
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMOF side chains as sources of supramolecular interactions: organic pollutant extraction from water†‡
Nizami
Israfilov
 a,
Karel
Soukup
b,
Benoît
Louis
*c and
Jean-Marc
Planeix
*a
a,
Karel
Soukup
b,
Benoît
Louis
*c and
Jean-Marc
Planeix
*a
aCNRS, CMC UMR 7140, Université de Strasbourg, 4 rue Blaise Pascal, F-67000, Strasbourg, France. E-mail: planeix@unistra.fr
bInstitute of Chemical Process Fundamentals of the Czech Academy of Sciences, Rozvojová 135, 16502, Prague 6, Czech Republic
cCNRS, ICPEES UMR 7515, Université de Strasbourg, 25 rue Becquerel, F-67087, Strasbourg, France. E-mail: blouis@unistra.fr
First published on 22nd April 2022
Abstract
Polyethylene glycol-modified terphenyl tetracarboxylic ligands were used for the preparation of copper-based MOFs: SUM-102 and SUM-103. The influence of side chains on the stability and adsorption capacities was investigated. Compared to the simpler analogue-NOTT-101, these MOFs, in particular SUM-103, exhibit high stability in water. Also, side chains do not affect the thermostability. The extraction capacity of organic cationic and anionic dyes was demonstrated by adsorption experiments carried out in water. SUM-103 showed a high Langmuir adsorption capacity of methylene blue in an aqueous solution (194 ± 4 mg g−1 at 30 °C).
The great variety of composition and structure and the continuous development of multiple synthetic pathways makes Metal–Organic Frameworks (MOFs) and their composites, as porous solids, are interesting materials for the capture and degradation of many types of chemicals such as organic pollutants.1–4 Although the driving forces in MOF developments are their price and simplicity of design, currently, two crucial characteristics dominate the generalization of the use of MOFs for substrate trapping and gas adsorption: their adsorption capacity and their stability, particularly in the presence of water or humidity.
Faced with the challenges of adsorption and trapping of organic molecules or gases, the functionalization of MOFs and more particularly the incorporation of active binding sites within their cavity appeared interesting.5,6 However, optimizing supramolecular interactions in MOFs can improve and broaden their applications.11–14
Since the pioneering work of Cram, Pedersen, and Lehn,7–9 the modulation of supramolecular interactions has been widely exploited to design more or less complex systems capable of strongly and selectively binding both ions and organic molecules of various sizes and shapes. The poly(ethyleneoxy) chains present in both crown ethers and cryptands have also been used to design various “open” versions of these compounds called podands (ex. Tripod) displaying good selectivity and phase transfer properties.10,11 Their ability to solubilize in water as well as in numerous organic solvents is one of their interesting characteristics.
Thus, all of these aforementioned points motivated us to graft similar side chains to the ligand in order to observe the behaviour of those groups inside the pores of MOF and test their potential applications.
The original MOFs prepared and studied here illustrate what can be achieved by the combination of a known MOF framework with sufficient porosity and the addition of lateral fragments designed to provide their recognition and supramolecular interaction properties.
As model molecules to test the adsorption of our MOFs, we have selected cationic and anionic dyes. As a consequence of the increasing demand for textile goods, papers, or plastic organic dyes,15 which are generally toxic to the environment and in some cases carcinogenic appear as a significant environmental challenge.15,16 Moreover, the structure and nature of methylene blue (MB) are similar to those of antidepressants such as imipramine and desipramine; therefore, the adsorption of dyes could open a possibility to use these MOFs further for drug elimination and also maybe for drug delivery.
First of terphenyl-tetracarboxylic backbone was used by Schroder et al.17 Later, different other groups worked on various functionalization of these ligands.18–21 An interesting approach developed by Hosseini et al.22 consisted to modify the backbone with chiral groups, for their use in enantiomeric separation.
In our study, we grafted glyme and diglyme moieties to the terphenyl tetracarboxylic skeleton. Ligands were identified by 1H, 13C NMR, LR-MS, and elemental analysis. The structure of the ligands and also simplified the synthesis scheme of MOFs are given in Fig. 1.
