are closely connected to their proton affinities,35 and (b) basicity also informs about energetics of hydrogen bonding which is important for drug recognition by target enzymes. In the present study we concentrate on HPMPA as a good representative of ANPs. Its dianion, HPMPA2−, is shown in Fig. 1 together with PMEA2− and AMP2−,36–41 the natural metabolite that HPMPA mimics. In the course of this study comparisons will be made between these compounds.
HPMPA2− can accept up to four protons, two at the phosphonate group and, as we know from studies with adenosine,42 two more at the adenine residue, i.e., at N1 and N7. Further protonation at N3 gives rise to an exceedingly acidic species, with pKa ≈ −4.0.42,43 Therefore, we consider here only the 4-fold protonated species, i.e., H4(HPMPA)2+. If we write the corresponding equilibria in a general form (see legend of Fig. 1) we obtain the following deprotonation steps:
| H4(NP)2+ ⇌ H3(NP)+ + H+ | (1a) |
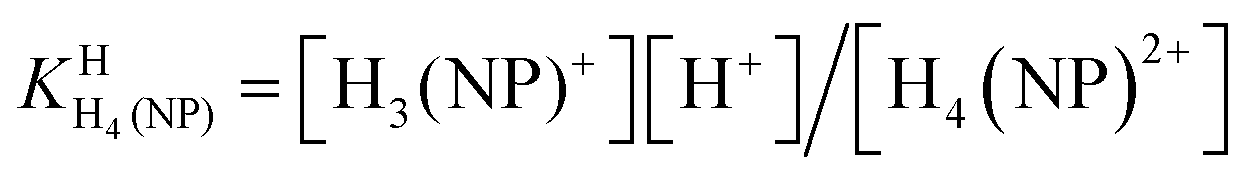 | (1b) |
| H3(NP)+ ⇌ H2(NP) + H+ | (2a) |
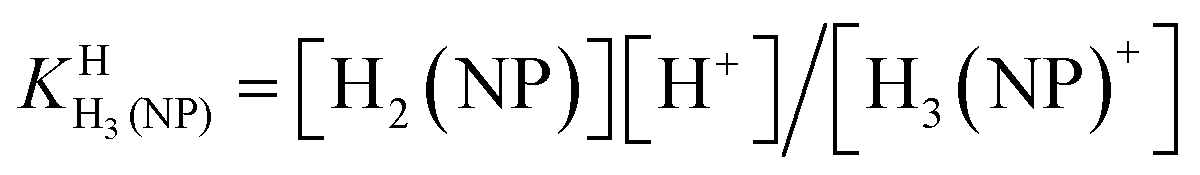 | (2b) |
| H2(NP) ⇌ H(NP)− + H+ | (3a) |
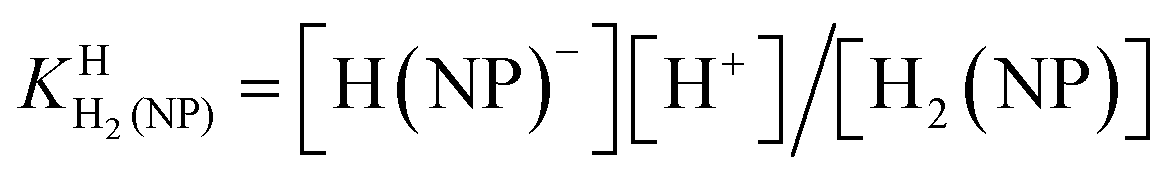 | (3b) |
| H(NP)− ⇌ (NP)2− + H+ | (4a) |
| KHH(NP) = [(NP)2−][H+]/[H(NP)−] | (4b) |
The first proton on (NP)2− will be bound to the most basic site, which is the phosphonate residue. In other words, the last proton to be released is from the monoprotonated phosphonate group with pKa ≈ 7 [eqn (4)] and the second to the last is from (N1)H+ with pKa ≈ 4 [eqn (3)].23,44–46 The release of the even more acidic protons [eqn (1) and (2)] from the P(O)2(OH)2 and (N7)H+ sites occurs with pKa values <2.36,45 From this follows that the acidity constants according to eqn (3) and (4) can be determined by potentiometric pH titrations;23,44–46 pKa values below 2 are not accessible by this method in an exact manner.36 Therefore, we decided to determine the pD dependence of the 1H NMR chemical shifts of the non-exchangeable aromatic and aliphatic hydrogens of HPMPA in D2O. This procedure will not only provide the acidity constants for the equilibria (1) through (4), but in addition may provide some information about the conformations of the species present in solution.
2. Experimental
The free acid of (S)-9-[3-hydroxy-2-(phosphonomethoxy)propyl]adenine, i.e., H2(HPMPA)± (Fig. 1) was prepared as described.47–49 The same lot had been used previously for studies of binary M(HPMPA) complexes29,44 (M2+ = Mg2+, Ca2+, Ni2+, Zn2+, etc.) as well as for those of ternary Cu(Arm)(HPMPA) complexes,28 where Arm = 2,2′-bipyridine or 1,10-phenanthroline.The buffers used for pH calibration (pH 4.64, 7.00, and 9.00)36 were from Metrohm AG, Herisau, Switzerland, and they were based on the U.S. National Institute of Science and Technology (NIST) scale (the apparatus was also from Metrohm).36 In addition, a buffer with pH 1.00 was also used (also NIST scale), which was obtained from Merck AG, Darmstadt, Germany (for further details see ref. 50). D2O (>99.8% D), NaOD ( > 99.9% D) and DNO3 (>99% D) were from Ciba-Geigy AG (Basel, Switzerland). All other reagents were the same as used before in related studies.27,29,36,50,51
The experiments with HPMPA were carried out exactly as described for PMEA and its derivatives,36 and this includes the evaluation methods. In brief, HPMPA was dissolved in 100% D2O, lyophilised, and then dissolved again in D2O, at a final concentration of 5 mM, in the presence of 0.1 M NaNO3 to control ionic strength (I). We note that at pD < 1 I is somewhat larger.36 The pD of samples was measured with a pH meter (Metrohm) that had been calibrated with the buffers in H2O mentioned above, and adjusted with μL quantities of NaOD or DNO3. To obtain pD values, 0.4 pH units were added.52 Tetramethyl ammonium nitrate (3.174 ppm) was used as chemical shift standard (1.8 mM), and 0.25 mM EDTA were added to prevent any line-broadening by paramagnetic impurities. Spectra were recorded on a Bruker AC-250 (operating at 250.134 MHz for 1H) using 16k datapoints and a spectral width of 3000 Hz.
