
Nanoparticulates reduce tumor cell migration through affinity interactions with extracellular migrasomes and retraction fibers†
Yuxi
Cheng
 ab,
Junji
Ren
ab,
Shumin
Fan
ab,
Peiyao
Wu
abc,
Wenshu
Cong
ab,
Yuxing
Lin
ab,
Shaojie
Lan
ab,
Siyang
Song
ab,
Bin
Shao
d,
Wenbing
Dai
ab,
Xueqing
Wang
ab,
Hua
Zhang
ab,
Bo
Xu
b,
Wenzhe
Li
b,
Xia
Yuan
b,
Bing
He
*ab and
Qiang
Zhang
*abc
ab,
Junji
Ren
ab,
Shumin
Fan
ab,
Peiyao
Wu
abc,
Wenshu
Cong
ab,
Yuxing
Lin
ab,
Shaojie
Lan
ab,
Siyang
Song
ab,
Bin
Shao
d,
Wenbing
Dai
ab,
Xueqing
Wang
ab,
Hua
Zhang
ab,
Bo
Xu
b,
Wenzhe
Li
b,
Xia
Yuan
b,
Bing
He
*ab and
Qiang
Zhang
*abc
aBeijing Key Laboratory of Molecular Pharmaceutics and New Drug Delivery Systems, School of Pharmaceutical Sciences, Peking University, Beijing, 100191, China. E-mail: zqdodo@bjmu.edu.cn; hebingmumu@bjmu.edu.cn
bState Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, Beijing 100191, China
cSchool of Pharmacy, Shenyang Pharmaceutical University, Shenyang 110016, China
dDepartment of Medical Oncology, Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education), Peking University Cancer Hospital, Beijing 100142, China
First published on 25th May 2022
Abstract
Nano–tumor interactions are fundamental for cancer nanotherapy, and the cross-talk of nanomedicines with the extracellular matrix (ECM) is increasingly considered essential. Here, we specifically investigate the nano–ECM interactivity using drug-free nanoparticulates (NPs) and highly metastatic cancer cells as models. We discover with surprise that NPs closely bind to specific types of ECM components, namely, retraction fibers (RFs) and migrasomes, which are located at the rear of tumor cells during their migration. This interaction is observed to alter cell morphology, limit cell motion range and change cell adhesion. Importantly, NPs are demonstrated to inhibit tumor cell removal in vitro, and their anti-metastasis potential is preliminarily confirmed in vivo. Mechanically, the NPs are found to coat and form a rigid shell on the surface of migrasomes and retraction fibers via interaction with lipid raft/caveolae substructures. In this way, NPs block the recognition, endocytosis and elimination of migrasomes by their surrounding tumor cells. Thereby, NPs interfere with the cell–ECM interaction and reduce the promotion effect of migrasomes on cell movement. Additionally, NPs trigger alteration of the expression of proteins related to cell-cell adhesion and cytoskeleton organization, which also restricts cell migration. In summary, all the findings here provide a potential target for anti-tumor metastasis nanomedicines.
New conceptsNano–tumor interaction is fundamental to understanding the principles of cancer nanotherapy. Furthermore, the cross-talk of nanomedicines with the extracellular matrix (ECM) is increasingly considered to be essential. In this work, we aim to explore the impact of nano–ECM interactivity on tumor cell metastasis. We find for the first time that nanoparticulates (NPs) can closely bind to specific types of lipid components in the ECM, namely, retraction fibers (RFs) and migrasomes, which are generated during tumor cell migration. Migrasomes, which are a new vesicular organelle discovered in recent years, are reported to mediate cell functions. We demonstrate that this nano–ECM interaction alters cell morphology, limits cell motion range, changes cell adhesion, and finally inhibits tumor cell metastasis in vitro and in vivo. Mechanically speaking, the NPs coat and form a rigid shell on the surface of migrasomes and RFs via interaction with the lipid raft/caveolae substructures. In this way, NPs block the recognition, endocytosis and elimination of migrasomes by their surrounding tumor cells. Thereby, NPs interfere with the cell–ECM interaction and reduce the promotion effect of migrasomes on cell movement. Additionally, NPs trigger alteration of the expression of proteins related to cell-cell adhesion and cytoskeleton organization, which also restricts cell migration. |
Introduction
The rapid development of nanomedicines in recent years provides a promising strategy for cancer therapy.1 By incorporating nanomaterials as carriers or directly using nanosized drugs, nanomedicines exhibit outstanding capability in improving therapeutic efficacy while reducing side effects.2 However, due to the high specific surface area and reactivity on the particle surface,3,4 nanomedicines always interact with a wide variety of biological ingredients with high affinity, making their in vivo delivery process more complicated than traditional preparations.5 The detailed mechanisms of delivery towards tumors for many nanomedicines have not been fully elucidated to date, and we do not understand clearly how they interact with different ingredients in the tumor microenvironment (TME).6,7The extracellular matrix (ECM) is one of the important components of the TME.8 ECM proteins including collagen, fibronectin and laminin regulate cell proliferation and differentiation, as well as tumor migration.8,9 Notably, recent studies have found that in addition to proteins and polysaccharides, tumor cells also excrete lipid components such as extracellular vesicles into the TME.10 These lipid components induce the recognition and internalization of surrounding cells and regulate multiple functions of tumors.11,12 Thus, broadly speaking, they can also be regarded as a specific ECM ingredient. In summary, the ECM forms a compact and complex barrier against the delivery of nanomedicines around the tumor.13,14 However, the incomplete understanding of nano–ECM interactions limits the efficiency of nano-delivery.15 In the R&D process in particular, the unclear understanding of nano–ECM interactions may even affect the ultimate clinical application of nanomedicines.16
In this study, we aimed at exploring the interactive characteristics between nanomedicines and ECM. In particular, we focused on a specific ECM ingredient, lipid components. Based on the close correlation of the ECM with tumor invasion, we mainly evaluated the influence of extracellular lipid structures on cell movement and migration. Polystyrene nanoparticles (NPs) with different diameters, PLGA nanoparticles (PLGA-NPs), SiO2 nanoparticles (SiO2-NPs) and liposomes were selected here as nanomedicine models, and the breast cancer cell line MDA-MB-231 and 4T1 cells were cultured and investigated in this study due to their highly metastatic behavior. Via classical migration assay and dynamic analysis of cell movement and morphology based on high-content imaging technology, we discovered the affinity of nanoparticles with specific kinds of lipid components, namely, retraction fibers and migrasomes. The latter, migrasomes, are a unique extracellular structure that has only been discovered in recent years17 and are receiving increasing attention following incremental studies on their functions. For the first time, we investigated the influence of their interaction with nanoparticles on cell migration, and demonstrated the retarding effect of nanoparticles on the role of migrasomes in accelerating tumor metastasis. We believe this finding may offer a new point of view for the improvement of the anti-tumor efficacy of nanomedicines.
