
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhoto-and thermoresponsive N-salicylideneaniline derivatives: solid-state studies and structural aspects†
Siya T.
Hulushe
 *a,
Frederick P.
Malan
b,
Eric C.
Hosten
c,
Kevin A.
Lobb
a,
Setshaba D.
Khanye
d and
Gareth M.
Watkins
a
*a,
Frederick P.
Malan
b,
Eric C.
Hosten
c,
Kevin A.
Lobb
a,
Setshaba D.
Khanye
d and
Gareth M.
Watkins
a
aDepartment of Chemistry, Rhodes University, P. O. Box 94, Makhanda 6139, South Africa. E-mail: hulushesiya@gmail.com
bDepartment of Chemistry, University of Pretoria, 02 Lynwood Road, Hatfield, Pretoria 0002, South Africa
cDepartment of Chemistry, Nelson Mandela University, Summerstrand, P. O. Box 77000, Gqeberha 6031, South Africa
dFaculty of Pharmacy, Rhodes University, P. O. Box 94, Makhanda 6139, South Africa
First published on 24th October 2022
Abstract
N-Salicylideneaniline (SA) and its derivatives are known to possess chromism upon exposure to external stimuli. Herein, we present mechanochemical synthesis of a series of photo-and thermoresponsive SA-derivatives and report on solid-state stabilisation of their tautomeric forms either by change in temperature or by photoirradiation. The influence of UV light on proton transfer between the enol-imine (EI) and keto-amine (KA) forms was investigated at λ1 = 254 and λ2 = 365 nm. Differential scanning calorimetry (DSC) measurements provided extra information on the thermodynamic relationship between the prototropic tautomers, and their exposition to liquid nitrogen, combined with variable temperature single-crystal X-ray diffraction (VT-SCXRD) and spectroscopic data, ascertained structural reasons for the intrinsic thermo-optical properties of the compounds. A series of structural determinations between 150 and 300 K further shed light on the thermomechanical behaviour exhibited by the thermoresponsive compounds. By virtue of calorimetry we were able to demonstrate proton transfer via the intramolecular O⋯N hydrogen bond over the temperature range 193–453 K. This present work demonstrates the importance of applying complementary analytical techniques and appropriate approaches for understanding the switching behaviour between the EI and KA forms. Furthermore, the assertion that it is predominantly the planarity (φ < 25°) that determines thermochromaticity is questioned.
Introduction
Manipulating molecular switching properties of switchable species by external stimuli opens up ample opportunities in crystal engineering and materials sciences. With the increased interest in switching materials, in particular N-salicylideneanilines, it is not surprising that the experimental methods (i.e. the use of external stimuli) utilised to stabilise and isolate or separate these species have evolved. In principle, switching among tautomers induced by external stimuli can be exploited for biological or material applications, making the stabilisation and isolation of such materials in recent times a centre of attention.1,2 These species have potential applications in optical storage, molecular switches, displays, signal, sensors, and optical data processing as well as numerous other photonic technologies.3–6 Switching devices have been an important area of focus owing to their interesting performance in photochromism and thermochromism. Among switching devices, N-salicylideneaniline (SA) derivatives are well-known to possess excellent reversible photo- and thermochromism phenomena.7–9 The mechanism of colour changes for SA-derivatives, to this point, has been carefully investigated and their generally recognised thermo- and photochromic processes are illustrated in Scheme 1.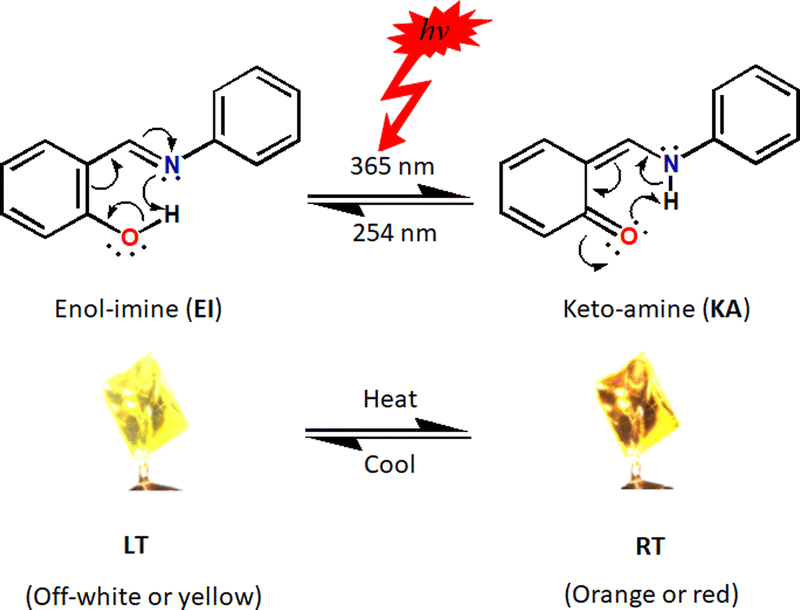 | ||
| Scheme 1 Illustration of photochromism and thermochromism in a tautomeric N-salicylideneaniline. | ||
When studying switching materials, the first two essential factors to look at are relative energies of their two (or more) forms as well as their structural differences. In the case of crystalline materials not only their respective molecular geometries (intramolecular interactions) but also the crystal packing, which characterise the spacing between the molecules and thus their intermolecular contacts, determine their switching properties.10–14 The conventional mechanism for thermochromism is a temperature-dependent ground-state enol–keto tautomerisation via intramolecular proton transfer between the enol-imine (EI) and keto-amine (KA) forms.15 Essentially, in solution, tautomers exist in rapid equilibrium with one another resulting in the formation of components that show similar physicochemical properties.16 Additionally, tautomeric equilibria in solution are often manipulated by solvent polarity and proticity, pH and so forth.17–20 Unlike in solution, in which different tautomeric forms coexist, usually only one tautomer emerges in solid-state. Few cases have been reported in which more than one tautomer appears in solid-state and these commonly include stoichiometric21–23 or non-stoichiometric24–27 mixtures displaying the same crystal. Even more scarce are examples in which both the enol–keto tautomeric forms are isolated in solid-state. However, the existence of a particular tautomer in the crystal depends, for the most part, on the parent molecule and the nature of the N-aryl substituent.28 In addition, the electron-donor or acceptor ability of the N-aryl substituents, their size and position, as well as D–H⋯A (D = donor, A = acceptor) hydrogen bond characteristics, can facilitate stabilisation of one tautomer over the other.29–32 Moreover, electron withdrawing substituents in the phenolic ring as well as an electron donating substituents in the aniline ring are known to stabilise the KA forms.28 Generally, it has been suggested that thermochromic imines are planar (dihedral angle between aromatic subunits φ < 25°) and photochromic ones are non-planar (φ > 25°).33–44 In this work, we have taken a step further by investigating this criterion in-depth.