Because the applied pH/pD scale is based on proton activity (as is common in biochemistry), the acidity constants determined are so-called practical, mixed, or Brønsted constants.53,54 The negative logarithms of these constants given for aqueous solution (25 °C; I = 0.1 M, NaNO3) can be converted into the corresponding concentration constants by subtracting 0.02 from the pKa values listed.54
3. Results and discussion
3.1 1H NMR spectra of HPMPA at different pD values and site attribution of the resonance signals
The upper part of Fig. 2 shows the full 1H NMR spectrum of HPMPA at pD 11.5, where the fully deprotonated species HPMPA2− dominates. The underlay in gray between 3.3 and 4.6 ppm highlights the region that encompasses the aliphatic protons, and is enlarged in the middle part of Fig. 2. For all three methylene groups, their two protons appear as two separate resonances for most of the pD range explored; we distinguish these by single and double primes (H1′, H1′′, H3′, H3′′, P-CH′, and P-CH′′; also see the HPMPA structure in Fig. 1). In the case of the CH2(1′) and CH2(3′) protons, this magnetic inequivalence is expected, as they are adjacent to the chiral C2′ carbon and hence diastereotopic. Thus, instead of a single doublet (due to coupling with H2′) for the pair, two doublets of a doublet (i.e. eight peaks) are observed, due to the additional splitting in consequence of the geminal coupling.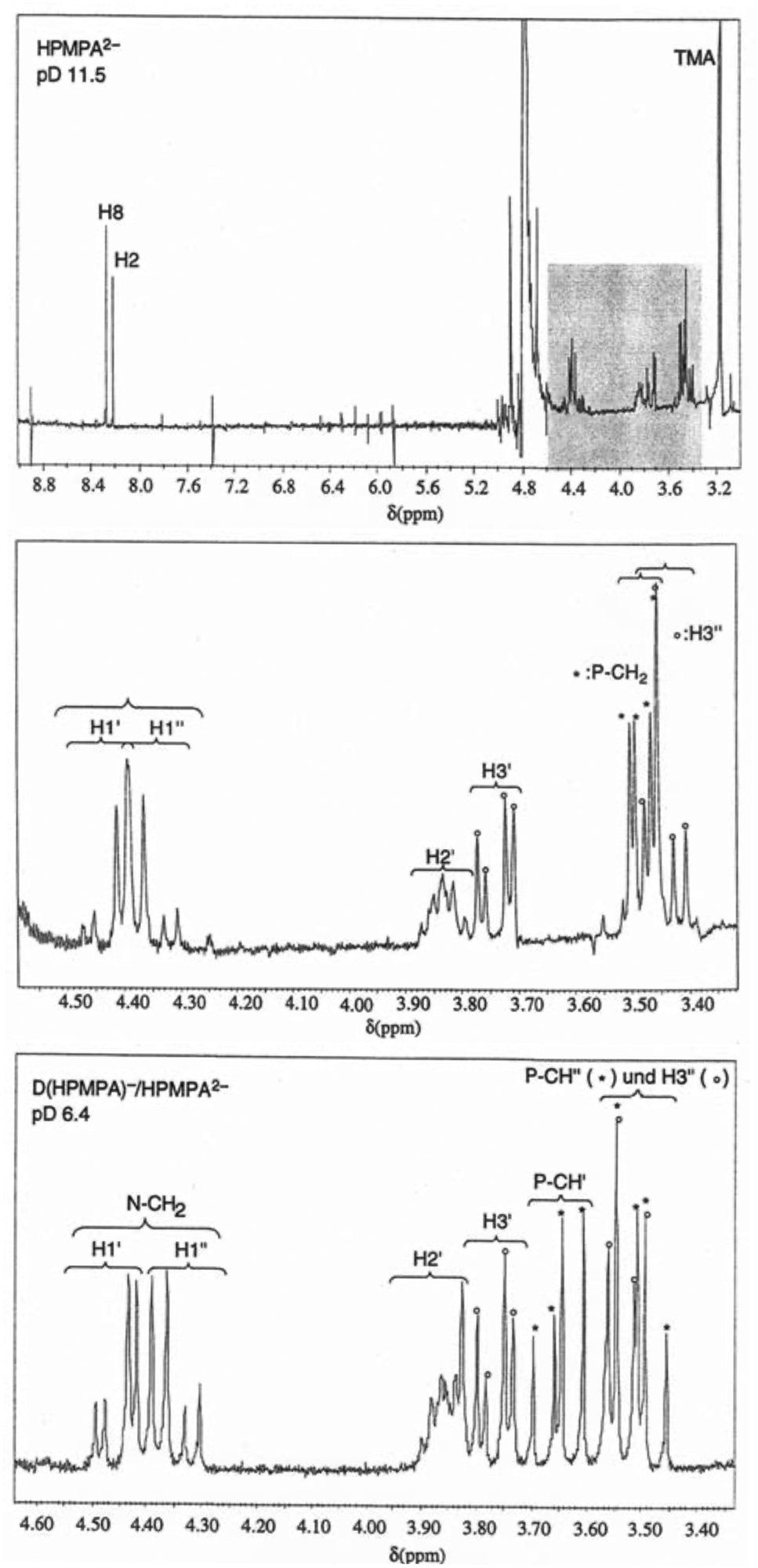 | ||
| Fig. 2 Top: 1H NMR spectrum of HPMPA2− in D2O at pD 11.5 (25 °C; I = 0.1 M, NaNO3. TMA = tetramethylammonium). Middle: Aliphatic region, that is, the expanded segment underlaid in grey in the above spectrum, showing all resonance assignments. Bottom: Aliphatic region of the 1H NMR spectrum of HPMPA at pD 6.4 where the species D(HPMPA)− and HPMPA2− are in fast equilibrium. All resonance assignments are indicated. The difference to the spectrum seen in the middle part consists in the splitting pattern of the methylene protons neighboring the phosphorus atom (P-CH′ and P-CH′′; two doublets-of-a-doublet, as opposed to the single doublet-of-a-doublet observed at pD 11.5). In addition, the splitting pattern (also two doublets-of-a-doublet) for the two CH2(1′) protons is also better recognised; at pD 11.5, the two innermost peaks are nearly coinciding. | ||
Because the signals of the CH2(1′) protons neighboring N9 are more downfield than the other aliphatic protons, the splitting pattern is easily discernible for these. At pD 11.5, the two doublets-of-doublets for the CH2(1′) protons overlap with each other, giving rise to an apparent septet, but at pD 6.4, all eight peaks for the two CH2(1′) protons of HPMPA are well resolved, as is seen in the lower part of Fig. 2. The larger splitting is due to the geminal coupling (2J = 15 Hz), and the smaller splittings refer to 3-bond coupling to H2′ (3J(H1′) = 4.5 Hz, 3J(H1′′) = 7 Hz). The same pattern is expected for the H3′ protons, but is slightly more difficult to observe because the signals of the various protons (H2′, H3′, H3′′, P-CH2) are partly overlapping. In addition, both H1′/H1′′ and H3′/H3′′ spin systems show second-order effects; this leads to asymmetries in line intensities, sometimes referred to as “roofing effect”. Together with analysis of coupling constants, these patterns allowed, with few exceptions, unambiguous assignments of all peaks, and ultimately the determination of the chemical shift for each individual proton in each spectrum.
At pD > 8 the methylene protons neighboring the phosphonate group (P-CH2) are magnetically equivalent and appear as one doublet-of-doublets (see Fig. 2, middle), with the larger splitting due to coupling to 31P (2J = 9.4 Hz), and the smaller splitting due to 4-bond coupling to H2′ (4J = 2.2 Hz). At pD < 8, the two protons become magnetically inequivalent, and give rise to two doublets-of-a-doublet (see Fig. 2, bottom), with the larger splitting due to geminal coupling (2J = 15 Hz), and the smaller due to coupling to 31P (2J = 9.4 Hz). The 4-bond coupling to H2′ is no longer observed. The pD at which the two protons begin to differ coincides with protonation of the phosphonate group; the significance of this observation will be discussed further below.
It may be added that the assignments of all aliphatic protons in Fig. 2 (middle and bottom) agree with those given in the literature for 1-deaza-HPMPA.55 The assignment of the signals of the two aromatic protons is based on comparisons with PMEA and 5′-AMP (Fig. 1) and the dependence of the signals on pD.36,38,56 Note that the signal of H8 is downfield from that of H2.
3.2 Chemical shifts in dependence on pD
Fig. 3 shows the chemical shifts of all protons of HPMPA in dependence on pD. The solid curves represent the computer-calculated best fits of the experimental data,36,38 and are based on the premise that there is fast exchange on the NMR timescale between the various species in equilibrium, giving average chemical shifts for each hydrogen at each pD value, as indeed observed in the spectra shown in Fig. 2.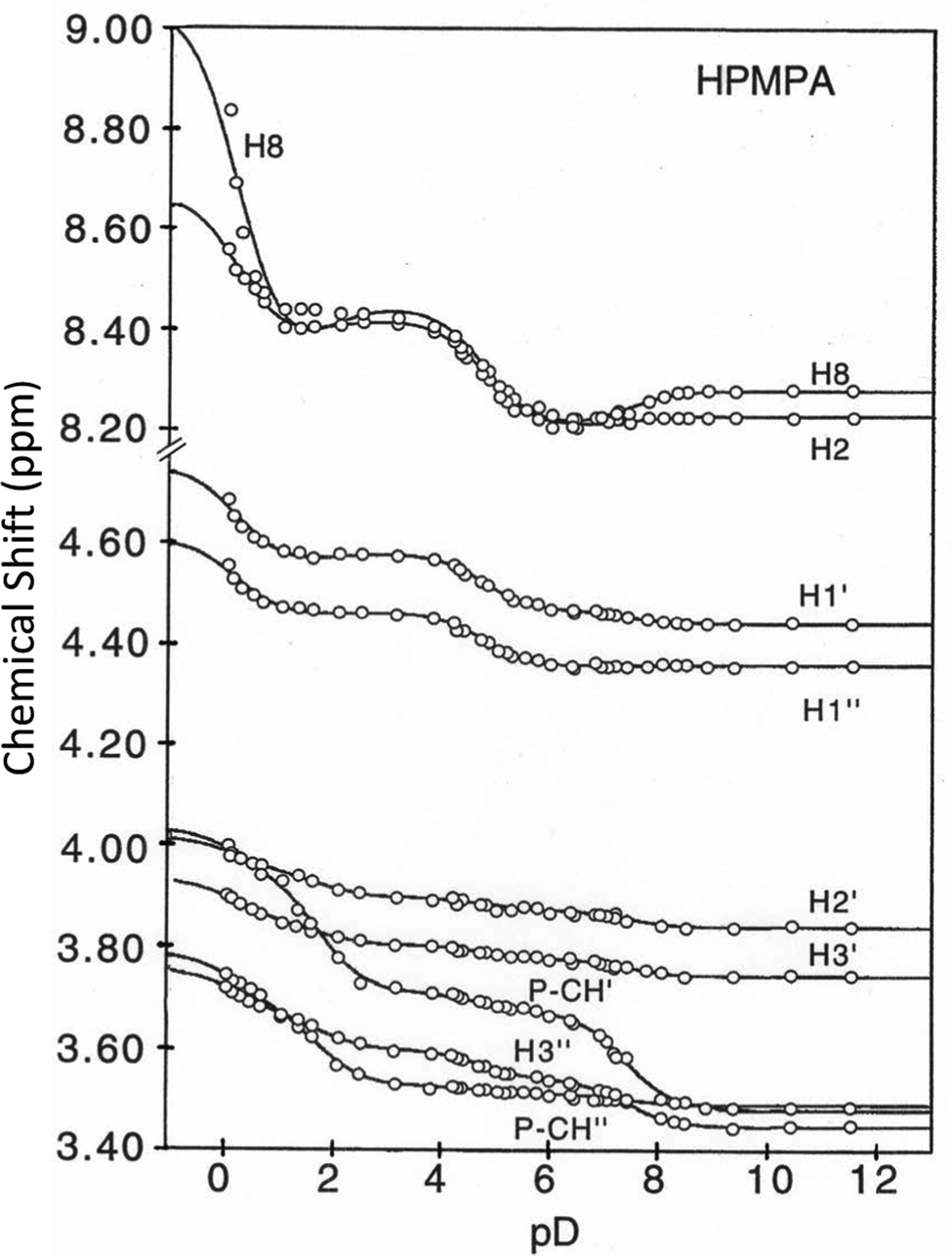 | ||
| Fig. 3 Variation of the chemical shifts (ppm) for all non-exchangeable aromatic and aliphatic protons of HPMPA (see Fig. 1) in dependence on pD (25 °C; I = 0.1 M, adjusted with NaNO3 when needed; at pD < 1 I is somewhat larger).36 The solid lines represent the computer-calculated best fits36,38 of the experimental data points by using the averaged pKa/av values given in Table 1; the resulting shifts are listed in Table 2 (see also Section 2 and text in Section 3.2). | ||
Like with PMEA,36 and as expected, also for HPMPA four protonation reactions are discernible from the combined plots (Fig. 3). Inflection points are at pD ca. 0, 2, 5, and 7.5, with different protons responding to a different extent to the four (de-)protonation reactions. With the exception of H8, for all protons curve fits for all four pKa values defined in eqn (1) to (4) could be carried out (Table 1).