1. NPs attenuate cell migration via altering cell morphology and limiting cell motion range
Polystyrene nanoparticles (NPs) were selected as the main model nanomedicine for the overall investigation in this study. We prepared suspensions of NPs with an average diameter of 70 nm, 200 nm and 500 nm. All three types of NPs possessed a relatively narrow size distribution range, with a PDI lower than 0.2 (Fig. S1A and B, ESI†). Based on the size distribution curves, the three NPs could be clearly distinguished from each other (Fig. 1A). Importantly, the model NPs obtained here could cover the size range of most nanomedicines involved in the field (30–1100 nm),18,19 ensuring that the size-dependent biological effects of NPs could be fully illuminated. The morphological uniformity and roundness of the NPs were further confirmed (Fig. 1B, Fig. S1C, ESI†). Then, various assays of cell viability showed that none of the three NPs generated obvious cytotoxicity, enabling the subsequent evaluation of biological effects (Fig. S1D–G, ESI†). Investigations of the surface chemistry further demonstrated that the three NPs had similar structure and composition on the particle surface (Fig. S2, ESI†). In addition, when the three NPs were dispersed in a complete culture medium, they exhibited the same characteristics of protein corona (Fig. S3, ESI†). It suggested that this series of properties remained consistent among the three types of NPs and would not interfere in the comparison study based on particle size.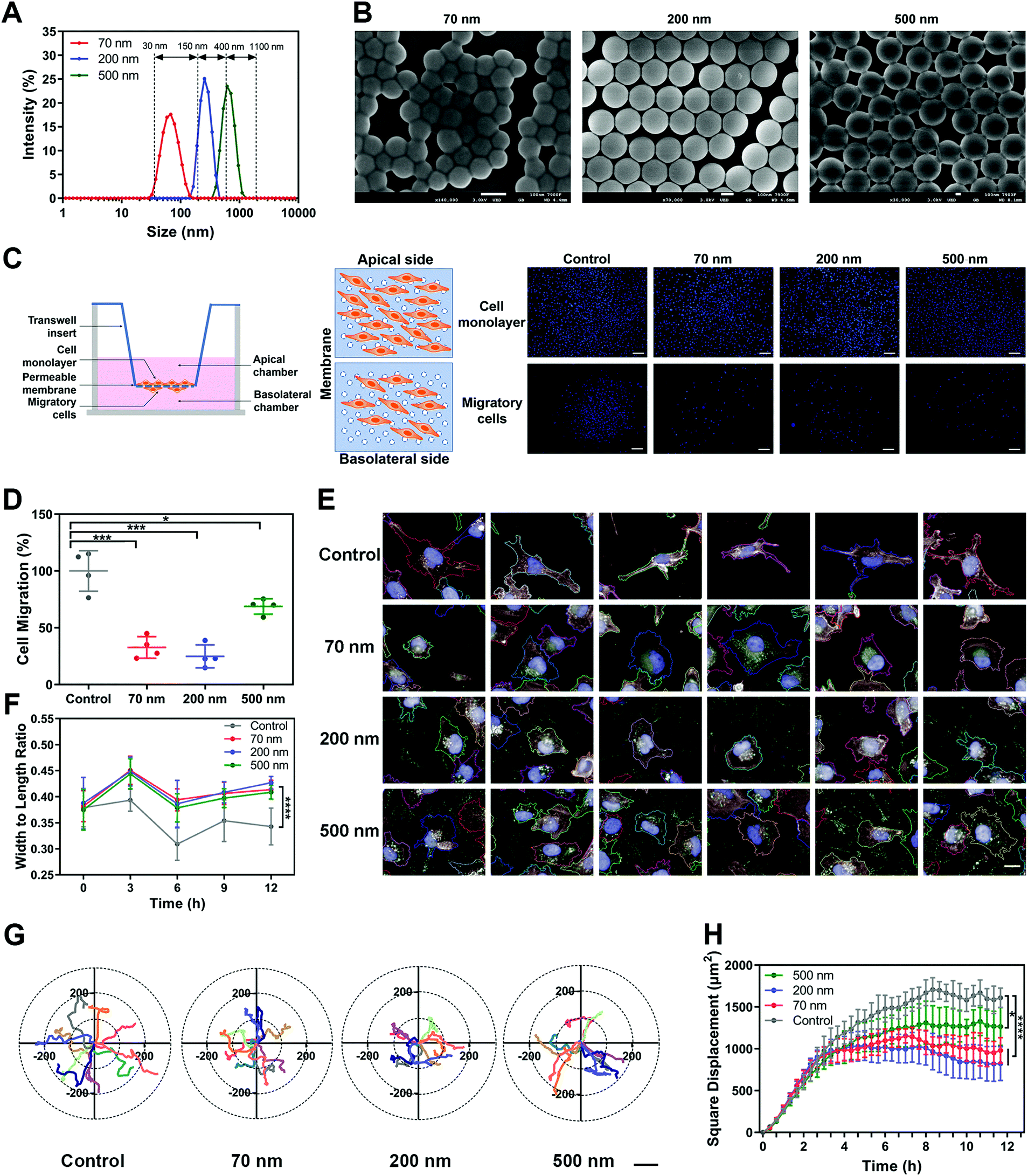 | ||
Fig. 1 NPs attenuate the migration of MDA-MB-231 cells via altering cell morphology and limiting cell motion range. (A) Intensity size distribution of the polystyrene nanoparticles (NPs). (B) Representative SEM images of NPs with different particle sizes. Scale bar, 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) nm. (C) Left, scheme of Transwell assay. Right, representative images of the apical and basolateral sides of the Transwell membrane. Scale bar, 200μm. (D) Migration ratio of MDA-MB-231 cells after being incubated with NPs, as tested using the Transwell assay (n = 4). (E) Representative images showing the morphological profiles of MDA-MB-231 cells with or without incubation with NPs. Scale bar, 20μm. (F) Width to length ratio of MDA-MB-231 cells calculated using a high content analysis system (n = 51). (G) Coordinate systems illustrating representative movement tracks of MDA-MB-231 cells. Scale bar, 200μm. (H) Square displacement of MDA-MB-231 cells incubated with NPs at each time point as calculated using the high content analysis system (n = 5). (Data are presented as mean ± SD. Statistical significances were calculated using Student's t-test: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.). nm. (C) Left, scheme of Transwell assay. Right, representative images of the apical and basolateral sides of the Transwell membrane. Scale bar, 200μm. (D) Migration ratio of MDA-MB-231 cells after being incubated with NPs, as tested using the Transwell assay (n = 4). (E) Representative images showing the morphological profiles of MDA-MB-231 cells with or without incubation with NPs. Scale bar, 20μm. (F) Width to length ratio of MDA-MB-231 cells calculated using a high content analysis system (n = 51). (G) Coordinate systems illustrating representative movement tracks of MDA-MB-231 cells. Scale bar, 200μm. (H) Square displacement of MDA-MB-231 cells incubated with NPs at each time point as calculated using the high content analysis system (n = 5). (Data are presented as mean ± SD. Statistical significances were calculated using Student's t-test: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.). | ||
Subsequently, we investigated the influence of NPs on cell migration using classical Transwell and wound healing assays. As shown in Fig. 1C and D, cell counting revealed that the three NPs significantly attenuated cell migration, with the 200 nm group showing the strongest impact. Additionally, we verified that the NPs themselves did not block the membrane pores during incubation (Fig. S4A, ESI†). The wound-healing assay demonstrated the same fact (Fig. S4B and C, ESI†). To clarify whether the effect of the NPs was composition-related, PLGA-NPs were prepared and tested accordingly (Fig. S4D–F, ESI†). Notably, they also reduced the cell migration significantly (Fig. S4G–H, ESI†). In fact, similar observations were also found in the study with SiO2-NPs (Fig. S6A–E, ESI†). Next, we prepared soft liposomes and performed the same experiment as for the hard NPs (polystyrene NPs, PLGA-NPs, and SiO2-NPs) to test the effect of particle stiffness.20–23 Interestingly, the addition of liposomes could not inhibit cell movement (Fig. S4I–M, ESI†). Thus, the attenuation of cell migration induced by nanoparticles might be a general fact, but it was dependent on the particle rigidity.
Cell morphology has been widely confirmed to affect cell migration.24,25 Here, we used a high-content analysis system to evaluate the cell morphology during nanoparticle incubation. As shown in Fig. S5A–D (ESI†), the three NPs significantly enhanced cell roundness and decreased cell length, but did not change the cell area or width. Namely, the NPs caused cell contraction (Fig. 1E and F). Compared to the round shape, a spindle-like morphology endows cells with greater migration capability.26,27 Thus, the alteration in cellular morphology induced by NPs was closely related to the attenuated cell migration.
Furthermore, we studied the cellular movement dynamics by depicting the motion tracks of individual cells after nanoparticle incubation (Movie 1, ESI†). As shown in Fig. 1G, all three NPs limited the cell motion range. Dynamic quantification of the square displacement of cell movement further confirmed this finding (Fig. 1H).28 Additionally, NPs decreased the accumulated distance and total displacement of cellular motion, but did not down-regulate the average speed of cell movement (Fig. S5E–H, ESI†). Both PLGA-NPs and SiO2-NPs showed a similar effect to that of polystyrene NPs on cell movement (Fig. S6A–E, ESI†). Additionally, we investigated the effect of bigger nanoparticles. Interestingly, 1000 nm NPs did not limit cell migration, but even promoted the motion of cells (Fig. S7, ESI†). Although the mechanism behind this is unclear, this result suggested that the inhibitory effect on cell migration was restricted to the nanoscale range of particles. In summary, these findings demonstrated that nanoparticles attenuated cell migration by limiting the cellular motion range.
