DOI:
10.1039/D2NJ00088A
(Paper)
New J. Chem., 2022,
46, 5593-5605
Environmentally friendly catechol-based synthesis of dibenzosultams†
Received
6th January 2022
, Accepted 24th February 2022
First published on 25th February 2022
Abstract
Sulfonamides, such as cyclic sultams, are widely used structural blocks in drug design. Here, an environmentally friendly route of synthesis is reported for sultams containing a catechol function in the secondary aromatic ring (ring B), making use of water as a solvent. The reaction can also be paired with THF for enhanced solubility. It occurs by the application of a base and heat and proceeds through what is presumably an oxidation of the catechol function followed by nucleophilic addition by the nitrogen of the sulfonamide. The reaction is robust and reproducible, as demonstrated by the successful synthesis of eight different sultams with significant variation possible in both the primary aromatic ring (ring A) and in the group attached to the sultam nitrogen, with isolated yields of 43–86% after purification.
Introduction
Sulfonamide derivatives, including the cyclic variants known as sultams, are known to exhibit a variety of biological activities, making them important building blocks in the study of multi-target drugs. They are widely used in drug design, such as in commercially available drugs from the oxicam family—a class of non-steroidal anti-inflammatory drugs,1 each of which contain a six-membered benzosultam.2,3 Because of the important role of sultams in medicinal chemistry, numerous pathways for the synthesis of bicyclic, tricyclic, tetracyclic and pentacyclic sultams have been developed, including sulfonylation reactions, alkylation reactions, Diels-Alder reactions and transition metal-catalysed reactions, among others.4,5
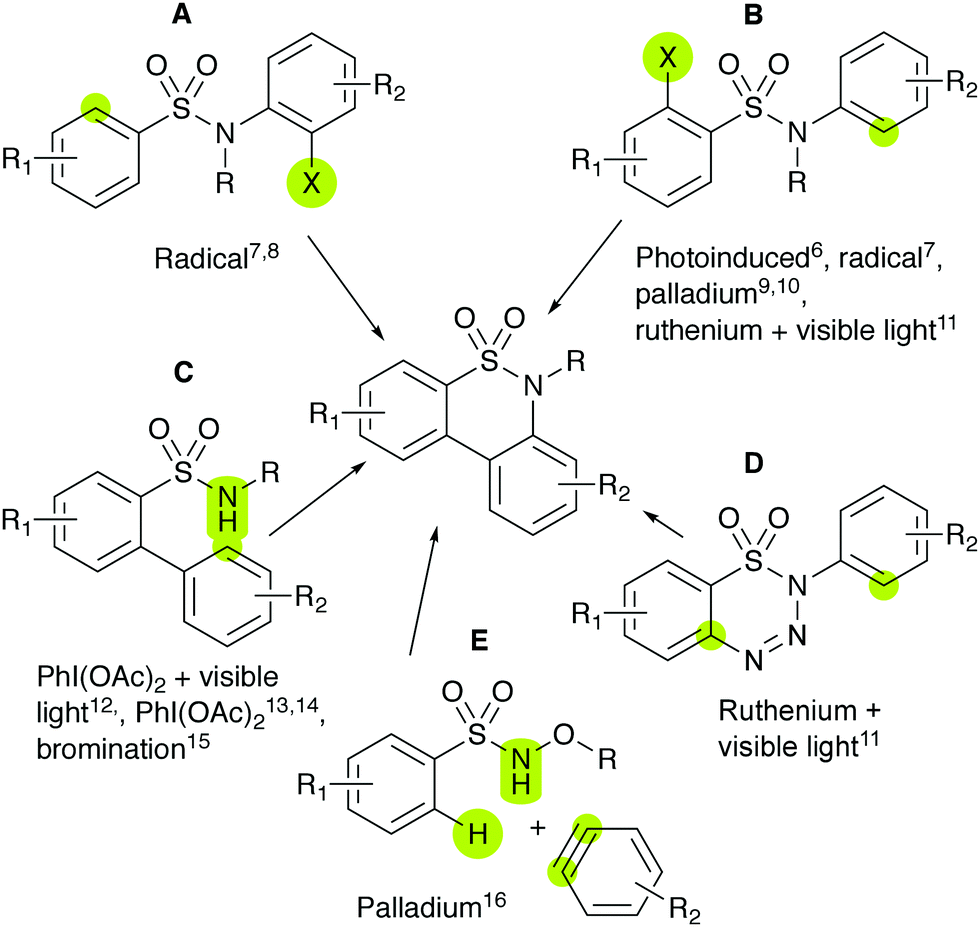
Recently, bicyclic sultams, such as benzosultams, have seen extensive method development. For tricyclic sultams, such as dibenzosultams, the synthetic approaches are less developed;5,6 some representative examples are shown in Scheme 1.
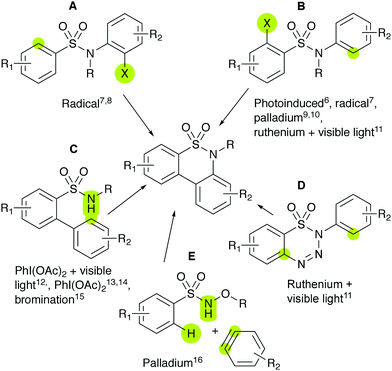 |
| | Scheme 1 Different published routes for dibenzosultam synthesis. Coloured sites are attached to form products. | |
Sultams were derived from structure A via radical cyclisation (X = Br)7 (X = I, R = Me).8 Structure B was reacted through photoinduced intramolecular arylation (X = Cl, Br),6 radical cyclisation (X = Br),7 palladium-catalysed cyclisation (X = Br)9 (X = R = H)10 and visible-light-promoted ruthenium-catalysed cyclisation (X = NH2).11 Structure C was cyclised to form sultams through either iodine-based oxidative C–H amination with (R = Me)12 or in the absence of visible light,13 tandem C–H amination and bromination with iodine-based oxidation14 or dibromohydantoin,15 thereby obtaining both the monobrominated and dibrominated variants. Structure D was reacted by intramolecular visible-light-promoted ruthenium-catalysed cyclisation,11 whilst structure E by intramolecular palladium-catalysed annulation with benzyne and N–O bond cleavage, leaving the N–H dibenzosultam.16
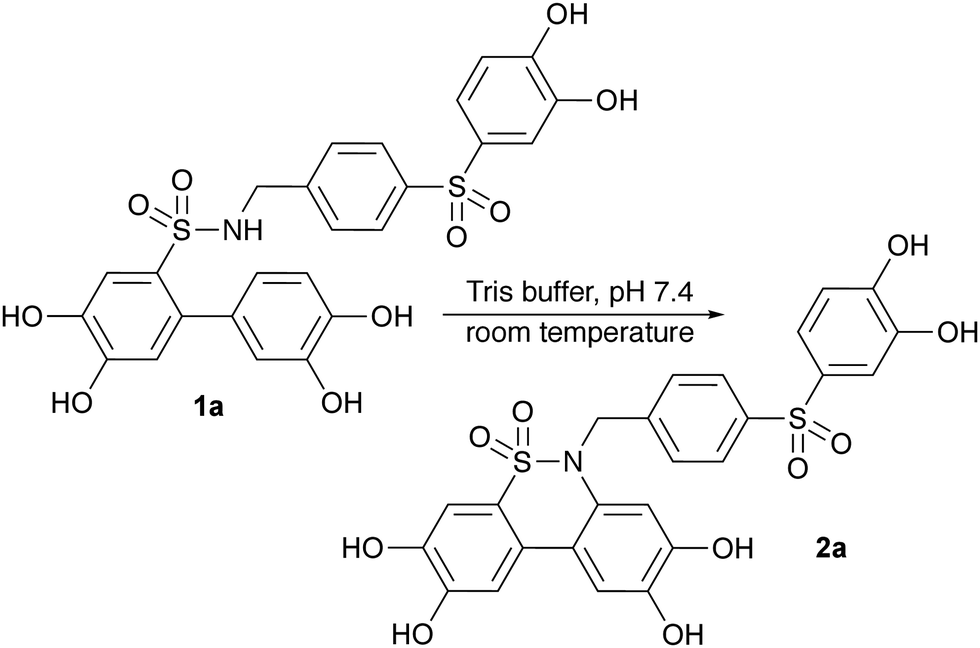
None of these methodologies have shown effectiveness for phenol- and catechol-containing substances or call for the use of ether protecting groups.7,8,10–16 Some reactions also require costly materials, such as transition metal catalysts.9–11,16 In this work, a new method is presented for synthesising dibenzosultams, which makes use of catechol functionalities as initiators for the cyclisation of sulfonamides under very mild conditions, in either an organic or aqueous solution. The reaction was discovered while determining the stability of biologically active compounds 1a and 2a in Tris buffer solution, prompting further exploration.
Results and discussion
The cyclisation reaction was initially observed with 10 nmol of 1a in a 1 mL solution of Tris buffer of 50 mM Tris–HCl, 15 mM KCl and 5 mM MgCl2 in MQ H2O, adjusted with K2CO3 to pH 7.4 at 19 °C (Scheme 2).
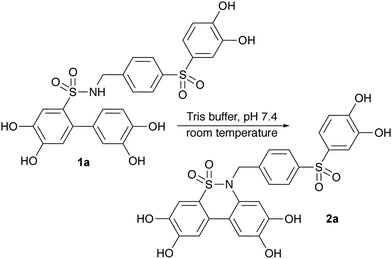 |
| | Scheme 2 Initially observed reaction with 1a in Tris buffer with pH 7.4 at 19 °C. | |
From here, the effects of buffer composition, the pH and the temperature were investigated. We designated 1g as the model substrate to screen different conditions. The contribution of different buffer components to the efficiency of the reaction was investigated by removing one component from the original buffer system to generate each composition, using HPLC to monitor each reaction progression by measuring the fraction of starting material at different time points (Fig. 1A).
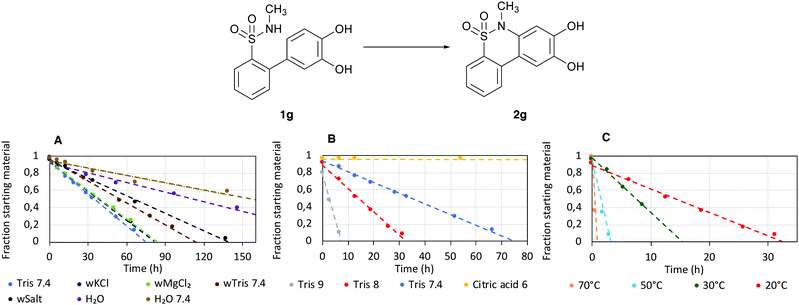 |
| | Fig. 1 Reaction progression for different conditions, (w = without). The reactions were made on a 100 μM scale and were monitored by HPLC. The buffers used are Tris 9, 8 and 7.4: MQ H2O, 50 mM Tris–HCl, 15 mM KCl, 5 mM MgCl2, buffer pH adjusted to pH 9, 8 and 7.4 with K2CO3, Citric acid 6: MQ H2O, 50 mM citric acid, 15 mM KCl, 5 mM MgCl2, buffer pH adjusted to pH 7.4 with NaOH, wTris 7.4: MQ H2O, 15 mM KCl, 5 mM MgCl2, buffer pH adjusted to pH 7.4 with K2CO3, wMgCl2: MQ H2O, 50 mM Tris–HCl, 15 mM KCl, buffer pH adjusted to pH 7.4 with K2CO3, wKCl: MQ H2O, 50 mM Tris–HCl, 5 mM MgCl2, buffer pH adjusted to pH 7.4 with K2CO3, wSalt: MQ H2O, 50 mM Tris–HCl, buffer pH adjusted to pH 7.4 with K2CO3. H2O: MQ H2O (assumed pH 7), H2O 7.4: MQ H2O, pH adjusted to pH 7.4 with K2CO3. (A) The reaction progressions for TRIS 7.4, wTris 7.4, wMgCl2, wKCl, wSalt, H2O and H2O 7.4 at 19 °C. (B) The reaction progressions for Tris 7.4, 8, 9 and Citric acid 6 at 19 °C. (C) The reaction progressions for Tris 8 at different temperatures. | |
This was done for all the following reactions on this scale. Then, the effects of the pH and temperature were investigated in a similar fashion (Fig. 1B and C).