| Hb |
|
|
|
pKDD(HPMPA) |
|---|---|---|---|---|
| a The evaluations for the individual protons (pKa/ind) are listed (I = 0.1 M, adjusted with NaNO3 when needed; at pD < 1 I was somewhat larger);36 all values result from the free variation of the pKa values and chemical shifts of the various species (Table 2), unless indicated otherwise (see footnote c). The error limits given with pKa/ind are the standard deviations resulting from the curve-fitting procedure (Fig. 3).36,38 The average pKa values, pKa/av, are the weighted means of the individual results; their errors correspond to twice the standard deviation. b The various protons are defined in Fig. 2 (see also Fig. 1). c Estimated value; see text in Section 3.2. | ||||
| H8 | −0.05 ± 0.4c | — | 4.610 ± 0.131 | 7.700 ± 0.488 |
| H2 | 0.358 ± 0.196 | 1.581 ± 0.417 | 4.738 ± 0.053 | 6.384 ± 0.294 |
| H1′ | −0.012 ± 0.272 | 1.724 ± 0.824 | 4.722 ± 0.070 | 7.296 ± 0.237 |
| H1′′ | −0.306 ± 0.924 | 1.013 ± 2.100 | 4.755 ± 0.066 | 6.326 ± 2.282 |
| H2′ | −0.313 ± 0.972 | 1.888 ± 0.255 | 4.746 ± 0.215 | 7.602 ± 0.144 |
| H3′ | 0.006 ± 0.321 | 1.770 ± 0.204 | 4.635 ± 0.148 | 7.404 ± 0.087 |
| H3′′ | −0.532 ± 0.564 | 1.730 ± 0.081 | 4.725 ± 0.052 | 7.411 ± 0.028 |
| P-CH′ | −0.514 ± 2.391 | 1.659 ± 0.088 | 4.812 ± 0.264 | 7.374 ± 0.040 |
| P-CH′′ | 0.481 ± 0.255 | 1.780 ± 0.102 | 5.358 ± 0.209 | 8.439 ± 0.198 |
| Weighted means for pKa/av | 0.18 ± 0.28 | 1.74 ± 0.11 | 4.74 ± 0.08 | 7.41 ± 0.11 |
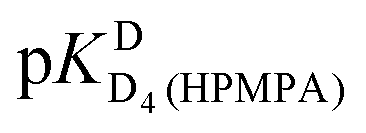
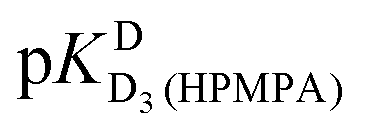
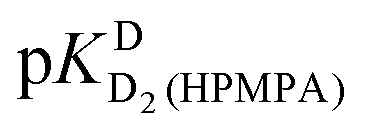
In the case of H8 fits did not converge to give values for 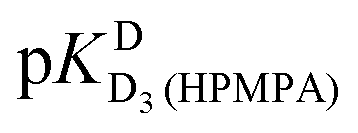 [eqn (2)] and
[eqn (2)] and 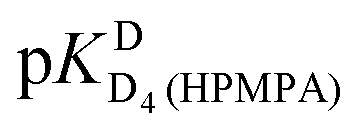 [eqn (1)], therefore for this proton, initially datapoints for the pD range of 3 to 11.5 only were used to provide values for
[eqn (1)], therefore for this proton, initially datapoints for the pD range of 3 to 11.5 only were used to provide values for 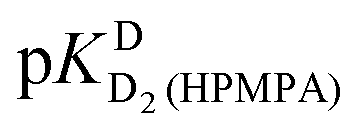 and pKDD(HPMPA). Next, the weighted means for pKDD(HPMPA) and
and pKDD(HPMPA). Next, the weighted means for pKDD(HPMPA) and 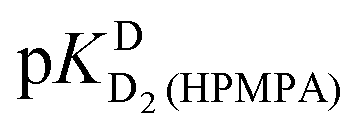 from all nine curve fits were calculated, as well as a value for
from all nine curve fits were calculated, as well as a value for 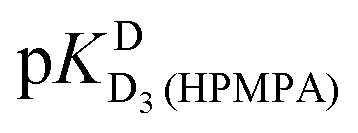 from the eight curves that allowed a fit (i.e., without that for H8). These three values (Table 1, last line) were now kept constant and a value for
from the eight curves that allowed a fit (i.e., without that for H8). These three values (Table 1, last line) were now kept constant and a value for 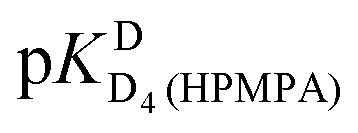 for the H8 proton chemical shift dependence was estimated by systematically varying the constant. The “best” value (−0.05 ± 0.4) was selected based on two criteria, namely, the error square sum of the fit and a reasonable shift difference (i.e. <1.5 ppm). The final weighted means from the process described are given as pKa/av in the terminating row of Table 1. The four pKa values are similar to those found for PMEA,36 which is unsurprising as the hydroxymethyl group is not expected to have a significant influence on any of the four protonation reactions.
for the H8 proton chemical shift dependence was estimated by systematically varying the constant. The “best” value (−0.05 ± 0.4) was selected based on two criteria, namely, the error square sum of the fit and a reasonable shift difference (i.e. <1.5 ppm). The final weighted means from the process described are given as pKa/av in the terminating row of Table 1. The four pKa values are similar to those found for PMEA,36 which is unsurprising as the hydroxymethyl group is not expected to have a significant influence on any of the four protonation reactions.
In order to determine the chemical shifts (δ) for all nine protons in the five differently protonated HPMPA species, the weighted means pKa/av (Table 1) were set as constants in a second round of curve fitting. Results of this analysis are listed in Table 2; the solid lines in Fig. 3 are those resulting from these fits. For the aromatic protons, the chemical shifts are similar to those of PMEA,36 again in line with expectations, as the hydroxymethyl group is not expected to have a significant influence on the chemical shifts of these protons.
