
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carboxamide carbonyl-ruthenium(II) complexes: detailed structural and mechanistic studies in the transfer hydrogenation of ketones†
Robert T.
Kumah
 ,
Paranthaman
Vijayan
and
Stephen O.
Ojwach
*
,
Paranthaman
Vijayan
and
Stephen O.
Ojwach
*
School of Chemistry and Physics, University of KwaZulu-Natal, Pietermaritzburg Campus, Private Bag X01, Scottsville, Pietermaritzburg 3209, South Africa. E-mail: ojwach@ukzn.ac.za; Fax: +27 (33) 260 5009; Tel: +27 (33) 260 5239
First published on 25th January 2022
Abstract
Reactions of N-(benzo[d]thiazol-2-yl)pyrazine-2-carboxamide (HL1) and N-(1H-benzo[d]imidazol-2-yl)pyrazine-2-carboxamide (HL2) ligands with Ru(PPh3)3ClH(CO) and Ru(PPh3)3(CO)2H2 precursors afforded the respective organo-carboxamide ruthenium(II) complexes [Ru(II)(CO)Cl(PPh3)2L1] (Ru1), [Ru(II)(CO)H(PPh3)2L1] (Ru2), [Ru(II)(CO)Cl(PPh3)2L2] (Ru3), and [Ru(II)(CO)H(PPh3)2L2] (Ru4). The Ru(II) complexes were characterised by NMR, FT-IR spectroscopies, mass spectrometry, micro-analyses, and single X-ray crystallography. The solid-state structures of complexes Ru1, Ru2, and Ru4 confirm distorted octahedral geometries around the Ru(II) atoms, containing one bidentate anionic carboxamidate ligand and four auxiliary ligands (PPh3/CO/H/Cl). All the complexes (Ru1–Ru4) displayed moderate catalytic activities in the transfer hydrogenation of a broad spectrum of ketones, giving a maximum turnover number (TON) of 990 within 6 h. The catalytic activities of the Ru(II) complexes were dependent on both the carboxamidate and auxiliary ligands. 31P{1H) NMR spectroscopy studies aided in proposing a monohydride pathway for the transfer hydrogenation reaction of ketones.
1. Introduction
The development of organometallic based complexes as catalysts in organic reactions is a major research field.1–3 To date, a considerable number of novel catalysts have been designed in attempts to meet the high demands of valuable industrial and fine chemical products.4 In the development of a catalyst for homogeneous reactions, an increasing impetus has been placed on ligand designs, which have the ability to modulate the electronic and steric parameters around the metal centre. This ultimately regulates the catalytic activity, stability and chemoselectivity in the organic transformation of interest.5,6 As a result, many transition metal complexes supported on various ligands bearing well-defined donor atoms have been tailored to achieve the desired catalytic properties. N-Heterocyclic carbenes (NHC),7 imino-based, amino-phosphines,8 imino-phosphines,9 carboxamides,10,11 thiosemicarbazone,12 amino-alcohols,13 phosphoramidites,14,15 selenium-based ligands16 among others17,18 are among the common ligands, which have been utilised in the past few years.A number of transition metals including first and second-row transition metals complexes19,20 have been exploited for their possible catalytic properties in the transfer hydrogenation (TH) of ketones with varied outcomes. Notably, Ru(II),2 Ir(II/III),21 Os(II), Rh(I/II),22 Fe(II),23 Ni(II)24 and Mn(I)25,26 represent the broad spectrum of transition metals that have been studied for their prospective catalytic properties. Ruthenium(II) complexes are extensively studied and have proven to be most active catalysts in the transfer hydrogenation of ketones compared to Rh(I/II), Ir(I/III), and Os(II) complexes.2,27,28 The chemistry and applications of carboxamide ligands have gained appreciable attention.29 Carboxamide ligands are less expensive and easily synthesized using simple condensation methods compared to the well-established but expensive phosphine based ligand systems.8–10 Neutral and anionic based carboxamide ligands have also been shown to improve the catalytic activities and stability of the resultant catalysts in the TH reactions.11,30
Recently, a number of Ru(II) carboxamide based complexes have been developed and utilised as potential catalysts in some organic transformations including transfer hydrogenation reactions.31 Notable among them is the Gupta's recently reported Ru(II)–phosphine–carboxamide complexes which demonstrated catalytic activity, TON (Turnover number) up to 99 in TH of ketones.10 Do and co-workers have also reported half-sandwich Ru(II) complexes bearing the pyridine–carboxamide backbone for TH of ketones and aldehydes and attained turn over number (TON) up to 200.32
Heterocyclic such as benzo[d]-thiazole and benzimidazole are known to alter the electron density at the metal center, which could have a significant impact on catalytic properties.33 Benzo-[d]-thiazole and benzo-imidazole heterocyclic groups have been integrated with pyridine moieties and their influence on the corresponding metal complexes in terms of catalytic reactions have been studied. For example, Ru(II/III)-pyridyl benzo-imidazole/benzo-thiazole complexes and their propensity in TH of ketones have been reported with moderate catalytic activity (TON up 540) by our group.34 Therefore, the combination of pyrazine and benzo-[d]-imidazole/benzo-[d]-thiazole groups with carboxamide functional group could impact greatly on the catalytic properties of the corresponding Ru(II) complexes. For these reasons, the development of Ru(II) complexes of pyrazine-benzo-[d]-thiozole/benzo-[d]-imidazole carboxamide has ignited our interest to prepare potential catalysts in the TH of ketones. Herein, we report the synthesis of new pyrazine-benzo-[d]-thiozole/benzo-[d]-imidazole carboxamide Ru(II) complexes with co-ligands; PPh3, Cl, CO, and hydride (H) and their potential applications as catalysts in the TH reaction of ketones. Detailed structural characterization and mechanistic studies in the transfer hydrogenation of ketones have been carried out and are discussed.
