
Emerging single-atom iron catalysts for advanced catalytic systems
Baisong
Chang
 *a,
Shaolong
Wu
a,
Yang
Wang
b,
Taolei
Sun
*a and
Zhen
Cheng
*c
*a,
Shaolong
Wu
a,
Yang
Wang
b,
Taolei
Sun
*a and
Zhen
Cheng
*c
aState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan 430070, P. R. China. E-mail: chang@whut.edu.cn; suntl@whut.edu.cn
bDepartment of Medical Technology, Suzhou Chien-shiung Institute of Technology, Taicang 215411, P. R. China
cState Key Laboratory of Drug Research, Molecular Imaging Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, P. R. China. E-mail: zcheng@simm.ac.cn
First published on 30th August 2022
Abstract
Due to the elusive structure–function relationship, traditional nanocatalysts always yield limited catalytic activity and selectivity, making them practically difficult to replace natural enzymes in wide industrial and biomedical applications. Accordingly, single-atom catalysts (SACs), defined as catalysts containing atomically dispersed active sites on a support material, strikingly show the highest atomic utilization and drastically boosted catalytic performances to functionally mimic or even outperform natural enzymes. The molecular characteristics of SACs (e.g., unique metal–support interactions and precisely located metal sites), especially single-atom iron catalysts (Fe-SACs) that have a similar catalytic structure to the catalytically active center of metalloprotease, enable the accurate identification of active centers in catalytic reactions, which afford ample opportunity for unraveling the structure–function relationship of Fe-SACs. In this review, we present an overview of the recent advances of support materials for anchoring an atomic dispersion of Fe. Subsequently, we highlight the structural designability of support materials as two sides of the same coin. Moreover, the applications described herein illustrate the utility of Fe-SACs in a broad scope of industrially and biologically important reactions. Finally, we present an outlook of the major challenges and opportunities remaining for the successful combination of single Fe atoms and catalysts.
1. Introduction
In the production of chemicals, catalysts play an indispensable role. For example, the industrial production of ammonia requires the use of Fe catalysts to boost the reaction rates. In recent years, catalysts have undergone rapid developments from bulk to nano levels, and as the size of the catalyst decreases, their catalytic performance in terms of activity and selectivity substantially improve. In this context, what happens when the size of the catalyst reaches the atomic level is a highly fascinating topic although technologically very challenging.In 1996, Xu and colleagues prepared a five-coordination titanium single-site heterogeneous catalyst for the catalytic epoxidation of cycloolefins.1 At this time, single-atom catalysts (SACs) did not attract the attention of researchers. It was not until 2011 that Zhang et al.2 proposed the concept of SACs and synthesized SACs with a single platinum (Pt) atom anchored on the surface of FeOx nanocrystals for the oxidation of CO. Since then, owing to the superior catalytic activity and selectivity with utmost atomic usage efficiency, SACs have arguably become the most active frontier the heterogeneous catalysis.
SACs, which are characterized by atomically dispersed active sites, interestingly show precisely located metal centers, unique metal–support interactions and an adjustable coordination environment.3,4 Hence, SACs inherit the advantages of homogeneous and heterogeneous catalysts.5 To date, many SACs based on various metals, such as Ru, Pt, Er, Ag, Au and Pd, have been developed and display excellent catalytic performances.6–8 However, despite their potential, the current examples highlighting the further practical application of noble metal catalysts have been limited to some extent. Two of the main obstacles are related to their (1) relatively high cost and (2) relatively low abundance in the Earth.9 Interestingly, transition metals hold vast potential to replace precious metal catalysts. Researchers have developed non-precious metal-based SACs (Fe, Co, Ni, Cu, Mo, etc.), resulting in a booming of the library of SACs.10–12 Among the non-precious metals, Fe is abundant in the Earth and displays excellent catalytic properties with good stability and recyclability. Besides, the facile scalable route for the synthesis of Fe-SACs ensures their large-scale application in industrial production.13–16 Today, Fe-SACs are extensively used in a wide range of research fields (Fig. 1). For example, in the energy storage and conversion fields, Fe-SACs have impressive electrochemical catalytic properties in fuel cells and metal–air cells, which can highly reduce the energy barriers of reaction pathways including but not limited to the nitrogen reduction reaction (NRR), hydrogen evolution reaction (HER), oxygen evolution reaction (OER), oxygen reduction reaction (ORR) and carbon dioxide reduction reaction (CO2RR). Importantly, Fe-SACs are demonstrated to have natural enzyme-like catalytic activities, which can functionally mimic or even outperform some metalloproteases, and hence they have been recently explored in the biological catalysis field for biosensing, wound disinfection and cancer treatment. In this review, initially, we present an overview of different support materials, focusing on how to achieve precise control of high Fe loading and single-atomic dispersion on the support surface. Subsequently, the typical characterization tools and applications of Fe-SACs in biocatalysis, energy storage and conversion are introduced. To appreciate this progress,the summary and future opportunities in the rapidly growing studies of Fe-SACs are finally discussed.
 | ||
| Fig. 1 Schematic illustration of single-atom iron catalysts for a broad scope of critical applications from biocatalysis to energy storage and conversion. | ||
2. Synthesis of Fe-SACs using different support materials
Fe-SACs possess superior catalytic activities owing to their abundant exposed sites and unsaturated coordination environment. Notably, single atoms anchored on the surface of a support have a high surface energy. Thus, during their preparation and catalysis processes, these atoms tend to agglomerate and form clusters or even nanoparticles, which will deteriorate their catalytic effect or fail. Accordingly, strong interactions between the single metal atoms and the substrate not only can prevent the agglomeration of single Fe atoms but also adjust the geometry and electronic structures of Fe-SACs, improving their catalytic performance.17 Therefore, the rational design of appropriate support materials and suitable synthesis processes are particularly important for the catalytic performance of Fe-SACs.2.1. Metal–organic frameworks (MOF) and their derivative-supported Fe-SACs
The unique advantage of MOFs is their designability, e.g., adjustable chemical compositions, structure, pore, particle size and shape, which can provide a high surface area, porosity (about 90% of the free volume), more exposed active sites and abundant pore positions.18–20 As anticipated, MOFs and their derivatives have become ideal support materials for anchoring an atomic dispersion of Fe and other single metal atoms.MOFs are composed of evenly distributed metal nodes and organic linkers.21 Among the MOF materials, the zeolite imidazole framework (ZIF) is most commonly utilized to construct SACs.22 One typical ZIF material is ZIF-8, which is composed of Zn atoms as metal nodes. Owing to the low boiling point of Zn atoms, ZIF-8 is extensively used as a support material for the synthesis of Fe-SACs via the spatial confinement strategy.23–25 Fe atoms are confined in a certain space to prevent their agglomeration or migration. This strategy requires that the size of the metal precursor be small enough to enter the “cage”, but not be too small, where its size must be larger than half of the cavity in the MOF material. This can guarantee that only one molecule of metal precursor enters one “cage”, preventing its agglomeration during the next pyrolysis treatment.18 By classifying the relevant literature, several representative size parameters can be derived, providing a critical guide for the design of Fe-SAC:ZIF-8 with a cavity diameter of ∼11.6 Å and the metal precursors of Fe(acac)3 and ferrocene with the molecular size of 9.7 and 6.4 Å, respectively. Li et al.26 used a cage-encapsulated-precursor pyrolysis strategy to prepare a highly reactive isolated single-atom Fe/N-doped porous carbon catalyst (Fe/ZIF-SAC) with an Fe loading of 2.16 wt% (Fig. 2a). During the synthesis process, molecular-scale cages were formed together with the assembly of Zn2+ and 2-methylimidazole. Importantly, one Fe(acac)3 molecule, in principle, was trapped in one cage, resulting from the size-matching effect. After pyrolysis at 900 °C under an Ar atmosphere, ZIF-8 was transformed into an N-doped porous carbon framework. Meanwhile, the Fe(acac)3 molecule in the cage was reduced by carbonization of the organic linker, leading to the production of isolated single Fe atoms anchored on nitrogen species. To verify the above-mentioned size-matching effect, it should be investigated what will happen if an Fe precursor with a larger molecular size than the ZIF-8 cavity diameter is selected to synthesize Fe-SACs. Recently, Fe(II) phthalocyanine (FePc) has been identified to be an excellent metal precursor with a relatively large molecular size of about 14.6 Å.14 Accumulating evidence indicates that FePc will destroy the microcavity of ZIF-8, causing it to rupture.27,28 In the subsequent pyrolysis process, ZIF-8 was converted into N-doped carbon and FePc was simultaneously reduced in situ to produce edge-dispersed Fe–N sites.26 However, a drawback inherent to the nature of this control experiment was that excessive FePc could form Fe2O3 nanoparticles. Thus, acid etching treatment was required to remove impurities, but clearly this process caused the loss of Fe atoms.
 | ||
| Fig. 2 Synthesis of Fe-SACs using different support materials. (a) Schematic illustration of the synthesis of isolated single iron atoms anchored on N-doped porous carbon. Reprinted with permission from ref. 26. Copyright 2017, John Wiley and Sons, Inc. (b) Schematic illustration of the synthesis of atomically dispersed iron-nitrogen active sites in porphyrinic triazine-based frameworks. Reprinted with permission from ref. 42. Copyright 2018, the American Chemical Society. (c) Schematic illustration of the synthesis of atomically dispersed iron-nitrogen species. Reprinted with permission from ref. 52. Copyright 2017, John Wiley & Sons, Inc. (d) Schematic illustration of the synthesis of porous dual-metal doped g-C3N4 patches attached to carbon nanotube bundles. Reprinted with permission from ref. 61. Copyright 2018, The Royal Society of Chemistry. (e) Schematic illustration of synthesis of SACs from metal foam (metal = Fe, Co, Ni, and Cu) by strong bonding from the dangling bonds of GO support. Reprinted with permission from ref. 4. Copyright 2019, John Wiley & Sons, Inc. | ||
In the case of MOFs and their derivatives as a support, another method used for the preparation of Fe-SACs is the coordination design strategy.29 This strategy requires that the organic linking group in the MOF material has one or more atoms (e.g., N, O and S) containing lone pairs of electrons. These atoms can serve as a “claw” to trap Fe atoms, thereby anchoring the dispersed Fe atoms on the surface of MOF and their derivatives.30,31
As a support material, we reiterate that the biggest advantage of MOFs and their derivatives is their designability, but this is just two sides of the same coin. MOFs and their derivatives support can be beneficial or detrimental for the preparation of SACs. The low chemical stability and weak electrical conductivity of MOFs and their derivatives make it difficult to transfer their charge for electrocatalytic applications.20 Considering this, we need to further study an alternative approach to improve the conductivity of SACs using MOFs and their derivative materials as a support, for example, combining them with conductive agents (graphene, nickel foam, etc.) to form a composite nanomaterial.
2.2. Covalent organic frameworks (COF) and their derivative-supported Fe-SACs
The organic building blocks of covalent organic frameworks (COFs) are connected via covalent bonds to construct a porous framework with a periodic structure.32,33 Similar to MOF materials, COFs have a high surface area, low density, tunable pore size, skeleton, composition and porosity and a variety of structural topologies.34–37 These features make them attractive in wide areas, such as gas storage, catalysts, optoelectronics, separation, and proton conduction.38,39Actually, COFs and their derivatives under pyrolysis have been found to act as ideal precursors for the production of porous carbon materials. When COFs and their derivatives are employed as a support, Fe2+/3+ can facilely diffuse into their cavity owing to the strong coordination interaction between Fe2+/3+ and N position in COFs and their derivatives. The periodic structures of the separation unit in COFs and their derivatives will remarkably inhibit the agglomeration of Fe atoms during the pyrolysis process. Based on the connectivity and geometry of their optimized molecular building blocks, COFs can be separated into two-dimensional (2D) layered structures and three-dimensional (3D) network structures.34 In general, 2D layered structures are widely used in the field of catalysts. 2D layered COFs are stacked face-to-face with the largest π orbital overlap, thereby promoting the charge migration along the framework and improving their conductivity. In addition, covalent bonds with extended π conjugation are connected to the skeleton, which can endow COFs with better physical and chemical stability.34,40
Atom-dispersed Fe metal sites can be introduced in COFs and their derivatives through two different paths. The first path is the direct use of Fe metal macrocyclic clusters as building blocks, such as Fe-based porphyrins and Fe phthalocyanine.41 Recently, Cao et al.42 prepared a highly stable atom-dispersed Fe–N4 species on porous porphyrin triazine-based frameworks (Fe/PTF-SACs) via a direct ionothermal method (Fig. 2b). Under the catalysis of molten ZnCl2, 5,10,15,20-tetrakis(4-cyanophenyl)porphyrinato-Fe(III) chloride (Fe-TPPCN) was transformed to Fe/PTF-SACs through a trimerization reaction in an evacuated ampule, followed by removal of ZnCl2 using diluted HCl. Taking a closer look at above synthetic processes, the contribution of molten ZnCl2 was the most prominent. It not only could catalyze the trimerization reaction of the Lewis acid, but also act as a pore-forming agent to produce mesopores. Consequently, the atomically dispersed Fe–N4 active sites were more exposed to the highly dispersed reactive species, which was conducive for the transport of substances and maximized the atom utilization. Meanwhile, materials under high-temperature pyrolysis will inevitably undergo partial graphitization, hence accelerating the electron transfer and improving the electrocatalytic activity of Fe-SACs.
Thus far, more than 200 2D layered COF materials have been developed. However, these COF materials usually exist in the form of insoluble, cross-linked powders or films, which highly limit their applications. Hence, it is particularly critical to develop water-soluble COFs as support materials.43,44 Since the crosslinking event is mainly stopped under swelling conditions rather than dissolution conditions, grafting hydrophilic groups is not feasible. Peng et al.45 designed and constructed iron quasi-phthalocyanine into conjugated a 2D network with a rigid structure and heterogeneously charged the coordinated single-atom centers to synthesize 2D COF materials. The strong interactions between the negative hydroxyl groups and positively charged quasi-phthalocyanine centers enabled the rigid structures of the COF to be easily solvated. Thus, the robust conjugation systems and Fe–N coordination centers of the COF materials caused them to display outstanding ORR properties. The factors affecting the catalytic performance of Fe-SACs included the Fe loading and the role of the support. A high N content in the support can anchor more Fe atoms, thereby increasing the metal loading, while a high specific surface area in the support can improve the mass transfer process in the catalytic process. Therefore, when using COF derivatives as a support, increasing their specific surface area becomes the main problem. Wang et al.46 synthesized an imine COF thin film at room temperature through an electrochemical exfoliation synthesis method. The COF thin film possessed high crystallinity and a hierarchical porous structure, which made it display relatively excellent mass transfer performances. Recently, Xiong et al.47 synthesized a hollow COF with high crystallinity and uniform morphology. These COF materials were demonstrated to exhibit a large specific surface area, radially oriented nanopore channels, uniform morphology and tunable particle size. Cooper and colleagues prepared a highly crystalline and stable COF material with good CO2 adsorption ability via the pre-assembly of reversible covalent bonds and post-synthesis framework reconstruction into irreversible covalent bonds.48 The above-mentioned COF materials all displayed a high specific surface area, and thus their catalytic activity will be greatly improved when they are used to prepare Fe-SACs.
2.3. Carbon nanotube (CNT)-supported Fe-SACs
CNTs are mainly composed of carbon atoms arranged in a hexagonal shape to form a coaxial circular tube with single to dozens of layers.36 The carbon atoms in CNTs are dominated by sp2 hybridization. Simultaneously, the hexagonal grid structure has a certain degree of bending to form a spatial topology, yielding a certain sp3 hybrid bond. Thus, the chemical bond formed has a mixture of sp2 and sp3 hybrid states, and these p orbitals will overlap outside the graphene sheet of CNTs, showing a highly delocalized large π bond. Consequently, CNTs have good electrical conductivity. At high temperatures, because the Fe atoms are flowing, they tend to migrate and aggregate to produce inactive nanoparticles, especially at a high content of Fe precursors. Thus, to improve the activity of the catalyst, the selection of the appropriate support to anchor Fe atoms is an effective route to increase the single-atom Fe loading for the preparation of Fe-SACs.49 CNTs provide abundant exposed Fe–Nx active sites, good chemical stability, high mechanical properties and unique porosity. Moreover, the excellent electrical conductivity of CNTs accelerates the electron transfer efficiency.9,36,50,51 Thus, all these features enable the use of CNTs as a support with vast potential for anchoring Fe atoms.According to whether the CNTs are simultaneously formed in the process of synthesizing Fe-SACs, their synthesis routes can be divided into two categories. One is the direct use of commercially available CNTs as a support. This synthesis method generally involves three steps, i.e., adsorption of Fe3+ cation (sometimes Na+ is also required), pyrolysis in an inert gas (sometimes NH3 is needed) to obtain atomically dispersed Fe–Nx active sites, and use of the acid leaching strategy to remove the nanoparticles or iron oxide generated by Fe agglomeration during pyrolysis. Accordingly, single-atom Fe anchored on CNT catalysts with high catalytic performances will be achieved.52–54 For example, Chen et al.52 proposed an effective method involving S to achieve atomically dispersed Fe–Nx species anchored on N, S co-decorated CNTs (Fig. 2c), generating new carbon-based bifunctional electrocatalysts. FeCl3 as the iron source and KSCN as the sulfur source were added to synthesize the N,S co-doped single atomic catalysts with a unique hierarchical structure and rich atom dispersed Fe–Nx species. The obtained Fe/CNT-SACs had a higher specific surface area (241.5 m2 g−1) compared with the SACs without S salt (173.9 m2 g−1). The increased surface area and pore volume of the catalyst facilitated the acid leaching. In addition, the addition of S salts can prevent the formation of iron-based carbides, which are not easily removed by acid leaching.
The other type of synthesis relies on the application of different carbon sources to synthesize CNTs during the preparation of Fe-SACs. Likewise, this synthetic route roughly involves three steps. The first is assembly polymerization, in which various Fe and carbon sources are introduced. Secondly, high-temperature pyrolysis is carried out in an inert atmosphere such as N2 or NH3. Finally, acid leaching is performed to remove other unwanted metal species,9,55 which will lead to the loss of Fe–Nx active sites, thereby reducing its catalytic performance.
By comparing the last step for the synthesis of CNT-supported Fe-SACs, acid etching treatment is usually needed and essential, regardless of the synthetic methods, to eliminate the nanoparticles or iron oxides generated from the agglomeration of Fe during pyrolysis. We advocate that the acid leaching strategy plays an increasingly crucial role in accelerating the exposure of Fe–Nx active sites, but it has to be delicately considered in future studies. Clearly, excessive acid leaching will also be irreversible to remove atomically dispersed single-atom Fe species, which possibly make the as-synthesized Fe-SACs fail in satisfying catalytic goals. Recently, the metal (Zn) atomic isolation strategy and silicon protective layer-assisted strategy have emerged as exciting alternatives to avoid Fe agglomeration in the synthesis of CNT-supported Fe-SACs, entering the development spotlight.54,56
2.4. Graphitic carbon nitride (g-C3N4)-supported Fe-SACs
Graphitic carbon nitride (g-C3N4) is a graphene analogue consisting of periodically linked tris-s-triazine units, which is produced via the thermal condensation of organic monomers, including urea, melamine and dicyandiamide.57–60 Considering its molecular structure, g-C3N4 can be regarded as N-doped graphene. Furthermore, the g-C3N4 framework contains rich and uniformly distributed pyridine N components with abundant lone pairs of electrons and large N coordination cavity, making it an impressive support for anchoring Fe atoms for the synthesis of Fe-SACs.41There are usually two routes for the fabrication of Fe-SACs using g-C3N4 as a support. The first is a direct in situ bottom-up synthetic method, which involves the direct heating and polymerization of mixtures of Fe salt and organic monomers to obtain Fe-SACs with high catalytic activities.41,57 For example, Sang et al.58 synthesized Fe-coordinated g-C3N4via the thermal condensation of dicyandiamide and FeCl2 in an N2 atmosphere. Notably, when g-C3N4 alone was selected as the supporting material, the catalytic effect of the as-prepared Fe-SACs was not very good due to its poor conductivity. Thus, to solve this issue, CNTs or crystalline carbon structure serving as a conductive substrate can be introduced in the g-C3N4 framework. Hybrid compositions not only facilitate charge transfer, but also suppress the strong accumulation of g-C3N4 layers, hence enabling a high Fe loading.41,57 For example, Song et al.61 adopted a high-temperature polymerization method to develop a structure composed of g-C3N4 nanopatch-enveloped CNTs, where isolated Fe atoms were embedded in the tri-s-triazine unit by forming a metal–Nx structure (Fig. 2d). The scheme showed that NH4Cl served as a dynamic gas template, which could generate ammonia gas during the pyrolysis process, and consequently adjust the porous structure of g-C3N4. The loosely packed g-C3N4 segment adhered to the CNT framework was conducive for electron transfer and increasing the exposed active sites.
Significantly, the majority of g-C3N4 materials are prepared via the thermal polymerization of melamine, cyanamide or dicyandiamide. However, these precursors are expensive and toxic. Accordingly, for large-scale industrial production in the future, we still need to select other precursors (e.g., urea) to produce g-C3N4-supported Fe-SACs.58,62–64
Another popular strategy to synthesize Fe-SACs using g-C3N4 as a support is the post-synthesis method (also called wet chemical method).57 This method involves impregnating the target metal salt in the liquid phase on the surface or edge defects of g-C3N4, and then reducing the metal ions using a reducing agent such as NaBH4 or low temperature heat treatment.41 The main advantage of this method is that the reaction conditions are relatively mild, which will not destroy the original basic structure of the support g-C3N4 during the synthesis of Fe-SACs. Moreover, the diffusion of the target metal ions will be more complete. There is a large number of coordination sites on g-C3N4, facilitating the uniform distribution of Fe ions on the surface of the g-C3N4 support.41,57 For example, Cao et al.65 proposed a surfactant-assisted wet chemical method to synthesize single-atom Fe supported on N-doped g-C3N4 (denoted as SA-Fe/NG), creating unique Fe-pyrrolic-N active species with an Fe atom and eight C atom active sites next to pyrrolic N.
2.5. Graphene-supported Fe-SACs
Graphene is a hexagonal 2D material composed of sp2 hybridized carbon.66,67 It is common in nature, but difficult to peel off the single-layer structure from the graphite widely used in daily life. The arrangement of carbon inside graphene is consistent with a monoatomic layer of graphite bonded by sp2 hybrid orbitals, in which each carbon atom has an unbonded electron in the p orbital. The p orbital of adjacent carbons is perpendicular to the plane direction formed by the δ bond and forms π-type bonds with a semi-full state, endowing graphene with excellent electrical conductivity and heat conduction. Compared with CNTs, both graphene and graphene nanosheets (GN) exhibit a high specific surface area and superior mechanical property, which are crucial requirements of support materials to anchor Fe atoms.66,68,69However, the interaction between graphene and iron atoms is relatively weak. The single Fe atoms are prone to aggregate, which deteriorates the catalytic activities of Fe-SACs. Thus, to address this issue, N doping and vacancy defects are two commonly employed modification methods.70 Defective graphene with vacancies that can firmly anchor metal atoms, and hence prevent their agglomeration, is often used as a support for the synthesis of Fe-SACs. Generally, to form graphene vacancies (monovacancy and divacancy graphene), a specific area is irradiated with a focused electron beam. The formed vacancies play multiple roles in the adsorption of single Fe atoms and prevent their agglomeration, thereby leading to highly dispersed single-atom active sites.71–73
Graphene oxide (GO), a derivative of graphene, is a single layer of graphite peeled from graphite oxide.67 Compared with graphene, which is composed of sp2 hybridized carbon, GO contains unoxidized aromatic regions (sp2 carbon atoms) and fatty six-membered ring regions (sp3 carbon atom) formed by oxidation damage to the lattice. Importantly, its surface and edges have more O-containing groups, e.g., –CH–(O)–CH–, –OH, –COOH and –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, which can complex with metal ions, especially those with multivalence.74 Moreover, due to its high specific surface energy, good hydrophilicity and mechanical property, GO has become an ideal support for the preparation of SACs.67 Qu et al.4 modified GO with a large amount of O-containing dangling bonds, which can provide anchoring sites for Fe atoms (Fig. 2e). In the synthetic process, the slurry of GO was mixed with the metal foam of iron. At this time, a large amount of iron and the suspended oxygen groups on the surface of graphene oxide were in close contact, and the iron transferred electrons to the oxygen groups. Consequently, a large number of Feδ+ (0 < δ < 3) species was formed. Besides, owing to the coordination, each iron atom and the four oxygen dangling bonds on the surface of GO formed Fe–O bonds. Finally, the iron atoms were pulled out of the metal foam under the action of ultrasound to yield their natural isolation, which prevented the iron atoms from agglomerating on the surface of GO.
O, which can complex with metal ions, especially those with multivalence.74 Moreover, due to its high specific surface energy, good hydrophilicity and mechanical property, GO has become an ideal support for the preparation of SACs.67 Qu et al.4 modified GO with a large amount of O-containing dangling bonds, which can provide anchoring sites for Fe atoms (Fig. 2e). In the synthetic process, the slurry of GO was mixed with the metal foam of iron. At this time, a large amount of iron and the suspended oxygen groups on the surface of graphene oxide were in close contact, and the iron transferred electrons to the oxygen groups. Consequently, a large number of Feδ+ (0 < δ < 3) species was formed. Besides, owing to the coordination, each iron atom and the four oxygen dangling bonds on the surface of GO formed Fe–O bonds. Finally, the iron atoms were pulled out of the metal foam under the action of ultrasound to yield their natural isolation, which prevented the iron atoms from agglomerating on the surface of GO.
Notably, the use of GO as a support for anchoring single metal atoms has certain drawbacks that cannot be circumvented. For example, under pyrolysis treatment, GO is prone to undergo irreversible accumulation. This phenomenon, which is an inherent nature of GO, causes a series of destructive consequences, including deterioration of the accessible surface area, blocking of catalytically active centers, and obstruction of mass diffusion/transfer, ultimately having a negative effect on the electrocatalytic performance. Thus, the design and fabrication of GO-supported Fe-SACs remain a grand challenge. Only recently, Tang et al.69 proposed a sophisticated and universal template-engaged method to synthesize 3D GO hollow nanospheres, which can exhibit improved mechanical strength, accelerated reaction kinetics and enhanced catalytic performance when employed as a catalyst support. From a positive point of view, 3D GO materials are expected to be an ideal platform to immobilize isolated single metal atoms.
2.6. Non-carbon-based material-supported Fe-SACs
The derivatives of MOFs and COFs, g-C3N4, CNTs and graphene are implicated as carbon-based supports, while non-carbon-based materials (MoS2, MoSe2, WS2, WSe2 and other transition metal sulfides) also deserve to be studied to support single Fe atoms. The basal plane of the crystal surface of transition metal chalcogenides is controlled by introducing multiple defect sites, enabling single atoms to be anchored to form high-loading SACs. In a typical experiment, Zheng et al.75 employed thiourea-coordinated Fe atoms as a precursor to synthesize atomically dispersed Fe supported on ultra-thin 2D transition metal chalcogenides. Notably, this method was not only suitable for metallic Fe, but could also be applied to anchor other metals, such as Co, Ni, Cu, Pt, Pd and Ru. Impressively, the single metal loading reached an astonishing 10%. Taking the MoS2-supported Fe as an example, n-BuLi was initially used to treat transition metal chalcogenides such as MoS2, and then they were peeled into ultra-thin monolayers. There were abundant S vacancies in these monolayers, which self-assembled with Fe–thiourea complexes to yield nuclei under hydrothermal environments. Finally, a single atomic Fe catalyst with MoS2 as a carrier was formed through H2 reduction at high temperature. At 2022, Li et al.76 constructed Fe single-atom catalytically active sites coordinated with S on mesoporous TiO2via a lattice confinement strategy. The spectroscopy analysis indicated that Fe atoms were anchored in the TiO2 lattice in the form of FeS2O2 coordination structures, which served as the active centers for the electrocatalytic nitrogen reduction reaction. It should be noted that the lattice confinement strategy is versatile and can be employed to synthesize single-atom catalysts, including Ni1S@TiO2, Co1Sx@TiO2, and Mo1Sx@TiO2. In a typical experiment, Zhang et al.77 synthesized 3D single-atom Fe-doped macroporous/mesoporous TiO2–SiO2 photocatalysts via an evaporation-driven self-assembly route. In this work, polystyrene spheres were arranged to produce polystyrene opal as a hard template for macropores and P123 micelles were used as a soft template for mesopores. Hydrolysis and calcination were performed to remove polystyrene and P123, leading to the formation of macroporous/mesoporous structures with the presence of massive single Fe atoms.Besides, the surface of transition metals contains a large amount of hydroxide groups, hence providing suitable anchoring sites for the preparation of SACs to coordinate with metal atoms via M–O(OH) interactions. Recently, He et al.78 constructed single Pt atoms anchored on a CuO support by utilizing the electron transfer between Pt atoms and CuO. Zhang et al.79 synthesized atomically dispersed Pt on RuO2via an impregnation-adsorption method, which exhibited excellent catalytic activity for methanol oxidation under alkaline conditions. The above-mentioned examples are associated with single Pt atoms supported by transition metal oxides because electron transfer could easily occur between Pt and transition metal oxides. Likewise, it needs to be further investigated if it also possible to synthesize Fe-SACs through interactions between single Fe atoms and other transition metal oxides.
3. Characterization of SACs
The information of active species (e.g., atomic structures, oxidation states and coordination chemistry) is closely related to the catalytic activity of SACs. With the development of advanced characterization techniques, an atomically precise understanding of SACs becomes possible, which can have quantifiable benefits for catalytic performance.3.1 Electron microscopy
Regarding high-resolution transmission electron microscopy (HRTEM), its detection limit is at the nanometer or sub-nanometer levels. Thus, it is impossible to clearly observe the distribution of single atoms on a support by HRTEM. Fortunately, sub-angstrom-resolution aberration-corrected scanning transmission electron microscopy (AC-STEM) can image individual atoms dispersed on different support materials.17 To outline the whole picture of SACs, the combination of HRTEM and AC-STEM is a widely accepted and indispensable characterization method. Firstly, the use of the HRTEM technique can observe the entire support with a nanoscale imaging resolution (Fig. 3a and b). For example, Jiang et al.14 utilized ZIF-8 and FePc precursors to synthesize atom-dispersed Fe–N4 species anchored on N-doped carbon, exhibiting abundant mesopores with a dodecahedron shape (Fig. 3b). Impressively, the imaging resolution will reach the atomic scale under AC-STEM characterization. The red circle in Fig. 3c represents a single scattered Fe atom. Meanwhile, auxiliary characterizations, e.g., energy-dispersive X-ray spectroscopy (EDS) and electron energy loss spectroscopy (EELS), together with electron microscopy can be employed to analyze the structure and elemental distribution of single metal atoms.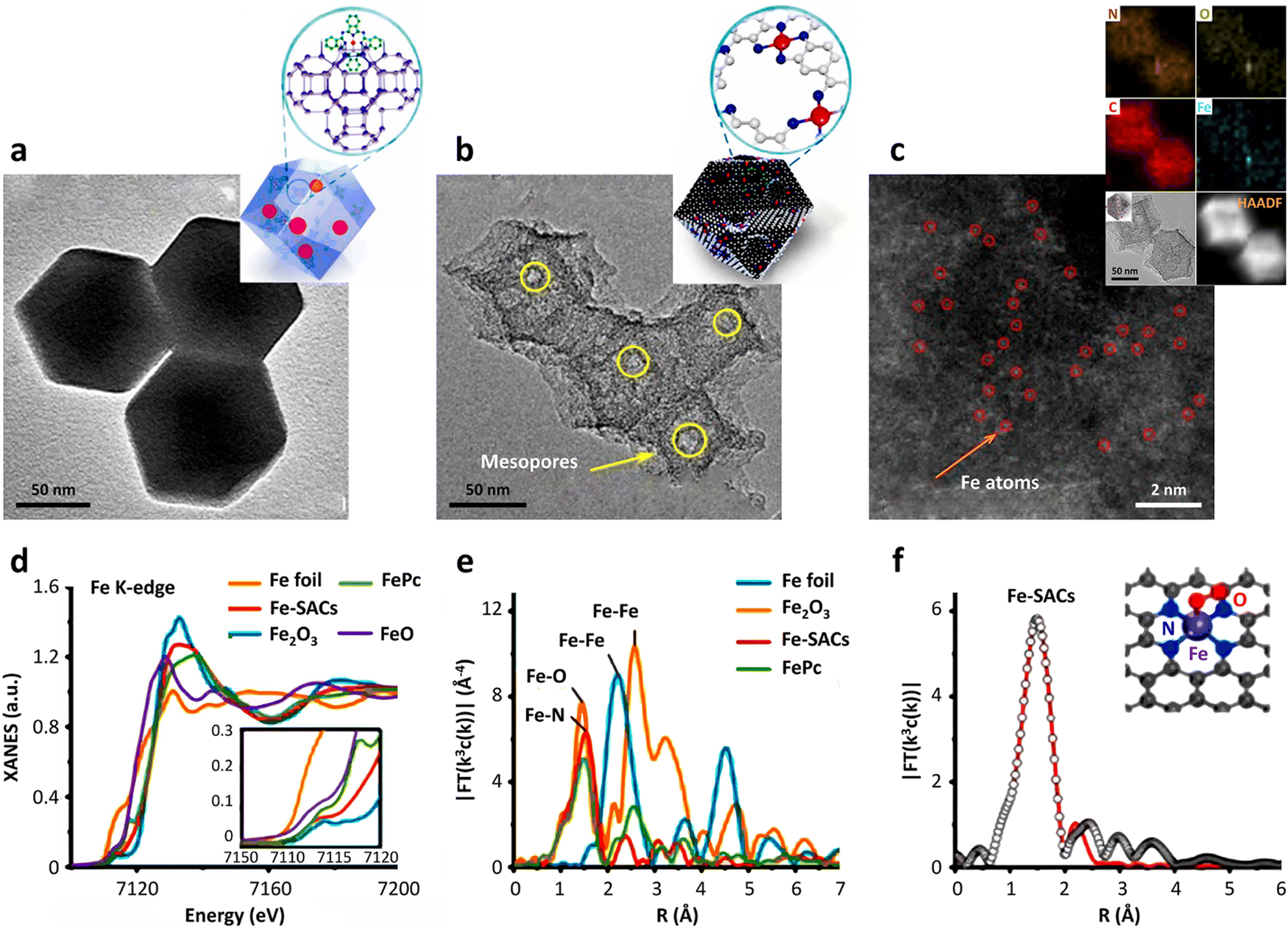 | ||
| Fig. 3 Characterization of SACs. (a) TEM image of FePc molecules encapsulated in the cavities of ZIF. (b) TEM image and (c) HAADF-STEM image of hierarchically porous Fe-SACs. (d) Fe K-edge XANES (inset was the magnified image) and (e) FT k3-weighted EXAFS spectra of Fe-SACs and the reference samples. (f) Corresponding FT-EXAFS fitting curves of Fe-SACs. Reprinted with permission from ref. 14. Copyright 2018, the American Chemical Society. | ||
3.2 Spectroscopy analysis
In addition to the use of the aforementioned electron microscopy technology to characterize the distribution of individual metal atoms on the support surface, it is also necessary to identify the catalytic property-associated coordination information such as electronic and atomic structures of SACs. The most commonly utilized spectroscopy technology is X-ray absorption fine structure spectroscopy (XAFS). According to the mechanism and peak shapes, XAFS can be divided into two categories,80i.e., X-ray absorption near edge spectroscopy (XANES) and extended X-ray absorption fine structure spectroscopy (EXAFS).The XANES technique provides the valence state, charge transfer and other information of the single metal atoms in SACs. Interestingly, due to the presence of the “fingerprint effect”,81,82 XANES has the capability of establishing the type of element. It is important to note that considering the definition of SACs, there should be no metal–metal bond (e.g., Fe–Fe bond) in the active sites on the surface of support. EXAFS analysis can clarify this atomically structural information, a decisive criterion in evaluating the successful synthesis of SACs. If EXAFS analysis shows that that there is no information on metal–metal bonds, it is basically inferred that only a single metal atom exists in SACs. Besides, some other characterizations of the active centers can be elucidated by EXAFS technology, including the type of atom coordinated with the central atom, the coordination number and the distance between the coordinated atoms and central atoms.17 As shown in Fig. 3d, taking Fe foil, FePc, FeO and Fe2O3 as references, the Fe K-edge XANES results indicated that the oxidation state of the Fe atoms in Fe-SACs were between the Fe(II) and Fe(III) states.14 According to the Fourier transform curve of EXAFS (Fig. 3e), it can be seen that there was only one main peak at 1.5 Å, corresponding to the Fe–N species, with the absence of Fe–O or Fe-Fe bonds, together revealing that a single Fe atom was coordinated with the N atoms on the support. After linearly fitting the Fourier transform curve of EXAFS, the number of N atoms coordinated with Fe atoms was calculated to be about 4 (Fig. 3f), indicating that the active sites of the Fe-SACs were atomically dispersed Fe–N4 species.14
Here, we stress that although the above-described modern characterization techniques have become powerful tools for studies at the atomic scale, the common spectroscopy tools can also offer key parameters of SACs in detail, e.g., mutual interactions between isolated metal and supports and atomically local structural information. This analysis data may allow common spectroscopy tools to complement or extend current advanced approaches for atomic catalysis research.62 In fact, as early as the 1970s, Yates et al.83 used infrared spectroscopy (IR) to confirm the presence of a single Rh atom in a catalyst. The underlying principle is monitoring the interaction between the adsorbed molecules (molecular probe) and the support of SACs.84 The variation in vibration frequency and intensity of the probe could be utilized to extract the characteristics of the active centers by calibration. Likewise, nuclear magnetic resonance (NMR) spectroscopy also contributes to clarifying the single-atom species and binding ligands in SACs, e.g., monoatomic Pt catalysts with a very low loading.84 However, thus far, NMR spectroscopy has not been applied for the characterization of Fe-SACs.85
4. Applications of Fe-SACs
Due to the excellent catalytic properties of Fe-SACs, their application fields are impressively wide, especially in energy storage and biocatalysis. In the case of energy storage, Fe-SACs play key roles in electrocatalytic, thermocatalytic and photocatalytic processes. In the biocatalysis field, Fe-SACs show excellent multi-enzymatic activities, making them suitable therapeutics in biosensing, wound disinfection, oxidative stress cell protection and cancer treatment.86–894.1. Fe-SACs for energy storage
To satisfy the large energy demand, the development of highly efficient catalysts is particularly critical for sustainable energy technologies.90 From a practical point of view, endowing catalysts with the capability to reduce the relatively large energy barriers of certain reactions has not yet been fully achieved owing to the inadequate design strategies and techniques available for the preparation of highly active catalysts.5,91 Consequently, Fe-SACs have currently entered the development spotlight for energy storage. According to the type of energy supply, catalysis can be divided into electrocatalysis, photocatalysis and thermal catalysis.57 Therefore, below we introduce the application of Fe-SACs in the above-mentioned three fields.4.1.1.1. ORR process. Metal–air batteries and fuel cells are ideal clean energy conversion equipment owing to their advantages of high energy density, low cost and zero emission of pollution gas.52,55 However, despite this promise, because of the slow kinetics of the ORR, there are several challenges (e.g., low energy conversion efficiency, poor stability in corrosive electrolytes and unsatisfactory lifespan) in their progression for energy storage.101,102 The ORR process has multi-step reaction pathways, which can be roughly divided into three categories (Fig. 4). One is the 4e− reaction pathway (formulas (1)–(4)), as follows: O2 + 2H2O + 4e− → 4OH−. The second is the 2e− reaction pathway (formulas (5) and (6)), as follows: O2 + H2O + 2e− → HO2− + OH−. The third is a series of reactions, where the 2e− and the 4e− pathways coexist.