The cyclisation reaction was accelerated by the presence of potassium and magnesium chloride salts, as well as by a buffered system (Fig. 1A). In addition, the pH of the reaction mixture had a significant effect on the reaction rate. Under slightly acidic conditions (pH 6), hardly any reaction occurred. Under slightly basic conditions (pH 7.4), the reaction was considerably faster, completing within 75 min (Fig. 1B). However, degradation of the product was observed at pH 9 (data not shown). Elevated temperature also increased the reaction speed, achieving a reaction time of 1 h for compound 1g (Fig. 1B). Furthermore, at 90 °C, severe degradation of the product was observed (data not shown). The optimal conditions for 1g were thus found to be in Tris buffer with pH 8 at 70 °C (a chromatogram of the reaction progression is shown in Fig. S1, ESI†).
Thereafter, the scope of the cyclisation reaction was explored by exposing suitable starting materials to the conditions identified in the section above. The compounds were selected to determine the effect of three different factors: (1) the effect of the substituent attached to the sulfonamide nitrogen, (2) the effects of substituents on ring A and (3) the effects of substituents on ring B (Scheme 3).
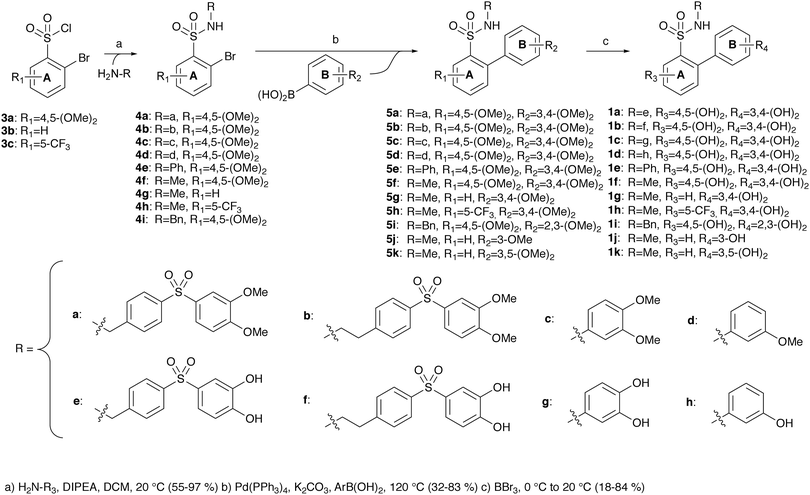 |
| | Scheme 3 Routes of synthesis for target compounds (1a–k). | |
The target compounds were synthesised through a three-step process (Scheme 3). Sulfonyl chlorides 3 were reacted with various amines (synthesis of the amines for R = a, b in Scheme S1, ESI†) to generate sulfonamides 4. Subsequently, Suzuki couplings using boronic acid derivatives were used to introduce ring B, furnishing compounds 5. Finally, the methoxy groups were converted to phenols through treatment with boron tribromide. The target compounds 1 were all obtained in moderate to good yields.
The reaction scope was firstly explored on a small scale of 0.1 μmol. Each target compound was exposed to the previously specified conditions. The reaction progression was monitored using HPLC, and the time for complete conversion of starting material into product was measured (Scheme 4).
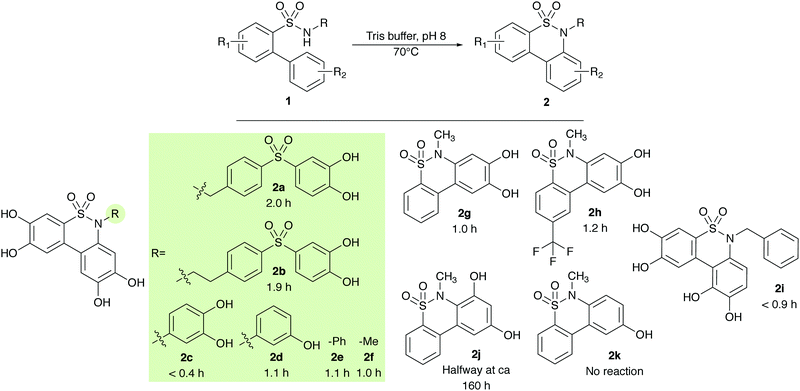 |
| | Scheme 4 Time for full conversion of starting material to product when exposed to Tris buffer at 70 °C and pH 8. | |
The reaction proceeded to completion for all R-substituents (2a–f), and the type of substituent used was found to effect the reaction speed. Compound 2c yielded the shortest reaction time of less than 0.4 h; compounds 2d–f were produced at similar speeds, with reaction times of approximately 1 h; and compounds 2a–b exhibited the longest reaction times of approximately 2 h (Scheme 4). The same was observed for variations in ring A (2f–h). The data indicated slightly increased reaction times for electron-withdrawing substituents and reduced reaction times for electron-donating substituents. In ring B, the substitution was determined to be of major importance, with only the catechol function allowing for short reaction times of around 1 h on this scale (2g, i). The resorcinol derivative (2j) reacted but at a very slow rate, with only about 50% conversion of the starting material after 160 h. With only one phenol group (2k), no reaction was observed. The reaction scope was therefore assumed to be limited to a catechol functionality in ring B.
To determine whether the reaction was scalable, the reaction conditions were further optimised. As many organic compounds are poorly soluble in H2O, the addition of other solvents was investigated by adding water miscible solvents to the Tris buffer with pH 8 (Fig. 2).
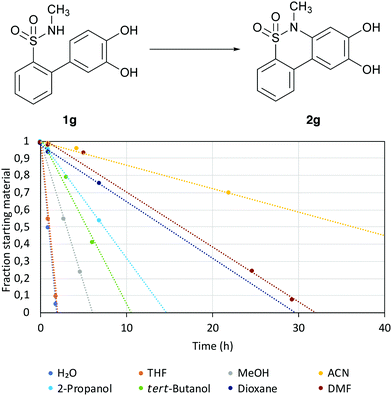 |
| | Fig. 2 Reaction progression in different solvents. The reactions were made at a 100 μM scale with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of Tris 8 to the solvent at 70 °C. :1 ratio of Tris 8 to the solvent at 70 °C. | |
Each solvent was mixed with an equal amount of Tris buffer with pH 8, followed by the addition of compound 1g. As a baseline, H2O was also included as one of the solvents, since the pH and salt concentrations in the solution would change with the additional solvent. The solvent with the highest effectiveness was determined to be THF, followed by MeOH.
To investigate if the reaction would proceed in THF, 0.1 μmol of compound 1g in DMSO stock solution was diluted in THF and monitored. At 70 °C, full conversion of 1g to 2g was observed in under 45 h. This shows that the reaction can also be run in an organic solvent.
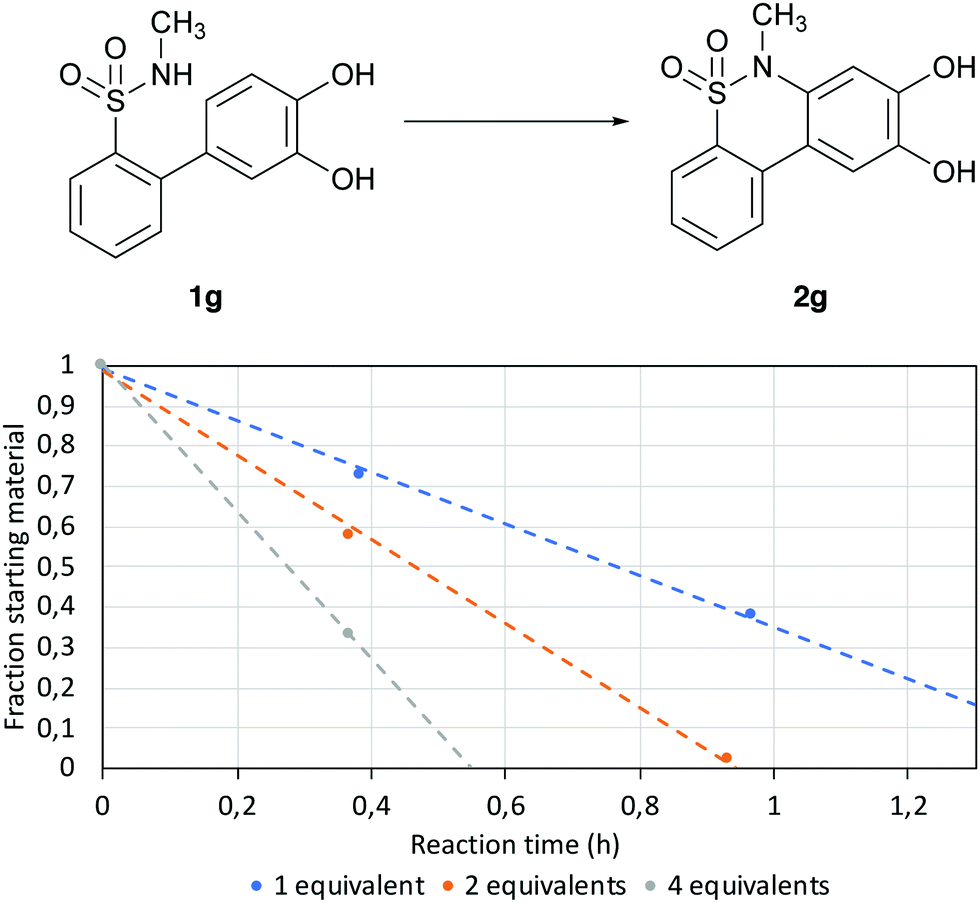
To avoid the use of large amounts of buffered solution it was investigated whether K2CO3 in MQ H2O could yield similar reaction times as Tris 8. This was performed by exposing 1g to different equivalents of K2CO3 in MQ H2O (Fig. 3).
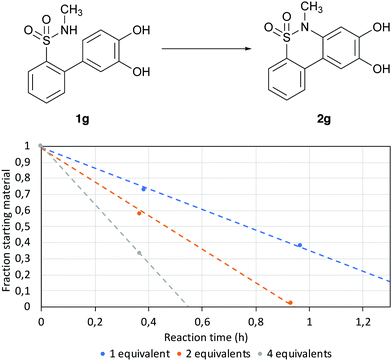 |
| | Fig. 3 Reaction progression for different equivalents of K2CO3. The reactions were made with 100 μM of 1g in H2O at 70 °C with different equivalents of K2CO3. | |
The results showed that the reaction speed was increased by adding higher amounts of base to the solution, achieving a reaction time of approximately 30 min when using four equivalents of K2CO3; thus, these conditions were then used for all the following experiments.
Compounds 1a, c–i were cyclised in H2O under microwave radiation at 70 °C at a scale of 0.027–0.11 mmol (9.9–41 mg), generating compounds 2a, c–i in 43–65% yield after acidic work up and purification (Table 1).
Table 1 Time of reaction and isolated yields for cyclisation reaction in H2O with 4 equivalents K2CO3 and heating by microwave irradiation at 70 °C
|
|
| Entry |
Substrate |
Time (h) |
Product |
| Number |
Isolated yielda (%) |
|
Isolated yields after acidic workup and purification by preparative HPLC.