2. NPs inhibit cell movement by binding to ECM, interfering with cell–ECM interaction and altering cell adhesion
To clarify the underlying mechanism behind the restriction effect of NPs on cell movement, we monitored the real-time dynamic processes of cellular motion (Movie 2, ESI†). Surprisingly, the NPs obviously adhered to the trajectories left after cell movement (Fig. 2A). This binding phenomenon became increasingly obvious with prolonged incubation time, leading to a clear presentation of the movement tracks of cells. Notably, the 200 nm NPs demonstrated the strongest binding ability among all the NPs, which was consistent with their having the strongest inhibitory effect on cell migration (Fig. 1D), suggesteding that the interaction between NPs and ECM might be the key factor affecting cell movement.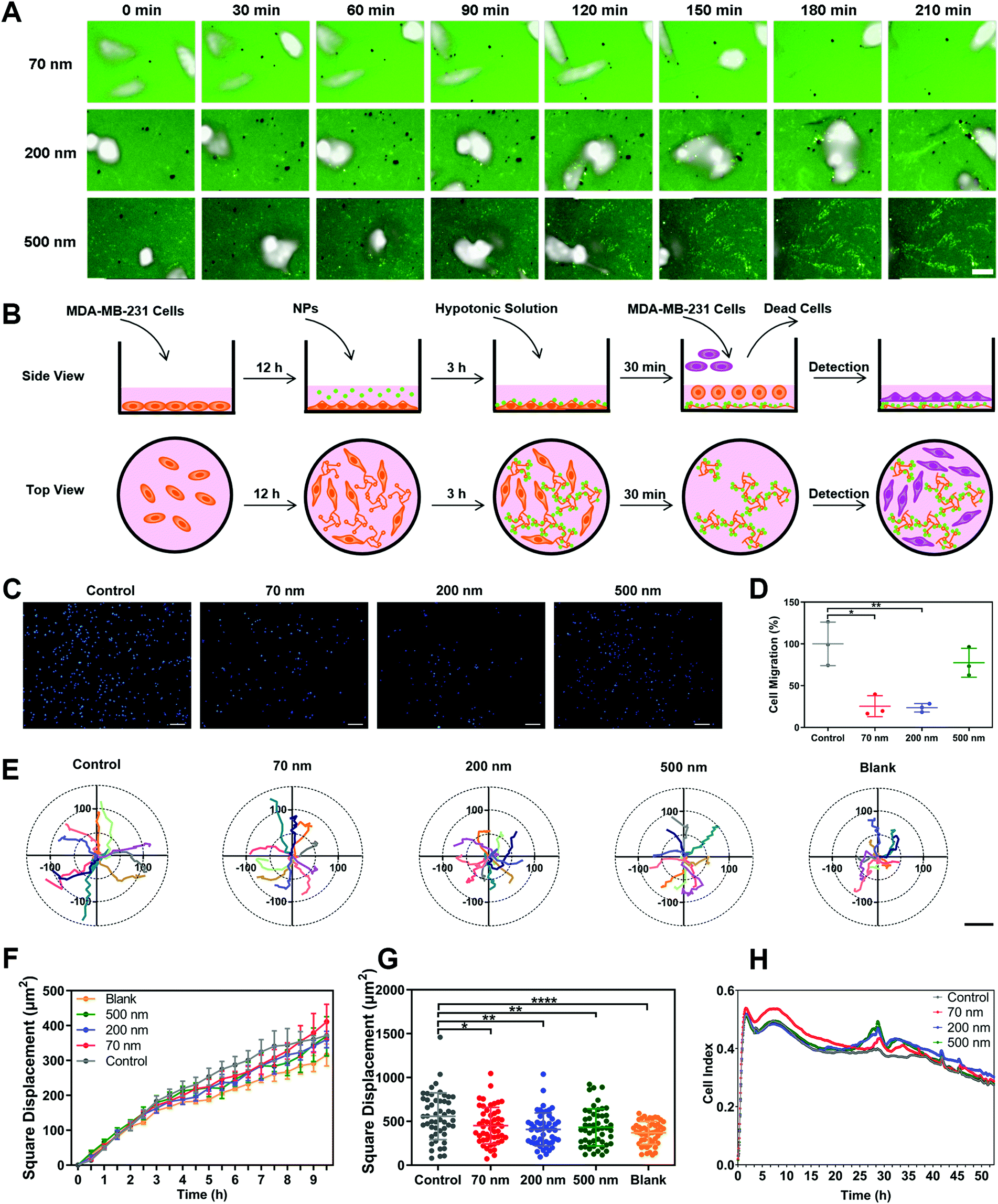 | ||
| Fig. 2 NPs inhibit the movement of MDA-MB-231 cells by binding to the ECM, interfering with cell–ECM interaction and altering cell adhesion. (A) Cell movement trajectories generated by the binding of polystyrene nanoparticles (NPs) at different time points. Scale bar, 20μm. (B) Scheme of the experiment investigating the role of the nano–ECM interaction in cell migration. (C) Representative images of the basolateral side of the Transwell membrane. Scale bar, 200μm. (D) Migration ratio of MDA-MB-231 cells incubated on the ECM coated with NPs tested by the Transwell assay (n = 3). (E) Coordinate systems illustrating representative cell movement tracks on the ECM coated with NPs. Scale bar, 200μm. (F) Square displacement of MDA-MB-231 cells incubated on the ECM coated with NPs at each time point calculated using a high content analysis system (n = 3). (G) Mean square displacement over 10 h of MDA-MB-231 cells incubated on the ECM coated with NPs calculated using a high content analysis system (n = 51). (H) Cell index of MDA-MB-231 cells incubated on the ECM coated with NPs at each time point measured using an RTCA system (n = 3). (Data are presented as mean ± SD. Statistical significances were calculated using Student's t-test: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.). | ||
To verify this hypothesis, we designed an experiment to elucidate the roles of nano–ECM interaction in cell migration. As shown in Fig. 2B, we first cultured cells in dishes and incubated the cells with different NPs for a period to induce the formation of the ECM and to promote the interaction between NPs and the ECM. Subsequently, the complete medium was replaced with a hypotonic solution, which caused the cells to burst and float, but the ECM still remained at the bottom of the dishes. The medium containing cell debris was then replaced by a fresh culture medium containing living cells. Finally, the movement of newly added cells was detected using a high-content analysis system (Movie 3, ESI†). As presented in Fig. 2C and D, the pre-binding of 70 nm and 200 nm NPs on the ECM significantly reduced cell migration. 500 nm NPs also displayed a slight inhibitory effect. In addition, the square displacements of cellular movement in all the nanoparticle groups were reduced due to the nano–ECM interactions (Fig. 2E to G). Other parameters, including accumulated distance, total displacement, and average speed, all decreased after the addition of NPs (Fig. S8A–C, ESI†). Interestingly, the cells showed the slowest movement when they were directly seeded in dishes without an ECM coating. This revealed that the ECM promoted cell movement via cell–ECM interaction.29,30 More importantly, NPs might interfere with this interaction via their pre-binding with ECM components.
Next, we tested the effect of nano–ECM interactions on cell adhesion using a real-time cell analysis (RTCA) system (Fig. S8D, ESI†). A higher index indicates greater cell adhesion and stronger restriction on cellular movement.31Fig. 2H reveals that the pre-binding of NPs on the ECM enhanced the adhesive kinetics of cells. The 70 nm NPs induced greater cell adhesion initially, while 200 nm and 500 nm NPs showed a stronger effect at the late stage. This might be attributed to differences in cellular interactions such as uptake among the three NPs (Fig. S8E, ESI†). In any case, these findings demonstrated that NPs could enhance cell adhesion and reduce cellular movement through interaction with the ECM.
