DOI:
10.1039/D2NA00467D
(Paper)
Nanoscale Adv., 2022,
4, 5330-5342
Cobalt doped titania-carbon nanosheets with induced oxygen vacancies for photocatalytic degradation of uranium complexes in radioactive wastes†
Received
19th July 2022
, Accepted 5th October 2022
First published on 12th October 2022
Abstract
The photocatalytic degradation of uranium complexes is considered among the most efficient techniques for the efficient removal of uranium ions/complexes from radioactive wastewater. Described here is a nanostructured photocatalyst based on a cobalt-doped TiO2 composite with induced oxygen vacancies (Co@TiO2-C) for the photocatalytic removal of uranium complexes from contaminated water. The synergy between oxygen vacancies and Co-doping produced a material with a 1.7 eV bandgap, while the carbon network facilitates electron movement and hinders the e–h recombination. As a result, the new photocatalyst enables the decomposition of uranium–arsenazo III complexes (U–ARZ3), followed by photocatalytic reduction of hexavalent uranium to insoluble tetravalent uranium. Combined with the nanosheet structure's high surface area, the photocatalytic decomposition, reduction efficiency, and kinetics were significantly enhanced, achieving almost complete U(VI) removal in less than 20 minutes from solution with a concentration as high as 1000 mL g−1. Moreover, the designed photocatalyst exhibits excellent stability and reusability without decreasing the photocatalytic performance after 5 cycles.
1. Introduction
The use of nuclear-powered electricity for diagnosing and treating diseases and other purposes has increased the possibility of radioactive species being leaked into the environment.1,2 Due to its radiochemical and toxicological effects, particular concern has been given to uranium, among other radioactive waste components.3 Uranium forms complexes in waste solutions with existing chelating agents such as tributyl phosphate, Alizarin red S, ethylenediaminetetraacetic acid (EDTA), xylenol orange, arsenazo III, and chlorophosphonazo.4–6 Such complexes are absorbed into the human body until they reach the liver and kidneys. Several methods have been used or suggested to remove uranium and other heavy metal ions from a nuclear waste solution, such as chemical precipitation, adsorption, membrane filtration, and ion exchange.7–14 However, photocatalytic degradation is receiving increasing interest for removing radionuclide complexes from contaminated solutions due to the low cost of operation, simplicity, and environmental friendliness.15–19 Recent work has revealed that photo-generated electrons can convert uranium into an insoluble form to reduce the soluble hexavalent state to its insoluble quaternary state.20,21
Titanium-based nanostructures are commonly used as photocatalysts to remove a wide range of organic and inorganic compounds. Coupled with its abundance, the excellent chemical and thermal stability of TiO2 make it one of the best candidates for photocatalysts.22 TiO2 exists in three different polymorphs (i.e., anatase, rutile, and brookite). These polymorphs exhibit different physical characteristics, such as the differences in their bandgap energies (i.e., 3.0 eV for rutile and 3.2 eV for both anatase and brookite).23 The photocatalysis mechanism exerted by TiO2 can be carried out by forming free electrons and holes in the conduction and valence band region.16,22
Uranyl ion reduction on TiO2 by ultraviolet (UV) irradiation was reported for the first time by Amadelli et al.24 Several studies have since looked at the photocatalytic reduction of U(VI) on TiO2 photocatalysts.25–49 Kim et al. demonstrated that TiO2 electrodes could photoelectrochemically reduce and eliminate U(VI) spikes in natural seawater.35 Li et al. also investigated TiO2 photocatalysis in uranium recovery from seawater. The photocatalytic approach demonstrated high efficiency for reducing and extracting U(VI) under different circumstances, with the reduced uranium deposits on TiO2 easily recovered by using Na2CO3 solution.46 Li et al. used TiO2/Fe3O4 and TiO2/graphene/Fe3O4 composites as photocatalysts to boost the photocatalytic activity of TiO2 for U(VI) reduction.41 The photo-generated e−in the TiO2 conduction band (CB) was partially transferred to the Fe3O4 surface, reducing Fe(II) and Fe(III) in the structural Fe3O4 phases when exposed to UV light. Therefore, U(VI) ions are reduced by both the generated Fe species and the photoelectrons, significantly enhancing the U(VI) removal. Although Li et al. reported that the photo-reactivity decreased when graphene was added, the graphene layer inhibits the photo-dissolution of Fe3O4, making the ternary composite considerably more stable. Therefore, designing a stable photocatalyst that can efficiently reduce U(VI) without contaminating the wastewater with the reaction by-product is essential for the wider application of photocatalytic treatment of nuclear waste. More importantly, most of the studies published so far have considered standard U(VI) solutions and ignored the organic matter that commonly exists in real waste solutions. These organic species influence the photocatalytic reduction of U(VI) by two mechanisms; (a) the adsorption of U(VI) on solid surfaces is altered by the competing diverse organics,50,51 and (b) U(VI) forms complex ions with a lot of this organic matter, which completely changes the nature of the uranium ions in the solutions.4–6
The present study introduces a nanostructured TiO2-based photocatalyst (Co@TiO2-C) to efficiently remove uranium complexes from contaminated water. Co@TiO2-C promotes two photocatalytic processes: decomposing the complexes, such as U(VI)–ARZ3, and reducing U(VI) to insoluble U(IV). We carefully designed porous nanostructures of connected 2D nanosheets of cobalt-doped anatase with induced oxygen vacancies. The synergy between the oxygen vacancies and the cobalt distortion results in a material with a low bandgap of 1.7 eV, facilitating photoelectron transfer. The oxygen vacancies were introduced via partial carbothermal reduction during nanophotocatalyst preparation. The uranium complex removal mechanism was also studied using several ex situ spectroscopic techniques. A satisfactory U(VI) removal can be achieved even at low concentrations. Notably, the photoreduction efficiency of 99.6% remains achievable by U(VI)–ARZ3 to form (UO2)O2·2H2O. The photocatalysis method using Co@TiO2-C was demonstrated to be a promising approach for complex radioactive waste remediation.
2. Materials and methods
2.1. Chemicals
Titanium isopropoxide Ti[OCH(CH3)2]4, cobalt acetate (CH3CO2)2Co, potassium chloride (KCl), ethanol (C2H5OH), glucose (C6H12O6), arsenazo III ((HO)2C10H2(SO3H)2(N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NC6H4AsO3H2)2), and uranyl nitrate UO2(NO3)2·6H2O were Sigma-Aldrich products (Germany). Deionized water (DIW) used in all experimental steps has an electric conductivity of 0.7 μS cm−1.
NC6H4AsO3H2)2), and uranyl nitrate UO2(NO3)2·6H2O were Sigma-Aldrich products (Germany). Deionized water (DIW) used in all experimental steps has an electric conductivity of 0.7 μS cm−1.
2.2. Synthesis of the Co/C doped TiO2 photocatalyst
About 0.5 mL of aqueous KCl (0.1 M), 0.88 g of cobalt acetate, and 1.5 mL of titanium isopropoxide were added to 100 mL ethanol and stirred for 4 h. The solution was then transferred to a 100 mL stainless steel autoclave lined with Teflon and heated at 170 °C for 30 h before being air-cooled to room temperature. The resultant product was collected and thoroughly cleaned with ethanol before being dried in an oven at 60 °C. To obtain Co@TiO2, the dried powder was sintered at 500 °C for 3 h. To prepare Co@TiO2-C, 0.42 g of the obtained Co@TiO2 composite was dispersed in 75 mL of DIW containing 0.019 g glucose. The liquid was stirred for 2 h before being transferred to a 100 mL Teflon-lined stainless steel autoclave. The autoclave was kept at 170 °C for 20 h before being air-cooled to ambient temperature. The resultant product was collected and thoroughly washed with water several times before being calcined in an oven at 500 °C for 3 h to generate an amorphous Co@TiO2-C photocatalyst.
2.3. Photocatalyst performance
Photodegradation/reduction of the U(VI)–ARZ3 complex and free ARZ3 and U(VI) in an aqueous solution was carried out under UV irradiation. The batch experiments were conducted in a photo-reactor at 25 ± 1 °C under ambient conditions. 10 mg of the photocatalyst was added into 10 mL of 100 mg L−1 of U(IV)–ARZ3 (S/L ratio 1:![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1) at different pH values. A similar experiment was carried out without ARZ3 for comparison. After a certain time, an aliquot (5 mL) of the solution was taken out and filtered for analysis. The concentration of U(VI) was determined using ICP. The degradation of U(VI)–ARZ3 complexes was determined at 450 and 650 nm using UV-vis spectrometry. The removal rate of all samples was calculated using the equation: R% = [(C0 − Ct)/C0] × 100; where C0 is the initial concentration and Ct is the concentration at time t.
1) at different pH values. A similar experiment was carried out without ARZ3 for comparison. After a certain time, an aliquot (5 mL) of the solution was taken out and filtered for analysis. The concentration of U(VI) was determined using ICP. The degradation of U(VI)–ARZ3 complexes was determined at 450 and 650 nm using UV-vis spectrometry. The removal rate of all samples was calculated using the equation: R% = [(C0 − Ct)/C0] × 100; where C0 is the initial concentration and Ct is the concentration at time t.