| Hb | δ D4(HPMPA) | δ D3(HPMPA) | δ D2(HPMPA) | δ D(HPMPA) | δ (HPMPA) |
|---|---|---|---|---|---|
| a The error limits correspond to twice the standard deviation;50 see Table 1 and also text in Section 3.2. b The labels of the various protons follow from Fig. 1 and 2. | |||||
| H8 | 9.045 ± 0.306 | 8.340 ± 0.064 | 8.484 ± 0.030 | 8.210 ± 0.021 | 8.279 ± 0.020 |
| H2 | 8.668 ± 0.111 | 8.381 ± 0.026 | 8.419 ± 0.014 | 8.207 ± 0.009 | 8.229 ± 0.006 |
| H1′ | 4.754 ± 0.076 | 4.556 ± 0.016 | 4.578 ± 0.008 | 4.465 ± 0.005 | 4.438 ± 0.004 |
| H1′′ | 4.606 ± 0.064 | 4.453 ± 0.014 | 4.461 ± 0.008 | 4.354 ± 0.005 | 4.356 ± 0.004 |
| H2′ | 4.014 ± 0.039 | 3.948 ± 0.012 | 3.897 ± 0.004 | 3.873 ± 0.004 | 3.841 ± 0.004 |
| H3′ | 3.933 ± 0.033 | 3.848 ± 0.009 | 3.803 ± 0.003 | 3.777 ± 0.003 | 3.744 ± 0.003 |
| H3′′ | 3.755 ± 0.033 | 3.670 ± 0.009 | 3.598 ± 0.004 | 3.541 ± 0.005 | 3.447 ± 0.004 |
| P-CH′ | 4.032 ± 0.046 | 3.941 ± 0.027 | 3.710 ± 0.009 | 3.675 ± 0.011 | 3.480 ± 0.010 |
| P-CH′′ | 3.788 ± 0.044 | 3.683 ± 0.018 | 3.525 ± 0.006 | 3.511 ± 0.003 | 3.489 ± 0.004 |
Based on the chemical shift values (δ) listed in Table 2 one may calculate shift differences (Δδ) between different protonated species. These values are helpful in ascertaining the sites to which the various equilibria (1) to (4) pertain. In a first approximation, we expect that the hydrogens that are closest to the site of protonation will experience the largest changes in chemical shift. Thus, both Fig. 3 and Table 3 furnish information about the site at which a particular acid–base reaction occurs: In the fully deprotonated HPMPA2− species (Fig. 1) the most basic site is the −PO32− residue which readily accepts a proton.23,36 This is confirmed by the P-CH′ hydrogen experiencing the largest shift difference upon the first protonation (Table 3, column 5); hence, the value given in Table 1 (bottom line) for pKDD(HPMPA) is firmly attributed to the deprotonation of –P(O)2(OD)− [eqn (4)].
| Hb | Δδ4 = δD4(HPMPA) − δD3(HPMPA) | Δδ3 = δD3(HPMPA) − δD2(HPMPA) | Δδ2 = δD2(HPMPA) − δD(HPMPA) | Δδ1 = δD(HPMPA) − δ(HPMPA) |
|---|---|---|---|---|
| a The error limits correspond to twice the standard deviation; they were calculated according to the error propagation after Gauss by taking into account the error limits given in Table 2 (see also bottom line of Table 1). b The protons are labeled according to Fig. 1 and 2. c The underlined numbers correspond to the largest shift differences within columns, indicating vicinity to the protonation site. | ||||
| H8 | ![[0 with combining low line]](https://www.rsc.org/images/entities/char_0030_0332.gif) . .![[7 with combining low line]](https://www.rsc.org/images/entities/char_0037_0332.gif) ![[5 with combining low line]](https://www.rsc.org/images/entities/char_0035_0332.gif) ± . ± .![[3 with combining low line]](https://www.rsc.org/images/entities/char_0033_0332.gif) ![[1 with combining low line]](https://www.rsc.org/images/entities/char_0031_0332.gif) c c |
–0.104 ± 0.071 | .![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif) ![[4 with combining low line]](https://www.rsc.org/images/entities/char_0034_0332.gif) ± .c ± .c |
–0.069 ± 0.029 |
| H2 | .![[8 with combining low line]](https://www.rsc.org/images/entities/char_0038_0332.gif) ± .c ± .c |
–0.038 ± 0.030 | . ± .c |
–0.022 ± 0.011 |
| H1′ | 0.198 ± 0.078 | –0.022 ± 0.018 | 0.113 ± 0.009 | 0.027 ± 0.006 |
| H1′′ | 0.153 ± 0.066 | –0.008 ± 0.016 | 0.107 ± 0.009 | –0.002 ± 0.006 |
| H2′ | 0.066 ± 0.041 | 0.051 ± 0.013 | 0.024 ± 0.006 | –0.002 ± 0.006 |
| H3′ | 0.085 ± 0.034 | 0.045 ± 0.010 | 0.026 ± 0.004 | –0.033 ± 0.004 |
| H3′′ | 0.085 ± 0.034 | 0.072 ± 0.010 | 0.057 ± 0.006 | 0.094 ± 0.006 |
| P-CH′ | 0.091 ± 0.053 | . ± .c |
0.035 ± 0.014 | .![[9 with combining low line]](https://www.rsc.org/images/entities/char_0039_0332.gif) ± .c ± .c |
| P-CH′′ | 0.105 ± 0.048 | . ± .c |
0.014 ± 0.007 | 0.022 ± 0.005 |
The next protonation step occurs at N1 and therefore significant downfield shifts are observed for H2 and H8 upon protonation of D(HPMPA)− (Table 3, column 4), which leads to the zwitterionic form D2(HPMPA)± [eqn (3)]. That H8 is also sensitive to acid–base reactions occurring at N1 is not surprising as electron density changes are easily dispersed through the π system of the purine base, as is also observed with other adenine derivatives.38,57
Further protonation leads to D3(HPMPA)+, and this reaction occurs again at the phosphonate residue, as the marked downfield shifts for both hydrogens of P–CH2 demonstrate (Table 3, column 3); the 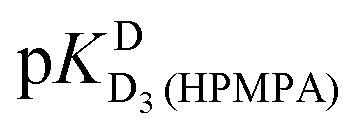 value close to 1.7 (Table 1, column 3) is due to the loss of the first proton from –P(O)(OD)2 [eqn (2)]. Finally, the formation of D4(HPMPA)2+ involves N7 as is demonstrated by the pronounced downfield shift for H8 (Table 3, column 2), together with a smaller downfield shift for H2. This conclusion agrees with results obtained for related systems.36,43,57
value close to 1.7 (Table 1, column 3) is due to the loss of the first proton from –P(O)(OD)2 [eqn (2)]. Finally, the formation of D4(HPMPA)2+ involves N7 as is demonstrated by the pronounced downfield shift for H8 (Table 3, column 2), together with a smaller downfield shift for H2. This conclusion agrees with results obtained for related systems.36,43,57
N3 is never protonated in systems discussed up to now, which agrees with previous experience.42,43 Hence, one may conclude that the basicity of the various ring nitrogens of the adenine residue decreases in the order N1 > N7 > N3.36
Generally, protonation of a site in the vicinity is expected to lead to downfield shifts, as a reduction in electron density will normally reduce electronic shielding of the proton of interest. The data collected in Table 3 show some interesting deviations from this general principle. Firstly, the first protonation of the phosphonate group gives rise to “wrong-way” (i.e. upfield upon protonation) shifts for the aromatic protons, most clearly observed for H8 (−0.069 ± 0.029; Table 3, column 5). Interestingly, H3′ also experiences a wrong-way shift upon this protonation reaction (–0.033 ± 0.004 ppm; Table 3, column 5). In all cases, such wrong-way shifts are thought to be a consequence of changes in the dominant conformation in dependence on protonation state, combined with anisotropy of the magnetic properties of the phosph(on)ate group.58
Secondly, the chemical shift trends for the P-CH2 protons (Table 3 and Fig. 3) also show some peculiar behaviour: whereas P-CH′ is – as expected – strongly affected by the (de)protonation of the neighboring phosphonate group at pD 7.4 (Δδ = 0.195 ± 0.015 ppm), P-CH′′ barely senses this protonation step (Δδ = 0.022 ± 0.005 ppm). Together with the observation that the first protonation of the phosphonate group also renders the two protons magnetically inequivalent, this suggests that this protonation reaction leads to the predominance of a conformation in which one of the P-CH2 protons either senses little from the change in electron density due to the protonation reaction, or, more likely, two opposing effects cancel each other out. It is also noteworthy that the pKa values for the two first protonation steps (8.439 ± 0.198 and 5.358 ± 0.209) determined from the curve fitting for P-CH′′ deviate significantly from the overall averages; this is due to both transitions being ill defined for this proton.
Either way, the seemingly paradoxical chemical shift trends, i.e. wrong-way shifts of H8, H2 and H3′ and the behaviour of the P-CH′′ proton, are in each case manifestations of the same concept: the change in charge and bound H+ due to (de)protonation of the phosphonate group leads to a change in conformational preferences and dynamics, similar to the situation in the parent nucleotide.59 Fully deprotonated purine nucleotides are known to favour anti conformations, in which H8 faces the phosphate group, whilst protonation diminishes the prevalence of anti conformations.38,59 These conformational preferences, together with anisotropic effects of the phosph(on)ate group,58 are thought to be responsible for the chemical shift trends for H8 in 5′-AMP.38,60 The magnitude of the wrong-way shifts for either PMEA36 or HPMPA is smaller than that for 5′-AMP, which is probably owed to the larger conformational flexibility of the acyclic analogues. Nonetheless, these similarities in conformational preferences with their parent compounds may contribute to their effectiveness as substrate analogues for both kinases and nucleic acid polymerases.