Herein we wish to demonstrate that although containing φ < 25° for most of the compounds examined here, they are however non-thermochromic. In this work, we present the syntheses of a series of 5-nitrosalicylideneaniline derivatives, Fig. 1, with a few exhibiting reversible photochromic and/or thermochromic properties. The synthesised compounds were characterised by various analytical and spectroscopic techniques. Thermochromic properties of compounds 1–7 were elucidated by variable temperature single-crystal X-ray diffraction (VT-SCXRD). Enol–keto phase transitions were demonstrated by differential scanning calorimetry (DSC) experiments. The aim of the present work was to investigate the solid-state switching properties of the enol–keto tautomeric forms in response to external stimuli in the form of light and heat. In addition, a series of different electron-rich and electron-deficient aromatic substituents were selected to probe the role of the electron-acceptor strength on the physicochemical properties of prototropic tautomers – isomers that readily interconvert by the exchange of a proton preceded by the switch of a single and the conjugated double bond system.
 | ||
| Fig. 1 ORTEP view and atom numbering scheme of compounds 1–7 with displacement ellipsoids drawn at 50% probability level. Hydrogen atoms are shown as off-white spheres with 0.2 Å radius. | ||
Results and discussion
Synthesis and single-crystal X-ray diffraction
Mechanochemical syntheses of compounds 1–7 are discussed in the Experimental section (Fig. S1, ESI†). X-Ray crystallography analyses reveal that 1, 4 and 7 crystallise in monoclinic P21/n (No. 14) space group, while both 3 and 6 crystallise in space group P21/c (No. 14); 2 and 5 crystallise in monoclinic systems with space groups C2/c (No. 12) and C2/m (No. 15), respectively. It is noteworthy to mention that the compounds are all non-centrosymmetric. Contrary to the rest of the structures, the keto-tautomeric form in 2 at 150 K (2_LT) and 298 K (2_RT) is stabilised by two water molecules as solvent of crystallisation. Compounds 1, 2 and 3a crystallise into the KA tautomeric forms and this is deduced from electron density mapping (Fig. S2, ESI†). For comparison, microscope images and superposition of similar compounds (employing the salicylaldimino moiety) are presented in Fig. 2 and 3. In contrast, 3b, 4_LT, 4_RT, 5_LT and 5_RT are all enols, again deduced from electron density mapping (Fig. S2 and S3, ESI†) and their bond lengths (dO1–H1) vary between 0.810(4) and 0.900(4) Å. Strong intramolecular hydrogen bonds are found between the N1 and O1 atoms with dN1⋯H1 bond lengths 1.830(2)–1.890(4) Å,45 while the respective N⋯O distances are 2.582(8)–2.616(3) Å; the values for the ∠N1⋯H1–O1 angles are 146.50(4)° to 51.11(4)° (Table S1, ESI†). The bond lengths of the fragment N1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C1–C11C12–O1 are indicative of electronic delocalisation (i.e. the C12–O1 bond lengths) with bond distances between 1.317(1) and 1.340(3) Å, and are also similar to the values observed for non-conjugated C–O bonds.37
C1–C11C12–O1 are indicative of electronic delocalisation (i.e. the C12–O1 bond lengths) with bond distances between 1.317(1) and 1.340(3) Å, and are also similar to the values observed for non-conjugated C–O bonds.37
 | ||
Fig. 2 Microscope image of single-crystals of compounds 1 (top left), 2 (top right), 3 (bottom left) and 4 (bottom right) obtained by recrystallisation from ethanol, water/methanol (0.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5 v/v ratio) binary mixture, ethyl acetate/methanol (0.5:0.5 v/v ratio) and acetonitrile, respectively. :0.5 v/v ratio) binary mixture, ethyl acetate/methanol (0.5:0.5 v/v ratio) and acetonitrile, respectively. | ||
 | ||
| Fig. 3 Microscope images of compounds 5 (top left), 6 (top centre) and 7 (top right) obtained by recrystallisation from methanol, methanol/hexane mixture (0.5:0.5 v/v ratio) and chloroform, respectively, and the view of the molecular overlay of 1_LT (purple) and 1_RT (yellow) {bottom left}, 3a (green) and 3b (red) {bottom centre}, 6_LT (red), AYUGUX46 (blue) and AYUGUX0147a (green) {bottom right}. | ||
The C1–C11 bond lengths are slightly shorter than the values observed for the corresponding bond in the KA tautomers, while the Csp2–Csp2 (C11–C12) bond length values are 1.269(1)–1.412(2) Å. Finally, Carom–CN–(C1N1) bond lengths show values between 1.269(1) and 1.282(3) Å corresponding to CN groups (1.301(11)) Å)2. Looking at the dihedral angles (φ > 25°) displayed by 4_LT, 4_RT, 6_LT and 6_RT, the compounds are anticipated to possess photochromic properties. This, however, shows that the so-called “rule”: closed packing-only thermochromic is not obeyed. The first examples showing the “rule” breaking was exemplified in ref. 41–43.
Contrary to AYUGUX,46AYUGUX0147a and MOSLUC,47b crystallographic analysis indicates that the proton (H1 atom) attached to iminic N1 is disordered (by electron density mapping) with a small partial occupation factor of the second hydrogen bond position near the nitrogen atom in both 6 and 7. Additionally, unlike 6_RT, 7_LT and 7_RT, SCXRD data for 6_LT show that the H1 seems to interact more with oxygen O1 atom rather than with the nitrogen N1 atom. In confirmation, and the O1–H1 or N1–H1 bond lengths are significantly shorter and longer with distances varying from 0.775(6) to 1.459(2) Å. Such intermediate values are consistent with average values between the corresponding bond lengths in EI and KA forms. Strong intramolecular hydrogen bonds between the N1 and O1 atoms with dN1⋯H1 bond lengths, the respective N⋯O distances and the values for the ∠N1⋯H1–O1 angles are similar to those described above.
The Tα, Tβ and torsion angles (Scheme S1, ESI†) for compounds 1–7 vary significantly while their packing indices are somewhat close. Packing index could not be determined for compound 5_RT which exhibits a typical disorder in the F1, F2 and F3 atoms of the trifluoromethyl group. However, compounds 3a and 3b showed significantly different packing indices confirming the existence of these two tautomeric forms. Although compounds 1–4, 6 and 7 display strong intramolecular hydrogen bonds that “fix” planarity in their corresponding salicylaldimino moieties, owing to the non-aromatic nature of the substituent at the imino nitrogen, the compounds are non-planar and of course compound 5 (completely planar) is an exception (see dihedral angles in Table S2, ESI†). The crystal packing in 1–7, Fig. S4, ESI,† is locked by various strong intermolecular hydrogen bonds between adjacent molecules generating different supramolecular synthons.