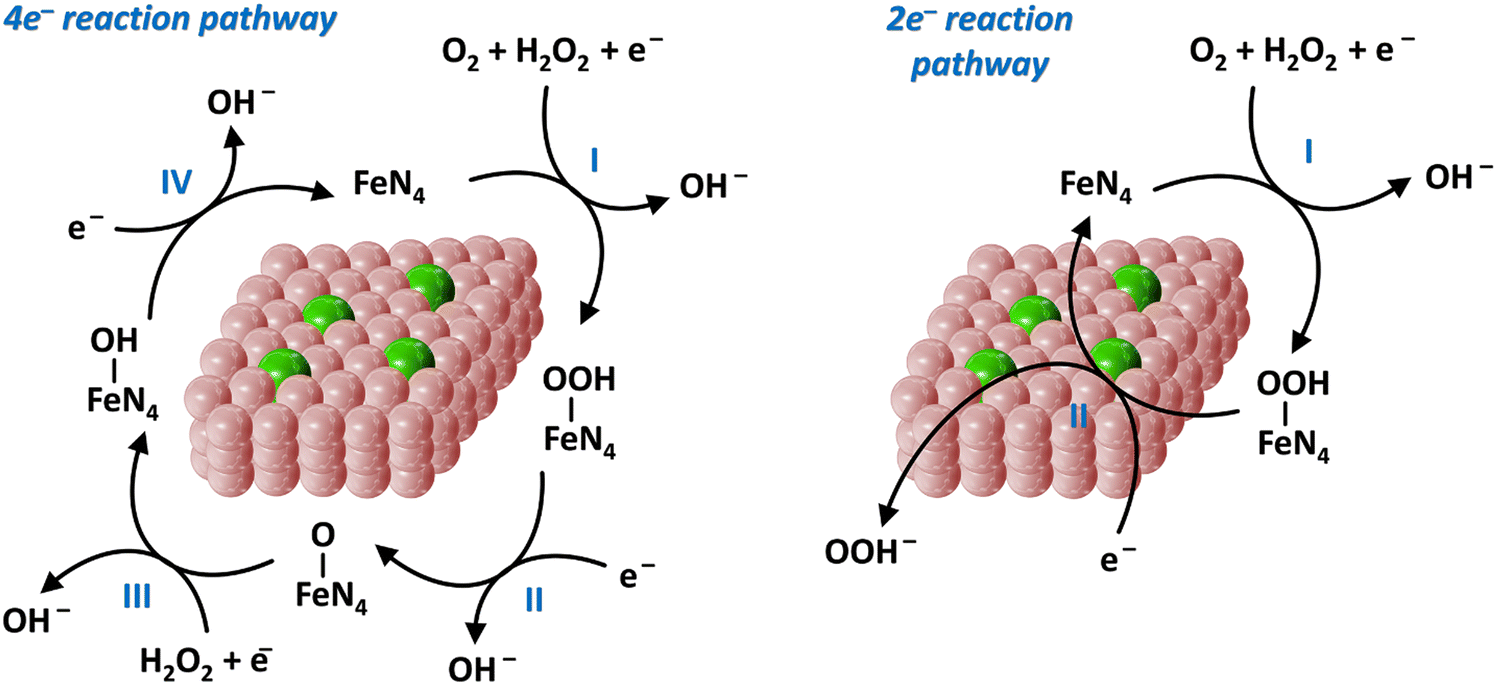 | ||
| Fig. 4 4e− reaction pathway and 2e− reaction pathway on single-atom iron catalysts. | ||
The 4e− reaction pathway is as follows:
| O2 + H2O + e− + * → OOH* + OH−. | (1) |
| OOH* + e− → O* + OH− | (2) |
| O* + H2O + e− → OH* + OH− | (3) |
| OH* + e− → * + OH− | (4) |
| O2 + H2O + e− + * → OOH* + OH−. | (5) |
| OOH* + e− → OOH− | (6) |
| Fe2+ + H2O2 → Fe3+ + OH− + ˙OH | (7) |
In the ORR process, many reaction intermediates (O*, OOH*, OH*, etc.) are produced and the adsorption of these intermediates on the surface of the electrode is vital to the ORR kinetics. Thus, an ideal ORR catalyst should show a suitable binding energy with the reaction intermediates.103 When Fe-SACs catalyze the ORR process following the 2e− reaction pathway, H2O2 will be generated, and then Fe reacts with the H2O2 to yield the strong oxidizing ˙OH (formula (7)). Notably, ˙OH results in the electrical oxidation of the carbon surface and the proliferation of micropores, which together oxidize the active sites to inactivate them.104 Consequently, the stability of the battery system will be deteriorated. Besides, because the energy transfer efficiency of the 2e− pathway is relatively low, it should be prevented as much as possible. As a comparison, the 4e− pathway is the optimal electron pathway to improve the ORR activity, which is required by the battery cathode.55
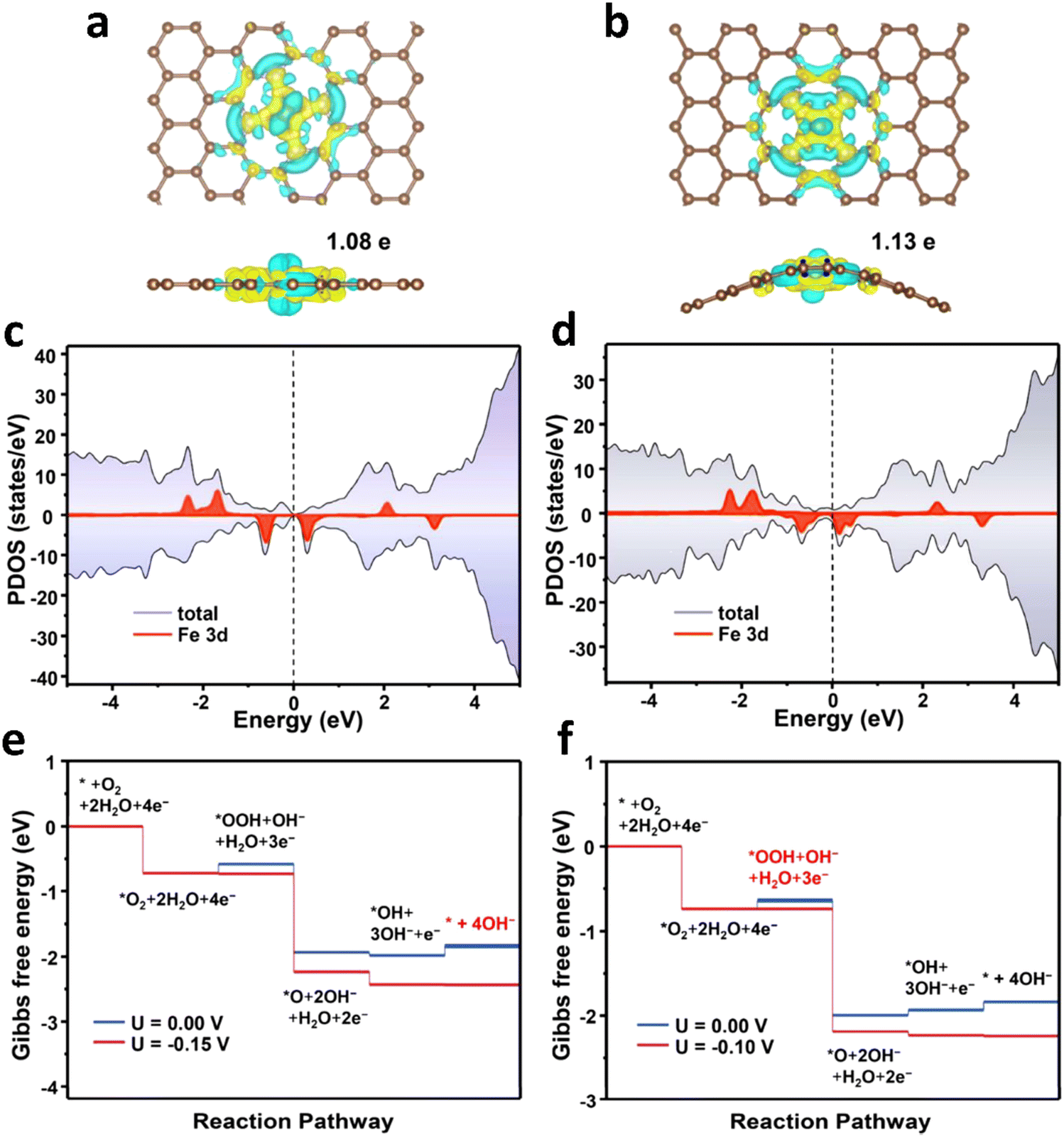
In fact, not all the Fe sites in Fe-SACs have a catalytic effect, such as the Fe atoms inside the support. Only the exposed active sites contribute to the catalytic properties. In this context, it is an efficient and direct way to improve the ORR activity by increasing the utilization of the metal atoms. Multiple lines of evidence reveal that the ORR process mainly occurs at the “catalyst–electrolyte–oxygen” three-phase interface. Guided by this discovery, recent studies attempted to increase the density of the Fe active sites exposed on the three-phase interface by adjusting the morphology of the catalysts.105–109 The reactants and products are transported in large quantities between the catalyst layers, and the utilization rate of the Fe active sites is improved. In a typical experiment, Hou et al.110 fabricated Fe-SACs with an overhang-eave structure, enabling more stretched edges, which can act as the three-phase interface of the ORR reaction. Also, the mass transfer of the ORR reaction substances between the catalyst layers was remarkably promoted, and the Fe active sites were exposed to the maximum extent, resulting in excellent ORR activity. In an alkaline solution, the detected onset potential and half-wave potential were 1.0 and 0.85 V, respectively, which are comparable to that of the commercial Pt/C catalysts (Fig. 5a–c). Designing and controlling the local structure of the metal sites are key to achieving efficient catalysis by single-atom metal catalyst sites. Generally, in the synthesized Fe-SACs, the plane of the active site Fe–N4 is straight. If the plane of the active site is adjusted, will it affect its catalytic activity? In a typical experiment, Wu et al.111 tuned the catalytic activity of Fe–N sites of surface single atoms through a stress-modulating method and observed enhanced ORR activities. In the synthetic process, the self-assembled structure of chiral surfactant molecules was used as a template, and then pyrolysis was performed to generate polypyrrole with a helical structure. Consequently, the active sites of Fe–N4 were rich in compressive stress. A large number of isolated non-planar curved Fe–N4 right-handed single Fe atoms was distributed on the helical hollow carbon fibers during the pyrolysis process. The electrochemical test results showed that the Fe-SACs had a higher onset potential, half-slope potential and kinetic current density compared with other types of catalysts in both acidic and basic conditions. DFT calculation was further utilized to determine the planar and curved Fe–N4 structures. The charge density difference diagrams demonstrated that the charges of the Fe atoms can transfer to the adjacent N atoms, irrespective of a flat or curved Fe–N4 structure (Fig. 6a and b). The Bader charge of Fe in the curved Fe–N4 structure was larger than that of the planar one, indicating that the curved structure facilitated the charge transfer. Moreover, compared with the planar Fe–N4 structure, the d-band center of the curved Fe–N4 structure shifted down. Consequently, the adsorption energy with oxygen-containing intermediates was reduced, thereby improving the ORR activity (Fig. 6c and d). The Gibbs free energy diagram results revealed that for the planar Fe–N4 structure, the potential determination step was *OH + 3OH− + e− → * + 4OH−, and the onset potential was determined to be about −0.15 V. The potential determination step of the curved Fe–N4 structure was *O2 + 2H2O + 4e− → *OOH + OH− + H2O + 3e−, and the onset potential was −0.10 V (Fig. 6e and f). The above-mentioned data indicated that the curved Fe–N4 structure was more active than the planar Fe–N4 structure.
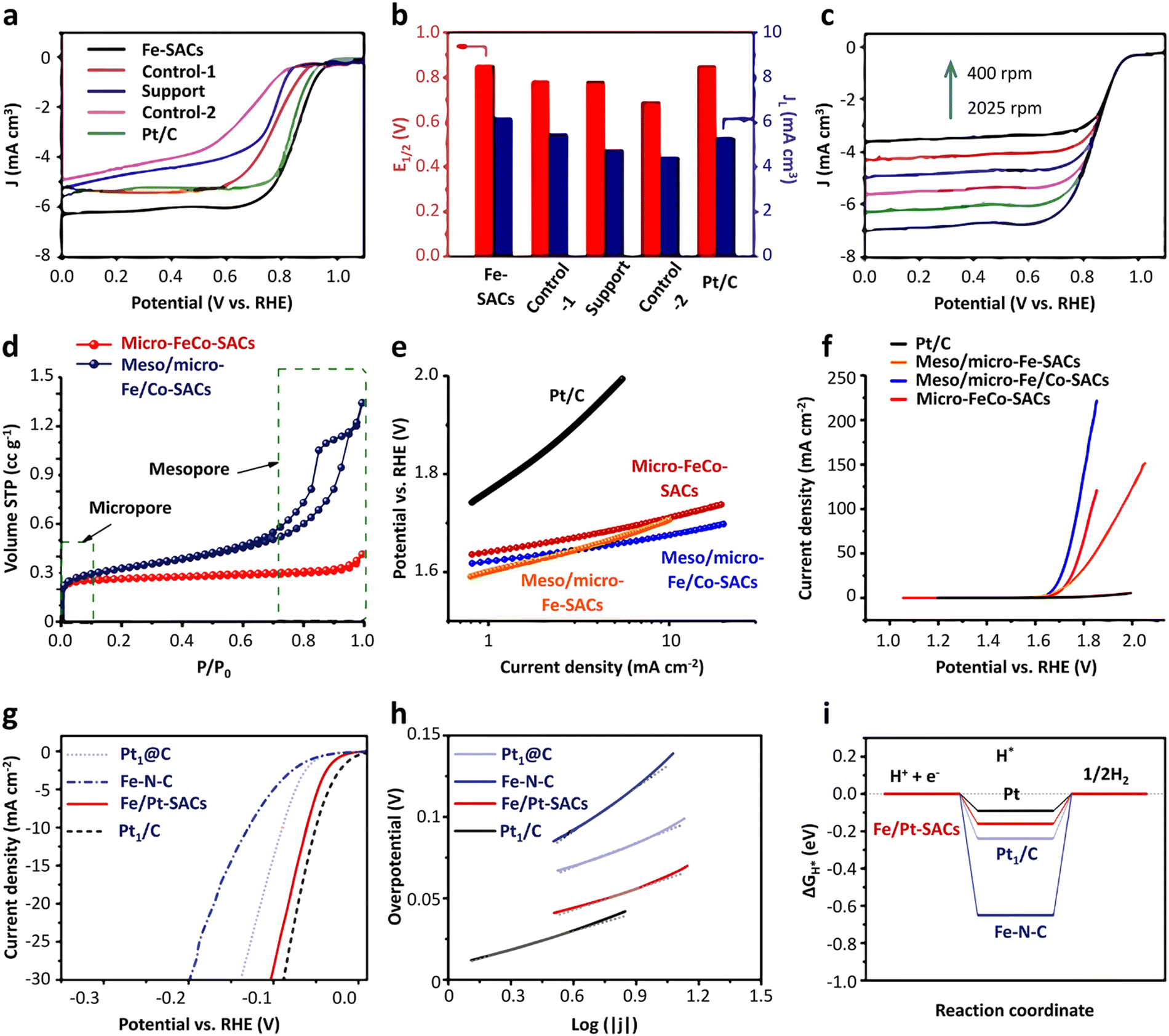 | ||
| Fig. 5 Applications of Fe-SACs in energy storage. (a) ORR polarization curves and (b) corresponding E1/2 and JL values of Fe-SACs. (c) LSV curves of Fe-SACs at different rotation rates. Reprinted with permission from ref. 110. Copyright 2020, John Wiley & Sons, Inc. (d) N2 adsorption/desorption isotherms of meso/micro-Fe/Co-SACs and micro-Fe/Co-SACs. (e) Tafel plot and (f) RDE polarization curves of meso/micro-Fe/Co-SACs and reference samples for the OER. Reprinted with permission from ref. 161. Copyright 2018, John Wiley & Sons, Inc. (g) HER polarization curve and (h) Tafel plot of catalyst Fe/Pt-SACs and reference samples in 0.5 M H2SO4. (i) DFT calculation of free energy ΔGH* of H* adsorption for the indicated catalysts in 1 M KOH. Reprinted with permission from ref. 104. Copyright 2018, John Wiley & Sons, Inc. | ||
 | ||
| Fig. 6 (a and b) Charge density difference diagrams (yellow: electron accumulation and cyan: electron depletion), (c and d) projected density of states, (e and f) ORR Gibbs free energy changes of planar Fe–N4 (k, m, and o) and curved Fe–N4 (l, n, and p). Reprinted with permission from ref. 111. Copyright 2021, John Wiley & Sons, Inc. | ||
The electrolyte pH also affects the ORR activities of Fe-SACs. In acidic media, the ORR activity of Fe-SACs will be seriously damaged because of the acid leaching, causing a large overpotential (40–400 mV) and extremely poor stability.112 Previous studies demonstrated that pyridine N, which plays an decisive contribution in regulating the ORR activity, will be coupled with H+ in acidic media.55 This complexation reduces the charge density of carbon atoms adjacent to the Fe–N4 active sites, largely inhibiting the ORR activity. Besides, a second and likely more fundamental reason for the slow ORR kinetics of Fe-SACs is that the binding energy between the Fe–N4 active sites and O2 or the reaction intermediates is not optimal. This guideline helps efforts to focus on identifying a way to optimize the adsorption energy between Fe and O2 and reduce the bond energy of the Fe–O bond. The difference in the coordination environment of atom-dispersed Fe active centers results in a variation in charge density around them, which will in principle affect the adsorption energy and the final ORR activity.36 Doping with oxygen atoms during the synthesis of Fe-SACs by forming specific local structures (e.g., carbonyl groups) can facilitate the production of OOH* and the reduction of OH* species. This synergistic effect drastically decreases the overpotential of Fe-SACs, and thus boost their ORR activity.113 N doping is the most widely used route in the design and synthesis of Fe-SACs. In the pyrolysis process, the formation of carbon defects and an increase in the polarity of the surface of carbon atoms adjacent to N will occur, thereby promoting the rapid transfer of electrolyte and electrons and increasing the ORR activity.114–116 N has a higher electronegativity (χ = 3.07) than that of C (χ = 2.55), and thus for N-doped carbon formed by pyrolysis, the N atoms exhibit strong ability to attract electrons.36,41 Density functional theory (DFT) indicated that the active site of Fe–N4 formed by co-doping of Fe and N is very similar to Pt, which can absorb oxygen, and subsequently break the OO bonds during the ORR reaction.117 Taking a closer look at Fe-SACs, N-doped carbon has three structures, namely pyrrole N, pyridine N (affording coordination sites for Fe atoms in the form of Fe–Nx) and graphite N (mainly affecting the geometry and electronic structure of carbon skeleton in Fe-SACs). Studies have shown that pyridine N and graphite N are advantageous species for ORR activities. Specifically, pyridine N can improve the onset potential of the ORR, while graphite N increases the kinetic current and the limiting diffusion current density of Fe-SACs, and thus an increase in both N contents in the catalyst is beneficial for the ORR activity.117–119
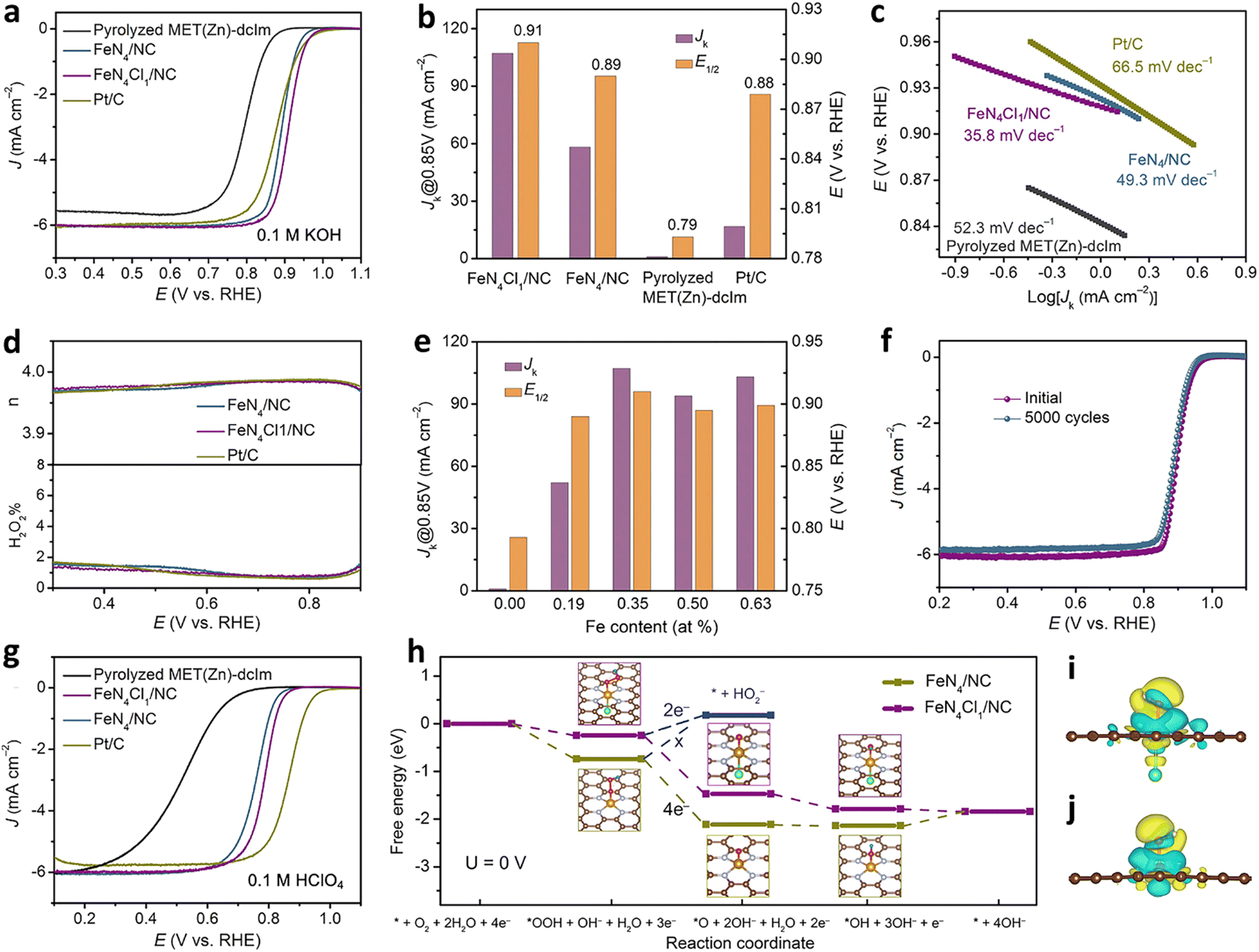
In addition to O and N doping, S, P, C, B and F doping is also possible.120 In a typical experiment, Hu et al. synthesized a single atom catalyst (FeN4Cl1/NC) with the FeN4Cl1 configuration with a metal triazole salt (MET) skeleton as the precursor and chlorine secondary ligand 4,5-dichloroimidazole (dcIm) as the raw material. The catalyst had a high iron content (2.78 wt%) and a relatively excellent void/volume ratio of 0.92. In FeN4Cl1/NC, because the Cl atom had a lower electronegativity, the d-band delocalization will be enhanced. Consequently, the electronic structure of the atomic iron changed, showing an optimal free energy of adsorption with the reaction intermediate *OH during the ORR process. Under alkaline conditions (0.1 M KOH), the measured half slope potential was 0.91 V, which was greater than that of the FeN4/NC catalyst (E1/2 = 0.89V) without Cl doping. Also, the Tafel slope (35.8 mV dec−1) was smaller than that of FeN4/NC (49.3 mV dec−1) and Pt/C (66.5 mV dec−1) (Fig. 7a–c). The results of the RRDE test revealed that the average electron transfer number of FeN4Cl1/NC in the ORR process was 3.97, and the yield of H2O2 was lower than 2%. The data suggested that the ORR process followed a four-electron transfer route (Fig. 7d). Importantly, the content of iron had a direct influence on the ORR performance. When the content of iron was 0.35%, FeN4Cl1/NC had the largest E1/2 and jk (Fig. 7e). Simultaneously, FeN4Cl1/NC exhibited longer stability. After 5000 consecutive potential cycles, E1/2 showed a slight negative shift (Fig. 7f). Under acidic conditions (0.1 M HClO4), its half slope potential was 0.79 V, which was greater than that of FeN4/NC (E1/2 = 0.76 V), and it had a lower Tafel slope (47.4 mV dec−1) (Fig. 7g). The DFT calculation results are shown in Fig. 7h. The two FeN4Cl1/NC and FeN4/NC catalysts followed the four-electron transfer path during the ORR process. For the FeN4/NC catalyst, the rate-determining step was the last step, and it can be seen that the FeN4 active site had a strong adsorption of *OH, which made the final desorption process extremely difficult, thereby inhibiting the ORR rate. For the FeN4Cl1/NC catalyst, all the electron transfer steps of the adsorption free energy ΔG were negative, indicating that due to the coordination of Cl atoms, *OH was easy to desorb from the FeN4 active site in the final process. The result of the electron density difference when the *OH was adsorbed on the active site showed that the axial electron transfer between the iron atom and the chlorine atom will affect the charge of the iron atom, thereby reducing the free energy of adsorption between the iron atom and *OH, thus contributing to the ORR process (Fig. 7i and j).121
 | ||
| Fig. 7 Electrocatalytic ORR performance of FeN4Cl1/NC. ORR polarization curves (a) and comparison of Jk at 0.85 V and E1/2 (b) and Tafel plots (c) of various catalysts. (d) Electron transfer number (n, top) and H2O2 yield (bottom) versus potential for various catalysts. (e) Comparison of Jk at 0.85 V and E1/2 of FeN4Cl1/NC samples with different Fe loadings. (f) ORR polarization curves of FeN4Cl1/NC before and after 5000 cycles. (g) LSV polarization curves for various catalysts. (h) Free energy diagram of the ORR on FeN4/NC and FeN4Cl1/NC (U = 0). Calculated electron density difference for OH* adsorbed on (i) FeN4Cl1/NC and (j) FeN4/NC. Reprinted with permission from ref. 121. Copyright 2021, John Wiley & Sons, Inc. | ||
The S atom has strong ability to attract electrons, with an electronegativity of ∼2.58. Thus, the co-doping of S and N dramatically alters the electron density around the Fe active sites of Fe-SACs. Consequently, the C atoms appended to the Fe active sites display more a positive charge, which is conducive to the adsorption of O2, and thus improve the ORR activity.52 Generally, the application scope of S doping is relatively narrow and it is preferentially doped at the edges and defects of graphene, whereas P atoms can be randomly doped in carbon-based materials.122 Recently, Yu et al.123 proposed a new conceptual strategy to reset the local coordination environment of Fe–Nx active sites by controlling the dynamic transfer of Fe–S bonds in Fe-SACs catalysts. The spectral and theoretical results showed that the selective cleavage of Fe–S bonds caused the incorporation of electron-withdrawing sulfur oxide on the Fe–Nx active site, thereby promoting the desorption of oxygen-containing intermediates from the active site. Cumulative evidence showed that the doping of P atoms increased the contents of pyridine N in Fe-SACs, adjusting the electronic structure of the Fe active site.124 Accordingly, the adsorption of OH* intermediates was weakened, increasing the ORR activity. The doping of B atoms not only can provide electron-deficient sites (improving the electron transfer at the Fe–Nx–C sites), but also enhances the interactions of oxygen-containing species, thereby enhancing the ORR activity.118,124 Doping with F atoms can increase the adsorption capacity for O2 and enhance the ORR activity due to the synergistic effect between nitrogen and fluorine.120
In summary, the coordination environment of Fe–Nx active sites is determined by the doping of heteroatoms and the number of N atoms coordinated with Fe atoms. It can be found that for the majority of Fe-SACs, the number of N atoms coordinated to the central Fe atom is 4, and other coordination numbers of 2, 3, and 5 were also reported.125–127 According to DFT analysis, the coordination states of the Fe active sites, especially the number of coordination atoms, will have a huge impact on the catalytic activity by affecting the binding energy between the O2/active intermediates (OH*, OOH*, O*, etc.) and the central Fe atom.128 In a typical experiment, Lai et al.125 synthesized an Fe–N/C electrocatalyst with five-coordinated Fe–Nx active sites through the host–guest chemistry strategy. According to DFT calculations, the five-coordinated Fe–Nx active site can increase the ORR reaction rate in acidic medium by reducing the reaction barrier and the adsorption energy of the OH* reaction intermediate. Importantly, reports validated that the introduction of other transition metal elements M (Co, Mn, Cu, Zn, Ni, etc.) in Fe-SACs to generate a bimetallic site catalyst with N3Fe–MN3 structures is beneficial for strengthening the binding capability with O2.129 To a certain extent, the bimetallic SACs will increase the bond length of the OO double bonds, and accordingly weaken the energy of these double bonds, thus facilitating their cleavage. Similarly, single-atom catalysts with isolated diatomic metal sites can also enhance their catalytic effects by adjusting the electronic states of adjacent metal atoms. For example, Han et al. synthesized atomically dispersed platinum with iron anchored on a nitrogen-doped carbon support (Fe–N4/Pt–N4@NC), where the Pt–N4 molecule was adjacent to the Fe–N4 active site. The DFT calculation and electrochemical analysis results showed that Pt–N4 molecules can activate the O2 adsorbed on the Fe–N4 active sites, and also re-hybridize the 3d orbitals of Fe and 2p orbitals of O, thereby optimizing the adsorption of oxygen intermediates and accelerating the entire ORR dynamics process. Simultaneously, as for comparison, Co–N4/Pt–N4@NC and Mn–N4/Pt–N4@NC catalysts were also prepared. The results showed that the existence of Pt–N4 molecules had no effect on the ORR process of Co–N4/Pt–N4@NC, but inhibited the ORR process of the Mn–N4/Pt–N4@NC catalyst. The main reason for this was due to the existence of Pt–N4 molecules. There was a large free energy of adsorption between the active site of Mn–N4 and *OH, which made it difficult to desorb from the active site and reduced the ORR catalytic performance.130 These characteristics enabled the bimetallic SACs to follow the 4e− pathways during the ORR reaction, representing an enhanced catalytic performance (Table 1).112,131,132
| Support | Active site | E 1/2 (V) | E onset (V) | Ref. |
|---|---|---|---|---|
| The symbol “—” indicates no results were reported in the study. | ||||
| N-Doped carbon | Fe–Nx | 0.84 | 0.96 | 119 |
| N,S co-doped carbon | Fe–S4N2 | 0.90 | — | 15 |
| N,S co-doped carbon | Fe–Nx | 0.97 | — | 336 |
| N,P co-doped carbon | Fe–N4 | 0.86 | — | 122 |
| N,S,P co-doped carbon | Fe–N4 | 0.91 | — | 124 |
| N,P co-doped carbon | Fe–N3P | 0.87 | 0.94 | 337 |
| N,Cl co-doped carbon | Fe–Cl1N4 | 0.92 | — | 338 |
| N,B co-doped carbon | Fe–Nx | 0.84 | 0.97 | 118 |
| N,F co-doped carbon | Fe–Nx | 0.82 | 0.90 | 120 |
| N-Doped carbon | OFeN4–O–FeN4O | 0.83 | 0.93 | 113 |
| N-Doped carbon | Fe–N4 | 0.85 | — | 117 |
| N-Doped carbon | Fe–N5 | 0.74 | 0.86 | 125 |
| Multiwalled CNTs | Fe–N5 | 0.92 | — | 126 |
| N,F co-doped porous carbon | Fe–N5 | 0.88 | 0.99 | 127 |
| N-Doped carbon | Fe–N2 | 0.86 | 1.05 | 339 |
| N doped carbon | Fe–N2 | 0.93 | 0.99 | 340 |
| N,S co-doped carbon | Fe–N3S | 0.84 | 0.96 | 341 |
| N-Doped porous carbon | N3Fe–CoN3 | 0.86 | 1.06 | 112 |
| N-Doped carbon | Co–N4, Fe–N4 | 0.86 | 0.96 | 114 |
| N-Doped carbon | N3Fe–CoN3 | 0.95 | 0.95 | 132 |
| N-Doped carbon | Fe–N4 | 0.89 | 1.01 | 342 |
| N-Doped carbon | Fe–N4, Mn–N4 | 0.90 | — | 343 |
| N,S co-doped carbon | Fe–N4, Mn–N2S2 | 0.90 | — | 344 |
| N-Doped carbon | Fe–N4, Co–N4 | — | 0.93 | 345 |
| N-Doped carbon | Fe–N4 | 0.86 | — | 346 |
| N-Doped carbon | Fe–N4 | 0.91 | — | 14 |
| N-Doped carbon | Fe–N4 | 0.87 | 1.01 | 52 |
| N-Doped carbon | Fe–N4 | 0.91 | 1.05 | 347 |
| N-Doped carbon | Fe–N4 | 0.88 | — | 65 |
| N-Doped carbon | Fe–Nx | 0.82 | — | 54 |
| N-Doped carbon | Fe–Nx | 0.87 | 1.05 | 69 |
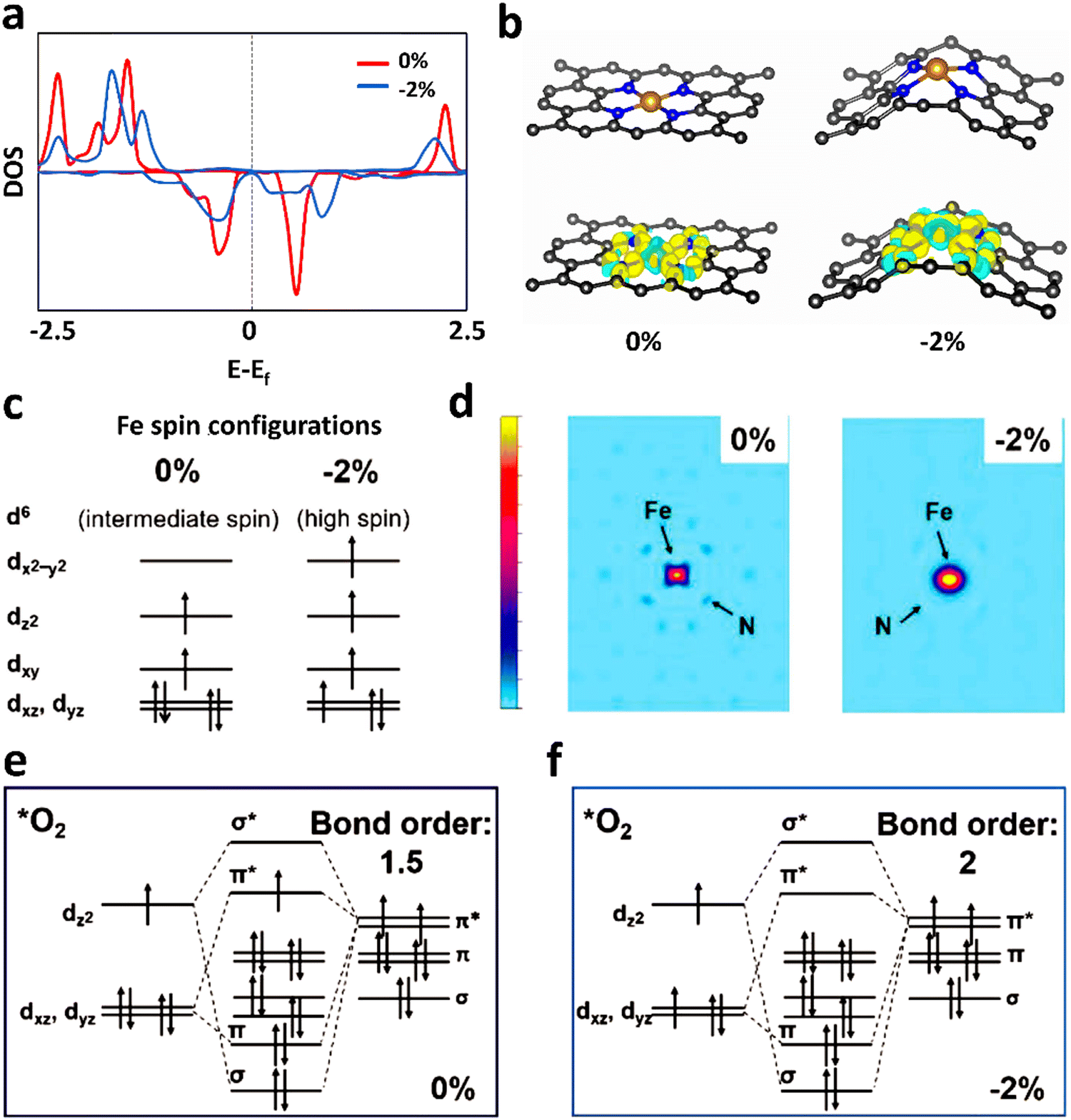
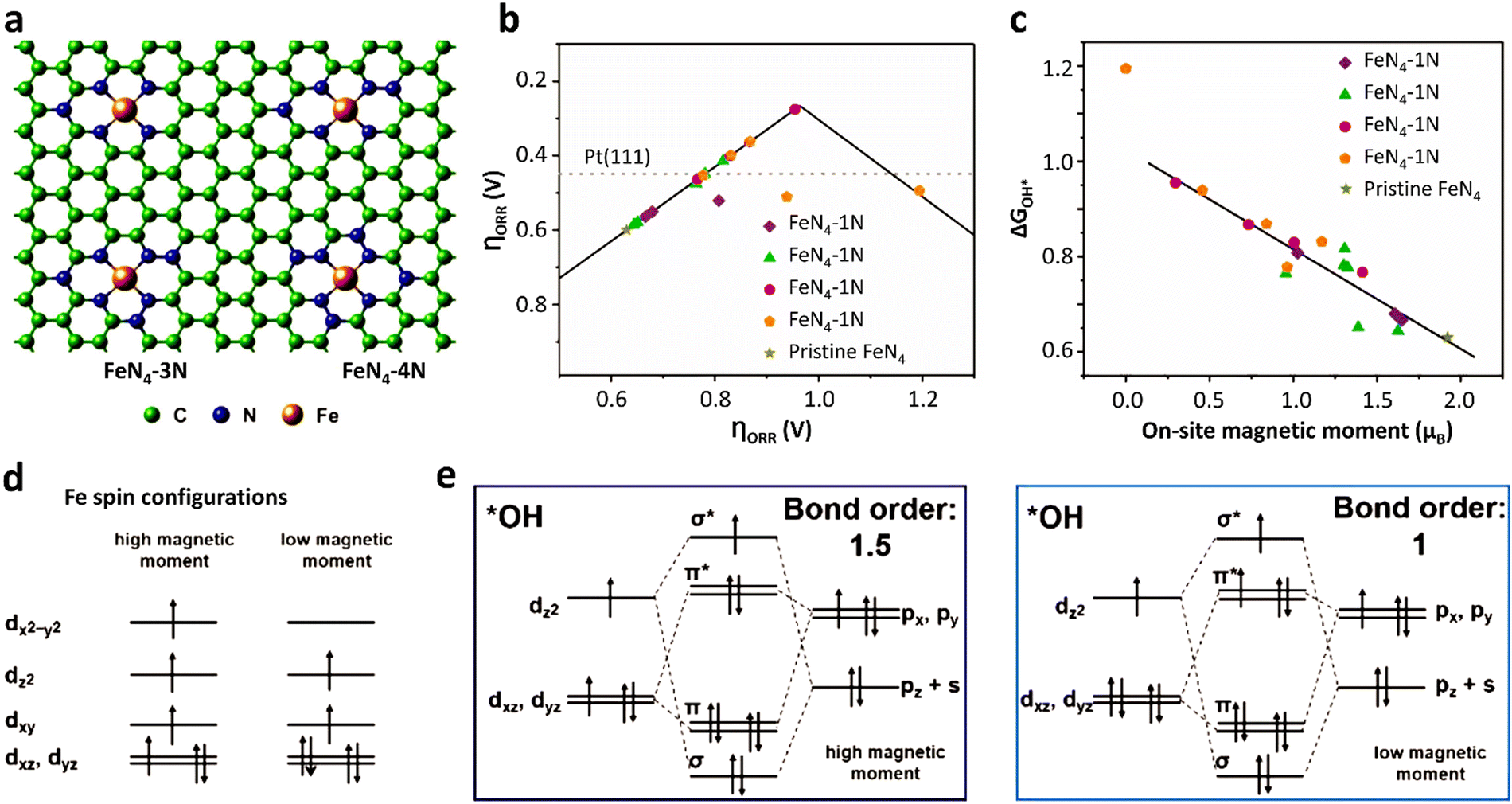
Due to the paramagnetic properties of ground-state oxygen in oxygen-catalyzed reactions, its production, electron transport during reduction reactions, and orbital interactions between intermediates and catalysts are closely related to the spin state of the catalyst materials, and thus the kinetics and thermodynamics of the reaction are also very sensitive to the spin configuration. In a typical experiment, Wu et al.133 found that applying a 2% shrinkage stress to FeN4 could move the 3d orbital of Fe in the center of the Fe–N bond shrinkage, promote the adsorption capacity of FeN4 for O2 molecules, and then improve the ORR reaction (Fig. 8a and b). Using the local spin configuration to analyze the ORR performance on FeN4, it can be seen from the DOS diagram that after applying stress, the self-selected density increased and the Fe–N bond contracted. Besides, the iron ion changed from an intermediate spin state to a high spin state (Fig. 8c), resulting in a wider spin channel (Fig. 8d) and a larger adsorption bond order for O2 (Fig. 8e). All these factors contributed to both the kinetic and thermodynamic aspects of the ORR.134 Kang et al.135 found that the magnetic moment of FeN4 and N-rich FeN4 materials had a linear relationship with the ORR reaction descriptor (ΔG*OH), and the magnetic moment of 0.25 μB corresponded to the optimal ΔG*OH, while the magnetic moment value of the original FeN4 site was close to 2 μB, corresponding to a too low value of ΔG*OH. Consequently, its ORR performance was relatively inferior (Fig. 9a–c). From the perspective of spintronics, the reduction of the magnetic moment revealed that the spin state of the iron site changed from a high-spin state to a low-spin state. After analyzing the spin–orbit interaction between the iron site and *OH, it was found that the FeN4 material with a smaller magnetic moment will show a smaller bond level for the adsorption of *OH, and thus the adsorption energy of *OH will also be weaker. The value of ΔG*OH will also increase, which corresponds to a better ORR performance (Fig. 9d and e).134
 | ||
| Fig. 8 Spin-related charge transfer and orbital interactions of the ORR on FeN4 with and without 2% Fe–N bond contraction. (a) Density of states (DOS) of the 3d orbitals of the central Fe cation in the two FeN4 sites. 0% and −2% denote pure FeN4 site and FeN4 site with 2% Fe–N bond contraction, respectively. (b) Adsorption configuration of O2 adsorption on two FeN4 sites. (c) Possible Fe spin configurations in FeN4. (d) Spin channel of Fe site in FeN4. (e) Orbital interactions between Fe sites and the triplet O2 molecule. (a and b) Reprinted with permission from ref. 132. Copyright 2019, John Wiley & Sons, Inc. (c–e) Reprinted with permission from ref. 133. Copyright 2020, John Wiley & Sons, Inc. | ||
 | ||
| Fig. 9 Spin-related orbital interactions on pristine FeN4 and spin-engineered FeN4. (a) Representative configurations of FeN4 with different amounts of surrounding graphitic N atoms. (b) Overpotential (ηORR) as a function of *OH adsorption free energy (ΔG*OH). (c) Linear relationship between ΔG*OH and on-site magnetic moments (namely, the spin) of the Fe center in FeN4 sites. (d) Possible spin state of the Fe center with high and low magnetic moment. (e) Orbital interactions between Fe center and *OH intermediate. (a–c) Reprinted with permission from ref. 135. Copyright 2019, John Wiley & Sons, Inc. (d and e) Reprinted with permission from ref. 134. Copyright 2020, John Wiley & Sons, Inc. | ||
Transition metals (M = Mn, Fe, Co, Ni, etc.) possess 3d unoccupied orbitals, which can accommodate foreign electrons and reduce the bonding strength between OOH* and O*/OH* intermediates.136 In the ORR, the activity of the catalyst is mainly affected by its electronic structure. The catalyst boosted the formation of M(m+1)+–O22− bonds on the its surface, ensuring the rapid replacement of O2−/OH− and the regeneration of OH−.137 Based on the coordination environment, Fe(III) possessed multiple spin states, including low-spin t52g e0g, medium-spin t42g e1g, and high-spin t32g e2g.138 The low-spin electron configuration was d2xy d2yz d1xz. No electrons occupied the antibonding orbitals, which resulted in a strong Mm+/O2 interaction and stable M(m+1)+–O22− bond, making it difficult to realize the M(m+1)+–O22−/Mm+–OOH transition.139 The electron configuration of high spin was d1xy d1yz d1xz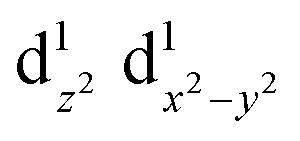 , and the high eg filling
, and the high eg filling 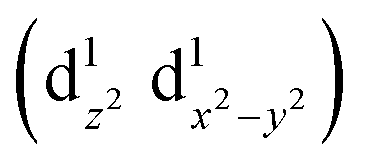 caused poor adsorption capacity and poor performance.140 The mid-spin electron configuration was dxy2 d1yz d1xz
caused poor adsorption capacity and poor performance.140 The mid-spin electron configuration was dxy2 d1yz d1xz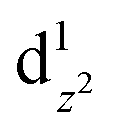 and the single dz2 electron in the mid-spin state could easily penetrate the anti-bonding π orbital of oxygen, showing high ORR activity.141 Therefore, by improving the surrounding chemical environment of transition metals and modulating their electron spin states, the electronic structure of the catalytic site can be precisely regulated, and high-performance ORR catalysts with tunable activity are expected to be obtained. For example, Zhang et al.142 prepared Fe, Mn/N–C electrocatalysts with dispersed Fe, Mn bimetallic atoms for the ORR process. Magnetic measurements showed that the introduction of Mn–N structures led to the delocalization of FeIII electrons and the spin state transition of FeIII from low spin (t52g e0g) to medium spin (t42g e1g), which can facilitate the penetration of the antibonding π orbital of oxygen. DFT calculation revealed that the Mn–N groups dispersed by adjacent atoms can effectively activate the Fe(III) site through the spin state transition and electronic regulation. Consequently, the Fe, Mn/N–C catalyst could moderately interact with oxygen with an optimized bond length and adsorption energy, which promoted the kinetic process of the ORR reaction (Fig. 10).