3. Results and discussion
3.1. Characterization of the Co@TiO2-C photocatalyst
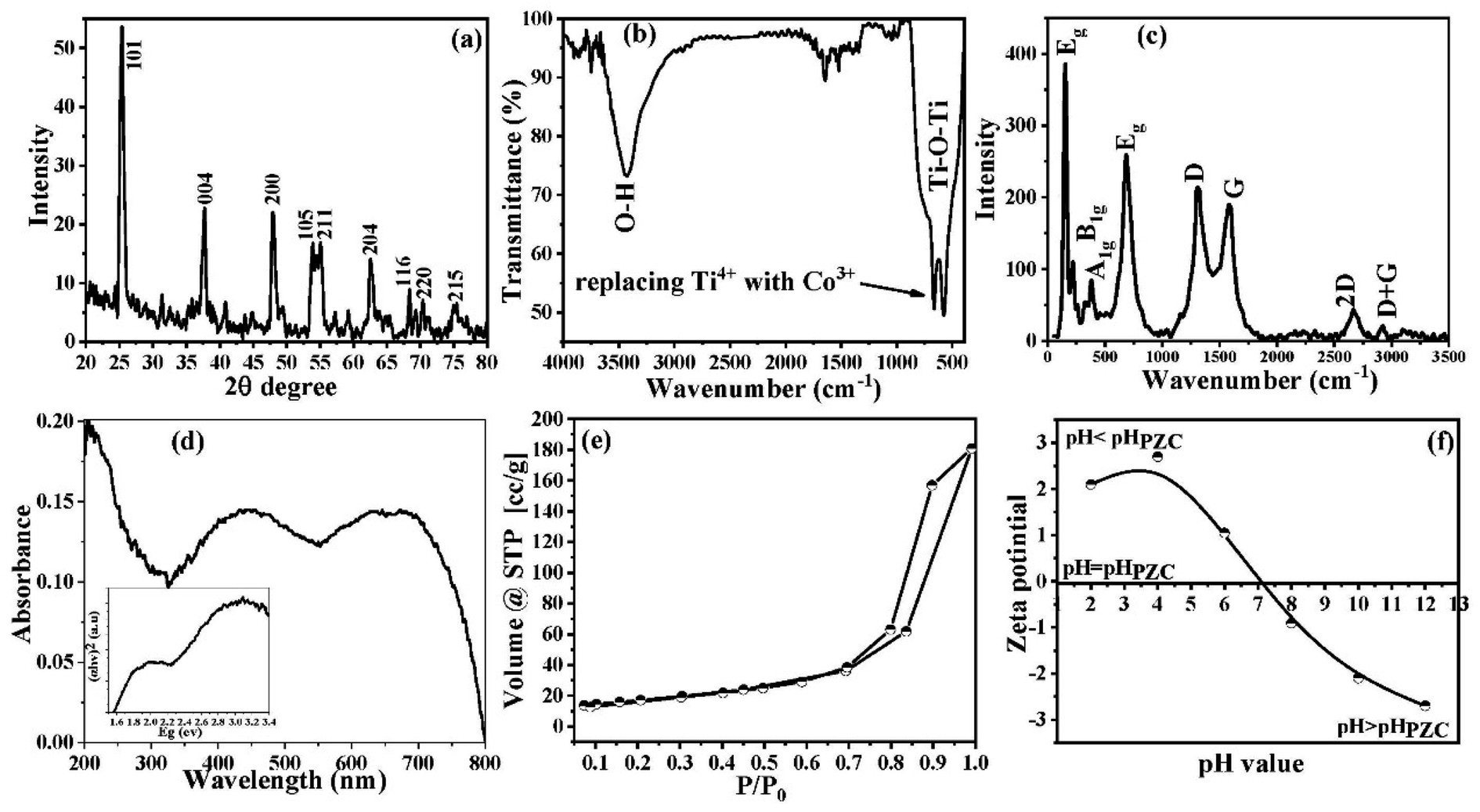
The phase characterization of the as-obtained Co@TiO2-C nanostructure was first determined by the XRD technique. The XRD pattern for the Co@TiO2-C nanostructure is presented in Fig. 1a. In the XRD pattern, the characteristic diffraction peaks (101), (004), (200), (105), (211), (204), (116), (220) and (215) of pure TiO2 can be detected. The peak for the 101 plane slightly shifts from 2θ = 25.28° for pure anatase vs. 2θ = 25.44° for Co@TiO2-C.52 This shift is caused by changes in the local structure around Ti4+ following Co2+ substitution and oxygen vacancy creation.53 The crystal size of the Co@TiO2-C nanocomposite sample is calculated using Scherrer's equation to be 11.38 nm. It should be noted that the XRD spectrum of the composite showed some minor peaks of sub-oxides and Magneli phases (the unlabeled peaks in Fig. 1a), which can be attributed to the partial reduction of the oxides. The percentage of the sub-oxides and Magneli phases was calculated to be around 9%. It is well known that these phases are conductive and therefore increase the overall conductivity of the composite.54,55 We believe that these nonstoichiometric phases have contributed to photocatalytic activities.
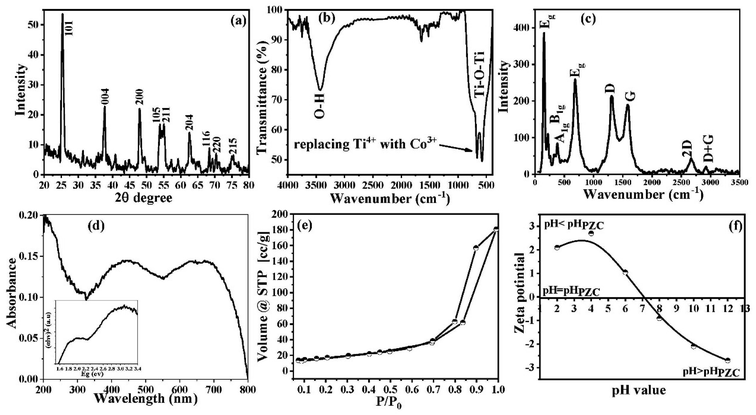 |
| | Fig. 1 (a) XRD patterns, (b) FTIR spectrum, (c) Raman spectrum, (d) UV-vis spectroscopy, (d inset) the energy bandgap, (e) N2 adsorption/desorption isotherm, and (f) zeta potential in 0.01 M KCl as a function of pH of the synthesized Co@TiO2-C photocatalyst. | |
The surface chemistry of the Co@TiO2-C photocatalyst is investigated using the FTIR spectrum (Fig. 1b). The FTIR spectrum revealed a high-intensity vibration band at 3425 cm−1 attributed to the O–H group's stretching vibration.56 The vibration peak for the adsorbed water molecules on the surface of the nanoparticles is at 1643 cm−1 (bending vibration mode). The vibration band at ∼1396 cm−1 is associated with the bending vibration of the C–H bond. High-intensity deformation bands between 400 and 800 cm−1 correspond to the Ti–O–Ti vibrational mode.57,58 This deformation is associated with replacing Ti4+ with Co3+ within the anatase network.
The Raman spectrum acquired for the Co@TiO2-C sample is illustrated in Fig. 1c. Peaks at 151, 390, 510, and 627 cm−1 are ascribed to anatase phase modes Eg, B1g, A1g, and Eg, respectively.59 The strength of the B1g, A1g, and Eg peaks decreased after Co doping and carbothermal reduction (Fig. S1a†). This result suggests that the Co did not entirely transform the anatase phase and implies considerable deformation in the long-order crystallite, consistent with the XRD and FTIR results. The detection peaks at the D and G band locations (1300 cm−1 and 1850 cm−1, respectively) demonstrates the existence of carbon despite the calcination in air with Ti–Co oxides.60,61 The peak intensity ratio ID/IG is 1.33, indicating that the carbon is most likely amorphous.
UV-vis spectroscopy confirms the substitution of Ti4+ with the Co dopant. The absorption peak of pure anatase at 363 nm corresponds to the intrinsic band–band transition.62 The Co@TiO2-C spectrum shows two peaks at 495 and 582 nm (Fig. 1d). The peak at a lower wavelength is associated with the 6A1g(S) to 4A1g(G), 4Eg(G), and A1g(S) to 4T2g(G) transitions, whereas the peak at 582 nm is for the Co2+ in octahedral coordination 6A1g(S) to 4T1g(G). Each Ti4+ in TiO2 is coordinated with six oxygen atoms. When Co2+ substitutes Ti4+, the electrons in the d-orbital of Co2+ experience repulsion, resulting in the splitting of Co2+ d-orbitals, showing that Co2+ is inside the lattice.
The energy band gap of Co@TiO2-C can be determined from the linear portion of the Tauc plot, which was fitted and extrapolated for zero absorbance, i.e., F[R] = 0.63 The energy bandgap of Co@TiO2-C is 1.6 eV decreased from 3.2 eV in pure anatase (inset of Fig. 1d). Such energy levels indicate a shift in the local distribution of Ti4+ or partial conversion of Ti4+ to Ti3+ and the formation of empty oxygen sites. The change in the bandgap of Co@TiO2-C can be explained by the reduced particle size and the synergy between the Co substitution and the oxygen vacancies caused by the regulated carbothermal process.
The N2 adsorption/desorption technique was used to investigate the porosity structure of the Co@TiO2-C nanocomposite. As shown in Fig. 1e, the Co@TiO2-C isotherm exhibits typical characteristics for type IV isotherms, according to the IUPAC classification.64 At P/Po ≥ 0.7, the isotherm displays a distinguished hysteresis, indicating a large number of mesopores (Fig. 1e). The isotherm also exhibits an increasing trend at low pressure, corresponding to adsorption at micropores. Because of the hierarchal porous structure with nanochannels between the oxide particles, it is possible to deduce that there is a multilayer range of physisorption isotherms. Co@TiO2-C has an estimated SBET of 60.946 m2 g−1. As a result of co-doping and partial carbothermal reduction, certain new pores are introduced into the structure. One explanation for the increase in SBET might be that oxygen or COx gases are exiting the oxide structures, which relates to creating oxygen vacancies. Furthermore, the nanoparticles are arranged into 2D nanosheets, as will be discussed with the SEM images, reducing aggregation and creating additional surfaces. The point of zero charges (pzc) of the synthesized Co@TiO2-C photocatalyst was found to be 7.1 (Fig. 1f). This high pHPZC value resulted from cobalt and oxygen vacancies, and it is virtually consistent with the FTIR spectrum data since it verifies the increase in the absorption of the hydroxyl group at 3425 cm−1.65
The SEM photos clearly show the morphology of the Co@TiO2-C nanostructure (Fig. 2a). The morphology has a definite 2D structure, indicating that TiO2 nanoparticles tend to cluster in the form of sheets. The nanosheet structure is maintained following cobalt additions, and partial carbothermal reduction implies preferential development directions. The 2D nature of the morphology is most likely due to the increase in the density of states perpendicular to the C-axes caused by Co doping of the Ti site and the partial elimination of oxygen by carbon. The porous zig-zag structure is formed by loosely linking the 2D sheets together. More information may be gleaned from the TEM pictures (Fig. 2b), which revealed that the material is made up of nanoparticles with parallel facets and an average size of about 15 nm that shows a uniform distribution with pore ordering over graphite sheets (Fig. 2c and d).