Solid-state studies
The infrared (IR) spectra of compounds 3b, 4, and 5 displayed typical O–H, CN, and C–O stretching vibrations for enol-imines.48–52 In contrast to the above assignment, the medium-strong bands in the lower wavelength range 3300–2500 cm−1 correspond to N–H stretching frequencies for compounds 1, 2 and 3a. Nonetheless, there was not enough evidence that could be deduced from the IR spectra substantiating that compounds 6 and 7 exist as either a KA or EI. Among the chemical signals which aided us in distinguishing the tautomeric forms, the most sensitive in solid-state (SS) 13C NMR spectra is the shift related to the carbon atom (here tagged C12) bearing the keto or enol functionality.
Ideally, for the KA form this signal appears at ca 180 ppm, whereas for the EI form it is expected to shift to ca 160 ppm.53,54 Solid-state 13C cross-polarisation/magnetic angle spinning (CP/MAS) NMR experiments (Fig. 4) showed a significantly different set of signals especially for C1 and C12 (included in the intramolecular OH⋯N/NH⋯O hydrogen bond). The chemical signals were assigned by means of cross-polarisation from 1H to 13C (hC.CP) SS NMR method. As with previous research groups,53,54 the 13C (CP/MAS) NMR signals belonging to C1 (imine carbon) and C12 carbon atoms appear at 167.5 and 178.5 ppm in 3b while the signals in 3a are observed at 173.5 and 216.4 ppm, respectively. These outcomes, together with the SCXRD studies, clearly demonstrated that compound 3b exists as an EI while 3a is its KA tautomer.
 | ||
| Fig. 4 Solid-state 13C (CP/MAS) NMR spectra of (a) compound 3a and (b) compound 3b. | ||
Fig. 5 depicts differential scanning calorimetry (DSC) measurements for 3b, 4 and 5 at the temperature ranging from 193 K to 453 K. Enol–keto (3b → 3a) and keto–enol (3a → 3b) were observed by a small endotherm at 374 K (single-step phase transition) and, exotherms at 354 K and 375 K (two-step phase transformation; highlighted pink in Fig. 5a), respectively, under heat-cool conditions. The melting curve for 3b show a temperature arrest at 415 K (region highlighted orange) and there exist a slow solidification process in the DSC cooling trace at 396 K (highlighted light-blue).
 | ||
| Fig. 5 Heating-cooling truncated DSC curves (Exotherm Up) for compounds (a) 3b, (b) 4 and (c) 5. | ||
Compound 4 and 5 exhibited similar enol → keto → enol phase transformations, slow solidification and ‘melts’ certainly with different thermal events. On heating, 4 and 5 demonstrated exothermic peaks at 420 K and 376 K, respectively, with corresponding endotherms at 306 K and 270 K on subsequent cooling.
All the above experiments were performed multiple times with identical results (reversible conversions were observed infinite times through consecutive heating and cooling cycles using closed aluminium caps).
Furthermore, DSC profiles using open caps (ESI†) revealed shallow exotherms in the range 441–573 K which were assigned to melting points of the compounds. Among the samples, 1 showed the highest melting point (onset: 573 K), with 3b and 7 displaying almost identical ‘melts’ (onset: 471 K), whilst the melting points of the other samples indicated stronger/weaker intermolecular interactions stabilising their crystal structures (onsets: 2 490 K; 4 503 K; 5 441 K; 6 461 K).
Photoirradiation
Upon single-crystal-to-single-crystal (SCSC) ultraviolet (UV) light irradiation (at 365 nm for 48 hours), significant dissimilarities were observed in the solid-state UV-visible absorption spectra, in particular for 3a and 3b. Before UV light irradiation, the spectrum of 3b showed a small peak at 480 nm, and no profound absorption at ca. 500 nm. Essentially, a new peak (accompanied by colour change from orange to light yellow) emerged at about 500 nm for the photoinduced crystals of 3b suggesting enol → keto structural transformation to form 3a. However, UV-visible spectra did not display significant changes in the absorption bands for the other imines. This implies that compound 3 is photochromic while the rest are non-photochromic (Table 1).Thermochromism
The thermochromic properties of the compounds were investigated by repeated exposure to a temperature change from room to liquid-nitrogen temperature. The powdered forms of the imines were placed in test tubes and submerged into liquid nitrogen (at 77 K), while the thermal responsive single-crystals of compounds 3b, 4 and 5 were studied at 150, 180, 210, 240, 270 and 300 K. The results for crystalline and powdered forms (Fig. S5, ESI†) show that these compounds are reversibly thermochromic. The crystal colours of 3b and 5 both changed from orange to pale orange upon heating from 150 to 300 K (Fig. 6), while their powdered forms became pale yellow and yellow at 77 K, respectively. On heating from 150 up to 300 K, the colour of the crystal of 4 changed from brown to yellow-brown, while the colour of its powder was brown-green at RT and became paler upon cooling to 77 K. | ||
| Fig. 6 Photograph images of thermochromic crystals of (a) 3b, (b) 4 and (c) 5 at 150–300 K. | ||
Crystallographic details of compounds 3b, 4 and 5 from 100 K, 140 K, 150–300 K are presented in Tables S3–S7 (ESI†). Analysis of the diffraction patterns showed that the lengths of a-, b- and c-axes along with the β angle increase with temperature in the case of 3b. For 4, β angle contraction is accompanied by volume expansion upon heating while the unit cell edges experience linear positive thermal expansion (PTE). In compound 5, large PTE along c- and b-axes occurs with increasing temperature, whereas a- shows an uncommon behaviour of contracting (uniaxial negative thermal expansion; NTE). The thermal expansion along the a-axis is negative, but small in comparison to the expansion along b- and c-, therefore globally the volume increases with temperature. We established that these changes are only reversible in the range 150–240 K for 3b, as confirmed by unit cell determinations at this temperature range. The trend of unit cell parameter changes from 150 to 300 K is plotted in Fig. 7 for compounds 3b, 4 and 5. For 3b, 4 and 5 in the 150–300 K range, principal axes and linear expansion coefficient were calculated utilising the programme PASCal.55 Thermal expansion coefficients of compounds 3b, 4 and 5 within the temperature range 150–300 K are summarised in Table S8 (ESI†). Fig. 8 illustrates how the principal axes lengths change with increasing temperature for 3b, 4 and 5 along with the expansivity/compressibility plots for the principal axes X1, X2 and X3. The PTE of the a-, b- and c-axes in 3b are relatively small—the coefficients of linear thermal expansion (in MK−1) over the temperature range 150–300 K are 13.0; 35.0 and 149 for X1, X2 and X3, respectively. The volume-thermal-expansion coefficient αv (102.7 MK−1 on average) for 3b is about 1¼ times larger than for ice, but almost 5 times smaller than for methanol monohydrate.56 The major part of the expansion in 4 is along the c-axis with thermal expansion coefficients αa = 21.8; αb = 53.2 and αa = 92.7 MK−1 along the X1, X2 and X3, respectively. The coefficient of volumetric thermal expansion is 169.4 MK−1. A thermal expansion study of 5, aimed at characterising its mechanical anisotropy, revealed a large difference in the lattice coefficients of thermal expansion: −6.4, 56.5 and 181.4 MK−1 for X1, X2 and X3, respectively, while the coefficient of volumetric thermal expansion is 233.9 MK−1. The low value along the a-axis indicates the “hard” direction of the crystal, along which the intermolecular interactions are strongest, highlighting the presence of hydrogen bonds along a; thus, in this direction, molecules experience strong van der Waals forces. The hydrogen-bond structure in 3b, 4 and 5 is preserved, because no phase change has been observed over the range 150–300 K; essentially, it is presumed that there exist a concerted movement of molecules to optimise the van der Waals forces along the crystallographic a-axis.