and the single dz2 electron in the mid-spin state could easily penetrate the anti-bonding π orbital of oxygen, showing high ORR activity.141 Therefore, by improving the surrounding chemical environment of transition metals and modulating their electron spin states, the electronic structure of the catalytic site can be precisely regulated, and high-performance ORR catalysts with tunable activity are expected to be obtained. For example, Zhang et al.142 prepared Fe, Mn/N–C electrocatalysts with dispersed Fe, Mn bimetallic atoms for the ORR process. Magnetic measurements showed that the introduction of Mn–N structures led to the delocalization of FeIII electrons and the spin state transition of FeIII from low spin (t52g e0g) to medium spin (t42g e1g), which can facilitate the penetration of the antibonding π orbital of oxygen. DFT calculation revealed that the Mn–N groups dispersed by adjacent atoms can effectively activate the Fe(III) site through the spin state transition and electronic regulation. Consequently, the Fe, Mn/N–C catalyst could moderately interact with oxygen with an optimized bond length and adsorption energy, which promoted the kinetic process of the ORR reaction (Fig. 10).
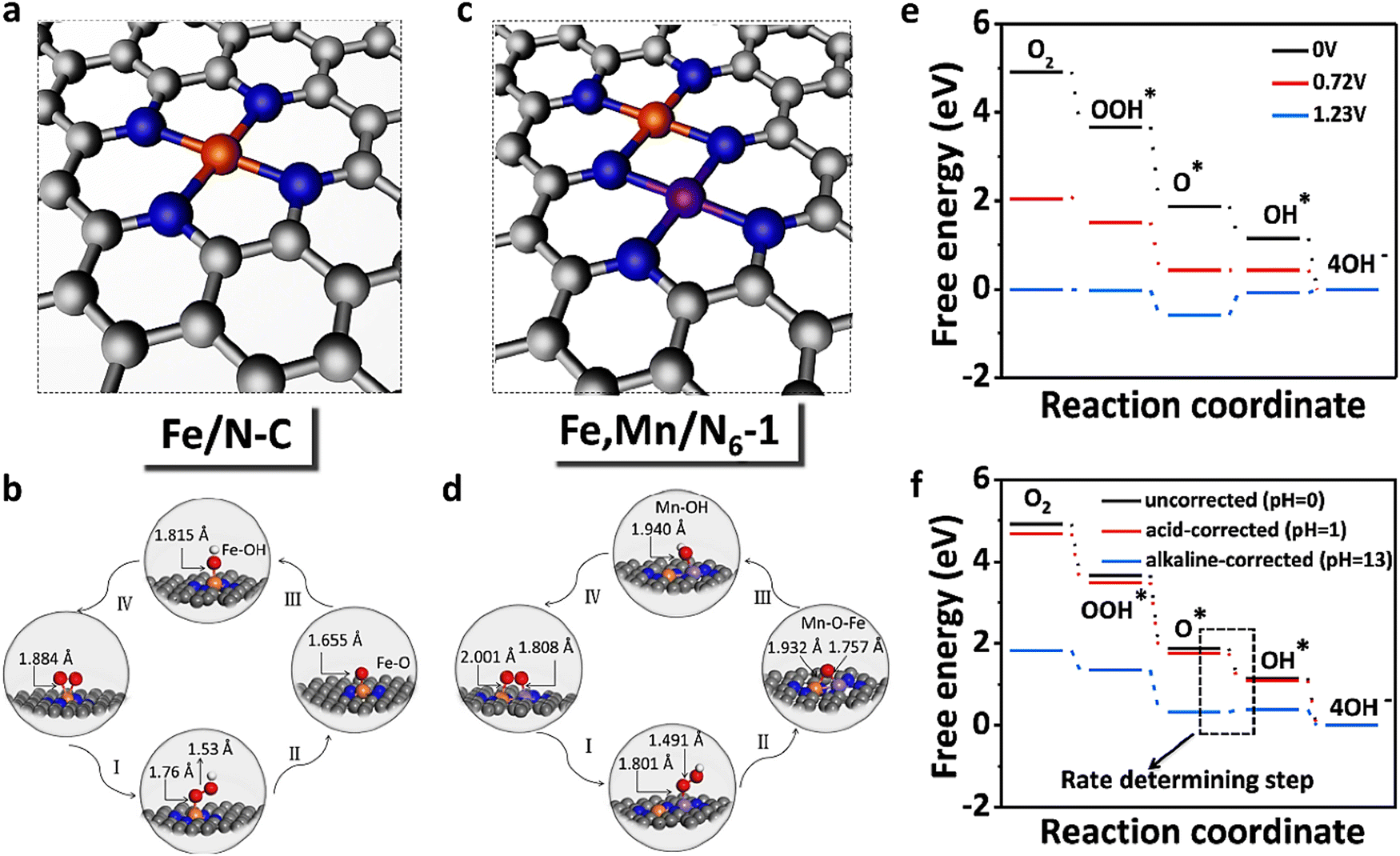 | ||
| Fig. 10 Regulating Fe-spin state by atomically dispersed Mn–N in Fe-SACs with high oxygen reduction activity. Optimized structure of (a) Fe/N–C and (c) Fe, Mn/N–C. Optimized atomic structures for the main process of the ORR: (b) Fe/N–C and (d) Fe, Mn/N–C. (e) Pathways for Fe, Mn/N–C are summarized at U = 0 V, 0.72 V, and 1.23 V, respectively. (f) pH-corrected free energy diagram of Fe, Mn/N6−1. Reprinted with permission from ref. 142. Copyright 2021, Springer Nature Limited. | ||
Fe-SACs have been widely used for the ORR due to their abundant atomically dispersed active sites (Fe–N4).143,144 However, although they exhibit high ORR activity, they are prone to demetallization, surface carbon oxidation and carbon corrosion caused by Fenton reactions under acidic conditions, which reduce their stability.145–148 Therefore, developing ORR catalysts with high activity and stability in acidic media remains a significant challenge.
4.1.1.2. OER process. In contrast to the ORR process, the OER process is its reverse reaction.149 It mainly occurs in water splitting or the rechargeable process of metal–air batteries. Given that the OER process involves 4e− transfer steps, its kinetics are sluggish, where a large overpotential is generally required, resulting in a low energy utilization rate.150
For the OER process, which electrochemical pathway will occur depends on the medium conditions. In a neutral and alkaline media, the overall reaction equation is as follows: 4OH− → O2 + 2H2O + 4e− (formulas (8)–(11)). In acidic medium, the reaction equation is 2H2O → O2 + 4H+ + 4e− (formulas (12)–(15)).151 The following steps involve multiple proton-coupled electron transfers. In general, the OER process requires an overpotential of 1.23 V.17
In a neutral or alkaline medium (Fig. 11):
| OH− + * → OH* + e−. | (8) |
| OH* + OH− → O* + H2O + e− | (9) |
| O* + OH− → OOH* + e− | (10) |
| OOH* + OH− → * + O2 + H2O + e− | (11) |
| H2O + * → OH* + H+ + e−. | (12) |
| OH* → O* + H+ + e− | (13) |
| O* + H2O → OOH* + H+ + e− | (14) |
| OOH* → * + O2 + H+ + e− | (15) |
 | ||
| Fig. 11 4e− reaction pathways of the OER processes on single-atom iron catalysts in neutral or alkaline medium. | ||
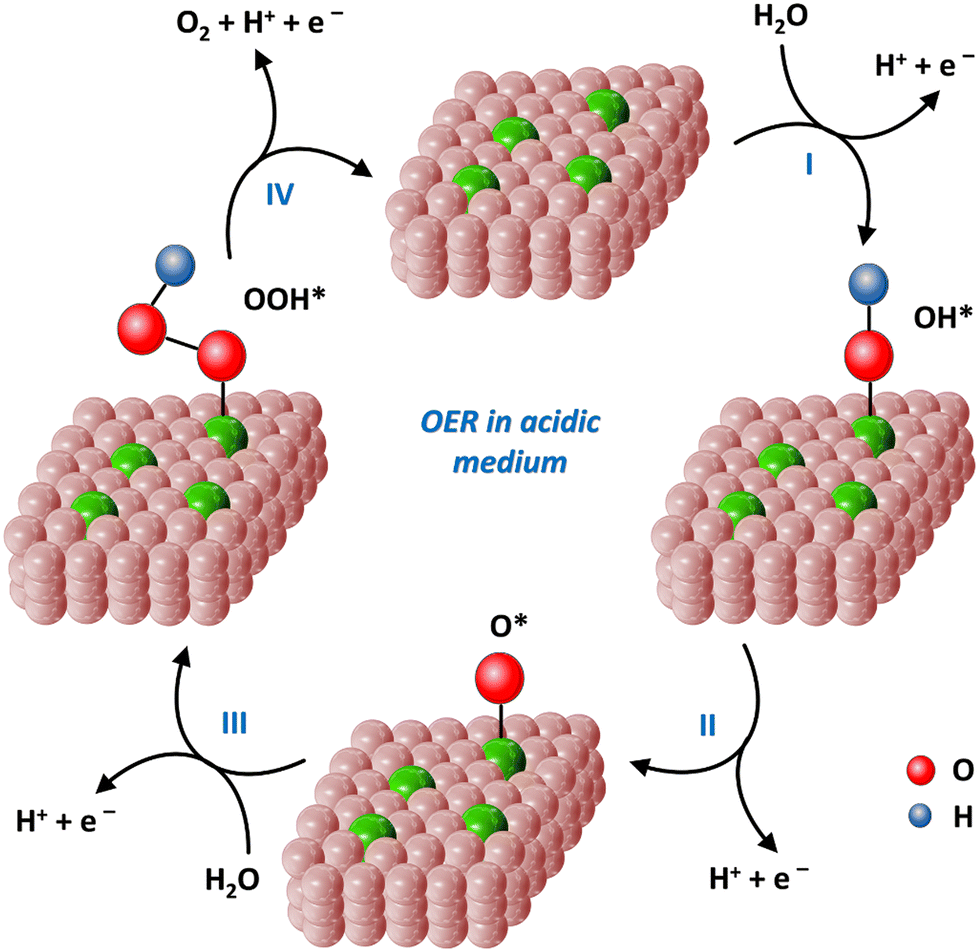 | ||
| Fig. 12 4e− reaction pathways of the OER processes on the single-atom iron catalysts in acidic medium. | ||
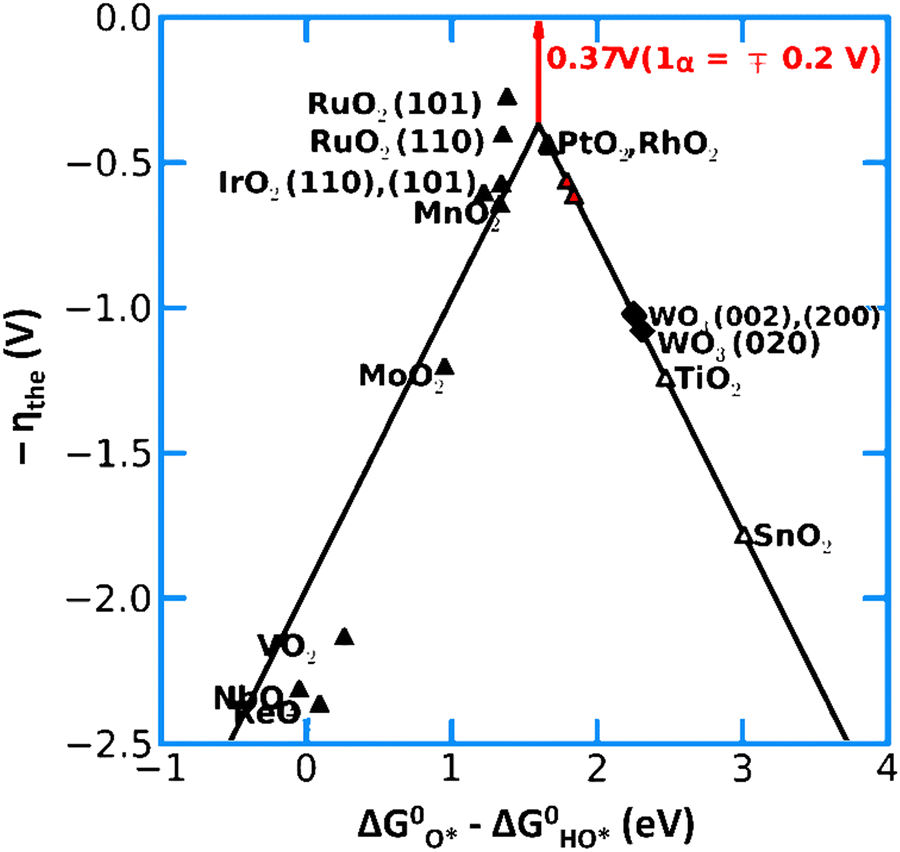 | ||
| Fig. 13 Activity trends for the OER on various metal oxide catalysts. The negative value of the theoretical overpotential is plotted against the descriptor for the OER (the standard free energy of HO* oxidation). Reprinted with permission from ref. 152. Copyright 2012, The Royal Society of Chemistry. | ||
Under alkaline conditions, there are generally two variables for the OER test of the catalyst. The first is the overpotential. The smaller the overpotential of a catalyst is, the higher its OER performance. The other is the Tafel slope. When studying the OER dynamics, the Tafel plots is obtained from the corresponding polarization curve at low overpotential, and then the Tafel equation can be obtained by linear fitting.61 Studies have shown that the RDS can be determined based on the Tafel slope. If the Tafel slopes of two catalysts are approximately the same, they have a similar RDS in the OER process. When the Tafel slope is less than about 80 mV dec−1, this indicates that in the specific path of the OER, the step after the first electron transfer is the RDS, which is OH* + OH− → O* + H2O + e−. The OH surface species are rearranged by surface reaction. When the Tafel slope is greater than 100 mV dec−1, the RDS is the formation of the OH* intermediate in the first electron transfer step.61,155,156
Research results showed that when Fe, Co, and Ni are used as catalysts, the OER catalytic effect follows the order of Ni > Co > Fe. According to DFT calculation, it was found that N doping will cause the redistribution of the charge of the carbon material.157 In the N-doped carbon catalyst, the C atom close to the N atom became the preferential binding site of the oxygen intermediate in the OER process, rather than the doped N atom itself. Therefore, for SACs, the M–Nx metal center and the C atom close to the N atom may act as active sites. Oxygen intermediates such as OH*, O*, OOH* and O2* will be generated in the OER process. Different intermediates may have different adsorption positions in the catalyst. Some of them are adsorbed on the M–Nx metal active center, while others will be adsorbed on the C atoms close to the N atom. This adsorption ability is mainly related to the difference in the adsorption energy between the oxygen intermediate and the two positions. In this context, whether the C atom will become the adsorption site of the oxygen intermediates in the OER process largely depends on the number of d electrons of the metal. For the Fe and Co transition metals, their 3d electron numbers are 6 and 7, respectively. Therefore, all the oxygen intermediates are more strongly bound to the M–Nx active site in the metal center than the C site. In the case of SACs synthesized from Fe and Co metals, the two intermediates (O* and OH*) preferentially combine with the C atom close to the N atom, and the OOH* intermediate preferentially adsorbs on the metal centers (Fig. 14a and b). The limit barrier of the OER reaction is a critical parameter to evaluate catalytic activity (Fig. 14c). According to the DFT calculation results, for Fe-SACs, the RDS is O* + OH− → OOH* + e−, with a limit barrier of 0.97 eV. The large limit barrier indicates that the chemical adsorption between the oxygen intermediate and the active site is strong, thereby increasing the activation barrier during the reaction. When the current density was 10 mA cm−2, the overpotentials of the Fe, Co, and Ni catalysts were 488 mV, 402 mV, and 331 mV, respectively (Fig. 14d). The Tafel slope can be using the Tafel plot. Because the overpotential and Tafel slope of metal catalysts both show the trend of Fe > Co > Ni, the OER activity of the single-atom catalysts follow the trend of Ni > Co > Fe (Fig. 14e). Fe will yield a semiconducting Fe–OOH* species with the O2 intermediate of OOH* in the OER reaction, hence reducing the electron transfer rate. Moreover, the bonding strength between the Fe active sites and OOH* causes the deactivation of these active sites, which directly inhibit the OER activity.158 Therefore, to improve the catalytic performance of Fe-SACs, the most effective method is the doping of Ni or Co atoms. It is widely acknowledged that the doping of Co atoms is beneficial to improve the conductivity of catalysts and can also activate the inactive metal atoms. Thus, the OER activity is increases obviously.149,159–161 Typically, Thomas’ group synthesized hierarchically mesoporous/microporous carbon nanosheets with highly dispersed Fe/Co bimetallic centers by heteroatom doping (Fe/Co–N–C).161 This synthesis method employed FeCl3·6H2O as the active salt and SiO2 as a template to adjust the porosity and thickness of the carbon nanosheets. A large number of mesoporous/microporous structures was generated with a specific surface area of 1321 m2 g−1 (Fig. 5d), which was beneficial for the diffusion of electrolyte and gas during the ORR/OER process, thereby enhancing the catalytic activity. The authors measured the ORR and OER activity of the catalyst meso/micro-Fe/Co–N–C by rotating disk electrode technology in alkaline media. It had a high onset potential (Eonset = 0.954 V) and half-wave potential (E1/2 = 0.886 V). Moreover, due to the rich mesoporous/microporous structure, this catalyst showed a high limit current density, reaching 6.3 mA cm−2, which was higher than that of the commercial Pt/C catalyst (5.6 mA cm−2). It can be concluded from the OER polarization curve that meso/micro-Fe/Co–N–C had a lower potential of 1.67 V when the current density was 10 mA cm−2. According to the Tafel plot corresponding to the OER diagram, the catalyst was demonstrated to show a lower voltage gap value (Egap = 0.78 V) at a current density of 10 mA cm−2, indicating the good ORR/OER bifunctional activity of meso/micro-Fe/Co–N–C (Fig. 5e and f). As is known, if a catalyst has high OER activity, it will have a moderate affinity with the OH intermediate and can improve the adsorption of hydroxide and the equilibrium adsorption rate of the catalyst surface. Consequently, the last step of the OER path generation of oxygen becomes a rate-determining step, which ultimately reduces the Tafel slope. Obviously, in addition to the introduction of Co atoms in Fe-SACs, Ni atoms can also be co-doped, which is conducive to the formation, association and dissociation of OOH*.162
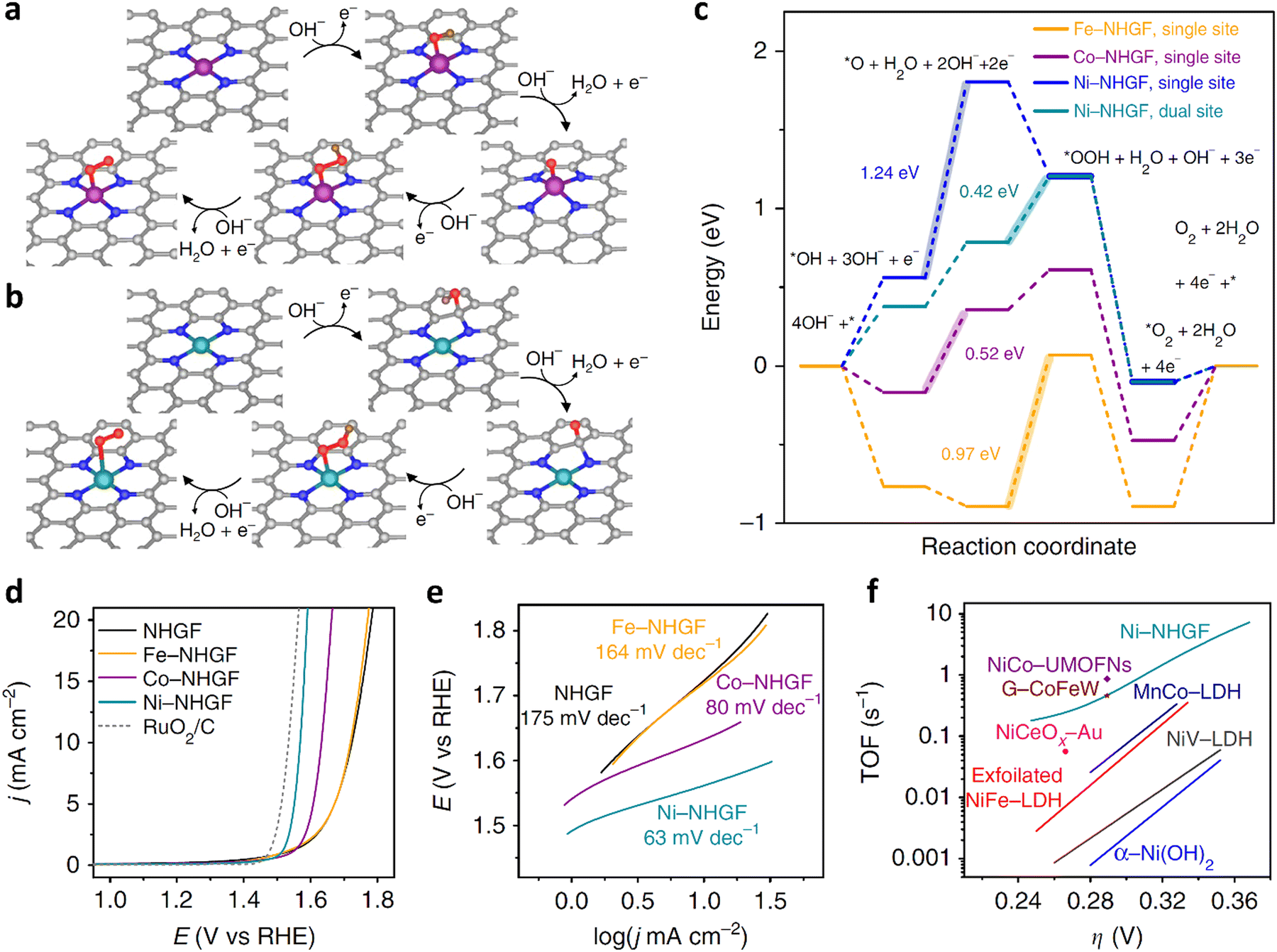 | ||
| Fig. 14 (a) Possible reaction schemes for M–N4 active site during the OER process. (b) Possible reaction scheme of the dual-center mechanism for the catalyst with MN4 active site in the OER process. (c) Free energy diagrams of Fe-NHGF, Co-NHGF and Ni-NHGF and other single-atom catalysts in the OER process. (d) Evaluation diagram of OER activity of NHGF, Fe-NHGF, Co-NHGF and Ni-NHGF in 1 M KOH electrolyte. (e) Tafel plots of the corresponding catalysts shown in d. (f) TOF value of Ni-NHGF catalyst and other metal catalysts. Reprinted with permission from ref. 157. Copyright 2018, Springer Nature Limited. | ||
In metal–air batteries or fuel cells, dual-functional electrocatalysts are usually required to drive multiple electrochemical reactions simultaneously, but a single atom active center with only a single metal component is difficult to satisfy high selectivity and activity for different electrocatalytic reactions. As can be seen from the above discussion, among the SACs of Fe, Co, and Ni, the Fe-SACs (Fe > Co > Ni) are the most catalytically effective for the ORR process, and the Ni-SACs (Ni > Fe > Co) are the best for the OER process. Therefore, it is a direct way to develop dual-functional electrocatalysts by integrating two single-atom catalysts on a support. In principle, the catalysts of the respective atoms correspond to their respective catalytic processes, making them not only have excellent ORR catalytic effect, but also have excellent OER catalytic effect. Recently, Chen et al.163 embedded Ni atoms and Fe atoms in the inner and outer walls of graphene hollow nanospheres through a step-by-step self-assembly strategy. Given that the two active centers of Ni–N4 and Fe–N4 were distributed on different sides of the graphene hollow nanospheres in the catalyst, the obtained SACs could balance the competition between the ORR and OER rate-limiting steps by reducing the mutual interference of ORR and OER active centers, showing higher OER and ORR catalytic activities. According to electrochemical tests and density functional calculations, the outer Fe–N4 active center had a higher catalytic performance for the ORR process, while the inner Ni–N4 active center could boost the OER reactions. During the OER process, the RDS was the first discharge step, corresponding to the formation of the OH* intermediate (Fig. 15a). Moreover, the Egap was measured to be about 0.79 V, suggesting the excellent activity and electrode reversibility as a bifunctional oxygen catalyst. According to the d-band center theory, the center value of the d-band farther from the Fermi level indicated that the adsorption capacity was weaker.164 By measuring the projected density of states (PDOS) of the single atom sites of Ni and Fe, the binding between the Ni–N4 active center and oxygen was much weaker than that with the Fe–N4 active center, which resulted in the weaker ORR activity of the Ni–N4 active center, but its OER activity was better than that of the Fe–N4 active center (Fig. 15b and c). After the catalyst was assembled into a zinc–air battery, it showed excellent electrode efficiency and cycle stability.
 | ||
| Fig. 15 (a) DFT-optimized adsorption configurations of the reaction adsorbates. (b) DFT-calculated reaction free energies of oxygen electrocatalytic reactions (U = 0 V) and (c) PDOS of the single-atom sites of Fe–N4 and Ni–N4. The black dashed line in (c) indicated the Fermi level. The red and blue vertical bars represent the calculated d-band centers of Fe and Ni, respectively. Reprinted with permission from ref. 161. Copyright (2020), John Wiley & Sons, Inc. | ||
Due to the excessively high spin state of iron oxides (filling amount is about 2.0), the binding strength of iron oxides to the reaction intermediates is inhibited, resulting in unsatisfactory OER catalytic activity. Thus, to solve this issue, Shen et al.140 anchored Fe(III) atoms on ultrathin TiO2 nanoribbons through an adsorption-oxidation strategy (the surface of ultrathin TiO2 nanoribbons was rich in –OH as sites to anchor Fe and prevent aggregation), thereby synergistically reducing the spin state of Fe (1.08). The formed σ bond with oxygen-containing intermediates was stronger during the catalytic OER process, which could enhance the adsorption of oxygen-containing intermediates (Fig. 16a–c). Meanwhile, orbital hybridization of Fe 3d and Ti 3d with electron delocalization boosted the electrical conductivity of the nanoribbons, thereby reducing the ohmic loss. All these factors together resulted in excellent OER activity (Fig. 16d–f).