3. Nano–ECM interaction is reflected by the affinitive adhesion of NPs with retraction fibers
Due to the high-affinity adhesion of NPs to the movement trajectories behind the cells shown in Fig. 2A, it was speculated that the nano–ECM interaction might be related to the residual structures after cell movement. In fact, these structures are referred as retraction fibers (RFs), and are specific for most tumor cells with high metastatic capacity.32 RFs are a type of special plasma membrane structures and usually trail the rear of cells along with cell movement (Fig. 3A, red and blue boxes).12 Based on the lipid compositions of RF, we used the lipid-specific fluorescent probe DiD to label the RFs (Fig. 3B) and investigated the dynamic interaction between the NPs and RFs using confocal laser scanning microscopy (CLSM).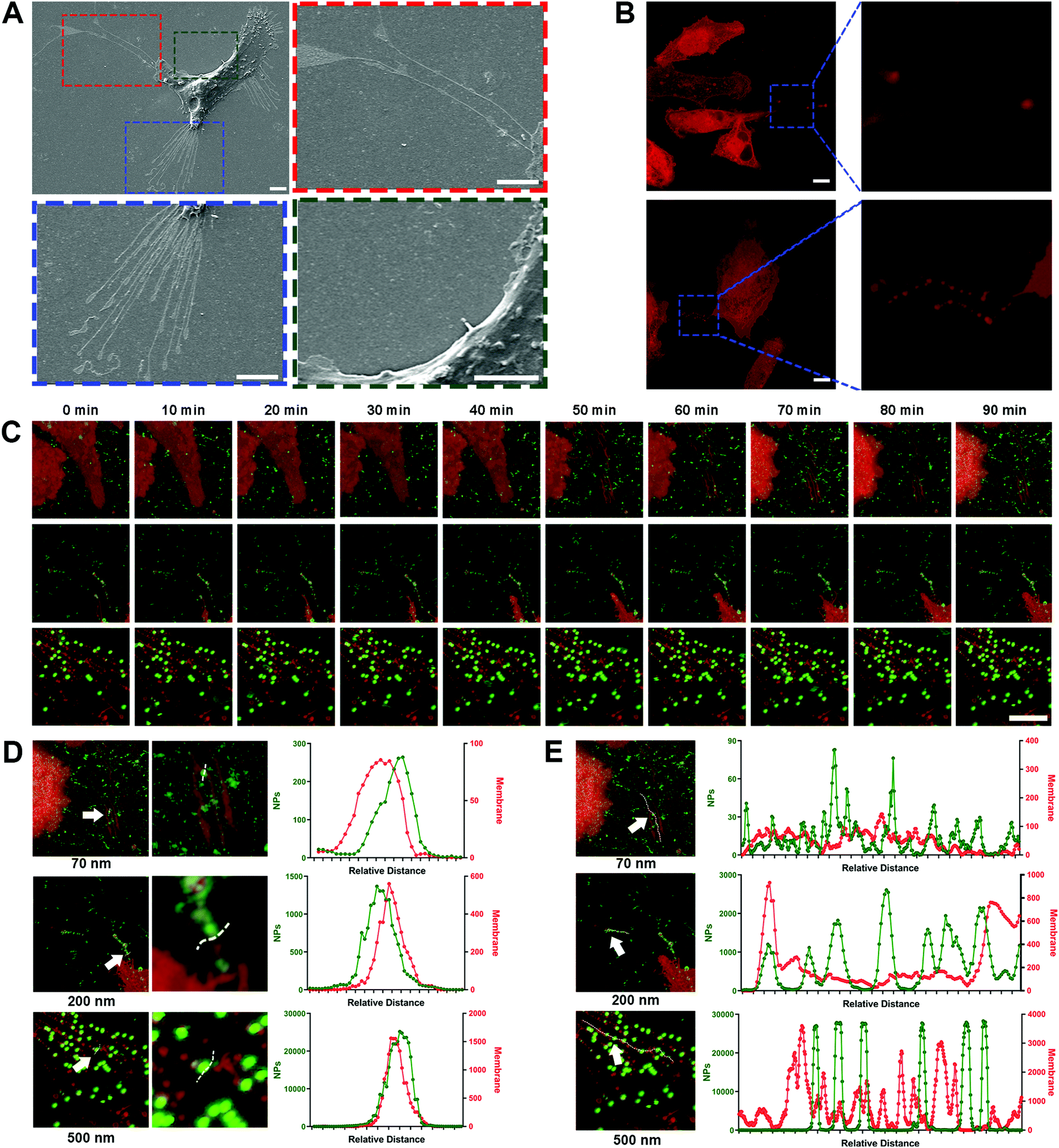 | ||
| Fig. 3 Nano–ECM interaction is reflected by the adhesion affinity of NPs with retraction fibers. (A) Representative SEM images showing the morphological characteristics and RFs of MDA-MB-231 cells. Red and blue boxes, cellular leading edge; green box, cellular trailing edge. Scale bar, 10μm. (B) Representative CLSM images showing the morphological characteristics and RFs of the cells. Scale bar, 10μm. (C) Representative CLSM images showing the dynamic combination between RFs and 70 nm, 200 nm and 500 nm polystyrene nanoparticles (NPs) at different time points. Scale bar, 10μm. (D) Co-localization analysis of NPs and membrane structures at different positions across RFs (white lines, indicated by arrows). (E) Co-localization analysis of NPs and membrane structures at different positions along RFs (white lines, indicated by arrows). | ||
Surprisingly, all three NPs, especially the 200 nm and 500 nm NPs, clearly bound to the RFs at the rear of living cells (Fig. 3C, Movie 4, ESI†). This phenomenon was confirmed via quantitative co-localization analysis (Fig. 3D). As the extension of incubation time, more NPs bound to the surface of the RFs, clustering as spheres and then forming bead-string structures along the RFs (Fig. 3E). Interestingly, PLGA-NPs and SiO2-NPs also presented similar behavior (Fig. S9A–D, ESI†). Generally, these findings fully indicated the strong interaction between NPs and RFs, which has never been reported before.
Next, we investigated the interactions between the NPs and ECM proteins such as fibronectin and collagen.33,34 Unlike RFs, fibronectin is usually located at the cell edge rather than in the intercellular area, and more importantly, the NPs did not show any apparent co-localization with fibronectin (Fig. S9E, ESI†). Pre-binding of NPs with collagen also failed to attenuate the cell movement (Fig. S9F–J, ESI†). These findings indicated that NPs might be more likely to interact with lipid components like RFs in the ECM. The interaction between the NPs and ECM proteins was not the core driving force for the inhibition of tumor cell movement.