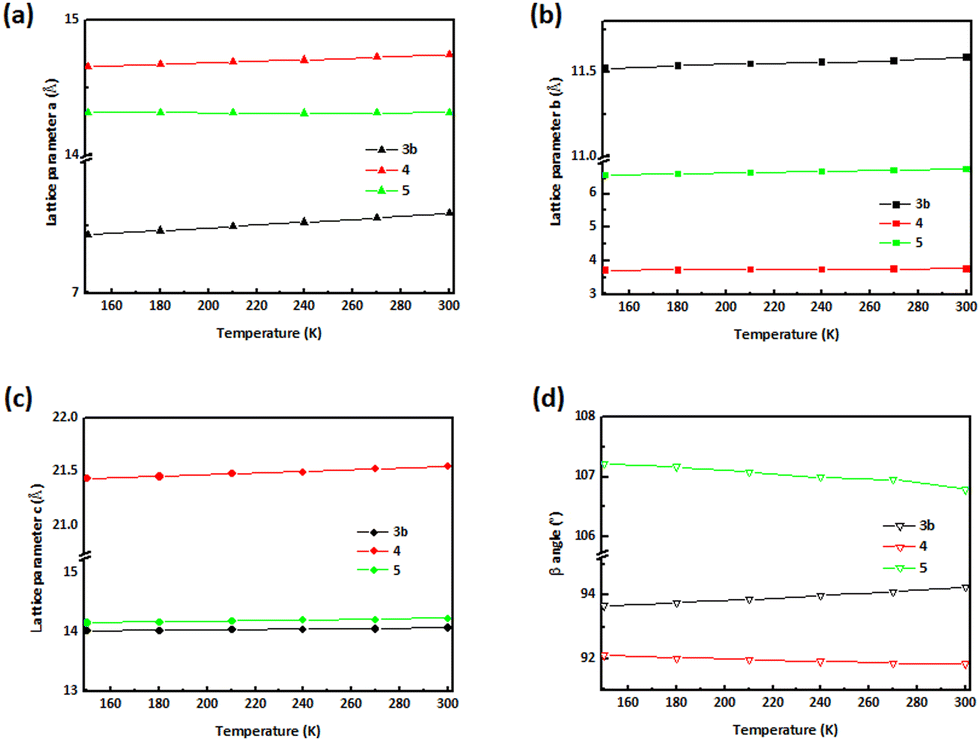 | ||
| Fig. 7 The effect of temperature on structural parameters. Structural determinations were repeatedly collected for the same crystal, starting at 150–300 K, heating the crystal by 30 K between successive data sets. Temperature dependences of (a) a-, (b) b-, (c) c-axes and (d) β angles of 3b, 4 and 5. | ||
 | ||
| Fig. 8 Percentage changes in principal axis lengths as a function of temperature along with the expansivity/compressibility plots for the principal axes X1, X2 and X3 for (a and b) 3b, (c and d) 4 and (e and f) 5. | ||
Conclusions
Herein we present solid-state insights into photo-and thermochromism in a series of selected N-salicylideneaniline derivatives. This work focused on an in-depth structural characterisation, combining differential scanning calorimetry (DSC), variable temperature single-crystal X-ray diffraction (VT-SCXRD) and spectroscopic data. The molecules studied here are almost planar (0 < dihedral angle (φ) < 8.31(9)°), of course compounds 4 and 6 are exceptions, and are presented as either enol-imines or keto-amines.In addition, the electron withdrawing character of the N-aryl substituents at 2-position facilitated stabilisation of keto-imines.
Formation of intermolecular hydrogen bonding and further aggregation of the hydrogen-bonded clusters participated as a stimulant for the temperature-induced shift to the tautomer that is less stable. A detailed structural study of the compounds has led us to the opinion that the thermochromic properties and enol–keto tautomerism are governed by supramolecular influences. Furthermore, ultraviolet-induced single-crystal-to-single-crystal (SCSC) transformation assisted enol-to-keto tautomerism in 3. In this regard, compound 5 exhibited partial negative thermal expansion (NTE) while both 3 and 4 demonstrated positive thermal expansion (PTE) together with thermochromism and, photochromism in the case of 3. However, there exist some examples where the so-called “rule”: closed packing-only thermochromic behaviour was not obeyed in the present account. It has been shown here that, with proper control, multiple responsive switching materials can be achieved. Essentially, SA-derivatives of this nature can be exploited as ultraviolet strength sensors for naked eye detection of UV radiation pollution. In addition, tuning of thermoresponsive materials holds promise in further applications such as non-expansive electronics and thermomechanical devices.
Experimental section
General
Materials; 5-nitrosalicylaldehyde, o-aminophenol, o-anisidine, o-bromoaniline and m-chloroaniline, m-(trifluoromethyl)-aniline, p-toluidine and p-anisidine were sourced from Sigma-Aldrich (Pty) Ltd and Merck (Pty) Ltd with >99% chemical purity. NMR solvents (acetone-d6, CDCl3 and DMSO-d6) were sourced from Sigma-Aldrich (Germany) with >98% chemical purity. Other solvents utilised; DMSO, ethanol, ethyl acetate and hexane were from Protea Chemicals (South Africa) while chloroform was obtained from BM Scientific/Parow Industria (South Africa). Methanol from Merck (Germany) and acetonitrile from Ranbaxy Fine Chemicals Ltd (India) of high-performance liquid chromatography (HPLC) grade. All these chemicals were commercially available and used without further purification.Synthesis of the compounds
Mechanochemical syntheses were performed at room temperature (RT; 298 K) and at 40–50% relative humidity. In all cases, equimolar amounts of 5-nitrosalicylaldehyde (1.0 mmol) and the respective anilines (1.0 mmol) were used in order to obtain the SA-derivatives. Neat grinding (NG) of 1–7 in an agate mortar led, in most cases, first to yellow/orange moist paste-like reaction mixtures, which started to solidify after 0–25 min and the crystalline solids were obtained in 57–70% yields. For each of the compounds, the simulated SCXRD patterns corresponded with that of PXRD which indicated that the Schiff bases were obtained as pure crystalline phases.Photoirradiation
The single-crystal samples were irradiated for 24 hours (UV lamp, long wavelength at λ1 = 365 nm and short wavelength at λ2 = 254 nm, 6.0 watt of full intensity).N), 1582, 1469 (s, CCarom), 1307 (s, N–O2), 1176 (s, C–O). UV-visible: λmax (nm) (ε, M−1 cm−1) (band assign.) (CHCl3): 446 (628) (n–π*), 366 (1234) (n–π*), 279 (6336) (π–π*), 241 (7710) (CT). 1H NMR (DMSO-d6, 400 MHz) δ (ppm): exp. 6.94–6.98 (4H, m, ArH), 7.23 (1H, m, ArH), 7.58–7.62 (1H, m, ArH), 8.17 (1H, dd, J = 9.4 Hz and J = 2.9 Hz, ArH), 8.61 (1H, d, J = 2.9 Hz, CHN), 9.30 (1H, s, O–H), 10.39 (1H, s, N–H); 15.72 (1H, s, O–H); 13C (DMSO-d6, 400 MHz) (δ ppm): 116.9, 117.06, 119.2, 120.3, 121.0, 129.2, 129.8, 130.2, 130.9, 137.2, 150.9 (ArC), 159.7 (PhCHN), 173.0 (ArC). HRMS (ESI) m/z calculated for C13H11N2O4 259.0719, found 259.0711 [M + H+]. Anal. calcd C, 60.47; H, 3.90; N, 10.9%. Found. C, 60.52; H, 3.88; N, 10.9%.