We note that the data in Table 3 also suggest that H8 appears to experience a wrong-way shift upon the second protonation of the phosphonate group (−0.104 ± 0.071 ppm; Table 3, column 3), but would caution that the datapoints for H8 deviate considerably from the fitted curve in this pH range (Fig. 3) – which of course coincides with the 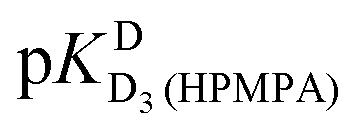 value for which no fitting was possible for this proton. The plot in Fig. 3 therefore does not support the existence of a wrong-way shift for this step.
value for which no fitting was possible for this proton. The plot in Fig. 3 therefore does not support the existence of a wrong-way shift for this step.
3.3 Acidity constants of D4/H4(HPMPA)2+ in D2O and H2O plus attribution of the protonation sites
In Table 429,36,44,61 in row (2) the pKa values for D4(HPMPA)2+ are collected. These values were transformed62 to water as solvent by applying eqn (5):| pKa/H2O = (pKa/D2O − 0.45)/1.015 | (5) |
| Row no. | NP/solvent/method |
|
|
|
pKDD(NP)b–PO2(OH)− |
|---|---|---|---|---|---|
| a (25 °C; I = 0.1 M, adjusted with NaNO3 when needed; see also legend for Fig. 3). The error limits of the NMR data are twice and those of the potentiometric pH titrations are three times the standard deviation. b For rows (3) and (4) D needs to be replaced by H. c The values in row (1) are from Table 2 in ref. 36. d These values are from the bottom line of Table 1. e These pKa values for H2O as solvent were calculated with eqn (5) from those valid for D2O and given in row (2); here the same error limits apply as given for the corresponding constants valid for D2O as solvent. f This value is an estimate based on differences between acidity constants and on the information given in ref. 61 (see also ref. 29). g These constants were determined by potentiometric pH titrations44 and they were confirmed recently by further potentiometric pH titrations.29 | |||||
| (1)c | PMEA/D2O/1H NMR | 0.1 ± 0.5 | 1.69 ± 0.13 | 4.72 ± 0.09 | 7.66 ± 0.36 |
| (2)d | HPMPA/D2O/1H NMR | 0.18 ± 0.28 | 1.74 ± 0.11 | 4.74 ± 0.08 | 7.41 ± 0.11 |
| (3)e | HPMPA/H2O/1H NMR | –0.27 (±0.28) | 1.27 (±0.11) | 4.23 (±0.08) | 6.86 (±0.11) |
| (4) | HPMPA/H2O/pot. | (1.16 ± 0.10)f | 4.14 ± 0.01g | 6.84 ± 0.01g | |
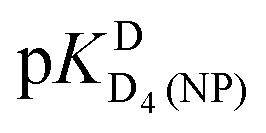
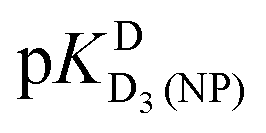
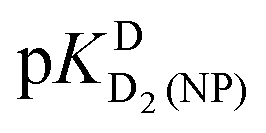
Comparison of the acidity constants for D4(PMEA)2+ [row (1)] with those for D4(HPMPA)2+ [row(2)] indicate, as expected, that the presence of the –CH2-OH substituent (see Fig. 1) has no marked effect on the acid–base properties. This conclusion is confirmed for the constants that have been determined for water as solvent.29
Thanks to the information summarised in Table 3 the protonation sites can be identified as is given in Table 4: In H2O as solvent [row (3)] the first protonation occurs at the phosphonate group (pKHH(HPMPA) = 6.86) and the second one at N1 of the adenine residue  . The next protonation reaction takes place again at the phosphonate group
. The next protonation reaction takes place again at the phosphonate group  and the final one at N7 of the adenine residue
and the final one at N7 of the adenine residue 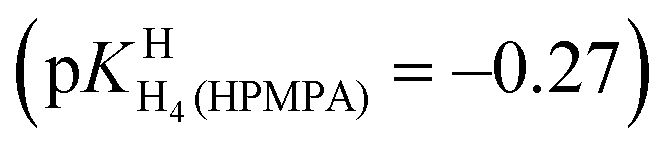 .
.
The values for the acidity constants pKHH(HPMPA) and 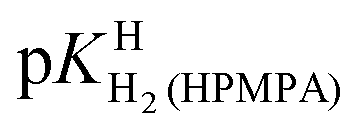 [Table 4, row (4)] have also been determined by potentiometric pH titrations. These constants agree well with the data obtained by 1H NMR measurements [row (3)] and this very satisfying result demonstrates the usefulness of these two independent methods which complement each other.
[Table 4, row (4)] have also been determined by potentiometric pH titrations. These constants agree well with the data obtained by 1H NMR measurements [row (3)] and this very satisfying result demonstrates the usefulness of these two independent methods which complement each other.
Finally, it is interesting to consider the first two deprotonation steps of H4(PMEA)2+ in aqueous solution. From the constants in row (1) of Table 4 and the application of eqn (5)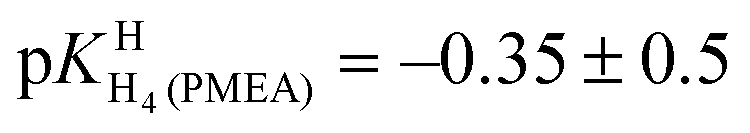 and
and 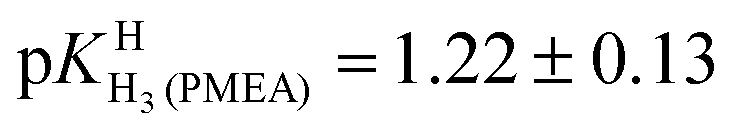 follow. These results fit excellently into the picture defined in row (3) of Table 4 by the constants for H4(HPMPA)2+ and H3(HPMPA)+, confirming again the close relationship between PMEA and HPMPA (Fig. 1).
follow. These results fit excellently into the picture defined in row (3) of Table 4 by the constants for H4(HPMPA)2+ and H3(HPMPA)+, confirming again the close relationship between PMEA and HPMPA (Fig. 1).
3.4 Intrinsic basicity and development of a microconstant scheme
For H2(HPMPA)± the constants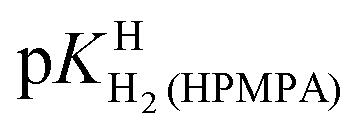 [=4.23; Table 4, row (3)] and pKHH(HPMPA) (=6.86) are more than 2.5 log units apart, and therefore, there is practically no overlap between equilibria (3) and (4). Moreover, and this is of equal significance, the (de)protonation reactions occur at different parts of HPMPA, that is, at the phosphonate group and the adenine moiety (Fig. 1) and the protonation sites will sense only little from each other due to the distance between them. This is different for the acid–base reactions that involve N1 and N7; both sites are at the adenine residue and therefore a proton at N1 will affect the properties of N7, as there is repulsion between these two positively charged sites – even more so since they are part of the same π system. In other words, a positive charge “at N1” will facilitate deprotonation at N7(H)+.
[=4.23; Table 4, row (3)] and pKHH(HPMPA) (=6.86) are more than 2.5 log units apart, and therefore, there is practically no overlap between equilibria (3) and (4). Moreover, and this is of equal significance, the (de)protonation reactions occur at different parts of HPMPA, that is, at the phosphonate group and the adenine moiety (Fig. 1) and the protonation sites will sense only little from each other due to the distance between them. This is different for the acid–base reactions that involve N1 and N7; both sites are at the adenine residue and therefore a proton at N1 will affect the properties of N7, as there is repulsion between these two positively charged sites – even more so since they are part of the same π system. In other words, a positive charge “at N1” will facilitate deprotonation at N7(H)+.
To learn the true proton affinity of N7 we therefore must consider the situation in a species which has a free, unprotonated N1 site; we write this species as +H·N7(HPMPA)N1 (or even shorter as +H·N7–N1). The consequence of this is that there must also be species in which N1 is protonated, namely +H·N7(HPMPA)N1·H+ (or shorter +H·N7–N1·H+). This in turn means that there are two ways from N7(HPMPA)N1 to form the species with a diprotonated adenine residue, beginning protonation either with N1 and followed by that of N7, or vice versa. If we ignore for now the phosphonate residue, we can summarise the situation regarding the two N sites as given in the equilibrium scheme shown in the upper part of Fig. 4.