2. Results and discussion
2.1 Synthesis and characterisation of compounds
The two carboxamide ligands, N-(benzo[d]thiazol-2-yl)pyrazine-2-carboxamide (HL1) and N-(1H-benzo[d]imidazol-2-yl)pyrazine-2-carboxamide (HL2) were prepared following literature procedures.35,36 Detailed synthetic protocol and spectroscopic data of the ligands are provided in the ESI†). The reactions of synthons HL1 and HL2 with Ru(PPh3)3ClH(CO) and Ru(PPh3)3(CO)2H2 precursors afforded air-stable Ru(II) compounds (Ru1–Ru4) in good to high yields (65–88%) as shown in Scheme 1.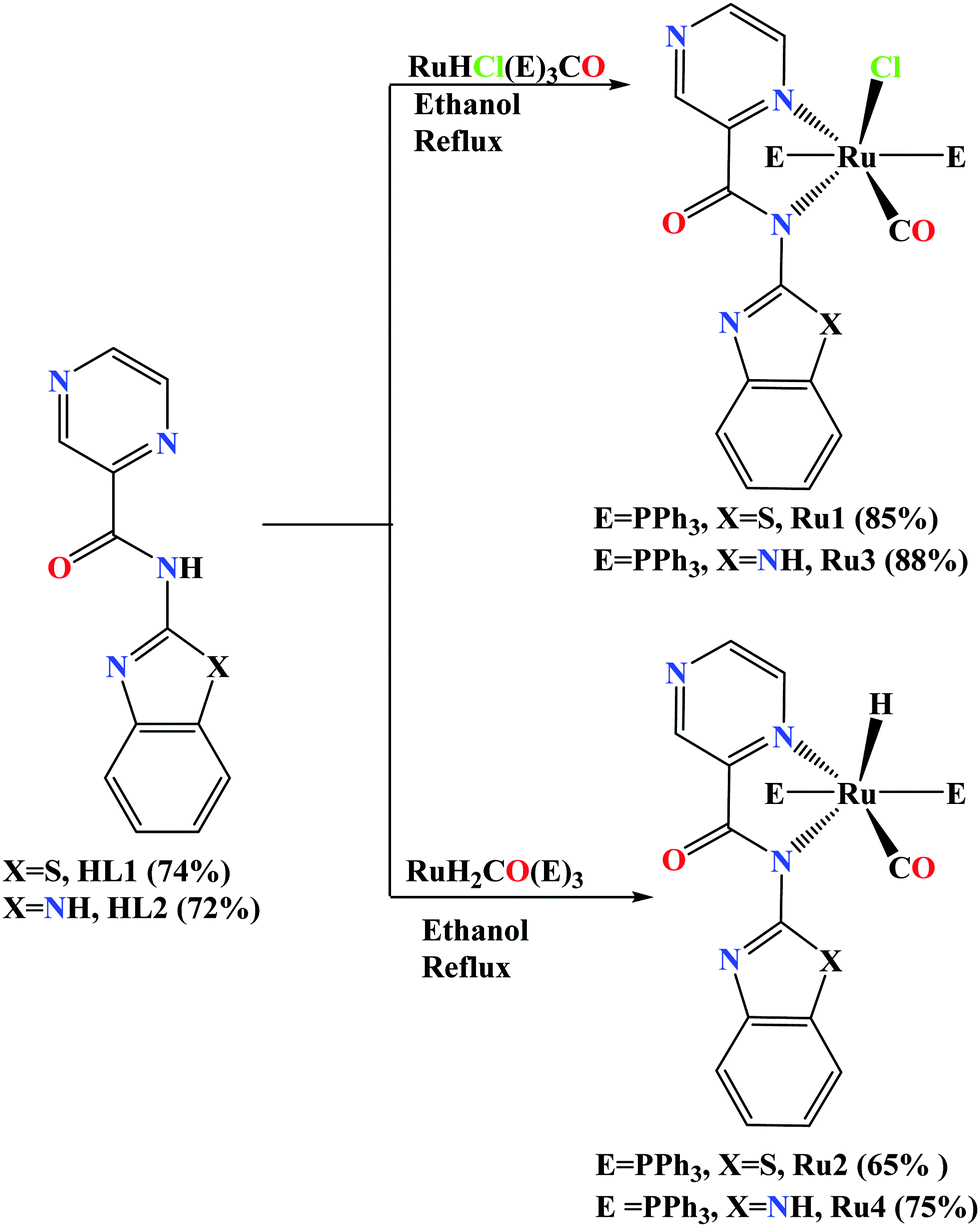 | ||
| Scheme 1 Synthesis of carboxamide Ru(II) complexes Ru1–Ru4. | ||
The Ru(II) compounds were characterised by 1H NMR, 13C NMR and FT-IR, spectroscopies, mass spectrometry, elemental analyses, and single-crystal X-ray analyses. The signals for the amide proton (N–H) in the 1H NMR spectra of the carboxamide ligands HL1 and HL2 were instrumental in the determination of the formation of the Ru(II) complexes (Fig. S1–S6, ESI†). For instance, the 1H NMR spectrum of the carboxamide ligands, HL1 showed the signal for the N–Hamide proton at 12.68 ppm (Fig. S1, ESI†) and upon the formation of Ru1, this signal disappeared (Fig. S3, ESI†). This trend was observed in the 1H NMR spectra of the other complexes Ru2–Ru4 (Fig. S4–S6, ESI†). The absence of the amide proton signal in the 1H NMR spectra of the Ru(II) complexes established the N–H deprotonation upon coordination as depicted in Scheme 1.36,37 In addition, the new triplet signals at δH −13.08 ppm and −13.26 ppm, assigned to the Ru–H protons were observed (Fig. S4 and S6, ESI†) in the 1H NMR spectra of complexes Ru2 and Ru4 respectively and assigned to the P–H coupling (t, 2JH–P = 20.3 Hz, 1HRu–H).36 13C{1H} NMR spectroscopy was also relevant in establishing the successful formation of complexes Ru1–Ru4. For example, 13C{1H} NMR spectrum of complex Ru1 showed slight shifts in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O(amidate) signals (166.8 ppm) in relation to the free ligands HL1 at 161.3 ppm (Fig. S7–S12, ESI†). While the carbonyl O atom is expected to be non-coordinating (see molecular structures in Fig. 1), these slight shifts could be attributed to electron flow from the HL1 ligand to the Ru atom, causing a deshielding effect in the carbonyl moiety. Furthermore, the presence of the coordinated pi acceptor carbonyl ligand is expected to favour sigma-donation from the HL1 ligand to the Ru atom.36,37 Indeed, this argument is supported by the lower (–Ru–C
O(amidate) signals (166.8 ppm) in relation to the free ligands HL1 at 161.3 ppm (Fig. S7–S12, ESI†). While the carbonyl O atom is expected to be non-coordinating (see molecular structures in Fig. 1), these slight shifts could be attributed to electron flow from the HL1 ligand to the Ru atom, causing a deshielding effect in the carbonyl moiety. Furthermore, the presence of the coordinated pi acceptor carbonyl ligand is expected to favour sigma-donation from the HL1 ligand to the Ru atom.36,37 Indeed, this argument is supported by the lower (–Ru–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O) signals observed in the 13C NMR spectra, for instance, at 198.1 ppm (typical range for terminal CO ligand is 200–210 ppm) for complex Ru3 (Fig. S11, ESI†).31 We also made use of 31P{1H} NMR spectroscopy to establish the identity of complexes Ru1–Ru4 (Fig. S13–S16, ESI†). For example, the 31P{1H} NMR spectra displayed sharp singlets at δP 29.4 ppm (Ru1), 48.2 ppm (Ru2), 48.2 (Ru3) and 23.2 ppm (Ru4), consistent with the existence of two magnetically equivalent PPh3 groups in trans configurations.36