Only purified by acidic workup.
|
| 1 |
1a
|
3.7 |
2a
|
86b |
| 2 |
1c
|
2.7 |
2c
|
59 |
| 3 |
1d
|
0.38 |
2d
|
63 |
| 4 |
1e
|
2.2 |
2e
|
43 |
| 5 |
1f
|
2.2 |
2f
|
59 |
| 6 |
1g
|
2.3 |
2g
|
65 |
| 7 |
1h
|
2.8 |
2h
|
55 |
| 8 |
1i
|
0.83 |
2i
|
58 |
All the reactions were observed to proceed to completion, and the variation in yield was most likely affected by workup and purification. This can be supported by the yield for entry 1 (Table 1) which was marginally higher, from only being worked up and not purified by preparative HPLC.
Compound 1g was also cyclised in a solvent mixture of THF and H2O in a ratio of 20:1 by microwave radiation at 70 °C, producing 2g in 50% yield after purification; this condition is applicable for chemicals that are less readily solvable in H2O. It was also cyclised at a scale of 1.0 mmol (279 mg), in H2O and THF in a ratio of 4:1 under microwave radiation at 70 °C, with the aid of air bubbled through the solution. This produced 2g in a yield of 78% after 8 h, which shows that the reaction is further scalable.
Cyclisation of sulfonamides to sultams has previously been reported to proceed through radical oxidation.12–15 To determine if our sulfonamides cyclise though a radical mechanism, radical scavengers (TEMPO, BHT and DABCO) were added to the reaction. However, none of the three radical scavengers had any effect on the reaction rate (data not shown), which indicates that the mechanism is not radical-based.
Considering this, the reaction is most probably mediated through a Michael-style addition of the sulfonamide nitrogen to the o-quinone. Catechol-containing compounds such as dopamine are well-reported in literature to undergo auto-oxidation in the presence of O2 to form the corresponding o-quinones.17–19 These are then able to react with nucleophiles such as amines, either condensing into imines at the ipso-position of the carbonyls or adding via 1,4-addition at the unsaturated carbons and re-aromatisation driven by proton abstraction to reform the catechol, which is favoured by basic conditions.17 The latter mode of reaction has been utilised for purposes such as regio-selective addition of guanosine residues to estrogen-3,4-quinones18 and spontaneous ring-closing and subsequent oxidation of dopamine to afford 5,6-dihydroxyindole in a Tris buffer system.19
This suggested mechanism was further investigated in the present research, using cyclic voltammetry, to study the redox behaviour of model compound 1g. The results indicated a clear anodic peak, corresponding to oxidation, but no cathodic peak (Fig. S2, ESI†). This is a well-known feature of catechol systems and has been reported several times when the nucleophile concentration is equal to that of the catechol.20,21 In this case, since the reaction is intramolecular, the compound could quickly cyclise, which would reduce the amount of available o-quinone and thus the extent of the backward reaction.
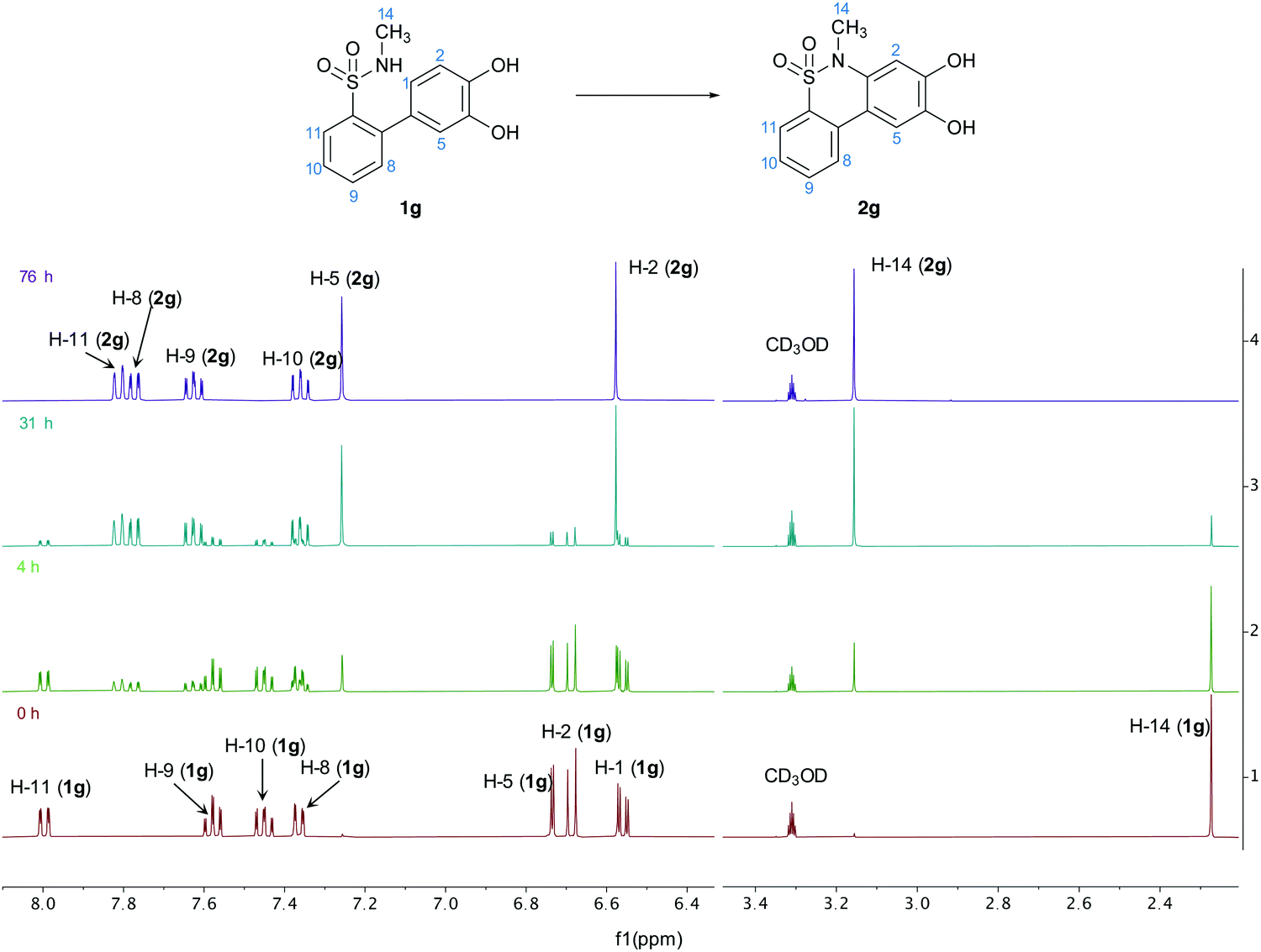
To support this, the reaction was run in deuterated methanol with KOH and monitored using NMR (Fig. 4).
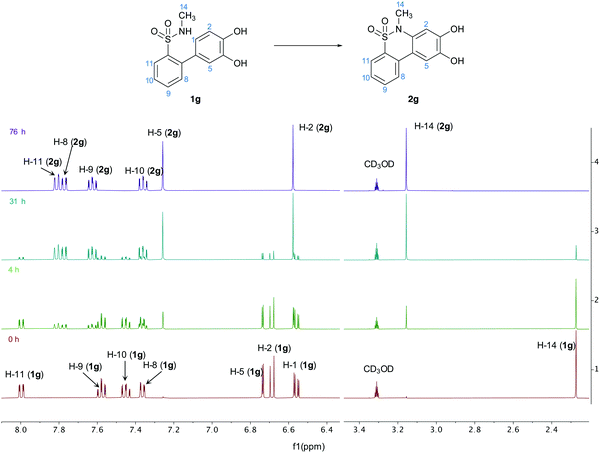 |
| | Fig. 4 Complete 1H-NMR spectra of the cyclisation reaction of 1g in deuterated methanol at different time points, with the peaks assigned for both structures 1g and 2g. | |
The spectra indicate that no intermediate was accumulated during the reaction and that the reactant was steadily and directly converted to the product. This further indicates that the mechanism occurs though a one-step oxidation reaction, in accordance with the results from the cyclic voltammetry.
Since the proposed mechanism is aided by oxygen gas, its effect on the reaction speed was investigated. To determine whether the reaction was promoted by dissolved oxygen gas, the reaction speed was measured for both the system in air and for the system purged with nitrogen (Fig. 5).
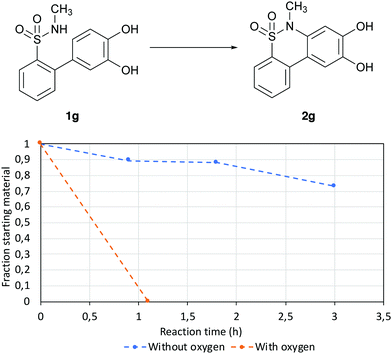 |
| | Fig. 5 Reaction progression for reaction with oxygen (air) and without oxygen (solution degassed and nitrogen atmosphere). | |
It was found that the presence of oxygen gas greatly increased the speed of the reaction, further indicating that the reaction proceeds in the proposed manner.
Conclusions
We report an environmentally friendly route for the synthesis of dibenzosultams in H2O (or THF if required for solubility), with potassium carbonate as a base. This reaction was demonstrated to work on a synthetically useful scale of up to 0.11 mmol (41 mg) on eight compounds; a scale of 1 mmol (279 mg) was used for model compound 1g, with the aid of air bubbled through the solution, to show further scalability. While ring B is limited to a catechol function, ring A and the chain attached to the sulfonamide nitrogen can be widely varied, making the reaction applicable for a large variety of molecules.
Experimental section
General information
Unless otherwise specified, all reagents were purchased from commercial sources and used as supplied. Microwave reactions were performed in a Biotage Initiator Reactor using single mode irradiation with temperature and pressure control and with fixed hold time on. Reactions were monitored by LC-MS (PerkinElmer Series 200; waters symmetry C8 column 3.5 μm, 4.6 × 50 mm; H2O/ACN (0.1% formic acid)), by thin-layer chromatography (TLC) on silica-gel-coated aluminium foils (silica gel 60 F254, Merck) and by analytical HPLC (Waters 2690 Separations Module; Atlantis® T3 5 μm column, 4.6 × 250 mm; H2O/ACN (0.1% TFA)). The TLC plates were visualized by UV light (λ = 254 nm). Purification by flash column chromatography was performed on a Biotage Isolera or Selekt instrument using prefabricated silica columns. In some cases, preparative HPLC (Waters 600 Controller; Atlantis® Prep T3 5 μm; H2O/ACN (0.1% TFA)) was used for purification. Melting points were determined with a Büchi B-545 apparatus. NMR spectra were recorded on a Varian 400 MHz NMR spectrometer at 25 °C. All chemical shifts are reported in parts per million (δ) relative to the residual solvent peak. The following abbreviations are used to denote signal patterns: (s) singlet, (d) doublet, (t) triplet, (q) quartet, (m) multiplet, and (br) broad, unless otherwise noted. Coupling constants (J) are reported in Hertz (Hz). High resolution mass spectra (HRMS) were recorded on an Agilent 1290 infinity LC system tandem to an Agilent 6520 Accurate Mass Q-TOF spectrometer.
Reaction optimization.
In a sealed 1.5 mL vial, 50 μL of a 2 mM stock solution of 1g in DMSO was mixed with 950 μL of each buffer and was heated using a heat block for the temperatures above 19 °C. The progress of the reaction was monitored by HPLC.
Aqueous buffers used.
Tris 9, 8 and 7.4.
MQ H2O, 50 mM Tris–HCl, 15 mM KCl, 5 mM MgCl2, buffer pH adjusted to pH 9, 8 and 7.4 with K2CO3.
Citric acid 6
.
MQ H2O, 50 mM citric acid, 15 mM KCl, 5 mM MgCl2, buffer pH adjusted to pH 7.4 with NaOH.
wTris 7.4
.