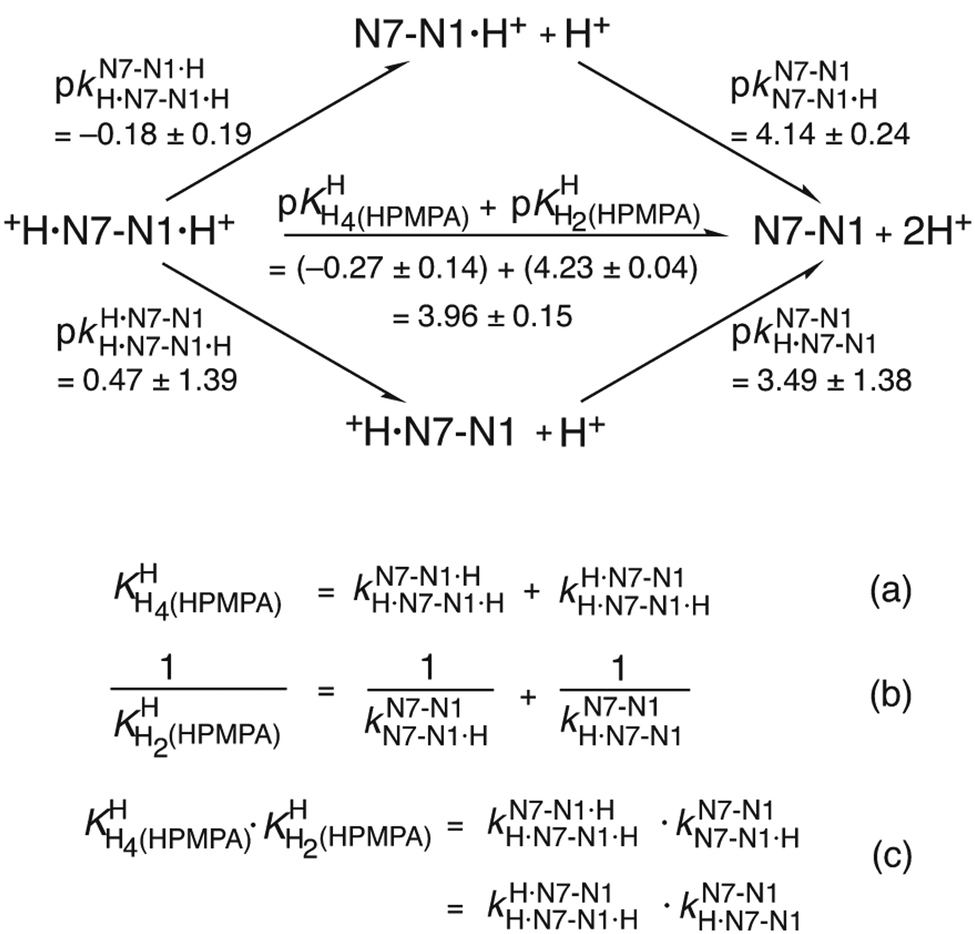 | ||
| Fig. 4 Equilibrium scheme for HPMPA (Fig. 1 and Table 4) defining the micro acidity constants (k) and showing their interrelation with the macro acidity constants (K) and the connection between N7–N1·H+ and +H·N7–N1 and the other species present. In N7–N1·H+ and +H·N7–N1 the proton is bound to N1 or to N7, respectively. The arrows indicate the direction for which the acidity constants are defined. Use of the value 3.49 ± 1.38 determined for the micro acidity constant pkN7–N1H·N7–N1 (see text in Section 3.5) permits calculation of the other micro acidity constants (see text). The error limits of the various constants were calculated according to the error propagation after Gauss; they correspond to one standard deviation (see also Table 4; footnote a). | ||
This scheme defines the reactions for the micro acidity constants, and in the lower part of Fig. 4 the interrelations are given between these microconstants and the measured macro acidity constants. The definitions provided in the lower part of Fig. 4 follow known routes.63Fig. 4 shows that there are four unknown micro acidity constants, but only three independent equations interrelating them with the macroconstants; thus, one of the microconstants must be obtained independently.
3.5 Estimation of a micro acidity constant for the N7-protonated adenine residue of HPMPA
How can we obtain or estimate one of the microconstants? Several years ago64 we had studied the acid–base properties of several guanine and hypoxanthine derivatives by determining their macro acidity constant as well as the micro acidity constant, pkN7–N1H·N7–N1, which defines the intrinsic acidity of the N7(H)+ site of these compounds under conditions where N1 is deprotonated and carries a negative charge. Interestingly, a plot of these microconstants pkN7–N1H·N7–N1versus the macro acidity constants of the N1(H) sites, pKa/N1(H), resulted in a straight line (eqn (6); correlation coefficient R = 0.976) [see Fig. 6 and eqn (10) in ref. 64]:| pkN7–N1H·N7–N1 = (2.43 ± 0.31)pKa/N1(H) − (17.77 ± 2.87) | (6) |
The data points for 9-methyladenine (pkHH(9MeA) = pKa/N1(H) = 4.10)64 and adenosine (pkHH(Ado) = pKa/N1(H) = 3.61)64 fall on their own straight line (the y-axis being defined by pkN7–N1H·N7–N1) which is parallel to that for the guanine/hypoxanthines. Thus, from the information given in ref. 64 follows eqn (7) which is valid for adenine derivatives; as error limits we now use one standard deviation (see Table 4):
| pkN7–N1H·N7–N1 = (2.43 ± 0.31)pKa/N1(H) − (6.79 ± 0.43) | (7) |
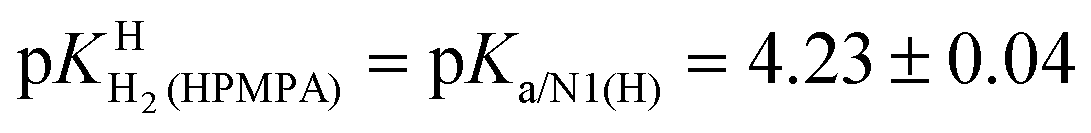 of H2(HPMPA)± [Table 4; row (3)] to eqn (7) provides the micro acidity constant pkN7–N1H·N7(HPMPA)N1 = 3.49 ± 1.38. Use of this value allows to complete the lower circle of Fig. 4 by providing pkH·N7–N1H·N7–N1·H = 0.47 ± 1.39. Application of this latter value to eqn (a) in the lower part of Fig. 4 together with
of H2(HPMPA)± [Table 4; row (3)] to eqn (7) provides the micro acidity constant pkN7–N1H·N7(HPMPA)N1 = 3.49 ± 1.38. Use of this value allows to complete the lower circle of Fig. 4 by providing pkH·N7–N1H·N7–N1·H = 0.47 ± 1.39. Application of this latter value to eqn (a) in the lower part of Fig. 4 together with 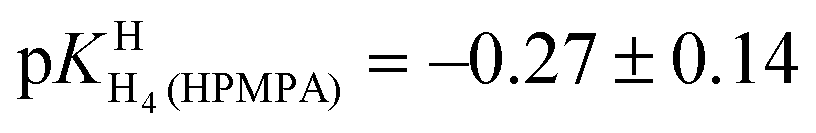 results in pkN7–N1·HH·N7–N1·H = −0.18 ± 0.19 and now also the upper circle of the scheme in Fig. 4 can be completed. Unfortunately, the large error limits prevent a further detailed evaluation, e.g., of the formation degrees of the isomeric species having the proton in one tautomer at N1 and in the other at N7 [eqn (8)]:
results in pkN7–N1·HH·N7–N1·H = −0.18 ± 0.19 and now also the upper circle of the scheme in Fig. 4 can be completed. Unfortunately, the large error limits prevent a further detailed evaluation, e.g., of the formation degrees of the isomeric species having the proton in one tautomer at N1 and in the other at N7 [eqn (8)]: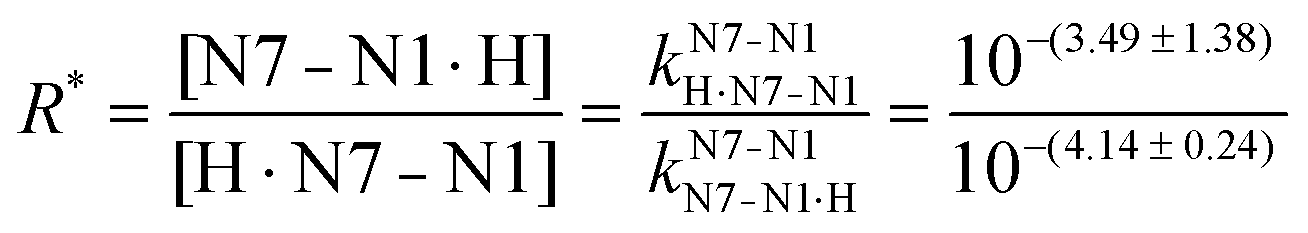 | (8) |
3.6 Further attempts to estimate the intrinsic proton affinity of N7
As the preceding result is not very satisfying, we aimed at a further independent evaluation, based on the reasoning that a proton at N1 is expected, due to charge repulsion, to facilitate the deprotonation of N7(H)+. This charge effect, ΔpKa, of N1(H)+ on N7(H)+ can be calculated for 9-methyladenine (9MeA) by using the results from Fig. 5 in ref. 64; that is pkN7–N1H·N7(9MeA)N1 − pkN7–N1·HH·N7–N1·H = (2.96 ± 0.10) − (−0.61 ± 0.06) = (3.57 ± 0.12). The same effect can also be quantified for adenosine (Ado) from Fig. 2 in ref. 42, that is, pkN7–N1H·N7(Ado)N1 − pkN7–N1·HH·N7–N1·H = (2.15 ± 0.15) − (−1.52 ± 0.08) = (3.67 ± 0.17). Clearly, the charge effect observed for 9-methyladenine and adenosine is within the error limits identical, despite the significant difference between the pKa values of 9MeA and Ado. This fact gives confidence in the further application of these data and we use therefore the average of the two values, that is, ΔpKa = 3.62 ± 0.10, as the charge effect that also holds for HPMPA, and then one obtains pkN7–N1·HH·N7(HPMPA)N1·H = pkN7–N1H·N7–N1 − ΔpKa = (3.49 ± 1.38) − (3.62 ± 0.10) = −0.13 ± 1.38.These derived values, namely −0.13 ± 1.38 and also 3.49 ± 1.38 (from Section 3.5) can be inserted into the upper and lower circles, respectively, of the microconstant scheme given in Fig. 5. The other microconstants in the scheme can now be calculated, making it complete.