 |
| | Fig. 2 SEM (a) image and TEM image (b–d) of the synthesized Co@TiO2-C photocatalyst. | |
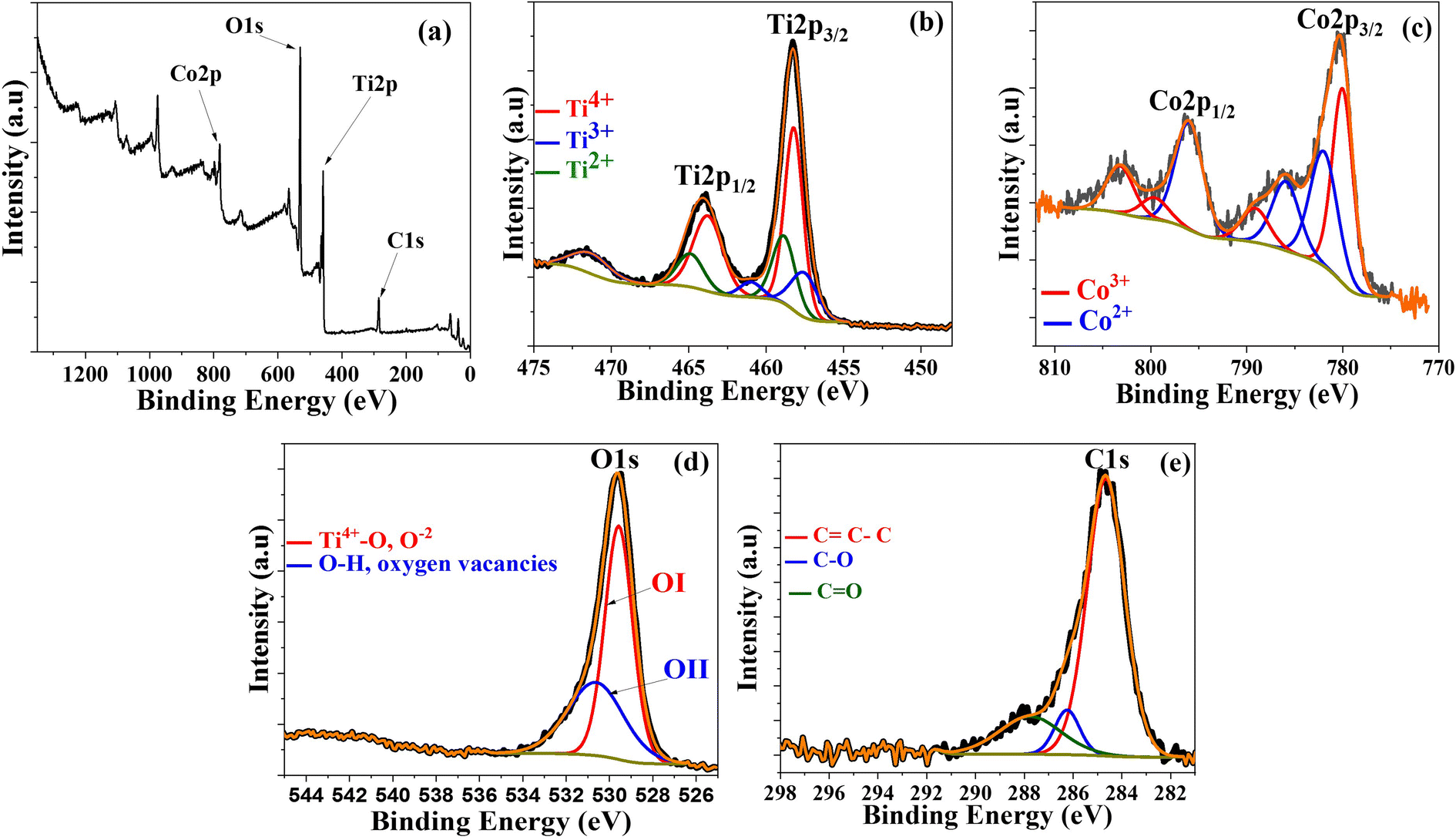
XPS was performed on Co@TiO2-C at room temperature to further estimate the possibility of secondary phases, oxygen deficiency, and oxidation states of Ti and Co in the near-surface region, as shown in Fig. 3. The peaks in the survey (Fig. 3a) at 459.17 eV, 781.5 eV, 530.8 eV, and 285.82 eV can be attributed to Ti 2p, Co 2p, O 1s, and C 1s elements, respectively.66–68 The high-resolution Ti 2p spectrum is shown in Fig. 3b. The presence of a pair of symmetric spin-orbit doublets with binding energies corresponding to Ti 2p3/2 and Ti 2p1/2 for the composite is verified by two large peaks at 458.24 eV and 463.99 eV, corresponding to Ti4+ in the TiO2 phase.66–69 Some peaks evidence the existence of the Ti3+ and Ti2+ oxidation states, together with Ti4+, proving the Co-doping and the partial reduction in the anatase crystals. The peaks at 457.78 eV and 460.25 eV correspond to Ti 2p3/2 and Ti 2p1/2 of Ti3+.69,70 Also, the peaks at 458.76 eV and 464.07 eV are for Ti 2p3/2 and Ti 2p1/2 of Ti2+.71,72 More evidence of doping can be obtained from the Co 2p spectrum (Fig. 3c). The spectrum is best fitted to two spin–orbit doublets characteristic of Co2+ and Co3+, where two peaks of Co 2p3/2 and Co 2p1/2 are located at 780.01 eV (Co3+) and 796.01 eV (Co2+). The two conspicuous shake-up satellite peaks are located at 785.92 eV and 803.22 eV, revealing the co-existence of Co2+ and Co3+. The other less intense peaks that appeared for Co 2p3/2 and Co 2p1/2 are located at 781.98 eV (Co2+) and 799.64 eV (Co3+).72 The co-existence of different states of cobalt suggests that carbothermal partial reduction has also taken place in the cobalt sites in the crystals. The high-resolution O 1s spectra (Fig. 3d) display two peaks at 529.57 eV and 530.62 eV. The low energy peak is attributed to O atoms bonded to metals (Ti4+–O), whereas the high-energy one can be attributed to oxygen vacancies or surface contamination by hydroxyl species.69–74 The C 1s spectra revealed three distinct peaks at 284.66, 286.24, and 287.76 eV (Fig. 3e). The first characteristic peak, at 284.66 eV, is for the C–C bond of the graphitic carbon on the surface of Co/TiO2. The other two peaks, at 286.24 and 287.76 eV, are for the C–O–C and CO bonds, suggesting that the carbon surface layer is bonded to Co@TiO2via oxide functional groups.66
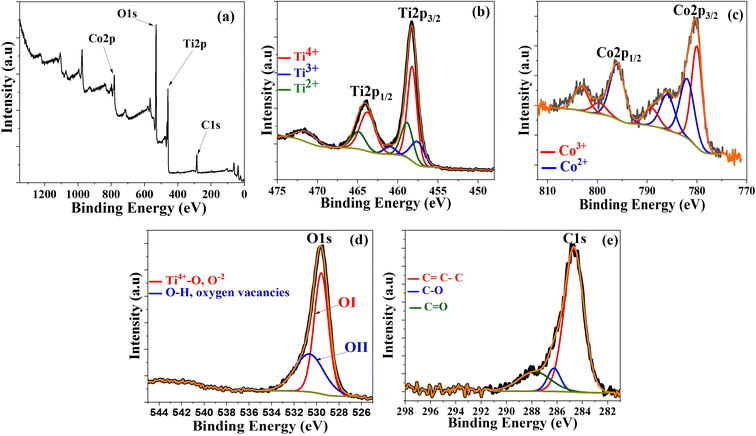 |
| | Fig. 3 XPS spectra of Co@TiO2-C, (a) Survey spectrum, (b) Ti 2p, (c) Co 2p, (d) O 1s and (e) C 1s core-levels of the Co@TiO2-C. | |
3.2. Photocatalytic performance of Co@TiO2-C catalyst
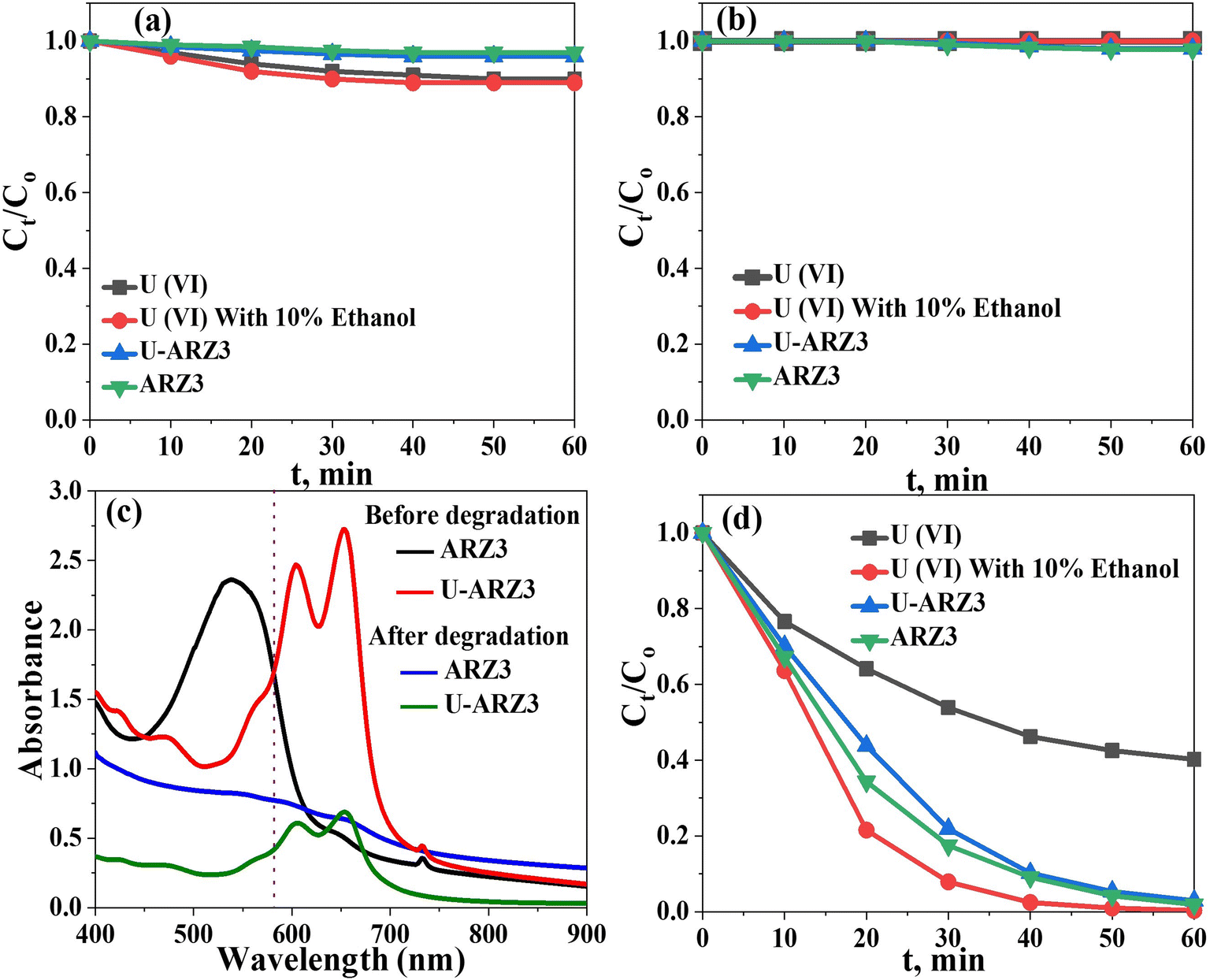
Uranium–ARZ3 complex degradation/reduction experiments using the Co@TiO2-C photocatalyst were carried out under UV irradiation. First, we tested the adsorption capability of Co@TiO2-C by stirring uranium nitrate or the U(VI)–ARZ3 complex with a concentration of 0.1 mM in the dark. The synthesized catalyst showed weak U(VI) adsorption in the dark (Fig. 4a). We then tested the photo-stability of the uranium nitrate, ARZ3, and U(VI)–ARZ3 complex solutions by exposing them to UV light without a photocatalyst. No noticeable changes in the concentrations can be observed, indicating good photo-stability (Fig. 4b). It is only when the solutions are irradiated with UV light in the presence of Co@TiO2-C that we start to observe a noticeable reduction in the uranium concentrations. The decrease in the peak intensities at λmax = 450 nm and λmax = 650 nm in the ARZ3 dye and U(VI)-ARZ3 complex, respectively, can be attributed to the breaking of the bond NN present in their chemical structure (Fig. 4c).50 For the U(VI)–ARZ3 solution, the N–N bond breaking is also associated with a decrease in the uranium concentration. These results imply a reduction in soluble hexavalent uranium U(VI) to insoluble tetravalent uranium U(IV) via a two-step photocatalytic process.18 Complementary to this, U(IV) nitrate in water/ethanol (10% V/V) was used to evaluate the photocatalytic activity of the synthesized photocatalyst towards fee U(IV) ions. The concentration of U(VI) decreased sharply in the first few minutes after irradiation, showing that the photocatalysis process was crucial for the conversion of U(VI) to an insoluble product (U(IV)) (Fig. 4d).