MQ H2O, 15 mM KCl, 5 mM MgCl2, buffer pH adjusted to pH 7.4 with K2CO3.
wMgCl2
.
MQ H2O, 50 mM Tris–HCl, 15 mM KCl, buffer pH adjusted to pH 7.4 with K2CO3.
wKCl
.
MQ H2O, 50 mM Tris–HCl, 5 mM MgCl2, buffer pH adjusted to pH 7.4 with K2CO3.
wSalt
.
MQ H2O, 50 mM Tris–HCl, buffer pH adjusted to pH 7.4 with K2CO3.
H2O
.
MQ H2O (assumed pH 7).
H2O 7.4
.
MQ H2O, pH adjusted to pH 7.4 with K2CO3.
The effect of buffer compositions was investigated using buffers Tris 7.4, wTris 7.4, wMgCl2, wKCl, wSalt, H2O and H2O 7.4 at 19 °C, the effect of the pH of the buffer using Tris 9, 8, 7.4 and Citric acid 6 at 19 °C and the effect of temperature using Tris 8 at 19 °C, 30 °C, 50 °C and 70 °C.
Substrate scope.
In a sealed 1.5 mL vial, 50 μL of a 2 mM stock solution of each compound in DMSO was mixed with 950 μL of Tris 8 and was heated to 70 °C. The progress of the reaction was monitored by HPLC.
Mix of aqueous buffer and organic solvents.
In a sealed 1.5 mL vial, 50 μL of a 2 mM stock solution of 1g in DMSO was mixed with 475 μL of Tris 8 and 475 μL of each solvent (H2O, THF, MeOH, ACN, 2-propanol, tert-butanol, dioxane and DMF) and was heated to 70 °C. The progress of the reaction was monitored by HPLC.
THF without buffer.
In a microwave vial 1.8 mL of THF and 100 μL of 2 mM stock solution of 1g in DMSO was added and stirred at 70 °C. The progress of the reaction was monitored by HPLC (reaction aliquot:ACN, 1:2).
Water with K2CO3.
In a sealed 1.5 mL vial, 50 μL of a 2 mM stock solution of 1g in DMSO was mixed with 25 μL, 50 μL and 100 μL of 4 mM K2CO3 in MQ H2O, and 925 μL, 900 μL and 850 μL respectively of MQ H2O was added for a total volume of 1 mL and was heated to 70 °C. The progress of the reaction was monitored by HPLC.
Radical inhibitors.
In a microwave vial 100 μL of 2 mM stock solution of 1g in DMSO was added to 100 μL, 200 μL of 4 mM TEMPO in THF, 100 μL, 10 μL of 4 mM BHT in THF and 100 μL of 4 mM DABCO in THF respectively and THF to bring the total volume up to 2 mL and stirred at 70 °C. The progress of the reaction was monitored by HPLC (reaction aliquot:ACN, 1:2).
With/without oxygen.
In the run with oxygen 1.8 mL of THF, 100 μL of 2 mM stock solution of 1g in DMSO and 100 μL of 8 mM K2CO3 stock solution was added to a microwave vial and stirred at 70 °C using a heat block. The progress was measured by taking 200 μL of the solution and mixing it with 400 μL and running the sample on the HPLC. In the run without oxygen THF and 8 mM K2CO3 stock solution was degassed. To a 1.5 mL vial 50 μL 2 mM stock solution of 1g was added, the vial was sealed and purged with nitrogen three times, 900 μL of the THF and 50 μL of the K2CO3 stock solution was added, the cap was further sealed with parafilm, and heated at 70 °C. For each time point that was to be measured, a separate vial was prepared. The progress of the reaction was monitored by HPLC.
Cyclic voltammetry (CV).
CV was carried out with an IKA Electrasyn 2.0 potentiostat using a glassy carbon disk working electrode and a platinum plated copper counter electrode. Measurements were referenced against a silver wire reference electrode in an 0.01 M AgNO3 solution in acetonitrile. Analytes were dissolved in nitrogen purged acetonitrile with 0.1 M (nBu4)NPF6 as electrolyte. The solution was then purged with nitrogen again just prior to data collection.
A. General procedure for sulfonamide synthesis
To a solution of the selected sulfonyl chloride (3a–c) in DCM (5 mL), DIPEA (1.1 equiv.) and the selected amine was added. The reaction was stirred at 19 °C and monitored by TLC until complete reaction. The mixture was concentrated, dissolved in EtOAc (10 mL) and was extracted with 0.1 M HCl (3 × 10 mL), the combined aqueous phase was extracted with EtOAc (10 mL) and the combined organic phase was washed with brine (10 mL), dried over Na2SO4, gravity filtered and concentrated to afford the desired sulfonamide (4a–i) without further purification.
B. General procedure for Suzuki coupling
To a solution of the selected sulfonamide (4a–i) and boronic acid derivative (1.5 equiv.) in toluene/EtOH/H2O (5:2:1, 16 mL) in a microwave vial, K2CO3 (4 equiv.) and tetrakis(triphenyl-phosphine)palladium(0) (0.03 equiv.) was added, the vessel capped and purged with nitrogen three times. The reaction was stirred at 120 °C using a heat block, unless otherwise stated, and monitored until complete reaction by TLC or LCMS. The mixture was extracted three times with H2O (3 × 10 mL) and the combined aqueous phase was extracted with EtOAc (10 mL). The combined organic phase was washed with brine (10 mL), dried over Na2SO4, filtered through Celite, concentrated, mounted on silica and purified by flash chromatography to afford the desired compound (5a–k).
C. General procedure for demethylation by boron tribromide
To a stirred solution of the selected methoxy protected bicyclic sulfonamide (4a–k) in DCM (5 mL) at 0 °C boron tribromide was added dropwise and the solution was gradually allowed to heat back to 19 °C and monitored until complete deprotection by LCMS or TLC. The reaction was quenched by adding H2O dropwise with the reaction vessel at 0 °C, until fuming stopped and then an excess was added (10 mL). The organic layer was separated, concentrated, diluted with EtOAc (10 mL) and washed with H2O (2 × 10 mL). The combined aqueous phase was extracted with EtOAc (10 mL) and the combined organic phase was washed with brine (10 mL), dried over Na2SO4, gravity filtered, concentrated, mounted on silica and purified by flash chromatography to afford the desired sulfonamide (1a–k).
D. General procedure for cyclisation in H2O
To a solution of the selected bicyclic sulfonamide (1a, b–i) in H2O (4 mL), K2CO3 (4 equiv.) was added. The reaction mixture was stirred and heated using microwave irradiation at 70 °C and monitored until complete reaction by HPLC. After cooling, HCl (6 equiv.) was added, and the solid formed was dissolved with EtOAc. The organic layer was separated and the aqueous phase was extracted with EtOAc (10 mL). The organic phase was washed with H2O (10 mL) and brine (10 mL), dried over Na2SO4, gravity filtered and concentrated. The crude was purified by preparative HPLC to afford the desired dibenzosultam (2a, b–i).
2-Bromo-N-(4-((3,4-dimethoxyphenyl)sulfonyl)benzyl)-4,5-dimethoxybenzenesulfonamide (4a).
2-Bromo-4,5-dimethoxy-benzenesulfonyl chloride (3a) (680 mg, 2.15 mmol) was reacted with S4a (729 mg, 2.37 mmol, 1.1 equiv.) and DIPEA (0.82 mL, 4.74 mmol, 2.2 equiv.) in 5 mL DMF at 19 °C for 1 h. The reaction was stopped by adding 50 mL ice water. The solid was filtered, washed with H2O and dried to afford 1.1 g (87%) of the title compound as a colourless film; m.p.: 87–100 °C; 1H NMR (400 MHz, CDCl3) δ 7.79 (d, J = 8.0 Hz, 2H), 7.54 (s, 1H), 7.51 (d, J = 8.5 Hz, 1H), 7.37 (d, J = 8.1 Hz, 2H), 7.33 (s, 1H), 7.08 (s, 1H), 6.91 (d, J = 8.6 Hz, 1H), 5.69 (t, J = 6.4 Hz, 1H), 4.10 (d, J = 6.2 Hz, 2H), 3.92 (s, 3H), 3.89 (s, 6H), 3.87 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 153.24, 152.67, 149.41, 148.18, 141.85, 141.74, 132.85, 130.21, 128.59, 127.63, 122.02, 117.13, 113.90, 111.19, 110.99, 109.92, 56.71, 56.53, 56.39, 56.33, 46.67.
2-Bromo-N-(4-((3,4-dimethoxyphenyl)sulfonyl)phenethyl)-4,5-dimethoxybenzenesulfonamide (4b).
2-Bromo-4,5-dimethoxy-benzenesulfonyl chloride (3a) (900 mg, 2.31 mmol) was reacted with S4b (817 mg, 2.54 mmol, 1.1 equiv.) and DIPEA (0.88 mL, 5.08 mmol, 2.2 equiv.) in 10 mL DMF at 19 °C for 1 h. The reaction was stopped by adding 40 mL ice water. The solid was filtered, washed with H2O and dried to afford 760 mg (55%) of the title compound as a white fluffy solid after normal phase column chromatography (pentane/EtOAc, compound eluted at 70–80% EtOAc); m.p.: 94–103 °C; 1H NMR (400 MHz, CDCl3) δ 7.77–7.67 (m, 2H), 7.50–7.39 (m, 2H), 7.26 (dd, J = 16.5, 2.1 Hz, 1H), 7.19–7.11 (m, 2H), 6.95 (d, J = 16.4 Hz, 1H), 6.82 (dd, J = 16.4, 8.6 Hz, 1H), 4.98 (t, J = 6.2 Hz, 1H), 3.81 (ddd, J = 14.7, 5.9, 3.3 Hz, 12H), 3.08–2.97 (m, 2H), 2.80–2.69 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 153.18, 152.55, 149.38, 148.14, 143.41, 140.97, 133.10, 130.02, 129.75, 127.80, 121.98, 117.09, 113.86, 110.99, 109.93, 56.68, 56.55, 56.40, 56.32, 43.84, 35.65.
2-Bromo-N-(3,4-dimethoxyphenyl)-4,5-dimethoxybenzenesulfonamide (4c).
Following general procedure A, 2-bromo-4,5-dimethoxybenzenesulfonyl chloride (3a) (500 mg, 1.58 mmol) was reacted with 3,4-dimethoxyaniline (267 mg, 1.74 mmol) in 10 mL DCM for 5 h to afford 570 mg (83%) of the title compound as a white solid after normal phase column chromatography (pentane/EtOAc, compound eluded at 50–70% EtOAc); m.p.: 158–161 °C; 1H NMR (400 MHz, CDCl3) δ 7.38 (s, 1H), 7.09 (s, 1H), 7.04 (s, 1H), 6.77 (s, 1H), 6.65 (dd, J = 8.6, 2.2 Hz, 1H), 6.59 (dd, J = 8.5, 2.3 Hz, 1H), 3.89 (s, 3H), 3.78 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 152.61, 149.27, 148.03, 147.67, 129.59, 128.75, 116.89, 115.60, 114.52, 111.26, 111.22, 108.04, 56.62, 56.47, 56.05, 56.03.
2-Bromo-N-(3-methoxyphenyl)-4,5-dimethoxybenzenesulfonamide (4d).
Following general procedure A, 2-bromo-4,5-dimethoxybenzenesulfonyl chloride (3a) (500 mg, 1.58 mmol) was reacted with 3-methoxyaniline (215 mg, 1.74 mmol) for 2 h to afford 480 mg (75%) of the title compound as a solid after normal phase column chromatography (pentane/EtOAc, compound eluded at 50-70% EtOAc); m.p.: 132-137 °C; 1H NMR (400 MHz, CDCl3) δ 7.50 (s, 1H), 7.22 (s, 1H), 7.11 (d, J = 8.2 Hz, 1H), 7.07 (s, 1H), 6.73 (d, J = 1.9 Hz, 1H), 6.69 (d, J = 8.0 Hz, 1H), 6.61 (d, J = 8.3 Hz, 1H), 3.88 (s, 3H), 3.83 (s, 3H), 3.72 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 160.31, 152.69, 147.98, 137.20, 130.14, 129.50, 117.06, 114.56, 113.57, 111.41, 111.08, 107.37, 56.59, 56.53, 55.40.