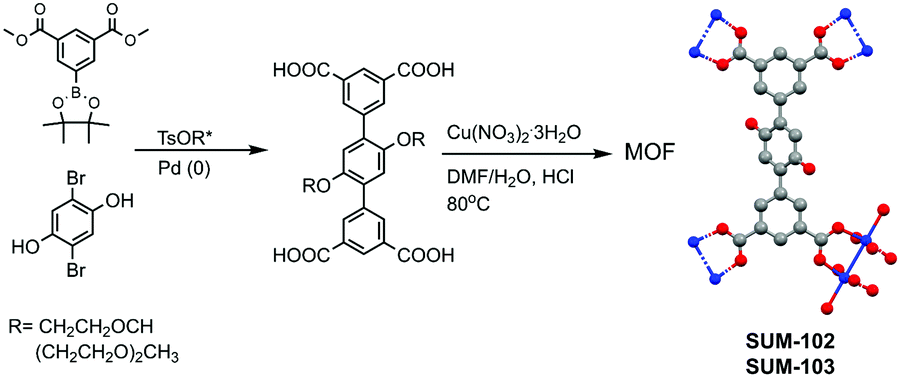 | ||
| Fig. 1 Simplified synthesis of ligands and MOFs. | ||
Detailed synthesis protocols and analyses could be found in ESI.‡ MOFs named SUM-102 (glyme) and 103 (diglyme), were obtained as microcrystalline powders and SUM-102 as a suitable crystal for single-crystal XRD. (SUM stands for Strasbourg University Materials). For these two MOFs, SCXRD and PXRD analysis show the same topology and connectivity mode of tetracarboxylic ligands as parent NOTT-101. However, the movement of large side chains made them impossible to be located. As expected, powder XRD of SUM-102 and SUM-103 showed an exact match with NOTT-101 (Fig. 2A).
 | ||
| Fig. 2 (A) Comparison of SUM-102 and SUM-103 to parent NOTT-101, also their stabilities in different conditions. (B) BET surface areas of SUM-102 and SUM-103. (C) SEM image of SUM-103. (D) Pore size distribution of SUM-103. Activation conditions: 8 h at 160 °C in vacuum. | ||
Furthermore, in Fig. 2A we compared water stability after heating under a vacuum of SUM-103 (8 h at 160 °C).
While stability in water or the presence of moisture is a strong limitation for the use of MOFs, we observe here that the presence of two ethyleneoxy units confers to the SUM-103, excellent water resistance. This effect is significantly weaker for SUM 102 for which the side chain consists of only one ethyleneoxy unit. Similarly, after heating in a vacuum for 8 h at 160 °C and re-exposure to ambient air, SUM-103 does not undergo any structural alteration observable on the powder diffractogram. In the case of SUM-102, a significant loss of crystallinity was observed. In stark contrast to SUM-103 (and to some extent to SUM-102), NOTT-101 crystallinity nearly completely vanished after activation (Fig. 2A). Schröder et al. for NOTT-101 and Zhou et al. for Cu2(TPTC-OMe) reported such degradation in the presence of water or humidity.17,18
Since the side chains could not be located by XRD, their presence was established by FT-IR analysis (see ESI‡). Indeed, C–H stretching vibrations (2890 cm−1) and the skeletal vibrations of aromatic rings or C–C–O chains (1200 and 1505 cm−1) demonstrated the presence of backbone and side chains.
The formation of MOFs can also be deduced from the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching band shifted to low energies due to the coordination of the carboxylate groups to Cu (1630 cm−1).
O stretching band shifted to low energies due to the coordination of the carboxylate groups to Cu (1630 cm−1).
Also, the Cu–O bond elongation band could be observed at 730 cm−1.23 A comparative analysis of the MOF and ligand shows a decrease or an absence of free carboxylate group linked peaks in MOF.
TGA analysis up to 450 °C under nitrogen of SUM-102 and SUM-103 (see ESI‡) are characterized by nearly the same pattern. The first phase of weight loss occurs in two waves of solvent evaporation, one below 50 °C for most volatile solvents and a second one between 70 °C and 200 °C for less volatile ones. The decomposition of MOFs started at 280 °C and 289 °C, respectively, in a comparable way with the parent NOTT-101. Therefore, the addition of oxygen-rich side chains does not influence the thermal stability of MOFs.
Their surface areas are obtained using Nitrogen and Argon adsorption–desorption isotherms measurements at 77 K (N2); 87 K (Ar). Before measurement, MOFs were activated at 160 °C in a vacuum for 8 h without solvent exchange.
Their nitrogen adsorption is displayed in Fig. 2B. Despite larger side chains of the H4L(diglyme)2 ligand, SUM-103 demonstrates a higher surface area. The BET surface area measured for SUM-102 is 869 m2 g−1 (N2) versus 846 m2 g−1 (Ar) and for SUM-103 is 1058 m2 g−1 (N2) versus 1016 m2 g−1 (Ar) and shows type I isotherm characteristic for purely microporous materials.
It can therefore be seen that the material with the longest side chains carrying two ethyleneoxy units has a greater adsorption capacity than the material with only one ethylenoxy unit in the side chains. A similar observation was explained by Zhou et al. as the pore splitting effect of large side chains, which creates an optimal “place” corresponding to the kinetic diameter of N2(Ar).18
The possible adaptation of the spatial arrangement within the pores due to the flexibility of these chains to optimize intermolecular interactions could be an explanation for this phenomenon.