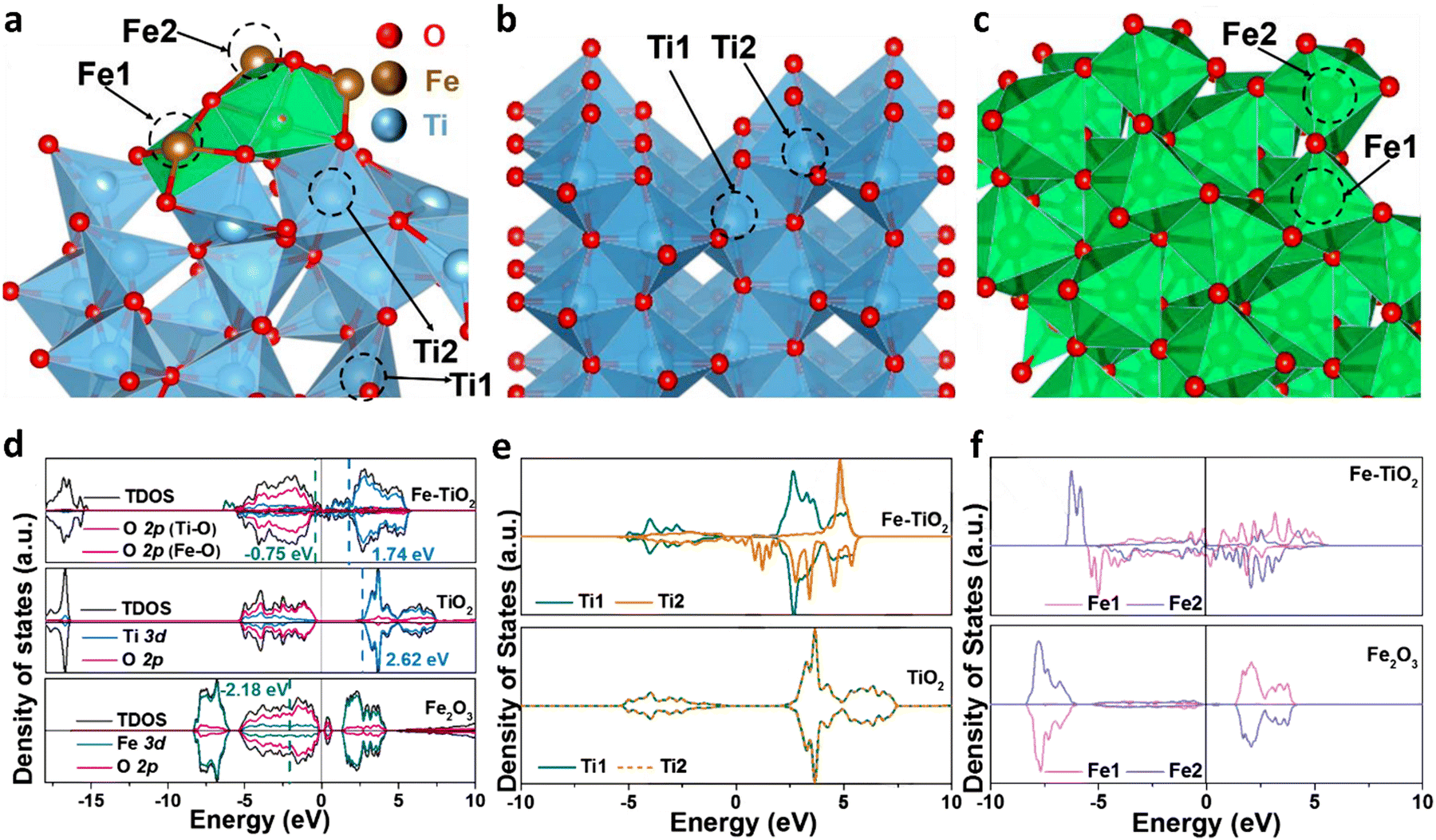 | ||
| Fig. 16 DFT calculations. (a–c) Side view model for DFT calculation of Fe-TiO2 (a), Fe2O3 (b) and TiO2 (c) models after structural optimization. (d) TDOS and projected DOS, and the dashed lines represent the d-band centers of Ti 3d and Fe 3d. (e and f) Projected Fe 3d (e) and Ti 3d (f) DOS of selected atoms in the models. Reprinted with permission from ref. 140. Copyright 2020, John Wiley & Sons, Inc. | ||
4.1.1.3. HER process. Because hydrogen energy (H2) can provide energy during the combustion process and emit zero CO2, it has become a substitute for fossil fuels to reduce the greenhouse effect and solve the energy crisis.165,166 Electrochemical water splitting is a clean and sustainable method for the preparation of hydrogen, which is the combination of two semi-reactions (Fig. 17), that is, the HER at the cathode and OER at the anode.17,41,167 Under standard conditions, the Gibbs free energy of water splitting is 273.2 kJ mol−1 with an equilibrium potential of 1.23 V.168 However, in practical applications, due to some kinetic obstacles, the equilibrium potential required for electrochemical water splitting is greater than 1.23 V, forming an overpotential. Thus, to accomplish water splitting, the use of catalysts is required to overcome this large equilibrium potential.166
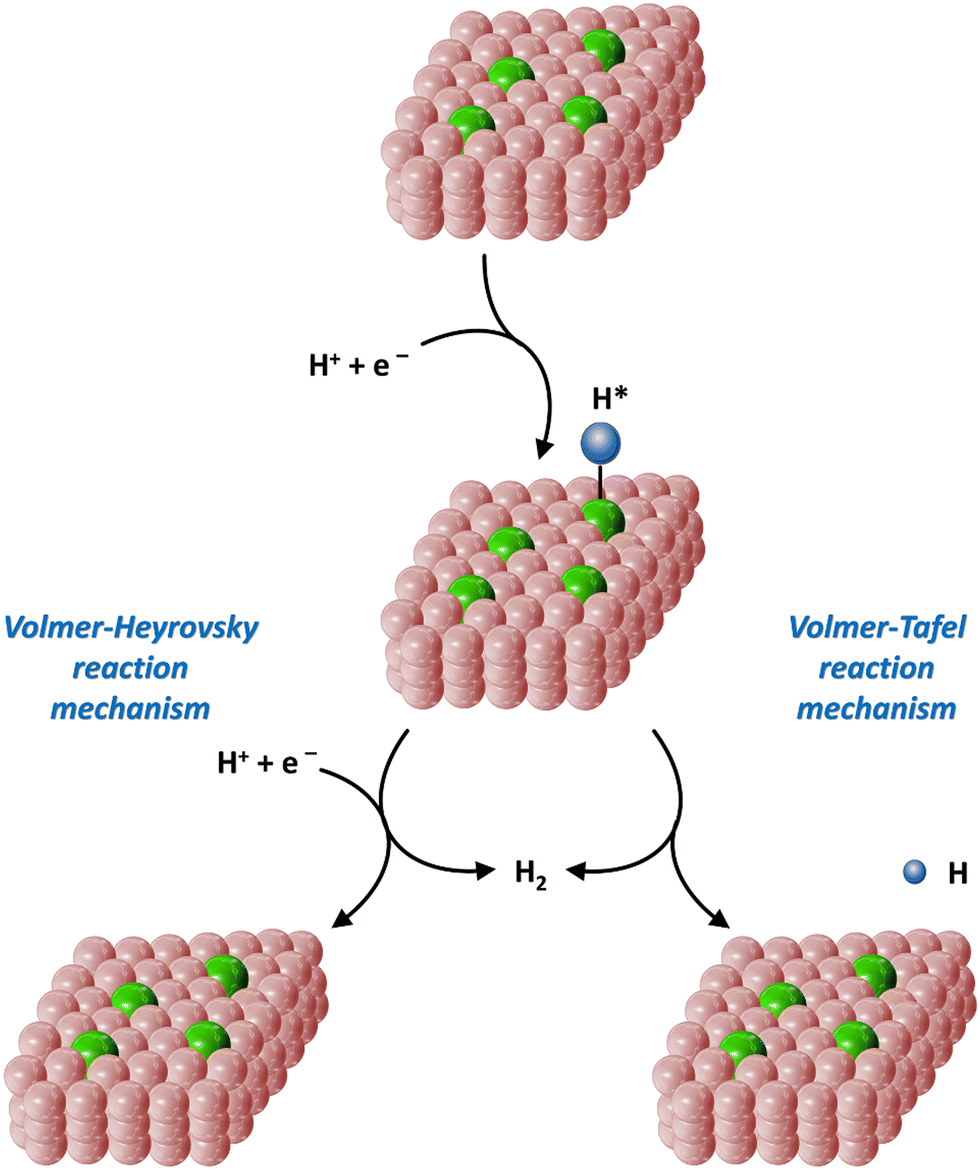 | ||
| Fig. 17 Possible e− reaction pathways for the HER processes on single-atom iron catalysts. | ||
The HER is a 2e− transfer reaction, which takes place on the electrode surface. The first step is proton-coupled electron transfer on the catalyst surface and form an H* intermediate at the active centers (Volmer–Heyrovsky or discharge reaction, formula (16)). The second step is divided into two pathways according to the amount of active H* intermediate on the electrode surface. If the number of H* is small, a single H* will combine with H+ and electrons to form H2 (formula (17)). Otherwise, if there are massive amounts of active H* intermediates on the electrode surface, two adjacent H* will recombine to form H2 (formula (18)).169,170
The Volmer–Heyrovsky reaction mechanism is as follows:
| H+ + e− + * → H* | (16) |
| H* + H+ + e− + * → H2 + * | (17) |
| H+ + e− + * → H* | (16) |
| 2H* + H2 + 2* | (18) |
Regardless of the reaction pathway, H* is the only chemical reaction intermediate. In this case, the free energy of H adsorption (ΔGH*) can be utilized as a scalar to evaluate the HER performances.17,41 Generally, catalysts with ΔGH* infinitely close to zero have the highest HER activity.165,171 To date, it is very difficult to clarify the specific working principles of different HER electrocatalysts. However, the Tafel slope can represent the rate-determining step (RDS), providing valuable insights into possible reaction pathways. Therefore, when measuring the HER performance of a catalyst, the Tafel slope is an indispensable parameter. Based on the Tafel slope, the reaction RDS can be determined, inferring the reaction mechanism of the catalyst used in HER catalysis. According to the Butler–Volmer kinetics, the RDS can be divided into three categories. The first type is that when the second step of the HER process, corresponding to the Tafel step (2H* → H2 + 2*), which is the RDS, the Volmer reaction is very fast. The resulting Tafel slope is 30 mV dec−1 at 25 °C. The second type is that when the Heyrovsky step (H* + H+ + e− + * → H2 + *) is the RDS, the reaction rate of the first step is also very fast, and its Tafel slope is 39 mV dec−1 at 25 °C. The third category is the Volmer reaction (H+ + e− + * → H*) as the RDS. At this time, the discharge of H+ is very slow, and hydrogen will be released through the recombination reaction step or the Tafel step. The resulting Tafel slope is 118 mV dec−1 at 25 °C.172
Multiple lines of evidence support that Fe-SACs have high catalytic activity for the ORR (refer to Section 4.1.1), but their catalytic performance for the HER process is still relatively mediocre.166 Thus, it is necessary to dope Fe-SACs with other transition metals to manipulate the electronic structures of the Fe active sites, thereby improving their catalytic activity for the HER process. Zeng et al.104 grafted a single Pt atom on the active site of Fe–N4 by bridging O2 to form a single atom catalyst with the structure of Pt1–O2–Fe1–N4 (Fig. 5g–i). Fe/Pt-SACs exhibited outstanding HER catalytic activity with an onset overpotential of about 15 mV, which was obviously less than that of the commercial Pt/C catalyst. Moreover, the Tafel slope of Fe/Pt-SACs was determined to be 42 mV dec−1, which is very close to that of the Pt/C catalyst (38 mV dec−1), revealing that HER process of Fe/Pt-SACs followed the Volmer–Heyrovsky reaction mechanism. Interestingly, Fe/Pt-SACs had a fast electron transfer rate based on the higher exchange current density of 0.039 mA cm−2 (Fig. 5h), boosting the HER kinetics. Grafting Pt1–O2 to the Fe–N4 active center afforded Fe with a spatial and electronic effect, which can facilitate the dissociation of water, thereby enhancing the HER dynamics. According to DFT calculation (Fig. 5i), the ΔGH* of Fe/Pt-SACs was about 0.16 eV, which is equivalent to that of the commercial Pt/C catalyst (∼0.09 eV).
When preparing Fe-SACs, the morphology and structure of the catalyst should be optimized. If the synthesized catalysts have a high specific surface area and density of active centers, more active catalytic centers will be accessible. Furthermore, the HER catalytic activity will be greatly improved by considering the following aspects: (1) suitable porosity between adjacent structures of the catalysts to achieve charge transport and electrolyte penetration into the deep active center of the catalyst surface and (2) higher crystallinity and appropriate topological defects to increase the electron density and conductivity of the material.
4.1.1.4. CO2RR process. The amount of CO2 in the air is increasing gradually, resulting from the massive consumption of fossil fuels, and the greenhouse effect-associated global warming has triggered a series of global issues. Luckily, CO2 can be reduced to various C1 products, including HCOOH, HCHO, CH3OH, CH4 and CO, under the catalysis of SACs.173,174
Experiments and theories show that the reaction mechanism of CO2 to form CO through the reduction process is as follows: CO2 produces a COOH* intermediate by a proton-coupled single electron transfer reaction (formula (19)), which, in turn is transformed to the CO* intermediate and H2O (formula (20)). The last step is the desorption of CO at the active sites (formula (21)).41,175 A key debate regarding the CO2RR mechanism is that the generation of the COOH* intermediate requires two steps, namely, the proton-uncoupled electron transfer mechanism.91,176
The proton-coupled electron transfer mechanism (Fig. 18) is as follows:
| CO2 + H+ + e− + * → COOH* | (19) |
| COOH* + H+ + e− → CO* + H2O | (20) |
| CO* → CO + * | (21) |
 | ||
| Fig. 18 Proton-coupled electron transfer mechanism of CO2RR processes on single-atom iron catalysts. | ||
Recent evidence demonstrated that the Fe in Fe-SACs carries a large positive charge with a valence state between 0 and +3, making it beneficial to effectively convert CO2 into CO during the CO2RR process. For example, Gu et al. used ZIF-8 as a support to synthesize Fe-SACs. The K-edge XANES spectra indicated that the Fe in the catalysts showed a +3 valence state. The CO2RR experiment was carried out in 0.5 M KHCO3 saturated with CO2. When the overpotential was 0.34 V, the partial current density reached 94 mA cm−2, and the Faraday efficiency of the cathode CO was calculated to be higher than 90% (Fig. 19a). Due to the presence of pyrrole N ligands, in the process of electrocatalysis, the Fe in the catalysts maintained the oxidation state of +3, and thus these Fe-SACs outperformed their counterparts in terms of catalytic activity and stability. To validate the influence of the oxidation state of Fe on the CO2RR catalytic effect, the Tafel slopes of Fe2+–N–C and Fe3+–N–C catalysts were determined to be about 117 and 71 mV dec−1, respectively (Fig. 19b), suggesting that the Fe3+ site had a faster CO2 adsorption rate than the common Fe2+ site. Moreover, when some deionized water or tap water was used to replace KHCO3, its Faraday efficiency and jCO almost remained constant, lasting at least 12 h (Fig. 19c).176 Furthermore, the type and number of N coordinated with the Fe atom have been shown to affect the catalytic activity of Fe-SACs.177,178
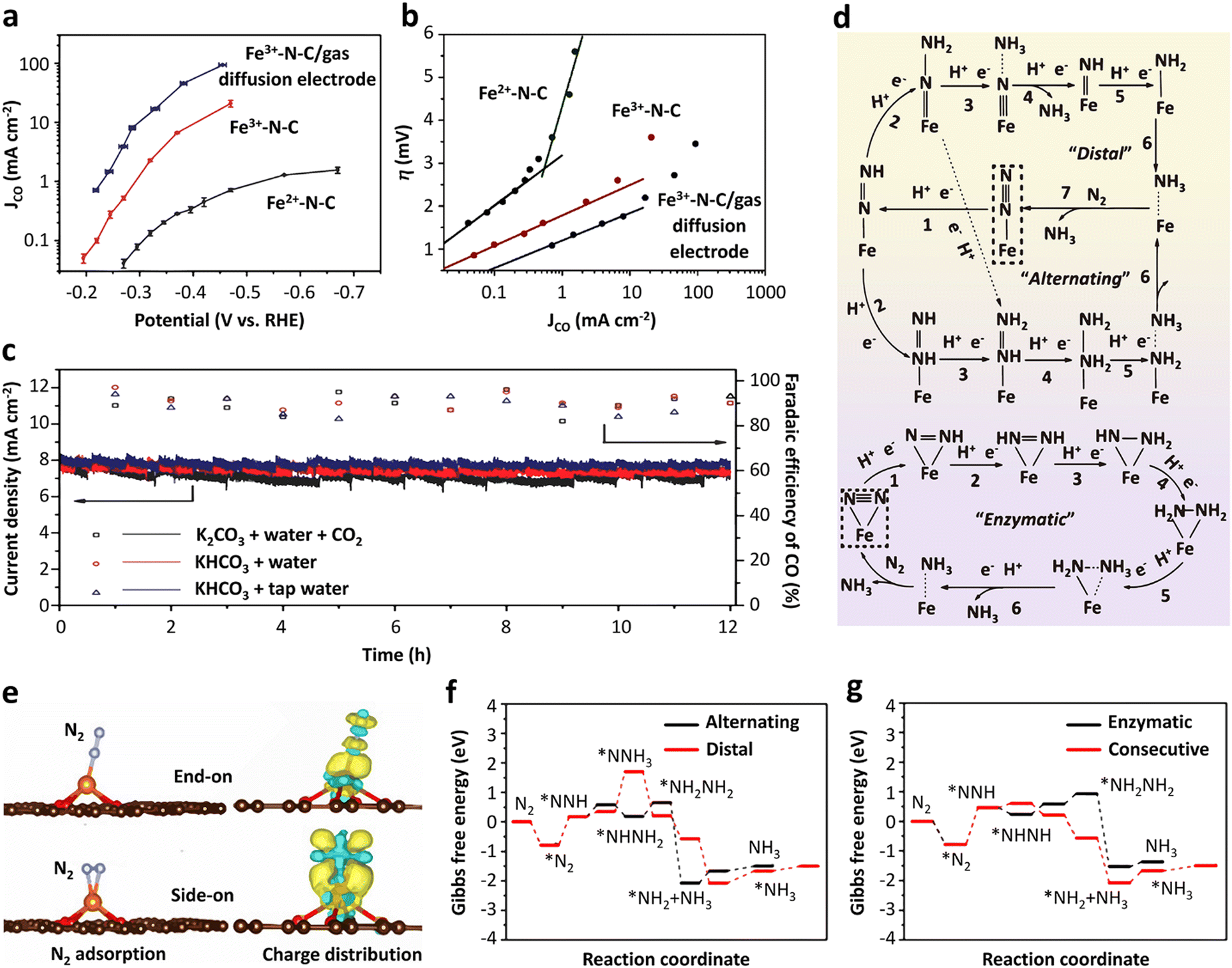 | ||
| Fig. 19 Applications of Fe-SACs in energy storage. (a) jCO and (b) Tafel plots of Fe3+–N–C in an H-cell (red) and on a gas diffusion electrode (blue), and of Fe2+–N–C in an H-cell (black), respectively. (c) Chronoamperometry curve and faradaic efficiency of CO production (dots) by Fe3+–N–C in H-cell at −0.37 V versus RHE. Reprinted with permission from ref. 176. Copyright (2019), the American Association for the Advancement of Science. (d) Mechanism diagrams of SACs with Fe–N4 active sites in the NRR process. Reprinted with permission from ref. 193. Copyright (2016), the American Chemical Society. (e) N2 adsorbed on the active site in an end-on and side-on structure and corresponding Bader charge distribution of *N2. (f) Gibbs free energy diagram of Fe–(O–C2)4 catalyst for the NRR process through distant and alternating pathways and (g) enzymatic and consecutive pathways. Reprinted with permission from ref. 95. Copyright (2020), John Wiley & Sons, Inc. | ||
Not only the oxidation state of iron in Fe-SACs will affect the CO2RR process, but also the reaction temperature in the electrochemical reduction of CO2 will affect the catalytic effect of Fe-SACs. In a typical experiment, Lin et al.179 separately synthesized atom-dispersed iron and nickel anchored on nitrogen-doped carbon supports and studied the temperature in the range of 303 K and 343 K. At a higher overpotential, increasing the reaction temperature increased the CO Faraday efficiency and current density of the Fe-SACs. When the temperature increased from 303 K to 323 K, the CO current density of the Fe-SACs increased rapidly from 114.4 mA cm−2 to 179.7 mA cm−2. With an increase in temperature to 343 K, the CO current density in this process increased slowly, and then plateaued. The final CO current density was 185.8 mA cm−2. In contrast, increasing the temperature inhibited the CO Faraday efficiency of Ni–N–C. When the temperature increased from 323 K to 343 K, the CO current density showed a downward trend from 252.2 mA cm−2 to 202.9 mA cm−2. The DFT calculation results indicated that an increase in temperature accelerated the reaction kinetics. Moreover, the difference in the CO2RR process of these two catalysts, i.e., Fe-SACs and Ni-SACs, was mainly due to the difference in the adsorption strength between the key reaction intermediates and the active sites. Therefore, the reaction temperature was also one of the main factors affecting the catalytic performance of Fe-SACs in the CO2RR process. However, Sun et al.180 introduced a single P atom in the form of a P–C bond in a nitrogen-doped carbon-supported single iron atom catalyst for the CO2RR process. Due to the addition of P atoms, the electron density of Fe in the active center increased. Accordingly, the formation of the *COOH intermediate was promoted, making it have a superior CO2RR catalytic performance under a lower overpotential. Under a low overpotential at 320 mV, the CO Faraday efficiency of Fe-SAC/NPC was maintained at 97% within 24 h. The current density of CO was stable at 5 mA cm−2 and it had a low Tafel slope (59 mV dec−1).
A good CO2RR catalyst should not only be able to balance the adsorption energy of the *COOH and *CO intermediate products, but also be beneficial for the desorption of CO. The coordination environment around the active center of Fe–Nx, e.g., the type of N coordinated with the Fe atom and the number of N atoms coordinated with the Fe atom, affected its catalytic activity. Studies have shown that the Fe–N3 active center had a high CO2RR catalytic activity and can effectively convert CO2 to CO. In a typical experiment, Wang et al.181 synthesized a single Fe atom uniformly dispersed N-doped porous carbon support via a gas diffusion strategy (Fe1NC/SX–Y, X and Y represented the particle size of ZIF-8 precursor and pyrolysis temperature, respectively). When the particle size of ZIF-8 was 100 nm and the temperature was 1000 °C, the prepared Fe1NC/S1-1000 catalyst had the best CO2RR catalytic activity (Fig. 20a). At −0.6 V, the partial current density of CO of Fe1NC/S1-1000 was 6.8 mA cm−2, which was about 1.2, 2.0 and 86 times that of Fe1NC/S1-800, Fe1NC/S1-900 and NC/S1-1000, respectively (Fig. 20b). At −0.65 V, the calculated turnover frequency was 2225 h−1, and its catalytic effect was comparable to that of Fe3+–N–C catalysts (Fig. 20c). To study the coordination environment of Fe atoms (type, content and number of coordinated N atoms), the content of various N and the corresponding FE of CO at different temperatures were measured. When the temperature was set as 800 °C, 900 °C and 1000 °C, the coordination atoms of the prepared catalysts were 3.8, 3.3 and 3.0, respectively. Moreover, the content of graphite N was 0.55, 1.11 and 1.32 wt%, respectively. Notably, the FE of CO was 81.8%, 90% and 95.6%, respectively. Simultaneously, the authors also constructed three structural models of FeN4, FeN3 (embedded in the graphene plane, and completely encapsulated by C atoms), and FeN3V (V represents vacancies) for density functional theory calculations (Fig. 20d and e). In the CO2RR process of the three models, only the process of converting *COOH to *CO was exothermic. Consequently, the catalytic activity of each model could be evaluated by calculating the two steps of CO2 conversion to *COOH and CO desorption from the active sites. Among the three models, the FeN3V model had the best catalytic activity and could balance the adsorption energy of the *COOH and *CO active intermediates, which was beneficial for the formation of CO. The above-mentioned experimental results and theories demonstrated that the synergy between graphite N and Fe–N3 species improved the CO2RR catalytic activity of the catalyst. The graphite N can redistribute the charge around the Fe atoms and promote the transport of electrons.
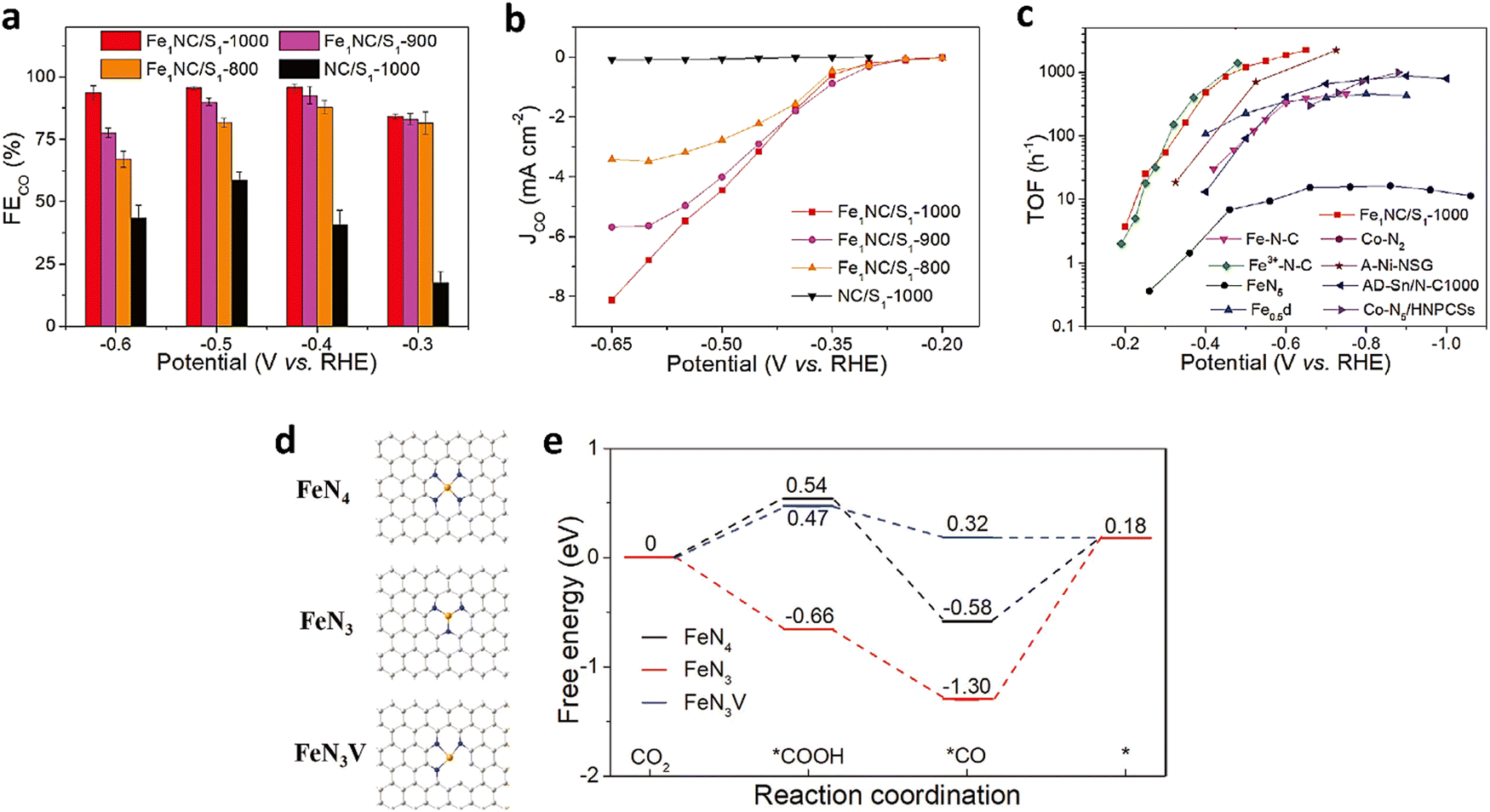 | ||
| Fig. 20 (a) FECO and (b) JCO of Fe1NC/S1-Y series and NC/S1-1000. (c) TOFs of Fe1NC/S1-1000 compared to that of other reported nonprecious metal/N-doped carbon-based SACs: Fe-SACs, Fe3+–N–C, Fe–N5, Fe0.5d, Co–N2, A-Ni-NSG, AD–Sn/N–C, and Co–N5/HNPCSs. (d) Top view of the optimized atomic structures for FeN4, FeN3, and FeN3V models embedded on graphene layer. The yellow, blue, and gray balls represent Fe, N, and C atoms, respectively. (e) Calculated free-energy diagrams for the CO2ER pathway of FeN4, FeN3, and FeN3V moieties. Reprinted with permission from ref. 181. Copyright 2020, John Wiley & Sons, Inc. | ||
Both experiments and theory show that in metal SACs, their catalytic performance for the CO2RR follows the order of Ni > Fe > Co, which is mainly related to the binding energy between each metal active site and the active intermediates during the CO2RR process.41,182 The onset potential in the CO2RR processes of the catalyst with Fe/Co–N4 active sites is relatively low, whereas it is difficult for the CO* active intermediate to desorb from metal atoms because of the high binding energy between them. The fatal consequence is the protonation of the CO* intermediates (or catalyst poisoning) and generation of other products, causing the Fe/Co–N4 catalysts to exhibit low CO selectivity. Considering SACs with Ni–N4 active centers, they have a higher current density, but when generating COOH* intermediates, their kinetics is slower, resulting in a decrease in the reaction rate.183–185 For metallic nickel, in the CO2RR process, its catalytic effect is greater than that of metallic iron. Therefore, the introduction of monoatomic nickel into a single atomic iron-based catalysts can improve their CO2RR catalytic activity. Recently, Jiao et al. adopted a zinc-assisted atomization strategy to dope single iron and zinc atoms in MOF-derived nitrogen-doped carbon (Fe1–Ni1–N–C), where the Fe–N4 and Ni–N4 active centers were adjacent to each other. Due to the interactions between the adjacent sites, the CO2RR catalytic activity was much larger than Fe1–N–C and Ni1–N–C of nitrogen-doped carbon supports modified with Fe and Ni single atoms. The CO2 electrochemical reduction test results showed that Fe1–Ni1–N–C had a higher current density and exhibited the largest Faraday efficiency (96.2%) at −0.5 V, which was better than that of Fe1–N–C and Ni1–N–C. Meanwhile, the CO current density of Fe1–Ni1–NC (jCO = 2.4 mA cm−2) was also greater than that of Fe1–N–C (2.1 mA cm−2), Ni1–N–C (0.1 mA cm−2) and N–C (0.7 mA cm−2). Fe1–Ni1–N–C had faster kinetics, and its Tafel slope (83 mV dec−1) was smaller than that of Fe1–N–C and Ni1–N–C. In the −0.5 V state, Fe1–Ni1–N–C could run continuously for 10 h and the Faraday efficiency and current density of CO remained unchanged, indicating that it had high stability. DFT calculation demonstrated that the coupling of adjacent Ni and Fe atoms in Fe1–Ni1–N–C was beneficial for the activation of CO2, thereby reducing the barrier for the formation of COOH* intermediates, ultimately improving the electrochemical reduction performance of Fe1–Ni1–N–C for CO2.186 Overall, designing bimetallic SACs may successfully negotiate the above-mentioned intrinsic barriers.175 Various electrochemical pathways can be performed on the optimal active centers and the catalytic activity of CO2RR will be improved by the synergistic effect.
4.1.1.5. NRR process. On the surface of the Earth, the content (∼78%, volume fraction) of N2 is extremely rich, but nitrogen basically exists in the form of gaseous dimolecular N2, which cannot be directly used by organisms or employed as industrial raw materials. Thus, nitrogen fixation technology has been rapidly developed to convert nitrogen to ammonia (biological way) and hydrazine (industrial way) or to oxidize nitrogen to form nitrogen oxide. Generally, the nitrogen fixation reaction refers to the reduction of N2 to ammonia with high chemical reaction activity. To date, the main method for industrial synthesis of ammonia is the Haber–Bosch reaction.187 However, the environmental impact (e.g., huge energy consumption and CO2 emission) associated with this reaction is not consistent with the concept of green chemistry and sustainable development.188,189 The electrochemical synthesis of NH3 under mild conditions was developed, in which renewable electricity and H2O as a hydrogen source are employed to directly convert N2 into NH3 by accepting electrons at the cathode (namely the NRR process, formula (22)), accompanying the OER reactions at the anode (formula (23)).
| Cathode: N2 + 6H+ + e− → 2NH3 | (22) |
Node: 3H2O−6e− → 6H+ + ![[/]](https://www.rsc.org/images/entities/char_e0ee.gif) O2 O2 | (23) |
There are three reaction mechanisms that are related to Fe as an electrocatalyst in the NRR reactions, namely, the distal mechanism, alternating mechanism and enzyme mechanism.190–192Fig. 19d shows the reaction pathways when Fe acts as the active center of the NRR process.193 Similar to the Fe–N complexes, which are effective NRR electrocatalysts, Fe-SACs also display great potential as NRR catalysts.194,195 In 2020, Zhang et al. designed Fe-SACs with an Fe–(O–C2)4 structure, in which an Fe atom was coordinated with 4 O atoms instead of common N atoms. An electrochemical test was performed in alkaline solution. When the applied potential was −0.1 V, the Faraday efficiency of NH3 reached the maximum of 29.3% with a yield of 32.1 μg h−1 mgcat.−1, showing outstanding NRR catalytic activity. N2 (bond length of 1.12 Å) adsorbs on the active center in two configurations, namely “side-on” (1.188 Å) and “end-on” (1.146 Å) (Fig. 19e). The various configurations will affect the amounts of electrons transferred from the Fe–(O–C2)4 structure to the adsorbed N2, leading to different catalytic properties. As shown in Fig. 19f and g, the “end-on” adsorption occurred via the remote and alternate reaction mechanism, and the “side-on” adsorption preferred to undergo the continuous hydrogenation reaction mechanism. It is vital to note that multiple reaction mechanisms possibly participate in the NRR process due to the acceptable energy input.95 Recently, Li et al.196 synthesized a new type of NRR single-atom catalyst. They used SiO2 nanospheres as a sacrificial template to anchor a single iron atom on N,O co-doped inverse opal carbon via high-temperature thermal addition. The structure had a high specific surface area (SBET = 361 m2 g−1), which was conducive to mass transfer and electron transportation. Among them, a single iron atom was coordinated with two nitrogen atoms and four oxygen atoms. The charge transfer between oxygen atoms and iron atoms not only can promote the transportation of nitrogen, but also effectively reduce the binding energy between the active sites and *N2. All these factors together can facilitate the desorption of NHx, increase the electron transfer rate, and effectively accelerate the rate determination step (*N2 → *NNH).