O) signals observed in the 13C NMR spectra, for instance, at 198.1 ppm (typical range for terminal CO ligand is 200–210 ppm) for complex Ru3 (Fig. S11, ESI†).31 We also made use of 31P{1H} NMR spectroscopy to establish the identity of complexes Ru1–Ru4 (Fig. S13–S16, ESI†). For example, the 31P{1H} NMR spectra displayed sharp singlets at δP 29.4 ppm (Ru1), 48.2 ppm (Ru2), 48.2 (Ru3) and 23.2 ppm (Ru4), consistent with the existence of two magnetically equivalent PPh3 groups in trans configurations.36
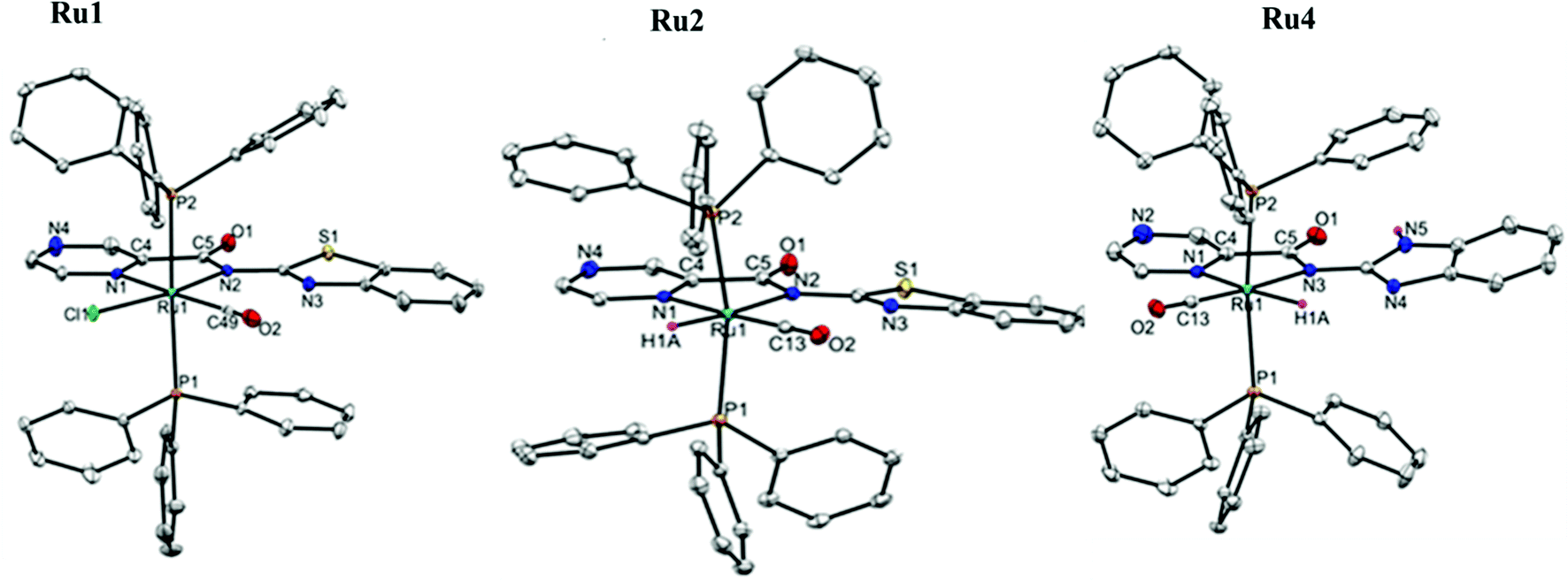 | ||
| Fig. 1 The ORTEP view of complexes Ru1, Ru2 and Ru4 with thermal ellipsoids at a 50% probability level. (Hydrogen atoms are omitted for clarity.) | ||
The successful coordination of the Ru(II) precursors to the carboxamide ligands HL1–HL2 was further supported by FT-IR spectroscopy. In the FT-IR spectra of complexes Ru1–Ru4, the ν(CO) absorption band shifted to lower frequencies between 1621–1629 cm−1 in the complexes with respect to the free ligands of 1691 cm−1 (HL1) and 1684 cm−1 (HL2). This observation could be attributed to resonance enhancement of the coordinated ligands leading to weakening of the carbonyl bond.38 In addition, the terminal carbonyl (–Ru–CO) signals were recorded in the region of 1935–1947 cm−1. These values are relatively lower compared to the values expected for terminal CO ligands (2000–2100 cm−1). The presence of sigma-donor spectator HLI and HL2 ligands may account for this observation.36 As reported in the 1H NMR spectral data, the signals corresponding to N–H(amidate) at 3251–3315 cm−1 in the free ligand disappeared upon metalation (Fig. S17–S22, ESI†).39,40 LC-MS spectra of the compounds gave the signals corresponding to their respective fragments; for example, m/z = 909.12 [M–Cl]+ (Ru1), 909.12 [M–H]+ (Ru2), 892.15 [M–Cl]+ (Ru3), and 892.15 [M–H]+ (Ru4) (Fig. S23–S28, ESI†). Furthermore, the experimental and calculated isotopic mass distribution were in good agreement as given in Fig. S23–S28 (ESI†). Notably, the experimental elemental analysis data compared favourably with the proposed empirical formulae, thus establishing both the empirical formulae and purity of the complexes.
2.2 Solid-state structure of complexes Ru1, Ru2, and Ru4
Molecular structures of the complexes Ru1, Ru2 and Ru4 were confirmed by single-crystal X-ray crystallography analyses. The perspective views of their solid-state structures are shown in Fig. 1, while the refinement data and selected bond parameters are presented in Tables S1 and S2 (ESI†) respectively. The solid-state structures of complexes Ru1, Ru2 and Ru4 have a distorted octahedral geometry around the Ru(II) centre, with the carboxamide ligand oriented nearly in the plane as defined by the Ru(II) centre and the two trans PPh3 co-ligands. The fifth and sixth coordination sites are occupied by either CO, Cl or H co-ligands to complete the octahedral geometry. The solid-state structures of complexes Ru1, Ru2, and Ru4 reveal that the Ru(II) atoms coordinate to the carboxamide ligands HL1 and HL2via the pyrazine and amidate N-atoms to form five-membered metallocycles; N(1)–C(4)–C(5)–N(2)–Ru(1). The average bond distance of Ru(1)–N(2)amidate (2.114(3) Å), Ru–Npyrazine (2.118(12) Å), Ru–P (2.400(11) Å), Ru–Cl (2.390(10) Å) and Ru–CO (2.421(11) Å) in complexes Ru1, Ru2 and Ru4 respectively are within the average bond lengths of Ru–Namidate (2.115(10) Å), Ru–Npyrazine (2.122(12) Å, Ru–P (2.383(12) Å), Ru–Cl (2.410(10) Å) and Ru–CO (1.819(15) Å) found in 8 similar Ru(II) structures deposited in the CCDC.41,42 The bite angles of the three-membered chelating ring N(1)–Ru–N(2): 77.84(13)° (Ru1), 76.13(5)° (Ru2), and 76.63(8)° (Ru4) are within the mean bite angles of 76.48(5)° found in 8 similar Ru(II) complexes deposited in CCDC file.41 The average P(1)–Ru–P(2) bond angles in complexes Ru1, Ru2 and Ru4 of 169.96(8)° deviate significantly from the ideal linearity, 180°. The slight deviation from the linearity of N(1)pyrz–Ru–C(13)carbonyl 176.46(14)° in the complexes Ru1, Ru2 and Ru4 was also observed and could originate from steric restrictions imposed by the bulkier PPh3 groups on the five-membered ruthenacycle. In addition, the average N(1)pyrz–Ru–Cl(1) bond angles of 89.68(8)° and N(1) pyrz–Ru–H, 92.80(2)° in compounds Ru2 and Ru4 show slight deviations from the ideal bond angle of 90°.2.3 Application of complexes Ru1–Ru4 in the transfer hydrogenation of ketones