:0.5 v/v ratio) mixture after 2 weeks. Yield 68%; M.P: 490 K, MW: 272.27 g mol−1. IR νmax (ATR, cm−1): 3500–3185 (br, O–H), 3445 (s, N–H), 3056 (s, C–H), 1617 (s, CN), 1554, 1463 (s, CCarom), 1312 (s, N–O2), 1178 (s, C–O). UV-visible: λmax (nm) (ε, M−1 cm−1) (band assign.) (CHCl3): 449 (1168) (n–π*), 350 (4366) (n–π*), 295 (15280) (π–π*), 238 (16314) (CT). 1H NMR (CDCl3, 400 MHz) δ (ppm): 3.88 (3H, s, OCH3), 6.95–7.00 (3H, m, ArH), 7.24–7.28 (2H, t, J = 8.2 Hz, ArH), 8.15–8.18 (1H, d, J = 12.0 Hz, ArH), 8.30–8.31 (1H, d, J = 2.8 Hz, CHN), 8.74 (1H, s, ArH), 15.31 (1H, s, O–H); 13C (CDCl3, 400 MHz) (δ ppm): 57.1 (PhOCH3), 112.3, 117.5, 118.8, 120.0, 122.1, 128.9, 129.7, 134.9, 139.9, 153.5, 160.1 (PhCHN), 169.7 (ArC). HRMS (ESI) m/z calculated for C14H13N2O4 273.0875, found 273.0873 [M + H+]. Anal. calcd C, 61.76; H, 4.44; N, 10.3%. Found. C, 61.66; H, 4.42; N, 10.4%.
N), 1561, 1472 (s, CCarom), 1378 (s, N–O2), 1738, 1173 (s, C–O). UV-visible: λmax (nm) (ε, M−1 cm−1) (band assign.) (MeOH): 361 (1871) (n–π*), 247 (2043) (n–π*), 207 (2248) (CT). 1H NMR (CDCl3, 400 MHz) δ (ppm): 7.21–7.25 (3H, m, ArH), 7.34–7.38 (4H, m, ArH), 10.3 (1H, s, CHN), 11.6 (1H, s, N–H); 13C (DMSO-d6, 400 MHz) (δ ppm): 107.9, 115.9, 117.8, 118.9, 122.6, 124.8, 128.7, 131.1, 132.5, 140.2, 146.2, 166.2 (PhCHN), 189.5 (ArCO). HRMS (ESI) m/z calculated for C13H10N2O3Br 320.9875, found 320.9872 [M + H+]. Anal. calcd C, 48.62; H, 2.83; N, 8.73%. Found. C, 48.65; H, 2.78; N, 8.76%.
N), 1550, 1470 (s, CCarom), 1354 (s, N–O2), 1176 (sh, C–O). UV-visible: λmax (nm) (ε, M−1 cm−1) (band assign.) (CHCl3): 448 (1242) (n–π*), 344 (9693) (n–π*), 271 (11334) (π–π*), 239 (16143) (CT). 1H NMR (CDCl3, 400 MHz) δ (ppm): 7.57 (1H, t, J = 7.7 Hz, ArH), 7.70 (1H, ddd, J = 7.8 Hz, J = 4.8 Hz and J = 1.4 Hz, ArH), 7.81 (1H, d, J = 8.0 Hz, ArH), 8.35–8.37 (2H, ddd, J = 9.2 Hz, J = 2.7 Hz and J = 0.8 Hz, ArH), 8.45–8.47 (1H, dd, J = 9.2 Hz and J = 2.8 Hz, ArH), 8.67 (1H, d, J = 2.7 Hz, CHN), 9.22 (1H, d, J = 8.5 Hz, ArH), 14.27 (1H, s, O–H). 1H NMR (acetone-d6, 400 MHz) δ (ppm): 7.06 (1H, d, J = 9.2 Hz, ArH), 7.13–7.25 (2H, m, ArH), 7.34–7.38 (1H, td, J = 7.9 Hz and J = 1.1 Hz, ArH), 7.63–7.65 (1H, dd, J = 7.9 Hz and J = 1.02 Hz, ArH), 8.20–8.23 (1H, dd, J = 9.2 Hz and J = 2.7 Hz, ArH), 8.35 (1H, d, J = 2.7 Hz, CHN), 8.64 (1H, s, ArH), 14.06 (1H, s, O–H); 13C (DMSO-d6, 400 MHz) (δ ppm): 118.5, 119.1, 119.9, 120.5, 129.0, 129.2, 129.6, 129.7, 133.5, 140.0, 145.5, 163.3 (PhCHN), 166.3 (ArC). HRMS (ESI) m/z calculated for C13H10N2O3Br 320.9875, found 320.9867 [M + H+]. Anal. calcd C, 48.62; H, 2.83; N, 8.73%. Found. C, 48.59; H, 2.78; N, 8.80%.
N), 1568, 1473 (s, CCarom), 1389 (s, N–O2), 1183 (s, C–O). UV-visible: λmax (nm) (ε, M−1 cm−1) (band assign.) (CHCl3): 453 (891) (n–π*), 354 (14758) (n–π*), 240 (16283) (CT). 1H NMR (CDCl3, 400 MHz) δ (ppm): 7.04–7.15 (2H, m, ArH), 7.26–7.32 (3H, m, ArH), 8.21–8.23 (1H, dd, J = 9.1 Hz and J = 2.4 Hz, ArH), 8.34–8.35 (1H, d, J = 2.5 Hz, CHN), 8.64 (1H, s, ArH), 13.9 (1H, s, O–H); 13C (CDCl3, 400 MHz) (δ ppm): 118.0, 118.4, 119.6, 121.5, 127.9, 128.6, 128.7, 130.7, 135.4, 148.3, 161.9 (ArC), 166.5 (PhCHN), 207.0 (ArCO). HRMS (ESI) m/z calculated for C13H10N2O3Cl 277.0379, found 277.0374 [M + H+]. Anal. calcd C, 56.43; H, 3.28; N, 10.1%. Found. C, 56.47; H, 3.21; N, 10.4%.
N), 1568, 1471 (s, CCarom), 1384 (s, N–O2), 1729, 1187 (s, C–O). UV-visible: λmax (nm) (ε, M−1 cm−1) (band assign.) (CHCl3): 448 (973) (n–π*), 361 (14950) (n–π*), 242 (16269) (CT). 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 7.15–7.23 (1H, t, J = 9.9 Hz, ArH), 7.69–7.76 (3H, m, ArH), 7.83 (1H, s, ArH), 8.67–8.66 (1H, dd, J = 9.2 Hz and J = 2.9 Hz, CHN), 8.69 (1H, d, J = 2.9 Hz, ArH), 9.18 (1H, s, ArH), 13.6 (1H, s, N–H); 13C (DMSO-d6, 400 MHz) (δ ppm): 118.5, 119.5, 124.2, 128.4, 129.1, 130.6, 130.9, 131.1, 140.0, 148.8, 163.3 (ArC), 166.2 (PhCHN), 189.5 (ArCO). HRMS (ESI) m/z calculated for C14H10N2O3F3 311.0643, found 311.0641 [M + H+]. Anal. calcd C, 54.20; H, 2.92; N, 9.03%. Found. C, 54.22; H, 2.89; N, 9.01%.
:0.5 v/v ratio) after 2 days. Yield 63%; M.P: 461 K, MW: 256.26 g mol−1. IR νmax (ATR, cm−1): 3500–3245 (m, O–H), 3455 (s, N–H), 3075 (sh, C–H), 1603 (s, CN), 1555, 1470 (s, CCarom), 1377 (s, N–O2), 1737, 1174 (s, C–O). UV-visible: λmax (nm) (ε, M−1 cm−1) (band assign.) (CHCl3): 445 (1087) (n–π*), 341 (13205) (n–π*), 309 (14248) (n–π*), 267 (13366) (π–π*), 240 (16250) (CT). 1H NMR (CDCl3, 400 MHz) δ (ppm): 2.33 (3H, s, CH3), 6.99–7.01 (1H, d, J = 9.5 Hz, ArH), 7.25–7.28 (2H, m, ArH), 8.16–8.19 (1H, dd, J = 9.2 Hz and J = 2.7 Hz, ArH), 8.30 (1H, d, J = 2.7 Hz, CHN), 8.63 (1H, s, ArH), 14.55 (1H, s, O–H); 13C (DMSO-d6, 400 MHz) (δ ppm): 21.1 (PhCH3), 118.8, 118.9, 121.7, 128.7, 129.1, 130.5, 138.0, 139.3, 143.8 (ArC), 161.2 (PhCHN), 167.9 (ArC). HRMS (ESI) m/z calculated for C14H13N2O3 257.0926, found 257.0924 [M + H+]. Anal. calcd C, 65.62; H, 4.72; N, 10.9%. Found. C, 65.63; H, 4.69; N, 11.2%.
N), 1555, 1472 (s, CCarom), 1365 (s, N–O2), 1733, 1179 (s, C–O). UV-visible: λmax (nm) (ε, M−1 cm−1) (band assign.) (CHCl3): 439 (916) (n–π*), 355 (11090) (n–π*), 274 (14507) (π–π*), 239 (16257) (CT). 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 3.81 (3H, s, CH3), 7.04–7.09 (3H, t, J = 8.6 Hz, ArH), 7.48–7.51 (2H, d, J = 8.8 Hz, ArH), 8.20–8.24 (1H, d, J = 12.04 Hz, ArH), 8.61 (1H, s, CHN), 9.15 (1H, s, ArH), 14.7 (1H, s, O–H); 13C (DMSO-d6, 400 MHz) (δ ppm): 55.9 (PhOCH3), 115.3, 118.79, 118.9, 123.3, 128.5, 128.9, 139.1, 139.3 (ArC), 159.6 (PhCHN), 159.8 (ArC), 167.7 (ArC). HRMS (ESI) m/z calculated for C14H13N2O4 273.0875, found 273.0873 [M + H+]. Anal. calcd C, 61.76; H, 4.43; N, 10.3%. Found. C, 61.79; H, 4.39; N, 10.3%.
Characterisation
°C min−1) over a wide temperature range of 80–250 °C. The difference in the power supplied to the furnaces of the sample and reference is recorded.
X-Ray data for 1_RT, 2_LT, 2_RT, 6_LT and 6_RT was collected at Rhodes University on a Bruker D8 VENTURE APEX4 AXS area detector diffractometer,61 equipped with a graphite monochromator and a Mo Kα (λ = 0.71073 Å, at LT (150 K) and RT (298 K) fine-focus sealed tube operated at 2.0 kW (50 kV, 40 mA). All reflections were emerged and integrated with the Bruker SAINT and XPREP software packages, respectively.62 Data were collected for absorption effects using the multi-scan techniques SADABS,63 and the structures were solved by the direct methods package SHELXT and refined using X-Seed software incorporating SHELXL.64,65 The final anisotropic full-matrix least-squares refinement was done on F2. The methyl and aromatic protons were placed in geometrically idealised positions (C–H = 0.93–0.98 Å) and constrained to ride on their parent atoms with Uiso(H) = 1.2Ueqv(C). SHELX constraints and restraints were used to model structures. The non-hydrogen atoms were refined with anisotropic displacement parameters.
Single-crystals of 1_LT, 4_LT, 4_RT, 5_LT, 5_RT, 7_LT and 7_RT and, variable temperature data sets were collected at University of Pretoria on a Rigaku XtaLAB Synergy R diffractometer equipped with either graphite monochromated Mo Kα radiation (λ = 0.71073 Å) or Cu Kα (λ = 1.54184 Å), with a rotating-anode X-ray source and a HyPix CCD detector. Data reduction and absorption were carried out using the CrysAlisPro (version 1.171.40.23a) software package.66 X-ray diffraction measurements were performed at LT (150 K) and RT (298 K) using an Oxford Cryogenics Cryostat. All structures were solved by an intrinsic phasing algorithm using SHELXTS57 and were refined by full-matrix least-squares methods based on F2 utilizing SHELXL.58 All non-hydrogen atoms were refined anisotropically while all hydrogen atoms were placed in idealised positions and refined using riding models.
CCDC No. 1899401, 1902658, 1966932, 1989369, 1989398, 1989403, 2014911, 2014914, 2019418, 2049690, 2049691, 2062423, 2157376, 2156456–2156467 and 2191107–2191119.74
Author contributions
Siya T. Hulushe: conceptualization, data curation, formal analysis, experimental investigation, methodology, validation, visualization, writing – manuscript; Frederick P. Malan: data curation, formal analysis, validation, review, editing manuscript; Eric C. Hosten: data curation, formal analysis, validation; Kevin A. Lobb: data curation, formal analysis, validation; Setshaba D. Khanye: resources, funding acquisition; Gareth M. Watkins: project administration, supervision, funding acquisition.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank Distinguished Professor Tebello Nyokong (DST Institute for Nanotechnology Innovation or DST-INI, Rhodes University – South Africa) for allowing us time to use solid-state ultraviolet-visible absorption spectrometer. We are grateful to Dr Xavier Siwe Noundou (Rhodes University, South Africa) for assisting us with solid-state 13C NMR experiments. The authors thank the anonymous reviewers of the New Journal of Chemistry (NJC), whose insightful comments aided us to improve the paper. This work was supported by the National Research Foundation (NRF), South Africa and Rhodes University Research Council.References
- E. Hadjoudis and I. M. Mavridis, Chem. Soc. Rev., 2004, 33, 579–588 CAS.
- (a) E. Hadjoudis, S. D. Chatziefthimiou and I. M. Mavridis, Curr. Org. Chem., 2009, 13, 269–286 CrossRef CAS; (b) A. Carletta, F. Spinelli, S. d’Agostino, B. Ventura, M. R. Chierotti, R. Gobetto, J. Wouters and F. Grepioni, Chem. – Eur. J., 2017, 23, 5317–5329 CrossRef CAS PubMed.
- B. Rösner, M. Milek, A. Witt, B. Gobaut, P. Torelli, R. H. Fink and M. M. Khusniyarov, Angew. Chem., Int. Ed., 2015, 54, 12976–12980 CrossRef.
- T. Mutai, H. Satou and K. Araki, Nat. Mater., 2005, 4, 685–687 CrossRef CAS PubMed.
- H. Akiyama, A. Sako, N. Tajima, M. Shizuma, R. Kuroda and Y. Imai, Tetrahedron, 2016, 72, 2109–2115 CrossRef CAS.
- P. T. Todorov, P. N. Peneva, S. I. Georgieva, R. I. Rusew, B. L. Shivachev and A. H. Georgie, New J. Chem., 2019, 43, 2740–2751 RSC.
- D. R. Kanis, M. A. Ratner and T. J. Marks, Chem. Rev., 1994, 94, 195–242 CrossRef CAS.
- N. Prasad and D. J. Williams, Introduction to Non-linear Optical Effects in Molecules and Polymers, Wiley, New York, 1991 Search PubMed.
- E. V. Shah and D. R. Roy, Comput. Mater. Sci., 2014, 88, 156–162 CrossRef CAS.
- H. Sun, J.-Y. Li, F.-F. Han, R. Zhang, B.-X. Miao and Z.-H. Ni, Dyes Pigm., 2019, 167, 143–150 CrossRef CAS.
- M. Ziółek, K. Filipczak and A. Maciejewski, Chem. Phys. Lett., 2008, 464, 181–186 CrossRef.
- S. D. Chatziefthimiou, Y. G. Lazarou, E. Hadjoudis, T. Dziembowska and I. M. Mavridis, J. Phys. Chem. B, 2006, 110, 23701–23709 CrossRef CAS PubMed.
- Z. Li, C. Zhang, S. Huang, S. Li, J. Yin and S. Hua Liu, Mol. Cryst. Liq. Cryst., 2012, 557, 84–89 CrossRef CAS.
- H. Houjou, H. Ikedo and I. Yoshikawa, Chem. Commun., 2017, 53, 10898–10901 RSC.
- J. Quertinmont, A. Carletta, N. A. Tumanov, T. Leyssens, J. Wouters and B. Champagne, J. Phys. Chem. C, 2017, 121, 6897–6908 CrossRef.
- K. Ogawa, Y. Kasahara, Y. Ohtani and J. Harada, J. Am. Chem. Soc., 1998, 120, 7107–7108 CrossRef CAS.
- K. Johmoto, T. Ishida, A. Sekine, H. Uekusa and Y. Ohashi, Acta Crystallogr., Sect. B: Struct. Sci., 2012, 68, 297–304 CrossRef CAS PubMed.
- J. Harada, T. Fujiwara and K. Ogawa, J. Am. Chem. Soc., 2007, 129, 16216–16221 CrossRef CAS PubMed.
- T. Fujiwara, J. Harada and K. Ogawa, J. Phys. Chem. B, 2004, 108, 4035–4038 CrossRef CAS.
- Y. Shinde, S. Sproules, L. Kathawate, S. Pal, V. B. Konkimalla and S. Salunke-Gawali, J. Chem. Sci., 2014, 126, 213–225 CrossRef CAS.
- F. H. Kamounah, L. Antonov, V. Petrov and G. J. van der Zwan, J. Phys. Org. Chem., 2007, 20, 313–320 CrossRef CAS.
- D. Nedeltcheva and L. Antonov, J. Phys. Org. Chem., 2009, 22, 274–281 CrossRef CAS.
- A. Afkhami, F. Khajavi and H. Khanmohammadi, Anal. Chim. Acta, 2009, 634, 180–185 CrossRef CAS PubMed.
- T. Steiner and G. Koellner, Chem. Commun., 1997, 1207–1208 RSC.
- H. Pizzala, M. Carles, W. E. E. Stone and A. Thevand, J. Mol. Struct., 2000, 526, 261–268 CrossRef CAS.
- B. D. Sharma and J. F. McConnell, Acta Crystallogr., 1965, 19, 797–806 CrossRef CAS PubMed.
- F. Betchel, J. Gaultier and C. Hauw, Cryst. Struct. Commun., 1973, 2, 469–472 Search PubMed.
- P. M. Bhatt and G. R. Desiraju, Chem. Commun., 2007, 2057–2059 RSC.
- M. Juribašić, N. Bregović, V. Stilinović, V. Tomišić, M. Cindrić, P. Šket, J. Plavec, M. Rubčić and K. Užarevic, Chem. – Eur. J., 2014, 20, 17333–17345 CrossRef PubMed.
- J. W. Ledbetter, Jr., J. Phys. Chem., 1968, 72, 4111–4115 CrossRef.
- T. Dziembowska, Pol. J. Chem., 1998, 72, 193–209 CAS.
- J. M. Fernandez-G, F. R. Portilla, B. Q. Garcia, R. A. Toscano and R. Salcedo, J. Mol. Struct., 2001, 561, 197–207 CrossRef CAS.
- M. D. Cohen and G. M. J. Schmidt, J. Phys. Chem., 1962, 66, 2442–2446 CrossRef CAS.
- J. Bergman, L. Leiserowitz and G. M. J. Schmidt, J. Chem. Soc., 1964, 2068–2085 RSC.
- M. D. Cohen, Y. Hirshberg and G. M. J. Schmidt, J. Chem. Soc., 1964, 2051–2059 RSC.
- M. D. Cohen and G. M. J. Schmidt, J. Chem. Soc., 1964, 2041–2051 RSC.
- M. Zbačnik, K. Pičuljan, J. Parlov-Vuković, P. Novak and A. Roodt, Crystals, 2017, 7, 1–22 CrossRef.
- E. Hadjoudis, M. Vittorakis and I. M. Mavridis, Tetrahedron, 1987, 43, 1345–1360 CrossRef CAS.
- V. S. Padalkar and S. Seki, Chem. Soc. Rev., 2016, 45, 169–202 RSC.
- M. D. Cohen, G. M. J. Schmidt and S. Flavian, J. Chem. Soc., 1964, 2041–2051 RSC.
- F. Robert, A. D. Naik, B. Tinant, R. Robiette and Y. Garcia, Chem. – Eur. J., 2009, 15, 4327–4342 CrossRef CAS PubMed.
- (a) D. A. Safin, K. Robeyns and Y. Garcia, CrystEngComm, 2012, 14, 5523–5529 RSC; (b) F. Robert, P.-L. Jacquemin, B. Tinant and Y. Garcia, CrystEngComm, 2012, 14, 4396–4406 RSC.
- D. A. Safin, M. Bolte and Y. Garcia, CrystEngComm, 2014, 16, 8786–8793 RSC.
- M. Zbačnik, I. Nogalo, D. Cinčić and B. Kaitner, CrystEngComm, 2015, 17, 7870–7877 RSC.
- M. C. Etter, J. C. MacDonald and J. Bernstein, Acta Crystallogr., Sect. B: Struct. Sci., 1990, 46, 256–262 CrossRef PubMed.
- M. N. Tahir, H. Ahmad Shad and R. H. Tariq, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, o2319 CrossRef CAS PubMed.
- (a) N. K. Kaynar, H. Tanak, S. Sahin, N. Dege, E. Ağar and M. Yavuz, Crystallogr. Rep., 2016, 61, 239–242 CrossRef CAS; (b) I. Killic, E. Agar, F. Ersahin and S. Isik, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, o737 CrossRef PubMed.
- A. Teimouri, A. N. Chermahini, K. Taban and H. A. Dabbagh, Spectrochim. Acta, Part A, 2009, 72, 369–377 CrossRef PubMed.
- H. A. Dabbagh, A. Teimouri, A. N. Chermahini and M. Shahraki, Spectrochim. Acta, Part A, 2008, 69, 449–459 CrossRef PubMed.
- G. Varasanyi, Assignments for Vibrational Spectra of Seven Hundred Benzene Derivatives, Adam Hilger, London, 1974 Search PubMed.
- H. Tanak, J. Phys. Chem. A, 2011, 115, 13865–13876 CrossRef CAS PubMed.
- Ç. Albayrak, G. Kaştaş, M. Odabaşoğlu and R. Frank, Spectrochim. Acta, Part A, 2011, 81, 72–78 CrossRef PubMed.
- (a) O. Domínguez, B. Rodríguez-Molina, M. Rodríguez, A. Ariza, N. Farfánc and R. Santillan, New J. Chem., 2011, 35, 156–164 RSC; (b) M. Jaworska, P. B. Hrynczyszyn, M. Wełniak, A. Wojtczak, K. Nowicka, G. Krasiński, H. Kassassir, W. Ciesielski and M. J. Potrzebowski, J. Phys. Chem. A, 2010, 114, 12522–12530 CrossRef CAS PubMed.
- (a) M. Rubčić, K. Uzžrević, I. Halasz, N. Bregović, M. Mališ, I. Đilović, Z. Kokan, R. S. Stein, R. E. Dinnebier and V. Tomišić, Chem. – Eur. J., 2012, 18, 5620–5631 CrossRef PubMed; (b) A. Makal, W. Schilf, B. Kamieński, A. Szady-Chelmieniecka, E. Grech and K. Wózniak, Dalton Trans., 2011, 40, 421–430 RSC; (c) J. Sitkowski, L. Stefaniak, I. Wawer, L. Kaczmarek and G. A. Webb, Solid State Nucl. Magn. Reson., 1996, 7, 83–84 CrossRef CAS PubMed; (d) S. R. Salman, J. C. Lindon, R. D. Farrant and T. A. Carpenter, Magn. Reson. Chem., 1993, 31, 991–994 CrossRef CAS; (e) H. Saitô, I. Ando and A. Ramamoorthy, Prog. Nucl. Magn. Reson. Spectrosc., 2010, 57, 181–228 CrossRef PubMed.
- M. J. Cliffe and A. L. Goodwin, PASCal: A Principal-Axis Strain Calculator for Thermal Expansion and Compressibility Determination., J. Appl. Crystallogr., 2012, 45, 1321–1329 CrossRef CAS.
- B. Nicolaï, I. B. Rietveld, M. Barrio, N. Mahé, J.-L. Tamarit, R. Céolin, C. Guéchot and J.-M. Teulon, Struct. Chem., 2013, 24, 279–283 CrossRef.
- Bruker, SAINT (Version 7.68A), Bruker AXS Inc., Madison, Wisconsin, USA, 2012 Search PubMed.
- Bruker, APEX2 (2011.4-1) and SADABS (Version 2008/1), Bruker AXS Inc., Madison, Wisconsin, USA, 2012 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- Bruker, APEX4, SAINT and SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2021 Search PubMed.
- Bruker, SAINT-Plus, Version 7.12 (Including XPREP), Bruker AXS Inc., Wisconsin, USA, 2004 Search PubMed.
- Bruker, SADABS, Version 2004/1, Bruker AXS Inc., Madison, Wisconsin, USA, 1998 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, SHELXL-97, Program for the refinement of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
- Rigaku Oxford Diffraction, Rigaku Corporation, Oxford, UK, 2018.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- A. L. Spek, PLATON, molecular geometry program, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- L. J. Barbour, X-Seed, J. Supramol. Chem., 2001, 1, 189–191 CrossRef CAS.
- L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., Sect. A: Found. Adv., 2015, A71, 59–75 CrossRef PubMed.
- K. Brandenburg, DIAMOND, Crystal Impact, Bonn, Germany, 2005 Search PubMed.
- F. H. Allen, O. Johnson, G. P. Shields, B. R. Smith and M. Towler, J. Appl. Crystallogr., 2004, 37, 335–338 CrossRef CAS.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1899401, 1902658, 1966932, 1989369, 1989398, 1989403, 2014911, 2014914, 2019418, 2049690, 2049691, 2062423, 2157376, 2156456–2156467 and 2191107–2191119. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d1nj03056f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2022 |