4. NPs interact with migrasomes in RFs via lipid rafts/caveolae substructures
To explore the mechanism of NP–RF interactions in detail, we investigated the location of nanoparticles in cellular substructures by labeling the lipid rafts/caveolae in the RFs and cell membrane using cholera toxin subunit B (CT-B). As shown in Fig. S10A–C (ESI†), the NPs were concentrated in the substructures that were rich in lipid rafts/caveolae.35Lipid rafts/caveolae have been widely reported to be involved in nano–bio interactions and to mediate the endocytosis of a variety of nanoparticles.36–38 These substructures are rich in cholesterol and sphingomyelin, making it easier for the lipid membrane to generate vesicles through invagination or budding.39 Interestingly, we found that these vesicles also formed in the RFs (Fig. 4A and B, arrows); these structures have been given the name ‘migrasomes’ and are mainly located at the ends or intersections of RFs.17 These migrasomes ranged from hundreds of nanometers to a few microns in size, and some of them presented irregular shapes, likely due to being dragged by the RFs. In fact, the structure of migrasomes is similar to that of exosomes, containing a variety of signal regulation proteins, including growth factors, chemokines, cytokines and morphogens, so as to regulate multiple functions of certain types of cells.40,41
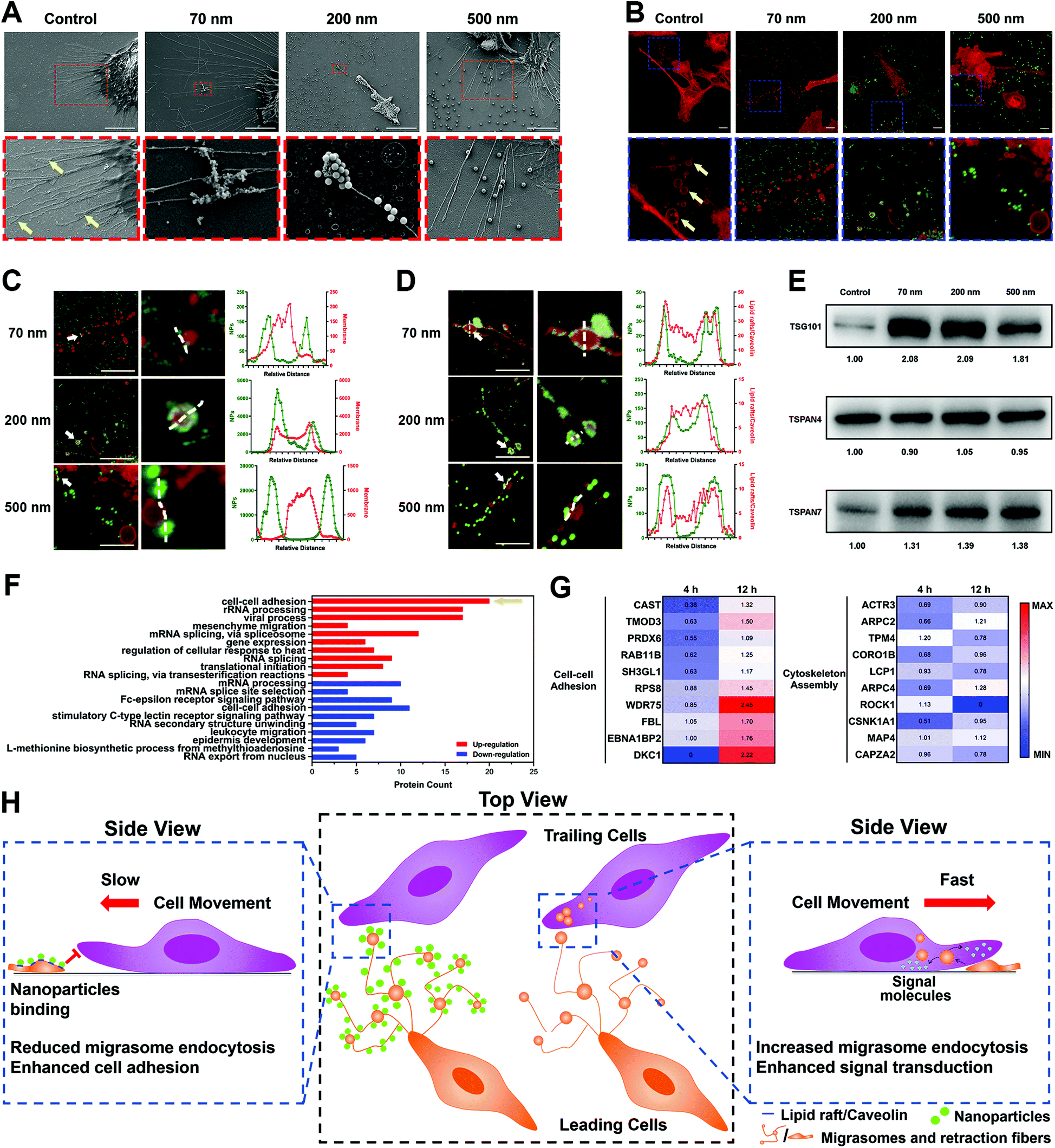 | ||
| Fig. 4 NPs inhibit tumor cell removal by blocking cell uptake of migrasomes via interaction with lipid rafts/caveolae and triggering alteration of the expression of motion-related proteins. (A) Representative SEM images showing the localization and characteristics of migrasomes, and their combination with NPs. Arrows indicate migrasomes. Scale bar, 10μm. (B) Representative CLSM images showing the localization and characteristics of migrasomes, and their combination with NPs. Arrows indicate migrasomes. Scale bar, 10μm. (C) Co-localization analysis of NPs and membrane structures at different positions across migrasomes (white lines, indicated by arrows). Scale bar, 10μm. (D) Co-localization analysis of NPs and lipid rafts/caveolae (labeled by CT-B) at different positions across migrasomes (white lines, indicated by arrows). Scale bar, 10μm. (E) Western blot analysis of TSPAN4, TSPAN7 and TSG101 expression in the NP-treated ECM. (F) Diagram illustrating the DAVID cluster analysis of cellular proteins after cells were cultured on NP-coated ECM for 12 h. (G) Heat maps illustrating the changes in the expression of proteins involved in cell–cell adhesion and cytoskeleton assembly after cells were cultured on NP-coated ECM for 4 h and 12 h. Data in the heat maps represent the change ratios of protein expression compared with the control group (cells cultured on the ECM without NPs). (H) Scheme of the inhibitory effect of nanoparticles on tumor migration through affinity interactions with extracellular migrasomes and retraction fibers. | ||
Surprisingly, when NPs were incubated with the ECM, they closely bound to migrasomes in the RFs (Fig. 4A–C). This was further demonstrated by separately labeling the lipid rafts/caveolae and cholesterol in migrasomes with CT-B and filipin, as well as by measuring the co-localization parameters of these markers with the NPs (Fig. 4D, Fig. S11A–C, ESI†).
In addition, we examined the influence of the NPs on the migrasomes by extracting ECM components and detecting the marker proteins of migrasomes using a western blot (WB) assay. The WB images of Tspan4 and Tspan7, both of which are specific structural proteins of migrasomes,40–42 as well as TSG101, which is a common component of extracellular vesicles,43 confirmed the presence of migrasomes (Fig. 4E). The corresponding grayscale analysis further illustrated that nanoparticle incubation resulted in an increase in the number of migrasomes (Fig. S11D, ESI†). Actually, this result could be attributed to two possibilities. One possible reason is that, the NPs might induce the generation of more migrasomes by promoting the secretion of ECM.12 Otherwise, NPs might interfere with the elimination of migrasomes by surrounding cells.
As a proof of concept, we used label-free quantitative (LFQ) proteomics to identify the secreted ECM proteins after nanoparticle treatment. A total of 1223 proteins were confirmed. The Venn diagram illustrated that the most high-abundance proteins coexisted in both the control and NP-treated groups (Fig. S12A, ESI†). Additionally, the correlation coefficient of protein expression between the control and NP-treated groups was almost the same as the data from the biological duplication (Fig. S12B, ESI†). Thus, the NPs did not obviously modulate the ECM excretion.
To test another possibility, we prepared DiD-labeled large multivesicular liposomes (LMVs) with a similar size to simulate migrasomes and settled them on dishes (Fig. S13A, ESI†). NPs were added and incubated in these dishes for a period to induce their binding on the LMVs as a simulation of nano–migrasome interaction (Fig. S13B, ESI†). Finally, living cells were seeded and cultured in the dishes, and the uptake of LMVs by these cells was detected using flow cytometry. As shown in Fig. S13C (ESI†), nanoparticle incubation significantly reduced the cellular uptake of LMVs. By inference, the NPs could protect migrasomes from being internalized by surrounding cells.