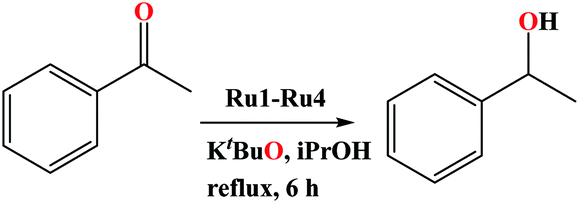
Preliminary investigations of the potential of the Ru(II) organo-carboxamide complexes (Ru1–Ru4) to catalyse the (TH) of ketones was carried using complex Ru3, acetophenone as the model substrate, isopropyl alcohol as a source of hydrogen and KOH as a base. The conversion of the substrates to their respective products was followed and determined using 1H NMR spectroscopy (Fig. S28–S44, ESI†). Percentage conversions of 53% was realised within 6 h at catalyst loadings of 0.10 mol% (Table 1, entry 1). In general, comparable percentage yields of crude products and conversions were realised, thus percentage yields have been adopted in the entire discussion.| Entry | Catalyst loading/mol% | Conversionb (%) | Yieldb (%) | TONc | TOFd/h−1 |
|---|---|---|---|---|---|
| a Reaction conditions: acetophenone, 1.00 mmol; KOH (0.100 mmol in 5 mL of iPrOH), anisole (1.00 mmol) was used as internal standard 82 °C time, 6 h. b Determined by 1H NMR spectroscopy (experiment repeated in triplicate to ensure reproducibility). c TON = moles of acetophenone converted per moles of Ru3). d TOF = (moles of acetophenone converted per moles of Ru3 per hour). | |||||
| 1 | 0.10 | 53 | 52 | 530 | 88 |
| 2 | 0.15 | 59 | 58 | 393 | 65 |
| 3 | 0.25 | 68 | 66 | 272 | 34 |
| 4 | 0.50 | 79 | 79 | 158 | 20 |
| 5 | 1.00 | 98 | 97 | 97 | 16 |
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Base | Catalyst | Base loading/mol% | Time/h | Conversionb/% | TOFc/h−1 |
| a Reaction condition: acetophenone, 1.00 mmol; [Ru3], 0.100 mol%; iPrOH, 5 mL; time, 6 h. b Determined by NMR spectroscopy internal standard (methoxybenzene). c TOF (turnover frequency) = moles of substrate converted per moles of catalyst per hour. d Reaction without a catalyst, reaction time, 24 h. e Base-free reaction using Ru4, time, 6. f Base-free reaction using Ru4, time, 18 h. g Base-free reaction using Ru3, reaction = 36 h (all experiments were conducted in triplicate to ensure reproducibility). | ||||||
| 1 | KOH | — | 10.0 | 6 | 5 | — |
| 2d | KOH | — | 10.0 | 36 | 16d | — |
| 3e | — | Ru4 | — | 6 | 39 | 65 |
| 4f | — | Ru4 | — | 18 | 99 | 28 |
| 5g | — | Ru3 | — | 36 | 93 | 51 |
| 6 | KOH | Ru3 | 2.5 | 6 | 24 | 40 |
| 7 | KOH | Ru3 | 5.0 | 6 | 61 | 102 |
| 8 | KOH | Ru3 | 7.5 | 6 | 72 | 120 |
| 9 | KOH | Ru3 | 10.0 | 6 | 86 | 143 |
| 10 | KOH | Ru3 | 100.0 | 6 | 97 | 164 |
| 11 | KtBuO | Ru3 | 10.0 | 6 | 91 | 152 |
| 12 | K2CO3 | Ru3 | 10.0 | 6 | 30 | 50 |
| 13 | NaOH | Ru3 | 10.0 | 6 | 42 | 70 |
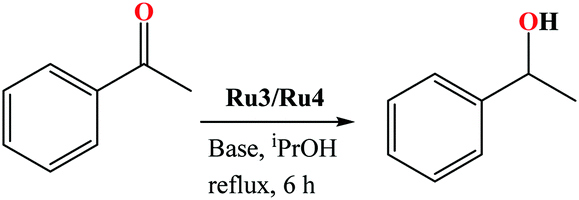
To establish the effects of the base loading on the rate of TH of acetophenone reaction, the concentration of KOH was varied from 0.025 mmol (2.5 mo1%) to 1.00 mmol (100 mol%) at constant catalyst loading (0.10 mol%) using complex Ru3 (Table 2, entries 6–10). From Table 2, it is evident that higher rates of the reactions were achieved at higher base loadings. For instance, TOF of up to 40 h−1 and 120 h−1 were obtained at base loadings of 2.5 mol% (0.025 mmol) and 7.5 mol% (0.075 mmol) as listed in Table 2, entries 6 vs. 8. The nature of the base in controlling the catalytic activity of the complexes in the TH of acetophenone was also investigated using K2CO3, NaOH and KtBuO (Table 2, entries 11–13). Expectedly, KtBuO gave the highest catalytic activity, and the observed order of KtBuO > KOH > NaOH > K2CO3 tally with the relative strengths of the bases.34