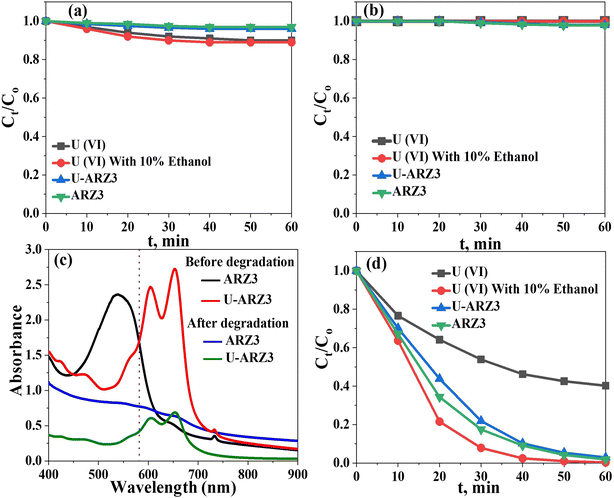 |
| | Fig. 4 (a) Effect of UV light on U(VI) and ARZ3 solutions without Co@TiO2-C catalyst. (b) Effect of Co@TiO2-C in the dark. (c) Effect of Co@TiO2-C in UV light. (d) Absorbance spectra of ARZ3 and the U(VI)–ARZ3 complex. | |
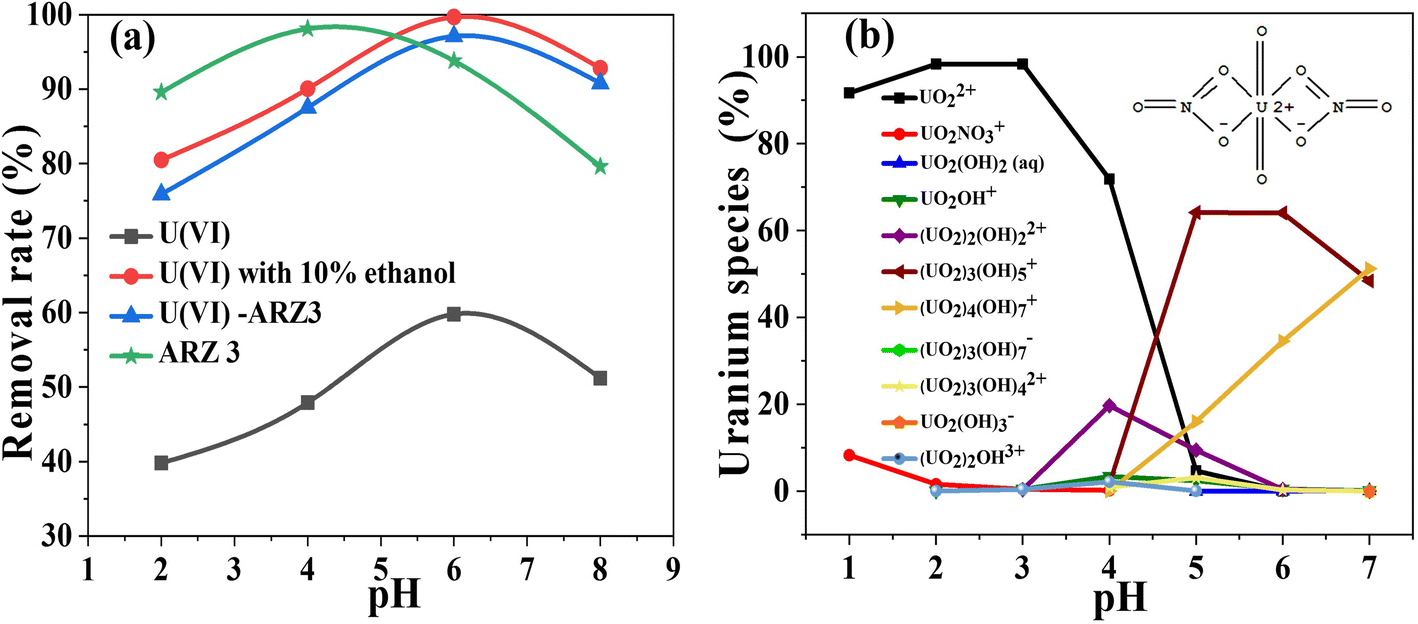
It is well known that the pH value strongly affects the photo-reaction process. For that reason, we studied the effect of pH on the removal of uranium from different solutions. As seen from Fig. 5a, all solutions' degradation occurs at pH values from 2 to 8. The removal efficiency achieves peak values at almost the same pH value, which is very close to the measured pHPZC of the solid surface of the Co@TiO2-C catalyst (about 7.1).
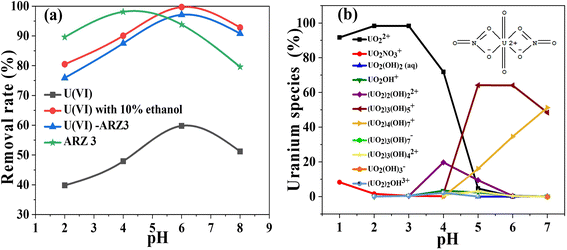 |
| | Fig. 5 (a) The effect of pH on the degradation/reduction of 50 mg L−1 U(VI)–ARZ3, free ARZ3 dye, and free U(VI) ions using the Co@TiO2-C photocatalyst for 60 min (solution volume; 10 mL, catalyst weight; 10 mg) and (b) speciation of uranium in aqueous solution at different pH values. | |
Without uranium in the solution, the highest rate of ARZ3 dye degradation was 98.1% at pH 4 (Fig. 5a). It should be mentioned that ARZ3 exists as an anion and gets a negative charge at low pH. The surface of Co@TiO2-C has positive charges in acidic media, resulting in repulsion forces with anionic ARZ3, and this hinders the degradation process.75 The difference in the symmetry of the ARZ3 bonds in acidic and basic media plays an important role in the degradation process. ARZ3 is found in the azo-form, and ionization of the acidic (–OH and –NH–N) or basic (–N2− and CO) groups leads to symmetry disruption and the appearance of quinone-hydrazone forms together with the azide.76 Fig. S1b† shows the structure of the arsenazo III and uranium/arsenazo III complex.
The best degradation efficiency of U(VI)–ARZ3 is about 97.1%. The best efficiency in aqueous solutions and the water/ethanol mixture is 59.8% and 99.69%, respectively. The peak in the photo-reduction efficiency of Co@TiO2-C at nearly the pHPZC is due to the presence of different uranium species, as displayed by Visual MINTEQ application (Fig. 5b). As a result of the forces of attraction and electrostatic repulsion between these species and the surface charge of Co@TiO2-C, protons are generated on the surface and hinder further adsorption of U(VI) ions (i.e., mainly with UO2+2). These protons also compete with the positive uranium ions to consume the photo-generated electron e− and thus reduce the photo-reaction activity. At pH ≥ 6, U(VI) exists in the form of neutrally and negatively charged ions (mainly UO2(OH)2 and UO2(OH)3−) which also generate electrostatic repulsion between them and the negative charges of the surface of Co@TiO2-C. As such, the photocatalytic activity also decreases at high pH.29,77–79 The low removal of uranium ions from the U(VI)–ARZ3 solution is likely due to the facile adsorption of the ARZ3 anions on the positively charged surface catalyst, leaving UO2+2 in the solution. On the other hand, the variation in photo-reduction efficiencies of free U(VI) ions in water compared to in the water/ethanol mixture is due to the ability of ethanol to scavenge the photo-hole h+ which are generated in the valence band (VB). These holes could re-oxidize U(IV) back to soluble U(VI).18 The re-oxidation of U(IV) into U(VI) could also take place by superoxide radical (i.e., ˙O2−) radicals, which are derived from the dissolved atmospheric O2. The concentration of these radicals significantly reduces when 10% ethanol is added to the solution.
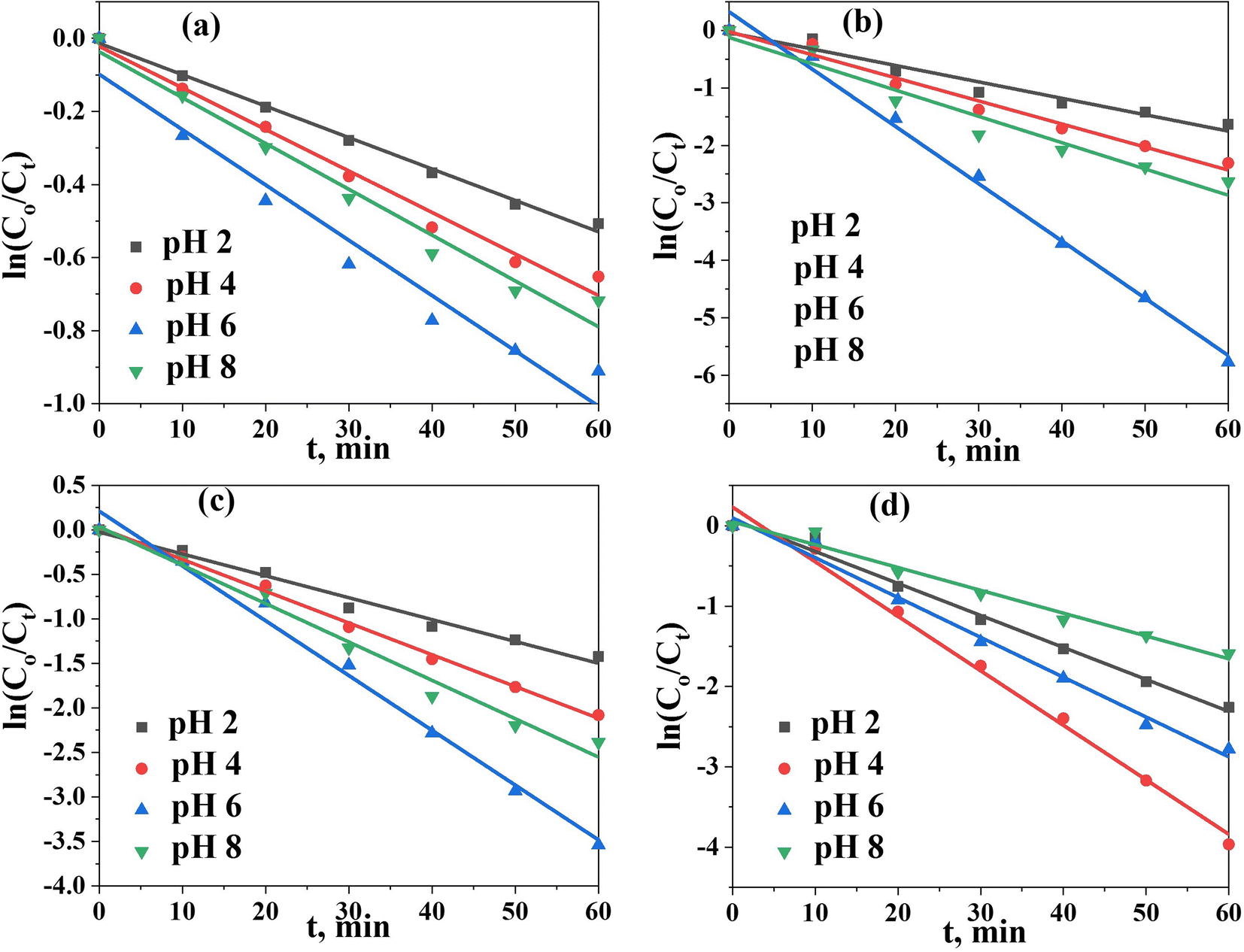
To shed more light on the kinetics of photodegradation/reduction of U(VI)–ARZ3, we first monitored the changes in absorption spectra of the degradation of the [U–ARZ3]n+ complex after the addition of the Co@TiO2-C catalyst as a function of time (Fig. S2†). Within the first 10 min, the amount of U(VI)–ARZ3 removed using the Co@TiO2-C catalyst is 29.8%, and equilibrium is acquired within 60 min. The photocatalytic removal data were applied to a pseudo-first-order model: ln(Ct/Co) = −Kt, where Co and Ct are the initial and final concentrations after time t of exposure to UV radiation, and K is the rate constant. The kinetic parameter was determined from the slope of plotting ln(Ct/Co) versus t (Fig. 6a–d). The regression coefficient (R2) values for the removal of U(VI)–ARZ3, free ARZ3, and U(VI) are calculated to be > 0.98 (Table S1†), indicating that the photodegradation/reduction processes can be well-fitted with the pseudo-first-order kinetic. At the optimum pH, Co@TiO2-C had the greatest apparent reaction rate constants of 6.155 × 10−2 min−1, 6.781 × 10−2 min−1, 9.97 × 10−2 min−1 and 1.514 × 10−2 min−1 for the U(VI)–ARZ3 complex, free ARZ3 dye, free U(VI) in water/ethanol and free U(VI) in aqueous media, respectively.
