
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcylselenoureas, selenosemicarbazones and selenocarbamate esters: Versatile ligands in coordination chemistry
Anja
Molter
,
Julia
Kuchar
 and
Fabian
Mohr
*
and
Fabian
Mohr
*
Anorganische Chemie, Fakultät für Mathematik und Naturwissenschaften, Bergische Universität Wuppertal, 42119 Wuppertal, Germany. E-mail: fmohr@uni-wuppertal.de
First published on 14th February 2022
Abstract
This article reviews the syntheses, structures, reactions and applications of metal complexes with both transition- and main-group-metals containing selenium compounds including acylselenoureas, selenosemicarbazones and selenocarbamate esters as ligands. Literature up to November 2021 has been covered.
Dr Anja Molter studied chemistry at the University of Braunschweig before joining the research group of professor Fabian Mohr at the University of Wuppertal for her PhD. Her research focused on metal complexes with various selenium compounds as ligands as well as the study of their biological properties. After completion of her PhD, Dr Molter became a chemistry and physics teacher at a high school in Solingen. |
 Julia Kuchar | Julia Kuchar completed her Bachelor's degree in chemistry in 2017 at the University of Wuppertal. After her Master's degree she commenced her PhD studies in 2019 at the same University in the research group of professor Fabian Mohr. During her undergraduate studies she worked on silver complexes with antimicrobial activity, as well as on the coordination chemistry of acylthioureas and acylselenoureas with coinage metals. Currently she is focusing on selenium-containing heterocycles and their biological activity. Julia is also treasurer of the JungChemikerForum Wuppertal-Hagen, a regional section of the youth organisation of the German Chemical Society. |
 Fabian Mohr | Professor Mohr carried out both his undergraduate studies and his PhD in chemistry at RMIT University in Melbourne, Australia. After postdoctoral stays at the University of Western Ontario and the University of Maryland, he moved to the University of Zaragoza in Spain with a Spanish research scholarship. In 2006 Fabian joined the University of Wuppertal as junior professor and became professor of inorganic chemistry there in 2014. Fabian is editor of a book on gold chemistry and has published more than 130 scientific articles mostly on the chemistry of precious metals and chalcogen ligands, especially those containing selenium. |
1. Introduction
There are a number of selenium compounds containing the Y–NHC(Se)–Z unit (Fig. 1). This moiety may in principle coordinate to a metal as either a neutral Se-donor or, upon deprotonation, as anionic Se- or N-donor or as chelating [Se,N]-ligand. If Y = ArC(O) and Z = NR2 the compounds are known as acylselenoureas, when Y = R2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and Z = NR2 they are selenosemicarbazones and when Y = Ar and Z = OR selenocarbamate esters (Fig. 1). As is evident, there is ample opportunity to introduce structural diversity and additional functionality in these compounds.
N and Z = NR2 they are selenosemicarbazones and when Y = Ar and Z = OR selenocarbamate esters (Fig. 1). As is evident, there is ample opportunity to introduce structural diversity and additional functionality in these compounds.
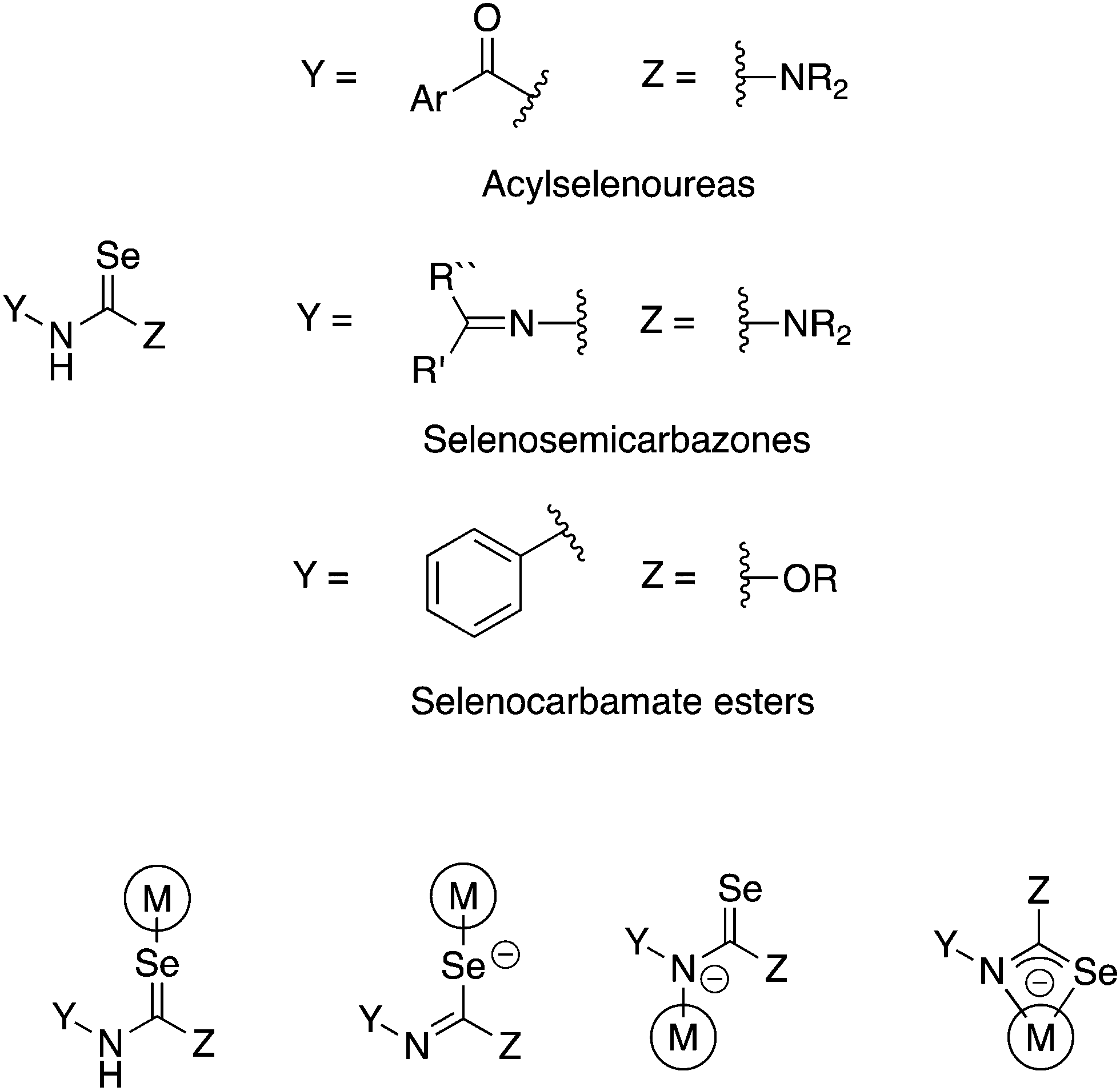 | ||
| Fig. 1 Schematic illustration of selenium compounds containing the –NHC(Se)– unit (top) and their possible coordination modes (bottom). | ||
The coordination chemistry of these three classes of selenium compounds (acylselenoureas, selenosemicarbazones and selenocarbamate esters) is subject of this article. Discussed are syntheses, structures, reactivity and applications of metal complexes with both transition metals and main group metals containing these selenium compounds as ligands. Although much of the work reviewed herein is older, in recent years new applications have been discovered, especially in the materials sciences and medicine, which have led to a renaissance in this area. In 2014 a review on the synthesis and biological applications of selenoureas was published, but focus here was the organic chemistry of this class of compounds.1 There also exists a review on the coordination chemistry of acylselenoureas published in Spanish in 19832 but, to the best of our knowledge, the coordination chemistry of selenosemicarbazones has to date not been reviewed. Given that the field has considerably advanced in the past years, we felt it was timely to provide an update and summary of both the historic, pioneering work and latest developments.
2. Acylselenoureas
Acylselenoureas of the type ArC(O)NHC(Se)NR2 were first reported by Douglass in 1937.3 These compounds can be prepared from the reaction of an acid chloride with KSeCN in acetone to form intermediate acylisoselenocyanates ArC(O)NCSe (amongst other oligomeric compounds4) which are generally not isolated and immediately reacted with a primary or secondary amine to give the desired acylselenoureas (Scheme 1).
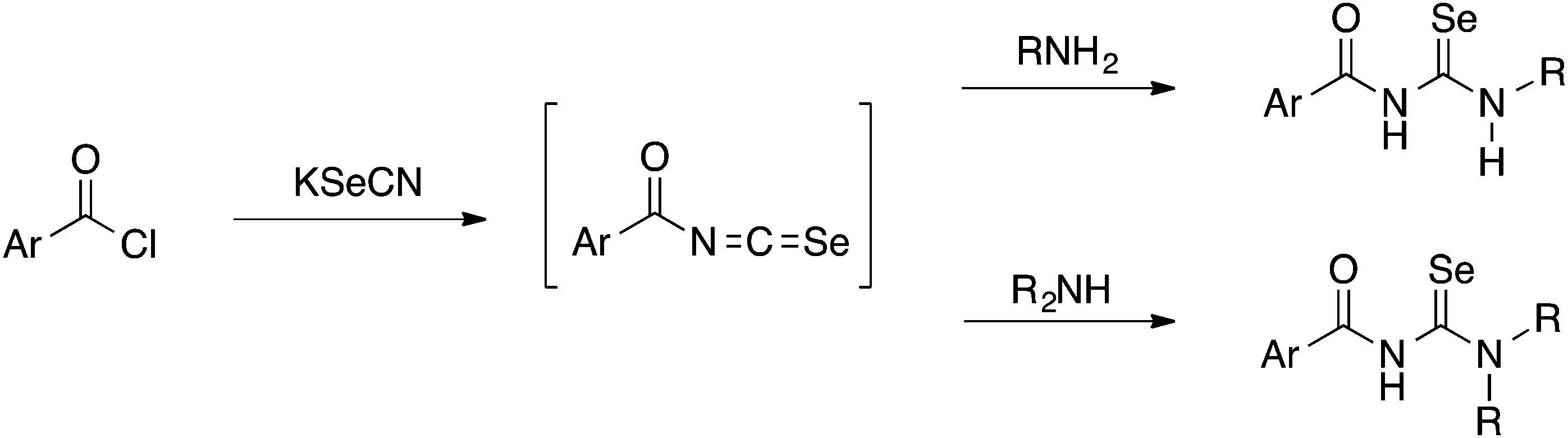 | ||
| Scheme 1 | ||
Although yields are extremely variable, this general procedure is still the method of choice for the synthesis of acylselenoureas. Subsequently in the 1960's and 1970's, the group of Bulka studied this reaction in some detail and (unsuccessfully) attempted to isolate the acylisoselenocyanates.5,6 The same procedure was later also used by Koketsu7 and Pazdera8 to prepare a variety of new selenourea derivatives. An alternative method using CH2Cl2 as solvent and polyethylene glycol 400 as a phase-transfer catalyst was reported by Zhang.9,10 Apart from the acylselenoureas derived from aromatic acid chlorides and anilines or dialkyl amines, there are also examples containing carbohydrate moieties (Fig. 2). These include the acylselenourea prepared from benzoyl chloride and O-acetyl-protected glucosamine, as well as the acylselenourea obtained from the reaction of an O-acetyl-protected gluconyl isoselenocyanate with aniline.11,12
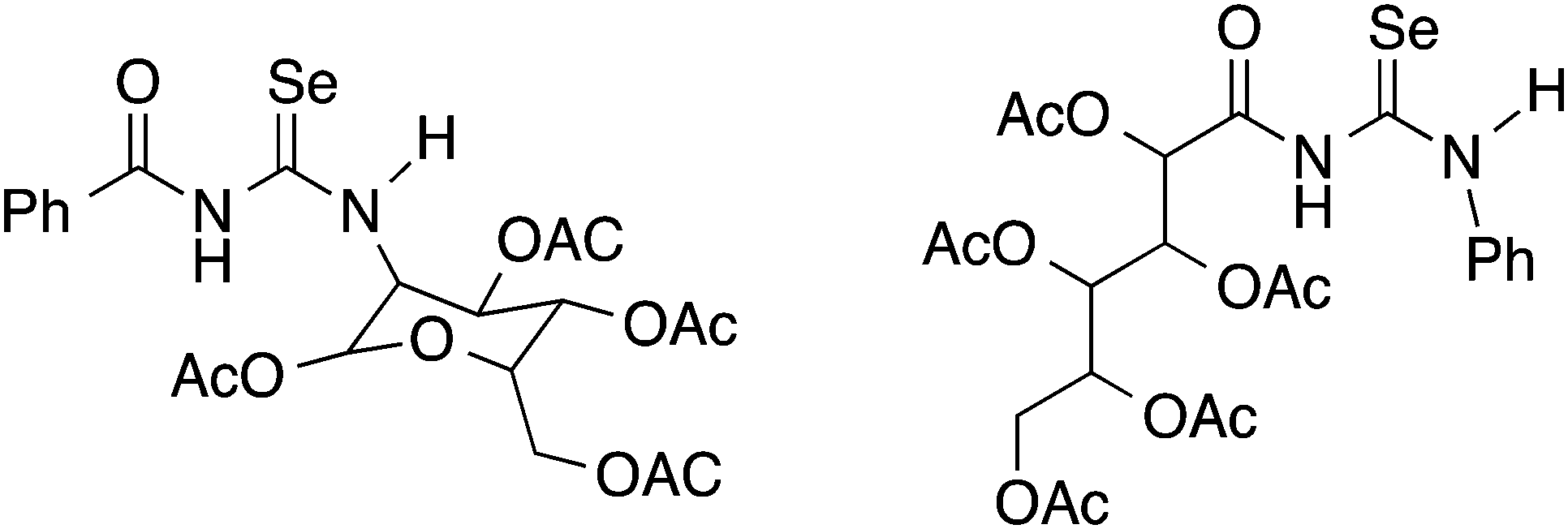 | ||
| Fig. 2 Acylselenoureas derived from carbohydrates. | ||
As will be shown below, the coordination chemistry of acylselenourea derivatives is so far restricted to those formed from aromatic acid chlorides and secondary amines. Table 1 lists these known acylselenoureas together with their yields and CCDC deposition codes for those compounds whose structures have been determined. The only metal-containing acylselenoureas derived from primary amines are the ferrocenyl-substituted derivatives ArC(O)NHC(Se)NHC6H4Fc which have been investigated in some detail by the group of Badshah.13–19
| Ar | NR2 | % yield | CCDC code | Ref. |
|---|---|---|---|---|
| Ph | NEt2 | 45, 39 | 622749 | 3 and 20 |
| Ph | N(1-naphthyl)2 | 58 | 3 | |
| Ph | NnBu2 | 57 | 677072 | 21 and 22 |
| Ph | NMe2 | No data | 22 | |
| Ph | NiBu2 | No data | 23 | |
| 4-MeC6H4 | morpholino | 64, 93 | 4,7 | |
| 4-MeC6H4 | NEt2 | 100, 96 | 7 and 24 | |
| 2,6-F2C6H3 | NiBu2 | 60 | 25 | |
| 2-FC6H4 | NEt2 | 96 | 248406 | 26 |
| 2-FC6H4 | NiBu2 | 68 | 248407 | 26 |
| 4-O2NC6H4 | NiBu2 | 29 | 685384 | 27 |
| 2-Naphthyl | NEt2 | 33 | 773721 | 28 |
| 4-MeC6H4 | NMePh | 64 | 791347 | 24 |
| Ph | NMePh | 55 | 791348 | 24 |
| Ph | Morpholino | 98 | 4 | |
| 4-MeOC6H4 | Morpholino | 91 | 4 | |
| 4-ClC6H4 | Morpholino | 85 | 4 | |
| 4-O2NC6H4 | Morpholino | 82 | 4 | |
| 4-O2NC6H4 | NEt2 | 33 | 852956 | 29 |
| 4-ClC6H4 | NiBu2 | 71 | 1586977 | 30 |
| 4-ClC6H4 | NnBu2 | 24 | 1586975 | 30 |
The first crystallographic study of two acylselenoureas is mentioned in a short note from 1963, in which only unit cell parameters and space groups are disclosed.31 The first full structure determination of an acylselenourea, that of PhC(O)NHC(Se)NHPh, was published by Hope in 1965,32 since then a total of 28 crystal structures of acylselenoureas have been deposited with the CCDC. A common feature in the structures derived from primary amines is the formation of a six-membered ring containing the NHC(Se)NHC(O) unit, which is stabilised through an intramolecular hydrogen bond between the N–H proton and the carbonyl oxygen atom (Fig. 3).