|
|
||||
|---|---|---|---|---|
| Entry | Complex | Conversionb [%] | TOFc/h−1 × 102 | k ob × 10−1 |
| a Condition: acetophenone, 1.00 mmol; KtBuO; [Ru], 0.100 mol%; iPrOH, 5 ml; 82 °C, time, 6 h. b Determined by NMR spectroscopy (mean values of two independent runs) by employing methoxybenzene was used as internal standard. c TOF (turn over frequency) = moles of acetophenone converted per moles of Ru3 per hour). d Base loading of 2.5 mol% (all experiments were carried out in triplicate to ensure reproducibility). | ||||
| 1 | Ru1 | 71 | 1.18 | 1.32(±0.01) |
| 2d | Ru2 | 94 | 1.46 | 2.02(±0.02) |
| 3 | Ru3 | 91 | 1.52 | 1.40(±0.04) |
| 4d | Ru4 | 99 | 1.52 | 2.08(±0.01) |
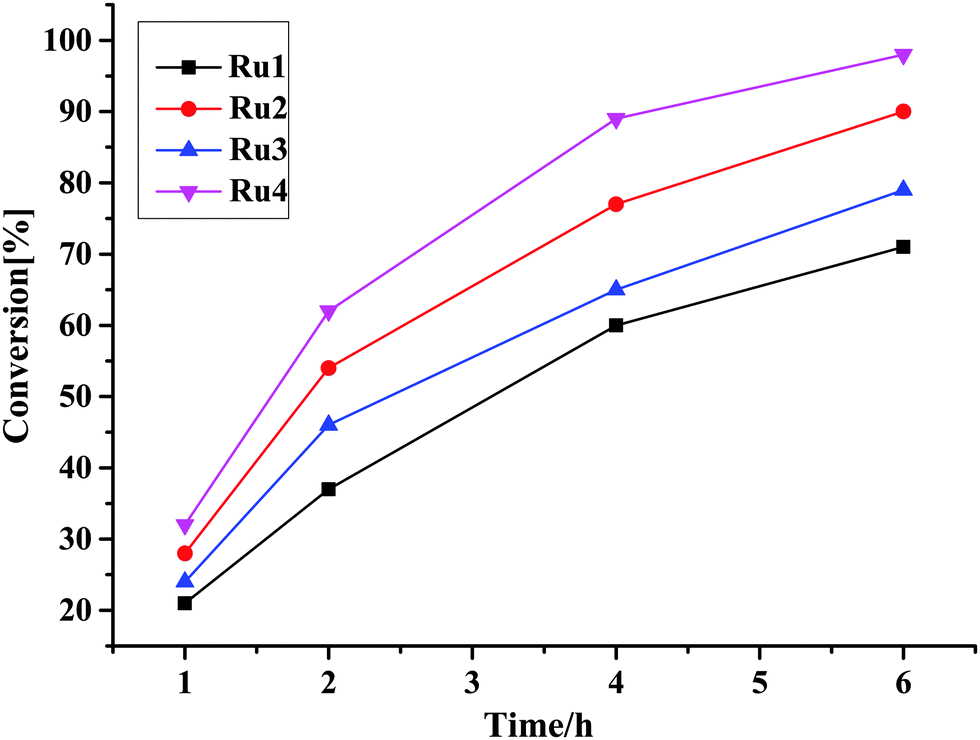 | ||
| Fig. 2 Plots of percentage conversion vs. time showing the effects of catalyst structure on the catalytic activity of TH of acetophenone reaction using complexes Ru1–Ru4 as a catalyst. | ||
It is also instructive to note that the Ru–H complexes Ru2 and Ru4 required lower base loadings of 2.5 mol% in comparison to the 10 mol% used for the non-hydride complexes Ru1, and Ru3 (Table 3, entries 2 and 4). In comparison to literature reports, the catalytic activities achieved by Ru1–Ru4 fall within the range of other related Ru(II)–PPh3 complexes of TOF up to 1.0 × 102 h−1.49–57 However, the catalytic activities of complexes Ru1–Ru4 are lower compared to some of the highly active ruthenium(II) complexes where the TOFs between 1.0 × 104–1.0 × 105 h−1 were achieved.58–61
| Entry | Substrate | Yieldb (%) | Entry | Substrate | Yieldb (%) |
|---|---|---|---|---|---|
| a General reaction conditions: substrate, 1.00 mmol (1.10 mL): internal standard methoxybenzene, 1.0 mmol (1.12 mL), [Ru4], 0.10 mol% (0.93 mg); KtBuO, 2.5 mol% (0.025 mmol) in isopropyl alcohol, temperature 82 °C. reaction time; 6 h. b Determined by 1H NMR spectroscopy (all experiments were conducted in triplicate to ensure reproducibility). Methoxy benzene was used as an internal standard. | |||||
| 1 |
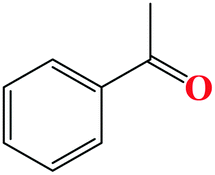
|
91 | 9 |
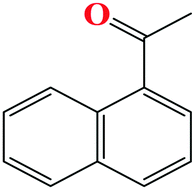
|
86 |
| 2 |
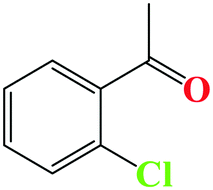
|
95 | 10 |
|
74 |
| 3 |
|
97 | 11 |
|
47 |
| 4 |
|
90 | 12 |
|
97 |
| 5 |
|
58 | 13 |
|
69 |
| 6 |
|
68 | 14 |
|
55 |
| 8 |
|
70 | 15 |
|
69 |
The TH of fused acetophenone substrates as in 1′-acetonaphthone and 2′-acetonaphthone were also accomplished, albeit with lower percentage conversions of 86% and 74% respectively. Notably, complex Ru4 was capable of reducing even the less reactive heterocyclic substrates (Table 4, entries 11–14). Interestingly, 1′-acetyl imidazole gave high percentage conversions of 97% (Table 4, entry 12). In contrast, the lower yields of, 2′-acetyl pyrazine and 1-(2-thienyl)-1-propanone of 55% (Table 4, entries 13 and 14) could be ascribed to possible irreversible coordination of the nitrogen and sulphur atoms to the active sites of the catalyst.65,66 The reactivity of the aliphatic ketones was also investigated using 2-propanone (70%) and 3-methyl-2-cyclohexanone (69%) substrates. The lower percentage conversions observed in comparison to acetophenone (91%) under similar reaction conditions (Table 4, entries 1, 8 and 15) could be ascribed to the electron-rich nature of the aliphatic ketones, thus reducing their reactivities.67
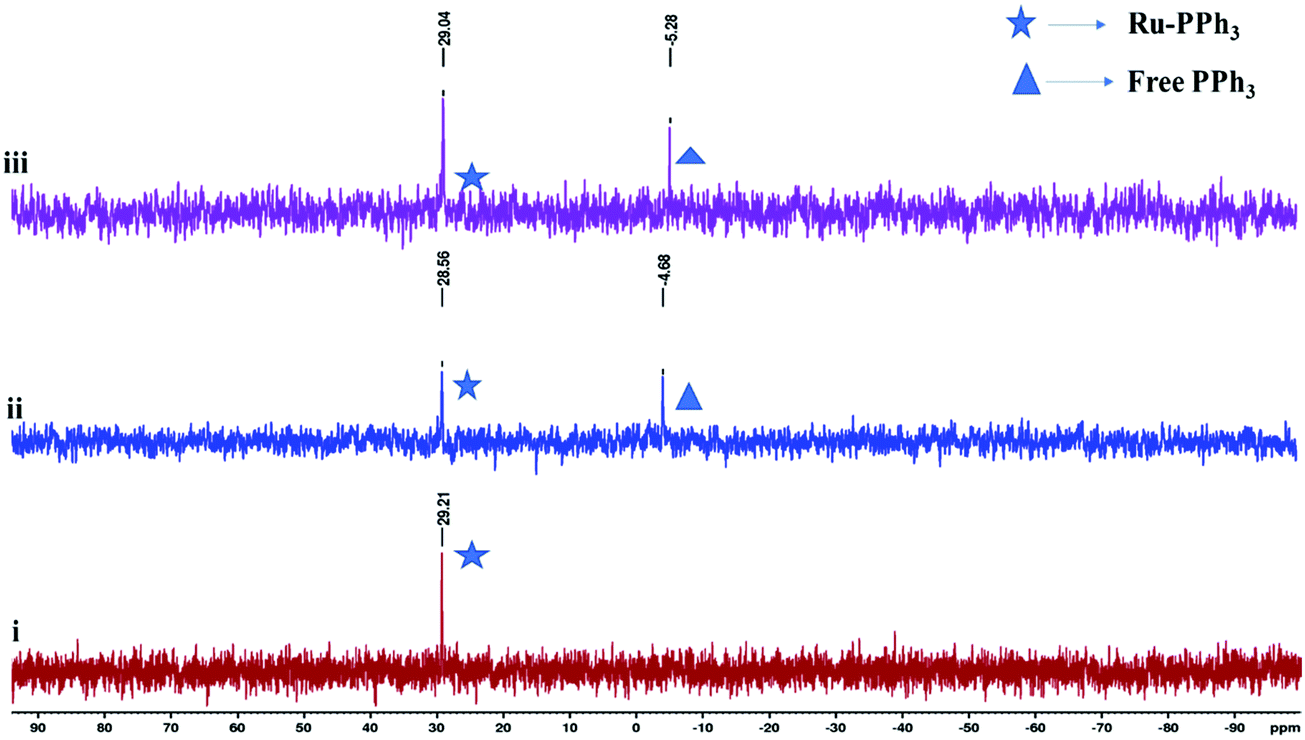 | ||
| Fig. 3 31P{1H} NMR spectral of complex [RuHCO(L4)(PPh3)2], Ru4 in the presence of acetophenone, iPrOH and KtBuO for 6 h. Spectrum, (i) shows a signal at δ 23.2 ppm corresponding to two equivalent PPh3 groups in the complex at t = 0. Spectra (ii) and (iii) show signals for the free PPh3 at δ −4.7 and 5.28 ppm after 4 h and 6 h respectively. | ||