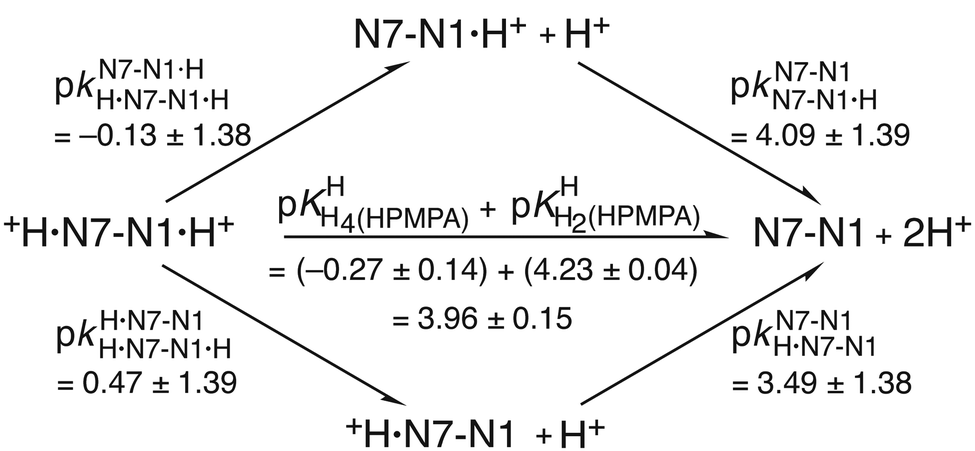 | ||
| Fig. 5 Micro acidity constant scheme for HPMPA. The interrelation between the macro acidity constants (K) and the micro acidity constants (k) are given in the lower part of Fig. 4. The main difference between Fig. 4 and this Fig. 5 is that now two micro acidity constants have been estimated, that is, pkN7–N1H·N7–N1 = 3.49 ± 1.38 (see Fig. 4 and text in Section 3.5) and pkN7–N1·HH·N7–N1·H = −0.13 ± 1.38 (see text in Section 3.6). Consequently, for a completion regarding the missing constants in the upper and lower circles in the scheme none of the equations (a), (b) and (c) of Fig. 4 are needed (see text in Section 3.6). | ||
Unfortunately, the large error of 3.49 ± 1.38 (= pkN7–N1H·N7–N1) propagates to pkN7–N1·HH·N7–N1·H = −0.13 ± 1.38. Despite this, things are overall conclusive because the previous result (Section 3.5) for the same micro acidity constant was −0.18 ± 0.19 (Fig. 4); in other words, the two rather different evaluation methods lead to the same result. This also holds for R* [eqn (8)] which with 100.60 (= 4.0) gives a formation degree of about 20% for the H·N7(HPMPA)N1± tautomer, in excellent agreement with the previous 18% (Section 3.5).
4. Conclusions and a further revealing observation
There is a further most interesting observation: for 9-methyladenine (9MeA) the following two micro acidity constants have been determined,64 that is, pkH·N7–N1H·N7–N1·H = 0.50 ± 0.08 and pkN7–N1N7–N1·H = 4.07 ± 0.08 (see Fig. 5 of ref. 64). These values are identical within their low error limits with the corresponding ones for HPMPA, which are pkH·N7–N1H·N7–N1·H = 0.47 (±1.39) and pkN7–N1N7–N1·H = 4.09 (±1.39) (see Fig. 5). In other words, 9MeA is a good mimic for the adenine part of HPMPA and therefore it is of relevance to note that the +H·N7(9MeA)N1 tautomer occurs with a formation degree of 7%.64It may therefore be concluded that the H·N7(HPMPA)N1± tautomer is certainly a minority species, but that it is definitely formed, the formation degree being between about 5 and 20%. Furthermore, the intrinsic basicity of N7 is quantified by the acidity constant pkN7–N1H·N7–N1 ≈ 3.5 (Fig. 4 and 5). This result is of relevance for the formation of macrochelates of phosph(on)ate-coordinated metal ions with N7.65–69 However, one has to be aware that these N7 interactions are weak and therefore easily affected by other weak metal ion interactions, e.g., with the ether oxygen, the ether oxygen in combination with N3, or the hydroxyl group of the –CH2–OH residue in M(HPMPA) (cf. ref. 29) and related complexes.45
Such very weak interactions provide pitfalls: To mention just one, which follows from the above observation29 that metal ions may interact with N3, but only in combination with the supporting ether oxygen–metal ion interaction, forming thus fused 5- and 7-membered chelate rings (cf.Fig. 1). This means, the phosphonate group of HPMPA2− is the primary binding site to which the metal ion is coordinated, followed by the ether oxygen and N3. In this context it is interesting to note that the acidic properties of N3(H)+ in the +H·N3(9MeA)N1,N7 tautomer have been estimated42 as pkN3–N1,N7H·N3(9MeA)N1,N7 ≈ 2.45. Using 9MeA as a mimic for HPMPA (see above) one may assume in a first approximation that its (N3)H+ acidity is approximately described by the mentioned micro acidity constant. Overall, the basicity of the adenine nitrogens decreases clearly in the order N1 > N7 > N3.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The support of this study by the Departments of Chemistry of the University of Basel and the University of Warwick is gratefully acknowledged.References
- In Interactions of Metal Ions with Nucleotides, Nucleic Acids, and Their Constituents, ed. A. Sigel and H. Sigel, Metal Ions in Biological Systems, Dekker, New York, 1996, vol. 32, pp. 1–814 Search PubMed.
- In Structural and Catalytic Roles of Metal Ions in RNA, ed. A. Sigel, H. Sigel and R. K. O. Sigel, Metal Ions in Life Sciences, Royal Society of Chemistry, Cambridge, 2011, vol. 9, pp. 1–391 Search PubMed.
- In Interplay between Metal Ions and Nucleic Acids, ed. A. Sigel, H. Sigel and R. K. O. Sigel, Metal Ions in Life Sciences, Springer, Dordrecht, 2012, vol. 10, pp. 1–353 Search PubMed.
- In Metallo-Drugs: Development and Action of Anticancer Agents, ed. A. Sigel, H. Sigel, E. Freisinger and R. K. O. Sigel, Metal Ions in Life Sciences, De Gruyter, Berlin, 2018, vol. 18, pp. 1–546 Search PubMed.
- R. L. Thompson, The effect of metabolites, metabolite antagonists, and enzyme inhibitions on the growth of vaccinia virus in Maitland type tissue cultures, J. Immunol., 1947, 55, 345–352 Search PubMed.
- R. L. Thompson, M. L. Price, S. A. Minton, Jr., G. B. Elion and G. H. Hitchings, Effects of purine derivatives and analogs on the multiplication of vaccinia virus, J. Immunol., 1950, 65, 529–534 CAS.
- I. Tamm, J. Bacteriol., 1956, 72, 42–53 CrossRef CAS PubMed.
- I. Tamm, K. Folkers and C. H. Shunk, J. Bacteriol., 1956, 72, 59–64 CrossRef CAS PubMed.
- A. Diwan, C. N. Gowdy, R. K. Robins and W. H. Prusoff, J. Gen. Virol., 1968, 3, 393–402 CrossRef CAS PubMed.
- S. R. Jenkins, F. W. Holly and R. K. Robins, J. Med. Chem., 1968, 11, 910 CrossRef CAS PubMed.
- A. Holý, E. De Clercq and I. Votruba, ACS Symp. Ser., 1989, 401, 51–71 CrossRef.
- A. Holý, Il Farmaco, 1991, 46(Suppl. 1), 141–146 Search PubMed.
- E. De Clercq, Pure Appl. Chem., 1998, 70, 567–577 CrossRef CAS.
- E. De Clercq, Collect. Czech. Chem. Commun., 1998, 63, 449–479 CrossRef CAS PubMed.
- S. Botros, S. William, L. Hammam, Z. Zidek and A. Holý, Antimicrob. Agents Chemother., 2003, 47, 3853–3858 CrossRef CAS PubMed.