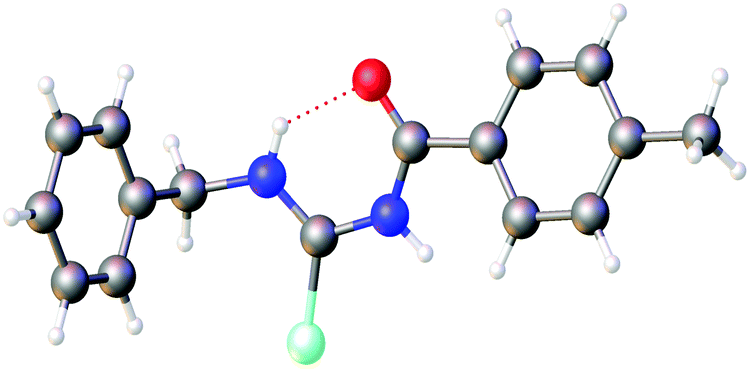 | ||
| Fig. 3 Six-membered ring structure formed by an intramolecular NH⋯O hydrogen bond in the molecular structure of p-TolC(O)NHC(Se)NBz. Atom colours: mint – selenium, blue – nitrogen, red – oxygen, grey – carbon, white – hydrogen. | ||
In the solid-state structures of acylselenoureas derived from secondary amines (Table 1), the intramolecular hydrogen bond can of course not be formed, thus the angle between the CO and CSe moieties varies from compound to compound.
It was the group of Beyer and Kirmse who first showed that PhC(O)NHC(Se)NEt2 reacts with Ni(II)-, Pd(II)- or Co(III)-acetate to give good yields of the square planar bis(chelate) complexes [M{PhC(O)NC(Se)NEt2}2] (M = Ni, Pd) or the octahedral tris(chelate) [Co{PhC(O)NC(Se)NEt2}3], in which the deprotonated acylselenourea acts as an O,Se-chelate ligand (Scheme 2).33 The proposed structures of the bis(chelate) complexes, in which the acylselenoureato ligands are mutually cis, was initially deduced from single-crystal EPR spectroscopy of the Cu(II) complex [Cu{PhC(O)NC(Se)NiBu2}2];34 the first X-ray crystal structures of such metal bis(selenoureato) complexes appeared only in the 1990's (see below).
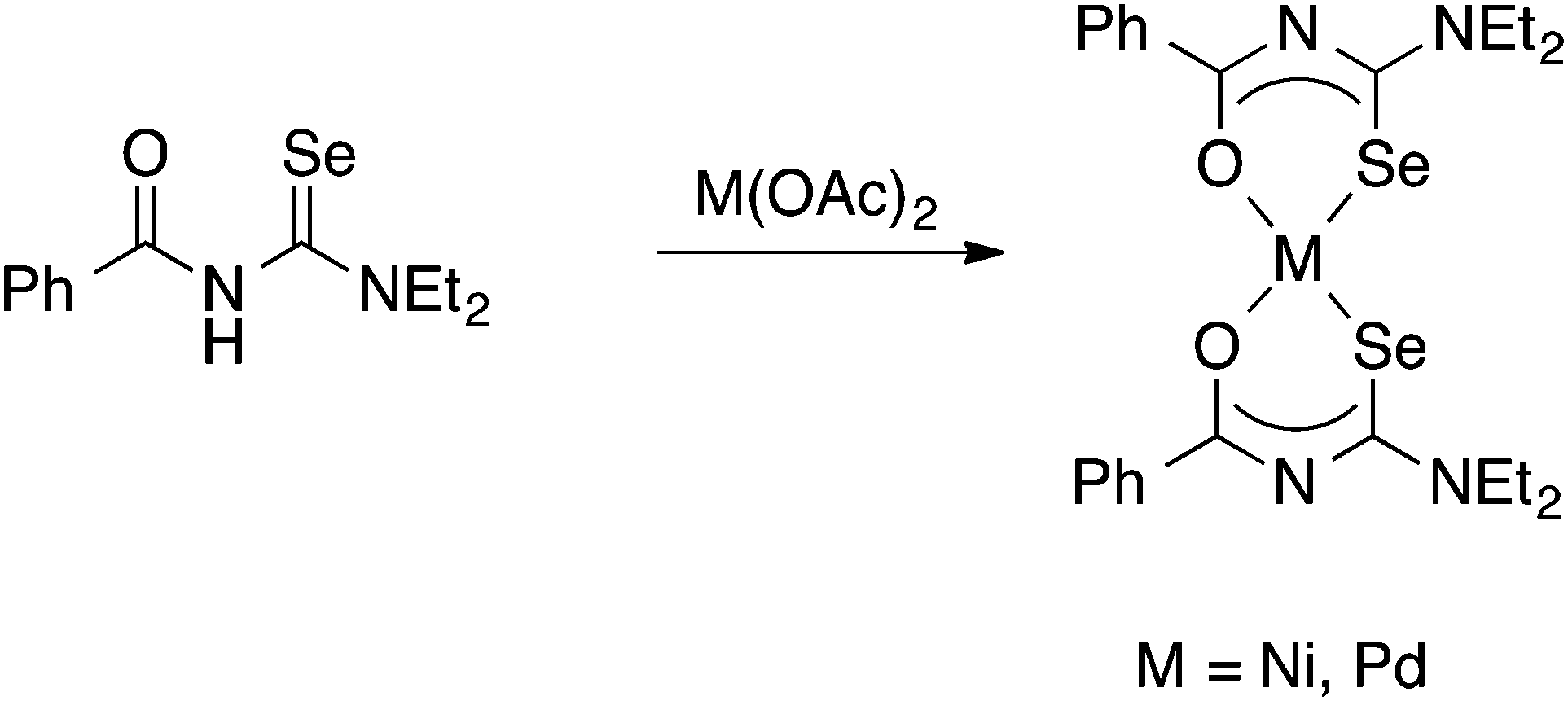 | ||
| Scheme 2 | ||
Subsequently, the same group reported detailed studies of mainly Ni(II) and Cu(II) bis(selenoureato) complexes. These investigations focused on EPR spectroscopy,34–39 ESCA spectroscopy,40,41 NMR spectroscopy42–44 as well as mass spectrometry.45,46 A summary of these results from the Beyer group was published in a review in 1983.2 In the mid-1980's the group of König began investigating chromatographic metal separation using acylselenoureas as chelating agents and found that good separation with very low sensitivities (in the ng range) could be achieved.22,47,48 Even earlier, a Spanish group utilised the coloured products formed by the reaction of acylselenoureas with palladium or osmium salts for the spectrophotometric determination of these metals. Using the sarcosine derived acylselenourea PhC(O)NHC(Se)NHCH2C(O)OEt, palladium(II) could be determined in acidic solutions over a linear range of 4–16 ppm.49 Os(VI) was also detected over a similar concentration range using the same acylselenourea.50 Similar results were also obtained using PhC(O)NHC(Se)NHPh.51 A further example of the application of acylselenoureas in metal analysis is the spectrophotometric determination of Ru(III) and Os(VI) using PhC(O)NHC(Se)NMePh.52 In all these examples however, the exact nature of the coloured species formed by interaction of the acylselenoureas with the metal ions was not further investigated.
In more recent times, various groups have explored the use of acylselenoureato metal complexes as single-source precursors for various metal selenide nanomaterials or thin films. The cadmium complex [Cd{PhC(O)NC(Se)NEt2}2] was used by Koch for the preparation of cubic-phase CdSe nanoparticles via a controlled thermolysis method.20 Similarly, the lead complexes [Pb{PhC(O)NC(Se)NEt2}2], [Pb{4-O2NC6H4C(O)NC(Se)NiBu2}2], [Pb{2-napC(O)NC(Se)NEt2}2] as well as the Pd(II) derivative [Pd{2-napC(O)NC(Se)NEt2}2] were used as precursors for the deposition of PbSe or PdSe thin films.27,28,53–57 The same group also used the tris(chelate) iron(III) compound [Fe{2-napC(O)NC(Se)NEt2}3] to obtain FeSe nanocrystals and thin films.58 The ferrocenyl-substituted selenourea [3-FcC6H4NHC(Se)NHC(O)-4-MeC6H4] was used as a single-source precursor to deposit FeSe nanoparticles on carbon nanotubes for photocatalytic applications.17 Our group has also studied the thermal behaviour of lead selenoureato bis(chelates) and the microwave-assisted formation of PbSe nanoparticles in ionic liquids.30
Limited data on the reactivity of bis(selenoureato) metal complexes is available. The Ni(II) complex [Ni{PhC(O)NC(Se)NEt2}2] reacts with diphosgene in benzene to afford a heterocyclic diselenazolium salt (Scheme 3), which was structurally characterised.59
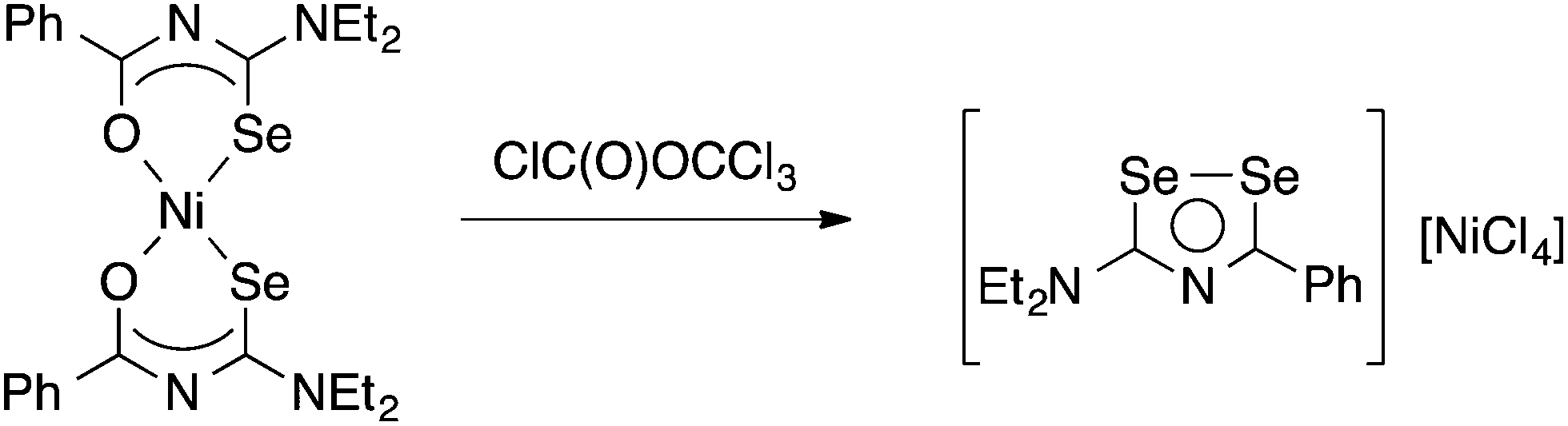 | ||
| Scheme 3 | ||
Although the preparation of N-(thiocarbamoyl)benzimidoylchlorides by reaction of the Ni(II) bis(thioureato) complexes [Ni{PhC(O)NC(S)NR2}2] with SOCl2 proceeds in good yields, the analogous N-(selenocarbamoyl)benzimidoylchlorides cannot be prepared by this method; they are however readily accessible from direct reaction of the acylselenoureas with thiophosgene (Scheme 4).60 Such compounds can however be much easier prepared by reacting an imidoyl chloride with KSeCN and subsequent treatment with an amine (Scheme 5).61,62
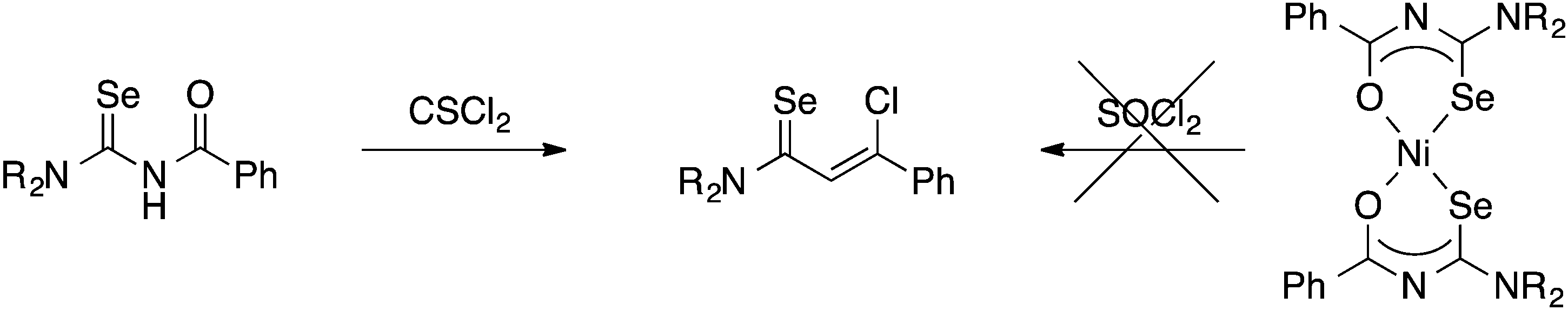 | ||
| Scheme 4 | ||
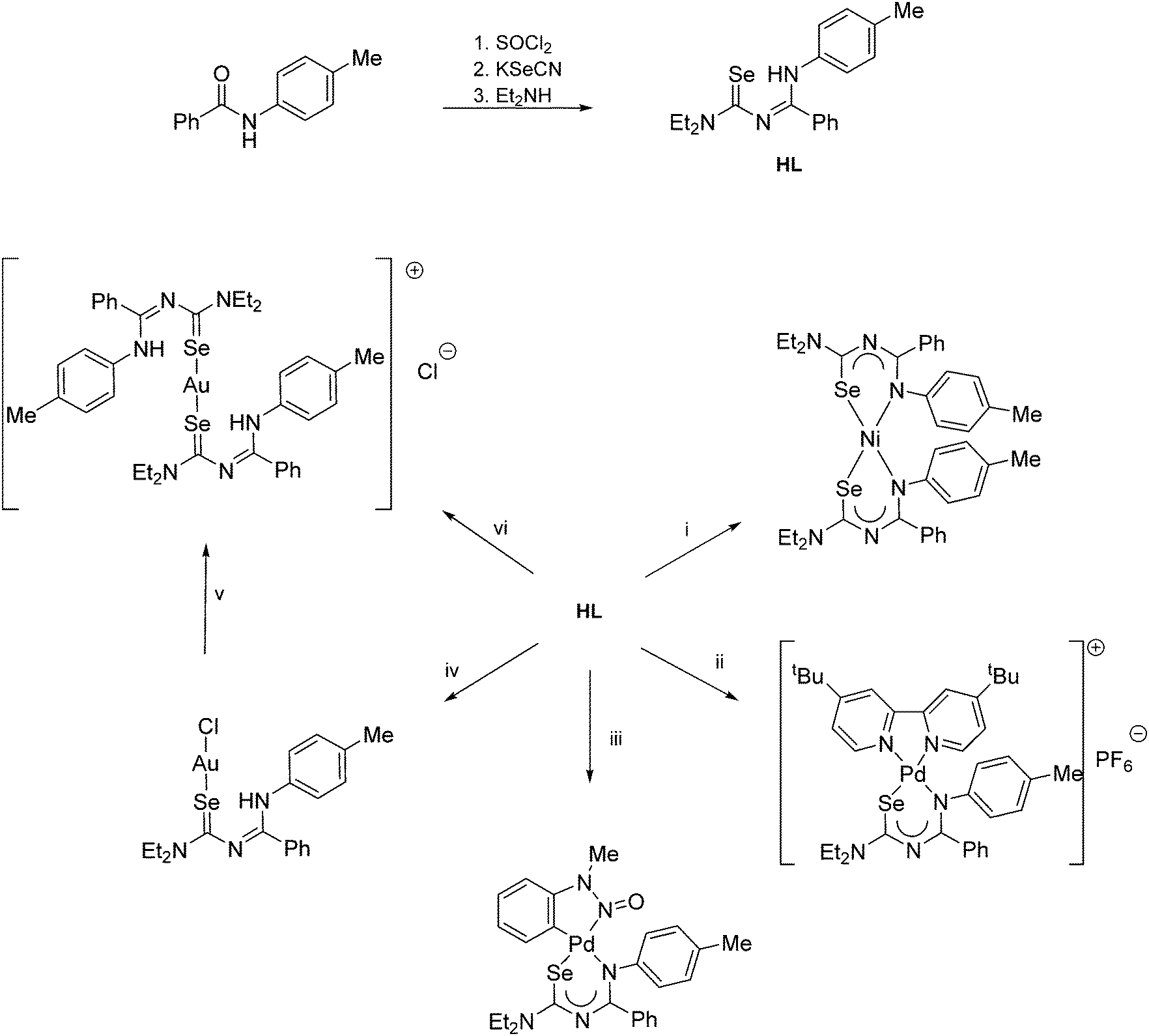 | ||
| Scheme 5 (i) Ni(OAc)2. (ii) [PdCl2(tBu2bipy)], Et3N, NH4PF6. (iii) [Pd(OAc){C6H4N(Me)NO}]2. (iv) 0.5 eq. H[AuCl4] or [AuCl(SMe2)]. (v) Standing in CDCl3. (vi) 0.5 eq. [AuCl(SMe2)]. | ||
The coordination chemistry of the diethyl derivative (HL in Scheme 5) has been investigated with various transition metals.62 Whilst its reactivity is generally very similar to that of the acylselenoureas, the structures of the chelate rings are considerably different. Whereas the acylselenoureato complexes feature almost planar chelate rings, the N-imidoselenoylcarbamato complexes display a boat-like conformation of the chelate rings. This difference has been attributed to a lesser degree of conjugation of the negative charge along the ligand backbone of the imodoyl derivatives, which was supported by DFT calculations.