2-Bromo-4,5-dimethoxy-N-phenylbenzenesulfonamide (4e).
Following general procedure A, 2-bromo-4,5-dimethoxybenzenesulfonyl chloride (3a) (300 mg, 0.95 mmol) was reacted with aniline (0.10 mL, 1.05 mmol, 1.1 equiv.) was reacted for 64 h to afford 289 mg (78%) of the title compound as a brownish white solid after normal phase column (pentane/EtOAc, eluded at 50% EtOAc); m.p.:180–182 °C; 1H NMR (400 MHz, CDCl3) δ 7.46 (s, 1H), 7.24–7.19 (m, 3H), 7.15–7.12 (m, 2H), 7.11–7.07 (m, 2H), 3.88 (s, 3H), 3.81 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 152.66, 148.00, 136.00, 129.55, 129.43, 125.82, 121.86, 117.04, 114.52, 111.35, 56.59, 56.52.
2-Bromo-4,5-dimethoxy-N-methylbenzenesulfonamide (4f).
Following general procedure A, 2-bromo-4,5-dimethoxybenzenesulfonyl chloride (3a) (1.000 g, 3.17 mmol) was reacted with methylamine solution, 2.0 M in THF (3.96 mL, 7.92 mmol, 2.5 equiv.) for 5 min to afford 955 mg (97%) of the title compound as a white crystalline solid; m.p.: 163–165 °C; 1H NMR (400 MHz, CDCl3) δ 7.61 (s, 1H), 7.14 (s, 1H), 5.03 (q, J = 5.9, 5.4 Hz, 1H), 3.94 (d, J = 3.7 Hz, 6H), 2.60 (d, J = 5.4 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 152.44, 148.10, 129.15, 117.00, 114.43, 110.99, 56.59, 56.39, 29.44.
2-Bromo-N-methylbenzenesulfonamide (4g).
Following general procedure A, 2-bromobenzenesulfonyl chloride (3b) (300 mg, 1.17 mmol) was reacted with methylamine solution, 2.0 M in THF (1.47 mL, 2.94 mmol, 2.5 equiv.) for 5 min to afford 286 mg (97%) of the title compound as a yellow oil; 1H NMR (400 MHz, CDCl3) δ 8.15 (d, J = 7.6 Hz, 1H), 7.75 (d, J = 7.9 Hz, 1H), 7.49 (t, J = 7.6 Hz, 1H), 7.46–7.41 (m, 1H), 5.06 (s, 1H), 2.61 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 137.54, 135.02, 133.80, 132.12, 127.87, 119.60, 29.27. NMR data were in agreement with those reported in the literature.22
2-Bromo-N-methyl-5-(trifluoromethyl)benzenesulfonamide (4h).
Following general procedure A, 2-bromo-4-(triflouromethyl)benzenesulfonyl chloride (3c) (297 mg, 0.918 mmol) was reacted with methylamine solution, 2.0 M in THF (1.61 mL, 3.21 mmol, 3.5 equiv.) for 30 min to afford 256 mg (88%) of the title compound as an off white see-through crystalline solid; m.p.: 39–42 °C; 1H NMR (40 0 MHz, CDCl3) δ 8.23 (d, J = 7.3 Hz, 1H), 7.95 (s, 1H), 7.72 (d, J = 7.5 Hz, 1H), 5.45 (q, J = 5.2 Hz, 1H), 2.63 (d, J = 5.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 141.42, 135.31 (q, J = 33.8 Hz), 132.44, 132.12 (q, J = 3.6 Hz), 126.48–118.17 (m), 29.19.
N-Benzyl-2-bromo-4,5-dimethoxybenzenesulfonamide (4i).
2-Bromo-4,5-dimethoxybenzenesulfonyl chloride (3a) (10.0 g, 31.69 mmol) was reacted with benzylamine (4.15 mL, 38.03 mmol, 1.2 equiv.) and DIPEA (16.45 mL, 95.07 mmol, 3.0 equiv.) in DCM (30 mL) for 1 h while warming from 0 °C to 19 °C. The reaction was quenched with H2O (30 mL) and extracted with DCM (3 × 30 mL). The organic phase was washed with 1 M HCl (3 × 50 mL), dried over Na2SO4 and evaporated to afford 10.41 g (85%) of the title compound as white crystalline solid after recrystallisation from hot MeOH; m.p.: 114-116 °C; 1H NMR (400 MHz, (CD3)2SO) δ 8.24 (s, 1H), 7.38 (s, 1H), 7.26 (s, 1H), 7.25–7.15 (m, 5H), 4.08 (s, 2H), 3.84 (s, 3H), 3.76 (s, 3H); 13C NMR (101 MHz, (CD3)2SO) δ 151.71, 147.34, 137.55, 131.45, 128.04, 127.61, 127.03, 117.43, 113.25, 110.56, 56.34, 55.78, 46.06.
N-(4-((3,4-Dimethoxyphenyl)sulfonyl)benzyl)-3′,4,4′,5-tetramethoxy-[1,1′-biphenyl]-2-sulfonamide (5a).
Following general procedure B, 4a (580 mg, 0.99 mmol) and 3,4-dimethoxybenzeneboronic acid (270 mg, 1.48 mmol) was reacted in 8 mL toluene/EtOH/H2O (5:2:1) for 1 h in the MW to afford 530 mg (83%) of the title compound as a fluffy white solid after normal phase column chromatography (pentane/EtOAc, the compound eluted at 80% EtOAc); m.p.: 102–110 °C; 1H NMR (400 MHz, CDCl3) δ 7.78 (d, J = 8.4 Hz, 2H), 7.61 (s, 1H), 7.51 (dd, J = 8.5, 2.2 Hz, 1H), 7.33 (d, J = 2.1 Hz, 1H), 7.22 (d, J = 8.4 Hz, 2H), 7.11 (d, J = 2.0 Hz, 1H), 6.90 (dd, J = 8.4, 7.6 Hz, 2H), 6.83–6.79 (m, 2H), 5.29 (s, 1H), 3.97 (s, 3H), 3.94 (s, 3H), 3.90 (s, 6H), 3.84 (d, J = 2.0 Hz, 6H), 3.79 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 153.26, 151.80, 149.43, 149.29, 148.45, 148.02, 141.86, 141.84, 133.55, 132.90, 130.86, 129.46, 128.40, 127.65, 122.02, 121.20, 114.72, 113.23, 112.25, 110.98, 110.88, 109.91, 56.52, 56.43, 56.39, 56.34, 56.14, 56.06, 46.63.
N-(4-((3,4-Dimethoxyphenyl)sulfonyl)phenethyl)-3′,4,4′,5-tetramethoxy-[1,1′-biphenyl]-2-sulfonamide (5b).
Following general procedure B, 4b (740 mg, 1.23 mmol) and 3,4-dimethoxybenzeneboronic acid (387 mg, 2.13 mmol, 1.73 equiv.) was reacted with K2CO3 (784 mg, 5.67 mmol, 4.61 equiv.) for 1 h in the MW to afford 380 mg (47%) if the title compound as a white foam after normal phase column chromatography (pentane/EtOAc, product not pure, pentane/DCM/THF, 9:9:2, isocratic separation); m.p.: 102–109 °C; 1H NMR (400 MHz, CDCl3) δ 7.79 (d, J = 8.3 Hz, 2H), 7.58 (s, 1H), 7.52 (d, J = 8.5 Hz, 1H), 7.34 (s, 1H), 7.15 (d, J = 8.2 Hz, 2H), 7.07 (s, 1H), 6.91 (d, J = 8.6 Hz, 1H), 6.83 (s, 2H), 6.76 (s, 1H), 3.96 (s, 3H), 3.93 (s, 3H), 3.90 (s, 9H), 3.86 (s, 3H), 2.81 (d, J = 5.9 Hz, 2H), 2.62 (t, J = 7.1 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 153.16, 151.62, 149.38, 149.30, 148.45, 147.94, 143.58, 140.85, 133.50, 133.12, 130.97, 129.61, 129.28, 127.70, 121.97, 120.86, 114.66, 113.41, 112.26, 110.96, 110.79, 109.92, 56.49, 56.38, 56.32, 56.21, 56.15, 43.89, 35.76.
N-(3,4-dimethoxyphenyl)-3′,4,4′,5-tetramethoxy-[1,1′-biphenyl]-2-sulfonamide (5c).
Following procedure B, 4c (670 mg, 1.55 mmol) and 3,4-dimethoxybenzeneboronic acid (423 mg, 2.32 mmol) was reacted for 1 h in the MW at 100 °C to afford 420 mg (55%) of the title compound as a white solid after reverse phase column chromatography (H2O/ACN, 10–42% ACN); m.p.: 119–122 °C; 1H NMR (400 MHz, CDCl3) δ 7.57 (s, 1H), 6.98 (s, 1H), 6.91 (s, 2H), 6.70 (s, 1H), 6.58 (d, J = 8.6 Hz, 1H), 6.37 (s, 1H), 6.20 (d, J = 8.6 Hz, 1H), 5.66 (s, 1H), 3.91 (s, 3H), 3.89 (s, 3H), 3.85 (s, 3H), 3.82 (s, 3H), 3.74 (s, 3H), 3.70 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 151.63, 149.05, 149.02, 148.08, 147.60, 146.23, 134.11, 131.10, 129.68, 129.46, 121.97, 114.88, 113.60, 112.64, 112.19, 111.19, 110.54, 105.23, 56.38, 56.21, 55.99, 55.98, 55.95, 55.81.
3′,4,4′,5-Tetramethoxy-N-(3-methoxyphenyl)-[1,1′-biphenyl]-2-sulfonamide (5d).
Following general procedure B, 4d (300 mg, 0.75 mmol) and 3,4-dimethoxybenzeneboronic acid (204 mg, 1.12 mmol) was reacted in 8 mL toluene/EtOH/H2O (5:2:1) for 1 h in the MW to afford 140 mg (41%) of the title compound as a white solid after reverse phase column chromatography (H2O/ACN, compound eluted at 45% ACN); m.p.: 154-166 °C; 1H NMR (400 MHz, CDCl3) δ 7.67 (s, 1H), 7.03 (t, J = 8.2 Hz, 1H), 6.94 (s, 2H), 6.89 (s, 1H), 6.70 (s, 1H), 6.53 (s, 1H), 6.32 (d, J = 2.0 Hz, 1H), 6.25 (s, 1H), 5.61 (s, 1H), 3.97 (s, 6H), 3.87 (d, J = 0.8 Hz, 3H), 3.83 (s, 3H), 3.68 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 160.18, 151.83, 149.17, 148.17, 147.75, 137.81, 134.27, 131.06, 129.86, 129.47, 122.07, 114.96, 113.43, 112.82, 110.99, 110.59, 109.47, 104.71, 56.59, 56.35, 56.16, 56.00, 55.31.
3′,4,4′,5-Tetramethoxy-N-phenyl-[1,1′-biphenyl]-2-sulfonamide (5e).