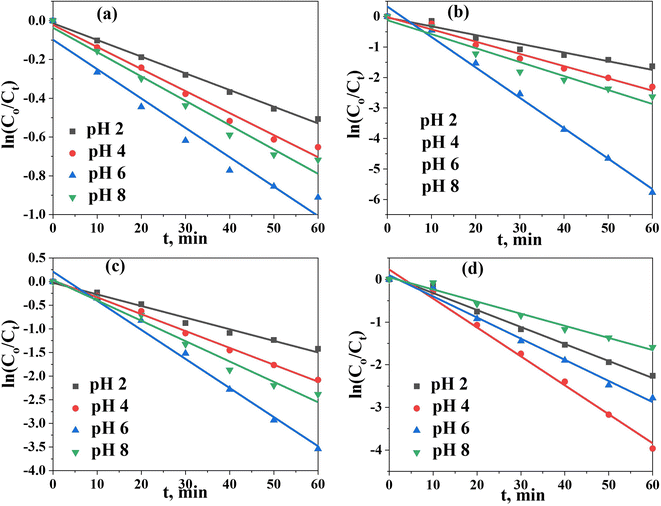 |
| | Fig. 6 Pseudo-first-order kinetic model. (a) U(VI) in aqueous solution, (b) U(VI) in aqueous solution (10% ethanol), (c) the U(VI)–ARZ3 complex, and (d) free ARZ3. | |
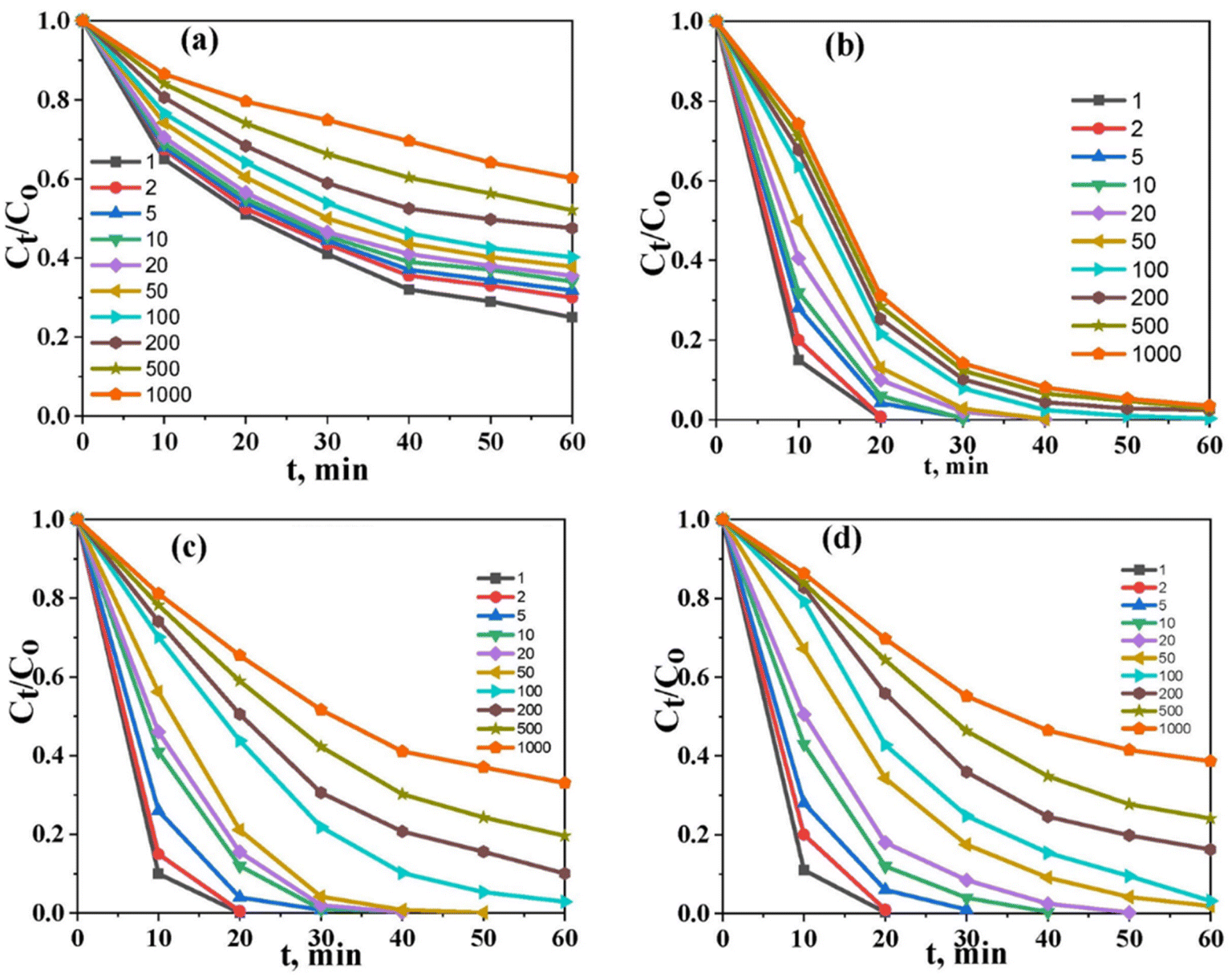
The initial concentration of the solution is an important factor in determining the performance of the photocatalysts. Therefore, the removal of various ions is investigated in solutions with concentrations ranging between 1 and 1000 mg L−1 under the optimal experimental conditions (i.e., pH 6, contact time; 60 min, S/L; 1 g L−1, and temp. 25 °C). As shown in (Fig. 7a–d), Co@TiO2-C appeared to remove the free ARZ3 dye at concentrations ≤ 100 mg L−1 with an efficiency of up to 97%. By increasing the concentrations, the efficiency of the photocatalyst increases and then gradually decreases until it reaches 61% at a concentration of 1000 mg L−1. For free U(VI) in pure water, the photocatalytic process cannot fully remove the uranium ions, even at a very low concentration. A significant improvement could be observed when 10% ethanol was added to the water. Full removal at a low concentration can be achieved within the first few minutes of photoexcitation. The high-concentration solution shows a removal efficiency of 96.8% after 60 min. The degradation efficiency of U(VI)–ARZ3 complexes with a concentration of up to 100 mg L−1 approaches almost 100% in a few minutes. The removal efficiency decreases with an increase in the concentration, reaching 66.9% at a concentration of 1000 mg L−1. Overall, the catalyst's efficiency is high at low concentrations and decreases with an increase in the concentration. We believe this is because of crowding the surface of the photocatalyst by new adsorbed ions and the need to remove the photodegraded product materials.