During the NRR reaction, a competitive reaction occurs simultaneously, i.e., the HER. The NRR is a 6-proton-coupled electron transfer process, while the HER is a 2-proton-coupled electron transfer process. Moreover, only a small overpotential is required to initiate the HER process, and thus it is the most common competitive side reaction for the NRR process. Accordingly, the redesign of electrocatalysts exhibiting a weak binding energy with hydrogen is needed to break this inevitable barrier.197 Therefore, to achieve a higher binding strength between Fe-SACs and N2 than that of hydrogen, systematic material-design methods have been recently developed for the fabrication of Fe-SACs, such as doping with heteroatoms (B, O, S, P, etc.), varying the coordination number of the Fe center atom, carbon support defect, S or N vacancies, active sites of bimetallic atoms, and others.198,199 Recently, Wang et al.200 investigated the effect of the electronic state of the Fe-SAC active center on the NRR process. The NRR process occurred at the active site of FeN4, instead of MoN4. The MoN4 adjacent to the FeN4 active site can tune the spin state of the Fe atom from a high spin state 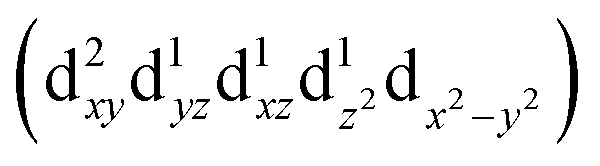 to an intermediate spin state
to an intermediate spin state 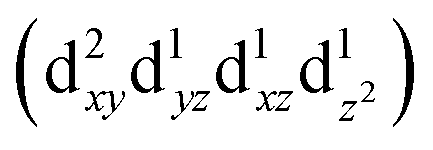 . The unpaired electrons of Fe were reduced, which was favorable for the reverse contribution of π and promoted bonding with the unpaired electrons of *N2. Thereby, it could further accelerate the first-step hydrogenation process (Fig. 21a–c). Simultaneously, the change in spin state caused an increase in the charge density of the Fe sites, which promoted the adsorption of N2. Meanwhile, the H intermediates adsorbed on the Mo sites can assist N hydrogenation via hydrogen flooding. The separated d-electrons and empty d-orbitals were more favorable for the overlapping of the Fe 3d and N 2p orbitals, which significantly lowered the energy barrier for the rate-determining step (Fig. 21d–g). In the NRR process, most scientists employ N2 as a raw material to convert it into ammonia. A few of them use nitrate as a raw material to synthesize ammonia, mainly because this process involves 8 electron transfer steps and many reaction pathways produce more by-products (NO2, NO2−, NO, N2O, N2, NH2OH, NH3, and NH2NH2), and reducing NO3− to N2 involves an N–N coupling step. This step generally requires two adjacent active sites for catalysis. Impressively, single-atom catalysts possess these characteristics. Wu et al. synthesized a single-atom iron catalyst for the synthesis of ammonia using NO3− as the raw material, and proved that the FeN4 active sites have excellent activity and selectivity in the process of converting NO3− to NH3.201
. The unpaired electrons of Fe were reduced, which was favorable for the reverse contribution of π and promoted bonding with the unpaired electrons of *N2. Thereby, it could further accelerate the first-step hydrogenation process (Fig. 21a–c). Simultaneously, the change in spin state caused an increase in the charge density of the Fe sites, which promoted the adsorption of N2. Meanwhile, the H intermediates adsorbed on the Mo sites can assist N hydrogenation via hydrogen flooding. The separated d-electrons and empty d-orbitals were more favorable for the overlapping of the Fe 3d and N 2p orbitals, which significantly lowered the energy barrier for the rate-determining step (Fig. 21d–g). In the NRR process, most scientists employ N2 as a raw material to convert it into ammonia. A few of them use nitrate as a raw material to synthesize ammonia, mainly because this process involves 8 electron transfer steps and many reaction pathways produce more by-products (NO2, NO2−, NO, N2O, N2, NH2OH, NH3, and NH2NH2), and reducing NO3− to N2 involves an N–N coupling step. This step generally requires two adjacent active sites for catalysis. Impressively, single-atom catalysts possess these characteristics. Wu et al. synthesized a single-atom iron catalyst for the synthesis of ammonia using NO3− as the raw material, and proved that the FeN4 active sites have excellent activity and selectivity in the process of converting NO3− to NH3.201
 | ||
| Fig. 21 Magnetic susceptibility and 57Fe Mössbauer spectra of the catalysts. (a) Simplified schematic of N2 bonding to transition metals. (b) Magnetic susceptibility of FePPc. (c) Room-temperature 57Fe Mössbauer spectrum of FeMoPPc. The data (scattered dots) are shown with the best fit (black dash line) and the deconvolution of two components, as indicated by the legend. DFT calculations of the NRR activity on FeMoPPc catalysts. (d) Different free-energy diagrams for the NRR on Fe atoms of FeMoPPc and Mo atoms of FeMoPPc. (e) Different free-energy diagrams for the HER on Fe atoms of FeMoPPc and Mo atoms of FeMoPPc. (f) Charge density difference calculations of N2 bonded to Fe atoms of FePPc and FeMoPPc (the electron excess area and electron deficiency area are represented by yellow and blue-green, respectively). (g) Charge density difference calculations of the N2 bonded to Mo atoms of MoPPc and FeMoPPc. Reprinted with permission from ref. 200. Copyright 2021, John Wiley & Sons, Inc. | ||
Similar to the CO2RR, the NRR is a reaction involving multiple reaction intermediates (including N2 adsorption/activation and NH3 desorption), and the HER acts as a major competing reaction, making selectivity a formidable challenge.202–204 In addition, the relatively poor catalytic activity due to the slow adsorption of nitrogen and the high cracking energy of the N–N triple bond (940.95 kJ mol−1) is also an urgent problem to be solved.205 Therefore, improving the catalytic activity and selectivity of Fe-SACs during the NRR process has become the main research topic. Studies have shown that metal nitrides have higher activity for the NRR at low onset potentials, while inhibiting the HER, mainly because they have a stronger binding energy for nitrogen than hydrogen.206,207 Therefore, the combined application of metal nitrides and Fe-SACs may be a good approach to catalyze the NRR. Meanwhile, understanding of the details of the interface mechanism will also provide the necessary guidance for the implementation of more targeted strategies to design better catalysts.208,209
4.1.1.6. H2O2 production. As a type of reactive oxygen species (ROS), H2O2 is widely used in various industries (food, biology, chemistry, environmental science, pharmaceutical preparation, papermaking, etc.) due to its strong sterilization effect and non-toxic and harmless decomposition products.210–212 Especially in the food production process, H2O2 is the widely used because its use can make food beautiful, whiten it, and increase its shelf life.213
Currently in industry, the most common method for the synthesis of H2O2 is the hydrogenation of anthraquinone with hydrogen, followed by oxidation in an organic medium.214,215 However, this method has several disadvantages. The first is that a large amount of reaction by-products and waste will be produced. Also, a large amount of energy will be consumed. Secondly, the generated H2O2 has a high concentration, which is easy to decompose.216 Therefore, the electrochemical synthesis of H2O2 has become an emerging technological method to replace the traditional method to prepare H2O2.217,218 The electrochemical synthesis of H2O2 mainly has the following advantages: (1) mild reaction conditions, less requirements for temperature and environmental pressure, (2) electricity as an energy source does not require the combustion of fossil fuels and (3) use of water and air as raw materials and a green source. The electrochemical production of H2O2 mainly relies on the 2e− process of the ORR, with the reaction equations shown in eqn (24) and (25).219 H2O2 acts as a by-product in the 4e− reaction process of the ORR, and thus during the reaction process, the 4e− process needs to be inhibited. Therefore, it is very important to choose a suitable catalyst.
| In alkaline medium: O2 + 2H2O + 2e− → H2O2 + 2OH− | (24) |
| Acidic medium: O2 + 2H+ + 2e− → H2O2 | (25) |
4.1.2.1. Oxidation of CO. To address the increasingly serious environmental pollution, the conversion of CO in automobile exhaust and industrial waste gas into non-toxic gas at room temperature or low temperature has become an urgent problem.229 The catalytic oxidation of CO is one of the main and vital prototype reactions in the heterogeneous catalysis process.230 Li et al.231 investigated the mechanism of the catalytic oxidation of CO by atomically dispersed Fe embedded on graphene with first-principle computation. By carefully comparing the reaction between the adsorbed O2 and CO by the Langmuir–Hinshelwood (LH) and Eley–Rideal (ER) mechanisms, the results showed that the Fe-SACs had better catalytic activity for the catalytic oxidation of CO through the ER mechanism (Fig. 22a). Firstly, CO combines with an O2 molecule adsorbed on the surface of Fe-SACs to form the CO3* intermediate state (CO + O2* → CO3*). The intermediate CO3* will combine with another CO molecule to release two CO2 molecules (CO + CO3* → 2CO2) (Fig. 22b). Similarly, Liu et al.225 also used first-principles calculations to calculate the catalytic mechanism of single-atom Fe-doped graphene nanosheets in the catalytic oxidation of CO. The N atoms coordinating with Fe atoms made single Fe atoms have more positive charges, thereby adsorbing more reactive gases. The introduction of Fe can adjust the electronic structure and magnetic properties of the graphene system, thereby improving its catalytic activity. As illustrated in Fig. 23, the LH and ER mechanisms were used to study the catalytic oxidation of CO. The results revealed that the LH mechanism as the starting state was more advantageous than the ER mechanism during the reaction process because the formation of reaction intermediates had a smaller energy barrier, and then the reaction intermediates generated CO2 through the ER reaction.
 | ||
| Fig. 22 (a) Atomic configurations of the initial state (IS), transition state (TS), intermediate state (MS), and final state (FS) for CO oxidation on Fe-embedded graphene. (b) Energy profiles for CO oxidation over Fe on defective graphene. Reprinted with permission from ref. 231. Copyright 2010, the American Chemical Society. | ||
 | ||
| Fig. 23 Minimum energy profiles and configurations of the different states for CO oxidation on Fe–GN4 by (a) LH mechanism and (b) ER mechanism. Black, blue, red and green balls represent the C, N, O and Fe atoms, respectively. Reprinted with permission from ref. 225. Copyright 2017, The Royal Society of Chemistry. | ||
As a typical magnetic transition metal, the spin state of the Fe single-atom active center undoubtedly plays an important role in regulating the catalytic performance of various chemical processes. Therefore, theoretical calculations and multiscale simulations are also effective means to determine the working mechanism of Fe-SACs. The underlying mechanisms of various Fe-SACs spin degrees of freedom with high performance can be resolved through theoretical calculations and simulations. In a typical experiment, Zhang et al.232 proposed that the 2D ferromagnetic organometallic framework Mn2C18H12 can be used as an ideal platform to efficiently realize the activation of the spin triplet O2 and the oxidation of CO based on the basic principles of quantum mechanics. The physical mechanism of the two-dimensional ferromagnetic organometallic framework single-atom catalyst system to efficiently excite O2 was a new “charge-spin dual cooperative mechanism” (Fig. 24a). O2 achieved a transition from a spin triplet state to a spin singlet state when the O2 molecule was adsorbed on a certain Mn single-atom active site on the ferromagnetic 2D MOF structure. Importantly, the single-atom active site of the catalyst and its neighboring single atoms had a very different balance of labor and cooperation in terms of charge and spin degrees of freedom. During O adsorption, the charge transfer was mainly provided by the adsorption active site, and its adjacent Mn atoms are weakly related to the active site in terms of charge degrees of freedom. However, according to the Wigner spin conservation selection rule, the adjacent Mn atoms have a significant synergistic response in the spin degrees of freedom, revealing that these single atoms accommodate most of the spin magnetic moment transferred from the O2 molecule. Consequently, the efficient excitation of O2 from the spin triplet state to the spin singlet state can be realized. The quantum synergistic mechanism effectively reduced the rate-determining step barrier of the reaction process, thereby promoting the catalytic oxidation of CO.233,234 This novel quantum effect of “charge-spin synergistic catalysis” has been further extended to various magnetic X2C18H12 (X = Mn, Fe, Co, and Ni) systems. Interestingly, a clear linear scaling relationship was found between the degree of chemical activity of these M-SACs for O2 excitation and the spin excitation energy (Fig. 24b–e).
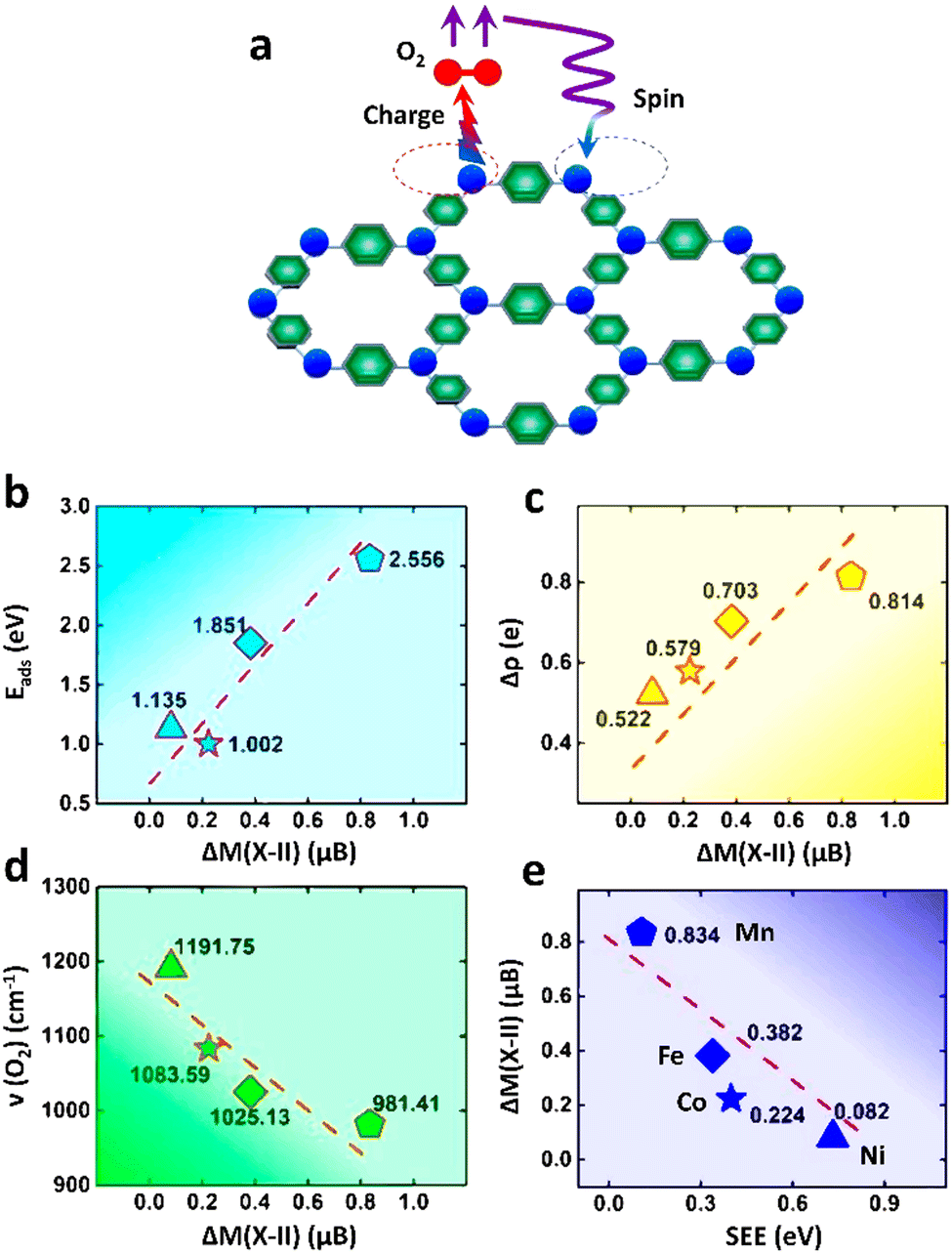 | ||
| Fig. 24 (a) Schematic diagram of the “charge-spin synergistic catalysis” mechanism of O2 activation by magnetic single-atom sites. (b) Adsorption energy Eads, (c) charge transfer Δρ and (d) O–O stretching vibrational frequency ν of the O2 molecule on the X-I sites with respect to the quantity of spin accommodated on X-II site ΔM(X-II) of 2D X2C18H12 MOF structures (X = Mn, Fe, Co, and Ni). (e) ΔM(X-II) as a function of spin excitation energy (SEE). Reprinted with permission from ref. 232. Copyright 2022, the American Chemical Society. | ||
4.1.2.2. Activation of C–H bond. Given that the C–H bond (409 kJ mol−1) in the benzene molecule is relatively stable, it is very difficult to oxidize benzene to phenol.235 To date, a widely employed route is the use of transition metals as catalysts for oxidation catalysis at high temperature (50–140 °C); however, this method generally consumes significant energy and shows poor catalytic property.236 Also, the catalytic oxidation of benzene to phenol at room temperature is an important barrier to be addressed. Deng et al.237 used FePc as the iron source and GNs as the carbon source to anchor atomically dispersed Fe–N4 species. In the presence of H2O2 oxidant, the FeN4/GN-x catalyst (x represents the Fe content in the catalyst) could catalytically oxidize benzene to phenol at room temperature. It can be seen from Fig. 25a that when the iron content was 2.7%, the corresponding catalyst had the highest catalytic activity. The conversion rate of benzene reached 23.4%, showing a yield of phenol of over 18% (Fig. 25b). DFT was used to calculate the oxidation process of benzene (Fig. 25c–e). FeN4/GN-2.7 used as a catalyst to catalyze the oxidation of benzene, which involved five steps. Firstly, H2O2 is decomposed into H2O2 and O atoms, while O atoms are adsorbed on the active site Fe–N4 to form Fe
O species, and then a second H2O2 molecule is dissociated again. The dissociated O atoms combine with Fe on the other side of the FeO species to form OFeO species. The O atom in the active center of OFeO will adsorb benzene molecules to form a C–H bond, with an energy barrier of 0.59 eV. The Fe–O bond will break to form phenol and FeO species, and then cyclically perform oxidation reactions. Meanwhile, the Bader charge analysis results showed that FeN4/GN-2.7 had 0.14 more electrons than FePc, which made it easier for FeN4/GN-2.7 to combine with O to form an Fe–O bond, thereby enhancing the catalytic oxidation activity of benzene. Moreover, Liu et al.238 used nano-MgO as a sacrificial template and synthesized monoatomic Fe-based catalysts containing different amounts of FeNx (x = 4–6) species by adjusting different pyrolysis temperatures. When the pyrolysis temperature was 700 °C, the catalytic effect was the best. The obtained Fe-SACs contained 28.3% high-spin FeN6, 53.8% low-spin FeN6 and 17.9% medium-spin FeN5 species. The most important species for C–H bond oxidation was FeN5 species, and its activity was about 10 times higher than that of the high/low-spin FeN6 species and about 3 times higher than that of the FeN4 species. The experiment results showed that the Fe-SACs could selectively oxidize C–H bonds and catalyze the oxidation of various substrates such as aromatic hydrocarbons, heterocyclic alkanes and aliphatic alkanes, with a selectivity of up to 99%.
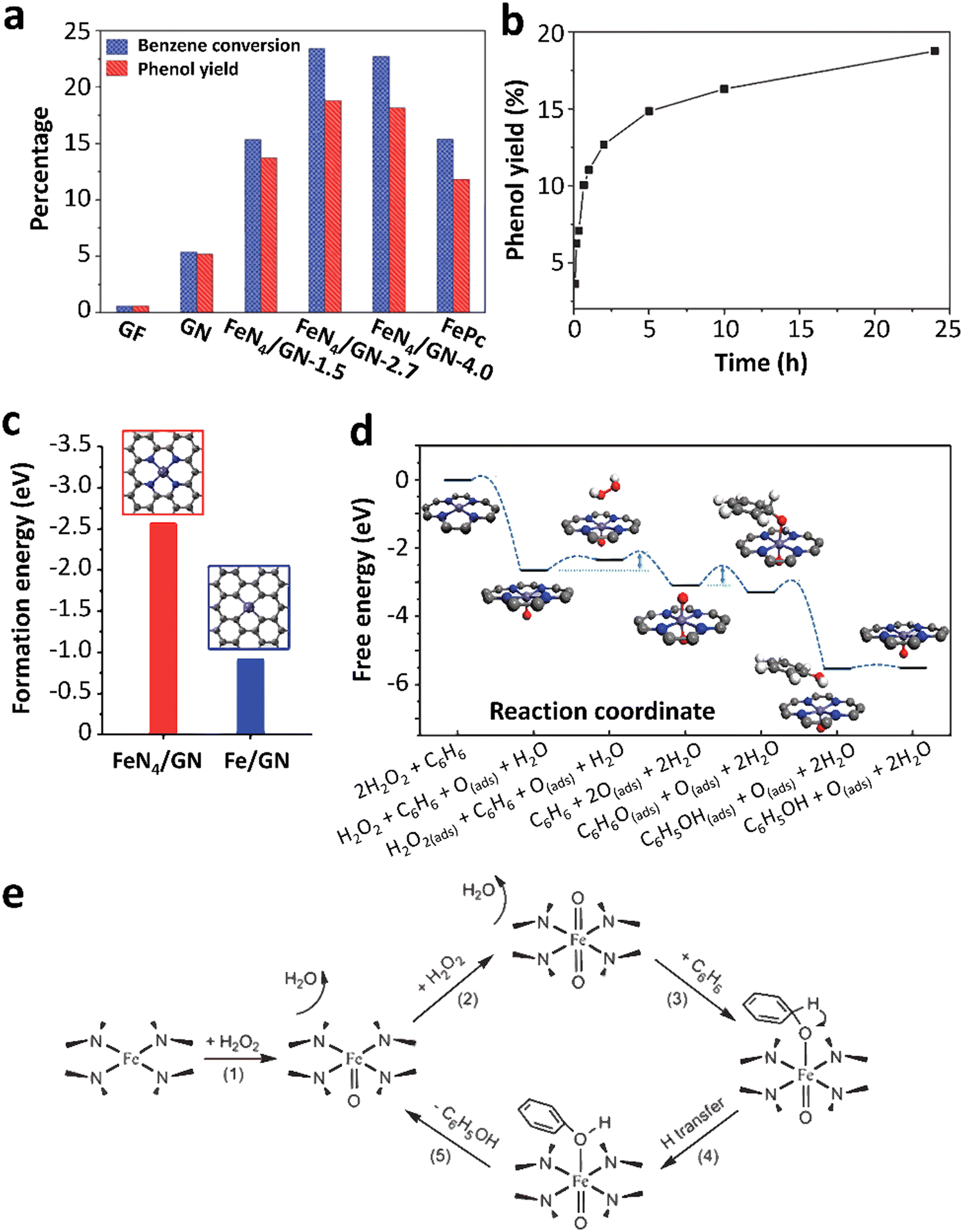 | ||
| Fig. 25 (a) Performance of the direct oxidation of benzene to phenol by FeN4/GN samples compared with GF, GN, and FePc. (b) Phenol yield of FeN4/GN-2.7 for the direct oxidation of benzene to phenol with different reaction times. (c) Formation energies of FeN4/GN and Fe/GN structures. The formation energy was calculated as follows: EFe-embedded − EFe-bulk − E(N)GN, where EFe-embedded and EFe-bulk are the total energies of FeN4/GN and the Fe/GN structure and an Fe atom in Fe bulk, respectively, and E(N)GN was the total energy of the optimized structure of FeN4/GN or Fe/GN with the Fe atom removed from the system. (d) Free energy diagram of the oxidation of benzene to phenol on FeN4/NG. The gray, blue, light blue, red, and white balls represent C, N, Fe, O, and H atoms, respectively. Proposed mechanism for C2H6 activation on the O–FeN4–O active site. (e) Scheme for the reaction mechanism of the oxidation of benzene to phenol on FeN4/NG. Reprinted with permission from ref. 237. Copyright 2015, the American Association for the Advancement of Science. | ||
The conversion of ethane into high value-added chemical products (such as C2H5OH, CH2CH2 and CH3COOH is also a critical step in industrial production.239,240 However, the difficulty of the cleavage of the C–H bond makes this conversion process more difficult and usually requires a high temperature (>400 °C), but its selectivity is poor. Meanwhile, some side reactions will occur, which may cause the product to over-oxidize or result in catalyst poisoning.241 Interestingly, Fe-SACs can convert ethane into C2 products through the catalytic oxidation of H2O2 at room temperature. In a typical experiment, Wang et al.242 synthesized atomic-grade Fe–N4 species uniformly dispersed on N-doped carbon. When H2O2 was used as an oxidant, the FeN4/GN catalyst could effectively convert ethane into CH3COOH, CH3CH2OOH, CH3CH(OH)2, CH2CH2 and other high value-added C2 products. As shown in Fig. 26a, consistent with the oxidation of benzene mentioned above, the activation of ethane was carried out according to the free radical mechanism. When ethylene is adsorbed on the O–Fe–O species, the C–H bond will be stretched to form a type of ˙CH2CH3 radical, and then form ethanol with the OH group or destroy another C–H bond to form CH2CH2. Notably, the produced ethanol can be used again as the reactant for oxidation catalysis. The C–H bond on the same carbon atom is activated, and finally CH3CHO is produced. The energy barriers of these three reactions are relatively low (Fig. 26b). Likewise, Cui et al.243 used Fe-SACs to convert methane into some high value-added C1 products (CH3OH, CH3OOH, HOCH2OOH, HCOOH, etc.) at room temperature. The conversion mechanism was roughly the same as that of ethane. It also first formed the O–Fe–O active intermediate, then activated methane through the free radical mechanism, and finally yielded the C1 product.
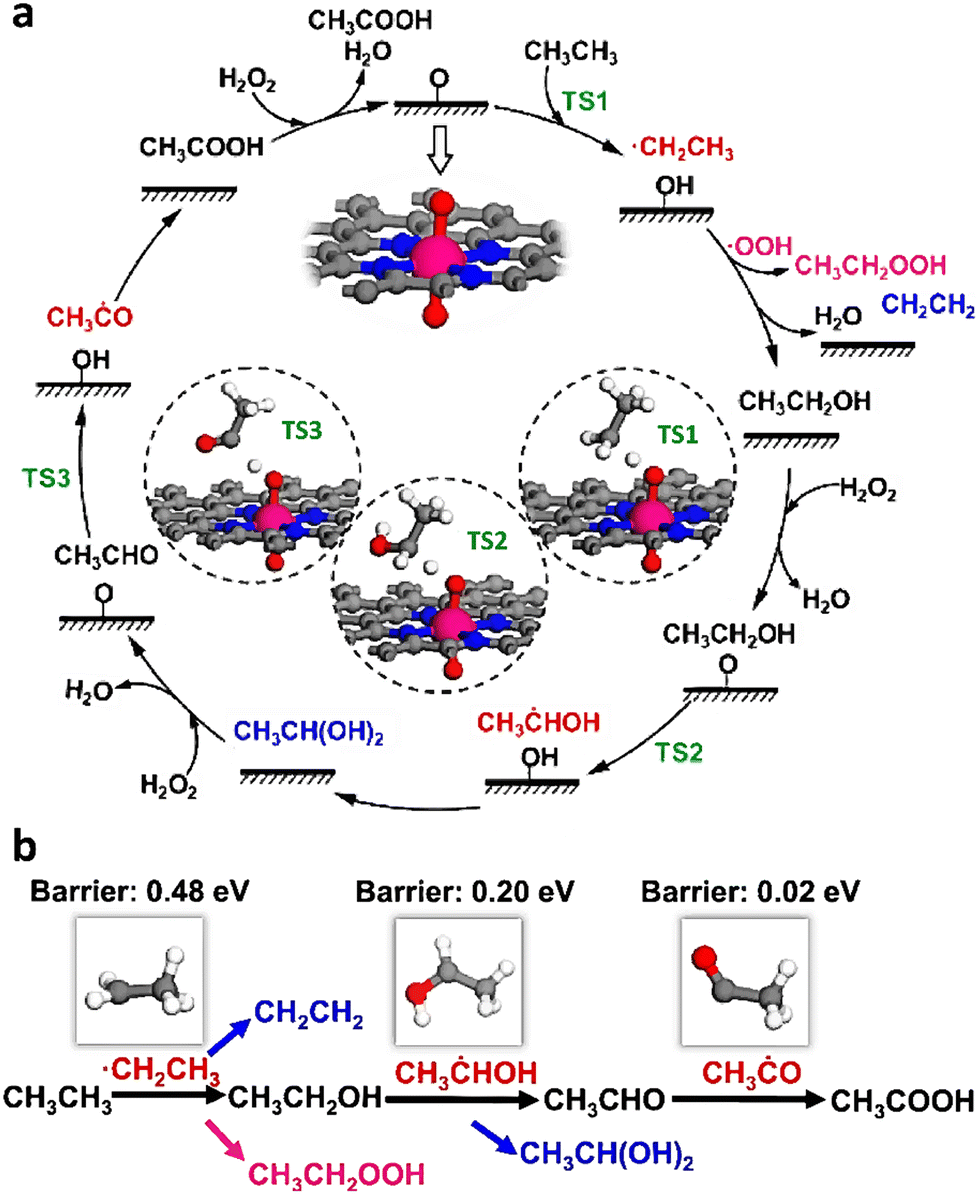 | ||
| Fig. 26 (a) Reaction cycle with the reactants (black), products (black, blue and pink), radical intermediates (red) and atomic structures of the active site and transition states TS1-TS3. (b) Reaction pathway with atomic structure of intermediates and the corresponding activation barrier. Reprinted with permission from ref. 242. Copyright 2019, Elsevier. | ||
4.1.2.3. Selective hydrogenation. Selective hydrogenation means that when the substrate contains two or more unsaturated functional groups, the catalyst selectively hydrogenates a specific functional group.244–246 Selective hydrogenation not only can improve the yield of the target product and reduce the yield of the by-products, but also can be performed in a mild manner, thereby reducing the energy consumption.246 It plays a pivotal role in the fine chemical, pharmaceutical, agrochemical and printing and dyeing industries.247–250 Due to the excellent catalytic activity of Fe-SACs, they can also be used to catalyze selective hydrogenation.
As a type of heterogeneous catalysis, the selective hydrogenation of unsaturated functional groups has become a focus of chemists, especially for the hydrogenation of nitro compounds. The mechanism is complex, and thus noble metal catalysts with excellent selectivity are required.251–255 Therefore, Yun et al.256 developed a new material of Fe single atoms and Fe2O3 clusters uniformly dispersed on N-doped carbon for the selective hydrogenation of nitro compounds (Fig. 27a). Using nitrobenzene as a model and hydrazine hydrate as a hydrogen source, the catalytic performance of Fe-SACs was tested. At room temperature, nitrobenzene could be completely hydrogenated to aniline within 12 min with a conversion rate of over 99%, which was greater than that of the FeN4 catalyst (50%) and iron nanoparticles (25%). Its TOF (1923 h−1) was higher than that of the FeN4 catalyst (748 h−1) and iron nanoparticles (75 h−1) alone. The DFT calculation results showed that the FeN4 catalyst was more active for the decomposition of hydrazine hydrate to produce hydrogen. The decomposition process of hydrazine hydrate followed the order of N2H4 → N2H4* → N2H3* → N2H2* → N2H* → N2* → N2 (Fig. 27b and c). The Fe2O3 cluster surface was more active for the reduction of aniline by nitrobenzene. The process of reduction and hydrogenation of nitrobenzene followed the sequence of PhNO2 →PhNO2* → PhNOOH* → PhN(OH)2* → PhNOH* → PhNHOH* → PhNH* → PhNH2*→ PhNH2 (Fig. 27d).257 Therefore, the results of the DFT calculations indicated that FeN4 can catalyze the decomposition of hydrazine hydrate to generate hydrogen.
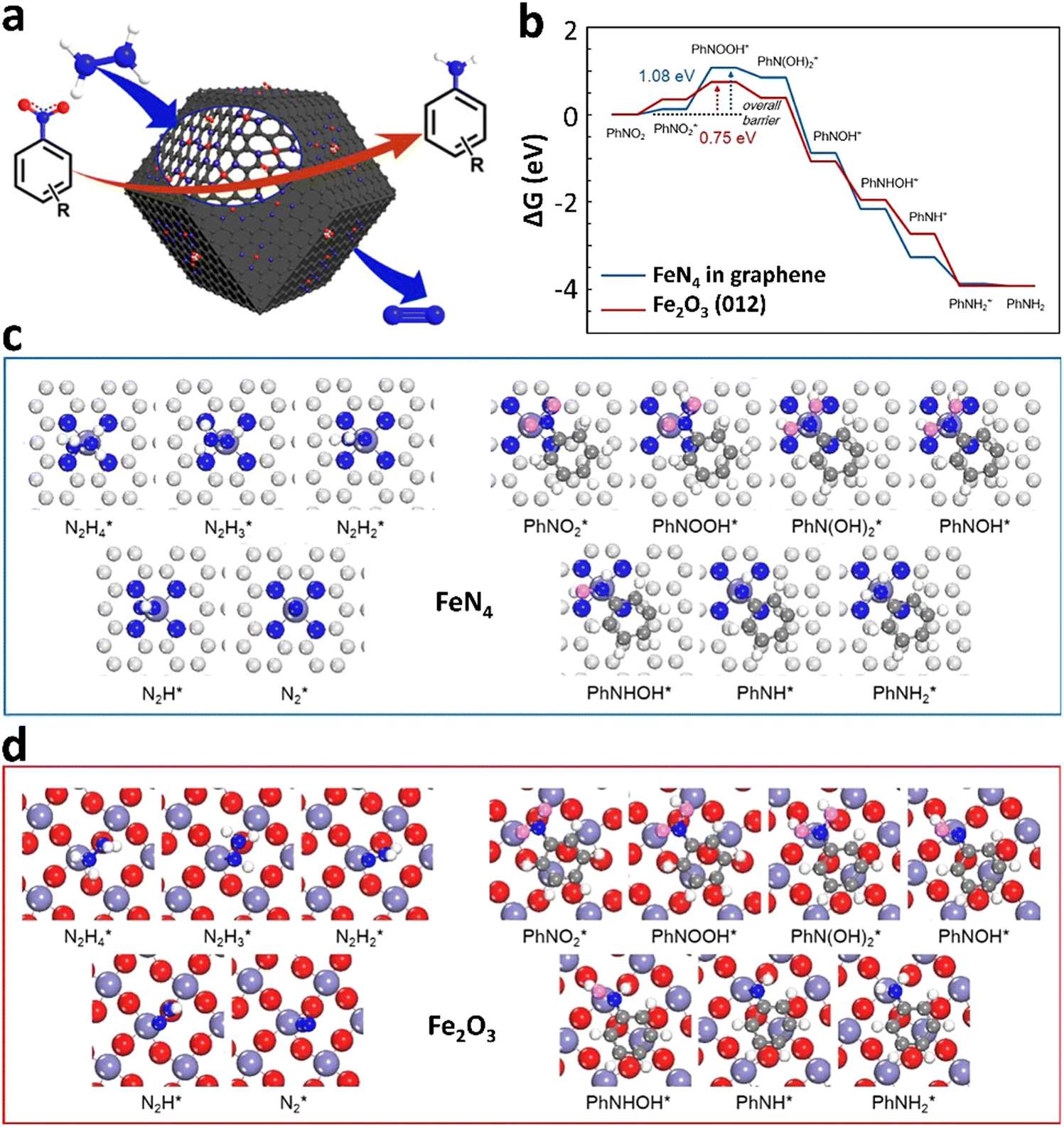 | ||
| Fig. 27 (a) Schematic diagram of the selective hydrogenation of nitrobenzene catalyzed by FeSAs/Fe2O3ACs/NPC. Gibbs free energy profiles for (b) nitrobenzene reduction processes on the FeN4 site embedded in graphene (lines in blue) and the Fe2O3(012) surface (lines in red), respectively. The dashed arrows indicate the overall barriers in the processes. Optimized structures of intermediates in hydrazine dehydrogenation and nitrobenzene reduction processes on the FeN4 site embedded in graphene (c) and Fe2O3(012) surface (d), respectively. Light gray: C in graphene; dark gray: C in molecules; white: H; red: O in Fe2O3; pink: O in molecules; blue: N; and purple: Fe. Reprinted with permission from ref. 256. Copyright 2020, the American Chemical Society. | ||
Similarly, Tian et al.258 synthesized atomically dispersed Fe metal sites supported on nitrogen-doped carbon for the selective hydrogenation and transfer hydrogenation of nitrobenzene. The catalytic performance for the selective hydrogenation and transfer hydrogenation of nitrobenzene to aniline was investigated using H2 and isopropanol as the hydrogen sources. When the temperature was set at 160 °C, the conversion and selectivity of nitrobenzene were higher than 99% with H2 as the hydrogen source. When the temperature was 220 °C, using isopropanol as the hydrogen source, the conversion and selectivity of nitrobenzene transfer hydrogenation both reached 99%. DFT calculations further revealed that the Fe-SACs exhibited an excellent catalytic performance for selective hydrogenation at lower temperature mainly owing to their efficient activation of reactants and intermediates. The increase in temperature can make Fe-SACs easily overcome the reaction energy barrier of isopropanol dehydrogenation, thereby improving the catalytic efficiency of its transfer hydrogenation.
The generation of H2 fuel by photocatalytic water splitting is a competitive and sustainable energy route.273,274 Generally, this process requires extremely active and stable light-harvesting semiconductors to meet the stringent requirements of suitable band positions and fast interfacial charge transfer processes. Accordingly, Zhang et al.267 synthesized atomically dispersed Fe integrated in porous coiled graphitic carbonitride (Fe@g-C3N4) for inducing ultra-high solar photon-driven hydrogen production. Fe@g-C3N4 had an excellent photocatalytic hydrogen production rate (3390 μmol h−1 g−1) and a good apparent quantum efficiency (6.89%) at 420 nm. The DFT calculation suggested that the presence of Fe atoms can optimize the electron/band structure of the g-C3N4 semiconductor, which helped to expand the light absorption range and shift the conduction band up to enhance the reducibility. Consequently, the separation efficiency of photoexcited electron–hole pairs improved. Simultaneously, g-C3N4 can shift its d orbital position to the Fermi level to affect the electronic structure of Fe, which not only can afford active sites, but also accelerate the interfacial charge transfer process, thereby improving its catalytic efficiency.
In addition to the above-mentioned nitrogen fixation methods, there are other ways to reduce N2 to NH3, such as N2 photo-fixation. Fu et al.275 reported the use of a combination of triphenylphosphine (PPh3) and sodium iodide (NaI) as photosensitizers for the catalysis of small organic molecules in the alkylation of silyl enol ethers. Inspired by this catalytic process, Hou et al.276 attempted to adsorb PPh3 on activated carbon and coupled it with a single-atom iron catalyst to form Fe1/C-PPh3/NaI for the heterogeneous catalysis of N2 photoimmobilization. Fe1/C-PPh3/NaI exhibited excellent catalytic activity in the heterogeneous catalysis process of N2 photoimmobilization, and the yield of NH3 was as high as 98 μmol (gcat h)−1, which was 1.5 times that of NH3 when [Ru(bpy)3]Cl2 was used as the photosensitizer.277 It can be found that under light conditions, PPh3/NaI as a photosensitizer can transfer hot electrons to Fe atoms through activated carbon, and then generated PPh3–I˙ radicals and oxidize H2O to O2. Meanwhile, hot electrons will reduce N2 to NH3 on Fe atoms (Fig. 28). NO3− as one of the most common pollutants can be used as a nitrogen source to synthesize NH3, which not only can treat environmental pollution, but also produce NH3, the most basic chemical product.278 It should be noted that the conversion of NO3− to NH3 involved multiple electronic reactions and reaction pathways, and thus understanding the reaction mechanism was extremely important for the selection of suitable catalysts.279 The porphyrin group of chlorophyll exhibits unique photosensitivity, and the Fe atom in nitrogenase can be used as a binding site to combine with N2 through an anti-bond. Inspired by this, Shang et al.280 synthesized a porphyrin-based metal–organic framework-based single-atom Fe catalyst for the photocatalytic synthesis of NH3. The NH3 yield (635 μg gcat.−1) and production rate (127 μg h−1 gcat.−1) increased by 82% and 50%, respectively. Alternatively, Wu et al.281 achieved the photocatalytic immobilization of nitrogen by coordinating photocatalytic N2 hydrogenation and water oxidation through a doping strategy. Both the experimental results and DFT results indicated that photoinduced hole-trapping polarons were formed on the Fe dopants, resulting in a high content of Fe(IV) species and promoting N2 hydrogenation on the adjacent oxygen vacancies.