5. NPs inhibit tumor cell removal by blocking cellular uptake of migrasomes and triggering alteration of the expression of motion-related proteins
Although we demonstrated the interaction of NPs with RFs and migrasomes, their impact on cell movement remained unclear. Here, we treated cells with methyl-β-cyclodextrin (MβCD), which damaged the lipid rafts/caveolae structures of the cellular membrane.40 This treatment triggered a significant reduction of fiber length and increase in RF fragments. However, the amount of migrasomes remained basically the same and the integral structures of the RFs and migrasomes were not destroyed (Fig. S14A–D, ESI†). However, tests based on multiple parameters showed that MβCD only affected the cell movement slightly (Fig. S14E–I, ESI†). Therefore, these findings suggested that the lipid rafts/caveolae of RFs and migrasomes might not be a critical factor for cell movement, although they were essential for the binding of NPs.It seemed reasonable to suppose that the internal contents of the RFs and migrasomes might be responsible. Therefore, we used RIPA lysis buffer to treat the ECM, which caused the complete destruction of the lipid RFs and migrasomes, and the release of their contents. As presented in Fig. S15A and B (ESI†), the square displacement of cellular movement was obviously reduced after RIPA treatment, possibly due to the release of the contents of the RFs and migrasomes. Other parameters also verified the reduction of cell motion (Fig. S15C–E, ESI†). Thus, it was possible that the biomolecules contained in the cavities of the RFs and migrasomes had the ability to promote cell movement. In fact, it has been reported that migrasomes are a kind of special extracellular vesicles, and their production is due to the higher rigidity at some regions in the RFs. When RFs are stretched during cell movement, migrasomes take shape automatically at these high-rigidity regions driven by physical factors.40 More importantly, a variety of biologically active molecules, including signal regulatory proteins, receptors, miRNAs, etc., are concentrated in migrasomes.44 Similar to the regulation principle of exosomes, migrasomes also must be recognized and internalized by surrounding cells to regulate cellular functions.17,45
It was demonstrated above that the cellular uptake of migrasomes was reduced owing to the binding of NPs. In order to clarify the molecular mechanisms, we used LFQ proteomics to examine the cellular response to the ECM and NPs (Fig. S16A and B, ESI†). The cluster analysis of DAVID function in Fig. 4F and Fig. S16C (ESI†) showed that when the cells were separately cultured with NP-coated ECM for different lengths of time, their functions were differently affected. Specifically, during the 4 h incubation, the binding of NPs to ECM induced the down-regulation of proteins relevant to cell–cell adhesion and actin assembly (Fig. S16C, ESI†). This meant that the cultured cells underwent a contraction phase during the initial stress response. Notably, during the 12 h incubation, many proteins in the cell–cell adhesion category were instead up-regulated, while the expression of cytoskeleton-associated proteins was not increased (Fig. 4F). This reflected the enhancement of cell adhesion and restriction of cellular motility. This double effect demonstrated that the cellular migration was continuously inhibited. The heat map of relevant representative proteins in Fig. 4G further confirmed the above facts.
In conclusion, all these findings revealed that NPs reduced cell movement through interaction with RFs and migrasomes. As illustrated in Fig. 4H, this interaction relied on the high-affinity interaction between NPs and the lipid rafts/caveolae substructures in RFs and migrasomes. By binding and forming a hard “protective shield” on the migrasome surface, the NPs blocked the cellular uptake of migrasomes, thereby reducing their promotion effect on cell movement. Compared with the “soft” lipid components in migrasomes, the “hard” texture of NPs caused the cells to express more proteins related to cell–cell adhesion to achieve strongly adhesion on the ECM (Fig. 4F). As a feedback, this finally restricted the movement of cells and their migration.
6. Peripheral NPs around a tumor inhibit tumor metastasis in vivo
Finally, to investigate whether the inhibitory mechanism on cell migration caused by the interaction between NPs and ECM could also work in vivo, we cultured mouse breast cancer cell line 4T1 transfected with luciferase and established a metastasis model in mice. It was found that 4T1 cells, as a classical hyper-metastatic cell line, also produced RFs and migrasomes during cellular movement (Fig. S17A, ESI†). These ECM-related structures also presented high affinity with NPs (Fig. S17B, ESI†). As shown in Fig. 5A, a modified in vivo tumor model was constructed. In brief, the obtained 4T1 tumor blocks were subcutaneously implanted in mice. Different nanoparticle suspensions were then administered surrounding these tumor blocks. As the aqueous solution in the suspensions was absorbed by the tissue, the NPs were retained in the peripheral space of the tumor. These local NPs interacted with the surrounding tissue and matrix, forming a nano–ECM network around the tumor. We believe that this strategy could basically simulate the impact of nano–ECM interaction on tumor metastasis.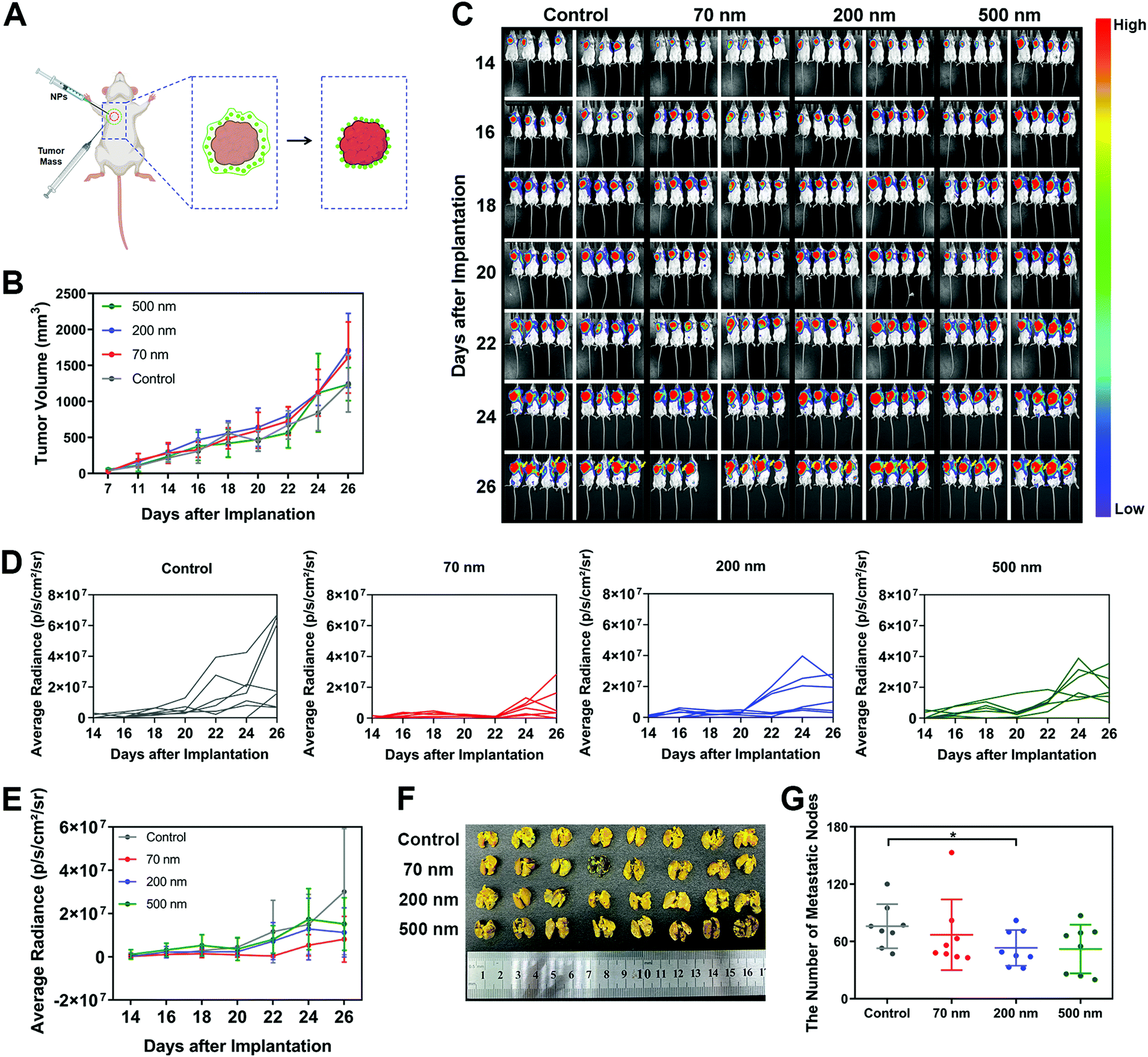 | ||
| Fig. 5 Peripheral NPs around a tumor inhibit tumor metastasis in vivo. (A) Scheme of the animal experiment. (B) Tumor volume growth curves for mice bearing 4T1 tumor cells treated with different NPs (n = 8). (C) Bioluminescence images showing metastatic regions of the mice at different time points. (D) Quantitative bioluminescence signals of metastatic regions in mice bearing 4T1 tumor cells treated with different NPs (shown as a single mouse). (E) Quantitative bioluminescence signals of metastatic regions in mice at different time points (n = 8). (F) Image of lungs with metastatic nodes obtained from mice treated with different NPs. (G) The number of lung metastatic nodes of mice treated with different NPs (n = 8). (Data are presented as mean ± SD. Statistical significances were calculated using Student's t-test: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.). | ||
In the results, no change in body weights was detected, indicating that the addition of NPs did not induce an obvious toxic effect (Fig. S17C, ESI†). Measurement of tumor size (Fig. 5B) revealed that the NPs did not weaken the cell growth at the implantation site. Next, we used an IVIS Lumina II Imaging System to continuously monitor the potential metastasis of tumor cells. According to the quantitative analysis of the bioluminescence signals shown in Fig. 5C to E, the implanted tumor began to metastasize towards the left lung as early as the 14th day. As time progressed, more mice with lung metastases were detected. Notably, all three NPs exhibited an inhibitory effect on tumor metastasis. As shown in Fig. 5D and E, the 70 nm NPs displayed the greatest inhibition on tumor metastasis regions. Additionally, based on the direct measurement of metastasis to different organs after sacrificing the mice on the 26th day, all three NP groups attenuated the extention of the tumor metastasis, especially the metastasis towards the lungs. Among all the treatments, only the 200 nm NPs demonstrated significant inhibition on the lung metastasis of the tumor (Fig. 5F and G), which was consistent with the results we found in vitro. We also detected a renal metastasis node in the control group but found nothing after NP treatment (Fig. S17D, ESI,† red circle). In addition, no metastasis to the liver or spleen was observed in any of the groups (Fig. S17E and F, ESI†). In summary, these results indicated that the NPs in the peripheral tissue of the tumor could retard tumor metastasis, which confirmed the significance of our findings on the specific interaction between NPs and ECM.