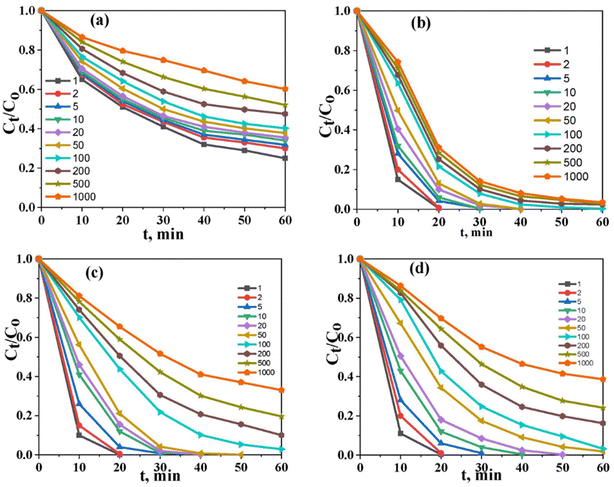 |
| | Fig. 7 The effect of the initial concentration under optimum conditions, (a) U(VI) in aqueous solution, (b) U(VI) in aqueous solution (10% ethanol), (c) the U(VI)–ARZ3 complex, and (d) free ARZ3. | |
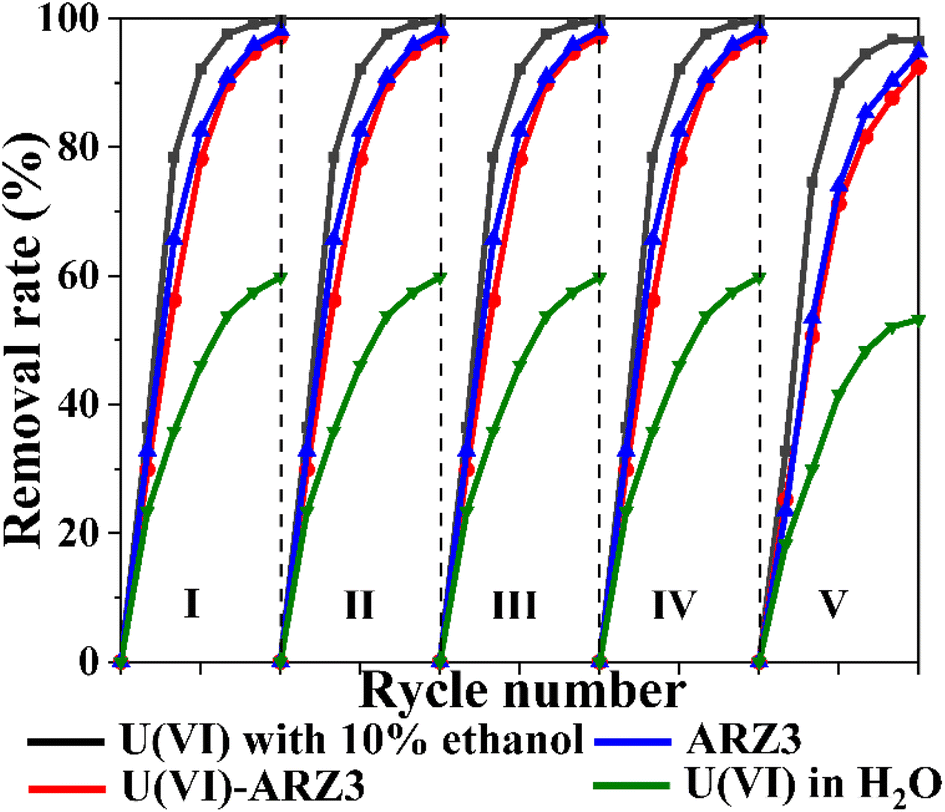
The reusability of catalysts is important from a practical point of view. Therefore, we have tested the Co@TiO2-C catalyst for five successive cycling runs using U(VI)–ARZ3 solution (Fig. 8). Following each cycle, the Co@TiO2-C was regenerated using 0.1 M HCl leaching, followed by water and ethanol washing. After the regeneration cycles, the degradation/reduction rate of uranium/ARZ3 remained almost consistent. The XRD spectra of Co@TiO2-C before and after 5 runs show almost no changes, suggesting the stability of the catalyst (Fig. 1a). These results demonstrate the catalyst's stability and the possibility of using it for several removal cycles.
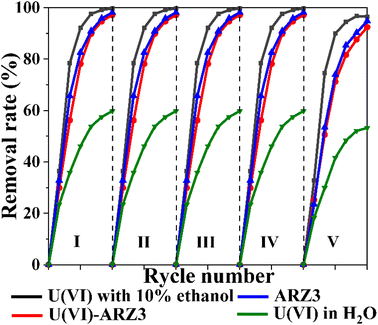 |
| | Fig. 8 Reusability studies of the Co@TiO2-C catalyst [10 mL of 100 mg L−1 concentration; 0.01 gm of catalyst; UV irradiation time, 60 min]. | |
A comparison was made between the efficiency of the Co@TiO2-C photocatalyst and some other catalysts that were used in the process of reducing dissolved U(VI) to insoluble U(IV) to remove nuclear pollutants from wastewater (Table. S2†).
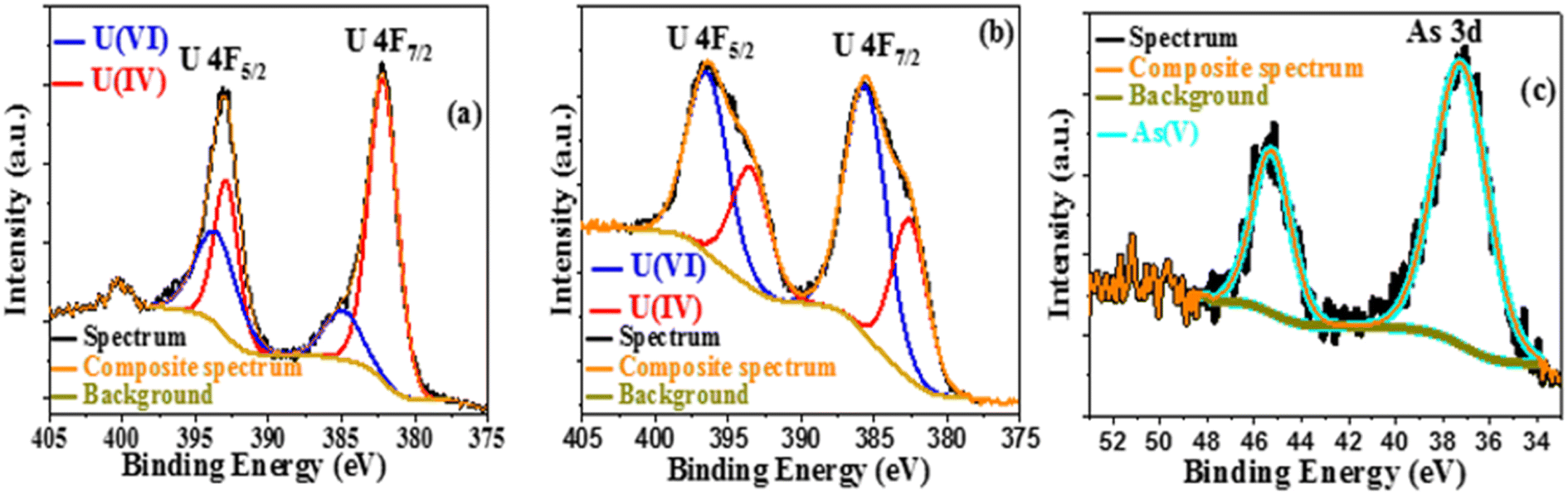
After excitation, the photocatalytic products were recovered by centrifugation, dried in the air, and studied using SEM and EDX. Uranium and arsenic are detected by elemental mapping in addition to the primary components of Co@TiO2-C (titanium, cobalt, carbon, and oxygen), as shown in Fig. S3–S5.† The XPS survey of the degraded product also confirmed the presence of arsenic and uranium in the precipitate after the photocatalytic process. Peaks for U f5/2, U f7/2, and As 3d with a decrease in the intensity of Ti 2p, Co 2p, O 1s, and C 1s (Fig. S6a and S7a†) can be observed. The high-resolution U 4f spectrum of the photocatalytic product in water/ethanol (Fig. 9a) shows two peaks at 393.54 eV and 384.95 eV, corresponding to U 4f5/2 and U 4f7/2 of U(VI),42 as well as two peaks at 392.89 eV and 382.81 eV for U 4f5/2 and U 4f7/2 of U(IV).42 The same peaks can be observed for the product collected from the U(VI)–ARZ3 solution (Fig. 9b). Finally, Fig. 9c shows two peaks at 37.25 eV and 45.28 eV that can be ascribed to As 3d of As(V).42 The existence of hierarchical forms of arsenic oxide is visible in the SEM image, while uranium oxide appeared as short rod-like particles distributed across the surface of the Co@TiO2-C catalyst (Fig. 10).80
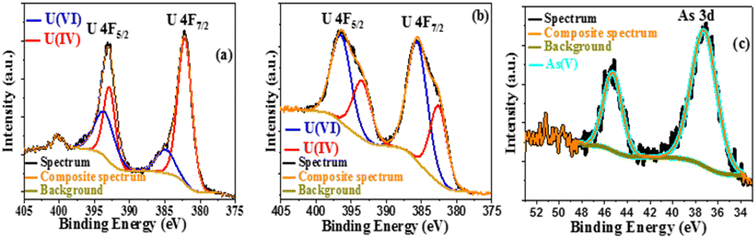 |
| | Fig. 9 (a) U 4f XPS spectra in 10% ethanol and (b and c) U 4f and As 3d XPS spectra of the U(VI)-AZR3 complex. | |
 |
| | Fig. 10 (a and b) SEM and TEM images of the ARZ3 degraded product. (c and d) SEM and TEM images of uranium and the arsenazo III degraded product. (e and f) SEM and TEM images of the uranium degraded product in ethanol media. | |
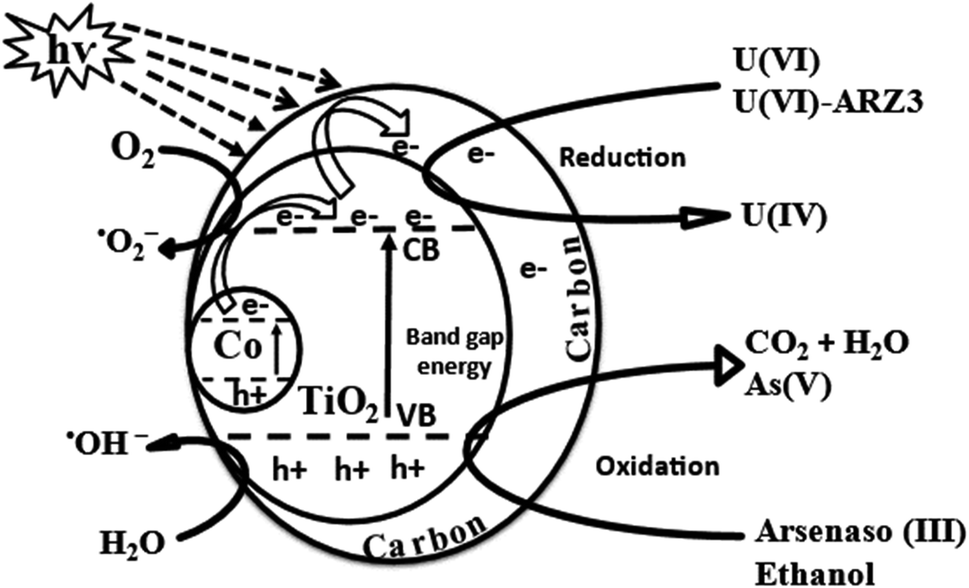
3.3. Photocatalytic mechanism
As discussed earlier, the crystal structure of anatase TiO2 is distorted by Co and oxygen vacancies. Ti4+ is replaced by Co3+ and loaded onto graphite sheets, which in addition to the oxygen vacancies, produces a bandgap of 1.7 eV. Thus, upon irradiation, excitation occurs at the bandgap of Co/TiO2, so the photo-generated electrons (e−) move from the valence band (VB) to the conduction band (CB), leaving positive holes (h+) in their place on the VB, and then this e− moves freely along the network of graphitic sheets. The graphitic sheets hinder electron and hole recombination,81 which facilitates charge separation and thus increases Co@TiO2-C efficiency.82 On the surface of the graphitic sheets, the photo-generated electrons interact with O2 to form ˙O2−, which can then react with H2O to form ˙OH/H2O2.83 In addition, h+ produced in the VB may react with H2O in the medium to form ˙OH. This free radical ˙O2−/˙OH and photo-generated e−/h+ can oxidize ARZ3 and reduce U(VI) to U(IV). The effects of free radicals on photocatalytic U(VI) reduction were further verified via radical trapping experiments, where ethanol and ARZ3 were added as scavengers for free radical ˙OH and photo-holes h+. Comparing the results in Fig. 4c indicates that in the absence of ethanol and ARZ3, U(VI) reduction has been reduced, while the presence of ethanol and U(VI)-ARZ3 promotes the removal of U(VI). These results demonstrated that ˙O2−radicals acted as the key reductive species for U(VI) reduction, while ˙OH radicals had negative effects on the reduction of U(VI).84 As a result, arsenic oxide and uranium oxides precipitate out of the solution, in agreement with the EDX results. This reaction mechanism can be described by using eqn (1) through (15) and is schematically summarised in Fig. 11.| | | Co/TiO2(1.7 Ev) + hν → e−CB + hVB+ | (1) |
| | | C + e−CB + hVB+ → C(e−) + hVB+ | (2) |
| | | ˙O2− + C + H2O → ˙OH/H2O2 + C | (4) |
| | | Co/TiO2(hVB+) + OH− → ˙OH + Co/TiO2 | (5) |
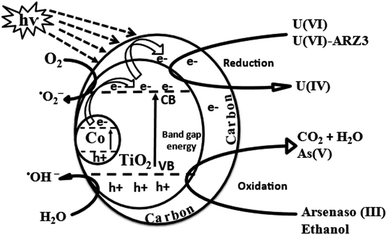 |
| | Fig. 11 Photocatalytic reduction of the U(VI)–ARZ3 mechanism using C@TiO2-C under UV light. | |
H2O
| | | H2O + hVB+ → ˙OH(ads) + H+(ads) | (6) |
ARZ3
| | | C22H18As2N4O14S2 + ˙OH → CO2 + H2O + organics intermediate | (8) |
| | | Organics intermediate + hVB+ → NH4++ SO42− + NO3− + As3+/As5+ | (9) |
| | | As3+/As5+ + eCB− + O2 → As2O3/As2O5 | (10) |
Ethanol
| | | C2H5OH + hVB+ → organics intermediate + H+ | (11) |
| | | Organics intermediate + O2 → CO2 +H2O/H2O2 | (12) |
U(VI)
| | | UO2+ + O2 → UO22+ +H2O2 | (14) |
| | | UO22+ + H2O2 + 2H2O → (UO2)O2·2H2O | (15) |
4. Conclusion
A carefully designed photocatalyst (Co@TiO2-C) was used to remove radioactive wastes under UV light. The photocatalytic performance of cobalt-doped titanium oxide was improved by inducing oxygen vacancies in the lattice and controlling the structure into 2D nanosheets. Hybridizing the oxide nanostructures with carbon during the hydrothermal process helped to prevent the recombination of the photo-generated holes and electrons and, in addition, to improve the catalytic efficiency, allowing the degradation of uranium complexes commonly associated with radioactive waste. Also, benefiting from the high surface area of the Co@TiO2-C catalyst, the photodegradation process was fast, with the ability to remove most of the uranium complex within 20–30 minutes of the process. The reaction mechanism was also investigated, and the spectroscopic analysis confirmed the reduction of soluble U(VI) to insoluble U(IV). It was also found that adding ethanol to the aqueous solutions improves the photocatalytic process by working as a scavenger to the photo-generated hole and minimizing the formation of hydrogen gas bubbles on the surface of the catalysts. We believe that the photocatalysts used in this work, coupled with the simple and affordable synthesis method, could provide new strategies and insights for the photo-reduction of toxic ions from polluted water.