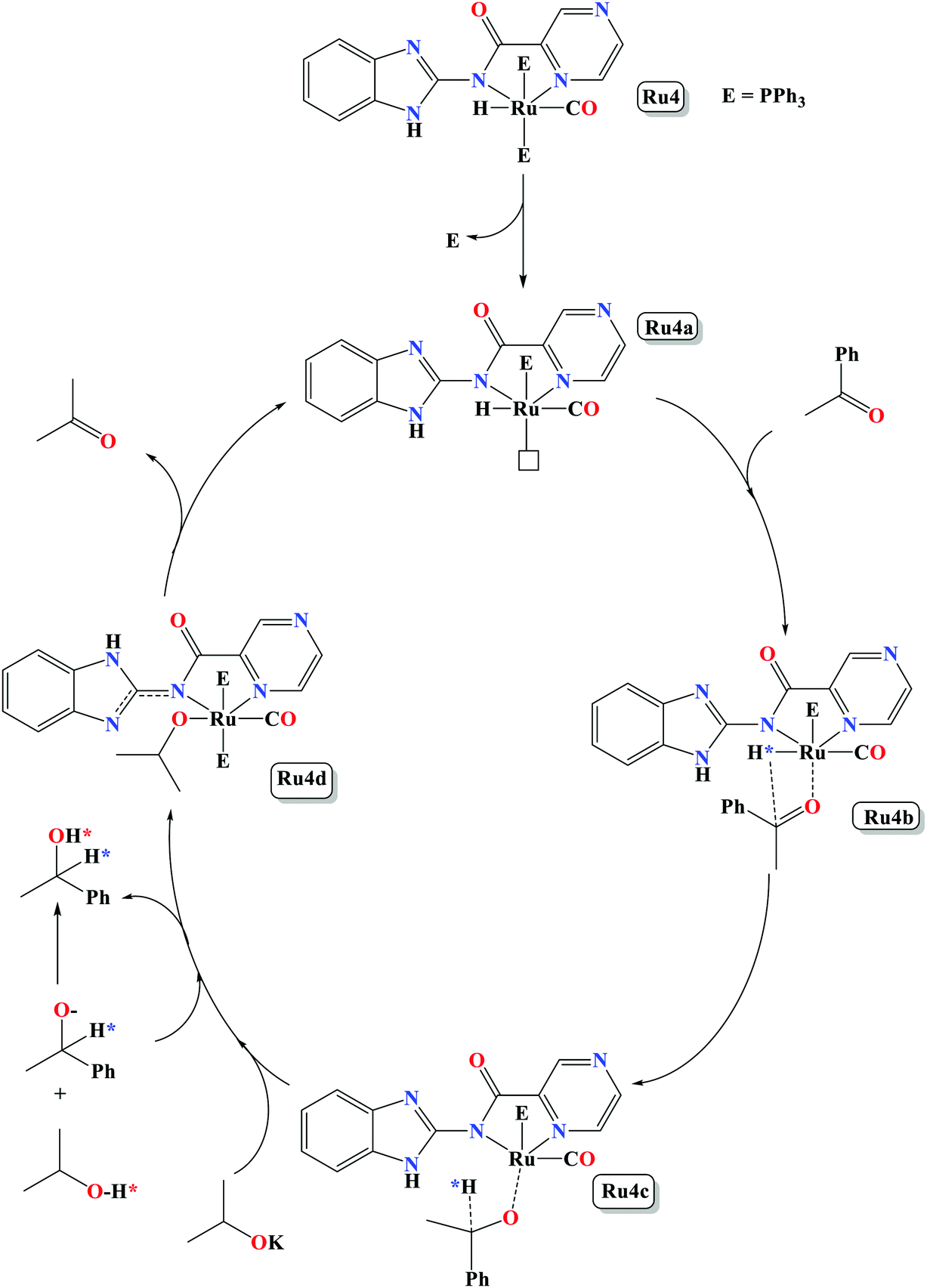 | ||
| Scheme 2 A proposed monohydride reaction pathway for transfer hydrogenation of ketone catalysed by Ru4 in the presence of isopropyl alcohol and KOH. | ||
3. Conclusions
In this work, we have successfully demonstrated the synthesis, structural studies of carboxamide organo-ruthenium(II/III) complexes bearing PPh3/CO/Cl/H co-ligands and their applications as catalysts in the TH of ketones. The coordination nature of the Ru(II) complexes was established to consist of one bidentate anionic ligand, and four co-ligands (PPh3/CO/Cl/H) to give distorted octahedral geometries. All the complexes showed good catalytic activities in the TH of wide range of ketone substrates. More importantly, the Ru–H complexes are capable of promoting the TH reactions under base free conditions. The catalytic activities of the Ru(II) complexes were regulated by the ligand backbone and identity of the auxiliary ligands. In general, Ru–H complexes displayed higher catalytic activities compared to the corresponding Ru–Cl analogues. An inner sphere monohydride mechanism, involving dissociation of one PPh3 group, was proposed as derived from in situ31P{1H} NMR spectroscopy studies.4. Experimental section
4.1 Materials and instrumentation
All the reagents were purchased from Sigma Aldrich and used without further purification. Standard procedures were followed in the purification and drying of solvents.71 All reactions were performed under an oxygen-free environment unless stated otherwise. The prefabricated ruthenium precursors [Ru(CO)(PPh3)3HCl] and [RuH2(PPh3)3(CO)] and were synthesised by adopting a modified procedure.72–74 The elemental analysis (C, H, N and S) data were recorded on the Thermal Scientific Flash 2000 instrument. The FT-IR spectra of all compounds were recorded on PerkinElmer spectrometer Zn–Se ATR in the range 4000–600 cm−1. All nuclear magnetic resonance, NMR (1H, 13C, 31P) spectra were obtained using a Bruker Avance spectrometer equipped with a Bruker magnet (9.395 T) with chemical shifts relative to tetramethylsilane (1H, 13C) and ortho-phosphoric acid (31P). The mass spectra of the compound were recorded on micro-mass LC premier micro-mass spectrometer. The single-crystal X-ray crystallography data were collected on a Bruker Apex II Duo diffractometer coupled with Oxford Instruments.4.2 X-Ray crystal structure determination
Single crystals suitable for X-ray crystallography analysis were mounted on a glass fibre held with epoxy cement. The single crystals were resized to fit the cross-section diameter of the collimator. The crystallography data of the complexes were collected on Bruker Apex-II CCD at 100 K and graphite monochromated Mo-Kα radiation (λ = 0.71073 Å). The structures were refined using direct methods programmes (SIR-92) and further refined by using a full-matrix least-squares approach on F2 using SHELXL-2018 and all fundamental calculations were done with the WinGX-2018 crystallographic software.75 HFIX fixed all hydrogen atoms in ideal places were incorporated in the refinement process using an isotropic thermal model.4.3 Synthesis and characterization of ruthenium complexes (Ru1–Ru4)
O)Ru–CO, 1629 (νCO)amidate, and 1565 (νCN)pyrazine. LC-MS: m/z: calcd 944.08; found 912.15 (M+–Cl). Anal. calcd for: C49H37ClN4O2P2RuS: C, 64.51; H, 4.09; N, 6.14; S, 3.40%. Found: C, 64.26; H, 4.01; N, 6.13; S, 3.36%.
Complexes Ru2–Ru4 were synthesised following the protocol described for Ru1.
O)Ru–CO, 1626 (νCO)amidate, and 1567 (νCN)pyrazine. LC-MS, m/z: calcd 912.12; found 911.08 (M+–H). Anal. calcd for: C49H38N4O2P2RuS: C, 64.68; H, 4.21; N, 6.16; S, 3.52%. Found: C, 64.41; H, 4.30; N, 6.01; S, 3.46%.
CO), 168.2(Cpyrazine), 165.5(Cbenzoimidazole), 149.7(Cpyrazine), 148.7(Cpyrazine), 148.4(Cpyrazine), 147.2 (Cbenzoimidazole), 146.9 (Cbenzoimidazole), 133.8(Cbenzoimidazole), 126.0(CPPh3), 123.1(CPPh3), 121.8(CPPh3), 120.4(CPPh3). 31P{1H} NMR (162 MHz, CDCl3) δ: 42.7 (s). FT-IR spectrum (Zn–Se ATR, cm−1): 1935 (νCO)Ru–CO, 1629 (νCO)amidate, 1562 (νCN)pyrazine. LC-MS, m/z: calcd 927.12; found 894.09 [M+–Cl]. Anal. calcd for: C49H38ClN5O2P2Ru: C, 63.47; H, 4.13; N, 7.55. Found: C, 63.96; H, 4.07; N, 6.65.
O)Ru–CO, 1622 (νCO)amidate, 1569 (νCN)pyrazine. LC-MS, m/z: calcd 893.11; found 892.12 (M+–H). Anal. calcd for: C49H38ClN5O2P2Ru: C, 63.46; H, 4.13; N, 7.55. Found: C, 62.91; H, 6.84; N, 6.37.
4.4 Typical transfer hydrogenation of ketone procedure
A typical procedure was followed, acetophenone (0.12 mL, 1.00 mmol), a solution of KOH (5.4 mg, 0.100 mmol, 10 mol%) in isopropyl alcohol (5 mL) and complex Ru3 (0.93 mg, 0.001 mmol, 0.1 mol%) to give molar ratio for Ru/KOH/substrate of 1/100/1000 and refluxed at 82 °C for 6 h under nitrogen atmosphere in two necked round-bottomed flask. Anisole (0.11 ml, 1.00 mmol) was introduced in the reaction flask as an internal standard. An aliquot of 0.05 mL of reaction crude was withdrawn using a syringe at regular time intervals, cooled, diluted with dried CHCl3 (0.25 mL), filtered, and the percentage conversions and yields were determined using 1H NMR spectroscopy. The percentage conversions and crude yields were calculated by comparing the integral value of CH3 of the methoxybenzene (internal standard), substrate (i.e. acetophenone) and the 1-phenylethanol product.76,77 For the mechanistic study, acetophenone (0.11 mL, 1.00 mmol), 0.10 ml of a solution of 2.5 mol% KtBuO in isopropyl alcohol and complex Ru4 (0.1 mol%) were mixed in (d8-toluene) and was analyzed using 31P{1H} NMR spectroscopy at 65 °C for 6 h.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge the University of Kwa-Zulu Natal for financial support and Mr Sizwe Zamisa for solving the structures of the complexes.References
- J. Louie, C. W. Bielawski and R. H. Grubbs, J. Am. Chem. Soc., 2001, 123, 11312–11313 CrossRef CAS PubMed.
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed.
- J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 198, 317–341 CrossRef CAS.
- S. Díez-González and S. P. Nolan, Angew. Chem., Int. Ed., 2008, 47, 8881–8884 CrossRef PubMed.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS PubMed.
- H. C. Abbenhuis, Chem. – Eur. J., 2000, 6, 25–32 CrossRef CAS.
- V. Dragutan, I. Dragutan, L. Delaude and A. Demonceau, Coord. Chem. Rev., 2007, 251, 765–794 CrossRef CAS.
- D. Milstein, Top. Catal., 2010, 53, 915–923 CrossRef CAS.
- W.-Y. Chu, C. P. Richers, E. R. Kahle, T. B. Rauchfuss, F. Arrigoni and G. Zampella, Organometallics, 2016, 35, 2782–2792 CrossRef CAS.
- S. Yadav, P. Vijayan, S. Yadav and R. Gupta, Dalton Trans., 2021, 50, 3269–3279 RSC.
- S. Yadav, P. Vijayan and R. Gupta, Ruthenium complexes of N/O/S based multidentate ligands: Structural diversities and catalysis perspectives, J. Organomet. Chem., 2021, 954, 122081 CrossRef.
- R. Ramachandran, G. Prakash, P. Viswanathamurthi and J. Malecki, Inorg. Chim. Acta, 2018, 477, 122–129 CrossRef CAS.
- K. Everaere, A. Mortreux and J. F. Carpentier, Adv. Synth. Catal., 2003, 345, 67–77 CrossRef CAS.
- A. K. Widaman, N. P. Rath and E. B. Bauer, New J. Chem., 2011, 35, 2427–2434 RSC.
- D. Huber, P. A. Kumar, P. S. Pregosin, I. S. Mikhel and A. Mezzetti, Helv. Chim. Acta, 2006, 89, 1696–1715 CrossRef CAS.
- Q.-F. Zhang, F. K. Cheung, W.-Y. Wong, I. D. Williams and W.-H. Leung, Organometallics, 2001, 20, 3777–3781 CrossRef CAS.
- J. C. Riedel and U. C. Irvine, PhD thesis, Development and design of transition metal catalyzed transformation of macrocyclization and C–C bond formation, University of California Irvine, 2019 Search PubMed.
- J. Choi and G. C. Fu, Science, 2017, 356, eaaf7230 CrossRef PubMed.
- K. Ohno, Y. Kataoka and K. Mashima, Org. Lett., 2004, 6, 4695–4697 CrossRef CAS PubMed.
- P. Yan, C. Nie, G. Li, G. Hou, W. Sun and J. Gao, Appl. Organomet. Chem., 2006, 20, 338–343 CrossRef CAS.
- W. Rabten, T. Åkermark, M. D. Kärkäs, H. Chen, J. Sun, P. G. Andersson and B. Åkermark, Dalton Trans., 2016, 45, 3272–3276 RSC.
- L. He, J. Ni, L. C. Wang, F. J. Yu, Y. Cao, H. Y. He and K. N. Fan, Chem. – Eur. J., 2009, 15, 11833–11836 CrossRef CAS PubMed.
- Z. Zhang, N. A. Butt, M. Zhou, D. Liu and W. Zhang, Chin. J. Chem., 2018, 36, 443–454 CrossRef CAS.
- Y.-Y. Li, S.-L. Yu, W.-Y. Shen and J.-X. Gao, Acc. Chem. Res., 2015, 48, 2587–2598 CrossRef CAS PubMed.
- R. van Putten, G. A. Filonenko, A. Gonzalez De Castro, C. Liu, M. Weber, C. Müller, L. Lefort and E. Pidko, Organometallics, 2019, 38, 3187–3196 CrossRef CAS PubMed.
- M. Perez, S. Elangovan, A. Spannenberg, K. Junge and M. Beller, ChemSusChem, 2017, 10, 83–86 CrossRef CAS PubMed.
- J. Qin, Z. Zhou, T. Cui, M. Hemming and E. Meggers, Chem. Sci., 2019, 10, 3202–3207 RSC.
- T. Liu, H. Chai, L. Wang and Z. Yu, Organometallics, 2017, 36, 2914–2921 CrossRef CAS.
- N. Zotova, F. J. Roberts, G. H. Kelsall, A. S. Jessiman, K. Hellgardt and K. K. M. Hii, Green Chem., 2012, 14, 226–232 RSC.
- R. M. Alam and J. J. Keating, Synthesis, 2021, 4709–4722 CAS.
- P. Melle, Y. Manoharan and M. Albrecht, Inorg. Chem., 2018, 57, 11761–11774 CrossRef CAS PubMed.