Crystals of SUM-102 and SUM-103 are also observed via scanning electron microscopy. The micrograph in Fig. 2C shows a crystal size of 10–20 μm for SUM-103, as also observed for SUM-102.
The dye adsorption capacity of both MOFs was evaluated. In the case of SUM-103, both isothermal adsorption and kinetic studies were performed. The adsorption parameters have been determined in water (neutral pH) at 30 °C (for isotherm). Before adsorption tests, adsorbed DMF molecules were exchanged with water (24 h at room temperature). MOF was then filtered and dried under air for another 24 h.
Firstly, we compared both SUM-102 and SUM-103 under identical conditions. The values of dyes adsorption percentage of dye solutions (10 mL; 10 mg L−1) over 3 mg MOF after 24 h and at 30 °C are given in Table 1.
| SUM-102 (%) | SUM-103 (%) | |
|---|---|---|
| Alizarin yellow R | 64 | 77 |
| Methylene blue | 83 | 94 |
Those values show that methylene blue is more efficiently adsorbed than alizarin yellow R. Also, we observe a significant increase in adsorption while increasing the side chain length. The study of the adsorption of methylene blue (MB) on SUM-103 was extended by performing measurements of isothermal adsorption capacity and a kinetic study, which are available in ESI.‡ The adsorption protocol was carried out using the batch method by adding 20, 50, 100, and 200 ppm methylene blue solutions over 3 mg MOF in different vials (10 mL) and then placed in the oven at 30 °C. After the adsorption of MB, the colour of the MOFs changed from blue to deep blue. The adsorption of MB on MOFs was also assessed by the presence of characteristic strong bands of MB in the FT-IR spectrum of final materials (Fig. 3). After 24 h, the supernatant of the MB solution was analyzed using a UV-VIS spectrophotometer at 664 nm.
 | ||
| Fig. 3 (From left to the right, SUM-103) Adsorption kinetics, 2.5 mL, 15 mg L−1 MB solution after 24 h at room temperature, FT-IR comparison before and after adsorption. | ||
The isothermal adsorption capacities were modeled according to Langmuir and Freundlich models. It is observed that the quantity of methylene blue that binds to the material is 194 ± 4 mg g−1 (Langmuir) for SUM-103.
By comparison, SUM-103, therefore, appears as a MOF exhibiting a significant adsorption capacity while exhibiting an average specific surface area (Table 2).
The kinetic study of adsorption was carried out at room temperature. For kinetics studies, 3 mg SUM-103 was placed in a quartz cell, later 2.5 mL, 15 mg L−1 methylene blue solution was added. At defined time intervals, the concentration of methylene blue was measured by UV-vis spectroscopy.
Pseudo-second order kinetic model fits the experimental data suitably. This validates that the adsorption is mostly chemisorption, rather than physisorption in nature. As expected, at low methylene blue concentration, it is possible to achieve nearly quantitative adsorption rates. Thus, after 48 h at 25 °C, 99.8% adsorption was observed for 2.5 mL of MB solution with a concentration of 8.7 mg L−1 over 1.8 mg of MOF, which seems consistent with the use of the model Langmuir adsorption.
In conclusion, new series of MOFs, SUM as Strasbourg University Materials have been prepared using new original ligands. Designed for supramolecular interactions, ligand side-chains are composed of ethylene glycol moieties. These MOFs exhibit useful properties without damageable reduction of their adsorption capacity. A single crystal study shows that the “primary structure” of NOTT MOFs is maintained. X-Ray diffraction patterns and TGA analysis shows their stability in air and water.
The amphiphilic character of the pores introduced by the ethylene glycol chains gave birth to specific stability to these solids in water and thus a strong potential of use for extracting organic pollutants from water.
As a consequence SUM-103 could be used in water to extract organic molecules such as methylene blue (cationic) or alizarin yellow R (anionic). Also, the comparative study of SUM-102 and SUM-103 showed higher values with the latter (83 vs. 94%, under the same conditions), thus highlighting the importance of supramolecular interactions of the side chains. Compared to other MOFs, and taking into account their surface area, the methylene blue uptake demonstrates that functionalization by fragments capable of supramolecular interactions with a substrate could be an interesting way for MOF adsorption modulation. Further studies of gas adsorption are currently under investigation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We express our gratitude to the Ministry of Foreign Affairs of France for providing the scholarship for the PhD project. Also, we would show appreciation to Nathalie Gruber for providing support for XRD studies and to Audrey Fluck for her support. Finally, we thank Pr Mir Wais Hosseini for his valuable advice.Notes and references
- S. Rojas, J. A.-R. Navarro and P. Horcajada, Dalton Trans., 2021, 50, 2493 RSC.
- K. A. Adegoke, O. S. Agboola, J. Ogunmodede, A. O. Araoye and O. S. Bello, Mater. Chem. Phys., 2020, 253, 123246 CrossRef.