Conflicts of interest
There are no conflicts to declare.
References
- R. C. Ewing, W. J. Weber and F. W. Clinard Jr, Radiation effects in nuclear waste forms for high-level radioactive waste, Prog. Nucl. Energy, 1995, 29(2), 63–127 CrossRef CAS.
- T. Xu,
et al., Review of the destruction of organic radioactive wastes by supercritical water oxidation, Sci. Total Environ., 2021, 799, 149396 CrossRef CAS PubMed.
-
AC08018188, A., Management of radioactive wastes produced by users of radioactive materials, Internat. Atomic Energy Agency, 1985 Search PubMed.
- A. Rufus, M. Dhanesh and S. Velmurugan, Dissolution of synthetic uranium dibutyl phosphate (U-DBP) in sodium EDTA and sodium carbonate based formulations, Prog. Nucl. Energy, 2017, 100, 373–379 CrossRef CAS.
- M. H. Khan, P. Warwick and N. Evans, Spectrophotometric determination of uranium with arsenazo III in perchloric acid, Chemosphere, 2006, 63(7), 1165–1169 CrossRef CAS PubMed.
- J. Du Preez, H. Rohwer and D. Morris, The chemistry of uranium. Part 31. The kinetics of the Reaction between Ce (IV) and U (IV) in acetone, Inorg. Chim. Acta, 1980, 43, 65–67 CrossRef CAS.
- A. Sabarudin,
et al., Functionalization of chitosan with 3, 4-dihydroxybenzoic acid for the adsorption/collection of uranium in water samples and its determination by inductively coupled plasma-mass spectrometry, Anal. Chim. Acta, 2007, 581(2), 214–220 CrossRef CAS PubMed.
- I. G. Alhindawy,
et al., Fabrication of mesoporous NaZrP cation-exchanger for U (VI) ions separation from uranyl leach liquors, Colloids Interfaces, 2019, 3(4), 61 CrossRef CAS.
- H. Parab,
et al., Uranium removal from aqueous solution by coir pith: equilibrium and kinetic studies, Bioresour. Technol., 2005, 96(11), 1241–1248 CrossRef CAS PubMed.
- L. Zhou,
et al., Immobilization of in situ generated Fe0-nanoparticles in tripolyphosphate-crosslinking chitosan membranes for enhancing U(VI) adsorption, J. Radioanal. Nucl. Chem., 2017, 311(1), 779–787 CrossRef CAS.
- H. Shakur,
et al., Selective removal of uranium ions from contaminated waters using modified-X nanozeolite, Appl. Radiat. Isot., 2016, 118, 43–55 CrossRef CAS PubMed.
- M.-L. Feng,
et al., Efficient removal and recovery of uranium by a layered organic–inorganic hybrid thiostannate, J. Am. Chem. Soc., 2016, 138(38), 12578–12585 CrossRef CAS PubMed.
- H. Meng,
et al., Synthesis and characterization of surface ion-imprinted polymer based on SiO2-coated graphene oxide for selective adsorption of uranium(VI), RSC Adv., 2015, 5(83), 67662–67668 RSC.
- A. Belgacem,
et al., The removal of uranium(VI) from aqueous solutions onto activated carbon developed from grinded used tire, Environ. Sci. Pollut. Res., 2014, 21(1), 684–694 CrossRef CAS PubMed.
- Y. Liu,
et al., Synthesis of gC3N4/TiO2 nanostructures for enhanced photocatalytic reduction of U(VI) in water, RSC Adv., 2021, 11(8), 4810–4817 RSC.
- J. Leal,
et al., Brookite and anatase nanomaterial polymorphs of TiO2 synthesized from TiCl3, Inorg. Chem. Commun., 2017, 84, 28–32 CrossRef CAS.
- R. Raliya,
et al., Photocatalytic degradation of methyl orange dye by pristine titanium dioxide, zinc oxide, and graphene oxide nanostructures and their composites under visible light irradiation, Appl. Nanosci., 2017, 7(5), 253–259 CrossRef CAS.
- R. Asahi,
et al., Visible-light photocatalysis in nitrogen-doped titanium oxides, Science, 2001, 293(5528), 269–271 CrossRef CAS PubMed.
- X. An and C. Y. Jimmy, Graphene-based photocatalytic composites, RSC Adv., 2011, 1(8), 1426–1434 RSC.
- M. H. Omidi,
et al., Synthesis and characterization of Au-NPs supported on carbon nanotubes: application for the ultrasound assisted removal of radioactive UO22+ ions following complexation with Arsenazo III: spectrophotometric detection, optimization, isotherm and kinetic study, J. Colloid Interface Sci., 2017, 504, 68–77 CrossRef CAS PubMed.
- M. Qiu,
et al., The photocatalytic reduction of U(VI) into U(VI) by ZIF-8/g-C3N4 composites at visible light, Environ. Res., 2021, 196, 110349 CrossRef CAS PubMed.
- H. Tada and M. Tanaka, Dependence of TiO2 photocatalytic activity upon its film thickness, Langmuir, 1997, 13(2), 360–364 CrossRef CAS.
- J. Zhang,
et al., New understanding of the difference of photocatalytic activity among anatase, rutile and brookite TiO2, Phys. Chem. Chem. Phys., 2014, 16(38), 20382–20386 RSC.
- R. Amadelli,
et al., Photodeposition of uranium oxides onto TiO2 from aqueous uranyl solutions, J. Chem. Soc., Faraday
Trans., 1991, 87(19), 3267–3273 RSC.
- J. Chen,
et al., Photocatalyzed deposition and concentration of soluble uranium(VI) from TiO2 suspensions, Colloids Surf., A, 1999, 151(1–2), 339–349 CrossRef CAS.
- E. Selli,
et al., Effects of humic acids on the photoinduced reduction of U(VI) in the presence of semiconducting TiO2 particles, Environ. Sci. Technol., 2000, 34(17), 3742–3748 CrossRef CAS.
- C. J. Evans,
et al., Photochemical removal of uranium from a phosphate waste solution, Green Chem., 2004, 6(4), 196–197 RSC.
- M. Bonato, G. Allen and T. Scott, Reduction of U(VI) to U(IVIV) on the surface of TiO2 anatase nanotubes, Micro Nano Lett., 2008, 3(2), 57 CrossRef CAS.
- M. Bonato, K. Ragnarsdottir and G. Allen, Removal of uranium (VI), lead(II) at the surface of TiO2 nanotubes studied by X-ray photoelectron spectroscopy, Water, Air, Soil Pollut., 2012, 223(7), 3845–3857 CrossRef CAS.
- S. O. Odoh,
et al., Theoretical study of the reduction of uranium(VI) aquo complexes on titania particles and by alcohols, Chem.–Eur. J., 2012, 18(23), 7117–7127 CrossRef CAS PubMed.
- L. Zhang,
et al., Photocatalytic reduction of uranyl ions over anatase and rutile nanostructured TiO2, Chem. Lett., 2013, 42(7), 689–690 CrossRef CAS.
- G. Wang,
et al., Adsorption and photocatalytic reduction of U(VI) in aqueous TiO2 suspensions enhanced with sodium formate, J. Radioanal. Nucl. Chem., 2015, 304(2), 579–585 CrossRef CAS.
- V. N. Salomone,
et al., New insights in the heterogeneous photocatalytic removal of U(VI) in aqueous solution in the presence of 2-propanol, Chem. Eng. J., 2015, 261, 27–35 CrossRef CAS.
- V. N. Salomone, J. M. Meichtry and M. I. Litter, Heterogeneous photocatalytic removal of U(VI) in the presence of formic acid: U(III) formation, Chem. Eng. J., 2015, 270, 28–35 CrossRef CAS.
- Y. K. Kim,
et al., Solar conversion of seawater uranium(VI) using TiO2 electrodes, Appl. Catal., B, 2015, 163, 584–590 CrossRef CAS.