Tetradentate acylselenoureas derived from isophthaloyl- and terephthaloylchloride are known, but were reported to be quite unstable. However, their Ni(II) and Cu(II) complexes (Scheme 6) could be isolated and were characterised by mass spectrometry, X-ray crystallography and EPR spectroscopy.39,63,64
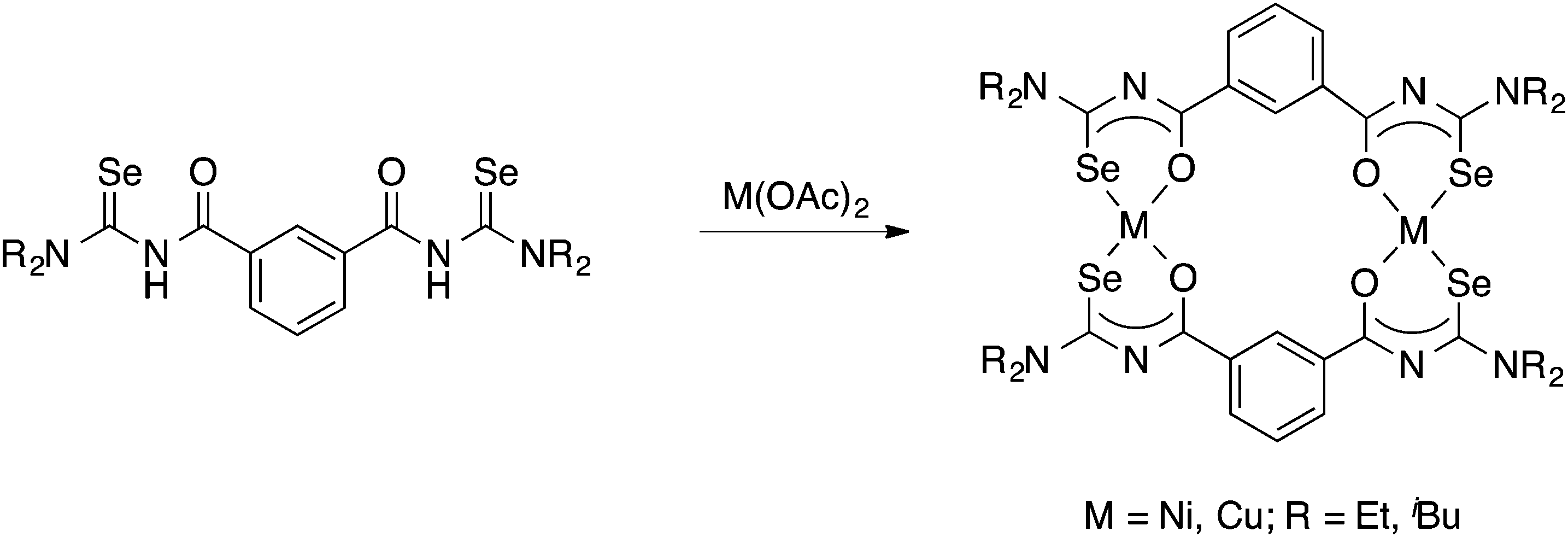 | ||
| Scheme 6 | ||
From the early 1990's a series of papers reporting the X-ray crystal structures of various metal complexes with PhC(O)NHC(Se)NEt2 appeared. These include the main group metal complexes [Tl{PhC(O)NC(Se)NEt2}]2,65 [In{PhC(O)NC(Se)NEt2}3]66 and [Pb{PhC(O)NC(Se)NEt2}2]67 as well as the transition metal complexes [Cd{PhC(O)NC(Se)NEt2}2],68 [Zn{PhC(O)NC(Se)NEt2}2],69 [Ni{PhC(O)NC(Se)NEt2}2]70 and [Co{PhC(O)NC(Se)NEt2}3].71 The structure of the Pd(II) bis(chelate) complex [Pd{PhC(O)NC(Se)NnBu2}2] as well as that of the selenourea itself was also communicated.21 Examples of selected structures are depicted in Fig. 4.
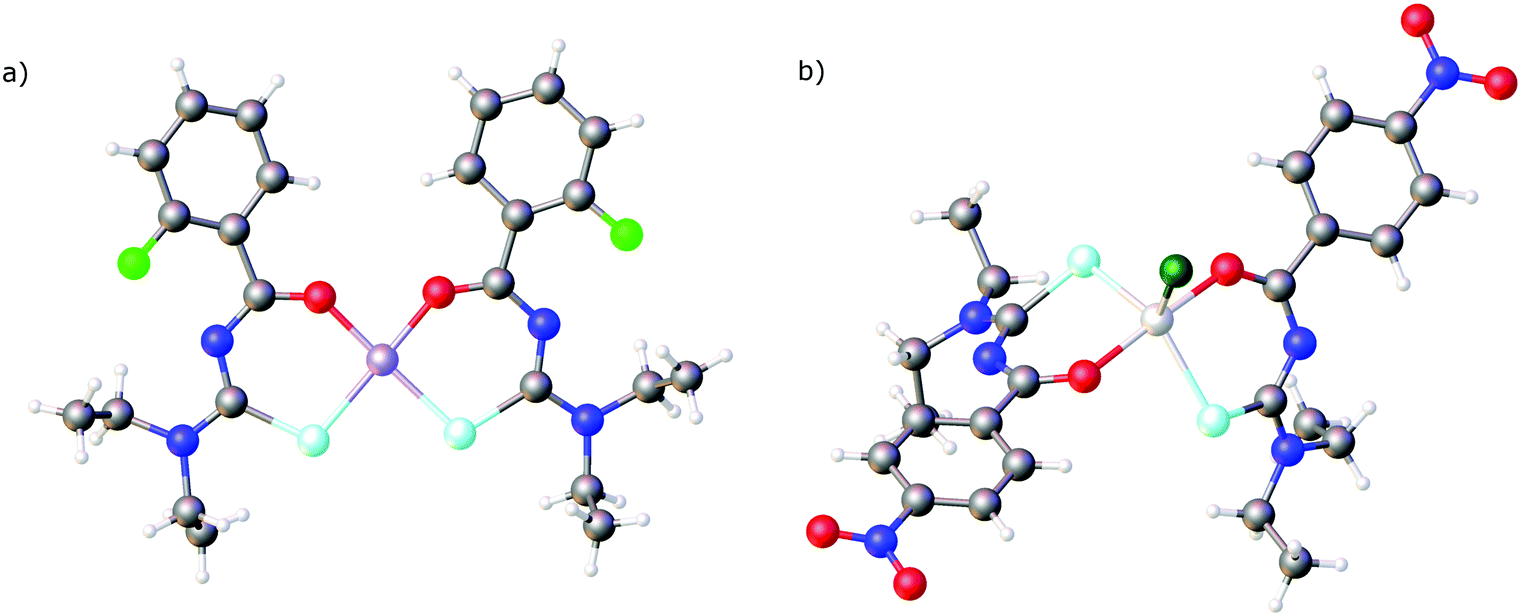 | ||
| Fig. 4 (a) Molecular structure of [Ni{(2-FC6H4)C(O)NC(Se)NEt2}]2. (b) Molecular structure of [GaCl{4-O2NC6H4C(O)NC(Se)NEt2}2]. Atom colours: mint – selenium, blue – nitrogen, red – oxygen, grey – carbon, lilac – nickel, light grey – gallium, light green – fluorine, dark green – chlorine, white – hydrogen. | ||
In all the structures mentioned above the deprotonated selenourea ligands adopt the cis O,Se-chelating coordination mode. To the best of our knowledge there is only one known compound in which two chelating acylselenoureas coordinate to a metal in a trans fashion: Crystals of the gallium(III)chlorido complex [GaCl{4-O2NC6H4C(O)NC(Se)NEt2}2] (Fig. 4b) were obtained during attempts to prepare the corresponding tris(chelate).72
Over the past years we also reported the first examples of metal complexes in which the acylselenoureato ligands coordinate to a metal only via selenium (Fig. 5). These include the mono- and dinuclear gold(I) complexes containing phosphines or N-heterocyclic carbene co-ligands.73,74 In the X-ray structures of these complexes the acylselenoureato moiety is rotated in such a way, that the hard oxygen atom points away from the soft metal centre. These gold(I) complexes were also amongst the first examples of heteroleptic acylselenourea complexes in which there are additional ligands present in the coordination sphere of the metal.
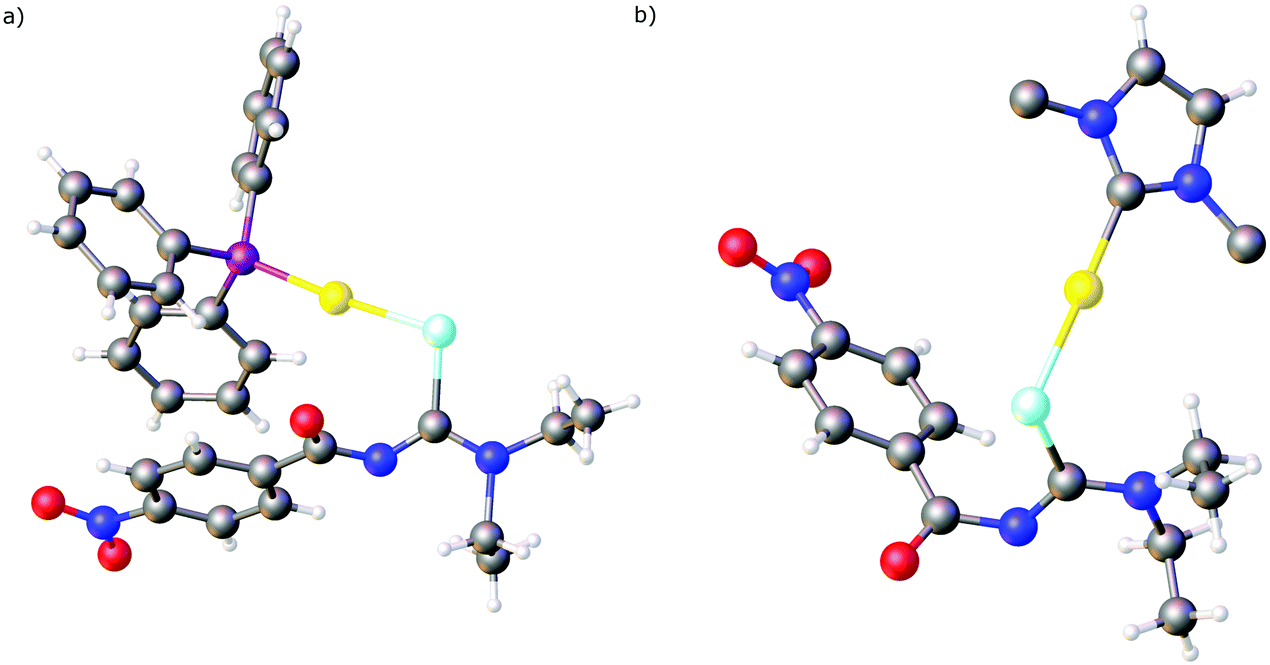 | ||
| Fig. 5 (a) Molecular structure of [Au(PPh3){4-O2NC6H4C(O)NC(Se)NEt2}]. (b) Molecular structure of [Au(IPr){4-O2NC6H4C(O)NC(Se)NEt2}]. Only the ipso-carbon atoms of the 2,6-(iPr)2C6H3 groups are shown. Atom colours: mint – selenium, blue – nitrogen, red – oxygen, magenta – phosphorous, grey – carbon, yellow – gold, white – hydrogen. | ||
We have subsequently reported the preparation and structural characterisation of organopalladium(II) compounds containing chelating acylselenourea ligands as well as monoanionic C–N or C–Se ligands by cleavage reactions of acetate- or chloro-bridged cyclopalladated species with the corresponding acylselenoureas (Scheme 7).75,76 Similarly, the polymeric organogold(III) compound [AuCl(tBu2Bip)]n containing a dicarbanionic biphenyl-derivative reacts with acylselenoureas in the presence of base, giving the corresponding chelate complexes (Scheme 7).77 It is important to note, that the carbanionic ligand stabilises the gold(III) oxidation state in these complexes. Reactions of acylselenoureas with gold(III) salts typically lead to reduction of the gold species and oxidation of the selenium compound. Cationic Pd(II) and Pt(II) complexes of the type [M{ArC(O)NC(Se)NR2}(L–L)]+ (M = Pd, Pt) featuring bidentate nitrogen- (bipyridine, phenanthroline) or phosphorus-ligands (dppe or PPh3) and monoanionic acylselenoureato ligands have been prepared and structurally characterised (Scheme 7).24,78 The reaction of [RuCl2(p-cym)]2 with acylselenoureas and Ph3P in the presence of NH4PF6 afforded good yields of the organoruthenium(II) cations [M{ArC(O)NC(Se)NR2}(p-cym)(PPh3)]+, which were fully characterised including several X-ray structures (Scheme 7).78 These complexes are chiral at the metal centre and feature four different ligating atoms (Se, O, P, and C) bound to ruthenium. In addition to structural data, we have collected detailed NMR spectroscopic data, in particular 77Se chemical shifts for both the acylselenoureas and the metal complexes. Studies of the biological activity of some of these substances against various tumour cell lines were also undertaken.29,78
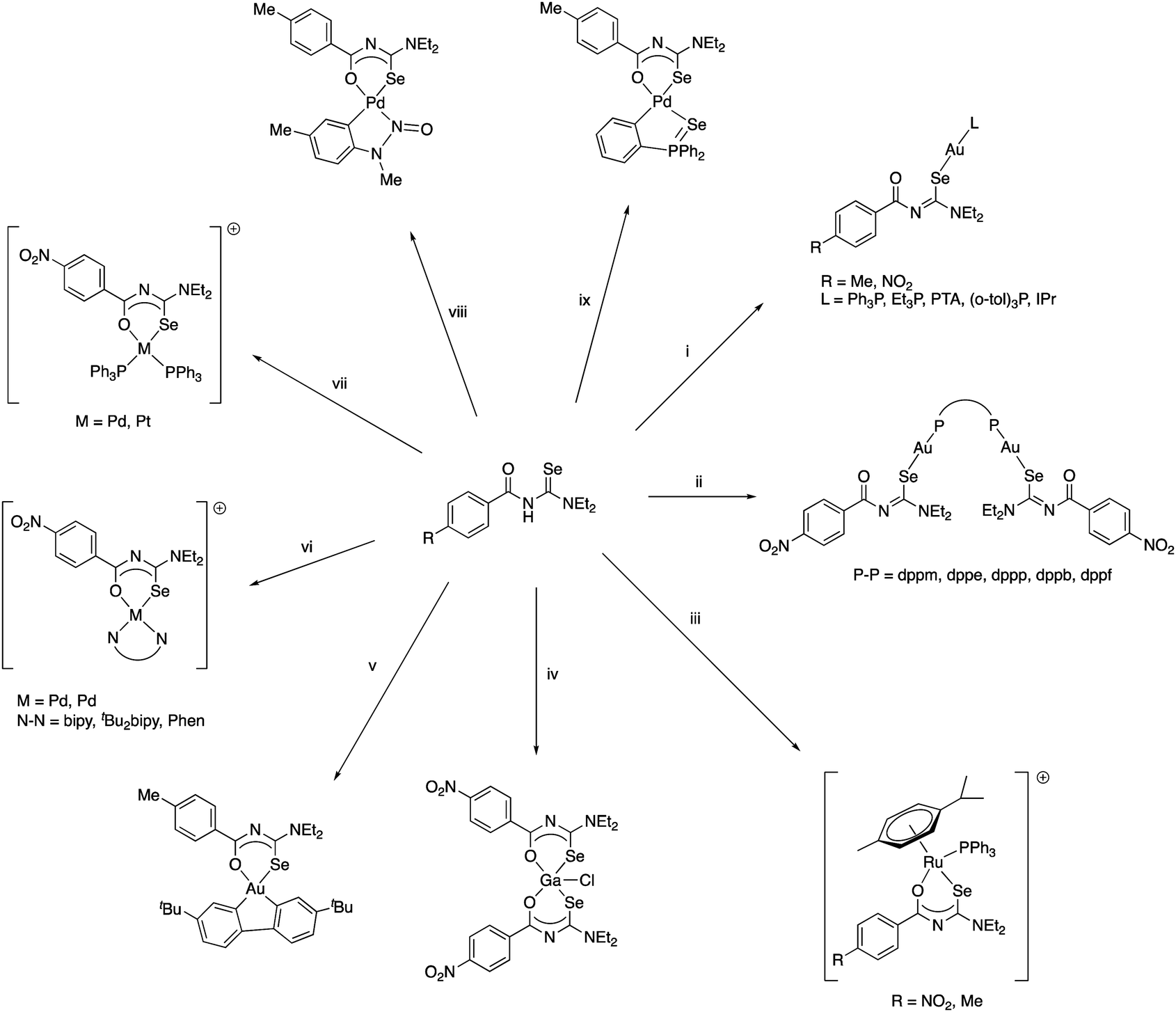 | ||
| Scheme 7 (i). [AuCl(L)] (L = Ph3P, Et3P, PTA, P(o-tol)3, IPr), base. (ii) [Au2Cl2(P–P)] (P–P = dppm, dppb, dppp, dppb, dppf), NaOAc. (iii) [RuCl2(p-cym)]2, PPh3, Et3N, NaBPh4. (iv) GaCl3, EtOH. (v) [AuCl(tBu2Bip)]n, NaOMe. (vi) cis-[MCl2(N–N)] (M = Pd, Pt); N–N = bipy. | ||
The results of a study of the reactivity of some acylselenoureas with the oxorhenium(V) salt nBu4N[ReOCl4] have also been reported.79 Depending on the reaction conditions, different metal complexes were isolated and structurally characterised. In most cases, the deprotonated acylselenourea adopts the typical O,Se-chelating mode. However, one complex was isolated in which two of the three acylselenoureato ligands are chelating whilst the third bonds to the ReO-core only through the selenium atom (Fig. 6).