Conclusion
In this study, we aimed to explore the impact of nano–ECM interaction on cell migration. This important and general scientific issue has been studied from different angles. Large-sized graphene oxide (LS-GO) was reported to inhibit cell migration by patching the cell surface and imposing spatial confinement on cells.46 A nanostructured substrate could also significantly influence how the adsorbed cells handled invading viruses through the cellular response to nano-topological parameters of the substrate materials, such as stiffness, morphology, etc.47,48 In contrast to these established mechanisms, we discovered for the first time that nanoparticles closely bound to specific types of lipid components in ECM, namely, retraction fibers (RFs) and migrasomes. These lipid structures are distributed at the rear of tumor cells and form in the wake of cell movement.17 In particular, migrasomes, which are a new vesicular organelle discovered in recent years, have been reported to mediate cellular functions by regulating the release of cytoplasmic contents.45 Here, we demonstrated that polystyrene NPs, PLGA-NPs and SiO2-NPs all exhibited high affinity with RFs and migrasomes. More importantly, based on dynamic monitoring of cell movement, we found that this common interaction characteristic caused a reduction of cell migration, whereas it made no difference to tumor proliferation. In this mechanism, by coating and forming a hard shell on the migrasome surface, the nanoparticles blocked the recognition and endocytosis of migrasomes by surrounding cells, finally inhibiting the intrinsic pro-migration effect of migrasomes on cells. Nanoparticles bound to migrasomes and RFs through intensive interactions with lipid rafts/caveolae substructures. Interestingly, these substructures are also utilized by many different nanoparticles to enter cells. Considering the widely established correlations between multiple nanoparticles and lipid rafts/caveolae,49–51 we believe that our findings in this study may represent a general mechanism, although this needs to be further confirmed in subsequent studies. In conclusion, our discovery of the interaction of nanoparticles with RFs and migrasomes provides a new insight into the complicated and incompletely known nano–tumor interactions. Furthermore, the nanoparticle-triggered inhibition effect on cell migration might represent a potential target and strategy for anti-metastasis nanotherapy against RFs and migrasomes.Author contributions
Yuxi Cheng: conceptualization, methodology, validation, formal analysis, investigation, resources, data curation, writing – original draft, visualization; Junji Ren: investigation; Shumin Fan: investigation; Peiyao Wu: investigation; Wenshu Cong: methodology; Yuxing Lin: investigation; Shaojie Lan: investigation; Siyang Song: methodology; Bin Shao: methodology; Wenbing Dai: writing – review & editing; Xueqing Wang: writing – review & editing; Hua Zhang: writing – review & editing; Bo Xu: investigation; Wenzhe Li: investigation; Xia Yuan: investigation; Bing He: conceptualization, methodology, formal analysis, writing – review & editing, supervision; Qiang Zhang: conceptualization, writing – review & editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Major State Basic Research Development Program of China (2017YFA0205600), Natural Science Foundation of Beijing Municipality (L212013), AI + Health Collaborative Innovation Cultivation Project (Z211100003521002), and National Natural Science Foundation of China (U20A20412, 81690264, 81821004, 82073786).References
- J. Li and D. J. Burgess, Acta Pharm. Sin. B, 2020, 10, 2110–2124 CrossRef CAS PubMed.
- Q. Dai, N. Bertleff-Zieschang, J. A. Braunger, M. Bjornmalm, C. Cortez-Jugo and F. Caruso, Adv. Healthcare Mater., 2018, 7, 1700575 CrossRef PubMed.
- A. E. Nel, L. Mädler, D. Velegol, T. Xia, E. M.-V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- L. Xu, M. Xu, R. Wang, Y. Yin, I. Lynch and S. Liu, Small, 2020, 16, 2003691 CrossRef CAS PubMed.
- Y. Wang, R. Cai and C. Chen, Acc. Chem. Res., 2019, 52, 1507–1518 CrossRef CAS PubMed.
- E. Chen, S. Han, B. Song, L. Xu, H. Yuan, M. Liang and Y. Sun, Int. J. Nanomed., 2020, 15, 6311–6324 CrossRef CAS PubMed.
- Z. Chen, H. Pan, Y. Luo, T. Yin, B. Zhang, J. Liao, M. Wang, X. Tang, G. Huang, G. Deng, M. Zheng and L. Cai, Small, 2021, 17, 2007494 CrossRef CAS PubMed.
- S. Napoli, C. Scuderi, G. Gattuso, V. Di Bella, S. Candido, M. S. Basile, M. Libra and L. Falzone, Cells, 2020, 9, 1151 CrossRef CAS PubMed.
- J. Cui, D. Dean, F. J. Hornicek, Z. Chen and Z. Duan, J. Exp. Clin. Cancer Res., 2020, 39, 178 CrossRef PubMed.
- Y. Chen, R. Koshy, E. Guirado and A. George, Acta Biomater., 2021, 120, 224–239 CrossRef CAS PubMed.
- R. Kalluri and V. S. LeBleu, Science, 2020, 367, 640 CrossRef PubMed.
- D. N. Wu, Y. Xu, T. L. Ding, Y. Zu, C. Yang and L. Yu, Cell Res., 2017, 27, 1397–1400 CrossRef CAS PubMed.
- J. H. Waknine-Grinberg, S. Even-Chen, J. Avichzer, K. Turjeman, A. Bentura-Marciano, R. K. Haynes, L. Weiss, N. Allon, H. Ovadia, J. Golenser and Y. Barenholz, PLoS One, 2013, 8, e72722 CrossRef CAS PubMed.
- X.-j Cai, Z. Wang, Y.-y Xu, G.-y Yang, R.-y Zhang and Y. Wang, Drug Delivery Transl. Res., 2020, 11, 1186–1197 CrossRef PubMed.
- B.-W. Huang and J.-Q. Gao, J. Controlled Release, 2018, 270, 246–259 CrossRef CAS PubMed.
- P. Henrich-Noack, D. Nikitovic, M. Neagu, A. O. Docea, A. B. Engin, S. Gelperina, M. Shtilman, P. Mitsias, G. Tzanakakis, I. Gozes and A. Tsatsakis, Nanomedicine, 2019, 17, 359–379 CrossRef CAS PubMed.
- L. Ma, Y. Li, J. Peng, D. Wu, X. Zhao, Y. Cui, L. Chen, X. Yan, Y. Du and L. Yu, Cell Res., 2015, 25, 24–38 CrossRef CAS PubMed.
- C. He, Y. Hu, L. Yin, C. Tang and C. Yin, Biomaterials, 2010, 31, 3657–3666 CrossRef CAS PubMed.
- H. I. Labouta, N. Asgarian, K. Rinker and D. T. Cramb, ACS Nano, 2019, 13, 1583–1594 CAS.
- H. R. Vutukuri, M. Hoore, C. Abaurrea-Velasco, L. van Buren, A. Dutto, T. Auth, D. A. Fedosov, G. Gompper and J. Vermant, Nature, 2020, 586, 52–56 CrossRef CAS PubMed.
- K. Gnanasekaran, H. Chang, P. J.-M. Smeets, J. Korpanty, F. M. Geiger and N. C. Gianneschi, Nano Lett., 2020, 20, 4292–4297 CrossRef CAS PubMed.
- A. Darvish, G. Goyal, R. Aneja, R. V. K. Sundaram, K. Lee, C. W. Ahn, K. B. Kim, P. M. Vlahovska and M. J. Kim, Nanoscale, 2016, 8, 14420–14431 RSC.
- E. Rideau, F. R. Wurm and K. Landfester, Small, 2020, 16, 1905230 CrossRef CAS PubMed.
- P. Li, N. J. Butcher and R. F. Minchin, Cell Adhes. Migr., 2020, 14, 1–11 CrossRef CAS PubMed.
- M. Ueta, K. Takaoka, M. Yamamura, H. Maeda, J. Tamaoka, Y. Nakano, K. Noguchi and H. Kishimoto, Mol. Med. Rep., 2019, 20, 4331–4339 CAS.
- S. Konar, P. Edwina, V. Ramanujam, A. Arunachalakasi and S. K. Bajpai, J. Biomed. Mater. Res., Part B, 2020, 108, 2368–2377 CrossRef CAS PubMed.
- R. B. Samuel Alkmin, H. Simon, D. Hinton, R. HGoldsmith, M. Patankar and P. J. Campagnola, presented in part at the Biophotonics Congress: Optics in the Life Sciences Congress 2019 (BODA,BRAIN,NTM,OMA,OMP), Tucson, Arizona United States, 14–17 April 2019, 2019.
- R. J. Petrie, A. D. Doyle and K. M. Yamada, Nat. Rev. Mol. Cell Biol., 2009, 10, 538–549 CrossRef CAS PubMed.
- L. Depau, J. Brunetti, C. Falciani, E. Mandarini, G. Riolo, M. Zanchi, E. Karousou, A. Passi, A. Pini and L. Bracci, J. Med. Chem., 2020, 63, 15997–16011 CrossRef CAS PubMed.
- L. A. Naleskina, L. M. Kunska and V. P. Chekhun, Exp. Oncol., 2020, 42, 252–262 CAS.
- C. M. Dowling, C. Herranz Ors and P. A. Kiely, Biosci. Rep., 2014, 34, 415–427 CrossRef CAS PubMed.
- A. C. Taylor and E. Robbins, Dev. Biol., 1963, 7, 660 CrossRef.
- H. Laronha and J. Caldeira, Cells, 2020, 9, 1076 CrossRef CAS PubMed.
- J. K. Mouw, G. Ou and V. M. Weaver, Nat. Rev. Mol. Cell Biol., 2014, 15, 771–785 CrossRef CAS PubMed.
- J. H.-R. Hetmanski, H. de Belly, I. Busnelli, T. Waring, R. V. Nair, V. Sokleva, O. Dobre, A. Cameron, N. Gauthier, C. Lamaze, J. Swift, A. Del Campo, T. Starborg, T. Zech, J. G. Goetz, E. K. Paluch, J. M. Schwartz and P. T. Caswell, Dev. Cell, 2019, 51, 460–475 CrossRef CAS PubMed e410.
- A. M. Bannunah, D. Vllasaliu, J. Lord and S. Stolnik, Mol. Pharmaceutics, 2014, 11, 4363–4373 CrossRef CAS PubMed.
- H. Akita, T. Fujiwara, S. Santiwarangkool, N. Hossen, K. Kajimoto, A. El-Sayed, Y. Tabata and H. Harashima, Small, 2016, 12, 1212–1221 CrossRef CAS PubMed.
- L. Ding, X. Zhu, Y. Wang, B. Shi, X. Ling, H. Chen, W. Nan, A. Barrett, Z. Guo, W. Tao, J. Wu and X. Shi, Nano Lett., 2017, 17, 6790–6801 CrossRef CAS PubMed.
- B. H. Sung, A. von Lersner, J. Guerrero, E. S. Krystofiak, D. Inman, R. Pelletier, A. Zijlstra, S. M. Ponik and A. M. Weaver, Nat. Commun., 2020, 11, 2092 CrossRef CAS PubMed.
- Y. W. Huang, B. Zucker, S. J. Zhang, S. Elias, Y. Zhu, H. Chen, T. L. Ding, Y. Li, Y. J. Sun, J. Z. Lou, M. M. Kozlov and L. Yu, Nat. Cell Biol., 2019, 21, 991 CrossRef CAS PubMed.
- D. Jiang, Z. Jiang, D. Lu, X. Wang, H. S. Liang, J. F. Zhang, Y. P. Meng, Y. Li, D. N. Wu, Y. W. Huang, Y. L. Chen, H. T. Deng, Q. Wu, J. W. Xiong, A. M. Meng and L. Yu, Nat. Cell Biol., 2019, 21, 966 CrossRef CAS PubMed.
- Y. Zhang, J. Wang, Y. Ding, J. Zhang, Y. Xu, J. Xu, S. Zheng and H. Yang, Front. Cell Dev. Biol., 2020, 8, 438 CrossRef PubMed.
- G. van Niel, G. D'Angelo and G. Raposo, Nat. Rev. Mol. Cell Biol., 2018, 19, 213–228 CrossRef CAS PubMed.
- X. Zhao, Y. Lei, J. Zheng, J. Peng, Y. Li, L. Yu and Y. Chen, Cell Discovery, 2019, 5, 27 CrossRef PubMed.
- M. Zhu, Q. Zou, R. Huang, Y. Li, X. Xing, J. Fang, L. Ma, L. Li, X. Yang and L. Yu, Cell Res., 2021, 31, 237–240 CrossRef CAS PubMed.
- T. Chen, Q. Zhang, Y. Song, A. N. Isak, X. Tang, H. Wang, Z. Ma, F. Sun, Q. Pan and X. Zhu, Nanoscale Horiz., 2021, 6, 979–986 RSC.
- C. J. Bettinger, R. Langer and J. T. Borenstein, Angew. Chem., Int. Ed., 2009, 48, 5406–5415 CrossRef CAS PubMed.
- Y.-h Jang, Y.-s Park, J.-s Nam, Y. Yang, J.-e Lee, K.-h Lee, M. Kang, A. Chialastri, H. Noh, J. Park, J. S. Lee and K.-i Lim, Nanoscale, 2019, 11, 5693–5704 RSC.
- G. Gu, Q. Hu, X. Feng, X. Gao, M. Jiang, T. Kang, D. Jiang, Q. Song, H. Chen and J. Chen, Biomaterials, 2014, 35, 8215–8226 CrossRef CAS PubMed.
- B. He, P. Lin, Z. Jia, W. Du, W. Qu, L. Yuan, W. Dai, H. Zhang, X. Wang, J. Wang, X. Zhang and Q. Zhang, Biomaterials, 2013, 34, 6082–6098 CrossRef CAS PubMed.
- D. Chen, N. A. Monteiro-Riviere and L. W. Zhang, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2017, 9, e1419 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and Fig. S1–S17 (PDF). Supplementary Movies 1–4. Western blot raw images (PDF). Original proteomics data. See DOI: https://doi.org/10.1039/d2nh00067a |
| This journal is © The Royal Society of Chemistry 2022 |