- A. H. Ngo and L. H. Do, Inorg. Chem. Front., 2020, 7, 583–591 RSC.
- B. Çetinkaya, I. Özdemir, C. Bruneau and P. H. Dixneuf, Eur. J. Inorg. Chem., 2000, 29–32 CrossRef.
- A. O. Ogweno, S. O. Ojwach and M. P. Akerman, Dalton Trans., 2014, 43, 1228–1237 RSC.
- R. Reddyrajula and U. Dalimba, Bioorg. Med. Chem. Lett., 2020, 30, 126846 CrossRef CAS PubMed.
- P. Vijayan, S. Yadav, S. Yadav and R. Gupta, Inorg. Chim. Acta, 2020, 502, 119285 CrossRef CAS.
- B. P. R. Aradhyula, W. Kaminsky and M. R. Kollipara, J. Mol. Struct., 2017, 1149, 162–170 CrossRef CAS.
- P. Vijayan, S. Yadav, S. Yadav and R. Gupta, Inorg. Chim. Acta, 2020, 502, 119285 CrossRef CAS.
- D. Bansal, G. Kumar, G. Hundal and R. Gupta, Dalton Trans., 2014, 43, 14865–14875 RSC.
- D. Bansal and R. Gupta, Dalton Trans., 2016, 45, 502–507 RSC.
- C. R. Groom and I. J. Bruno, Acta Crystallogr. Sect. B: Structural Sci., Crys. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr. Sec. B: Struct. Sci., 2002, 58, 389–397 CrossRef PubMed.
- V. Polshettiwar and R. S. Varma, Green Chem., 2009, 11, 1313–1316 RSC.
- G. K. Ramollo, I. Strydom, M. A. Fernandes, A. Lemmerer, S. O. Ojwach, J. L. Van Wyk and D. I. Bezuidenhout, Inorg. Chem., 2020, 59, 4810–4815 CrossRef CAS PubMed.
- A. M. Hall, P. Dong, A. Codina, J. P. Lowe and U. Hintermair, ACS Catal., 2019, 9, 2079–2090 CrossRef CAS.
- R. Noyori, M. Yamakawa and S. Hashiguchi, J. Org. Chem., 2001, 66, 7931–7944 CrossRef CAS PubMed.
- K. Abdur-Rashid, A. J. Lough and R. H. Morris, Organometallics, 2000, 19, 2655–2657 CrossRef CAS.
- S. E. Clapham, A. Hadzovic and R. H. Morris, Coord. Chem. Rev., 2004, 248, 2201–2237 CrossRef CAS.
- O. Altan and M. K. Yılmaz, J. Organomet. Chem., 2018, 861, 252–262 CrossRef CAS.
- C. S. Cho, B. T. Kim, T.-J. Kim and S. C. Shim, J. Org. Chem., 2001, 66, 9020–9022 CrossRef CAS PubMed.
- M. Aydemir and A. Baysal, Polyhedron, 2010, 29, 1219–1224 CrossRef CAS.
- V. Nagalakshmi, R. Nandhini, G. Venkatachalam and K. Balasubramani, J. Coord. Chem., 2020, 73, 206–216 CrossRef CAS.
- V. V. Matveevskaya, D. I. Pavlov, T. S. Sukhikh, A. L. Gushchin, A. Y. Ivanov, T. B. Tennikova, V. V. Sharoyko, S. V. Baykov, E. Benassi and A. S. Potapov, ACS Omega, 2020, 5, 11167–11179 CrossRef CAS PubMed.
- N. S. Chowdhury, C. GuhaRoy, R. J. Butcher and S. Bhattacharya, Inorg. Chim. Acta, 2013, 406, 20–26 CrossRef CAS.
- M. Bala, A. Ratnam, R. Kumar and K. Ghosh, J. Organomet. Chem., 2019, 880, 91–97 CrossRef CAS.
- P. Roy, D. Sarkar, P. Ghosh, C. K. Manna, N. Murmu and T. K. Mondal, J. Mol. Struct., 2020, 1204, 127524 CrossRef CAS.
- D. A. Cavarzan, C. B. Pinheiro and M. P. de Araujo, Trans. Met. Chem., 2015, 40, 117–123 CrossRef CAS.
- W. Baratta, J. Schütz, E. Herdtweck, W. A. Herrmann and P. Rigo, J. Organomet. Chem., 2005, 690, 5570–5575 CrossRef CAS.
- T. C. Johnson, W. G. Totty and M. Wills, Org. Lett., 2012, 14, 5230–5233 CrossRef CAS PubMed.
- W. Baratta, P. Da Ros, A. Del Zotto, A. Sechi, E. Zangrando and P. Rigo, Angew. Chem., Int. Ed., 2004, 43, 3584–3588 CrossRef CAS PubMed.
- J. Witt, A. Pöthig, F. E. Kühn and W. Baratta, Organometallics, 2013, 32, 4042–4045 CrossRef CAS.
- A. Maity, A. Sil and S. K. Patra, Eur. J. Inorg. Chem., 2018, 4063–4073 CrossRef CAS.
- B. Paul, K. Chakrabarti and S. Kundu, Dalton Trans., 2016, 45, 11162–11171 RSC.
- K. Li, J.-L. Niu, M.-Z. Yang, Z. Li, L.-Y. Wu, X.-Q. Hao and M.-P. Song, Organometallics, 2015, 34, 1170–1176 CrossRef CAS.
- T. Liu, K. Wu, L. Wang, H. Fan, Y.-G. Zhou and Z. Yu, Organometallics, 2019, 39, 93–104 CrossRef.
- R. T. Kumah, N. Tsaulwayo, B. A. Xulu and S. O. Ojwach, Dalton Trans., 2019, 48, 13630–13640 RSC.
- Z. Cao, H. Qiao and F. Zeng, Organometallics, 2019, 38, 797–804 CrossRef CAS.
- A. M. Kalsin, T. A. Peganova, I. S. Sinopalnikova, I. V. Fedyanin, N. V. Belkova, E. Deydier and R. Poli, Dalton Trans., 2020, 49, 1473–1484 RSC.
- T. Ohkuma, N. Utsumi, K. Tsutsumi, K. Murata, C. Sandoval and R. Noyori, J. Am. Chem. Soc., 2006, 128, 8724–8725 CrossRef CAS PubMed.
- J. Wettergren, E. Buitrago, P. Ryberg and H. Adolfsson, Chem. – Eur. J., 2009, 15, 5709–5718 CrossRef CAS PubMed.
- A. Vogel, A Text-Book of Practical Organic Chemistry, 846, ELBS and Longmann, London, 1989 Search PubMed.
- Y. Ogiwara, M. Miyake, T. Kochi and F. Kakiuchi, Organometallics, 2017, 36, 159–164 CrossRef CAS.
- J. Zhang, H. Wang, Z. Li and C. Wang, Trans. Met. Chem., 1995, 20, 118–119 CAS.
- M. L. Berch and A. Davidson, J. Inorg. Nucl. Chem., 1973, 35, 3763–3767 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- F. Zeng and Z. Yu, Organometallics, 2009, 28, 1855–1862 CrossRef CAS.
- D. Wang, A. Bruneau-Voisine and J.-B. Sortais, Catal. Commun., 2018, 105, 31–36 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional spectroscopic and mass spectral data (NMR, FT-IR and LC-MS spectroscopic data) and graphical representation. CCDC 2086532–2086534. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1nj05657c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2022 |