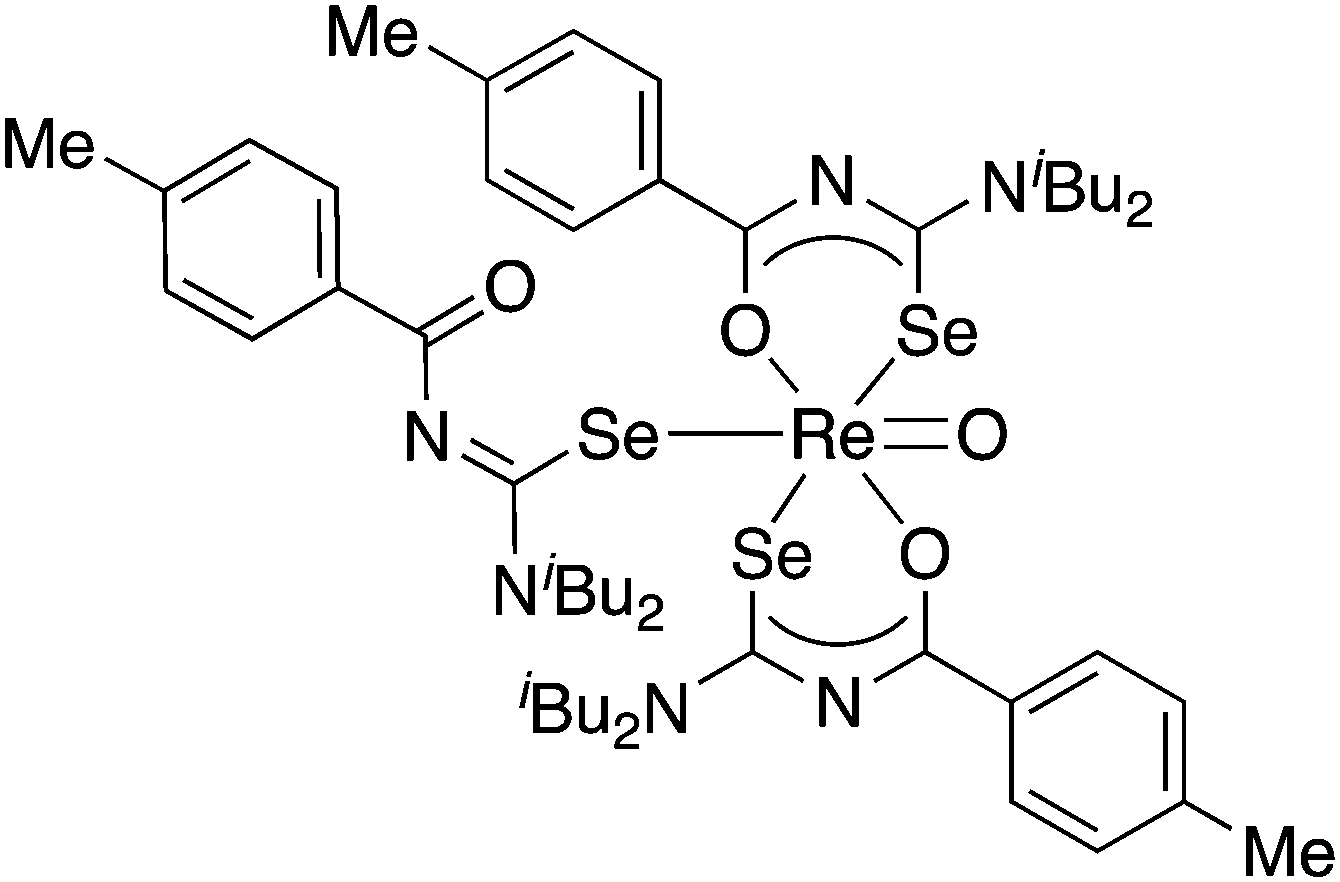 | ||
| Fig. 6 An oxorhenium(V) complex containing both anionic Se,O-chelating and neutral Se-bound acylselenourea ligands. | ||
More recently we found, that Ag2O dissolves in a solution of acylselenoureas, forming tetranuclear silver(I) clusters [Ag4{ArC(O)NC(Se)NEt2}4] containing the deprotonated acylselenoureato ligands (Fig. 7a).80 These clusters react with four or eight equivalents of a phosphine affording dinuclear Ag(I) compounds [Ag2{ArC(O)NC(Se)NEt2}2(P)2] or the mononuclear complexes [Ag{ArC(O)NC(Se)NEt2}(P)2] (P = Ph3P, PTA), respectively (Scheme 8).
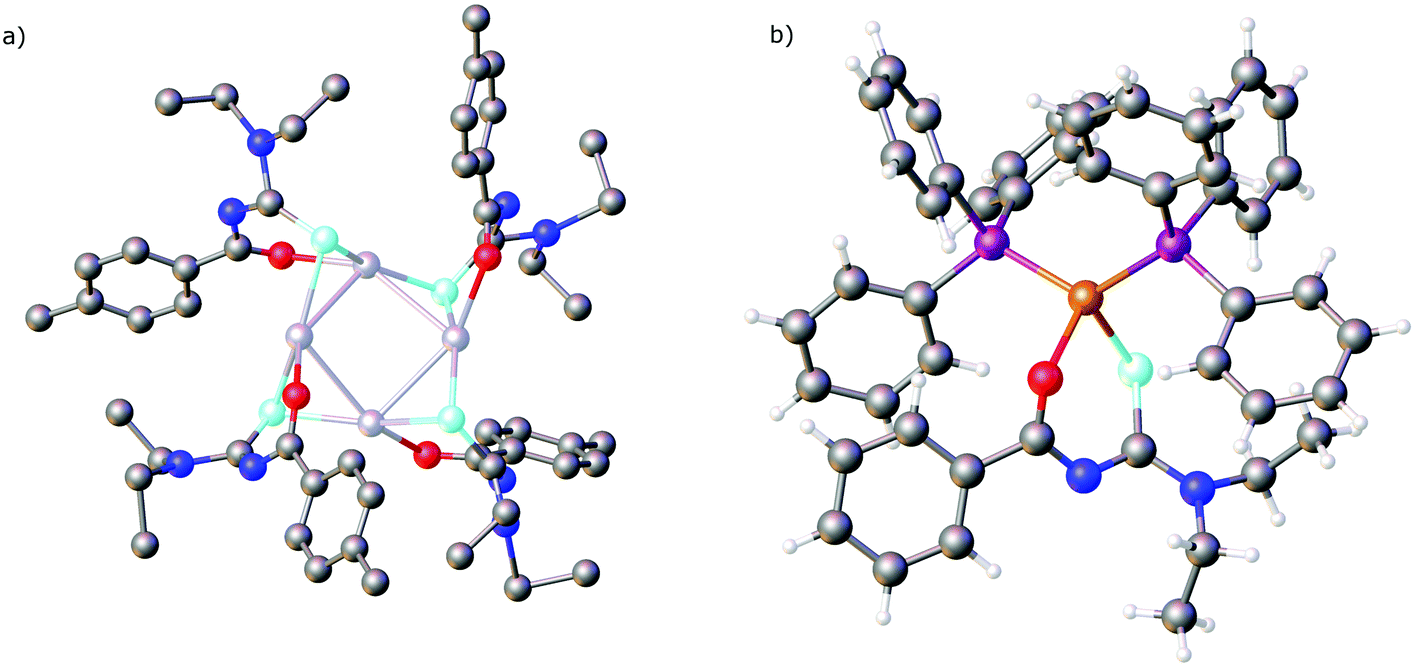 | ||
| Fig. 7 (a) Molecular structure of [Ag4{PhC(O)NC(Se)NEt2}4]. Hydrogen atoms have been omitted for clarity. (b) Molecular structure of [Cu(Ph3P)2{PhC(O)NC(Se)NEt2}]. Atom colours: mint – selenium, blue – nitrogen, red – oxygen, grey – carbon, magenta – phosphorous, light grey– silver, orange – copper, white – hydrogen. | ||
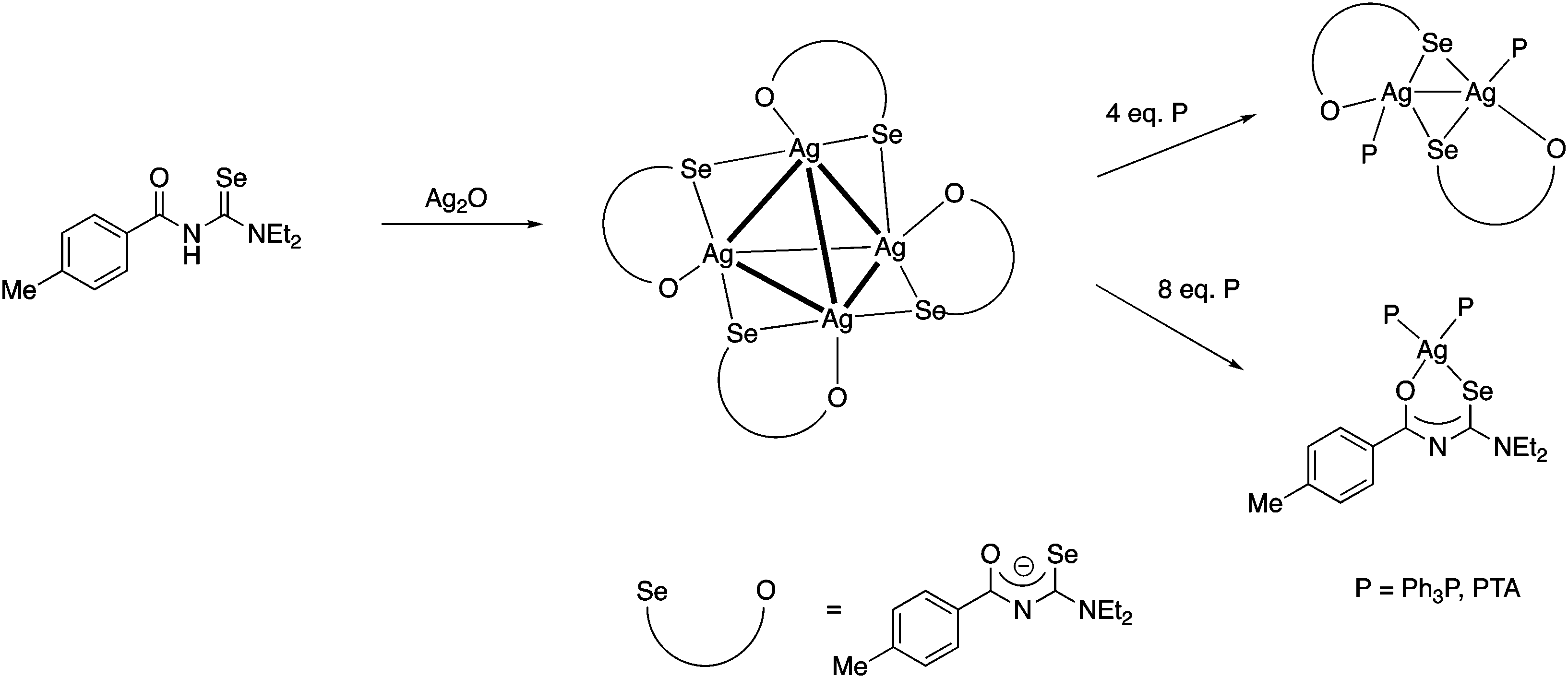 | ||
| Scheme 8 | ||
Although acylselenoureato complexes of copper(II) have long been known and are well-studied, their Cu(I) counterparts were only reported recently. The phosphine and N-heterocyclic carbene Cu(I) compounds [Cu{ArC(O)NC(Se)NEt2}(Ph3P)2] (Fig. 7b) and [Cu{ArC(O)NC(Se)NEt2}(IPr)] containing deprotonated chelating acylselenourea ligands are readily prepared from the acylselenoureas and the copper(I) compounds [CuCl(IPr)] or [Cu(Ph3P)2]NO3.81 The presence of the co-ligands is essential in this case to stabilise the copper(I) oxidation state.
3. Selenosemicarbazones
Selenosemicarbazones RR′CNNHC(Se)NH2 were first reported in the 1950's by Huls and Renson.82,83 By varying the nature of the substituents on the imine carbon atom, a large number of derivatives can be synthesised. Such selenosemicarbazones are accessible from the condensation of acetone selenosemicarbazone, cyclohexanone selenosemicarbazone or selenosemicarbazide with an aldehyde or ketone. Whilst selenosemicarbazide H2NC(Se)NHNH2 was commercially available, acetone selenosemicarbazone82–84 or the more stable cyclohexanone selenosemicarbazone85 can be prepared in reasonable yields from the reaction of KSeCN with hydrazine hydrate in the presence of the ketone under acidic conditions. We have published an improved, reproducible preparation for this synthon together with its X-ray crystal structure.86 If the substituent on the imine carbon carries a potential donor atom, a functionalised selenosemicarbazone is obtained. For the purpose of this article, a compound of the type D–CRNNHC(Se)NH2, where D represents a donor atom (N, O or P) shall be defined as a functionalised selenosemicarbazone. Thus, in the following discussion the selenosemicarbazones are grouped according to the type of donor atom.
3.1 Complexes of selenosemicarbazones containing no additional donor atoms
The very first report of a selenosemicarbazone as ligand in a metal complex dates back to 1968. The group of Gerbeleu showed that cyclohexanone selenosemicarbazone or benzaldehyde selenosemicarbazone act as neutral Se-donors in octahedral Co(III) complexes containing dimethylglyoximato ligands forming either complexes of the type [CoCl(DMG)2(HLSe)] or [Co(DMG)2(HLSe)2]+ (DMG = dimethylglyoximato; HLSe = neutral selenosemicarbazone), depending on the reaction stoichiometry.87 Acetone selenosemicarbazone or cyclohexanone selenosemicarbazone react with divalent nickel salts in the presence of base to afford diamagnetic, square planar complexes of the type [Ni(LSe)2] (LSe = monoanionic selenosemicarbazone).88 Here the deprotonated selenosemicarbazones coordinate to the metal as monoanionic [N,Se]− ligands. In the absence of a base, the chelate complexes [NiCl2(HSe)] or [ZnCl2(HSe)] containing neutral selenosemicarbazone ligands are formed with Ni2+ and Zn2+ ions, respectively.88 A series of octahedral nickel(II) and cobalt(II) complexes containing thio- or selenocyanato ligands as well as neutral, Se-bound selenosemicarbazones [M(NCE)2(HLSe)4] (M = Ni, Co; E = S, Se) was reported by Gerbeleu.89 Through detailed IR-spectroscopic studies it was established that the chalcogenocyanate ligands are N-bound to the metal. In 1983 three papers on the coordination chemistry of acetone selenosemicarbazone and furfural selenosemicarbazone with nickel(II) and cadmium(II) were published. In these compounds the ligands also act as monoanionic [N,Se]− ligands, forming bis(chelates) with the divalent metal centres. This structural assignment was however solely based on spectroscopic data.90–923.2 Complexes of selenosemicarbazones containing oxygen donor atoms
Oxygen functionalised selenosemicarbazones derived from salicylaldehyde, adipoin, 2-hydroxy-1-naphthaldehyde, pyruvic acid, phenylglyoxalic acid and diacetyl monoxime have been prepared and used as ligands in coordination chemistry (Fig. 8).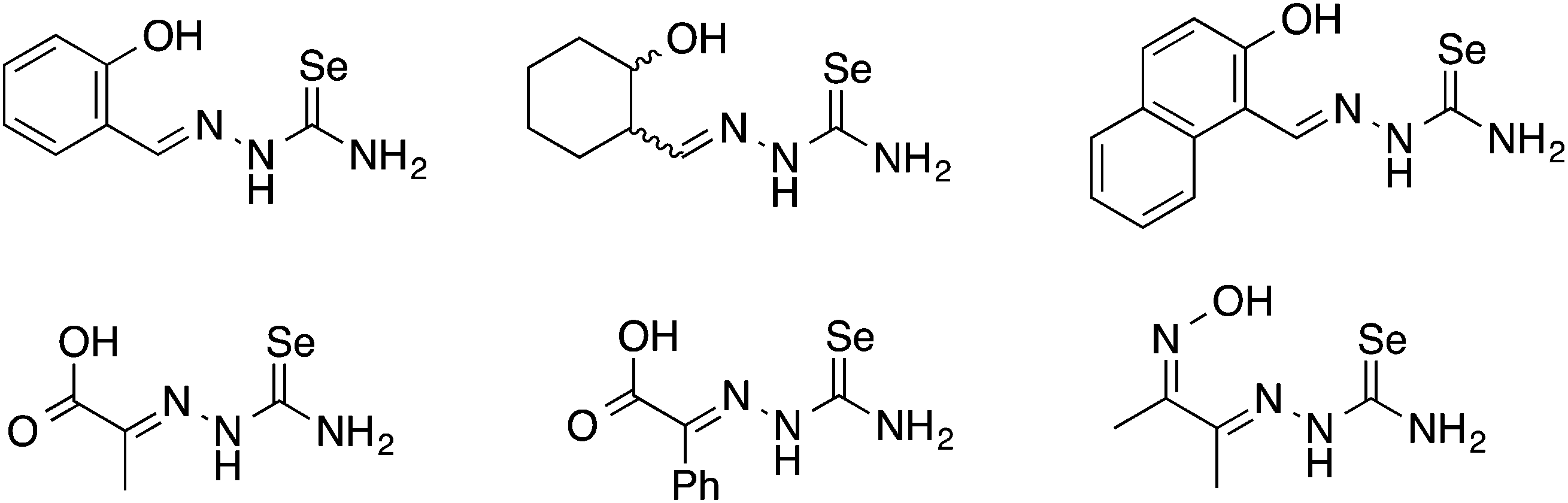 | ||
| Fig. 8 Examples of functionalised selenosemicarbazones containing oxygen donor atoms. | ||
Salicylaldehyde selenosemicarbazone (abbreviated here as H2SeLsal) reacts with Co3+, Cr3+ and Fe3+ ions in the presence of a base to form octahedral salts of the type [M(SeLsal)2]−, in which the salicylaldehyde selenosemicarbazone acts as a dianionic, tridentate [O,N,Se]2− ligand bound to the metal via the phenolic oxygen-, imine nitrogen- and selenium-atoms.93 Similarly, with CuCl2 in the presence of pyridine, the diamagnetic, square planar complex [Cu(SeLsal) (Py)] is obtained, in which [O,N,Se]2− coordination is also observed.94 The pyridine ligand can be substituted by bidentate N-donors such as 1,10-phenanthroline or 2,2′-bipyridine to give the five-coordinate complexes [Cu(SeLsal)(N–N)] (N–N = 1,10-phenanthroline or 2,2′-bipyridine). With vanadyl sulfate, salicylaldehyde selenosemicarbazone shows some unexpected reactivity: when ammonia or KOH are used as base and one additional equivalent of salicylaldehyde is present, an oxovanadium(IV) salt is formed, which contains the condensation product of salicylaldehyde selenosemicarbazone with salicylaldehyde (Fig. 9).95 In this case, the resulting tetradentate macrocycle coordinates to the metal only through the imine nitrogen atoms and the phenolic oxygen atoms. The selenium atom does not interact with the metal. The same template condensation process is also observed in complexes with Ni(II) and Cu(II) when NaOH or KOH are used as base.96
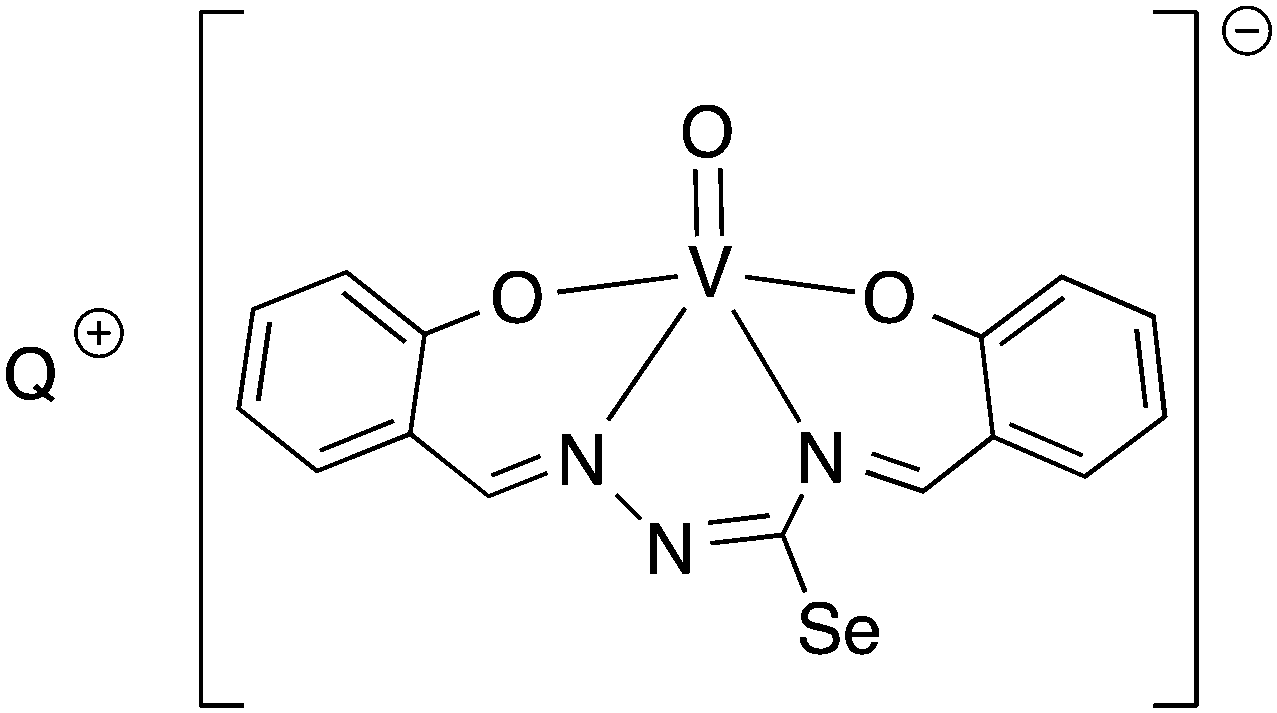 | ||
| Fig. 9 Vanadyl salt formed by template condensation of salicylaldehyde selenosemicarbazone with salicylaldehyde (Q = NH4, K). | ||
In the presence of pyridine or bidentate N-donors, six-coordinate V(IV) complexes with the [O,N,Se]2− coordination mode of salicylaldehyde selenosemicarbazone have been proposed.95 However, based on a crystal structure determination, we have observed that in such systems salicylaldehyde selenosemicarbazone in fact acts as a dianionic [O,N,N]2− ligand (Fig. 10), with the selenium atom not bound to the metal.97 Given that the vanadyl ion is classified as a hard base, it is perhaps not surprising, that the soft selenium atom does not coordinate to vanadium.
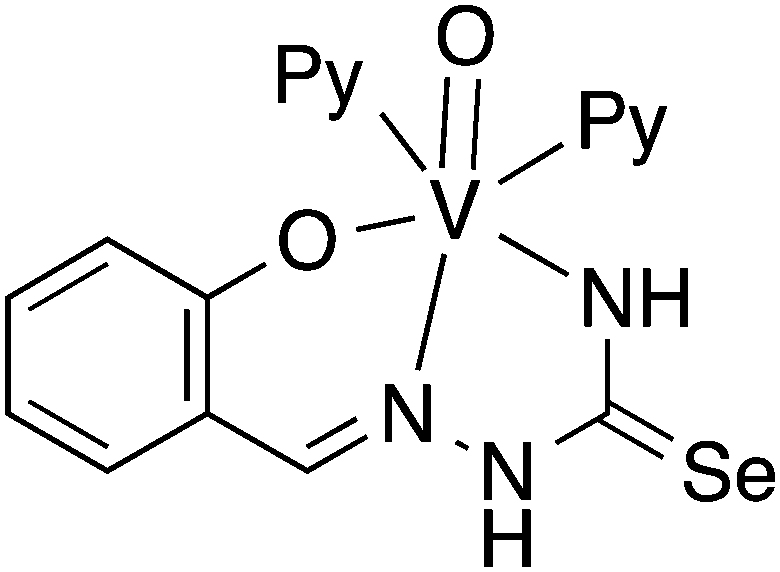 | ||
| Fig. 10 Salicylaldehyde selenosemicarbazone as a dianionic [O,N,N]2− ligand. | ||
The group of Gerbeleu also reported the first X-ray structure of a metal complex containing salicylaldehyde selenosemicarbazone, which confirmed that the compound indeed acts as a dianionic, tridentate [O,N,Se]2− ligand in the nickel(II) complex [Ni(SeLsal)(PPh3)] (Fig. 11).98,99
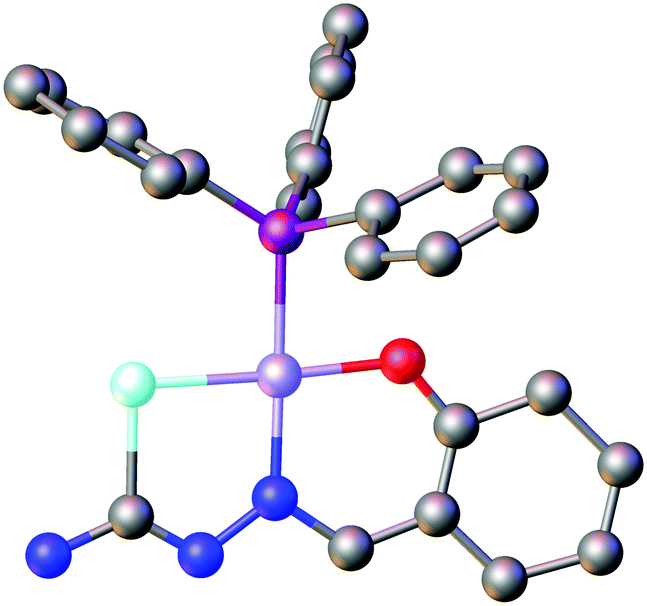 | ||
| Fig. 11 Molecular structure of [Ni(SeLsal)(PPh3)]. Hydrogen atoms have been omitted for clarity. Atom colours: mint – selenium, blue – nitrogen, red – oxygen, grey – carbon, magenta – phosphorous, lilac – nickel. | ||
Structurally related to salicylaldehyde selenosemicarbazone is adipoin selenosemicarbazone, which is reported to form analogous complexes with Co3+, Ni2+, Zn2+ and Cu2+ ions.100,101 These compounds however, were reported to be less stable than their salicylaldehyde selenosemicarbazone counterparts.
The selenosemicarbazones derived from pyruvic acid and phenylglyoxalic acid both contain a carboxylic acid functional group, which upon deprotonation may coordinate to a metal centre (in addition to the N- and Se-atoms of the selenosemicarbazone). Although no structural data has so far been reported, the group of Gerbeleu has studied the coordination chemistry of these selenosemicarbazones with various metals. The selenosemicarbazone derived from pyruvic acid forms oxovanadium(IV) complexes in the presence of pyridine or 3-picoline, similar to those with salicylaldehyde selenosemicarbazone.95 Other complexes containing pyruvic acid or phenylglyoxalic acid selenosemicarbazone with Co3+, Ni2+, Cu2+, Fe3+, Fe2+ and Cr3+ have also been investigated.102,103 In each case, in the presence of a base, the selenosemicarbazones act as dianionic, tridentate [O,N,Se]2− ligands (Fig. 12).
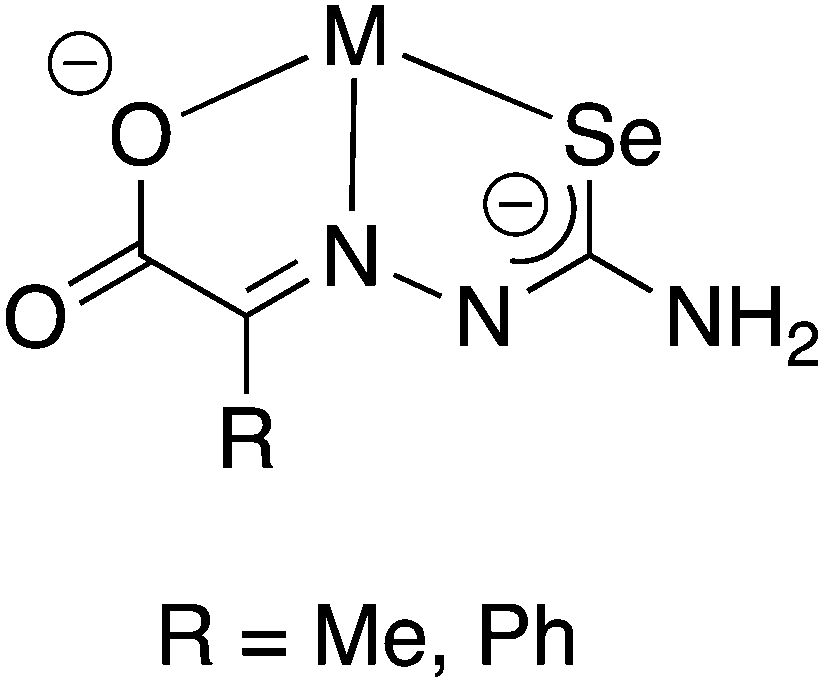 | ||
| Fig. 12 Schematic illustration of the proposed coordination mode of the doubly deprotonated selenosemicarbazones of pyruvic acid or phenylglyoxalic acid. | ||
Although the selenosemicarbazone derived from diacetyl monoxime MeC(NOH)C(Me)NNHC(Se)NH2 (abbreviated here as H2SeLOx) contains a hydroxy group, there is so far no evidence that the oxygen atom coordinates to a metal centre. In a series of papers, the group of Gerbeleu reported the coordination chemistry of this ligand with copper, nickel, cobalt and iron. The selenosemicarbazone reacts with copper(II) salts to give green, paramagnetic complexes of the type [CuX2(H2SeLOx)] (X = Cl, Br), in which the neutral ligand is bound to the metal through the nitrogen atoms of the oxime and imine and the selenium atom.104 In the presence of one equivalent of sodium acetate the hydroxy-group of the ligand is deprotonated and compounds formulated as [CuX(HSeLOx)] (X = Cl, Br) were isolated. With an excess of sodium acetate, a poorly soluble brown complex containing the doubly deprotonated ligand was obtained. With nickel(II) salts in the absence of base, brown paramagnetic products containing the octahedral cations [Ni(H2SeLOx)2]2+ are formed. If the reaction was carried out in aqueous ammonia a brown diamagnetic product formed, which was formulated as the square planar species [Ni(SeLOx)(NH3)].105 When nickel(II) acetate is used in the reaction, a poorly soluble diamagnetic material was isolated. In the latter two complexes, the doubly deprotonated selenosemicarbazone is bound to the metal through the nitrogen atoms of the oxime and imine and the selenium atom. Cobalt(II) salts react with the selenosemicarbazone under aerobic conditions to give the octahedral cobalt(III) salts [Co(HSeLOx)2]X (X = Cl, Br, I, NO3, NCS).106 Under strictly anaerobic conditions some Co(II) compounds could also be isolated, but their instability prevented detailed characterisation. Similar reactions were examined with iron(II) and iron(III) salts.106 With hydrated CrCl3 a material was isolated, which was formulated as the neutral octahedral species [Cr(SeLOx)(HSeLOx)], containing one monoanionic and one dianionic selenosemicarbazone ligand.107 In all these examples, the proposed structures are based on elemental analysis, molar conductivity, magnetic measurements and powder diffraction data. To date there are however no reported X-ray structures of complexes containing this selenosemicarbazone to confirm the structures of any of the products discussed above.
3.3 Complexes of selenosemicarbazones containing nitrogen donor atoms
Selenosemicarbazones containing additional nitrogen donor-atoms, which have been used as ligands, are derived from the condensation of selenosemicarbazide or its derivatives with 2-pyridinecarboxaldehyde, 2-acetylpyridine, 2-benzoylpyridine, 2-quinolinecarboxaldehyde or 8-quinolinecarboxaldehyde (Fig. 13).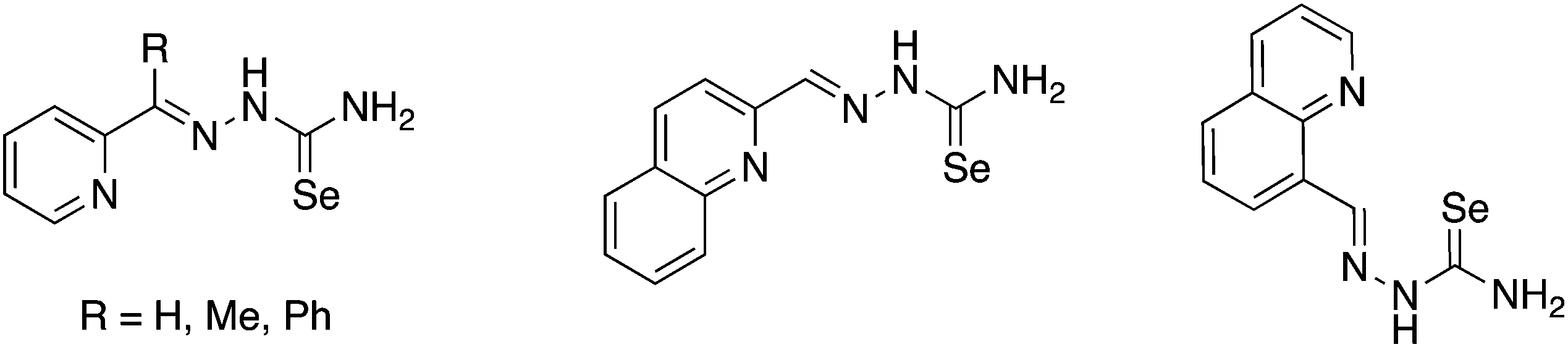 | ||
| Fig. 13 Examples of functionalised selenosemicarbazones containing nitrogen donor atoms. | ||
The first reported metal complexes with nitrogen-functionalised selenosemicarbazones date back to 1971 from the group of Gerbeleu. Reactions of the selenosemicarbazone derived from 8-quinolinecarboxaldehyde (abbreviated here as HSeL8quin) with Co(III), Ni(II) and Cu(II) salts was examined.108,109 The selenosemicarbazone can displace the water molecule in trans-[CoCl(DMG)2(H2O)] (DMG = dimethylglyoximato) to give the corresponding selenosemicarbazone complex in which the neutral ligand acts as a Se-donor.108 Interestingly, when the same reaction is carried out with two equivalents of the selenosemicarbazone both dimethylglyoximato ligands are displaced forming the octahedral cationic complex [Co(SeL8quin)2]Cl. In this compound, the deprotonated selenosemicarbazone binds to the metal through two nitrogen atoms (quinoline and imine) and the selenium atom. With nickel(II) salts paramagnetic octahedral salts [Ni(HSeL8quin)2]X2 (X = Cl, Br, I) and square planar diamagnetic compounds [NiX(SeL8quin)] (X = NCS, NO2) were isolated.108 The latter compound forms in the presence of base. With copper(II) salts products of the type [CuX2(HSeL8quin)] (X = Cl, Br, NO3) were formed but no reduction of the copper salt by the selenium compound was observed.109 The antibacterial and antifungal activity of these copper(II) complexes were subsequently studied, however neither the selenosemicarbazone nor its copper complexes were active.110 In 2016, the cationic cobalt(III) complex [Co(SeL8quin)2]ClO4 was prepared and fully characterised with modern spectroscopic methods.111 The X-ray structure unambiguously confirmed that the deprotonated selenosemicarbazone is bound to the metal through selenium- and two nitrogen-atoms. The complex showed considerable in vitro anticancer activity, and a variety of experiments (e.g. apoptosis induction, cell cycle changes and caspase activation) were carried out to determine possible mechanisms of action.111
The isomeric selenosemicarbazone derived from 2-quinolinecarboxaldehyde (abbreviated here as HSeL2quin) forms complexes of the type [MX(SeL2quin)] (M = Pt, Pt; X = Cl; M = Cd, X = OAc) as well as the octahedral bis(chelates) [M(SeL2quin)2] (M = Ni, Zn) and [Co(SeL2quin)2]ClO4.112–116 These compounds were tested for their anticancer activity and detailed experiments to elucidate their mode of action have been reported together with spectroscopic and structural data for several metal complexes and the selenosemicarbazone itself.
Several groups have studied the activity of 2-pyridyl-derived selenosemicarbazones as well as their metal complexes against Chagas disease and malaria.117–121 The coordination chemistry and biological activity of metal complexes containing selenosemicarbazones derived from 2-acetylpyridine (HSeLacpy) have been investigated in some detail over the years. Structural diversity can be increased by varying the substituents on the nitrogen atom at the 4-position. The synthesis of these compounds involves S-methylation of the corresponding thiosemicarbazones and subsequent selenation using in situ generated NaSeH (Scheme 9).119,121
 | ||
| Scheme 9 Synthesis of N4-disubstituted 2-acetylpyridine selenosemicarbazones. NR2 can be derived from dialkyl amines, cyclic amines or bicyclic amines. | ||
The octahedral iron(III) salt [Fe(LSeacpy)2][FeCl4] containing 2-acetylpyridine selenosemicarbazone substituted at the 4-position with the bicyclic amine 3-azabicyclo[3.2.2]nonane, was the first structurally authenticated metal complex containing an N-functionalised selenosemicarbazone.122 The same group also reported the corresponding square planar nickel(II) complex [NiCl(LSeacpy)].123 Other groups described Mn(II) as well as Ni(II) and Cu(II) complexes with the same selenosemicarbazone.120,124
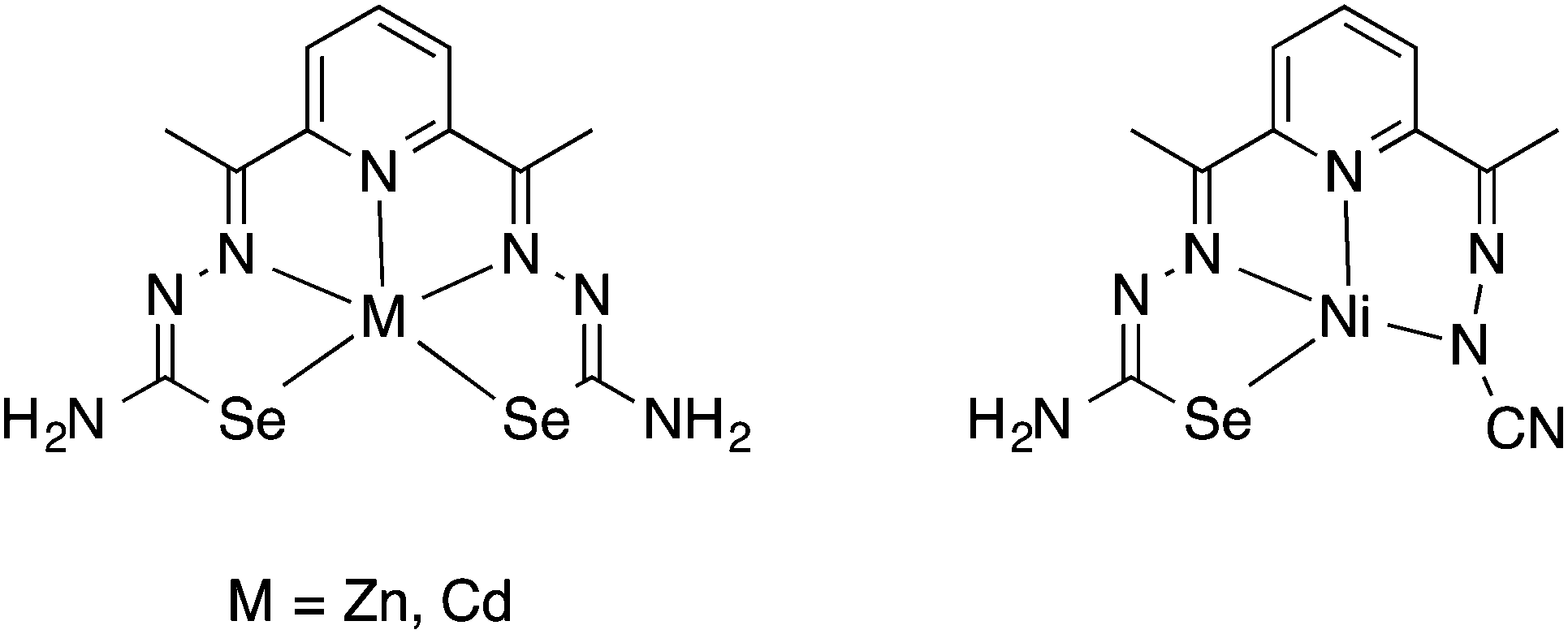
The N4-dimethyl derivative of 2-acetylpyridine selenosemicarbazone reacts with Ga(NO3)3 in the presence of NH4PF6 to afford the octahedral bis(chelate) salt [Ga(LSeacpy)2]PF6.125 X-ray structures for both the selenosemicarbazone and the gallium(II) complex were reported together with a study on the in vitro antitumor activity in two different cell lines. In a subsequent paper, the same group reported a more detailed study which also included the corresponding iron and ruthenium bis(chelate) salts.126 The authors demonstrated that the antitumor activity correlates with the atomic number of the chalcogen, the selenium compound thus being the most active compared to its sulfur and oxygen counterparts.
The mono- and dinuclear copper(II) complexes [Cu(OAc)(LSeacpy)] and [Cu2(LSeacpy)3]ClO4 were prepared from the reaction of the selenosemicarbazone with Cu(OAc)2 or Cu(ClO4)2 in the presence of Et3N, respectively.127 Both compounds were spectroscopically and structurally characterised. Furthermore, the copper complexes were shown to be involved in the generation of reactive oxygen species and target the lysosome to induce lysosomal membrane permeabilization.127 We reported a series of mono- and dinuclear gold(I) phosphine complexes containing derivatives of 2-acetylpyridine selenosemicarbazone.128 From X-ray diffraction experiments we could show that these are unique examples of compounds in which the potentially tridentate selenium ligands act as monoanionic Se-ligands towards the gold centre. Some of the compounds display antimalaria activity, although the activity of the corresponding sulfur compounds was higher.128
Five-coordinate complexes containing the main group metals In(III), Sb(III) and Bi(III) including [InBr2(LSeacpy)], [SbCl2(LSeacpy)] and [BiBr2(LSeacpy)] can be prepared from the corresponding metal trihalides and the selenosemicarbazone; the proton being eliminated as HX.129 The molecular structures were determined and the thermal behaviour of the indium compound was investigated by DSC/TGA. The complex melts at 262 °C and starts to lose more than 60% mass above 500 °C.129 There is thus potential for this type of complex as single-source precursor for InSe materials. Using the same synthetic strategy, we prepared a series of tin(IV) compounds starting from SnCl4 and the organotin halides [SnCl2R2] (R = Me, nBu, Ph).130 Depending on the tin compound used, different products were obtained and structurally characterised (Fig. 14). Whilst SnCl4 or [SnCl2(nBu)2] in all cases gave products of the type [SnCl(R)2(LSeacpy)] (R = Cl, nBu), [SnCl2(Ph)2] yielded product mixtures containing [SnCl2(Ph)(LSeacpy)] and [SnCl(Ph)2(LSeacpy)]. The former arises from elimination of PhH, whilst in the latter HCl is liberated during the reaction. Curiously, when using [SnCl2Me2], a salt was isolated containing the five-coordinate [SnMe2(LSeacpy)]+ cation. All of the compounds were characterised by 77Se and 119Sn NMR spectroscopy as well as by 119Sn Mössbauer spectroscopy.130
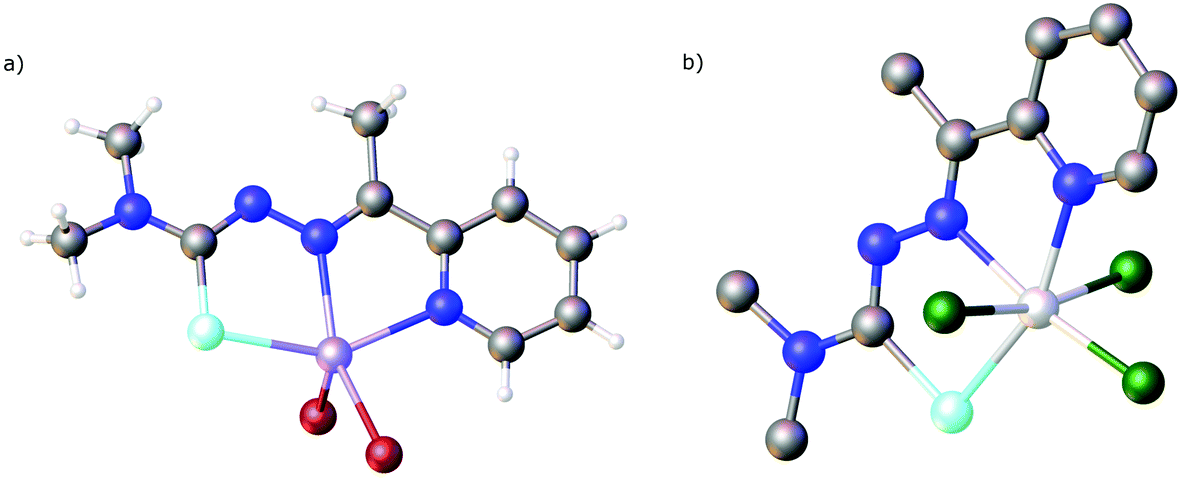 | ||
| Fig. 14 (a) Molecular structure of [InBr2(LSeacpy)]. (b) Molecular structure of [SnCl3(LSeacpy)]. Hydrogen atoms have been omitted for clarity. Atom colours: mint – selenium, blue – nitrogen, grey – carbon, blood red – bromine, lilac – indium, dark green – chlorine, light grey – tin, white – hydrogen. | ||
Based on the HSAB-principle, an anionic selenosemicarbazone is classified as a soft base, which would therefore tend to form stable complexes with soft metal ions. The vanadyl cation [VO]2+ is considered a hard metal centre and thus would not be expected to form complexes with soft bases such as a deprotonated selenosemicarbazone. Nevertheless, we could show that 2-acetylpyridine selenosemicarbazone readily reacts with [VO(acac)2] to give the green V(IV) complex [V(O)(acac)(LSeacpy)], containing both an acac-ligand and the deprotonated [N,N,Se]− coordinating selenosemicarbazone. This compound is oxidised in air to the yellow V(V) dioxo species [V(O)2(LSeacpy)] (Fig. 15).131 Together with the X-ray structures, we reported the EPR spectrum of the paramagnetic V(IV) complex.
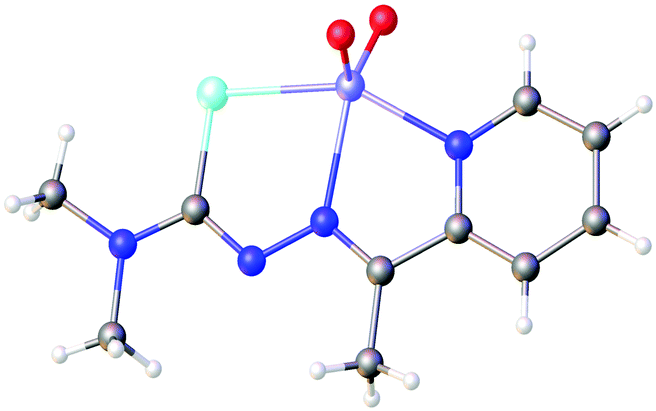 | ||
| Fig. 15 Molecular structure of [V(O)2(LSeacpy)]. Atom colours: mint – selenium, blue – nitrogen, red – oxygen, grey – carbon, lilac – vanadium, white – hydrogen. | ||
The reaction of 2-acetylpyridine selenosemicarbazone with [PdCl2(MeCN)2] or K2[PtCl4] gives the corresponding metal halide complexes [MCl(LSepy)] (M = Pd, Pt).132 In the presence of a silver salt and dppe, these complex reacts to afford the phosphine-bridged dinuclear dications [(LSepy)M(μ-dppe)M(LSepy)]2+ (M = Pd, Pt), which were isolated as their respective BPh4 salts. In the case of palladium, the same product can be obtained from the selenosemicarbazone and [Pd(OTf)2(dppe)] in the presence of base and NaBPh4.132
In a series of papers, the group of Andelkovic reported the preparation, spectroscopic and structural characterisation of metal complexes containing 2-pyridinecarboxaldehyde selenosemicarbazone. These include [CdCl2(HLSepy)] and [ZnCl2(HLSepy)], in which the neutral ligand adopts the [N,N,Se] coordination mode.133 The square planar [MCl(LSepy)] (M = Pd, Pt) and octahedral complexes [Co(LSepy)2]BF4 and [Ni(LSepy)2] were also reported.113 In the latter compounds, the selenosemicarbazone is deprotonated and binds to the metal centre via the selenium atom and two nitrogen atoms (pyridyl and imine). Detailed studies of the anticancer activity and the mode of action of these complexes have also been disclosed.113,133–135
A potentially pentadentate selenosemicarbazone was prepared by the condensation reaction of 2,6-diacetylpyridine with two equivalents selenosemicarbazide.136 Treatment of this bis(selenosemicarbazone) with various metal acetates (Cd, Zn and Ni) lead to the isolation of the corresponding complexes, in which the doubly deprotonated selenosemicarbazone is bound to the metal through two selenium- and two imine-nitrogen atoms as well as the nitrogen of the pyridyl group (Fig. 16 left). In the case of the nickel complex, elimination of hydrogen selenide occurs in one of the selenosemicarbazone-arms during complexation, leading to formation of a tetradentate [Se,N,N,N]-selenosemicarbazone (Fig. 16 right).136 The in vitro antitumor activity of the bis(selenosemicarbazone) and the three metal complexes were evaluated in several human cancer cell lines.137 Whilst the nickel complex was inactive, the bis(selenosemicarbazone) and its Zn(II) complex were the most active in all cell lines and were able to induce necrosis.
 | ||
| Fig. 16 Examples of complexes with multidentate selenosemicarbazone ligands. | ||
3.4 Complexes of selenosemicarbazones containing phosphorus donor atoms
There is only one reported example of a phosphine-containing selenosemicarbazone. Condensation of 2-(diphenylphosphino)benzaldehyde with selenosemicarbazide affords the corresponding selenosemicarbazone which was subsequently coordinated to a Ni(II) centre.138 In the resulting diamagnetic, square planar complex the ligand acts as a monoanionic [P,N,Se]− ligand.4. Selenocarbamate esters
The selenocarbamate ester PhNHC(Se)OEt, obtained from the reaction of PhNCSe with EtOH, was first reported in 1971 together with a series of Co(II) complexes in which the compound acts as a neutral Se-donor ligand (Scheme 10).139 Our group has reported the preparation and crystal structures of various mono- and dinuclear gold(I) phosphine complexes containing the deprotonated selenocarbamate ester [PhNC(Se)OMe]−, which coordinates to gold only via selenium (Scheme 10).140,141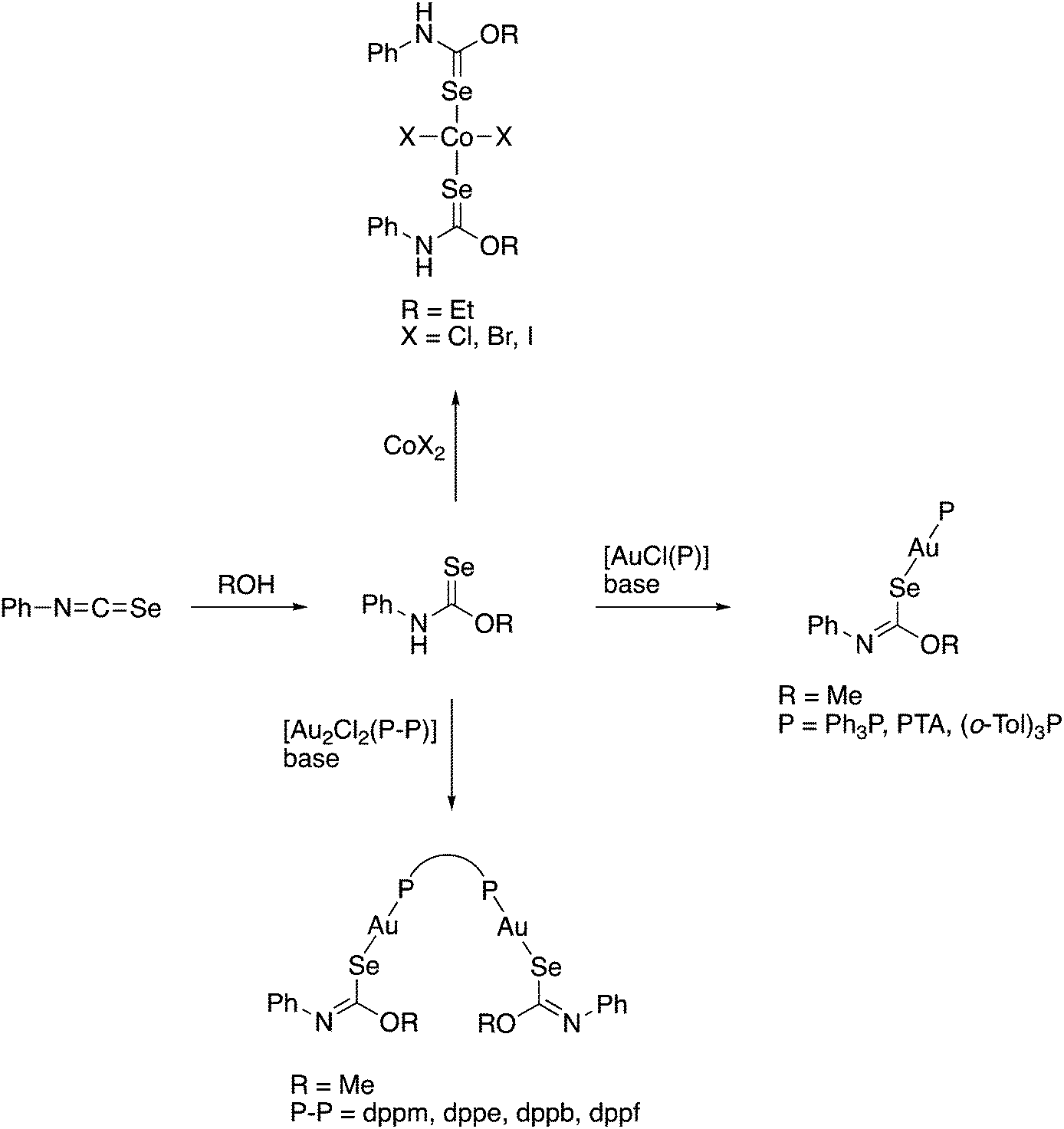 | ||
| Scheme 10 | ||
5. Conclusions
We hoped to illustrate in this article that although the origins of the coordination chemistry of the selenium-containing ligand classes comprising acylselenoureas, selenosemicarbazones and selenocarbamate esters date back more than forty years, there still remains much to explore. The development of modern analytical tools, especially X-ray diffraction and 77Se-NMR spectroscopy has allowed researchers to unambiguously elucidate structures of the compounds and to understand their reactivity. A variety of applications including analytical chemistry, materials chemistry (single-source precursors for a range of metal-selenide nanomaterials) as well as medicine (antimicrobial and anticancer activity) will continue to inspire future developments in this area of selenium-coordination chemistry.Abbreviations
| Ac | acetyl |
| acac | acetylacetonate |
| Bipy | 2,2′-bipyridine |
| t Bu2Bip | 4,4′-di(tert-butyl)biphenyl-2,2′-diyl |
| t Bu2bipy | 4,4′-di(tert-butyl)-2,2′-bipyridine |
| Bz | benzyl |
| p-cym | 1-methyl-4-isopropylbenzene |
| DMG | dimethylglyoximato |
| dppm | bis(diphenylphosphino)methane |
| dppe | 1,2-bis(diphenylphosphino)ethane |
| dppp | 1,3-bis(diphenylphosphino)propane |
| dppb | 1,4-bis(diphenylphosphino)butane |
| dppf | 1,1′-bis(diphenylphosphino)ferrocene |
| Fc | ferrocenyl |
| IPr | 1,3-di(2,6-diisopropylphenyl)imidazolylidene |
| 2-nap | 2-naphthyl |
| OTf | trifluoromethanesulfonate |
| Phen | 1,10-phenanthroline |
| PTA | 1,3,5-triaza-7-phosphaadamantane |
| o-Tol | 2-methylphenyl |
| p-Tol | 4-methylphenyl |
Conflicts of interest
There are no conflicts to declare.References
- R. A. Hussain, A. Badshah and A. Shah, Appl. Organomet. Chem., 2014, 28, 61–73 CrossRef CAS.
- L. Beyer, Rev. Colomb. Quim., 1983, 12, 69–85 CAS.
- I. B. Douglass, J. Am. Chem. Soc., 1937, 59, 740–742 CrossRef CAS.
- Y. Zhou and H. Heimgartner, Helv. Chim. Acta, 2000, 83, 539–553 CrossRef CAS.
- E. Bulka, K. D. Ahlers and E. Tuček, Chem. Ber., 1967, 100, 1459–1464 CrossRef CAS.
- E. Bulka, D. Ehlers and E. Storm, Z. Chem., 1970, 10, 403–404 CrossRef CAS.
- M. Koketsu, Y. Yamamura, H. Aoki and H. Ishihara, Phosphorus, Sulfur Silicon Relat. Elem., 2006, 181, 2699–2708 CrossRef CAS.
- J. Šibor, D. Žůrek, R. Marek, M. Kutý, O. Humpa, J. Marek and P. Pazdera, Collect. Czech. Chem. Commun., 1999, 64, 1673–1695 CrossRef.
- T. B. Wei, H. Wang, Q. Lin and Y. M. Zhang, Chin. J. Org. Chem., 2005, 28, 1565–1569 Search PubMed.
- T. B. Wei, H. Wang, Q. Lin and Y. M. Zhang, Chem. J. Chin. Univ., 2006, 27, 1680–1682 CAS.
- O. López, S. Maza, V. Ulgar, I. Maya and J. G. Fernández-Bolaños, Tetrahedron, 2009, 65, 2556–2566 CrossRef.
- Z. J. Witczak, Tetrahedron, 1985, 41, 4781–4785 CrossRef CAS.
- R. A. Hussain, A. Badshah, M. N. Tahir, B. Lal and I. A. Khan, Aust. J. Chem., 2013, 66, 626–634 CrossRef CAS.
- R. A. Hussain, A. Badshah, A. Sohail, B. Lal and A. A. Altaf, Inorg. Chim. Acta, 2013, 402, 133–139 CrossRef CAS.
- R. A. Hussain, A. Badshah, A. Sohail, B. Lal and K. Akbar, J. Mol. Struct., 2013, 1048, 367–374 CrossRef CAS.
- R. A. Hussain, A. Badshah, M. N. Tahir, T. Ul Hassan and A. Bano, J. Biochem. Mol. Toxicol., 2014, 28, 60–68 CrossRef CAS PubMed.
- R. A. Hussain, A. Badshah, S. Marwat, F. Yasmin and M. N. Tahir, J. Organomet. Chem., 2014, 769, 58–63 CrossRef CAS.
- R. A. Hussain, A. Badshah, F. Yasmin, M. D. Khan and M. N. Tahir, Aust. J. Chem., 2014, 68, 298–306 CrossRef.
- R. A. Hussain, A. Badshah, M. D. Khan, N. Haider, B. lal, S. I. Khan and A. Shah, Mater. Chem. Phys., 2015, 159, 152–158 CrossRef CAS.
- J. C. Bruce, N. Revaprasadu and K. R. Koch, New J. Chem., 2007, 31, 1647–1653 RSC.
- J. C. Bruce and K. R. Koch, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2008, C64, m1–m4 CrossRef PubMed.
- M. Schuster and K. H. König, Fresenius Z. Anal. Chem., 1987, 327, 102–104 CrossRef CAS.
- R. Herzschuh, B. Birner, L. Beyer, F. Dietze and E. Hoyer, Z. Anorg. Allg. Chem., 1980, 464, 159–168 CrossRef CAS.
- A. Molter, J. Rust, C. W. Lehmann and F. Mohr, ARKIVOC, 2011, vi, 10–17 Search PubMed.
- M. Kampf, R. Richter, J. Griebel, A. Weller and R. Kirmse, Z. Anorg. Allg. Chem., 2005, 631, 698–708 CrossRef CAS.
- M. Kampf, R. Richter, L. Hennig, A. Eidner, J. Baldamus and R. Kirmse, Z. Anorg. Allg. Chem., 2004, 630, 2677–2686 CrossRef CAS.
- J. Akhtar, M. A. Malik, S. K. Stubbs, P. O'Brien, M. Helliwell and D. J. Binks, Eur. J. Inorg. Chem., 2011, 2984–2990 CrossRef CAS.
- J. Akhtar, R. F. Mehmood, M. A. Malik, N. Iqbal, P. O'Brien and J. Raftery, Chem. Commun., 2011, 47, 1899–1901 RSC.
- A. Molter, S. Kathrein, B. Kircher and F. Mohr, Dalton Trans., 2018, 47, 5055–5064 RSC.
- K. Klauke, A. Schmitz, A. C. Swertz, B. B. Beele, B. Geisen, C. Schlüsener, C. Janiak and F. Mohr, New J. Chem., 2020, 44, 7719–7726 RSC.
- M. Perez Rodriguez, M. Cubero and A. Lopez Castro, Nature, 1963, 200, 1091 CrossRef.
- H. Hope, Acta Crystallogr., 1965, 18, 259–264 CrossRef CAS.
- L. Beyer, R. Kirmse and E. Hoyer, Z. Chem., 1975, 15, 197 CrossRef CAS.
- R. Kirmse, L. Beyer and E. Hoyer, Chem. Phys. Lett., 1977, 49, 544–546 CrossRef CAS.
- R. Kirmse, L. Beyer and E. Hoyer, Z. Chem., 1975, 15, 454–455 CrossRef CAS.
- J. Stach, R. Kirmse, A. Heinrich, W. Dietzsch, J. Hartung and L. Beyer, Z. Chem., 1983, 23, 453–454 CrossRef CAS.
- M. Kampf, R. Richter, L. Hennig, A. Eidner, J. Baldamus and R. Kirmse, Z. Anorg. Allg. Chem., 2004, 630, 2677–2686 CrossRef CAS.
- M. Kampf, R. Richter, J. Griebel, A. Weller and R. Kirmse, Z. Anorg. Allg. Chem., 2005, 631, 698–708 CrossRef CAS.
- A. Rodenstein, J. Griebel, R. Richter and R. Kirmse, Z. Anorg. Allg. Chem., 2008, 634, 1735–1741 CrossRef CAS.
- Y. Salyn, E. K. Zhumandilov, V. I. Nefedov, R. Scheibe, G. Leonhardt, L. Beyer and E. Hoyer, Z. Anorg. Allg. Chem., 1977, 432, 275–279 CrossRef CAS.
- V. I. Nefedov and E. K. Zhumandilov, Inorg. Chim. Acta, 1978, 30, L285–L286 CrossRef CAS.
- E. Kleinpeter and L. Beyer, J. Prakt. Chem., 1975, 317, 938–942 CrossRef CAS.
- L. Beyer, S. Behrendt, E. Kleinpeter, R. Borsdorf and E. Hoyer, Z. Anorg. Allg. Chem., 1977, 437, 282–288 CrossRef CAS.
- E. Kleinpeter, S. Behrendt and L. Beyer, Z. Anorg. Allg. Chem., 1982, 495, 105–114 CrossRef CAS.
- R. Herzschuh, B. Birner, L. Beyer, F. Dietze and E. Hoyer, Z. Anorg. Allg. Chem., 1980, 464, 159–168 CrossRef CAS.
- J. Stach, R. Herzschuh, R. Kirmse, L. Beyer and J. Hartung, Z. Anorg. Allg. Chem., 1984, 514, 223–230 CrossRef CAS.
- K. H. König, H. J. Pletsch and M. Schuster, Fresenius Z. Anal. Chem., 1986, 325, 621–624 CrossRef.
- M. Schuster and K. H. König, Fresenius Z. Anal. Chem., 1988, 331, 383–386 CrossRef CAS.
- D. P. Bendito, J. Casillas and F. Pino, Inf. Quim. Anal., 1966, 20, 69–75 CAS.
- D. P. Bendito and F. Pino, Inf. Quim. Anal., 1967, 21, 9–13 Search PubMed.
- F. Pino-Pérez, F. Burriel Matí and J. M. Balcells, An. R. Soc. Esp. Fis. Quim., Ser. B, 1959, 55B, 579–590 Search PubMed.
- S. C. Shome, M. Mazumdar and S. K. Das, J. Ind. Chem. Soc., 1980, 57, 69–72 CAS.
- J. Akhtar, J. C. Bruce, M. A. Malik, K. R. Koch, M. Afzaal and P. O’Brien, Mater. Res. Soc. Symp. Proc., 2009, 1148E, 1148-PP Search PubMed 1112-1108.
- J. Akhtar, M. Akhtar, M. A. Malik, P. O'Brien and J. Raftery, J. Am. Chem. Soc., 2012, 134, 2485–2487 CrossRef CAS PubMed.
- S. A. Saah, P. D. McNaughter, M. A. Malik, J. A. M. Awudza, N. Revaprasadu and P. O’Brien, J. Mater. Sci., 2018, 53, 4283–4293 CrossRef CAS.
- T. E. Ezenwa, P. D. McNaughter, J. Raftery, D. J. Lewis and P. O’Brien, Dalton Trans., 2018, 47, 16938–16943 RSC.
- S. A. Saah, M. D. Khan, P. D. McNaughter, J. A. M. Awudza, N. Revaprasadu and P. O’Brien, New J. Chem., 2018, 42, 16602–16607 RSC.
- M. Akhtar, J. Akhtar, M. A. Malik, F. Tuna, M. Helliwell and P. O'Brien, J. Mater. Chem., 2012, 22, 14970–14975 RSC.
- K. Rosenbaum, L. Beyer, R. Richter and E. Hoyer, Z. Anorg. Allg. Chem., 1992, 617, 89–92 CrossRef CAS.
- G. Weber, J. Hartung and L. Beyer, Tetrahedron Lett., 1988, 29, 3475–3476 CrossRef CAS.
- Y. Zhou, A. Linden and H. Heimgartner, Helv. Chim. Acta, 2000, 83, 1576–1598 CrossRef CAS.
- A. Bredenkamp, X. Zeng and F. Mohr, Polyhedron, 2012, 33, 107–113 CrossRef CAS.
- R. Köhler, L. Beyer, R. Richter, J. Sieler and J. Stach, Z. Anorg. Allg. Chem., 1991, 600, 73–81 CrossRef.
- A. Rodenstein, J. A. Odendal, R. Kirmse and K. R. Koch, Inorg. Chem. Commun., 2011, 14, 99–102 CrossRef CAS.
- W. Bensch and M. Schuster, Z. Anorg. Allg. Chem., 1993, 619, 1689–1692 CrossRef CAS.
- M. Schuster and W. Bensch, Z. Anorg. Allg. Chem., 1994, 620, 737–742 CrossRef CAS.
- M. Schuster and W. Bensch, Z. Naturforsch., B: J. Chem. Sci., 1994, 49b, 1615–1619 CrossRef.
- W. Bensch and M. Schuster, Z. Anorg. Allg. Chem., 1993, 619, 791–795 CrossRef CAS.
- W. Bensch and M. Schuster, Z. Anorg. Allg. Chem., 1993, 619, 786–790 CrossRef CAS.
- W. Bensch and M. Schuster, Z. Anorg. Allg. Chem., 1994, 620, 177–182 CrossRef CAS.
- W. Bensch and M. Schuster, Z. Anorg. Allg. Chem., 1994, 620, 1479–1482 CrossRef CAS.
- A. Molter and F. Mohr, Z. Naturforsch., B: J. Chem. Sci., 2013, 68, 91–94 CrossRef CAS.
- A. Molter, J. Rust, C. W. Lehmann and F. Mohr, Tetrahedron, 2012, 68, 10586–10591 CrossRef CAS.
- J. Kuchar, J. Rust, C. W. Lehmann and F. Mohr, New J. Chem., 2019, 10750–10754 RSC.
- F. Fuge, C. Lehmann and F. Mohr, J. Organomet. Chem., 2009, 694, 2395–2401 CrossRef CAS.
- A. Molter, J. Rust, C. W. Lehmann and F. Mohr, Inorg. Chem. Commun., 2016, 73, 69–71 CrossRef CAS.
- B. David, U. Monkowius, J. Rust, C. W. Lehmann, L. Hyzak and F. Mohr, Dalton Trans., 2014, 43, 11059–11066 RSC.
- A. Molter, PhD thesis, University of Wuppertal, 2011.
- V. D. Schwade, A. Hagenbach, E. Shulz Lang, K. Klauke, F. Mohr and U. Abram, Eur. J. Inorg. Chem., 2014, 1949–1954 CrossRef CAS.
- M. Dörner, J. M. Rautiainen, J. Rust, C. W. Lehmann and F. Mohr, Eur. J. Inorg. Chem., 2017, 789–797 CrossRef.
- J. Kuchar, J. Rust, C. W. Lehmann and F. Mohr, Eur. J. Inorg. Chem., 2018, 5215–5222 CrossRef CAS.
- R. Huls and M. Renson, Bull. Soc. Chim. Belg., 1956, 65, 511–522 CrossRef CAS.
- R. Huls and M. Renson, Bull. Soc. Chim. Belg., 1956, 65, 684–695 CrossRef CAS.
- J. M. Cano Pavon and F. Pino, Talanta, 1972, 19, 1659–1663 CrossRef CAS.
- D. J. Fry and P. J. Keogh, UK Pat., GB1326597, 1973 Search PubMed.
- P. Bippus, A. Molter, D. Müller and F. Mohr, J. Organomet. Chem., 2010, 695, 1657–1662 CrossRef CAS.
- A. V. Ablov, N. V. Gerbeleu and A. M. Romanov, Russ. J. Inorg. Chem., 1968, 13, 413–416 Search PubMed.
- A. V. Ablov, N. V. Gerbeleu, A. M. Romanov and V. M. Vlad, Russ. J. Inorg. Chem., 1971, 16, 718–719 Search PubMed.
- A. N. Vedyanu and N. V. Gerbeleu, Russ. J. Inorg. Chem., 1976, 21, 1828–1831 Search PubMed.
- D. Negoiu and I. Ghelase, Rev. Roum. Chim., 1983, 28, 225–234 CAS.
- D. Negoiu and I. Ghelase, Rev. Roum. Chim., 1983, 28, 355–364 CAS.
- I. Ghelase and D. Negoiu, Rev. Roum. Chim., 1983, 28, 463–470 CAS.
- A. V. Ablov, N. V. Gerbeleu and A. M. Romanov, Russ. J. Inorg. Chem., 1968, 13, 1558–1561 Search PubMed.
- N. V. Gerbeleu, M. D. Revenko and A. V. Ablov, Russ. J. Inorg. Chem., 1972, 17, 709–710 Search PubMed.
- N. V. Gerbeleu, M. D. Revenko and A. V. Ablov, Russ. J. Inorg. Chem., 1972, 17, 71–74 Search PubMed.
- N. V. Gerbeleu and M. D. Revenko, Russ. J. Inorg. Chem., 1972, 17, 1132–1135 Search PubMed.
- Unfortunately, the structure is of poor quality, however the atom connectivity can be unambiguously established.
- T. N. Tarkhova, L. E. Nikolaeva, M. A. Simonov, A. V. Ablov, N. V. Gerbeleu and A. M. Romanov, Dokl. Chem., 1974, 214, 138–141 Search PubMed.
- L. E. Nikolaeva, A. A. Shevyrev, T. N. Tarkhova and N. V. Belov, Sov. Phys. Crystallogr., 1974, 19, 320–322 Search PubMed.
- N. V. Gerbeleu and V. G. Bodyu, Russ. J. Inorg. Chem., 1972, 17, 857–859 Search PubMed.
- V. G. Bodyu and N. V. Gerbeleu, Russ. J. Inorg. Chem., 1972, 17, 1123–1125 Search PubMed.
- N. Y. Negryatse, A. V. Ablov and N. V. Gerbeleu, Russ. J. Inorg. Chem., 1972, 17, 65–67 Search PubMed.
- N. V. Gerbeleu and V. G. Bodyu, Russ. J. Inorg. Chem., 1973, 18, 1596–1598 Search PubMed.
- A. V. Ablov, N. V. Gerbeleu and N. Y. Negryatse, Russ. J. Inorg. Chem., 1969, 14, 515–517 Search PubMed.
- A. V. Ablov, N. V. Gerbeleu and N. Y. Negryatse, Russ. J. Inorg. Chem., 1970, 15, 61–62 Search PubMed.
- A. V. Ablov, N. V. Gerbeleu and N. Y. Negryatse, Russ. J. Inorg. Chem., 1971, 16, 568–571 Search PubMed.
- V. Y. Plotkin, M. G. Felin, N. A. Subbotina and V. V. Zelentsov, Russ. J. Inorg. Chem., 1983, 28, 825–827 Search PubMed.
- A. V. Ablov, N. V. Gerbeleu and B. T. Oloi, Russ. J. Inorg. Chem., 1971, 16, 379–382 Search PubMed.
- B. T. Oloi, N. V. Gerbeleu and A. V. Ablov, Russ. J. Inorg. Chem., 1971, 16, 1537–1538 Search PubMed.
- M. D. Revenko, V. I. Prisacari, A. V. Dizdari, E. F. Stratulat, I. D. Corja and L. M. Proca, Pharm. Chem. J., 2011, 45, 351–354 CrossRef CAS.
- N. R. Filipović, S. Bjelogrlić, G. Portalone, S. Pelliccia, R. Silvestri, O. Klisurić, M. Senćanski, D. Stanković, T. R. Todorović and C. D. Muller, MedChemComm, 2016, 7, 1604–1616 RSC.
- N. Gligorijević, T. R. Todorović, S. Radulović, D. M. Sladić, N. R. Filipović, D. Gođevac, D. Jeremić and K. K. Anđelković, Eur. J. Med. Chem., 2009, 44, 1623–1629 CrossRef PubMed.
- T. R. Todorović, A. Bacchi, D. M. Sladić, N. M. Todorović, T. T. Božić, D. D. Radanović, N. R. Filipović, G. Pelizzi and K. K. Anđelković, Inorg. Chim. Acta, 2009, 362, 3813–3820 CrossRef.
- N. Filipović, N. Polović, B. Rašković, S. Misirlić-Denčić, M. Dulović, M. Savić, M. Nikšić, D. Mitić, K. Anđelković and T. Todorović, Monatsh. Chem., 2014, 145, 1089–1099 CrossRef.
- N. R. Filipović, S. Bjelogrlić, A. Marinković, T. Ž. Verbić, I. N. Cvijetić, M. Senćanski, M. Rodić, M. Vujčić, D. Sladić, Z. Striković, T. R. Todorović and C. D. Muller, RSC Adv., 2015, 5, 95191–95211 RSC.
- I. S. Đorđevic, J. Vukašinović, T. R. Todorović, N. R. Filipović, M. V. Rodić, A. Lolić, G. Portalone, M. Zlatović and S. Grubišić, J. Serb. Chem. Soc., 2017, 82, 825–839 CrossRef.
- C. Pizzo, P. Faral-Tello, G. Salina, M. Fló, C. Robello, P. Wipf and G. Mahler, Med. Chem. Commun., 2012, 3, 362–368 RSC.
- C. Pizzo, P. Faral-Tello, G. Yaluff, E. Serna, S. Torres, N. Vera, C. Saiz, C. Robello and G. Mahler, Eur. J. Med. Chem., 2016, 109, 107–113 CrossRef CAS PubMed.
- J. P. Scovill, D. L. Klayman and C. F. Franchino, US Pat., US4657903, 1987 Search PubMed.
- Y. K. Bhoon, J. P. Scovill and D. L. Klayman, Indian J. Chem., 1983, 22A, 267–269 CAS.
- D. L. Klayman, J. P. Scovill, J. F. Bartosevich and C. J. Mason, Eur. J. Med. Chem., 1981, 16, 317–320 CAS.
- D. X. West, P. M. Ahrweiler, G. Ertem, J. P. Scovill, D. L. Klayman, J. L. Flippen-Anderson, R. Gilardi, C. George and L. K. Pannell, Transition Met. Chem., 1985, 10, 264–270 CrossRef CAS.
- D. X. West, J. P. Scovill, J. V. Silverton and A. Bavoso, Transition Met. Chem., 1986, 11, 123–131 CrossRef CAS.
- B. S. Garg, M. R. P. Kurup, S. K. Jain and Y. K. Bhoon, Transition Met. Chem., 1988, 13, 92–95 CrossRef CAS.
- C. R. Kowol, R. Eichinger, M. A. Jakupec, M. Galanski, V. B. Arion and B. K. Keppler, J. Inorg. Biochem., 2007, 101, 1946–1957 CrossRef CAS PubMed.
- C. R. Kowol, E. Reisner, I. Chiorescu, V. B. Arion, M. Galanski, D. V. Deubel and B. K. Keppler, Inorg. Chem., 2008, 47, 11032–11047 CrossRef CAS PubMed.
- Z. Al-Eisawi, C. Stefani, P. J. Jansson, A. Arvind, P. C. Sharpe, M. T. Basha, G. M. Iskander, N. Kumar, Z. Kovacevic, D. J. R. Lane, S. Sahni, P. V. Bernhardt, D. R. Richardson and D. S. Kalinowski, J. Med. Chem., 2016, 59, 294–312 CrossRef CAS PubMed.
- A. Molter, J. Rust, C. W. Lehmann, G. Deepa, P. Chiba and F. Mohr, Dalton Trans., 2011, 40, 9810–9820 RSC.
- A. Molter and F. Mohr, Dalton Trans., 2011, 40, 3754–3758 RSC.
- A. Molter, G. N. Kaluderovic, H. Kommera, R. Paschke, T. Langer, R. Poettgen and F. Mohr, J. Organomet. Chem., 2012, 701, 80–86 CrossRef CAS.
- A. Molter, E. Bill and F. Mohr, Inorg. Chem. Commun., 2012, 17, 124–127 CrossRef CAS.
- A. Molter and F. Mohr, Polyhedron, 2016, 120, 118–123 CrossRef CAS.
- S. Bjelogrlić, T. Todorović, A. Bacchi, M. Zec, D. Sladić, T. Srdić-Rajić, D. Radanović, S. Radulović, G. Pelizzi and K. Anđelković, J. Inorg. Biochem., 2010, 104, 673–682 CrossRef PubMed.
- T. Srdić-Rajić, M. Zec, T. Todorović, K. Anđelković and S. Radulović, Eur. J. Med. Chem., 2011, 46, 3734–3747 CrossRef PubMed.
- M. Zec, T. Srdic-Rajic, A. Konic-Ristic, T. Todorovic, K. Andjelkovic, I. Filipovic-Ljeskovic and S. Radulovic, Anti-Cancer Agents Med. Chem., 2012, 12, 1071–1080 CrossRef CAS PubMed.
- T. R. Todorović, A. Bacchi, G. Pelizzi, N. O. Juranić, D. M. Sladić, I. D. Brčeski and K. K. Andelkovića, Inorg. Chem. Commun., 2006, 9, 862–865 CrossRef.
- M. Zec, T. Srdic-Rajic, A. Krivukuca, R. Jankovic, T. Todorović, K. Andelkovic and S. Radulovic, Med. Chem., 2014, 10, 759–771 CrossRef CAS PubMed.
- I. D. Brčeski, V. M. Leovac, G. A. Bogdanović, S. P. Sovilj and M. Revenco, Inorg. Chem. Commun., 2004, 7, 253–256 CrossRef.
- P. Porta, T. Tarantelli, L. Gastaldi and C. Furlani, Inorg. Chim. Acta, 1971, 5, 616–622 CrossRef CAS.
- D. Gallenkamp, E. Tiekink and F. Mohr, Phosphorus, Sulfur Silicon Relat. Elem., 2008, 183, 1050–1056 CrossRef CAS.
- D. Gallenkamp, T. Porsch, A. Molter, E. R. T. Tiekink and F. Mohr, J. Organomet. Chem., 2009, 694, 2380–2385 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2022 |