- J. M. Meichtry,
et al., Mechanistic Features of the TiO2 Heterogeneous Photocatalysis of Arsenic and Uranyl Nitrate in Aqueous Suspensions Studied by the Stopped-Flow Technique, ChemPhysChem, 2016, 17(6), 885–892 CrossRef CAS PubMed.
- Y. Guo,
et al., Adsorption and photocatalytic reduction activity of uranium(VI) on zinc oxide/rectorite composite enhanced with methanol as sacrificial organics, J. Radioanal. Nucl. Chem., 2016, 310(2), 883–890 CrossRef CAS.
- C. Lu,
et al., Boron doped g-C3N4 with enhanced photocatalytic UO22+ reduction performance, Appl. Surf. Sci., 2016, 360, 1016–1022 CrossRef CAS.
- C. Lu,
et al., Photocatalytic reduction elimination of UO22+ pollutant under visible light with metal-free sulfur doped g-C3N4 photocatalyst, Appl. Catal., B, 2017, 200, 378–385 CrossRef CAS.
- S. Lee,
et al., Homogeneous photoconversion of seawater uranium using copper and iron mixed-oxide semiconductor electrodes, Appl. Catal., B, 2017, 207, 35–41 CrossRef CAS.
- Z.-J. Li,
et al., Enhanced photocatalytic removal of uranium (VI) from aqueous solution by magnetic TiO2/Fe3O4 and its graphene composite, Environ. Sci. Technol., 2017, 51(10), 5666–5674 CrossRef CAS PubMed.
- H. He,
et al., Simultaneous removal and recovery of uranium from aqueous solution using TiO2 photoelectrochemical reduction method, J. Radioanal. Nucl. Chem., 2017, 313(1), 59–67 CrossRef CAS.
- L. Hu,
et al., Integration of adsorption and reduction for uranium uptake based on SrTiO3/TiO2 electrospun nanofibers, Appl. Surf. Sci., 2018, 428, 819–824 CrossRef CAS.
- J. Feng,
et al., Photocatalytic reduction of Uranium(VI) under visible light with Sn-doped In2S3 microspheres, Chemosphere, 2018, 212, 114–123 CrossRef CAS PubMed.
- X.-H. Jiang,
et al., Simultaneous photoreduction of Uranium(VI) and photooxidation of Arsenic(III) in aqueous solution over g-C3N4/TiO2 heterostructured catalysts under simulated sunlight irradiation, Appl. Catal., B, 2018, 228, 29–38 CrossRef CAS.
- P. Li,
et al., Photoconversion of U(VI) by TiO2: an efficient strategy for seawater uranium extraction, Chem. Eng. J., 2019, 365, 231–241 CrossRef CAS.
- P. Li,
et al., Heterostructure of anatase-rutile aggregates boosting the photoreduction of U(VI), Appl. Surf. Sci., 2019, 483, 670–676 CrossRef CAS.
- H. Wang,
et al., Enhanced photoreduction of U(VI) on C3N4 by Cr(VI) and bisphenol A: ESR, XPS, and EXAFS investigation, Environ. Sci. Technol., 2019, 53(11), 6454–6461 CrossRef CAS PubMed.
- C. Cerrillos and D. F. Ollis, Photocatalytic reduction and removal of uranium from a uranium-EDTA solution, J. Adv. Oxid. Technol., 1998, 3(2), 167–173 CAS.
- Q. Fan,
et al., The adsorption behavior of U(VI) on granite, Environ. Sci.: Processes Impacts, 2014, 16(3), 534–541 RSC.
- J. Wang,
et al., Sorption of uranyl ions on TiO2: effects of pH, contact time, ionic strength, temperature and HA, J. Environ. Sci., 2019, 75, 115–123 CrossRef CAS PubMed.
- S. Yang,
et al., Exfoliated graphitic carbon nitride nanosheets as efficient catalysts for hydrogen evolution under visible light, Adv. Mater., 2013, 25(17), 2452–2456 CrossRef CAS PubMed.
- I. G. Alhindawy,
et al., Improving the photocatalytic performance of cobalt-doped titania nanosheets by induced oxygen vacancies for efficient degradation of organic pollutants, Nano-Struct. Nano-Objects, 2022, 31, 100888 CrossRef CAS.
- A. M. Abdelkader and E. El-Kashif, Calciothermic reduction of zirconium oxide in molten CaCl2, ISIJ Int., 2007, 47(1), 25–31 CrossRef CAS.
- A. M. Abdelkader and D. J. Fray, Synthesis of self-passivated, and carbide-stabilized zirconium nanopowder, J. Nanopart. Res., 2013, 15(12), 1–8 CrossRef.
- N. Fernando,
et al., Layer-by-layer electrode fabrication for improved performance of porous polyimide-based supercapacitors, Materials, 2021, 15(1), 4 CrossRef PubMed.
- B. Choudhury and A. Choudhury, Luminescence characteristics of cobalt doped TiO2 nanoparticles, J. Lumin., 2012, 132(1), 178–184 CrossRef CAS.
- T. M. H. Nguyen and C. W. Bark, Synthesis of cobalt-doped TiO2 based on metal–organic frameworks as an effective electron transport material in perovskite solar cells, ACS omega, 2020, 5(5), 2280–2286 CrossRef CAS PubMed.
- R. Lontio Fomekong and B. Saruhan, Synthesis of Co3+ doped TiO2 by co-precipitation route and its gas sensing properties, Front. Mater., 2019, 6, 252 CrossRef.
- L. Cançado,
et al., Geometrical approach for the study of G′ band in the Raman spectrum of monolayer graphene, bilayer graphene, and bulk graphite, Phys. Rev. B, 2008, 77(24), 245408 CrossRef.
- M. R. Islam,
et al., Fully printed and multifunctional graphene-based wearable e-textiles for personalized healthcare applications, iScience, 2022, 25(3), 103945 CrossRef CAS PubMed.
- M. Nasir,
et al., Characterization and activity of the Ce and N co-doped TiO2 prepared through hydrothermal method, Chem. Eng. J., 2014, 236, 388–397 CrossRef CAS.
- A. Merazga,
et al., Optical band-gap of reduced graphene oxide/TiO2 composite and performance of associated dye-sensitized solar cells, Mater. Sci. Eng. B, 2020, 259, 114581 CrossRef CAS.
- S.-S. Chang,
et al., Mesoporosity as a new parameter for understanding tension stress generation in trees, J. Exp. Bot., 2009, 60(11), 3023–3030 CrossRef CAS PubMed.
- E. A. Elshehy, Hexagonal Cylinder Mesoporous Sorbent for Separation of Uranium Ions from Nitrate Media, Chem. Eng. Technol., 2021, 44(8), 1470–1478 CrossRef CAS.
- Q. Xiao,
et al., Solar photocatalytic degradation of methylene blue in carbon-doped TiO2 nanoparticles suspension, Sol. Energy, 2008, 82(8), 706–713 CrossRef CAS.
- B. Gong,
et al., XPS study of cobalt doped TiO2 films prepared by pulsed laser deposition, Surf. Interface Anal., 2014, 46(10–11), 1043–1046 CrossRef CAS.
- G. Sadanandam,
et al., Cobalt doped TiO2: A stable and efficient photocatalyst for continuous hydrogen production from glycerol: Water mixtures under solar light irradiation, Int. J. Hydrogen Energy, 2013, 38(23), 9655–9664 CrossRef CAS.
- K. Lalitha,
et al., Continuous hydrogen production activity over finely dispersed Ag2O/TiO2 catalysts from methanol: water mixtures under solar irradiation: a structure–activity correlation, Int. J. Hydrogen Energy, 2010, 35(9), 3991–4001 CrossRef CAS.
- K. Lalitha,
et al., Highly stabilized and finely dispersed Cu2O/TiO2: a promising visible sensitive photocatalyst for continuous production of hydrogen from glycerol: water mixtures, J. Phys. Chem. C, 2010, 114(50), 22181–22189 CrossRef CAS.
- J.-G. Li,
et al., Cobalt-doped TiO2 nanocrystallites: radio-frequency thermal plasma processing, phase structure, and magnetic properties, J. Phys. Chem. C, 2009, 113(19), 8009–8015 CrossRef CAS.
- C. Huang,
et al., Room temperature ferromagnetism of Co-doped TiO2 nanotube arrays prepared by sol–gel template synthesis, Chem. Phys. Lett., 2006, 432(4–6), 468–472 CrossRef CAS.
- A. K. R. Police,
et al., Photocatalytic degradation of isoproturon pesticide on C, N and S doped TiO2, J. Water Resour. Prot., 2010, 2010 Search PubMed.
- H. Liu,
et al., Mesoporous cobalt-intercalated layered tetratitanate for efficient visible-light photocatalysis, Chem. Eng. J., 2013, 215, 396–403 CrossRef.
- I. Ahmed, R. F. Aglan and M. M. Hamed, Removal of Arsenazo III and Thorin from radioactive waste solutions by adsorption onto low-cost adsorbent, J. Radioanal. Nucl. Chem., 2017, 314(3), 2253–2262 CrossRef CAS.
- S. Savvin and E. Kuzin, Arsenazo III and its analogues-IV: The electronic structure of arsenzo III in media of different acidity, Talanta, 1968, 15(9), 913–921 CrossRef CAS PubMed.
-
Y. Sun, J. Li, and X. Wang, Retracted: the retention of uranium and europium onto sepiolite investigated by macroscopic, spectroscopic and modeling techniques, Elsevier, 2014 Search PubMed.
- Y. Sun,
et al., Highly efficient enrichment of radionuclides on graphene oxide-supported polyaniline, Environ. Sci. Technol., 2013, 47(17), 9904–9910 CrossRef CAS PubMed.
- L. Ping,
et al., The immobilization of U(VI) on iron oxyhydroxides under various physicochemical conditions, Environ. Sci.: Processes Impacts, 2014, 16(10), 2278–2287 RSC.
- K.-W. Kim,
et al., Evaluation of the stability of precipitated uranyl peroxide and its storage characteristics in solution, J. Nucl. Sci. Technol., 2016, 53(2), 263–270 CrossRef CAS.
- L. Sun,
et al., Anatase TiO2 nanocrystals with exposed {001} facets on graphene sheets via molecular grafting for enhanced photocatalytic activity, Nanoscale, 2012, 4(2), 613–620 RSC.
- D. C. Hurum,
et al., Explaining the enhanced photocatalytic activity of Degussa P25 mixed-phase TiO2 using EPR, J. Phys. Chem. B, 2003, 107(19), 4545–4549 CrossRef CAS.
- M. S. A. S. Shah,
et al., Single-step solvothermal synthesis of mesoporous Ag–TiO2–reduced graphene oxide ternary composites with enhanced photocatalytic activity, Nanoscale, 2013, 5(11), 5093–5101 RSC.
- J. Wang,
et al., Tunable mesoporous g-C3N4 nanosheets as a metal-free catalyst for enhanced visible-light-driven photocatalytic reduction of U(VI), Chem. Eng. J., 2020, 383, 123193 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence de,
Emad A.
Elshehy
de,
Emad A.
Elshehy