Following general procedure B, 4e (145 mg, 0.39 mmol) and 3,4-dimethoxybenzeneboronic acid (106 mg, 0.58 mmol) was reacted for 1 h to afford 53 mg (32%) of the title compound as an off-white solid after reverse phase column chromatography (H2O/ACN (eluted at 40–100% ACN)); m.p.: 209–212 °C; 1H NMR (400 MHz, CDCl3) δ 7.66 (s, 1H), 7.13 (t, J = 8.0 Hz, 2H), 7.00–6.92 (m, 3H), 6.88 (s, 1H), 6.69 (d, J = 5.8 Hz, 3H), 5.64 (s, 1H), 3.96 (s, 6H), 3.87 (s, 3H), 3.81 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 151.68, 149.04, 148.05, 147.64, 136.52, 134.08, 130.97, 129.37, 129.01, 123.93, 121.89, 118.64, 114.79, 114.74, 113.22, 112.66, 112.61, 110.52, 56.42 (d, J = 4.6 Hz), 56.19 (d, J = 5.7 Hz), 56.00 (d, J = 4.6 Hz), 55.85 (d, J = 6.1 Hz).
3′,4,4′,5-Tetramethoxy-N-methyl-[1,1′-biphenyl]-2-sulfonamide (5f).
Following general procedure B, 4f (226 mg, 0.73 mmol) and 3,4-dimethoxybenzeneboronic acid (199 mg, 1.09 mmol) was reacted for 1 h to afford 149 mg (56%) of the title compound as a white solid after reverse phase column chromatography (H2O/ACN, eluted at 40% ACN); m.p.: 176–177 °C; 1H NMR (400 MHz, CDCl3) δ 7.51 (s, 1H), 7.03 (s, 1H), 6.92 (d, J = 8.2 Hz, 1H), 6.86 (d, J = 8.2 Hz, 1H), 6.74 (s, 1H), 3.85 (t, J = 16.2 Hz, 12H), 3.44–3.39 (m, 1H), 2.24 (d, J = 6.7 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 151.28, 148.95, 148.13, 147.70, 133.33, 130.84, 128.38, 120.96, 114.39, 112.83, 112.29, 110.72, 56.19, 56.10, 55.86, 55.79, 29.06.
3′,4′-Dimethoxy-N-methyl-[1,1′-biphenyl]-2-sulfonamide (5g).
Following general procedure B, 4g (214 mg, 0.86 mmol) and 3,4-dimethoxybenzeneboronic acid (234 mg, 1.28 mmol) was reacted for 30 min to afford 136 mg (52%) of the title compound as a yellow solid after normal phase column chromatography (pentane/EtOAc, eluded at 50–100% EtOAc); m.p.: 187–189 °C; 1H NMR (400 MHz, CDCl3) δ 3.91 (d, J = 17.6 Hz, 7H), 3.44 (q, J = 5.4 Hz, 1H), 2.32 (d, J = 5.5 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 149.20, 148.34, 139.81, 136.97, 132.36, 132.23, 131.07, 129.86, 127.88, 121.01, 112.77, 110.82, 56.02, 55.93, 29.17.
3′,4′-Dimethoxy-N-methyl-4-(trifluoromethyl)-[1,1′-biphenyl]-2-sulfonamide (5h).
Following general procedure B, 4h (257 mg, 0.81 mmol) and 3,4-dimethoxybenzeneboronic acid (221 mg, 1.21 mmol) was reacted for 30 min to afford 196 mg (65%) of the title compound as a white solid after reverse phase column chromatography (ACN/H2O, 40–50% ACN, very flat gradient); m.p.: 110–112 °C; 1H NMR (400 MHz, CDCl3) δ 8.26 (dd, J = 15.3, 8.2 Hz, 1H), 7.74 (ddd, J = 15.6, 8.2, 2.0 Hz, 1H), 7.64–7.56 (m, 1H), 7.12–7.06 (m, 1H), 7.03–6.92 (m, 2H), 3.96–3.86 (m, 6H), 2.33 (dd, J = 15.6, 5.4 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 149.83, 148.71, 140.92, 140.81, 134.18 (q, J = 33.6 Hz), 130.71, 129.31 (t, J = 3.6 Hz), 124.92–124.76 (t, J = 3.4 Hz), 127.41–119.19 (q, J = 274 Hz), 112.68, 111.11, 56.22, 56.12, 29.27.
N-Benzyl-2′,3′,4,5-tetramethoxy-[1,1′-biphenyl]-2-sulfonamide (5i).
Following general procedure B, 4i (2.6 g, 6.6 mmol) and 2,3-dimethoxyphenylboronic acid (1.8 g, 9.9 mmol was reacted in 13 mL toluene/EtOH/H2O (5:2:1) for 1 h in the MW at 120 °C. The reaction mixture was separated from the aqueous phase with ethyl acetate. The organic phase was dried over Na2SO4, filtered through Celite and evaporated to afford 1.76 g (60%) of the title compound as a white solid after normal phase flash chromatography (pentane/EtOAc, gradient, 0–40% EtOAc, (Sfär Duo 50 g)) and (DCM/EtOAc gradient (Sfär Duo 50 g, 0–0.5% EtOAc through 10 CV –very slow)); m.p.: 129–131 °C; 1H NMR (400 MHz, (CD3)2SO) δ 7.61 (t, J = 6.2 Hz, 1H, NH), 7.43 (s, 1H, Ar), 7.32–7.18 (m, 5H, Ar), 7.06–6.99 (m, 2H, Ar), 6.81–6.72 (m, 2H, Ar), 3.98–3.86 (m, 2H, CH2), 3.83 (s, 3H, CH3), 3.81 (s, 3H, CH3), 3.79 (s, 3H, CH3), 3.57 (s, 3H, CH3); 13C NMR (101 MHz, (CD3)2SO) δ 152.06, 150.34, 147.47, 145.80, 138.06, 134.11, 130.97, 130.61, 128.19, 127.62, 127.09, 122.85, 122.78, 114.82, 112.22, 110.99, 59.85, 55.90, 55.77, 55.62, 46.05.
3′,5′-dimethoxy-N-methyl-[1,1′-biphenyl]-2-sulfonamide (5j).
Following general procedure B, 4g (163 mg, 0.65 mmol) and 3,5-dimethoxybenzeneboronic acid (177 mg, 0.98 mmol) was reacted for 30 min to afford 170 mg (88% purity) (75%) of the title compound as a white crystalline solid after reverse phase column chromatography (H2O/MeOH, product eluted at 68% MeOH) and was used directly in the synthesis of 41 without further purification.
3′-Methoxy-N-methyl-[1,1′-biphenyl]-2-sulfonamide (5k).
Following general procedure B, 4g (150 mg, 0.60 mmol) and 3-methoxybenzeneboronic acid (137 mg, 0.90 mmol) was reacted for 30 min to afford 142 mg (90% purity) (77%) of the title compound as a white solid like film after normal phase column chromatography (pentane/EtOAc, eluted at 67% EtOAc) and was used directly in the synthesis of 42 without further purification.
N-(4-((3,4-dihydroxyphenyl)sulfonyl)benzyl)-3′,4,4′,5-tetrahydroxy-[1,1′-biphenyl]-2-sulfonamide (1a).
Following general procedure C, 5a (260 mg, 0.40 mmol) was reacted with boron tribromide, 1 M in DCM (2.83 mL, 2.83 mmol, 7.0 equiv.) in DCM (10 mL) for 1.5 h to afford 190 mg (84%) of the title compound as brown oil that turns to a white fluffy solid after trituration with DCM; m.p.: 204–208 °C; 1H NMR (400 MHz, CD3OD) δ 7.77–7.73 (m, 2H), 7.46 (s, 1H), 6.87 (d, J = 8.3 Hz, 1H), 6.84 (d, J = 2.1 Hz, 1H), 6.72 (d, J = 8.1 Hz, 1H), 6.67 (s, 1H), 6.62 (dd, J = 8.1, 2.1 Hz, 1H), 3.81 (s, 2H); 13C NMR (101 MHz, CD3OD) δ 151.94, 150.07, 147.08, 146.30, 145.63, 145.20, 144.62, 142.82, 135.11, 132.74, 132.17, 129.75, 129.65, 128.24, 122.31, 121.61, 120.34, 118.20, 117.41, 116.48, 115.78, 115.29, 47.07; HRMS (ESI) calcd for C25H21NO10S2 [M + H]+ 560.0685, found, 560.0677.
N-(4-((3,4-Dihydroxyphenyl)sulfonyl)phenethyl)-3′,4,4′,5-tetrahydroxy-[1,1′-biphenyl]-2-sulfonamide (1b).
Following general procedure C, 5b (120 mg, 0.18 mmol) was reacted with boron tribromide, 1 M in DCM (1.28 mL, 1.28 mmol, 7.0 equiv.) in 10 mL DCM for 2 h to afford 60 mg (57%) of the title compound as a white solid after normal phase column chromatography (DCM/MeOH, 2–10% MeOH); m.p.: 196 °C (decomposition); 1H NMR (400 MHz, CD3OD) δ 7.76 (d, J = 8.5 Hz, 2H), 7.42 (s, 1H), 7.28 (dd, J = 8.3, 2.3 Hz, 1H), 7.25 (dd, J = 5.4, 3.2 Hz, 3H), 6.86 (d, J = 8.3 Hz, 1H), 6.81 (d, J = 2.1 Hz, 1H), 6.73 (d, J = 8.1 Hz, 1H), 6.66 (s, 1H), 6.63 (dd, J = 8.1, 2.1 Hz, 1H), 2.83 (t, J = 7.2 Hz, 2H), 2.64 (t, J = 7.2 Hz, 2H); 13C NMR (101 MHz, CD3OD) δ 151.81, 149.93, 146.97, 146.25, 145.69, 145.60, 145.16, 141.80, 134.84, 132.76, 132.08, 130.68, 129.45, 128.31, 122.10, 121.58, 120.19, 117.98, 117.25, 116.45, 115.78, 115.25, 44.55, 36.54; HRMS (ESI) calcd for C26H23NO10S2 [M + H]+ 574.0842, found, 574.0842.
N-(3,4-Dihydroxyphenyl)-3′,4,4′,5-tetrahydroxy-[1,1′-biphenyl]-2-sulfonamide (1c).
Following general procedure C, 5c (420 mg, 0.86 mmol) was reacted with boron tribromide, 1 M in DCM (6.86 mL, 6.86 mmol, 8.0 equiv.) for 2 h to afford 143 mg (41%) of the title compound as an off-white solid after reverse phase column chromatography (H2O/ACN, compound eluted at 15% ACN); m.p.: 136 °C (decomposition); 1H NMR (400 MHz, CD3OD) δ 7.41 (s, 1H), 6.78–6.75 (m, 2H), 6.62–6.59 (m, 2H), 6.55 (d, J = 8.5 Hz, 1H), 6.44 (d, J = 2.5 Hz, 1H), 6.18 (dd, J = 8.5, 2.6 Hz, 1H); 13C NMR (101 MHz, CD3OD) δ 149.93, 146.35, 146.00, 145.22, 144.85, 143.42, 135.78, 132.45, 130.77, 129.34, 122.65, 120.43, 118.41, 118.17, 116.11, 115.50, 113.88, 110.39; HRMS (ESI) calcd for C18H15NO8S [M + H]+ 406.0597, found, 406.0593.
3′,4,4′,5-Tetrahydroxy-N-(3-hydroxyphenyl)-[1,1′-biphenyl]-2-sulfonamide (1d).
Following general procedure C, 5d (70 mg, 0.15 mmol) was reacted with boron tribromide, 1 M in DCM (0.91 mL, 0.91 mmol, 6.0 equiv.) for 2 h to afford 12 mg (20%) of the title compound as a pale beige film after reverse phase column chromatography (H2O/MeOH, compound eluted at 37% MeOH); m.p.: 128-134 °C; 1H NMR (400 MHz, CD3OD) δ 7.52 (s, 1H), 6.93 (t, J = 8.2 Hz, 1H), 6.76 (d, J = 8.1 Hz, 1H), 6.72 (d, J = 2.1 Hz, 1H), 6.58 (s, 1H), 6.56 (dd, J = 8.1, 2.1 Hz, 1H), 6.41–6.35 (m, 2H), 6.25 (ddd, J = 8.1, 2.1, 0.9 Hz, 1H); 13C NMR (101 MHz, CD3OD) δ 158.83, 150.07, 146.03, 145.21, 144.91, 140.00, 135.86, 132.26, 130.49, 129.24, 122.54, 120.42, 118.31, 118.21, 115.50, 111.24, 111.11, 106.83; HRMS (ESI) calcd for C18H15NO7S [M + H]+ 390.0648, found, 390.0646.
3′,4,4′,5-Tetrahydroxy-N-phenyl-[1,1′-biphenyl]-2-sulfonamide (1e).
Following the general procedure C, 5e (86 mg, 0.20 mmol) was reacted with boron tribromide, 1 M in DCM (1.76 mL, 1.76 mmol, 8.8 equiv.) in 5 mL chloroform for 4 h to afford 22 mg (29%) of the title compound as a beige after reverse phase column chromatography (H2O:MeOH, compound eluted at 42% MeOH); m.p.: 160-170 °C; 1H NMR (400 MHz, CD3OD) δ 7.52 (s, 1H), 7.13 (t, J = 8.0 Hz, 2H), 6.93 (t, J = 7.4 Hz, 1H), 6.80 (d, J = 7.5 Hz, 2H), 6.75 (d, J = 8.1 Hz, 1H), 6.68 (s, 1H), 6.57 (s, 1H), 6.53 (d, J = 8.2 Hz, 1H); 13C NMR (101 MHz, CD3OD) δ 148.74, 144.67, 143.86, 143.56, 137.47, 134.49, 130.82, 128.36, 127.69, 122.79, 121.10, 119.04, 118.35, 116.89, 116.84, 114.10; HRMS (ESI) calcd for C18H15NO6S [M + H]+ 374.0698, found, 374.0699.
3′,4,4′,5-Tetrahydroxy-N-methyl-[1,1′-biphenyl]-2-sulfonamide (1f).
Following general procedure C, 5f (151 mg, 0.41 mmol) was reacted with boron tribromide, 1 M in DCM (1.81 mL, 1.81 mmol, 4.4 equiv.) for 2.5 h to afford 23 mg (18%) of the title compound as a beige soild after reverse phase column chromatography (H2O:MeOH, compound eluted at 28% MeOH); m.p.: 155–160 °C; 1H NMR (400 MHz, CD3OD) δ 7.43 (s, 1H), 6.84 (d, J = 2.1 Hz, 1H), 6.79 (d, J = 8.1 Hz, 1H), 6.72 (dd, J = 8.1, 2.1 Hz, 1H), 6.69 (s, 1H), 2.30 (s, 3H); 13C NMR (101 MHz, CD3OD) δ 149.93, 146.34, 145.65, 145.22, 135.00, 132.10, 128.32, 121.98, 120.16, 120.12, 117.81, 117.60, 117.58, 115.81, 29.17, 29.15; HRMS (ESI) calcd for C13H13NO6S [M + H]+ 312.0542, found, 312.0545.
3′,4′-Dihydroxy-N-methyl-[1,1′-biphenyl]-2-sulfonamide (1g).
Following the general procedure C, 5g (262 mg, 0.85 mmol) was reacted with boron tribromide, 1 M in DCM (0.79 mL, 0.79 mmol, 2.2 equiv.) for 20 min to afford 175 mg (74%) of the title compound as a beige solid/film after reverse phase column chromatography (H2O: MeOH, 35–60% eluted at 38% MeOH); m.p.: 130–134 °C. 1H NMR (400 MHz, CD3OD) δ 7.99 (d, J = 7.8 Hz, 1H), 7.55 (t, J = 7.5 Hz, 1H), 7.45 (t, J = 7.2 Hz, 1H), 7.28 (d, J = 6.8 Hz, 1H), 6.92 (s, 1H), 6.86 (d, J = 8.0 Hz, 1H), 6.75 (d, J = 8.1 Hz, 1H), 2.32 (s, 3H). 13C NMR (101 MHz, CD3OD) δ 146.35, 145.47, 142.07, 138.02, 133.76, 133.28, 131.81, 129.96, 128.40, 121.79, 117.48, 115.88, 29.05; HRMS (ESI) calcd for C13H13NO4S [M + H]+ 280.0644, found, 280.0639.
3′,4′-Dihydroxy-N-methyl-4-(trifluoromethyl)-[1,1′-biphenyl]-2-sulfonamide (1h).
Following the general procedure C, 5h (182 mg, 0.48 mmol) was reacted with boron tribromide, 1 M in DCM (2.13 mL, 2.13 mmol, 4.4 equiv.) for 30 min to afford 121 mg (72%) of the title compound as a beige solid/film after reverse phase column chromatography (H2O/MeOH, eluted at 56% MeOH); m.p.: 169-172 °C; 1H NMR (400 MHz, CD3OD) δ 8.20 (d, J = 8.3 Hz, 1H), 7.79 (d, J = 8.5 Hz, 1H), 7.59 (s, 1H), 6.93 (s, 1H), 6.87 (d, J = 8.1 Hz, 1H), 6.79 (d, J = 8.2 Hz, 1H), 2.37 (s, 3H); 13C NMR (101 MHz, CD3OD) δ 147.09, 145.89, 143.54, 142.57, 134.43 (q, J = 33.0 Hz), 131.18, 130.52 (dd, J = 7.1, 3.2 Hz), 128.87–120.48 (m), 117.47, 116.11, 28.99; HRMS (ESI) calcd for C14H12F3NO4S [M + H]+ 348.0517, found, 348.0520.
N-Benzyl-2′,3′,4,5-tetrahydroxy-[1,1′-biphenyl]-2-sulfonamide (1i).
Following the general procedure C, 5i (80 mg, 0.18 mmol) was reacted with boron tribromide, 1 M in DCM (0.79 mL, 0.79 mmol, 4.4 equiv.) for 3.25 h to afford 39 mg (56%) of the title compound as dark grey film/foam after reverse phase column chromatography (H2O:MeOH, compound eluted at 52% MeOH); m.p.: 124–133 °C; 1H NMR (400 MHz, CD3OD) δ 7.50 (s, 1H), 7.28–7.18 (m, 5H), 6.79 (d, J = 6.1 Hz, 1H), 6.73 (s, 1H), 6.68 (t, J = 7.7 Hz, 1H), 6.62 (d, J = 6.0 Hz, 1H), 4.00–3.86 (m, 2H); 13C NMR (101 MHz, CD3OD) δ 150.26, 146.52, 145.55, 143.75, 138.71, 131.18, 129.89, 129.39, 128.97, 128.53, 128.41, 123.35, 120.69, 120.23, 117.35, 115.76, 48.05; HRMS (ESI) calcd for C19H17NO6S [M + H]+ 388.0855, found, 388.0853.
3′,5′-Dimethoxy-N-methyl-[1,1′-biphenyl]-2-sulfonamide (1j).
Following the general procedure C, 5j (167 mg, 0.54 mmol) was reacted with boron tribromide, 1 M in DCM (2.39 mL, 2.39 mmol, 4.4 equiv.) for 3 h to afford 25 mg (17%) of the title compound as an off-white film/foam after normal phase column chromatography (DCM:MeOH, product eluted at 68% MeOH) (not pure) and preparative HPLC (H2O/ACN, 30–45% ACN in 60 min); m.p.: 108–111 °C; 1H NMR (400 MHz, CD3OD) δ 8.01 (d, J = 7.9 Hz, 1H), 7.61 (t, J = 7.5 Hz, 1H), 7.52 (t, J = 7.7 Hz, 1H), 7.34 (d, J = 7.4 Hz, 1H), 6.35 (s, 2H), 6.34 (s, 1H), 2.39 (s, 3H); 13C NMR (101 MHz, CD3OD) δ 157.75, 141.08, 141.01, 137.01, 131.98, 131.91, 128.74, 127.41, 107.59, 101.92, 27.76; HRMS (ESI) calcd for C13H13NO4S [M + H]+ 280.0644, found, 280.0648.
3′-Hydroxy-N-phenyl-[1,1′-biphenyl]-2-sulfonamide (1k).
Following the general procedure C, 5k (140 mg, 0.36 mmol) was reacted with boron tribromide, 1 M in DCM (1.11 mL, 1.11 mmol, 2.2 equiv.) for 2 h to afford 63 mg (47%) of the title compound as a white crystalline solid after reverse phase column chromatography (H2O/MeOH, compound eluted at 52% MeOH); m.p.: 157–159 °C; 1H NMR (400 MHz, CD3OD) δ 8.02 (d, J = 8.0 Hz, 1H), 7.62 (t, J = 7.5 Hz, 1H), 7.55–7.50 (m, 1H), 7.33 (d, J = 7.5 Hz, 1H), 7.26–7.21 (m, 1H), 6.92–6.82 (m, 3H), 4.90 (s, 1H), 2.37 (s, 3H); 13C NMR (101 MHz, CD3OD) δ 157.93, 142.38, 141.98, 138.58, 133.60, 133.30, 130.12, 130.07, 128.83, 121.41, 117.33, 116.05, 29.01; HRMS (ESI) calcd for C13H13NO3S [M + H]+ 264.0694, found, 264.0695.
6-(4-((3,4-Dihydroxyphenyl)sulfonyl)benzyl)-2,3,8,9-tetrahydroxy-6H-dibenzo[c,e][1,2]thiazine 5,5-dioxide (2a).
Following general procedure D, 1a (18.2 mg, 0.033 mmol) was reacted for 3.67 h to afford 15.6 mg (86%) of the title compound as a beige solid; m.p.: 205–215 °C; 1H NMR (400 MHz, CD3OD) δ 7.70 (d, J = 8.6 Hz, 2H), 7.32 (d, J = 8.6 Hz, 2H), 7.26–7.23 (m, 2H), 7.19 (s, 1H), 7.15 (s, 1H), 7.06 (s, 1H), 6.88 (d, J = 8.6 Hz, 1H), 6.58 (s, 1H), 4.84 (s, 2H); 13C NMR (101 MHz, CD3OD) δ 151.98, 151.15, 147.74, 147.05, 146.40, 145.39, 143.18, 143.08, 132.53, 132.08, 129.81, 128.11, 127.19, 126.62, 121.70, 119.74, 116.55, 115.27, 112.06, 111.83, 110.98, 109.72, 54.63; HRMS (ESI) calcd for C25H19NO10S2 [M + H]+ 558.0529, found, 558.0525.
6-(3,4-Dihydroxyphenyl)-2,3,8,9-tetrahydroxy-6H-dibenzo[c,e][1,2]thiazine 5,5-dioxide (2c).
Following general procedure D, 1c (28.6 mg, 0.071 mmol) was reacted for 23 min, neutralized, concentrated and separated from some of the K2CO3 by dissolving the solid in MeOH and filtering of the solid to afford 16.8 mg (59%) of the title compound as a brown film after preparative HPLC (H2O/ACN, 15-27% ACN in 30 min); m.p.: 210–217 °C; 1H NMR (400 MHz, CD3OD) δ 7.28 (d, J = 2.3 Hz, 2H), 7.16 (s, 1H), 6.72 (d, J = 8.4 Hz, 1H), 6.53 (d, J = 2.5 Hz, 1H), 6.49 (dd, J = 8.4, 2.5 Hz, 1H), 6.43 (s, 1H); 13C NMR (101 MHz, CD3OD) δ 151.31, 147.89, 146.71, 146.65, 146.42, 145.04, 134.40, 132.14, 127.51, 126.12, 121.02, 118.82, 116.42, 116.19, 112.14, 111.39, 110.11; HRMS (ESI) calcd for C18H13NO8S [M + H]+ 404.0440, found, 404.0446.