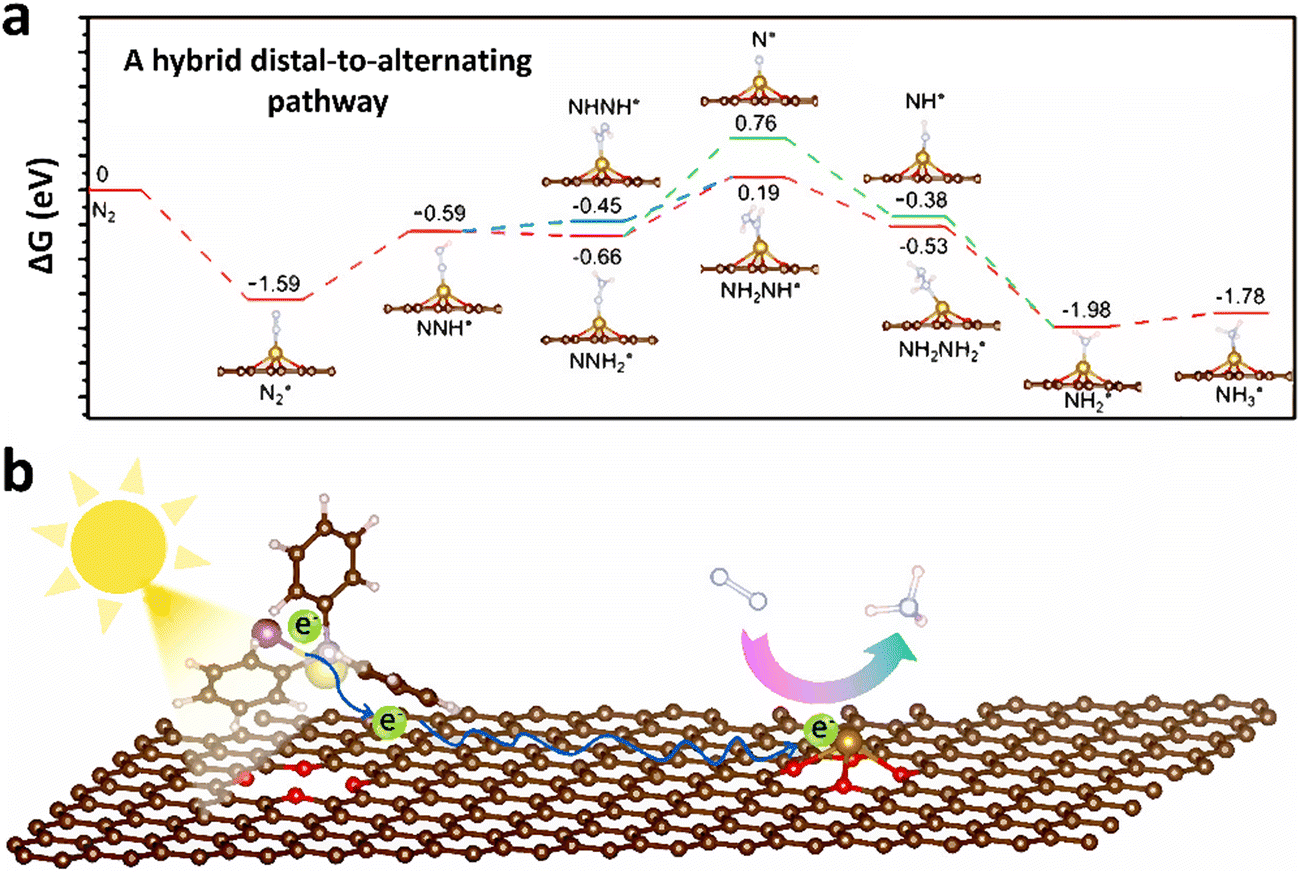 | ||
| Fig. 28 (a) Optimized reaction path for the dissociation of N2 to NH3 over Fe1O4/C. *Represents the adsorbed sites. (b) Schematic diagram of the process of N2 photofixation over Fe1/C-PPh3/NaI. Reprinted with permission from ref. 276. Copyright 2020, the American Chemical Society. | ||
Cumulative studies support that Fe-SACs can also be used for other photocatalytic reactions, such as organic synthesis, activation of C–H bonds and degradation of organic materials.282–285 In a typical experiment, Wen et al.283 synthesized a biomimetic photocatalyst with a single-atom iron site for organic synthesis by coupling carbon nitride with hemin via a chemical cross-linking method. Under the irradiation of visible light, it could sulfonate olefins to form β-ketosulfones (94% yield) under normal temperature and pressure. More interestingly, under near-infrared light, it can be used for the sulfonation reaction of androstenone. This was mainly because carbon nitride can enhance the separation ability of photogenerated electron pairs and holes, thereby improving the activity and stability of photocatalysts. Xiao et al.282 synthesized a single Fe atom aggregated on carbon nitride for the photocatalytic activation of O2 to generate the superoxide anion (O2˙−) to promote the activation of α-C–H bond of ethylbenzene to form acetophenone with higher conversion and selectivity than 99%. DFT calculations showed that a single Fe atom coordinates with 4 N atoms to form a low-spin Fe–N4 active center, which provides an adsorption site for O2 and allows the electrons generated in carbon nitride to transfer to the adsorbed O2, producing O2˙−. Zhao et al.285 synthesized a single-atom photocatalyst with Fe–N4 active sites and used it for the catalytic activation of peroxomonosulfate and sulfamethoxazole degradation. Under visible light, 98.7% of sulfamethoxazole with an initial concentration of 10 mg L−1 was degraded using 50 mg mL−1 of catalyst. The photogenerated electrons accelerated the cycle of triple bond Fe(II)/triple bond Fe(III), and the photogenerated holes further boosted the generation of 1O2.
4.2. Applications of Fe-SACs in biocatalysis
Enzymatic reactions are an indispensable process in metabolism, where enzymes play a crucial role as the catalyst.286 Essentially, enzymes possess the advantages of fast reaction rate under mild conditions, unique specificity and adaptability, making them widely used in disease diagnosis and treatment.287 Unfortunately, natural enzymes also have some inherent drawbacks, e.g., high cost, poor stability, easy to inactivate, difficulty in recovery and storage, and poor selectivity.89,288 In 2007, Perrett et al. first discovered the peroxidase (POD)-like activity of Fe3O4 nanoparticles, and since then a series of nanomaterials that can functionally mimic the catalytic properties of natural enzymes (called nanozymes) have been significantly developed (Fig. 29).289 Nanozymes have the characteristics of natural enzymes and nanomaterials including low cost, high stability, facile storage, adjustable activity and easy mass production, which enable them to display impressive application prospect.290,291 In fact, compared with natural enzymes, the selectivity and activity of nanozymes are not perfect, while their catalytic activity is not as high as expected owing to the low density of active sites.292 The uneven surface and geometric structures of nanozymes led to a complicated catalytic mechanism. It is difficult to establish the relationship between actual catalytically active sites and catalytic activity, further hindering the application of nanozymes.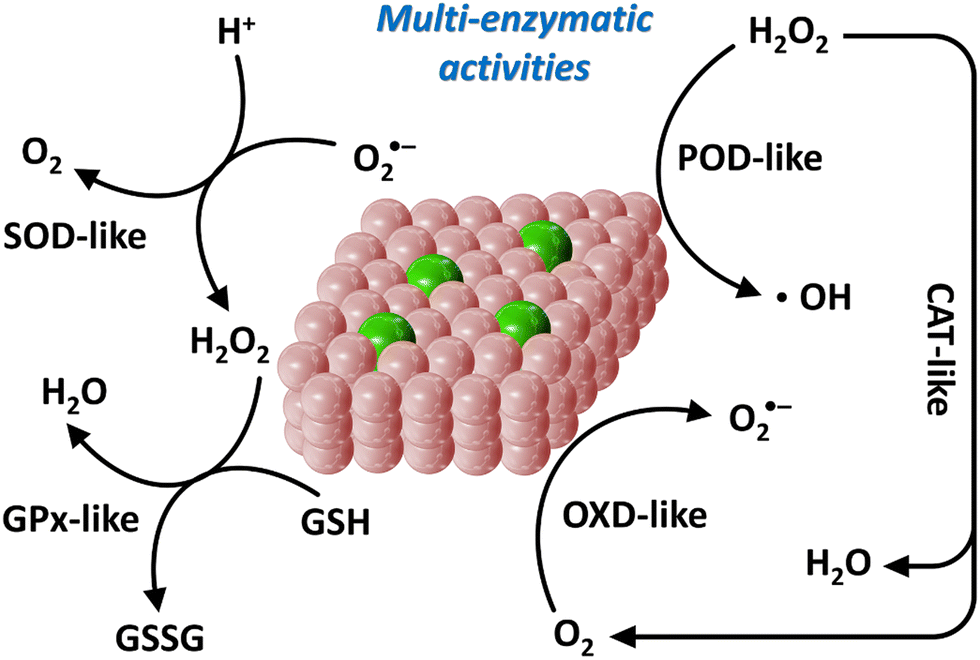 | ||
| Fig. 29 Single-atom iron catalysts functionally mimicking several different antioxidant enzymes. | ||
To bridge the huge gap between the catalytic performances natural enzymes and nanozymes, single-atom nanozymes (SAzymes) have been recently developed, showing very impressive catalytic activity and selectivity.293,294 More interestingly, the uniformity of the active sites and geometric structures are conducive to the accurate identification and characterization of active sites in catalytic reactions, and also helps to understand the catalytic mechanism of SAzymes.287 Experiments and theory suggest the similar active sites (M–Nx) of SAzymes to natural enzymes, especially in terms of geometric, chemical and electronic structures.292 For example, the active site of horseradish peroxidase (HRP) has a single heme b cofactor with a proximal ligand.295 Notably, this molecular structure is consistent with the structure of the Fe–N4 active centers in SAzymes. By clarifying the relationship between the structures and activity of natural enzymes, continued interdisciplinary efforts have been devoted to designing SAzymes with the same activity or even better activity than natural enzymes for large-scale production, showing a great application prospect in the field of biocatalysis.89,296,297
The working strategy underlying chemodynamic therapy is to initiate a Fenton reaction to convert H2O2 into a strong oxidizing reactive oxygen radical (˙OH), which then induces cell tumor apoptosis.302,303 Compared with general nanozymes, the higher atom utilization and selectivity of Fe-SACs enable them to more easily catalyze the oxidation of H2O2 to generate strong oxidative ˙OH, which is widely used in tumor therapy. In a typical experiment, He et al. studied the therapeutic effect of Fe-SACs on the activity of lung cancer cells. As shown in Fig. 30a, in the presence of oxygen, Fe-SACs could significantly reduce the number of lung cancer cells, which was observed by fluorescent imaging. The killing rate of Fe-SACs on lung cancer cells reached 88% (Fig. 30b and c). These results all proved that Fe-SACs can generate active oxygen by activating oxygen to induce cancer cell apoptosis. In the presence of oxygen, 3O2 can be adsorbed on the active Fe–N4 sites, thereby changing its electronic structure by activating 3O2 to produce 1O2. Then, the protons and electrons from the carrier surface were adsorbed to form HO2·−, and finally produce ROS to induce cancer cell apoptosis.86 Moreover, Shi's group applied Fe-SACs with POD-like activities to catalyze H2O2 to produce strong oxidizing ˙OH species under acidic condition. The in vivo and in vitro study demonstrated that Fe-SACs could not only kill tumor cells, but also induced the accumulation of lipid peroxides, hence causing tumor cell ferroptosis. The synergy of these two effects can play a key role in tumor treatment.304 Notably, Fe-SACs can also serve as catalase (CAT)-like enzymes and photosensitizers to yield reactive oxygen radicals under light irradiation, inducing the apoptosis of cancer cell.296 In meantime, the favorable biodegradability and biocompatibility of Fe-SACs guarantee their desirable biosafety both in vivo and in vitro. NOx is a transmembrane complex enzyme with 6 subunits, which is mainly expressed in immune cells. The intracellular subunit of the complex enzyme can convert NADPH to NADP+, transfer electrons to the gp91phox subunit of the cell membrane through the transmembrane region and use its heme active site to convert O2 molecules into superoxide free radicals. A lack of NOx may lead to immunodeficiency and cause chronic granulomatous disease (CGD). Recently, Wu et al. designed and synthesized Fe–N-doped two-dimensional graphene materials based on the structural characteristics of NOx active sites and electron transport (FeNGR). Their studies confirmed that FeNGR can efficiently convert NADPH to NADP+, with the generation of oxygen free radicals simultaneously. The conversion rate was as high as 93%. Compared with protease, FeNGR was more stable and could maintain 85% catalytic activity in extreme reaction systems (pH = 1–12, temperature 4–90 °C, and 0–100% organic solvent). In addition, FeNGR could replace the biological function of NOx and catalyze the generation of superoxide radicals in NOx-deficient cells, further activating the downstream immune signaling molecules.305
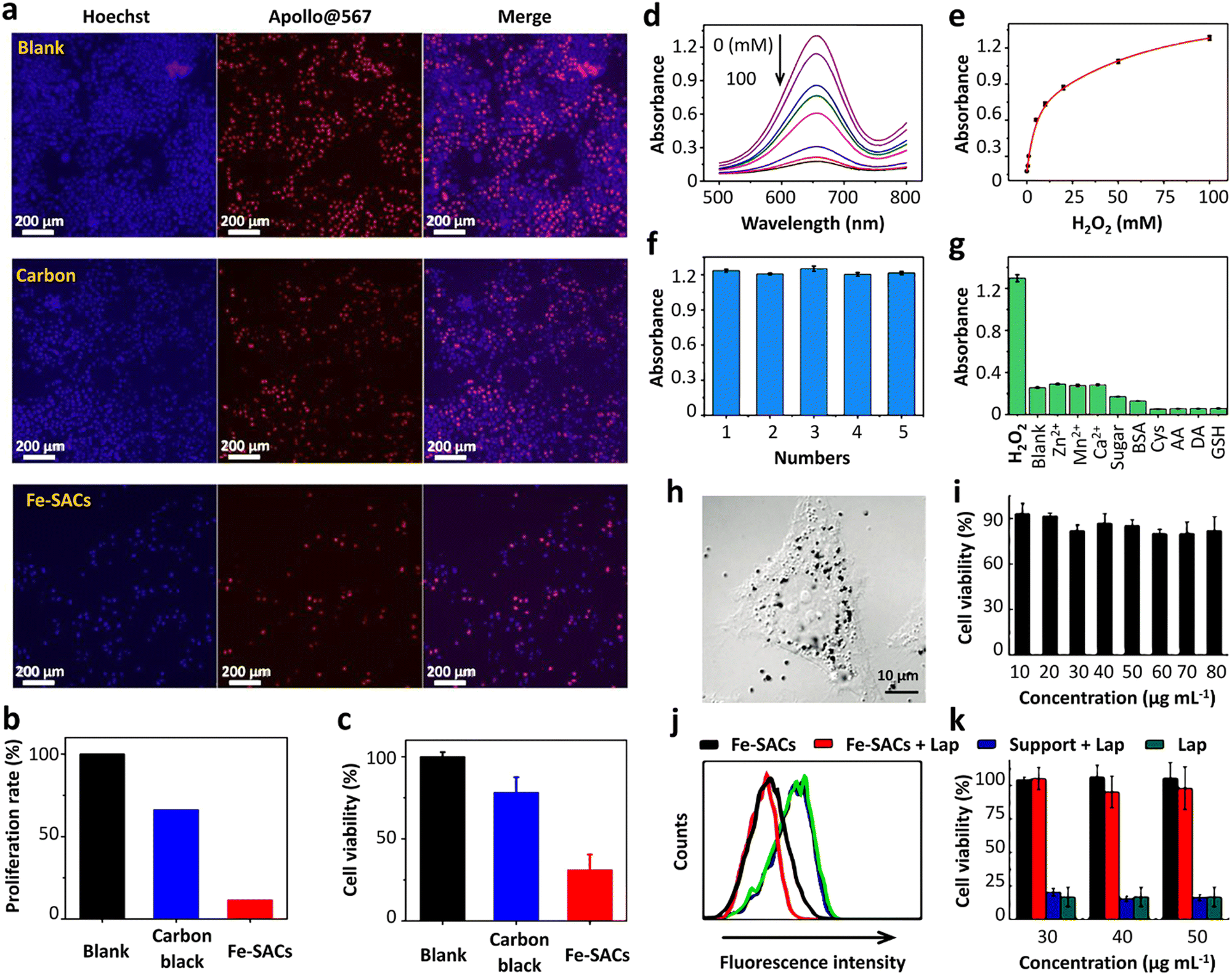 | ||
| Fig. 30 Applications of Fe-SACs in biocatalysis. (a) Fluorescence microscopy images of lung cancer cells treated with and without carbon black and Fe-SACs for 24 h. (b) Proliferation rate and (c) MTT assay of lung cell viability incubated with and without carbon black and Fe-SACs. Reprinted with permission from ref. 86. Copyright (2021), the American Chemical Society. (d) UV-vis absorption spectra of TMB oxidized by Fe-SACs with different concentrations of H2O2 and (e) corresponding calibration curve. (f) Reproducibility and (g) selectivity of H2O2 detection by Fe-SACs. Reprinted with permission from ref. 308. Copyright (2019), the American Chemical Society. (h) Confocal laser scanning microscopy image of HeLa cell with Fe-SACs. (i) Cytotoxicity of Fe-SACs. (j) Intracellular ROS scavenging by Fe-SACs in β-Lap-treated cancer cells using flow cytometry. (k) Effects of different concentrations of Fe-SACs on the viability of β-Lap-treated cancer cells. Reprinted with permission from ref. 286. Copyright (2019), The Royal Society of Chemistry. | ||
Recently, Fe-SACs have been reported to show POD-like activity comparable to that of HRP, which can catalyze H2O2 to yield ˙OH, and then oxidize the colorless substrate TMB to the blue oxidation product oxTMB.308,309 By elevating the H2O2 concentration, the absorbance of the product at 652 nm also gradually increased with a linear relationship (Fig. 30d and e). Interestingly, Fe-SACs also have excellent durability and anti-interference properties (Fig. 30f and g). Taking advantage of the POD-like catalytic activity of Fe-SACs, colorimetric sensing can be further extended to detect ascorbic acid, butyrylcholinesterase and acetylcholine.53,310,311 Of course, the oxidase-like activity of Fe-SACs is another working principle for colorimetric sensing. For example, Wu et al.312 confirmed the feasibility of Fe-SACs with oxidase-like activity for the indirect detection of acetylcholinesterase. Chen et al.313 employed the oxidase-like activity of Fe-SACs to evaluate the contents of alkaline phosphatase.
Besides the aforementioned colorimetric sensing by the enzyme-like activity of Fe-SACs, the detection of substances can be performed by using electrochemical, electrochemiluminescence and fluorescence methods.314 For example, Gu et al.315 introduced as-prepared Fe-SACs in an electrochemiluminescence system to improve the luminescence signal. Wang et al.316 used the fluorescence method to detect acetylcholinesterase by Fe-SACs with POD-like activity.
CAT catalyzes the decomposition of H2O2 to produce O2, hence eliminating ROS, and is used as an oxidative stress relief agent to protect normal tissues and cells. SOD can eliminate ROS by catalyzing the disproportionation of O2˙− to H2O2 and O2. Ma et al. reported that Fe-SACs showed both CAT- and SOD-like antioxidant activities, and further evaluated their ability to remove ROS in cells. HeLa cells exhibited the efficient cellular uptake and accumulation of Fe-SACs in the cytosol, as evidenced by the black punctate of Fe-SACs inside HeLa cells (Fig. 30h). Cell viability studies indicated the minimal cytotoxicity of Fe-SACs (Fig. 30i). To verify the ROS removal ability of Fe-SACs, β-lapachone (β-Lap), which can be bioactivated by enzymes overexpressed in HeLa cells to produce massive ROS in short time, was added to the cell culture medium. 2′,7′-Dichlorofluorescein diacetate (DCFDA) was utilized to fluorescently stain the in situ formed ROS. Compared with the cells without β-Lap treatment, the fluorescence intensity of the cells treated by β-Lap increased 6.8 times (Fig. 30j). Surprisingly, when HeLa cells were pretreated in Fe-SACs, and then treated with β-Lap, no obvious fluorescence was observed, indicating that the ROS produced by β-Lap were eliminated by Fe-SACs. The above-mentioned results all supported that Fe-SACs had the ability to eliminate ROS, thereby protecting cells from oxidative stress and damage (Fig. 30k).286 Moreover, Fe-SACs were also found to show CAT-like and GPx-like activity, which can effectively eliminate ROS and control the H2O2 level in cells.89 In summary, Fe-SACs with multiple antioxidant enzyme activities are more effective than a single antioxidant enzyme in reducing the oxidative stress response of cells.321
Typically, in 2020, Fan et al. designed hollow porous carbon spheres co-doped with N and Fe (Fe/N-HCNs), which exerted multienzyme mimicking activities, such as POD-, OXD-, CAT- and SOD-like catalytic activities. To determine the therapeutic effect of Fe/N-HCNs on infectious inflammation, animal experiments were performed for wound healing of animal skin infections. On the 5th day, the scars of skin infection wound in the Fe/N-HCNs + H2O2 group were obviously reduced (Fig. 31a and b). On the 11th day, the scars disappeared and the wounds healed completely. Due to the OXD-like catalytic activity of Fe/N-HCNs, the animal wound was protected from bacterial infection and inflammation. Fig. 31c indicates that Fe/N-HCNs had good biocompatibility. To further validate the therapeutic effect of Fe/N-HCNs on animal skin infection wounds, hematoxylin–eosin (H&E) staining of the wounds 11th day later was conducted (Fig. 31d). A large number of inflammatory cells appeared in the control group. Importantly, when Fe/N-HCNs + H2O2 synergistic treatment was performed, optimal treatment effects were observed. The inflammatory cells disappeared quickly, which is associated with the healed skin infection wound. The above-mentioned data shows that Fe/N-HCNs can serve as a good anti-infective inflammation drug, which effectively treat inflammation caused by bacterial infection and promote wound healing. Furthermore, based on the SOD-like activity of Fe/N-HCNs, the therapeutic effect on non-infectious inflammation was monitored. The experimental results of colitis induced by dextran sodium sulfate (DSS) as a model are shown in Fig. 31e and f. Colon shortening is the most important feature of colitis. The colon after DSS treatment was significantly shortened, but after treatment using Fe/NHCNs, the colon became longer, revealing that Fe/NHCNs had an excellent therapeutic effect on colitis. After DSS treatment, compared with the control group, the tissue mucosal structure of the mice was severely damaged. As anticipated, Fe/NHCNs could restore the tissue mucosal structures of the mice to a certain extent and inflammatory cells had also been highly reduced. Together, the prominent multi-enzyme activities of Fe/N-HCNs served as an anti-inflammatory alternative for both infectious and noninfectious inflammation.326 Recently, Li et al. synthesized a single-atom nanoenzyme with FeN3P as the active center. The precise coordination of N and P affects the electronic structure of the Fe atom in the active center, and thus the as-obtained SACs exhibited excellent-POD-like enzyme activity for tumor treatment. To identify the excellent enzyme activity of the FeN3P-SAzyme, the authors also prepared an FeN4-SAzyme with only N coordination and Fe3O4 nanoparticles for comparison. The measured specific activity showed that the enzyme activity of FeN3P-SAzyme (316 U mg−1) was 10 times that of FeN4-SAzyme (33.8 U mg−1) and 30 times that of the Fe3O4 nanoparticles (9.12 U mg−1). Furthermore, the inhibitory effects of FeN3P-SAzyme and FeN4-SAzyme on cells showed concentration and time dependence (Fig. 32a and b), respectively, while the Fe3O4 nanoparticles basically had no inhibitory effect on the cells. After FeN3P-SAzyme was endocytosed by the cells, it finally entered into the lysosome (Fig. 32c and d). H2DCFDA was used as a fluorescent probe to detect ROS in HepG2 liver cancer cells treated by FeN3P-SAzyme. After the addition of GSH, the intracellular ROS was reduced, indicating that FeN3P-SAzyme had POD-like activity and can be used to inhibit tumor cells (Fig. 32e). The mouse experiment is shown in Fig. 32f and g. After using FeN3P-SAzyme to treat mouse tumors, the tumor growth of the mice was significantly inhibited.327
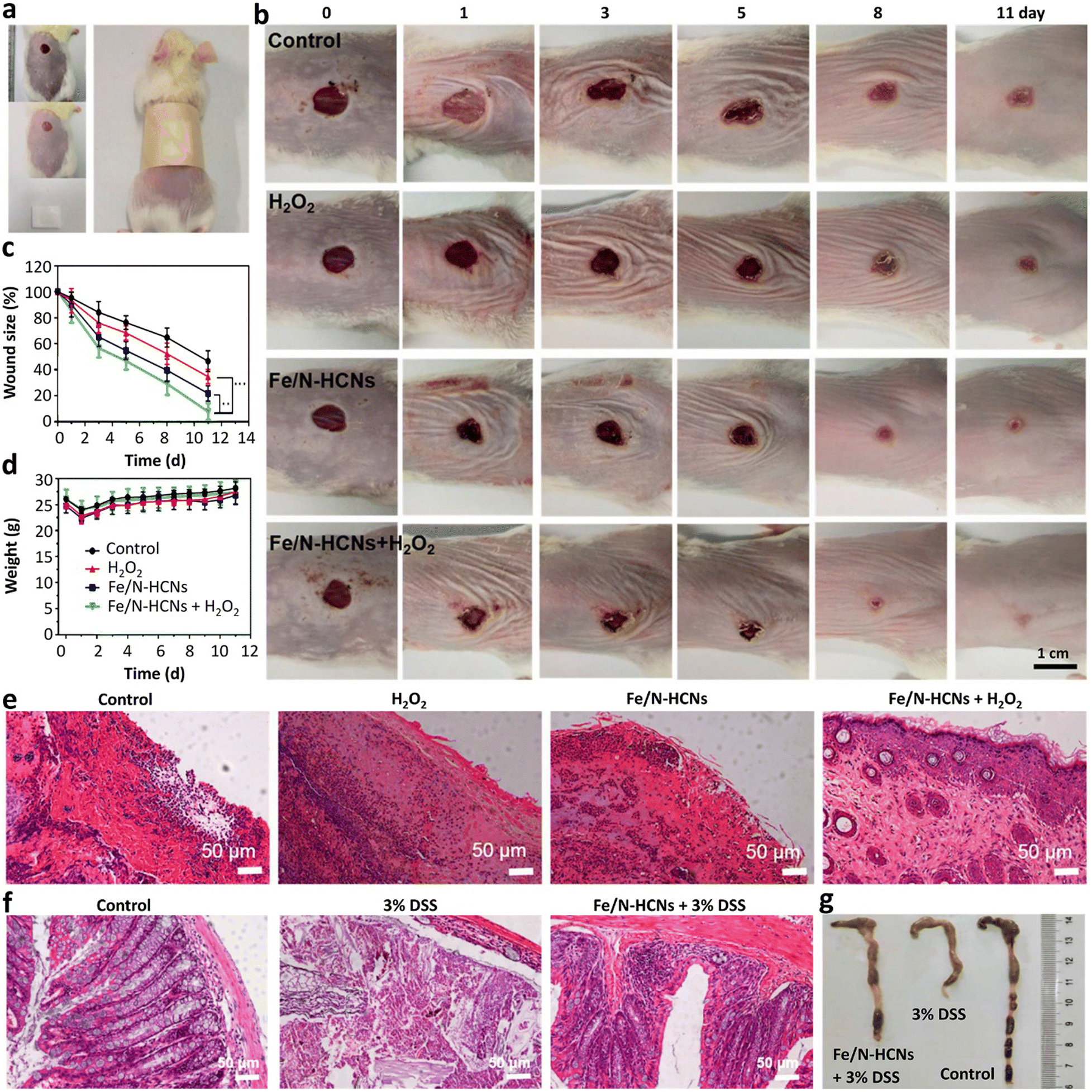 | ||
| Fig. 31 Applications of Fe-SACs for infected-wound healing. (a) Photographic images of the extent of wound healing. (b and c) Changes in wound size in the mice in each group on days 0, 1, 3, 5, 8, and 11. (d) Changes in body weight of the mice during wound healing in each group. (e) Hematoxylin and Eosin (H&E) staining wound sections from each group (n = 4). (f) H&E staining sections of colons. (g) Photo of the colons in each group. Reprinted with permission from ref. 326. Copyright (2020), the American Chemical Society. | ||
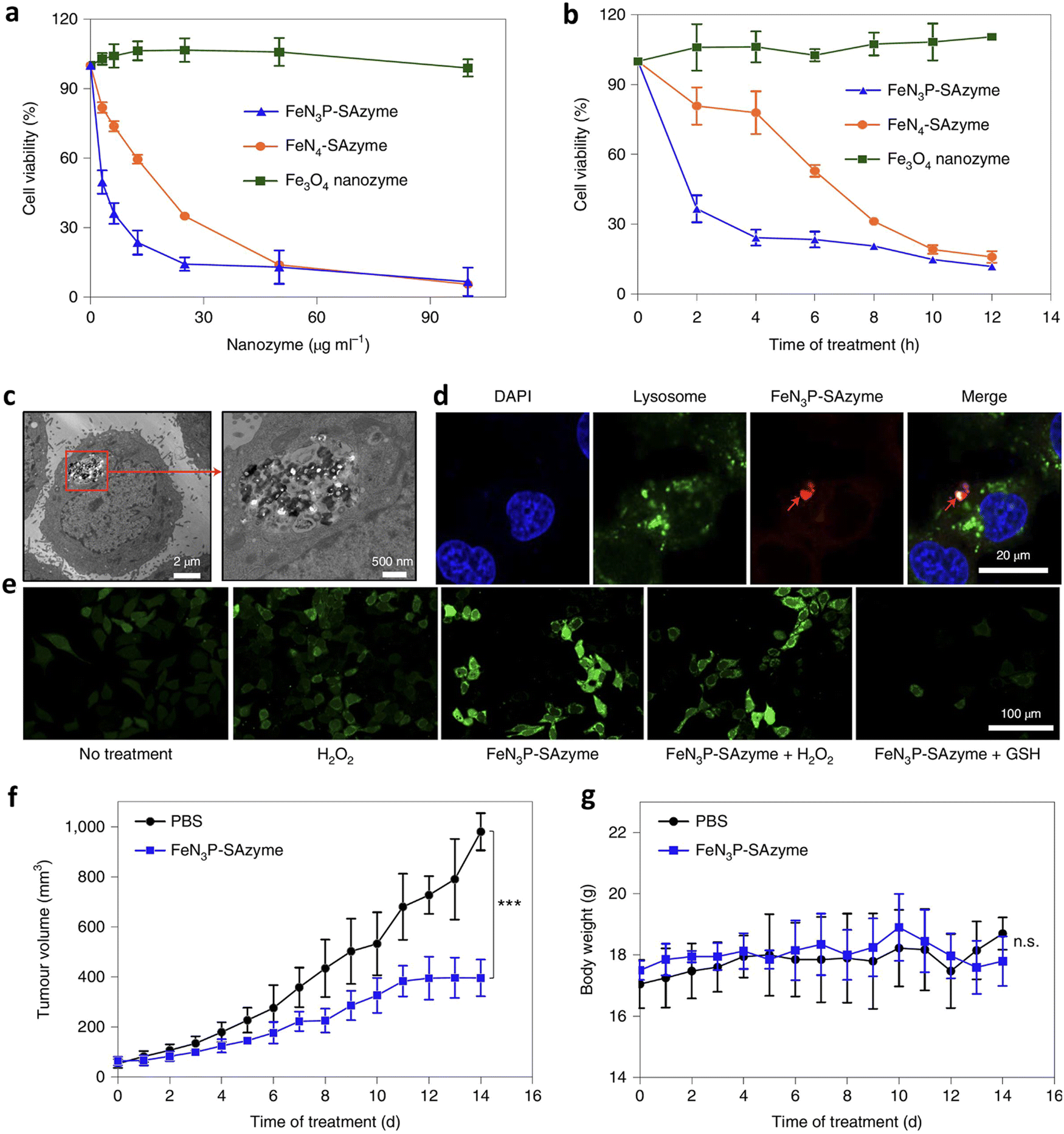 | ||
| Fig. 32 Applications of Fe-SACs for infected-wound healing. (a) Cell viability of HepG2 hepatoma cells after incubation with different concentrations of FeN3P-SAzyme, FeN4-SAzyme and Fe3O4. (b) SAzymes induced time-dependent cell death. (c) Visualization of the internalization of FeN3P-SAzyme in HepG2 tumour cells using TEM. (d) Confocal images of the intracellular uptake of FeN3P-SAzyme in lysosomes. (e) Intracellular oxidative species in HepG2 cells treated with FeN3P-SAzyme in the absence or presence of H2O2 or antioxidant glutathione (GSH). (f and g) Anti-tumour activity (f) and toxicity (g) of FeN3P-SAzyme. Reprinted with permission from ref. 327. Copyright (2020), Springer Nature Limited. | ||
The organic compounds in industrial wastewater pose a serious threat to humans. Hence, it is necessary to develop catalysts to degrade organic compounds. Among them, the most effective strategy is to use strong oxidants to remove difficult-to-degrade organics. ˙OH is an excellent strong oxidant, owing to its high oxidation potential of ∼2.8 V.303 Since Fe-SACs have been found to show POD-like activity, which catalyze H2O2 to generate a large amount of ˙OH, Fe-SACs in principle efficiently remove organic pollutants. For example, the Fe-SACs designed by An et al. in the presence of H2O2 had removal rates of 96%, 98% and 98% for methylene blue, rhodamine B and phenol, respectively.87 Fe-SACs also catalyzed the dehydrogenation of diphenylhydrazine to azobenzene with a selectivity of 100% and yield of 75.3%.86 Meanwhile, the POD-like activity of Fe-SACs enabled them to initiate Fenton reaction in the presence of H2O2, therefore effectively degrading phenol and other phenol aqueous solutions with a removal rate of ∼83%.329
In the production of fine chemicals, epoxidation occupies an important position, and selecting a suitable catalyst (e.g., single-atom catalysts) is particularly important. In a typical experiment, Tian et al.330 synthesized a highly dispersed Fe2 cluster via a wet-chemical strategy and used it for trans-stilbene epoxidation with a high conversion rate of 91% and selectivity of 93%. Wen et al.283 designed biomimetic photocatalysts with single-atom Fe active sites by chemical cross-linking. Under the irradiation of visible light or near-infrared light, the catalyst could promote the sulfonation of olefins to form β-ketosulfones with a yield of up to 94%. Obviously, Fe-SACs have other applications, such as air filtration systems, preventing the emission of volatile gases and electrochemical organic conversion.331 In the future development, Fe-SACs can be more widely applied in the fields mentioned above.
5. Conclusion and outlook
Since Zhang et al. proposed the concept of SACs in 2011, SACs have attracted considerable interest as industrial catalysts and therapeutic nanozymes.2 In recent years, the development of SACs, especially Fe-SACs, promises to inherit the main advantages of homogeneous catalysts (isolated active site), heterogeneous catalysts (good stability and facile to separate) and natural enzymes (excellent activity and selectivity). These advantages make Fe-SACs shine in the fields of electrochemical catalysis, in which the larger energy barriers for electrocatalytic reactions (e.g., ORR, OER, HER, CO2RR and NRR) will be clearly reduced. Interestingly, the application of Fe-SACs in catalytic biomedicine has emerged. The active center (e.g., Fe–Nx species) of Fe-SACs shows a similar structure to the catalytically active center of metalloprotease, thus offering huge potential for achieving superior catalytic activity and selectivity to functionally mimic or even outperform natural enzymes. Multiple lines of evidence support the multi-enzymatic (POD-, OXD-, CAT- and SOD-like) activities of Fe-SACs, which have a greater application prospect in the biomedical domains of biosensing, wound disinfection, cancer treatment and oxidative stress cytoprotection.However, despite the significant progress achieved, the majority of synthetic routes and application of Fe-SACs are still in the laboratory stage. In our opinion, there is still plenty of work to be done to overcome the major challenges in the design, synthesis, characterization and application of Fe-SACs (Fig. 33). These challenges are as follows:
 | ||
| Fig. 33 Prospects and future challenges of Fe-SACs, ranging from design, synthesis, and characterization to applications. | ||
(1) One inevitable technological challenge to engineer SACs towards practical application is developing cost-effective and green synthetic processes for SACs. For the preparation of Fe-SACs, the energy consumption is always large. Thus far, most Fe-SACs are ultimately prepared via high-temperature (700–1000 °C) pyrolysis treatment. This high-temperature procedure requires a high energy input, which makes the use of Fe-SACs difficult in large-scale industrial production and hinders their further applications.
(2) Engineering of the composition, crystal phase (if applicable) and architecture of support materials and manipulating the metal–support interactions are essential to optimize the catalytic properties of Fe-SACs. However, integrating all these parameters into a limited nano/micro-scale space of support materials has not yet to be fully achieved due to the currently inadequate design strategies and techniques available for the synthesis of Fe-SACs. Considering the accumulating evidence, in addition to the chemical compositions of support materials, their uniform cavity and ordered structure significantly contribute to the spatial distribution of single Fe atoms. Firstly, an ideal support material should possess a large specific surface area, making it more convenient to obtain charge transport and electrolyte penetration in the deep active centers of the catalyst surface. Secondly, the tailored topological defects and appropriate crystallinity of support materials will be greatly beneficial for increasing the electron density and conductivity of SACs, as well as further activating the inactive single Fe atoms. This is where natural organisms provide a rich source of inspiration for the innovative synthesis methods of support materials. Nature abounds with intricate hierarchical textures consisting of amorphous and crystalline hybrid phases, which synergistically intertwine to offer extraordinary functions, e.g., enhanced surface area, superior mechanical integrity and high transport rate.332,333 Notably, these features are an integrated embodiment for high-performance electrochemical catalytic pathway. Other examples of natural evolution-inspired strategies are the use of pre-formed solid intermediates as starting phases, which can enable the bottom-up creation of smart materials with robust functionality compared to the substances found in nature. To date, a representative example is that the magnetosome of magnetotactic bacteria always exploits solid, poorly crystalline ferrihydrite intermediates as precursors in the biomineralization of stoichiometric and structurally pure Fe3O4 crystals.334 The synthesis of bioinspired materials via the solid phase is an alternative approach to manufacture a variety of sophisticated support materials with precise control over their chemical composition, crystal structure, size and morphology by means of biomolecular templates.
(3) Functionally similar to natural enzymes, the design and synthesis of Fe-SACs enabling precise control over a high metal loading and single-atomic dispersion are a challenging yet highly rewarding endeavor, possibly resulting in long-awaited industrial breakthrough in utilizing catalysts. The catalytic activity of Fe-SACs is directly related to the loading amounts of single-atom Fe metals on the support material. However, Fe-SACs usually suffer from a low-loading of Fe atoms (generally less than 10%), which is inherent to the classical material-design dilemma for Fe-SACs, namely, the high Fe loading and single-atomic dispersion (the two typical characters) tend to be mutually exclusive. Because single metal atoms inherently display a huge specific surface energy, they easily undergo migration, agglomeration and sintering during their preparation and catalytic reactions, hence deteriorating the overall catalytic activities of Fe-SACs. However, Wang et al.335 recently synthesized a series of single-atom catalysts with a high loading by calcining coordination polymers at high temperature, and the synthesis reached the gram scale. Interestingly, this method was suitable for a variety of metal atoms, including Fe (30.0 wt%), Ni (21.6 wt%), Cu (22.4 wt%), Zn (21.1 wt%), Ru (13.5 wt%), Rh (3.5 wt%), Pd (3.8 wt%), Pt (3.2 wt%) and Ir (4.4 wt%). Therefore, this high loading is expected to shine in the field of industrial catalysis.