- W. Ren, J. Gao, C. Lei, Y. Xie, Y. Cai, Q. Ni and J. Yao, Chem. Eng. J., 2018, 349, 766–774 CrossRef CAS.
- L. Wang, H. Xu, J. Gao and Q. Zhang, Coord. Chem. Rev., 2019, 398, 213016 CrossRef.
- T. D. Duong, S. A. Sapchenko, I. da Silva, H. G.-W. Godfrey, Y. Cheng, L. L. Daemen, P. Manuel, M. D. Frogley, G. Cinque, A. J. Ramirez-Cuesta, S. Yang and M. Schröder, Chem. Sci., 2020, 11, 5339–5346 RSC.
- K. Shen, L. Qin and H.-G. Zheng, Dalton Trans., 2016, 45, 16205 RSC.
- J. M. Timko, R. G. Helgeson, M. Newcomb, G. W. Gokel and D. J. Cram, JACS, 1974, 96(22), 7097–7099 CrossRef CAS.
- J.-M. Lehn, Pure Appl. Chem., 1980, 52(11), 2441–2459 CAS.
- C. J. Pedersen, JACS, 1967, 89(26), 7017–7036 CrossRef CAS.
- G. Soula, J. Org. Chem., 1985, 50(20), 3717–3721 CrossRef CAS.
- A. Perrin, D. Myers, K. Fucke, O. M. Musa and J. W. Steed, Dalton Trans., 2014, 43, 3153–3161 RSC.
- J. Tian, Z.-Y. Xu, D.-W. Zhang, H. Wang, S.-H. Xie, D.-W. Xu, Y.-H. Ren, H. Wang, Y. Liu and Z.-T. Li, Nat. Commun., 2016, 7, 11580 CrossRef CAS PubMed.
- D. Balestri, P. P. Mazzeo, R. Perrone, F. Fornari, F. Bianchi, M. Careri, A. Bacchi and P. Pelagatti, Angew. Chem., Int. Ed., 2021, 60, 10194–10202 CrossRef CAS PubMed.
- G.-F. Hua, X.-J. Xie, W. Lu and D. Li, Dalton Trans., 2020, 49, 15548–15559 RSC.
- N. K. Gupta, Y. Ghaffari, S. Kim, J. Bae, K. S. Kim and M. Saifuddin, Sci. Rep., 2020, 10, 4942 CrossRef CAS PubMed.
- S. S. Auerbach, D. W. Bristol, J. C. Peckham, G. S. Travlos, C. D. Hébert and R. S. Chhabra, Food Chem. Toxicol., 2010, 48, 169–177 CrossRef CAS PubMed.
- X. Lin, J. Jia, X. Zhao, K. M. Thomas, A. J. Blake, G. S. Walker, N. R. Champness, P. Hubberstey and M. Scröder, Angew. Chem., Int. Ed., 2006, 45, 7358–7364 CrossRef CAS PubMed.
- T. A. Makal, X. Wang and H.-C. Zhou, Cryst. Growth Des., 2013, 13, 4760–4768 CrossRef CAS.
- H.-M. Wen, G. Chang, B. Li, R.-B. Lin, T.-L. Hu, W. Zhou and B. Chen, Cryst. Growth Des., 2017, 17, 2172–2177 CrossRef CAS.
- T. Xia, J. Cai, H. Wang, X. Duan, Y. Cui, Y. Yang and G. Qian, Microporous Mesoporous Mater., 2015, 215, 109–115 CrossRef CAS.
- Q. Zhang, J. Yu, J. Cai, R. Song, Y. Cui, Y. Yang, B. Chen and G. Qian, Chem. Commun., 2014, 50, 14455–14458 RSC.
- D. Asnaghi, R. Corso, P. Larpent, I. Bassanetti, A. Jouaiti, N. Kyritsakas, A. Comotti, P. Sozzani and M. W. Hosseini, Chem. Commun., 2017, 53, 5740 RSC.
- S. Lin, Z. Song, G. Che, A. Ren, P. Li, C. Liu and J. Zhang, Microporous Mesoporous Mater., 2014, 193, 27–34 CrossRef CAS.
- M. R. Azhar, H. R. Abid, H. Sun, V. Periasamy, M. O. Tadé and S. Wanga, J. Colloid Interface Sci., 2017, 490, 685–694 CrossRef CAS PubMed.
- E. Haque, V. Lo, A. I. Minett, A. T. Harris and T. L. Church, J. Mater. Chem. A, 2014, 2, 193 RSC.
- A. A. Mohammadi, A. Alinejad, B. Kamarehie, S. Javan, A. Ghaderpoury, M. Ahmadpour and M. Ghaderpoori, Int. J. Environ. Sci. Technol., 2017, 14, 1959–1968 CrossRef CAS.
Footnotes |
| † In memory of Guy Ourisson. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nj00273f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2022 |