2,3,8,9-Tetrahydroxy-6-(3-hydroxyphenyl)-6H-dibenzo[c,e][1,2]thiazine 5,5-dioxide (2d).
Following general procedure D, 1d (41 mg, 0.11 mmol) was reacted for 2.67 h, neutralised and the solvent evaporated to afford 25.5 mg (60%) of the title compound as a yellow film after preparative HPLC (H2O/ACN, 15–33% ACN for 30 min); m.p.: 235 °C (decomposition); 1H NMR (400 MHz, CD3OD) δ 7.29 (s, 2H), 7.17–7.10 (m, 2H), 6.71 (d, J = 7.5 Hz, 1H), 6.55 (d, J = 7.2 Hz, 1H), 6.51 (s, 1H), 6.45 (s, 1H); 13C NMR (101 MHz, CD3OD) δ 159.15, 151.37, 147.96, 146.47, 145.57, 142.00, 133.69, 130.74, 127.49, 126.00, 119.72, 115.77, 112.28, 112.20, 112.11, 111.47, 111.42, 110.30, 110.26; HRMS (ESI) calcd for C18H13NO7S [M + H]+ 388.0491, found, 388.0490.
2,3,8,9-Tetrahydroxy-6-phenyl-6H-dibenzo[c,e][1,2]thiazine 5,5-dioxide (2e).
Following general procedure D, 1e (9.9 mg, 0.027 mmol) was reacted for 2.17 h to afford 4.2 mg (43%) of the title compound as a yellow film after preparative HPLC (H2O/ACN, 40-60% ACN for 40 min); m.p.: 198–215 °C; 1H NMR (400 MHz, CD3OD) δ 7.35–7.26 (m, 5H), 7.12 (s, 1H), 7.07 (s, 1H), 7.05 (s, 1H), 6.41 (s, 1H); 13C NMR (101 MHz, CD3OD) δ 151.43, 148.04, 146.54, 145.71, 141.28, 133.66, 130.15, 128.77, 128.61, 127.52, 125.98, 119.97, 112.29, 112.26, 111.49, 110.31; HRMS (ESI) calcd for C18H13NO6S [M + H]+ 372.0542, found, 372.0544.
2,3,8,9-Tetrahydroxy-6-methyl-6H-dibenzo[c,e][1,2]thiazine 5,5-dioxide (2f).
Following general procedure D, 1f (17 mg, 0.055 mmol) was reacted for 2.17 h to afford 9.6 mg (59%) of the title compound as a brown film after preparative HPLC (H2O/ACN, 25–60% ACN for 30 min); m.p.: 215 °C (decomposition); 1H NMR (400 MHz, CD3OD) δ 7.23 (s, 1H), 7.21 (s, 1H), 7.20 (s, 1H), 6.76 (s, 1H), 3.13 (s, 3H); 13C NMR (101 MHz, CD3OD) δ 149.83, 146.51, 144.99, 143.44, 132.55, 125.80, 123.63, 116.93, 110.52, 110.45, 108.65, 108.55, 34.48; HRMS (ESI) calcd for C13H11NO6S [M + H]+ 310.0385, found, 310.0390.
8,9-Dihydroxy-6-methyl-6H-dibenzo[c,e][1,2]thiazine 5,5-dioxide (2g).
This compound was synthesized in two different ways and on two different scales: (i) Following general procedure D, 1g (14 mg, 0.050 mmol) was reacted for 2.25 h, concentrated, separated from the K2CO3 by dissolving the solid in acetone and filtering of the solid to afford 9 mg (65%) of the title compound as a yellow oil/film after preparative HPLC (H2O/ACN, 20–100% ACN for 30 min). (ii) To a solution of 1g (30 mg, 0.11 mmol) in THF/H2O (20:1, 4.2 mL), K2CO3 (4 equiv.) was added. The reaction mixture was stirred and heated by microwave irradiation at 70 °C and monitored until complete reaction by HPLC (3 h). The mixture was concentrated, and the product was separated from the K2CO3 by dissolving the solid in acetone and filtering of the solid to afford 15 mg (50%) of the title compound as a beige solid/film after preparative HPLC (H2O/ACN, 20-100% ACN for 30 min). (iii) Scale up: to a solution of 1g (279 mg, 1.0 mmol) in H2O/THF (4:1, 20 mL), K2CO3 (4 equiv.) was added. The reaction mixture was bubbled with air and then stirred and heated by microwave irradiation at 70 °C until complete reaction by HPLC (8 h). After cooling HCl (6 equiv.) was added, the solvent evaporated and the crude resolubilised in EtOAc (25 mL) and H2O (25 mL), the layers were separated and the aqueous phase was extracted with EtOAc (10 mL), the combined organic phase was washed with H2O (10 mL) and brine (10 mL), dried over sodium sulfate, gravity filtered, concentrated and mounted on silica to afford 216 mg (78%) of the title compound as a pearlescent foam after reverse phase flash chromatography (H2O/ACN, compound eluted at 33% ACN); m.p.: 209–211 °C; 1H NMR (400 MHz, CD3OD) δ 7.86–7.80 (m, 2H), 7.67 (t, J = 7.7 Hz, 1H), 7.47 (t, J = 7.6 Hz, 1H), 7.40 (s, 1H), 6.78 (s, 1H), 3.17 (s, 3H); 13C NMR (101 MHz, CD3OD) δ 149.27, 144.94, 134.51, 134.31, 133.88, 133.79, 128.22, 125.94, 123.70, 117.80, 112.49, 109.44, 35.37; HRMS (ESI) calcd for C13H11NO4S [M + H]+ 278.0487, found, 278.0484.
8,9-Dihydroxy-6-methyl-2-(trifluoromethyl)-6H-dibenzo[c,e][1,2]thiazine 5,5-dioxide (2h).
Following general procedure D, 1h (31 mg, 0.090 mmol) was reacted for 2.75 h to afford 17 mg (55%) of the title compound as a pearly solid after preparative HPLC (H2O/ACN, 40–60% ACN for 45 min); m.p.: 255–267 °C; 1H NMR (400 MHz, CD3OD) δ 8.15 (s, 1H), 8.04 (d, J = 8.2 Hz, 1H), 7.80 (d, J = 8.1 Hz, 1H), 7.46 (s, 1H), 6.85 (s, 1H), 3.25 (s, 3H); 13C NMR (101 MHz, CD3OD) δ 148.85, 143.78, 135.40, 134.29–133.28 (q, J = 32.8 Hz), 133.87, 133.36, 127.66–119.43 (q, J = 272.0 Hz), 123.55, 123.25 (q, J = 4.0 Hz), 121.53 (q, J = 4.1 Hz), 115.28, 110.99, 107.90, 33.88; HRMS (ESI) calcd for C14H10F3NO4S [M + H]+ 346.0355, found, 346.0349.
6-benzyl-2,3,9,10-tetrahydroxy-6H-dibenzo[c,e][1,2]thiazine 5,5-dioxide (2i).
Following general procedure D, 1i (19.2 mg, 0.050 mmol) was reacted for 50 min to afford 11 mg (58%) of the title compound as a grey-brown film after preparative HPLC (H2O/ACN, 25–33% ACN for 40 min); m.p.: 249–251 °C; 1H NMR (400 MHz, CD3OD) δ 7.97 (s, 1H), 7.23 (s, 1H), 7.15–7.06 (m, 3H), 6.99 (d, J = 5.8 Hz, 2H), 6.71 (d, J = 8.5 Hz, 1H), 6.57 (d, J = 8.6 Hz, 1H), 4.67 (s, 2H); 13C NMR (101 MHz, CD3OD) δ 149.60, 146.03, 144.80, 144.50, 136.85, 132.36, 129.50, 129.00, 128.53, 128.17, 125.53, 117.57, 116.64, 114.91, 109.99, 55.99; HRMS (ESI) calcd for C19H15NO6S [M + H]+ 386.0698, found, 386.0694.
Author contributions
Conceptualisation; SL, AN-P, ON, MG. Supervision; AN-P, ON, MG. Investigation and methodology; all authors. Writing – original draft; SL. Writing – review and editing; all authors. Funding acquisition, project administration and resources; MG.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge financial support from the Swedish Research Council (grant no. 2019-03991 to MG).
Notes and references
- S. Xu, C. A. Rouzer and L. J. Marnett, IUBMB Life, 2017, 66, 803–811 CrossRef PubMed.
- S. Apaydın and M. Török, Bioorg. Med. Chem. Lett., 2019, 29, 2042–2050 CrossRef PubMed.
- S. Debnath and S. Mondal, Eur. J. Org. Chem., 2018, 933–956 CrossRef CAS.
- V. A. Rassadin, D. S. Grosheva, A. A. Tomashevskii and V. V. Sokolv, Chem. Heterocycl. Compd., 2013, 49, 39–65 CrossRef CAS.
- K. C. Majumdar and S. Mondal, Chem. Rev., 2011, 111, 7749–7773 CrossRef CAS PubMed.
- W. D. Guerra, R. A. Rossi, A. B. Pierinioch and S. M. Barolo., J. Org. Chem., 2016, 81, 4965–4973 CrossRef CAS PubMed.
- D. Biswas, L. Samp and A. K. Ganguly, Tetrahedron Lett., 2010, 51, 2681–2684 CrossRef CAS.
- F. Ujjainwalla, M. L. E. N. da Mata, A. M. K. Pennell, C. Escolano, W. B. Motherwella and S. Vázquezc, Tetrahedron, 2015, 71, 6701–6719 CrossRef CAS.
- D. K. Rayabarapu, A. Zhou, K. O. Jeon, T. Samarakoon, A. Rolfe, H. Siddiqui and P. R. Hanson, Tetrahedron, 2009, 65, 3180–3188 CrossRef CAS PubMed.
- J. K. Laha, K. P. Jethava and N. Dayal, J. Org. Chem., 2014, 79, 8010–8019 CrossRef CAS PubMed.
- Y.-Y. Han, H. Wang and S. Yu, Org. Chem. Front., 2016, 3, 953–956 RSC.
- C. Martínez, A. E. Bosnidou, S. Allmendinger and K. Muñiz, Chem. – Eur. J., 2016, 22, 9929–9932 CrossRef PubMed.
- Y. Li, Q. Ding, G. Qiu and J. Wu, Org. Biomol. Chem., 2014, 12, 149–155 RSC.
- L. Li, Y. Li, Z. Zaho, H. Lou and Y.-N. Ma, Org. Lett., 2019, 21, 5995–5999 CrossRef CAS PubMed.
- Y.-N. Ma, Y. Gao, Y. Jing, J. Kang, J. Zhang and X. Chen, J. Org. Chem., 2020, 85, 2918–2926 CrossRef CAS PubMed.
- S. Feng, S. Li, J. Li and J. Wei, Org. Chem. Front., 2019, 6, 517–522 RSC.
- J. Yang, M. A. C. Stuart and M. Kamperman, Chem. Soc. Rev., 2014, 43, 8271–8298 RSC.
- D. E. Stack, G. Li, A. Hill and N. Hoffman, Chem. Res. Toxicol., 2008, 21, 1415–1425 Search PubMed.
- L. Wang, L. Hui and W. Su, Carbohydr. Polym., 2022, 275, 118710 CrossRef CAS PubMed.
- D. Lowinsohn, P. T. Lee and R. G. Compton, Int. J. Electrochem. Sci., 2014, 9, 3458–3472 Search PubMed.
- S. Kummer, W. Ruth and U. Kragl, Electroanalysis, 2016, 28, 1992–1999 CrossRef CAS.
- M. Arshad, W. Siddiqui, I. Khan and A. M. Asiri, J. Chil. Chem. Soc., 2014, 59, 2697–2700 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2nj00088a |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Amalyn
Nain-Perez
,
Amalyn
Nain-Perez