(4) From the point of view of practical applications, how to avoid the poisoning of Fe-SAC catalysts remains a significant challenge. Unlike common catalysts, SACs will be completely deactivated if the intermediates or by-products are firmly combined with SACs. Generally, it is difficult for active intermediates to desorb from single metal atoms, owing to the high binding energy between them. Therefore, it is worth further studying to regenerate the isolated metal active sites for the recycling use of Fe-SACs.
(5) The identification of the relationship between the internal structure and catalytic activity of Fe-SACs is particularly important for the rational synthesis of catalysts, which requires more advanced characterization tools to observe the atomic active sites. Beyond the modern characterization techniques that have been utilized, new-generation convergent probes with a quicker detection rate and better temporal and spatial resolution should be developed to record instantaneous variations in the atomic active centers on the carrier. Furthermore, advanced instruments in other research fields including cryogenic transmission electron microscopy, which has evolved into an indispensable method for the characterization of structural biology, can be introduced in SAC-related fields. These new techniques are pivotal to prevent the collapse of the support skeleton and original single Fe atom aggregation during the characterization of Fe-SACs.
Considering aforementioned fundamental and technical hurdles to be settled, Fe-SACs have recently emerged as an interdisciplinary research field from chemistry, physics, catalytic science and biomedicine. Notably, the research on Fe-SACs is still in its infancy and further development of this field calls for more efforts. We expect that significant and promising directions would continue to emerge well beyond the outlook described herein.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51803161), the Natural Science Foundation of Hubei Province, China (2021CFB344), the Young Top-notch Talent Cultivation Program of Hubei Province, China, and the Shanghai Municipal Science and Technology Major Project.References
- M. A. Roberts, G. Sankar, J. M. Thomas, R. H. Jones, H. Du, J. Chen, W. Pang and R. Xu, Nature, 1996, 381, 401–404 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- F. Li, G.-F. Han, H.-J. Noh, S.-J. Kim, Y. Lu, H. Y. Jeong, Z. Fu and J.-B. Baek, Energy Environ. Sci., 2018, 11, 2263–2269 RSC.
- Y. Qu, L. Wang, Z. Li, P. Li, Q. Zhang, Y. Lin, F. Zhou, H. Wang, Z. Yang, Y. Hu, M. Zhu, X. Zhao, X. Han, C. Wang, Q. Xu, L. Gu, J. Luo, L. Zheng and Y. Wu, Adv. Mater., 2019, 31, 1904496 CrossRef CAS PubMed.
- J. Li, H. Liu, M. Wang, C. Lin, W. Yang, J. Meng, Y. Xu, K. A. Owusu, B. Jiang, C. Chen, D. Fan, L. Zhou and L. Mai, Chem. Commun., 2019, 55, 334–337 RSC.
- X. Ge, P. Zhou, Q. Zhang, Z. Xia, S. Chen, P. Gao, Z. Zhang, L. Gu and S. Guo, Angew. Chem., Int. Ed., 2019, 59, 232–236 CrossRef PubMed.
- L. Kuai, Z. Chen, S. Liu, E. Kan, N. Yu, Y. Ren, C. Fang, X. Li, Y. Li and B. Geng, Nat. Commun., 2020, 11, 48 CrossRef PubMed.
- Q. Liu, J. Wang, J. Zhang, Y. Yan, X. Qiu, S. Wei and Y. Tang, Catal. Sci. Technol., 2020, 10, 450–457 RSC.
- C. Zhu, S. Fu, J. Song, Q. Shi, D. Su, M. H. Engelhard, X. Li, D. Xiao, D. Li, L. Estevez, D. Du and Y. Lin, Small, 2017, 13, 1603407 CrossRef PubMed.
- W. Chen, J. Pei, C.-T. He, J. Wan, H. Ren, Y. Zhu, Y. Wang, J. Dong, S. Tian, W.-C. Cheong, S. Lu, L. Zheng, X. Zheng, W. Yan, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 16086–16090 CrossRef CAS PubMed.
- C. Ling, X. Bai, Y. Ouyang, A. Du and J. Wang, J. Phys. Chem. C, 2018, 122, 16842–16847 CrossRef CAS.
- L. Han, X. Liu, J. Chen, R. Lin, H. Liu, F. Lue, S. Bak, Z. Liang, S. Zhao, E. Stavitski, J. Luo, R. R. Adzic and H. L. Xin, Angew. Chem., Int. Ed., 2019, 58, 2321–2325 CrossRef CAS.
- Y. Cheng, S. He, S. Lu, J.-P. Veder, B. Johannessen, L. Thomsen, M. Saunders, T. Becker, R. De Marco, Q. Li, S.-Z. Yang and S. P. Jiang, Adv. Sci., 2019, 6, 1802066 CrossRef PubMed.
- R. Jiang, L. Li, T. Sheng, G. Hu, Y. Chen and L. Wang, J. Am. Chem. Soc., 2018, 140, 11594–11598 CrossRef CAS PubMed.
- Q. Li, W. Chen, H. Xiao, Y. Gong, Z. Li, L. Zheng, X. Zheng, W. Yan, W.-C. Cheong, R. Shen, N. Fu, L. Gu, Z. Zhuang, C. Chen, D. Wang, Q. Peng, J. Li and Y. Li, Adv. Mater., 2018, 30, 1800588 CrossRef PubMed.
- F. Xiao, G.-L. Xu, C.-J. Sun, M. Xu, W. Wen, Q. Wang, M. Gu, S. Zhu, Y. Li, Z. Wei, X. Pan, J. Wang, K. Amine and M. Shao, Nano Energy, 2019, 61, 60–68 CrossRef CAS.
- J. Guo, J. Huo, Y. Liu, W. Wu, Y. Wang, M. Wu, H. Liu and G. Wang, Small Methods, 2019, 3, 1900159 CrossRef.
- A. Han, B. Wang, A. Kumar, Y. Qin, J. Jin, X. Wang, C. Yang, B. Dong, Y. Jia, J. Liu and X. Sun, Small Methods, 2019, 3, 1800471 CrossRef.
- Q.-L. Zhu, W. Xia, L.-R. Zheng, R. Zou, Z. Liu and Q. Xu, ACS Energy Lett., 2017, 2, 504–511 CrossRef CAS.
- J. Meng, X. Liu, C. Niu, Q. Pang, J. Li, F. Liu, Z. Liu and L. Mai, Chem. Soc. Rev., 2020, 49, 3142–3186 RSC.
- W.-H. Li, W.-H. Deng, G.-E. Wang and G. Xu, EnergyChem, 2020, 2, 100029 CrossRef.
- J. Wang, G. Han, L. Wang, L. Du, G. Chen, Y. Gao, Y. Ma, C. Du, X. Cheng, P. Zuo and G. Yin, Small, 2018, 14, 1704282 CrossRef PubMed.
- H. Huang, K. Shen, F. Chen and Y. Li, ACS Catal., 2020, 10, 6579–6586 CrossRef CAS.
- X. Chen, N. Wang, K. Shen, Y. Xie, Y. Tan and Y. Li, ACS Appl. Mater. Interfaces, 2019, 11, 25976–25985 CrossRef CAS PubMed.
- X. Wang, H. Zhu, C. Yang, J. Lu, L. Zheng and H.-P. Liang, Carbon, 2022, 191, 393–402 CrossRef CAS.
- Y. Chen, S. Ji, Y. Wang, J. Dong, W. Chen, Z. Li, R. Shen, L. Zheng, Z. Zhuang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 6937–6941 CrossRef CAS PubMed.
- C. Zhao, X. Dai, T. Yao, W. Chen, X. Wang, J. Wang, J. Yang, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 8078–8081 CrossRef CAS PubMed.
- V. Armel, S. Hindocha, F. Salles, S. Bennett, D. Jones and F. Jaouen, J. Am. Chem. Soc., 2017, 139, 453–464 CrossRef CAS PubMed.
- Y. Hou, M. Qiu, M. G. Kim, P. Liu, G. Nam, T. Zhang, X. Zhuang, B. Yang, J. Cho and M. Chen, Nat. Commun., 2019, 10, 1392 CrossRef PubMed.
- L. Jiao, G. Wan, R. Zhang, H. Zhou, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2018, 57, 8525–8529 CrossRef CAS PubMed.
- D.-L. Meng, C.-H. Chen, J.-D. Yi, Q. Wu, J. Liang, Y.-B. Huang and R. Cao, Research, 2019, 2019, 1768595 CAS.
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef.
- H. Wang, Z. Zeng, P. Xu, L. Li, G. Zeng, R. Xiao, Z. Tang, D. Huang, L. Tang, C. Lai, D. Jiang, Y. Liu, H. Yi, L. Qin, S. Ye, X. Ren and W. Tang, Chem. Soc. Rev., 2019, 48, 488–516 RSC.
- G. Lin, H. Ding, D. Yuan, B. Wang and C. Wang, J. Am. Chem. Soc., 2016, 138, 3302–3305 CrossRef CAS PubMed.
- X. Ao, W. Zhang, Z. Li, J.-G. Li, L. Soule, X. Huang, W.-H. Chiang, H. M. Chen, C. Wang, M. Liu and X. C. Zeng, ACS Nano, 2019, 13, 11853–11862 CrossRef CAS.
- J.-C. Li, P.-X. Hou and C. Liu, Small, 2017, 13, 1702002 CrossRef.
- S. M. J. Rogge, A. Bavykina, J. Hajek, H. Garcia, A. I. Olivos-Suarez, A. Sepulveda-Escribano, A. Vimont, G. Clet, P. Bazin, F. Kapteijn, M. Daturi, E. V. Ramos-Fernandez, F. X. Llabres i Xamena, V. Van Speybroeck and J. Gascon, Chem. Soc. Rev., 2017, 46, 3134–3184 RSC.
- X. Yan, H. Liu, Y. Li, W. Chen, T. Zhang, Z. Zhao, G. Xing and L. Chen, Macromolecules, 2019, 52, 7977–7983 CrossRef CAS.
- Y. Zeng, R. Zou, Z. Luo, H. Zhang, X. Yao, X. Ma, R. Zou and Y. Zhao, J. Am. Chem. Soc., 2015, 137, 1020–1023 CrossRef CAS PubMed.
- N. Huang, L. Zhai, D. E. Coupry, M. A. Addicoat, K. Okushita, K. Nishimura, T. Heine and D. Jiang, Nat. Commun., 2016, 7, 12325 CrossRef PubMed.
- Y. Zhu, W. Peng, Y. Li, G. Zhang, F. Zhang and X. Fan, Small Methods, 2019, 3, 1800438 CrossRef.
- J.-D. Yi, R. Xu, Q. Wu, T. Zhang, K.-T. Zang, J. Luo, Y.-L. Liang, Y.-B. Huang and R. Cao, ACS Energy Lett., 2018, 3, 883–889 CrossRef CAS.
- A. M. Evans, L. R. Parent, N. C. Flanders, R. P. Bisbey, E. Vitaku, M. S. Kirschner, R. D. Schaller, L. X. Chen, N. C. Gianneschi and W. R. Dichtel, Science, 2018, 361, 52–57 CrossRef CAS.
- T. Hasell and A. I. Cooper, Nat. Rev. Mater., 2016, 1, 16053 CrossRef CAS.
- P. Peng, L. Shi, F. Huo, S. Zhang, C. Mi, Y. Cheng and Z. Xiang, ACS Nano, 2019, 13, 878–884 CrossRef CAS PubMed.
- L. Wang, C. Xu, W. Zhang, Q. Zhang, M. Zhao, C. Zeng, Q. Jiang, C. Gu and Y. Ma, J. Am. Chem. Soc., 2022, 144, 8961–8968 CrossRef CAS PubMed.
- Z. Xiong, B. Sun, H. Zou, R. Wang, Q. Fang, Z. Zhang and S. Qiu, J. Am. Chem. Soc., 2022, 144, 6583–6593 CrossRef CAS PubMed.
- W. Zhang, L. Chen, S. Dai, C. Zhao, C. Ma, L. Wei, M. Zhu, S. Y. Chong, H. Yang, L. Liu, Y. Bai, M. Yu, Y. Xu, X.-W. Zhu, Q. Zhu, S. An, R. S. Sprick, M. A. Little, X. Wu, S. Jiang, Y. Wu, Y.-B. Zhang, H. Tian, W.-H. Zhu and A. I. Cooper, Nature, 2022, 604, 72–79 CrossRef CAS PubMed.
- J.-C. Li, M. Cheng, T. Li, L. Ma, X. Ruan, D. Liu, H.-M. Cheng, C. Liu, D. Du, Z. Wei, Y. Lin and M. Shao, J. Mater. Chem. A, 2019, 7, 14478–14482 RSC.
- Y. Li, W. Zhou, H. Wang, L. Xie, Y. Liang, F. Wei, J.-C. Idrobo, S. J. Pennycook and H. Dai, Nat. Nanotechnol., 2012, 7, 394–400 CrossRef CAS PubMed.
- Y. Ma, L. Sun, W. Huang, L. Zhang, J. Zhao, Q. Fan and W. Huang, J. Phys. Chem. C, 2011, 115, 24592–24597 CrossRef CAS.
- P. Chen, T. Zhou, L. Xing, K. Xu, Y. Tong, H. Xie, L. Zhang, W. Yan, W. Chu, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2017, 56, 610–614 CrossRef CAS PubMed.
- N. Cheng, J.-C. Li, D. Liu, Y. Lin and D. Du, Small, 2019, 15, 1901485 CrossRef CAS PubMed.
- J.-C. Li, Z.-Q. Yang, D.-M. Tang, L. Zhang, P.-X. Hou, S.-Y. Zhao, C. Liu, M. Cheng, G.-X. Li, F. Zhang and H.-M. Cheng, NPG Asia Mater., 2018, 10, e461–e461 CrossRef CAS.
- D. Liu, J.-C. Li, Q. Shi, S. Feng, Z. Lyu, S. Ding, L. Hao, Q. Zhang, C. Wang, M. Xu, T. Li, E. Sarnello, D. Du and Y. Lin, ACS Appl. Mater. Interfaces, 2019, 11, 39820–39826 CrossRef CAS PubMed.
- Y. J. Sa, D.-J. Seo, J. Woo, J. T. Lim, J. Y. Cheon, S. Y. Yang, J. M. Lee, D. Kang, T. J. Shin, H. S. Shin, H. Y. Jeong, C. S. Kim, M. G. Kim, T.-Y. Kim and S. H. Joo, J. Am. Chem. Soc., 2016, 138, 15046–15056 CrossRef CAS PubMed.
- Y. Wang, J. Mao, X. Meng, L. Yu, D. Deng and X. Bao, Chem. Rev., 2019, 119, 1806–1854 CrossRef CAS PubMed.
- Y. Oh, J. O. Hwang, E.-S. Lee, M. Yoon, V.-D. Le, Y.-H. Kim, D. H. Kim and S. O. Kim, ACS Appl. Mater. Interfaces, 2016, 8, 25438–25443 CrossRef CAS PubMed.
- X. Wang, X. Chen, A. Thomas, X. Fu and M. Antonietti, Adv. Mater., 2009, 21, 1609 CrossRef CAS.
- Y. Zheng, Y. Jiao, Y. Zhu, Q. Cai, A. Vasileff, L. H. Li, Y. Han, Y. Chen and S.-Z. Qiao, J. Am. Chem. Soc., 2017, 139, 3336–3339 CrossRef CAS PubMed.
- D. Liu, S. Ding, C. Wu, W. Gan, C. Wang, D. Cao, Z. U. Rehman, Y. Sang, S. Chen, X. Zheng, Y. Wang, B. Ge and L. Song, J. Mater. Chem. A, 2018, 6, 6840–6846 RSC.
- Y. Peng, X. Yu, W. Yin, W. Dong, J. Peng and T. Wang, ACS Appl. Bio Mater., 2020, 3, 59–67 CrossRef CAS.
- W.-J. Ong, L.-L. Tan, S.-P. Chai, S.-T. Yong and A. R. Mohamed, Nano Energy, 2015, 13, 757–770 CrossRef CAS.
- R. B. N. Baig, S. Verma, R. S. Varma and M. N. Nadagouda, ACS Sustainable Chem. Eng., 2016, 4, 1661–1664 CrossRef CAS.
- L. Yang, D. Cheng, X. Zeng, X. Wan, J. Shui, Z. Xiang and D. Cao, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 6626–6631 CrossRef CAS.
- Y. Tang, J. Zhou, Z. Shen, W. Chen, C. Li and X. Dai, RSC Adv., 2016, 6, 93985–93996 RSC.
- H. Amani, E. Mostafavi, H. Arzaghi, S. Davaran, A. Akbarzadeh, O. Akhavan, H. Pazoki-Toroudi and T. J. Webster, ACS Biomater. Sci. Eng., 2019, 5, 193–214 CrossRef CAS.
- C. Zhang, S. Yang, J. Wu, M. Liu, S. Yazdi, M. Ren, J. Sha, J. Zhong, K. Nie, A. S. Jalilov, Z. Li, H. Li, B. I. Yakobson, Q. Wu, E. Ringe, H. Xu, P. M. Ajayan and J. M. Tour, Adv. Energy Mater., 2018, 8, 1703487 CrossRef.
- X. Qiu, X. Yan, H. Pang, J. Wang, D. Sun, S. Wei, L. Xu and Y. Tang, Adv. Sci., 2019, 6, 1801103 CrossRef PubMed.
- Z.-y Gao, W.-j Yang, X.-l Ding, G. Lv and W.-p Yan, Phys. Chem. Chem. Phys., 2018, 20, 7333–7341 RSC.
- A. W. Robertson, B. Montanari, K. He, J. Kim, C. S. Allen, Y. A. Wu, J. Olivier, J. Neethling, N. Harrison, A. I. Kirkland and J. H. Warner, Nano Lett., 2013, 13, 1468–1475 CrossRef CAS PubMed.
- X. Zhao, X. Liu, B. Huang, P. Wang and Y. Pei, J. Mater. Chem. A, 2019, 7, 24583–24593 RSC.
- W. Yang, Z. Gao, X. Liu, X. Li, X. Ding and W. Yan, Catal. Sci. Technol., 2018, 8, 4159–4168 RSC.
- C. Zhang, S. Yang, J. Wu, M. Liu, S. Yazdi, M. Ren, J. Sha, J. Zhong, K. Nie, A. S. Jalilov, Z. Li, H. Li, B. I. Yakobson, Q. Wu, E. Ringe, H. Xu, P. M. Ajayan and J. M. Tour, Adv. Energy Mater., 2018, 8, 1703487 CrossRef.
- J. Zheng, K. Lebedev, S. Wu, C. Huang and S. Tsang, J. Am. Chem. Soc., 2021, 143, 7979–7990 CrossRef CAS PubMed.
- J. Chen, Y. Kang, W. Zhang, Z. Zhang, Y. Chen, Y. Yang, L. Duan, Y. Li and W. Li, Angew. Chem., Int. Ed., 2022, 61, e202203022 CAS.
- S. Wu, Z. Chen, W. Yue, S. Mine, T. Toyao, M. Matsuoka, X. Xi, L. Wang and J. Zhang, ACS Catal., 2021, 11, 4362–4371 CrossRef CAS.
- Z. Jiang, M. Tian, M. Jing, S. Chai, Y. Jian, C. Chen, M. Douthwaite, L. Zheng, M. Ma, W. Song, J. Liu, J. Yu and C. He, Angew. Chem., Int. Ed, 2022, 61, e202200763 CAS.
- Z. Zhang, J. Liu, J. Wang, Q. Wang, Y. Wang, K. Wang, Z. Wang, M. Gu, Z. Tang, J. Lim, T. Zhao and F. Ciucci, Nat. Commun., 2021, 12, 5235 CrossRef CAS PubMed.
- R. Nietuby, E. Sobczak and K. E. Attenkofer, J. Alloys Compd., 2001, 328, 126–131 CrossRef.
- J. Yang, B. Chen, X. Liu, W. Liu, Z. Li, J. Dong, W. Chen, W. Yan, T. Yao, X. Duan, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 9495–9500 CrossRef CAS PubMed.
- X. Li, W. Bi, L. Zhang, S. Tao and Y. Xie, Adv. Mater., 2016, 28, 2427–2431 CrossRef CAS PubMed.
- J. T. Yates, T. M. Duncan, S. D. Worley and R. W. Vaughan, J. Chem. Phys., 1979, 70, 1219–1224 CrossRef CAS.
- J. Liu, ACS Catal, 2017, 7, 34–59 CrossRef CAS.
- J. H. Kwak, J. Hu, D. Mei, C. W. Yi, D. H. Kim, C. H. Peden, L. F. Allard and J. Szanyi, Science, 2009, 325, 1670–1673 CrossRef CAS.
- F. He, L. Mi, Y. Shen, T. Mori, S. Liu and Y. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 35327–35333 CrossRef.
- S. An, G. Zhang, J. Liu, K. Li, G. Wan, Y. Liang, D. Ji, J. T. Miller, C. Song, W. Liu, Z. Liu and X. Guo, Chinese J. Catal., 2020, 41, 1198–1207 CrossRef CAS.
- M. Huo, L. Wang, H. Zhang, L. Zhang, Y. Chen and J. Shi, Small, 2019, 15, 1901834 CrossRef PubMed.
- M. Lu, C. Wang, Y. Ding, M. Peng, W. Zhang, K. Li, W. Wei and Y. Lin, Chem. Commun., 2019, 55, 14534–14537 RSC.
- B. Lu, Q. Liu and S. Chen, ACS Catal., 2020, 10, 7584–7618 CrossRef CAS.
- A. S. Varela, M. Kroschel, N. D. Leonard, W. Ju, J. Steinberg, A. Bagger, J. Rossmeisl and P. Strasser, ACS Energy Lett., 2018, 3, 812–817 CrossRef CAS.
- Q. Zhang and J. Guan, Adv. Funct. Mater., 2020, 30, 2000768 CrossRef CAS.
- X. Wang, Y. Jia, X. Mao, D. Liu, W. He, J. Li, J. Liu, X. Yan, J. Chen, L. Song, A. Du and X. Yao, Adv. Mater., 2020, 32, 2000966 CrossRef CAS PubMed.
- L. Bai, Z. Duan, X. Wen and J. Guan, J. Catal., 2019, 378, 353–362 CrossRef CAS.
- S. Zhang, M. Jin, T. Shi, M. Han, Q. Sun, Y. Lin, Z. Ding, L. R. Zheng, G. Wang, Y. Zhang, H. Zhang and H. Zhao, Angew. Chem., Int. Ed., 2020, 59, 13423–13429 CrossRef CAS PubMed.
- Y. Wu, J. Cai, Y. Xie, S. Niu, Y. Zang, S. Wu, Y. Liu, Z. Lu, Y. Fang, Y. Guan, X. Zheng, J. Zhu, X. Liu, G. Wang and Y. Qian, Adv. Mater., 2020, 32, 1904346 CrossRef CAS PubMed.
- T. N. Huan, N. Ranjbar, G. Rousse, M. Sougrati, A. Zitolo, V. Mougel, F. Jaouen and M. Fontecave, ACS Catal, 2017, 7, 1520–1525 CrossRef CAS.
- J. He, T. Zheng, D. Wu, S. Zhang, M. Gu and Q. He, ACS Catal, 2022, 12, 1601–1613 CrossRef CAS.
- X. Li and Z. Xiang, Nat. Commun., 2022, 13, 57 CrossRef CAS PubMed.
- C. C. Wang, D. W. Luo, B. X. Hou, M. Gholizadeh, Z. X. Tan and X. J. Han, J. Renewable Mater., 2022, 10, 1337–1348 CAS.
- Z. Yang, Y. Wang, M. Zhu, Z. Li, W. Chen, W. Wei, T. Yuan, Y. Qu, Q. Xu, C. Zhao, X. Wang, P. Li, Y. Li, Y. Wu and Y. Li, ACS Catal., 2019, 9, 2158–2163 CrossRef CAS.
- J.-C. Li, F. Xiao, H. Zhong, T. Li, M. Xu, L. Ma, M. Cheng, D. Liu, S. Feng, Q. Shi, H.-M. Cheng, C. Liu, D. Du, S. P. Beckman, X. Pan, Y. Lin and M. Shao, ACS Catal., 2019, 9, 5929–5934 CrossRef CAS.
- J. Zhang, Q.-A. Huang, J. Wang, J. Wang, J. Zhang and Y. Zhao, Chin. J. Catal., 2020, 41, 783–798 CrossRef CAS.
- X. Zeng, J. Shui, X. Liu, Q. Liu, Y. Li, J. Shang, L. Zheng and R. Yu, Adv. Energy Mater., 2018, 8, 1701345 CrossRef.
- L. Jiao and H.-L. Jiang, Chem, 2019, 5, 786–804 CAS.
- D.-S. Bin, Z.-X. Chi, Y. Li, K. Zhang, X. Yang, Y.-G. Sun, J.-Y. Piao, A.-M. Caoand and L.-J. Wan, J. Am. Chem. Soc., 2017, 139, 13492–13498 CrossRef CAS PubMed.
- Y.-G. Sun, J.-Y. Piao, L.-L. Hu, D.-S. Bin, X.-J. Lin, S.-Y. Duan, A.-M. Cao and L.-J. Wan, J. Am. Chem. Soc., 2018, 140, 9070–9073 CrossRef CAS PubMed.
- S.-N. Zhao, J.-K. Li, R. Wang, J. Cai and S.-Q. Zang, Adv. Mater., 2022, 34, 2107291 CrossRef CAS PubMed.
- K.-M. Zhao, S. Liu, Y.-Y. Li, X. Wei, G. Ye, W. Zhu, Y. Su, J. Wang, H. Liu, Z. He, Z.-Y. Zhou and S.-G. Sun, Adv. Energy Mater., 2022, 12, 2103588 CrossRef CAS.
- C.-C. Hou, L. Zou, L. Sun, K. Zhang, Z. Liu, Y. Li, C. Li, R. Zou, J. Yu and Q. Xu, Angew. Chem., Int. Ed., 2020, 59, 7384–7389 CrossRef CAS PubMed.
- J. Yang, Z. Wang, C.-X. Huang, Y. Zhang, Q. Zhang, C. Chen, J. Du, X. Zhou, Y. Zhang, H. Zhou, L. Wang, X. Zheng, L. Gu, L.-M. Yang and Y. Wu, Angew. Chem., Int. Ed., 2021, 60, 22722–22728 CrossRef CAS PubMed.
- J. Wang, Z. Q. Huang, W. Liu, C. R. Chang, H. L. Tang, Z. J. Li, W. X. Chen, C. J. Jia, T. Yao, S. Q. Wei, Y. Wu and Y. D. Lie, J. Am. Chem. Soc., 2017, 139, 17281–17284 CrossRef CAS PubMed.
- L. Gong, H. Zhang, Y. Wang, E. Luo, K. Li, L. Gao, Y. Wang, Z. Wu, Z. Jin, J. Ge, Z. Jiang, C. Liu and W. Xing, Angew. Chem., Int. Ed., 2020, 59, 13923–13928 CrossRef CAS PubMed.
- X. Jia, W. Gu, S. Zhang and E. Wang, ChemElectroChem, 2017, 4, 2005–2011 CrossRef CAS.
- T. Sharifi, E. Gracia-Espino, A. Chen, G. Hu and T. Wagberg, Adv. Energy Mater., 2020, 10, 1902084 CrossRef CAS.
- L. Zheng, Y. Zhao, H. Zhang, W. Xia and J. Tang, ChemSusChem, 2022, 15, e202102642 CAS.
- H. Zhang, S. Hwang, M. Wang, Z. Feng, S. Karakalos, L. Luo, Z. Qiao, X. Xie, C. Wang, D. Su, Y. Shao and G. Wu, J. Am. Chem. Soc., 2017, 139, 14143–14149 CrossRef CAS PubMed.
- K. Yuan, S. Sfaelou, M. Qiu, D. Lützenkirchen-Hecht, X. Zhuang, Y. Chen, C. Yuan, X. Feng and U. Scherf, ACS Energy Lett., 2018, 3, 252–260 CrossRef CAS.
- Y. Li, T. Liu, W. Yang, Z. Zhu, Y. Zhai, W. Gu and C. Zhu, Nanoscale, 2019, 11, 19506–19511 RSC.
- Y.-G. Lee and H.-J. Ahn, Appl. Surf. Sci., 2019, 487, 389–397 CrossRef CAS.
- L. Hu, C. Dai, L. Chen, Y. Zhu, Y. Hao, Q. Zhang, L. Gu, X. Feng, S. Yuan, L. Wang and B. Wang, Angew. Chem., Int. Ed, 2021, 60, 27324–27329 CrossRef CAS PubMed.
- J.-C. Li, H. Zhong, M. Xu, T. Li, L. Wang, Q. Shi, S. Feng, Z. Lyu, D. Liu, D. Du, S. P. Beckman, X. Pan, Y. Lin and M. Shao, Sci. China Mater., 2019, 63, 965–971 CrossRef.
- L. Yu, Y. Li and Y. Ruan, Angew. Chem., Int. Ed., 2021, 60, 25296–25301 CrossRef CAS PubMed.
- Y. Chen, S. Ji, S. Zhao, W. Chen, J. Dong, W.-C. Cheong, R. Shen, X. Wen, L. Zheng, A. I. Rykov, S. Cai, H. Tang, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Nat. Commun., 2018, 9, 5422 CrossRef CAS PubMed.
- Q. Lai, L. Zheng, Y. Liang, J. He, J. Zhao and J. Chen, ACS Catal., 2017, 7, 1655–1663 CrossRef CAS.
- P.-J. Wei, G.-Q. Yu, Y. Naruta and J.-G. Liu, Angew. Chem., Int. Ed., 2014, 53, 6659–6663 CrossRef CAS PubMed.
- Y.-M. Zhao, P.-C. Zhang, C. Xu, X.-Y. Zhou, L.-M. Liao, P.-J. Wei, E. Liu, H. Chen, Q. He and J.-G. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 17334–17342 CrossRef CAS PubMed.
- A. Zitolo, V. Goellner, V. Armel, M.-T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Nat. Mater., 2015, 14, 937 CrossRef CAS.
- S. Yang, T. Zhang, G. Li, L. Yang and J. Y. Lee, Energy Stor. Mater., 2017, 6, 140–148 CrossRef.
- A. Han, X. Wang, K. Tang, Z. Zhang, C. Ye, K. Kong, H. Hu, L. Zheng, P. Jiang, C. Zhao, Q. Zhang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 19262–19271 CrossRef CAS PubMed.
- Z. Lu, B. Wang, Y. Hu, W. Liu, Y. Zhao, R. Yang, Z. Li, J. Luo, B. Chi, Z. Jiang, M. Li, S. Mu, S. Liao, J. Zhang and X. Sun, Angew. Chem., Int. Ed., 2019, 58, 2622–2626 CrossRef CAS PubMed.
- S. Yang, X. Xue, J. Zhang, X. Liu, C. Dai, Q. Xu, J. Lian, Y. Zhao, G. Li, H. Li and S. Yuan, Chem. Eng. J., 2020, 395, 125064 CrossRef CAS.
- J. Li, H. Zhang, W. Samarakoon, W. Shan, D. A. Cullen, S. Karakalos, M. Chen, D. Gu, K. L. More, G. Wang, Z. Feng, Z. Wang and G. Wu, Angew. Chem., Int. Ed., 2019, 58, 18971–18980 CrossRef CAS.
- Y. Sun, S. Sun, H. Yang, S. Xi, J. Gracia and Z. J. Xu, Adv. Mater., 2020, 32, 2003297 CrossRef CAS PubMed.
- D. Xia, X. Yang, L. Xie, Y. Wei, W. Jiang, M. Dou, X. Li, J. Li, L. Gan and F. Kang, Adv. Funct. Mater., 2019, 29, 1906174 CrossRef CAS.
- H. Wang, R. Liu, Y. Li, X. Lü, Q. Wang, S. Zhao, K. Yuan, Z. Cui, X. Li, S. Xin, R. Zhang, M. Lei and Z. Lin, Joule, 2018, 2, 337–348 CrossRef CAS.
- C. Mu, J. Mao, J. Guo, Q. Guo, Z. Li, W. Qin, Z. Hu, K. Davey, T. Ling and S.-Z. Qiao, Adv. Mater., 2020, 32, 1907168 CrossRef CAS.
- Z. Li, Z. Zhuang, F. Lv, H. Zhu, L. Zhou, M. Luo, J. Zhu, Z. Lang, S. Feng, W. Chen, L. Mai and S. Guo, Adv. Mater., 2018, 30, 1803220 CrossRef PubMed.
- X.-T. Wang, T. Ouyang, L. Wang, J.-H. Zhong, T. Ma and Z.-Q. Liu, Angew. Chem., Int. Ed., 2019, 58, 13291–13296 CrossRef CAS PubMed.
- G. Shen, R. Zhang, L. Pan, F. Hou, Y. Zhao, Z. Shen, W. Mi, C. Shi, Q. Wang, X. Zhang and J.-J. Zou, Angew. Chem., Int. Ed., 2020, 59, 2313–2317 CrossRef CAS PubMed.
- T. Ouyang, X.-T. Wang, X.-Q. Mai, A.-N. Chen, Z.-Y. Tang and Z.-Q. Liu, Angew. Chem., Int. Ed., 2020, 59, 11948–11957 CrossRef CAS PubMed.
- G. Yang, J. Zhu, P. Yuan, Y. Hu, G. Qu, B.-A. Lu, X. Xue, H. Yin, W. Cheng, J. Cheng, W. Xu, J. Li, J. Hu, S. Mu and J.-N. Zhang, Nat. Commun., 2021, 12, 1734 CrossRef CAS PubMed.
- L. Gong, H. Zhang, Y. Wang, E. Luo, K. Li, L. Gao, Y. Wang, Z. Wu, Z. Jin, J. Ge, Z. Jiang, C. Liu and W. Xing, Angew. Chem., Int. Ed., 2020, 59, 13923–13928 CrossRef CAS PubMed.
- J. Han, X. Meng, L. Lu, J. Bian, Z. Li and C. Sun, Adv. Funct. Mater., 2019, 29, 1808872 CrossRef CAS.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- X. Xie, C. He, B. Li, Y. He, D. A. Cullen, E. C. Wegener, A. J. Kropf, U. Martinez, Y. Cheng, M. H. Engelhard, M. E. Bowden, M. Song, T. Lemmon, X. S. Li, Z. Nie, J. Liu, D. J. Myers, P. Zelenay, G. Wang, G. Wu, V. Ramani and Y. Shao, Nat. Catal., 2020, 3, 1044–1054 CrossRef CAS.
- F. Zhou, P. Yu, F. Sun, G. Zhang, X. Liu and L. Wang, J. Mater. Chem. A, 2021, 9, 6831–6840 RSC.
- J. Li, M. T. Sougrati, A. Zitolo, J. M. Ablett, I. C. Oğuz, T. Mineva, I. Matanovic, P. Atanassov, Y. Huang, I. Zenyuk, A. Di Cicco, K. Kumar, L. Dubau, F. Maillard, G. Dražić and F. Jaouen, Nat. Catal., 2021, 4, 10–19 CrossRef CAS.
- C.-Y. Su, H. Cheng, W. Li, Z.-Q. Liu, N. Li, Z. Hou, F.-Q. Bai, H.-X. Zhang and T.-Y. Ma, Adv. Energy Mater., 2017, 7, 1602420 CrossRef.
- L. Bai, C.-S. Hsu, D. T. L. Alexander, H. M. Chen and X. Hu, J. Am. Chem. Soc., 2019, 141, 14190–14199 CrossRef CAS PubMed.
- J. Wu, L. Xiong, B. Zhao, M. Liu and L. Huang, Small Methods, 2020, 4, 1900540 CrossRef CAS.
- Á. Valdés, J. Brillet, M. Grätzel, H. Gudmundsdóttir, H. A. Hansen, H. Jónsson, P. Klüpfel, G.-J. Kroes, F. Le Formal, I. C. Man, R. S. Martins, J. K. Nørskov, J. Rossmeisl, K. Sivula, A. Vojvodic and M. Zäch, Phys. Chem. Chem. Phys., 2012, 14, 49–70 RSC.
- P. Li, M. Wang, X. Duan, L. Zheng, X. Cheng, Y. Zhang, Y. Kuang, Y. Li, Q. Ma, Z. Feng, W. Liu and X. Sun, Nat. Commun., 2019, 10, 1711 CrossRef PubMed.
- L. An, J. Feng, Y. Zhang, Y.-Q. Zhao, R. Si, G.-C. Wang, F. Cheng, P. Xi and S. Sun, Nano Energy, 2019, 57, 644–652 CrossRef CAS.
- W. Zhai, S. Huang, C. Lu, X. Tang, L. Li, B. Huang, T. Hu, K. Yuan, X. Zhuang and Y. Chen, Small, 2022, 18, 2107225 CrossRef CAS PubMed.
- X. Zhang, Q. Zhang, J. Cui, J. Yan, J. Liu and Y. Wu, Nanoscale, 2022, 14, 3212–3223 RSC.
- H. Fei, J. Dong, Y. Feng, C. S. Allen, C. Wan, B. Volosskiy, M. Li, Z. Zhao, Y. Wang, H. Sun, P. An, W. Chen, Z. Guo, C. Lee, D. Chen, I. Shakir, M. Liu, T. Hu, Y. Li, A. I. Kirkland, X. Duan and Y. Huang, Nat. Catal., 2018, 1, 63–72 CrossRef CAS.