- E. Devries, J. G. Stam, F. F. J. Franssen, H. Nieuwenhuijs, P. Chavalitshewinkoon, E. De Clercq, J. P. Overdulve and P. C. Vandervliet, Mol. Biochem. Parasitol., 1991, 47, 43–50 CrossRef CAS.
- R. Kaminsky, C. Schmid, Y. Grether, A. Holý, E. De Clercq, L. Naesens and R. Brun, Trop. Med. Int. Health, 1996, 1, 255–263 CrossRef CAS PubMed.
- P. Potmesil, M. Krecmerova, E. Kmonickova, A. Holý and Z. Zidek, Eur. J. Pharmacol., 2006, 540, 191–199 CrossRef CAS PubMed.
- E. De Clercq and G. D. Li, Clin. Microbiol. Rev., 2016, 29, 695–747 CrossRef PubMed.
- E. De Clercq, Med. Res. Rev., 2013, 33, 1278–1303 CrossRef CAS PubMed.
- E. De Clercq, A. Holý, I. Rosenberg, T. Sakuma, J. Balzarini and P. C. Maudgal, Nature, 1986, 323, 464–467 CrossRef CAS PubMed.
- M. G. Maghami, S. Dey, A.-K. Lenz and C. Höbartner, Angew. Chem., Int. Ed., 2020, 59, 9335–9339 CrossRef PubMed.
- H. Sigel, D. Chen, N. A. Corfù, F. Gregáň, A. Holý and M. Strašák, Helv. Chim. Acta, 1992, 75, 2634–2656 CrossRef CAS.
- H. Sigel, Coord. Chem. Rev., 1995, 144, 287–319 CrossRef CAS (Invited contribution to this special ‘Bioinorganic Issue’ of CCR).
- H. Sigel, J. Indian Chem. Soc., 1997, 74, 261–271 CAS (P. Ray Award Lecture).
- A. Sigel, B. P. Operschall and H. Sigel, Coord. Chem. Rev., 2012, 256, 260–278 CrossRef CAS (Invited contribution to the special issue devoted to the ‘European Group of Thermodynamics of Metal Complexes’).
- R. B. Gómez-Coca, C. A. Blindauer, A. Sigel, B. P. Operschall, A. Holý and H. Sigel, Chem. Biodivers., 2012, 9, 2008–2034 CrossRef PubMed (Invited contribution in the context of ICBIC-15).
- C. A. Blindauer, R. Griesser, A. Holý, B. P. Operschall, A. Sigel, B. Song and H. Sigel, J. Coord. Chem., 2018, 71, 1910–1934 CrossRef CAS (JCC issue devoted to Dan Meyerstein on the occasion of his 80th birthday).
- C. A. Blindauer, A. Holý, B. P. Operschall, A. Sigel, B. Song and H. Sigel, Eur. J. Inorg. Chem., 2019, 3892–3903 CrossRef CAS.
- H. Sigel, C. A. Blindauer, A. Holý and H. Dvořáková, Chem. Commun., 1998, 1219–1220 RSC.
- H. Sigel, B. Song, C. A. Blindauer, L. E. Kapinos, F. Gregáň and N. Prónayová, Chem. Commun., 1999, 743–744 RSC.
- H. Sigel, Pure Appl. Chem., 1999, 71, 1727–1740 CAS.
- H. Sigel, Chem. Soc. Rev., 2004, 33, 191–200 RSC.
- H. Sigel, Pure Appl. Chem., 2004, 76, 375–388 CrossRef CAS.
- H. Sigel and L. E. Kapinos, Coord. Chem. Rev., 2000, 200–202, 563–594 CrossRef CAS.
- C. A. Blindauer, A. Holý, H. Dvořáková and H. Sigel, J. Chem. Soc., Perkin Trans. 2, 1997, 2353–2363 RSC.
- H. Schwalbe, W. Thomson and S. Freeman, J. Chem. Soc., Perkin Trans. 1, 1991, 1348–1349 RSC.
- R. Tribolet and H. Sigel, Eur. J. Biochem., 1987, 163, 353–363 CrossRef CAS PubMed.
- R. B. Martin and Y. H. Mariam, Met. Ions Biol. Syst., 1979, 8, 57–124 CAS.
- K. Aoki, Met. Ions Biol. Syst., 1996, 32, 91–134 CAS.
- K. Aoki and K. Murayama, Met. Ions Life Sci., 2012, 10, 43–102 CrossRef CAS PubMed.
- L. E. Kapinos, B. P. Operschall, E. Larsen and H. Sigel, Chem. – Eur. J., 2011, 17, 8156–8164 CrossRef CAS PubMed.
- R. L. Benoit and M. Fréchette, Can. J. Chem., 1984, 62, 995–1000 CrossRef CAS.
- B. Song, A. Holý and H. Sigel, Gazz. Chim. Ital., 1994, 124, 387–392 CAS (GCI issue dedicated to the ‘Memory of Professor Luigi Sacconi’).
- A. Fernández-Botello, B. P. Operschall, A. Holý, V. Moreno and H. Sigel, Dalton Trans., 2010, 39, 6344–6354 RSC.
- R. B. Gómez-Coca, A. Sigel, B. P. Operschall, A. Holý and H. Sigel, Can. J. Chem., 2014, 92, 771–780 CrossRef (CJC issue in honor of A. B. P. (Barry) Lever).
- H. Dvořáková, A. Holý and I. Rosenberg, Collect. Czech. Chem. Commun., 1994, 59, 2069–2094 CrossRef.
- A. Holý and I. Rosenberg, Collect. Czech. Chem. Commun., 1987, 52, 2775–2791 CrossRef.
- A. Holý, Collect. Czech. Chem. Commun., 1993, 58, 649–674 CrossRef.
- C. A. Blindauer, Untersuchungen zu Stabilität, Struktur und Reaktivität von Metallionenkomplexen einiger antiviraler Nucleotid-Analoga, PhD thesis, University of Basel, Switzerland, 1998.
- C. A. Blindauer, A. Sigel, B. P. Operschall, A. Holý and H. Sigel, Inorg. Chim. Acta, 2018, 472, 283–294 CrossRef CAS (Special ICA issue devoted to Imre Sóvágó on the occasion of his 70th birthday).
- P. K. Glasoe and F. A. Long, J. Phys. Chem., 1960, 64, 188–190 CrossRef CAS.
- M. Bastian and H. Sigel, J. Coord. Chem., 1991, 23, 137–154 (Special JCC issue to honor Arthur E. Martell on the occasion of his 75th birthday) Search PubMed.
- H. Sigel, A. D. Zuberbühler and O. Yamauchi, Anal. Chim. Acta, 1991, 255, 63–72 CrossRef CAS.
- H. Dvořáková, A. Holý and P. Alexander, Collect. Czech. Chem. Commun., 1993, 58, 1403–1418 CrossRef.
- A. Holý and I. Rosenberg, Collect. Czech. Chem. Commun., 1987, 52, 2801–2809 CrossRef.
- C. Meiser, B. Song, E. Freisinger, M. Peilert, H. Sigel and B. Lippert, Chem. – Eur. J., 1997, 3, 388–398 CrossRef CAS.
- A. Mucha, B. Knobloch, M. Jeżowska-Bojczuk, H. Kozłowski and R. K. O. Sigel, Chem. – Eur. J., 2008, 14, 6663–6671 CrossRef CAS PubMed.
- O. V. Shishkin, L. Gorb, O. A. Zhikol and J. Leszczynski, J. Biomol. Struct. Dyn., 2004, 22, 227–243 CrossRef CAS PubMed.
- R. B. Martin, Acc. Chem. Res., 1985, 18, 32–38 CrossRef CAS.
- A. Fernández-Botello, R. Griesser, A. Holý, V. Moreno and H. Sigel, Inorg. Chem., 2005, 44, 5104–5117 CrossRef PubMed.
- R. B. Martin, Science, 1963, 139, 1198–1203 CrossRef CAS PubMed.
- R. B. Martin, Met. Ions Biol. Syst., 1979, 9, 1–39 CAS.
- G. Kampf, L. E. Kapinos, R. Griesser, B. Lippert and H. Sigel, J. Chem. Soc., Perkin Trans. 2, 2002, 1320–1327 RSC.
- H. Sigel, S. S. Massoud and R. Tribolet, J. Am. Chem. Soc., 1988, 110, 6857–6865 CrossRef CAS.
- G. Liang and H. Sigel, Inorg. Chem., 1990, 29, 3631–3632 CrossRef CAS.
- H. Sigel, S. S. Massoud and N. A. Corfù, J. Am. Chem. Soc., 1994, 116, 2958–2971 CrossRef CAS.
- H. Sigel and B. Song, Met. Ions Biol. Syst., 1996, 32, 135–205 CAS.
- E. M. Bianchi, S. A. A. Sajadi, B. Song and H. Sigel, Chem. – Eur. J., 2003, 9, 881–892 CrossRef CAS PubMed.
Footnote |
| † Deceased. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2022 |