- S. L. Candelaria, N. M. Bedford, T. J. Woehl, N. S. Rentz, A. R. Showalter, S. Pylypenko, B. A. Bunker, S. Lee, B. Reinhart, Y. Ren, S. P. Ertem, E. B. Coughlin, N. A. Sather, J. L. Horan, A. M. Herring and L. F. Greenleette, ACS Catal, 2017, 7, 365–379 CrossRef CAS.
- M. Zeng, Y. Liu, F. Zhao, K. Nie, N. Han, X. Wang, W. Huang, X. Song, J. Zhong and Y. Li, Adv. Funct. Mater., 2016, 26, 4397–4404 CrossRef CAS.
- R. Gao, Y. Yin, F. Niu, A. Wang, S. Li, H. Dong and S. Yang, ChemElectroChem, 2019, 6, 1824–1830 CrossRef CAS.
- S. Li, C. Cheng, X. Zhao, J. Schmidt and A. Thomas, Angew. Chem., Int. Ed., 2018, 57, 1856–1862 CrossRef CAS PubMed.
- M. Ma, A. Kumar, D. Wang, Y. Wang, Y. Jia, Y. Zhang, G. Zhang, Z. Yan and X. Sun, Appl. Catal., B, 2020, 274, 119091 CrossRef CAS.
- J. Chen, H. Li, C. Fan, Q. Meng, Y. Tang, X. Qiu, G. Fu and T. Ma, Adv. Mater., 2020, 32, e2003134 CrossRef.
- B. Hammer and J. K. Norskov, Nature, 1995, 376, 238–240 CrossRef CAS.
- Y. Yang, Z. Lun, G. Xia, F. Zheng, M. He and Q. Chen, Energy Environ. Sci., 2015, 8, 3563–3571 RSC.
- T. He, C. Zhang and A. Du, Chem. Eng. Sci., 2019, 194, 58–63 CrossRef CAS.
- Q. Zhang and J. Guan, Adv. Funct. Mater., 2020, 30, 2000768 CrossRef CAS.
- J. Li and G. Zheng, Adv. Sci, 2017, 4, 1600380 CrossRef PubMed.
- J. Wang, F. Xu, H. Jin, Y. Chen and Y. Wang, Adv. Mater., 2017, 29, 1605838 CrossRef PubMed.
- X. Yu, J. Zhao, L.-R. Zheng, Y. Tong, M. Zhang, G. Xu, C. Li, J. Ma and G. Shi, ACS Energy Lett., 2018, 3, 237–244 CrossRef CAS.
- S. Sultan, J. N. Tiwari, A. N. Singh, S. Zhumagali, M. Ha, C. W. Myung, P. Thangavel and K. S. Kim, Adv. Energy Mater., 2019, 9, 1900624 CrossRef.
- X. Yu, J. Zhao, L.-R. Zheng, Y. J. Tong, M. Zhang, G. Xu, C. Li, J. Ma and G. Shi, ACS Energy Lett., 2018, 3, 237–244 CrossRef CAS.
- K. Li, S. Zhang, X. Zhang, S. Liu, H. Jiang, T. Jiang, C. Shen, Y. Yu and W. Chen, Nano Lett., 2022, 22, 1557–1565 CrossRef CAS PubMed.
- J. Chen, T. Wang, X. Wang, B. Yang, X. Sang, S. Zheng, S. Yao, Z. Li, Q. Zhang, L. Lei, J. Xu, L. Dai and Y. Hou, Adv. Funct. Mater., 2022, 32, 2110174 CrossRef CAS.
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS.
- J. Gu, C.-S. Hsu, L. Bai, H. M. Chen and X. Hu, Science, 2019, 364, 1091 CrossRef CAS PubMed.
- T. Wang, X. Sang, W. Zheng, B. Yang, S. Yao, C. Lei, Z. Li, Q. He, J. Lu, L. Lei, L. Dai and Y. Hou, Adv. Mater., 2020, 32, 2002430 CrossRef CAS PubMed.
- H. Zhang, J. Li, S. Xi, Y. Du, X. Hai, J. Wang, H. Xu, G. Wu, J. Zhang, J. Lu and J. Wang, Angew. Chem., Int. Ed., 2019, 58, 14871–14876 CrossRef CAS PubMed.
- L. Lin, H. Li, Y. Wang, H. Li, P. Wei, B. Nan, R. Si, G. Wang and X. Bao, Angew. Chem., Int. Ed, 2021, 133, 26786–26790 CrossRef.
- X. Sun, Y. Tuo, C. Ye, C. Chen, Q. Lu, G. Li, P. Jiang, S. Chen, P. Zhu, M. Ma, J. Zhang, J. H. Bitter, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 23614–23618 CrossRef CAS PubMed.
- T. Wang, X. Sang, W. Zheng, B. Yang, S. Yao, C. Lei, Z. Li, Q. He, J. Lu, L. Lei, L. Dai and Y. Hou, Adv. Mater., 2020, 32, 2002430 CrossRef CAS PubMed.
- F. Pan, H. Zhang, K. Liu, D. Cullen, K. More, M. Wang, Z. Feng, G. Wang, G. Wu and Y. Li, ACS Catal, 2018, 8, 3116–3122 CrossRef CAS.
- X. Qin, S. Zhu, F. Xiao, L. Zhang and M. Shao, ACS Energy Lett., 2019, 4, 1778–1783 CrossRef CAS.
- H. B. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- W. Ju, A. Bagger, G.-P. Hao, A. S. Varela, I. Sinev, V. Bon, B. Roldan Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Nat. Commun., 2017, 8, 944 CrossRef.
- L. Jiao, J. Zhu, Y. Zhang, W. Yang, S. Zhou, A. Li, C. Xie, X. Zheng, W. Zhou, S.-H. Yu and H.-L. Jiang, J. Am. Chem. Soc., 2021, 143(46), 19417–19424 CrossRef CAS PubMed.
- Z. Wang, Z. Yu and J. Zhao, Phys. Chem. Chem. Phys., 2018, 20, 12835–12844 RSC.
- Y. Huang, D. D. Babu, Z. Peng and Y. Wang, Adv. Sci, 2020, 7, 1902390 CrossRef CAS PubMed.
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Science, 2018, 360, eaar6611 CrossRef.
- J. Chatt, J. R. Dilworth and R. L. Richards, Chem. Rev., 1978, 78, 589–625 CrossRef CAS.
- L. Zhang, W. Zhao, W. Zhang, J. Chen and Z. Hu, Nano Research, 2019, 12, 1181–1186 CrossRef CAS.
- J. S. Anderson, G. E. Cutsail III, J. Rittle, B. A. Connor, W. A. Gunderson, L. Zhang, B. M. Hoffman and J. C. Peters, J. Am. Chem. Soc., 2015, 24, 7803–7809 CrossRef.
- X.-F. Li, Q.-K. Li, J. Cheng, L. Liu, Q. Yan, Y. Wu, X.-H. Zhang, Z.-Y. Wang, Q. Qiu and Y. Luo, J. Am. Chem. Soc., 2016, 138, 8706–8709 CrossRef CAS PubMed.
- J.-C. Liu, X.-L. Ma, Y. Li, Y.-G. Wang, H. Xiao and J. Li, Nat. Commun., 2018, 9, 1610 CrossRef PubMed.
- J. S. Anderson, J. Rittle and J. C. Peters, Nature, 2013, 501, 84–87 CrossRef CAS.
- Y. Li, J. Li, J. Huang, J. Chen, Y. Kong, B. Yang, Z. Li, L. Lei, G. Chai, Z. Wen, L. Dai and Y. Hou, Angew. Chem., Int. Ed., 2021, 60, 9078–9085 CrossRef CAS PubMed.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. N?Rskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed.
- M. Zafari, D. Kumar, M. Umer and K. S. Kim, J. Mater. Chem. A, 2020, 8, 5209–5216 RSC.
- Y. T. Liu, L. Tang, J. Dai, J. Yu and B. Ding, Angew. Chem., Int. Ed., 2020, 59, 13623–13627 CrossRef CAS PubMed.
- Y. Wang, W. Cheng, P. Yuan, G. Yang, S. Mu, J. Liang, H. Xia, K. Guo, M. Liu, S. Zhao, G. Qu, B.-A. Lu, Y. Hu, J. Hu and J.-N. Zhang, Adv. Sci, 2021, 8, 2102915 CrossRef CAS PubMed.
- Z.-Y. Wu, M. Karamad, X. Yong, Q. Huang, D. A. Cullen, P. Zhu, C. Xia, Q. Xiao, M. Shakouri, F.-Y. Chen, J. Y. Kim, Y. Xia, K. Heck, Y. Hu, M. S. Wong, Q. Li, I. Gates, S. Siahrostami and H. Wang, Nat. Commun., 2021, 12, 2870 CrossRef CAS.
- C. J. M. van der Ham, M. T. M. Koper and D. G. H. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC.
- K. Kugler, M. Luhn, J. A. Schramm, K. Rahimi and M. Wessling, Phys. Chem. Chem. Phys., 2015, 17, 3768–3782 RSC.
- V. Kordali, G. Kyriacou and C. Lambrou, Chem. Commun., 2000, 1673–1674 RSC.
- Y. Ma, T. Yang, H. Zou, W. Zang, Z. Kou, L. Mao, Y. Feng, L. Shen, S. J. Pennycook, L. Duan, X. Li and J. Wang, Adv. Mater., 2020, 32, 2002177 CrossRef CAS.
- Y. Abghoui, A. L. Garden, V. F. Hlynsson, S. Björgvinsdóttir, H. Ólafsdóttir and E. Skúlason, Phys. Chem. Chem. Phys., 2015, 17, 4909–4918 RSC.
- E. Skúlason, T. Bligaard, S. Gudmundsdóttir, F. Studt, J. Rossmeisl, F. Abild-Pedersen, T. Vegge, H. Jónsson and J. K. Nørskov, Phys. Chem. Chem. Phys., 2012, 14, 1235–1245 RSC.
- S. Licht, B. Cui, B. Wang, F. F. Li, J. Lau and S. Liu, Science, 2014, 345, 637–640 CrossRef CAS PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- N. Cheng, J.-C. Li, D. Liu, Y. Lin and D. Du, Small, 2019, 15, 1901485 CrossRef CAS.
- T. Zhang, Y. Xing, Y. Song, Y. Gu, X. Yan, N. Lu, H. Liu, Z. Xu, H. Xu, Z. Zhang and M. Yang, Anal. Chem., 2019, 91, 10589–10595 CrossRef CAS.
- L. Jiao, W. Xu, H. Yan, Y. Wu, C. Liu, D. Du, Y. Lin and C. Zhu, Anal. Chem., 2019, 91, 11994–11999 CrossRef CAS.
- Y. Peng, X. Yu, W. Yin, W. Dong, J. Peng and T. Wang, ACS Appl. Bio Mater., 2020, 3, 59–67 CrossRef CAS PubMed.
- K. Yuan, D. Lützenkirchen-Hecht, L. Li, L. Shuai, Y. Li, R. Cao, M. Qiu, X. Zhuang, M. K. H. Leung, Y. Chen and U. Scherf, J. Am. Chem. Soc., 2020, 142, 2404–2412 CrossRef CAS PubMed.
- R. Ciriminna, L. Albanese, F. Meneguzzo and M. Pagliaro, ChemSusChem, 2016, 9, 3374–3381 CrossRef CAS PubMed.
- H. Zhao and Z. Y. Yuan, ChemSusChem, 2021, 14, 1616–1633 CrossRef CAS PubMed.
- Y. Jiang, P. Ni, C. Chen, Y. Lu, P. Yang, B. Kong, A. Fisher and X. Wang, Adv. Energy Mater., 2018, 8, 1801909 CrossRef.
- K. Jiang, S. Back, A. J. Akey, C. Xia, Y. Hu, W. Liang, D. Schaak, E. Stavitski, J. K. Nørskov, S. Siahrostami and H. Wang, Nat. Commun., 2019, 10, 3997 CrossRef PubMed.
- W. Zhu and S. Chen, Electroanalysis, 2020, 32, 2591–2602 CrossRef CAS.
- X. Guo, S. Lin, J. Gu, S. Zhang, Z. Chen and S. Huang, ACS Catal, 2019, 9, 11042–11054 CrossRef CAS.
- Y. Sun, L. Silvioli, N. R. Sahraie, W. Ju, J. Li, A. Zitolo, S. Li, A. Bagger, L. Arnarson, X. Wang, T. Moeller, D. Bernsmeier, J. Rossmeisl, F. Jaouen and P. Strasser, J. Am. Chem. Soc., 2019, 141, 12372–12381 CrossRef CAS.
- Q. Gao, Ionics, 2020, 26, 2453–2465 CrossRef CAS.
- J. Zhang, H. Yang and B. Liu, Adv. Energy Mater., 2021, 11, 2002473 CrossRef CAS.
- K. Jiang, S. Back, A. J. Akey, C. Xia, Y. Hu, W. Liang, D. Schaak, E. Stavitski, J. K. Nørskov, S. Siahrostami and H. Wang, Nat. Commun., 2019, 10, 3997 CrossRef.
- Z. Liu, T. He, K. Liu, W. Chen and Y. Tang, RSC Adv., 2017, 7, 7920–7928 RSC.
- Y. Tang, J. Zhou, W. Chen, H. Chai, Y. Li, Z. Feng and X. Dai, Mol. Catal., 2019, 476, 110524 CrossRef CAS.
- E. H. Song, J. M. Yan, J. S. Lian and Q. Jiang, J. Phys. Chem. C, 2012, 116, 20342–20348 CrossRef CAS.
- H. Y. Zhuo, X. Zhang, J. X. Liang, Q. Yu, H. Xiao and J. Li, Chem. Rev., 2020, 120, 12315–12341 CrossRef CAS PubMed.
- C. J. Zhang and P. Hu, J. Am. Chem. Soc., 2001, 123, 1166–1172 CrossRef CAS.
- M. D. Ackermann, T. M. Pedersen, B. L. M. Hendriksen, O. Robach, S. C. Bobaru, I. Popa, C. Quiros, H. Kim, B. Hammer, S. Ferrer and J. W. M. Frenken, Phys. Rev. Lett., 2005, 95, 255505 CrossRef CAS PubMed.
- Y. Li, Z. Zhou, G. Yu, W. Chen and Z. Chen, J. Phys. Chem. C, 2010, 114, 6250–6254 CrossRef CAS.
- L. Zhang, X. Ren, X. Zhao, Y. Zhu, R. Pang, P. Cui, Y. Jia, S. Li and Z. Zhang, Nano Lett., 2022, 22, 3744–3750 CrossRef CAS PubMed.
- E. Wigner and E. E. Witmer, Z. Phys., 1928, 51, 859–886 CrossRef CAS.
- J. H. Moore, Phys. Rev. A, 1973, 8, 2359–2362 CrossRef CAS.
- G. Ding, W. Wang, T. Jiang, B. Han, H. Fan and G. Yang, ChemCatChem, 2013, 5, 192–200 CrossRef CAS.
- S.-i Niwa, et al. , Science, 2002, 295, 105–107 CrossRef CAS PubMed.
- D. Deng, X. Chen, L. Yu, X. Wu, Q. Liu, Y. Liu, H. Yang, H. Tian, Y. Hu and P. Du, Sci Adv., 2015, 1, e1500462–e1500462 CrossRef PubMed.
- W. Liu, L. Zhang, X. Liu, X. Liu, X. Yang, S. Miao, W. Wang, A. Wang and T. Zhang, J. Am. Chem. Soc., 2017, 139, 10790–10798 CrossRef CAS PubMed.
- L. Kong, J. Li, Q. Liu, Z. Zhao, Q. Sun, J. Liu and Y. Wei, J. Energy Chem., 2016, 25, 577–586 CrossRef.
- H. Zhu, D. C. Rosenfeld, D. H. Anjum, V. Caps and J.-M. Basset, ChemSusChem, 2015, 8, 1254–1263 CrossRef CAS PubMed.
- M. Peymani, S. M. Alavi and M. Rezaei, Int. J. Hydrogen Energy, 2016, 41, 19057–19069 CrossRef CAS.
- S. Wang, H. Li, M. He, X. Cui, L. Hua, H. Li, J. Xiao, L. Yu, N. P. Rajan, Z. Xie and D. Deng, J. Energy Chem., 2019, 36, 47–50 CrossRef.
- X. Cui, H. Li, Y. Wang, Y. Hu, L. Hua, H. Li, X. Han, Q. Liu, F. Yang, L. He, X. Chen, Q. Li, J. Xiao, D. Deng and X. Bao, Chem, 2018, 4, 1902–1910 CAS.
- M. Crespo-Quesada, F. Cárdenas-Lizana, A.-L. Dessimoz and L. Kiwi-Minsker, ACS Catal, 2012, 2, 1773–1786 CrossRef CAS.
- Q. Feng, S. Zhao, Q. Xu, W. Chen, S. Tian, Y. Wang, W. Yan, J. Luo, D. Wang and Y. Li, Adv. Mater., 2019, 31, 1901024 CrossRef PubMed.
- Z. Sun, S. Wang and W. Chen, J. Mater. Chem. A, 2021, 9, 5296–5319 RSC.
- T. He, C. Zhang, L. Zhang and A. Du, Nano Research, 2019, 12, 1817–1823 CrossRef CAS.
- E. M. Sulman, P. M. Valetsky, M. G. Sulman, L. M. Bronstein, A. I. Sidorov, V. Y. Doluda and V. G. Matveeva, Chem. Eng. Process, 2011, 50, 1041–1053 CrossRef CAS.
- S. R. More and G. D. Yadav, ACS Omega, 2018, 3, 7124–7132 CrossRef CAS PubMed.
- D. B. Bagal and B. M. Bhanage, Adv. Synth. Catal., 2015, 357, 883–900 CrossRef CAS.
- R. V. Jagadeesh, A. E. Surkus, H. Junge, M. M. Pohl, J. Radnik, J. Rabeah, H. Huan, V. Schünemann, A. Brückner and M. Beller, Science, 2013, 342, 1073–1076 CrossRef CAS PubMed.
- R. V. Jagadeesh, K. Murugesan, A. S. Alshammari, H. Neumann, M. M. Pohl, J. Radnik and M. Beller, Science, 2017, 358, 326–332 CrossRef CAS PubMed.
- Z. Zhang, C. Ma, Y. Tu, R. Si, J. Wei, S. Zhang, Z. Wang, J.-F. Li, Y. Wang and D. Deng, Nano Res., 2019, 12, 2313–2317 CrossRef CAS.
- L. Huang, Y. Lv, S. Liu, H. Cui, Z. Zhao, H. Zhao, P. Liu, W. Xiong, F. Hao and H. A. Luo, Ind. Eng. Chem. Res., 2020, 59, 1422–1435 CrossRef CAS.
- Y.-G. Ji, K. Wei, T. Liu, L. Wu and W.-H. Zhang, Adv. Synth. Catal., 2017, 359, 933–940 CrossRef CAS.
- R. Yun, F. Zhan, N. Li, B. Zhang, W. Ma, L. Hong, T. Sheng, L. Du, B. Zheng and S. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 34122–34129 CrossRef CAS.
- T. Sheng, Y.-J. Qi, X. Lin, P. Hu, S.-G. Sun and W.-F. Lin, Chem. Eng. J, 2016, 293, 337–344 CrossRef CAS.
- S. Tian, M. Hu, Q. Xu, W. Gong, W. Chen, J. Yang, Y. Zhu, C. Chen, J. He, Q. Liu, H. Zhao, D. Wang and Y. Li, Sci. China Mater., 2021, 64, 642–650 CrossRef CAS.
- Z.-H. Xue, D. Luan, H. Zhang and X. W. Lou, Joule, 2022, 6, 92–133 CrossRef CAS.
- S. Cestellos-Blanco, H. Zhang, J. M. Kim, Y.-x Shen and P. Yang, Nat. Catal., 2020, 3, 245–255 CrossRef CAS.
- P. Zhang and X. W. Lou, Adv. Mater., 2019, 31, 1900281 CrossRef PubMed.
- H. Tong, S. Ouyang, Y. Bi, N. Umezawa, M. Oshikiri and J. Ye, Adv. Mater., 2012, 24, 229–251 CrossRef CAS PubMed.
- B. Wang, H. Cai and S. Shen, Small Methods, 2019, 3, 1800447 CrossRef.
- Y. Li, Z. Wang, T. Xia, H. Ju, K. Zhang, R. Long, Q. Xu, C. Wang, L. Song, J. Zhu, J. Jiang and Y. Xiong, Adv. Mater., 2016, 28, 6959–6965 CrossRef CAS PubMed.
- C. Gao, J. Low, R. Long, T. Kong, J. Zhu and Y. Xiong, Chem. Rev., 2020, 120, 12175–12216 CrossRef CAS.
- L. Zeng and C. Xue, Nano Res., 2021, 14, 934–944 CrossRef CAS.
- W. Zhang, Q. Peng, L. Shi, Q. Yao, X. Wang, A. Yu, Z. Chen and Y. Fu, Small, 2019, 15, 1905166 CrossRef CAS.
- J. Li, P. Liu, Y. Tang, H. Huang, H. Cui, D. Mei and C. Zhong, ACS Catal, 2020, 10, 2431–2442 CrossRef CAS.
- W. Wu, E. Cui, Y. Zhang, C. Zhang, F. Zhu, C.-H. Tung and Y. Wang, ACS Catal, 2019, 9, 6335–6341 CrossRef CAS.
- C. Wang, A. Li, C. Li, S. Zhang, H. Li, X. Zhou, L. Hu, Y. Feng, K. Wang, Z. Zhu, R. Shao, Y. Chen, P. Gao, S. Mao, J. Huang, Z. Zhang and X. Han, Adv. Mater., 2019, 31, 1903491 CrossRef CAS PubMed.
- Z. Chen, S. Wu, J. Ma, S. Mine, T. Toyao, M. Matsuoka, L. Wang and J. Zhang, Angew. Chem., Int. Ed., 2021, 60, 11901–11909 CrossRef CAS PubMed.
- X. Ge, P. Zhou, Q. Zhang, Z. Xia, S. Chen, P. Gao, Z. Zhang, L. Gu and S. Guo, Angew. Chem., Int. Ed., 2020, 59, 232–236 CrossRef CAS PubMed.
- T. Hisatomi and K. Domen, Nat. Catal, 2019, 2, 387–399 CrossRef CAS.
- J. Liu, H. Wu, F. Li, X. Feng, P. Zhang and L. Gao, Adv. Sustainable Syst., 2020, 4, 2000151 CrossRef CAS.
- M. C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Science, 2019, 363, 1429–1434 CrossRef CAS PubMed.
- T. Hou, H. Peng, Y. Xin, S. Wang, W. Zhu, L. Chen, Y. Yao, W. Zhang, S. Liang and L. Wang, ACS Catal, 2020, 10, 5502–5510 CrossRef CAS.
- C. Gao, S. Chen, Y. Wang, J. Wang, X. Zheng, J. Zhu, L. Song, W. Zhang and Y. Xiong, Adv. Mater., 2018, 30, e1704624 CrossRef PubMed.
- Y. Wang, A. Xu, Z. Wang, L. Huang, J. Li, F. Li, J. Wicks, M. Luo, D.-H. Nam, C.-S. Tan, Y. Ding, J. Wu, Y. Lum, C.-T. Dinh, D. Sinton, G. Zheng and E. H. Sargent, J. Am. Chem. Soc., 2020, 142, 5702–5708 CrossRef CAS PubMed.
- G.-F. Chen, Y. Yuan, H. Jiang, S.-Y. Ren, L.-X. Ding, L. Ma, T. Wu, J. Lu and H. Wang, Nat. Energy, 2020, 5, 605–613 CrossRef CAS.
- S. Shang, W. Xiong, C. Yang, B. Johannessen, R. Liu, H.-Y. Hsu, Q. Gu, M. K. H. Leung and J. Shang, ACS Nano, 2021, 15, 9670–9678 CrossRef.
- S. Wu, Z. Chen, W. Yue, S. Mine, T. Toyao, M. Matsuoka, X. Xi, L. Wang and J. Zhang, ACS Catal, 2021, 11, 4362–4371 CrossRef CAS.
- X. Xiao, Z. Ruan, Q. Li, L. Zhang, H. Meng, Q. Zhang, H. Bao, B. Jiang, J. Zhou, C. Guo, X. Wang and H. Fu, Adv. Mater., 2022, 34, 2200612 CrossRef CAS PubMed.
- J. Wen, X. Yang, Z. Sun, J. Yang, P. Han, Q. Liu, H. Dong, M. Gu, L. Huang and H. Wang, Green Chem., 2020, 22, 230–237 RSC.
- Z. Zhao, W. Zhang, W. Liu, Y. Li, J. Ye, J. Liang and M. Tong, Chem. Eng. J, 2021, 407, 127167 CrossRef CAS.
- G. Zhao, W. Li, H. Zhang, W. Wang and Y. Ren, Chem. Eng. J., 2022, 430, 132937 CrossRef CAS.
- W. Ma, J. Mao, X. Yang, C. Pan, W. Chen, M. Wang, P. Yu, L. Mao and Y. Li, Chem. Commun., 2019, 55, 159–162 RSC.
- L. Jiao, H. Yan, Y. Wu, W. Gu, C. Zhu, D. Du and Y. Lin, Angew. Chem., Int. Ed., 2020, 59, 2565–2576 CrossRef CAS PubMed.
- Y. Wang, K. Qi, S. Yu, G. Jia, Z. Cheng, L. Zheng, Q. Wu, Q. Bao, Q. Wang, J. Zhao, X. Cui and W. Zheng, Nanomicro Lett., 2019, 11, 102 Search PubMed.
- H. Wei and E. Wang, Chem. Soc. Rev., 2013, 42, 6060–6093 RSC.
- J. Wu, X. Wang, Q. Wang, Z. Lou, S. Li, Y. Zhu, L. Qin and H. Wei, Chem. Soc. Rev., 2019, 48, 1004–1076 RSC.
- L. Huang, J. Chen, L. Gan, J. Wang and S. Dong, Sci. Adv., 2019, 5, eaav5490 CrossRef CAS PubMed.
- X. Zhang, G. Li, G. Chen, D. Wu, X. Zhou and Y. Wu, Coord. Chem. Rev., 2020, 418, 213376 CrossRef CAS.
- H. Zhang, X. F. Lu, Z. Wu and X. Lou, ACS Cent. Sci., 2020, 6, 1288–1301 CrossRef CAS PubMed.
- R. Yan, S. Sun, J. Yang, W. Long, J. Wang, X. Mu, Q. Li, W. Hao, S. Zhang, H. Liu, Y. Gao, L. Ouyang, J. Chen, S. Liu, X.-D. Zhang and D. Ming, ACS Nano, 2019, 13, 11552–11560 CrossRef CAS.
- X. Huang and J. T. Groves, Chem. Rev., 2018, 118, 2491–2553 CrossRef CAS PubMed.
- L. Wang, X. Qu, Y. Zhao, Y. Weng and S. Zhou, ACS Appl. Mater. Interfaces, 2019, 11, 35228–35237 CrossRef CAS PubMed.
- H. Xiang, W. Feng and Y. Chen, Adv. Mater., 2020, 32, 1905994 CrossRef CAS.
- M. Huo, L. Wang, Y. Chen and J. Shi, Nat. Commun., 2017, 8, 357 CrossRef PubMed.
- W. Zhang, J. Lu, X. Gao, P. Li, W. Zhang, Y. Ma, H. Wang and B. Tang, Angew. Chem., Int. Ed., 2018, 57, 4891–4896 CrossRef CAS PubMed.
- Z. Tang, Y. Liu, M. He and W. Bu, Angew. Chem., Int. Ed., 2019, 58, 946–956 CrossRef CAS PubMed.
- X. Chen, Y. Liu and W. Bu, Sci. Sin.: Chim., 2020, 50, 159–172 Search PubMed.
- C. Zhang, W. Bu, D. Ni, S. Zhang, Q. Li, Z. Yao, J. Zhang, H. Yao, Z. Wang and J. Shi, Angew. Chem., Int. Ed., 2016, 55, 2101–2106 CrossRef CAS PubMed.
- S. Zhai, X. Hu, Y. Hu, B. Wu and D. Xing, Biomaterials, 2017, 121, 41–54 CrossRef CAS PubMed.
- M. Huo, L. Wang, Y. Wang, Y. Chen and J. Shi, ACS Nano, 2019, 13, 2643–2653 CAS.
- D. Wu, J. Li, S. Xu, Q. Xie, Y. Pan, X. Liu, R. Ma, H. Zheng, M. Gao, W. Wang, J. Li, X. Cai, F. Jaouen and R. Li, J. Am. Chem. Soc., 2020, 142, 19602–19610 CrossRef CAS.
- X. Jiang, H. Wang, R. Yuan and Y. Chai, Anal. Chem., 2018, 90, 8462–8469 CrossRef CAS PubMed.
- B. Dou, J. Yang, R. Yuan and Y. Xiang, Anal. Chem., 2018, 90, 5945–5950 CrossRef CAS PubMed.
- L. Jiao, W. Xu, H. Yan, Y. Wu, C. Liu, D. Du, Y. Lin and C. Zhu, Anal. Chem., 2019, 91, 11994–11999 CrossRef CAS PubMed.
- L. Shen, M. A. Khan, X. Wu, J. Cai, T. Lu, T. Ning, Z. Liu, W. Lu, D. Ye, H. Zhao and J. Zhang, Biosens. Bioelectron., 2022, 205, 114097 CrossRef CAS PubMed.
- X. Niu, Q. Shi, W. Zhu, D. Liu, H. Tian, S. Fu, N. Cheng, S. Li, J. N. Smith, D. Du and Y. Lin, Biosens. Bioelectron., 2019, 142, 111495 CrossRef CAS PubMed.
- M. S. Kim, J. Lee, H. S. Kim, A. Cho, K. H. Shim, T. N. Le, S. S. A. An, J. W. Han, M. I. Kim and J. Lee, Adv. Funct. Mater., 2020, 30, 1905410 CrossRef CAS.
- Y. Wu, L. Jiao, X. Luo, W. Xu, X. Wei, H. Wang, H. Yan, W. Gu, B. Z. Xu, D. Du, Y. Lin and C. Zhu, Small, 2019, 15, 1903108 CrossRef CAS PubMed.
- Q. Chen, S. Li, Y. Liu, X. Zhang, Y. Tang, H. Chai and Y. Huang, Sens. Actuators, B, 2020, 305, 127511 CrossRef CAS.
- L. Yao, S. Gao, S. Liu, Y. Bi, R. Wang, H. Qu, Y. Wu, Y. Mao and L. Zheng, ACS Appl. Mater. Interfaces, 2020, 12, 6268–6275 CrossRef CAS.
- W. Gu, H. Wang, L. Jiao, Y. Wu, Y. Chen, L. Hu, J. Gong, D. Du and C. Zhu, Angew. Chem., Int. Ed., 2020, 59, 3534–3538 CrossRef CAS.
- M. Wang, L. Liu, X. Xie, X. Zhou, Z. Lin and X. Su, Sens. Actuators, B, 2020, 313, 128023 CrossRef CAS.
- B. C. Dickinson and C. J. Chang, Nat. Chem. Biol., 2011, 7, 504–511 CrossRef CAS PubMed.
- J. Zhao, W. Gao, X. Cai, J. Xu, D. Zou, Z. Li, B. Hu and Y. Zheng, Theranostics, 2019, 9, 2843–2855 CrossRef CAS PubMed.
- Z. Jiulong, C. Xiaojun, G. Wei, Z. Linlin, Z. Duowu, Z. Yuanyi, L. Zhaoshen and C. Hangrong, ACS Appl. Mater. Interfaces, 2018, 10, 26108–26117 CrossRef PubMed.
- J. Yao, Y. Cheng, M. Zhou, S. Zhao, S. Lin, X. Wang, J. Wu, S. Li and H. Wei, Chem. Sci., 2018, 9, 2927–2933 RSC.
- Y. P. Lee, S. H. Kim, J. W. Bang, H. S. Lee, S. S. Kwak and S. Y. Kwon, Plant Cell Rep., 2007, 26, 591–598 CrossRef CAS.
- S. Reuter, S. C. Gupta, M. M. Chaturvedi and B. B. Aggarwal, Free Radic. Biol. Med., 2010, 49, 1603–1616 CrossRef CAS PubMed.
- X. Zhuobin, Q. Zhiyue, L. Qi, H. Yixin, L. Dandan, S. Xinggui, F. Kelong, X. Juqun, G. Yunhao and T. Yan, Nat. Commun., 2018, 9, 3713 CrossRef PubMed.
- S. Levy, Nat. Med. Suppl., 2004, 10, S122 CrossRef CAS PubMed.
- H. Bai, H. Yuan, C. Nie, B. Wang, F. Lv, L. Liu and S. Wang, Angew. Chem., Int. Ed., 2015, 127, 13208–13213 CrossRef.
- L. Fan, P. Sun, Y. Huang, Z. Xu, X. Lu, J. Xi, J. Han and R. Guo, ACS Appl. Bio Mater., 2020, 3, 1147–1157 CrossRef CAS PubMed.
- S. Ji, B. Jiang, H. Hao, Y. Chen, J. Dong, Y. Mao, Z. Zhang, R. Gao, W. Chen, R. Zhang, Q. Liang, H. Li, S. Liu, Y. Wang, Q. Zhang, L. Gu, D. Duan, M. Liang, D. Wang, X. Yan and Y. Li, Nat. Catal., 2021, 4, 407–417 CrossRef CAS.
- X. Cui, H. Li, Y. Wang, H. Yuanli, L. Hua, H. Li, X. Han, Q. Liu, F. Yang, L. He, X. Chen, Q. Li, J. Xiao, D. Deng and X. Bao, Chem, 2018, 4, 1902–1910 CAS.
- C. Zhao, C. Xiong, X. Liu, M. Qiao, Z. Li, T. Yuan, J. Wang, Y. Qu, X. Wang, F. Zhou, Q. Xu, S. Wang, M. Chen, W. Wang, Y. Li, T. Yao, Y. Wu and Y. Li, Chem. Commun., 2019, 55, 2285–2288 RSC.
- S. Tian, Q. Fu, W. Chen, Q. Feng, Z. Chen, J. Zhang, W.-C. Cheong, R. Yu, L. Gu, J. Dong, J. Luo, C. Chen, Q. Peng, C. Draxl, D. Wang and Y. Li, Nat. Commun., 2018, 9, 2353 CrossRef PubMed.
- B. Singh, M. B. Gawande, A. D. Kute, R. S. Varma, P. Fornasiero, P. McNeice, R. V. Jagadeesh, M. Beller and R. Zbořil, Chem. Rev., 2021, 121, 13620–13697 CrossRef CAS PubMed.
- U. G. K. Wegst, H. Bai, E. Saiz, A. P. Tomsia and R. O. Ritchie, Nat. Mater., 2015, 14, 23–36 CrossRef CAS.
- Z. Chu and S. Seeger, Adv. Mater., 2015, 27, 7775–7781 CrossRef CAS PubMed.
- G. Mirabello, J. J. M. Lenders and N. A. J. M. Sommerdijk, Chem. Soc. Rev., 2016, 45, 5085–5106 RSC.
- Y. Xiong, W. Sun, P. Xin, W. Chen, X. Zheng, W. Yan, L. Zheng, J. Dong, J. Zhang, D. Wang and Y. Li, Adv. Mater., 2020, 32, 2000896 CrossRef CAS PubMed.
- E. Luo, M. Xiao, Y. Wang, J. Ge, C. Liu and W. Xing, ChemCatChem, 2018, 10, 3653–3658 CrossRef CAS.
- K. Yuan, D. Luetzenkirchen-Hecht, L. Li, L. Shuai, Y. Li, R. Cao, M. Qiu, X. Zhuang, M. K. H. Leung, Y. Chen and U. Scherf, J. Am. Chem. Soc., 2020, 142, 2404–2412 CrossRef CAS.
- Y. Han, Y. Wang, R. Xu, W. Chen, L. Zheng, A. Han, Y. Zhu, J. Zhang, H. Zhang, J. Luo, C. Chen, Q. Peng, D. Wang and Y. Li, Energy Environ. Sci., 2018, 11, 2348–2352 RSC.
- H. Shen, E. Gracia-Espino, J. Ma, H. Tang, X. Mamat, T. Wagberg, G. Hu and S. Guo, Nano Energy, 2017, 35, 9–16 CrossRef CAS.
- C. Zhu, Q. Shi, B. Z. Xu, S. Fu, G. Wan, C. Yang, S. Yao, J. Song, H. Zhou, D. Du, S. P. Beckman, D. Su and Y. Lin, Adv. Energy Mater., 2018, 8, 1801956 CrossRef.
- J. Zhang, M. Zhang, Y. Zeng, J. Chen, L. Qiu, H. Zhou, C. Sun, Y. Yu, C. Zhu and Z. Zhu, Small, 2019, 15, 1900307 CrossRef PubMed.
- Z. Wang, H. Jin, T. Meng, K. Liao, W. Meng, J. Yang, D. He, Y. Xiong and S. Mu, Adv. Funct. Mater., 2018, 28, 1802596 CrossRef.
- S. Gong, C. Wang, P. Jiang, L. Hu, H. Lei and Q. Chen, J. Mater. Chem. A, 2018, 6, 13254–13262 RSC.
- B. Wang, J. Zou, X. Shen, Y. Yang, G. Hu, W. Li, Z. Peng, D. Banham, A. Dong and D. Zhao, Nano Energy, 2019, 63, 103851 CrossRef CAS.
- T. Palaniselvam, V. Kashyap, S. N. Bhange, J.-B. Baek and S. Kurungot, Adv. Funct. Mater., 2016, 26, 2150–2162 CrossRef CAS.
- R. Ma, G. Lin, Q. Ju, W. Tang, G. Chen, Z. Chen, Q. Liu, M. Yang, Y. Lu and J. Wang, Appl. Catal., B, 2020, 265, 118593 CrossRef CAS.
- J. Han, X. Meng, L. Lu, J. Bian, Z. Li and C. Sun, Adv. Funct. Mater., 2019, 29, 1808872 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |