
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSugar-decorated carbon dots: a novel tool for targeting immunomodulatory receptors†
Oren
Cooper
a,
Mario
Waespy
b,
Dechao
Chen
 c,
Sørge
Kelm
b,
Qin
Li
cd,
Thomas
Haselhorst
*a and
Joe
Tiralongo
*a
c,
Sørge
Kelm
b,
Qin
Li
cd,
Thomas
Haselhorst
*a and
Joe
Tiralongo
*a
aInstitute for Glycomics, Gold Coast Campus, Griffith University Queensland, 4222 Australia. E-mail: t.haselhorst@griffith.edu.au; j.tiralongo@griifth.edu.au
bCentre for Biomolecular Interactions Bremen, Department of Biology and Chemistry, University of Bremen, 28334 Bremen, Germany
cSchool of Engineering and Built Environment, Nathan Campus, Griffith University, QLD 4111, Australia
dQueensland Micro- and Nanotechnology Centre, Australia, Nathan Campus, Griffith University, QLD 4111, Australia
First published on 3rd November 2022
Abstract
Interactions between sialic acid (Sia) and sialic acid-binding immunoglobulin-like lectins (siglecs) regulate the immune system, with aberrations contributing to pathologies such as autoimmunity, infectious disease and cancer. Over the last decade, several multivalent Sia ligands have been synthesized to modulate the Sia-binding affinity of proteins/lectins. Here, we report a novel class of multivalent siglec probes through the decoration of α(2,6)-sialyllactose ligands on inherently fluorescent carbon dots (CD). We show that the preference of α(2,3)-linked Sia for siglec-1 can be altered by increasing the multivalence of Sia ligands present on the CD, and that a locally high glycan concentration can have a direct effect on linkage specificity. Additionally, micromolar (IC50 ∼ 70 μM) interaction of α(2,6)-sialyllactose-CD (6-CD) with siglec-2 (CD22) revealed it was capable of generating a significant cytotoxic effect on Burkitt's Lymphoma (BL) Daudi B cells. This phenonomen was attributed to 6-CD's ability to form trans interactions with CD22 on masked BL Daudi cells as a direct result of clustering of the Sia moiety on the CD surface. Overall, our glycoengineered carbon dots represent a novel high affinity molecular probe with multiple applications in sialoglycoscience and medicine.
Introduction
The regulatory functions of sialic acid binding immunoglobulin-like lectins (siglecs) represent promising therapeutic strategies for a wide range of immunological disorders and cancer (immunoglyco-therapy).1,2 Siglecs are type I membrane proteins displaying an amino terminal V-set immunoglobulin domain that binds sialic acids (Sia) attached to terminal regions of cell surface glycoconjugates. Of these, siglec-2 (CD22) is of pivotal importance in B cell activation and as such an attractive target for therapies of autoimmune diseases and B cell derived non-Hodgkin's lymphoma. CD22 is a B cell-specific transmembrane protein of the Ig superfamily with seven Ig-like domains. Previous studies in mice, have demonstrated that CD22 has two distinct functions, firstly its association with the B cell receptors (BCR) followed by inhibition of the BCR signal (see ESI Fig. S1†).3–6 CD22 has known lectin properties and binds with high preference to α(2,6)-linked Sia in either cis configuration on the B cell surface or in trans configuration on the surface of other cells, soluble glycoproteins and cell-associated antigens.7–9 As periphery B cells usually display high levels of α(2,6)-linked Sia on the cell surface, CD22 will bind them with high affinity.10,11The majority of research targeting siglecs has been directed at antigen therapies,12–14 however as an alternative, multivalent sialoglycan ligands have emerged as promising tools targeting siglecs.15 Kiessling and co-workers for example constructed a copolymer that composed the BCR-binding epitope and the CD22-specific ligand. The authors found that trans-interactions between CD22 and Sia glycans are regulated by the activation of BCR. These copolymers enabled the simultaneous targeting of BCR and CD22 receptors, induced trans-interactions in the CD22 signaling, activating several pathways.16
The natural Sia ligand for CD22 typically occurs on the same cell (in cis) and/or on adjacent cells (in trans). cis ligands mask the binding of sialoside ligands and are thought to regulate the activity of CD22 as modulators of cell signalling.17 This has made it challenging to design targeted siglec monovalent sialosides.18–24 To address this, multivalent ligands of sufficient avidity have been shown to be able to compete with cis ligands, demonstrating a dynamic equilibrium of cis and trans ligand probes.25,26 The use of sialosides on nanoparticles as multivalent Sia ligands is limited.27–32 To date, the most suitable architectures for multivalent presentation of CD22 ligands are liposomes.33–37 Chen and co-workers have shown that doxorubicin loaded liposomes bearing ligands for CD22 are effective drug delivery vehicles for depletion of B cells in vivo.35 These studies highlight the importance of multivalent presentation of the Sia ligand for targeting of CD22-expressing cells.
Recent advances in glyco-nanotechnology has allowed the development of novel multivalent scaffolds that possess, at the nanoscale, inherent photonic,38 fluorescent,39 electronic,40 and magnetic41 properties to explore carbohydrate–lectin interactions in an unparalleled manner. Of these, quantum dots (QDs)42–45 and carbon dots (CDs)46,47 have been explored as suitable tools for cell targeting. QDs and CDs are zero-dimensional nano-architectures that are intrinsically fluorescent,48 which due to their high surface/volume ratios and abundant surface functional groups are capable of enhancing avidity, allowing researchers to exploit multivalent interactions. These platforms thus represent an ideal route for glycan-based cell targeting, for example as drug delivery systems or imaging agents and can be used as a diagnostic platform and biosensing tool making them highly promising theranostic devices.
In our previous work, we explored the use of CDs as multivalent scaffold for lactose.47 Here, self-assembled glycan monolayers (SAGM) were generated by functionalizing lactose with an epoxy linker, (3-glycidyloxypropyl)trimethoxysilane (GOPTS). Subsequent passivation of these SAGM to CDs provided us with a multivalent glycan coat to explore the effect lactose has on intracellular localization of CDs in cultured cells. Given our success with lactose, we have extended our approach to other glycan classes. To this end, we have engineered multivalent CDs using our SAGM method to target CD22 for BCR inhibition (see Fig. 1).
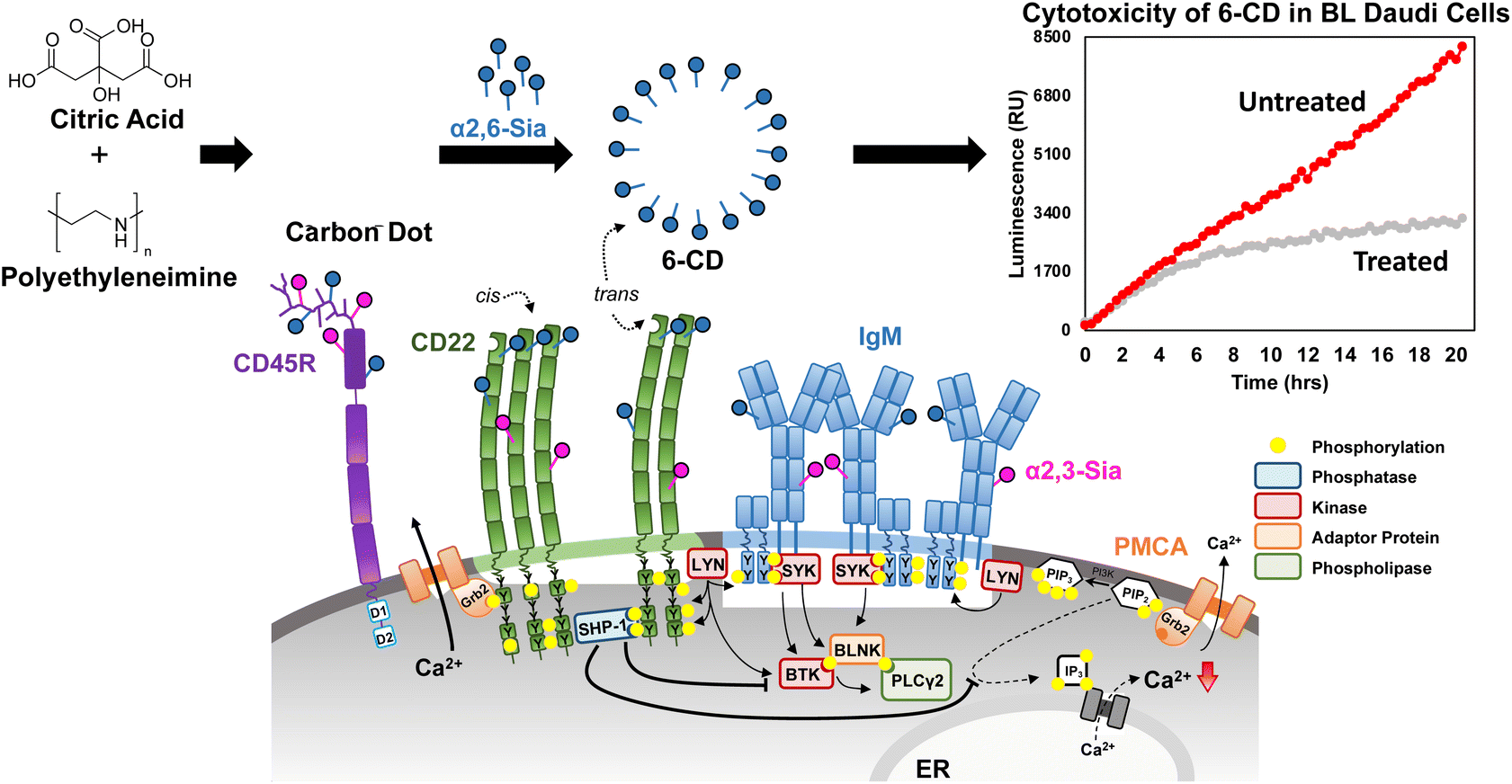 | ||
| Fig. 1 Targeting CD22 in Burkitts Lymphoma (BL) Daudi cells. The B cell receptor (BCR) is closed in resting B cells and CD22 forms homooligomers through cis-interactions with α(2,6)-linked Sia. For signal inhibition, CD22 needs to be recruited to the BCR which can occur through binding ligands of other cells or as illustrated here with carbon dots, via trans-interactions. Here, the proximity of CD22 to BCR leads to phosphorylation of the ITIMs of CD22 by LYN. Subsequent signaling cascades through SHP-1 dephosphorylation inhibit further Ca2+ release and negatively regulate the BCR signal. In our work, we demonstrate interaction of α(2,6)-sialyllactose-CD (6-CD) is capable of interacting with CD22 in trans leading to a significant cytotoxic effect. A more detailed insight into the regulation of the BCR is given in the ESI Fig. S1.† | ||
Experimental procedures
General procedures
All chemicals were purchased from Sigma-Aldrich and used without further purification. α2,3-Sialyllactose (2,3-SL) and α2,6-sialyllactose (2,6-SL) were purchased from Elicityl (France). Lactose (anhydrous) was purchased from Sigma-Aldrich. Glassware and syringes used for reactions requiring anhydrous solvents were dried in an oven (100 °C) for at least 2 h and allowed to cool in a desiccator. Liquid reagents, solutions or solvents were added via syringe through a rubber septum. Reactions were monitored through thin-layer chromatography (EtOAc/MeOH/H2O (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:0.2)) on TLC silica gel 60 F254 with UV light (365 nm) detection and by charring with 7% sulfuric acid in ethanol. Products were concentrated in vacuo using both a Büchi rotary evaporator R-114 (bath temperature set to 40 °C at a pressure of 15 mmHg) and a high vacuum line at room temperature. Water soluble compounds were freeze dried on a LDC 1 M Martin Christ Alpha 1–4. 1H NMR and 13C NMR spectra were measured in the solvent stated in a 400 MHz NMR spectrometer (Bruker) for the outlined reaction steps. The photoluminescence spectra of excitation and emission and absorption spectra of sample were recorded using Duetta (Horiba) fluorescence and absorbance spectrophotometer. 1H NMR spectra were obtained using a Bruker 400 MHz spectrometer. TEM was performed on a JEOL-2100 high-resolution transmission electron microscope with a voltage of 200 kV. FTIR spectral analysis was performed using a Bruker Alpha Fourier Transform Infra-Red spectrometer. Dynamic light scattering (DLS) and ζ analysis were carried out using a Nano ZS (Malvern Instruments). 384- and 96-well plate experiments were measured using an Infinite M200 Pro Multimodal Plate reader (Tecan) in various configurations (absorbance, fluorescence or luminescence) depending on the indicated readout.
:2:0.2)) on TLC silica gel 60 F254 with UV light (365 nm) detection and by charring with 7% sulfuric acid in ethanol. Products were concentrated in vacuo using both a Büchi rotary evaporator R-114 (bath temperature set to 40 °C at a pressure of 15 mmHg) and a high vacuum line at room temperature. Water soluble compounds were freeze dried on a LDC 1 M Martin Christ Alpha 1–4. 1H NMR and 13C NMR spectra were measured in the solvent stated in a 400 MHz NMR spectrometer (Bruker) for the outlined reaction steps. The photoluminescence spectra of excitation and emission and absorption spectra of sample were recorded using Duetta (Horiba) fluorescence and absorbance spectrophotometer. 1H NMR spectra were obtained using a Bruker 400 MHz spectrometer. TEM was performed on a JEOL-2100 high-resolution transmission electron microscope with a voltage of 200 kV. FTIR spectral analysis was performed using a Bruker Alpha Fourier Transform Infra-Red spectrometer. Dynamic light scattering (DLS) and ζ analysis were carried out using a Nano ZS (Malvern Instruments). 384- and 96-well plate experiments were measured using an Infinite M200 Pro Multimodal Plate reader (Tecan) in various configurations (absorbance, fluorescence or luminescence) depending on the indicated readout.
Carbon dot synthesis and glycan functionalization
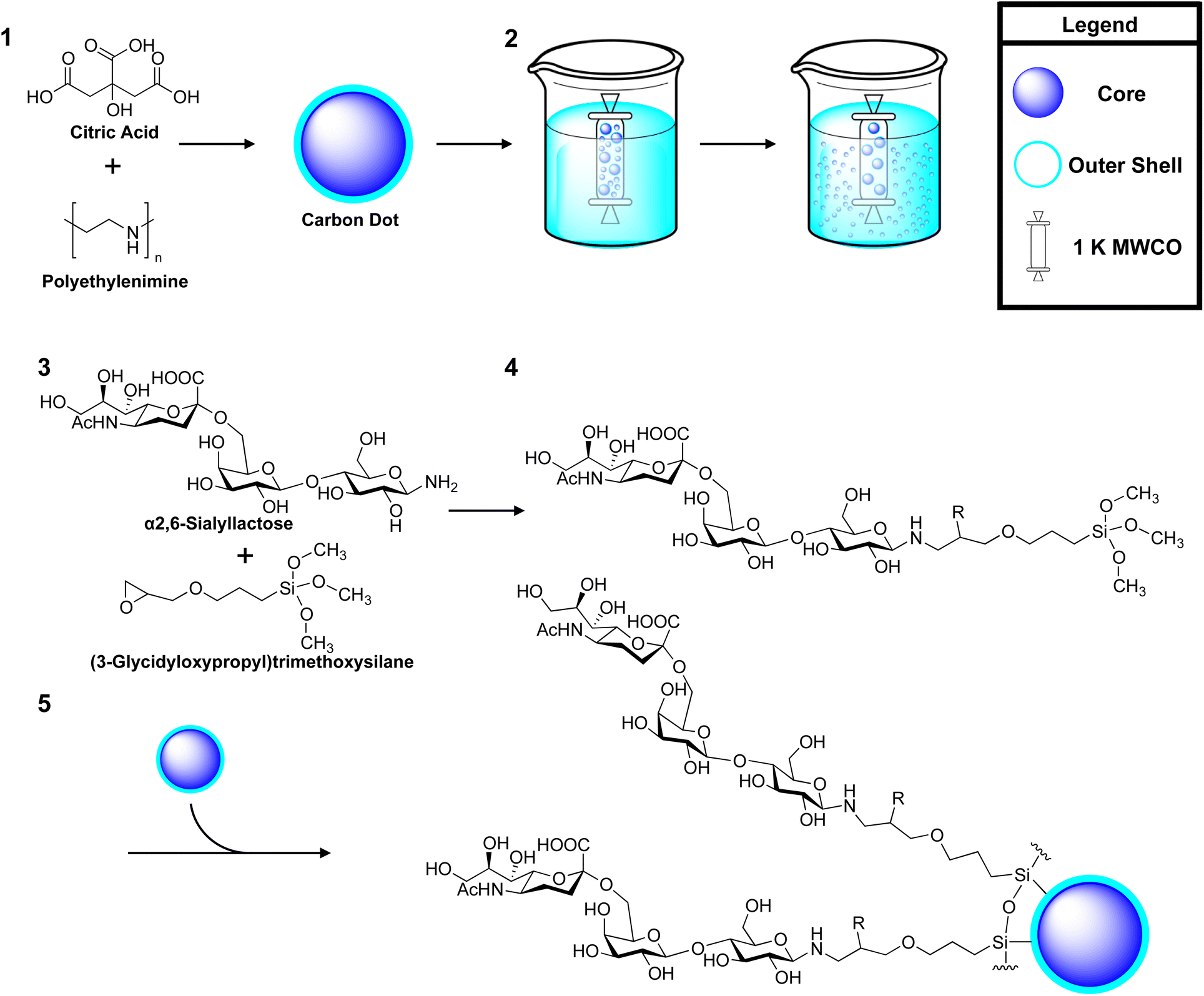 | ||
| Scheme 1 An overview of the synthesis of α2,6-sialyllactose (2,6-SL) coated carbon dots (CD). Synthesis reagents and conditions, 1 citric acid and polyethyleneimine (MW ∼ 800) (1:20) in 5 mL distilled water, treated in a Teflon-lined autoclave at 180 °C for 6 h; 2 dialysis in distilled water against 1 K MWCO to remove larger particles, only the dialysate was kept; 3 amination of 2,6-SL. Ammonium carbonate (1:5 molar excess) in ammonium hydroxide, 50 °C, 3 days; 4 silanization of 2,6-SL with GOPTS. Anhydrous DMSO:DMF (1:1), GOPTS in anhydrous methanol, 40 °C, overnight, acetic anhydride, DMAP, 4 °C, 15 min; 5 passivation to uCD. Anhydrous methanol, RT, overnight. R group indicates position of acetyl moiety after acetylation. | ||
:1). Anhydrous 3-(glycidyloxypropyl)trimethoxysilane (2%, v/v) was added to the mixture and allowed to react at 40 °C overnight to form either a lactose-GOPTS, 2,6-SL-GOPTS or 2,3-SL-GOPTS conjugate 4. The epoxy-ring was acetylated to prevent ring opening. Here, the product was cooled to 4 °C and neutralized with acetic anhydride and 4-dimethylaminopyridine (DMAP) (1:1:2, molar equivalents) for 15 min and stopped with the addition of methanol (1:1, v/v). The mixture was allowed to warm to RT and 10 mg of uCD 5 (in anhydrous methanol) was added and allowed to react overnight. The resulting products were first dialysed against 3.5 K MWCO (Spectrum Inc.) in MilliQ water for 16 h to remove larger aggregates of glyco-CDs. The particles <3.5 kDA were subsequently lyophilized and dialysed against 1 K MWCO (Spectrum Inc.) in 1 L of water for 16 h. Here, the particles >1 kDA were retained and lyophilized overnight to be used as test compounds. As all glyco-coated CDs were produced through self-assembly around uCD, they are all share a similar core identity only differing by their coat. As such they will be referred to hereafter as; 6-CD for 2,6-SL coated uCD, 3-CD for 2,3-SL coated uCD and Lac-CD for lactose coated uCD.
Biological verification and investigations
Results
Assembly and characterization of sialoglyco-CDs
In this study, four different CDs were employed to probe the interaction of multivalently displayed Sia with siglec. Here, SAGMs of lactose, α(2,6)-sialyllactose (2,6-SL) and α(2,3)-sialyllactose (2,3-SL) were generated using the epoxide functionality of GOPTS and employed to decorate CDs, thus imparting glycan functionality. Within this study, uncoated CD (uCD) and lactose-CD (Lac-CD) have both previously been extensively characterized, providing proof-of-principle for our synthesis as illustrated in Scheme 1.47,55 Subsequent generation of 6-CD and 3-CD was achieved through passivation to uCD with either a 2,6-SL or 2,3-SL SAGM, respectively. Here, amination of the anomeric carbonyl of glucose in 2,6-SL and 2,3-SL is performed to ensure the Sia moiety faces away from the CD core. Due to the higher reactivity of the amine group at the reducing end of glucose, the epoxy ring present in GOPTS affords us control in positioning the direction of the glycan ligands. The synthesis and conjugation of the glycan-silylate intermediate and SAGM passivation to uCD are outlined in Scheme 1. Importantly, self-assembly of the glycan monolayer occurs on the surface of the CD as the silanol hydrogen is far more electrophilic and therefore reactive when compared to the hydrogen of the carbinol moiety. Due to this difference in the electropositive silicon, a higher dipole moment for the silanol group occurs leading to greater hydrogen bonding to the CD.We have previously shown, that passivation of the glycan-silylate intermediates of SAGM on the CDs leads to a broader blue shift of the PL peak compared to uCD.47 This phenomenon was also observed for 6-CD and 3-CD with similar excitation peaks at 360 nm, indicating successful modification of uCD surface functional groups (see ESI, Fig. S2†). Of note, 3-CD also exhibited additional features, with lines of emission across several frequencies (415 and 430 nm) corresponding to excitation of 380 nm. This bimodal excitation has been similarly observed in uCD indicating there was still unreacted uCD present in this sample. Fourier transform infrared spectroscopy (FTIR) of both 3-CD and 6-CD (see ESI, Fig. S3†) confirmed the generation of the silicon network that forms the backbone of the SAGM. Here a sharp band at 980 cm−1 be ascribed to the Si–O–Si network. The small sharp peak positioned at 1100 cm−1 is likely due to the Si–C bond, and the large absorption band cantered at 875 cm−1 from the Si–N bond to PEI. Finally, we acquired the TEM for 6-CD and 3-CD (see ESI, Fig. S4†), which clearly shows a well-defined carbon core with a reduced area of density surrounding the core. DLS analysis (see ESI, Table S1†) illustrated a monodisperse population with size ranging from 70–90 nm, which is consistent with our previous findings.
To unambiguously confirm that uCD was decorated with our Sia-SAGMs, we employed 1H NMR spectroscopy. Consistent with our previous findings,47 the ring and anomeric protons of both 2,6-SL and 2,3-SL SAGM remain chemically unchanged through passivation to the uCD surface. 1H NMR spectra of 6-CD and 3-CD (see ESI Fig. S5 and S6†) revealed that the anomeric proton of the glucose moiety (GlcH-1) is predominantly covalently linked in the α-anomeric configuration to the GOPTS linker. Interestingly, this preference was absent in our previously characterized Lac-CD.47 To confirm that 2,6-SL and 2,3-SL glycans were conjugated correctly on 6-CD and 3-CD, we monitored the H3 equatorial and axial protons (H3eq/H3ax) of the Sia moiety as displayed in each panel of the 1H NMR spectra. The H3ax proton for 3-CD (∼1.82 ppm) and 6-CD (∼1.76 ppm) revealed that both CDs are uniquely distinct due to their glycosidic linkage. This is further supported by the H3eq protons of 3-CD (∼2.78 ppm) that appear as a pronounced doublet of doublet appearing upfield of the H3eq protons on 6-CD (∼2.75 ppm). The presence of protons resonating from the Sia moiety not only confirms successful passivation but more importantly, also pre-organisation and complete solvent exposure of the Sia residue.
The calculation of the molar concentration of glycosylated CDs was performed using the anthrone assay.56 Here, the molar concentration was revealed to be 356 ± 42.57 μM for Lac-CD, 382 ± 49.12 μM for 6-CD and 290 ± 55.56 μM for 3-CD. These values allowed direct comparison of concentrations used with monovalent glycans in subsequent experiments. Of note, the decreased molar concentration of 2,3-SL compared to Lac-CD and 6-CD supports the findings from the PL spectra of 3-CD which detected unreacted uCD. Using the already calculated molarity of the glycan coated CDs from the anthrone assay, the number of molecules per carbon dot can be derived from the below eqn (1):
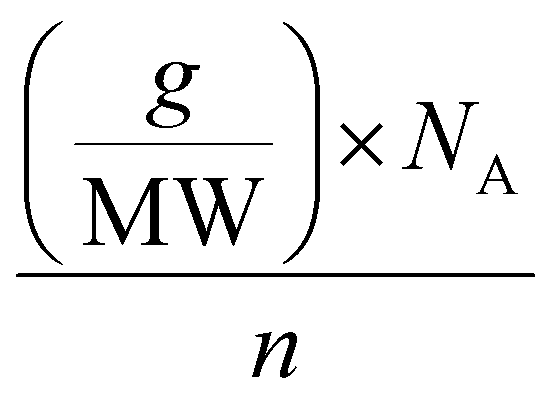 | (1) |
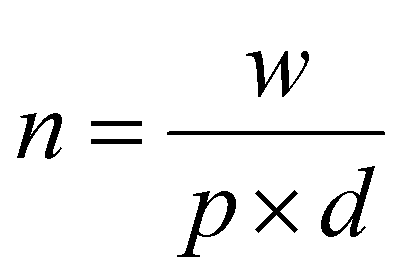 | (2) |
Hapten inhibition assays
In order to determine the relative inhibitory potential of 3-CD and 6-CD compared to their monovalent counterparts (2,3-SL and 2,6-SL), hapten inhibition assays with CD22 and siglec-1 were performed using immobilized fetuin as the Sia donor. Here, the equivalent micromolar concentrations of glycosylated-CDs, as calculated from the anthrone assay, were used to determine IC50 values for siglec-1 and CD22. Table 1 confirms similar inhibition of siglec activity as previously reported for 2,3-SL and 2,6SL,7,57–60 and Lac-CD (the “asialylated” control) was unable to inhibit either siglec-1 or CD22. As expected, 3-CD and 6-CD showed significant increases in inhibition against their respective siglec targets compared to their monovalent equivalent. For siglec-1, a 15-fold increase in inhibition was observed for 3-CD compared to monovalent 2,3-SL. Additionally, although 2,6-SL is not the preferred substrate for siglec-1, when displayed multivalently, as is the case with 6-CD, a 1.7-fold increase in rIC50 compared to monovalent 2,3-SL was also observed. This indicates that Sia linkage-specific binding to siglec-1 can be partially overcome by increasing the multivalency of Sia ligands. Moreover 6-CD displayed a 4.3-fold increase in rIC50 compared to its monovalent 2,6-SL parent compound against siglec-1. For CD22, as expected 3-CD was not shown to be inhibitory, while 6-CD demonstrated a 2.3-fold increase in rIC50 compared to 2,6-SL. Of note, the IC50 of 6-CD (67.9 μM) is comparable to that of the known CD22 sialoside inhibitor, methyl-α-9-N-(biphenyl-4-carbonyl)-amino-9-deoxy-Neu5Ac (BPC-Neu5Ac:IC50:35 μM).61 Taken together, this suggests that the multivalent display of Sia significantly increases avidity of simple glycans.| Compound | Siglec-1 | CD22 | ||
|---|---|---|---|---|
| IC50 (μM) | rIC50a | IC50 (μM) | rIC50b | |
| a rIC50 values were calculated against 2,3-SL (IC50 232.3 μM) as a reference compound. b rIC50 values were calculated against 2,6-SL (IC50 156.1 μM) as a reference compound. c Not inhibitory against 1.5 mM. Individual IC50 curves are presented in the ESI Fig. S7. | ||||
| 2,3-SL | 232.3 | 1 | n.ic | n.ic |
| 2,6-SL | 584.7 | 0.4 | 156.1 | 1 |
| 6-CD | 135.0 | 1.7 | 67.9 | 2.3 |
| 3-CD | 15.4 | 15.1 | n.ic | n.ic |
| Lac-CD | n.ic | n.ic | n.ic | n.ic |
BL daudi cell viability assays
To further explore the effect that the presentation of Sia moieties and multivalency has on siglec binding in more detail, we investigated the toxicity of our CDs on Burkitt's Lymphoma (BL) Daudi cells. Real time luminometry measurements (20 min intervals) was performed with cell viability being monitored for 20 h to provide a detailed examination of cell growth (Fig. 2). Interestingly, after 4 h of uCD treatment (Fig. 2, yellow), BL Daudi cells revealed a 2-fold increase in cell viability as measured through mitochondrial reductase activity. Compared to our control, which clearly do not divide as rapidly, these findings suggest that uCD may induce mitochondrial hyperactivation leading to increase metabolic viability and MTT reduction. This is apparent after 14 h, where the BL Daudi cell population suddenly declines. We have previously reported that following lysosomal escape, uCD is capable of co-localization within the mitochondria of HeLa cells.47 A similar mechanism may occur in BL Daudi cells, with uCD targeting the mitochondria leading to hyperactivation and increased MTT reduction. On the other hand, 6-CD (Fig. 2, grey) and Lac-CD (Fig. 2, blue) maintain comparable, albeit reduced metabolic activity, compared to the control (Fig. 2, red) up to 14 h. Beyond this point, however Lac-CD cytotoxicity is eventually overcome by the BL Daudi cells. This phenomenon was not apparent for 6-CD indicating that the Sia moiety is a strong determinant for sustained BL Daudi cytotoxicity. This was confirmed by the addition of 10 mM 2,6-SL, which abolished cell death (see bar graph insert in Fig. 2).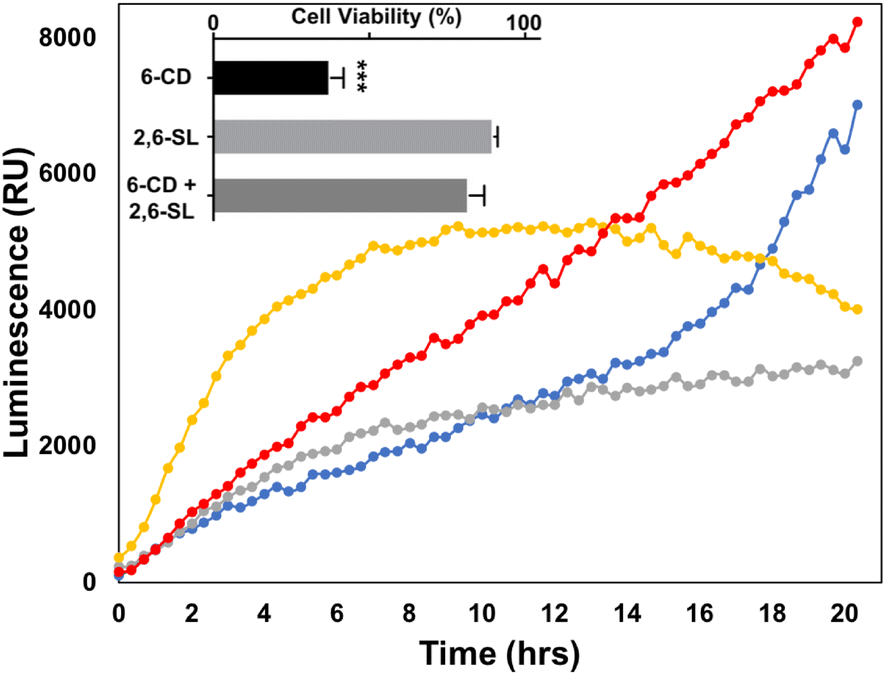 | ||
| Fig. 2 Realtime cell viability of BL Daudi cells with various carbon dots, 6-CD (grey); uCD (yellow); Lac-CD (blue). The bar graph insert is a 24 h endpoint reading of multivalent 6-CD, monovalent 2,6-SL and a mixture of 6-CD and 2,6-SL. RPMI control is indicated by *. | ||
Flow cytometric analysis of CDs
Flow cytometric analysis was performed to assess the extent to which 6-CD, Lac-CD and uCD can be recognized by surface receptors present on BL Daudi cells. CD22+ expression (Fig. 3A) on BL Daudi cells was confirmed before exploring whether cytotoxicity of 6-CD in BL Daudi cells is driven through interaction with CD22. Here, mild periodate treatment was performed to truncate endogenous linked Sia leading to unmasking of CD22.10 Initial probing of the Daudi cells with uCD and Lac-CD revealed contrasting interactions. Fig. 3B reveals an 8.5-fold increase in fluorescence for uCD compared to the control, likely due to the cationic nature of uCD leading to increased electrostatic interaction with the cell membrane and subsequent endocytosis.62 This is supported by the finding that Lac-CD exhibited only a 1.3-fold increase in fluorescence compared to the control, suggesting that the presence of the Lac coat is decreasing the non-specific electrostatic interaction associated with uCD. Fig. 3C demonstrates specific interaction of 6-CD with CD22, with an 8-fold increase in binding observed to the unlabelled Daudi population. Importantly, 6-CD bound with 6-fold higher affinity to BL Daudi cells compared to Lac-CD, highlighting that the α(2,6)-linked Sia linkage is the major determinant for binding of these CDs. Additionally, binding of 6-CD to BL Daudi cells could be inhibited by BPC-Neu5Ac, a known potent inhibitor of CD22,61 therefore unambiguously confirming 6-CD interaction with CD22. Our data clearly shows that the multivalent display of α(2,6)-linked Sia on CDs achieved using our SAGM approach permits specific targeting of CD22 on B cells.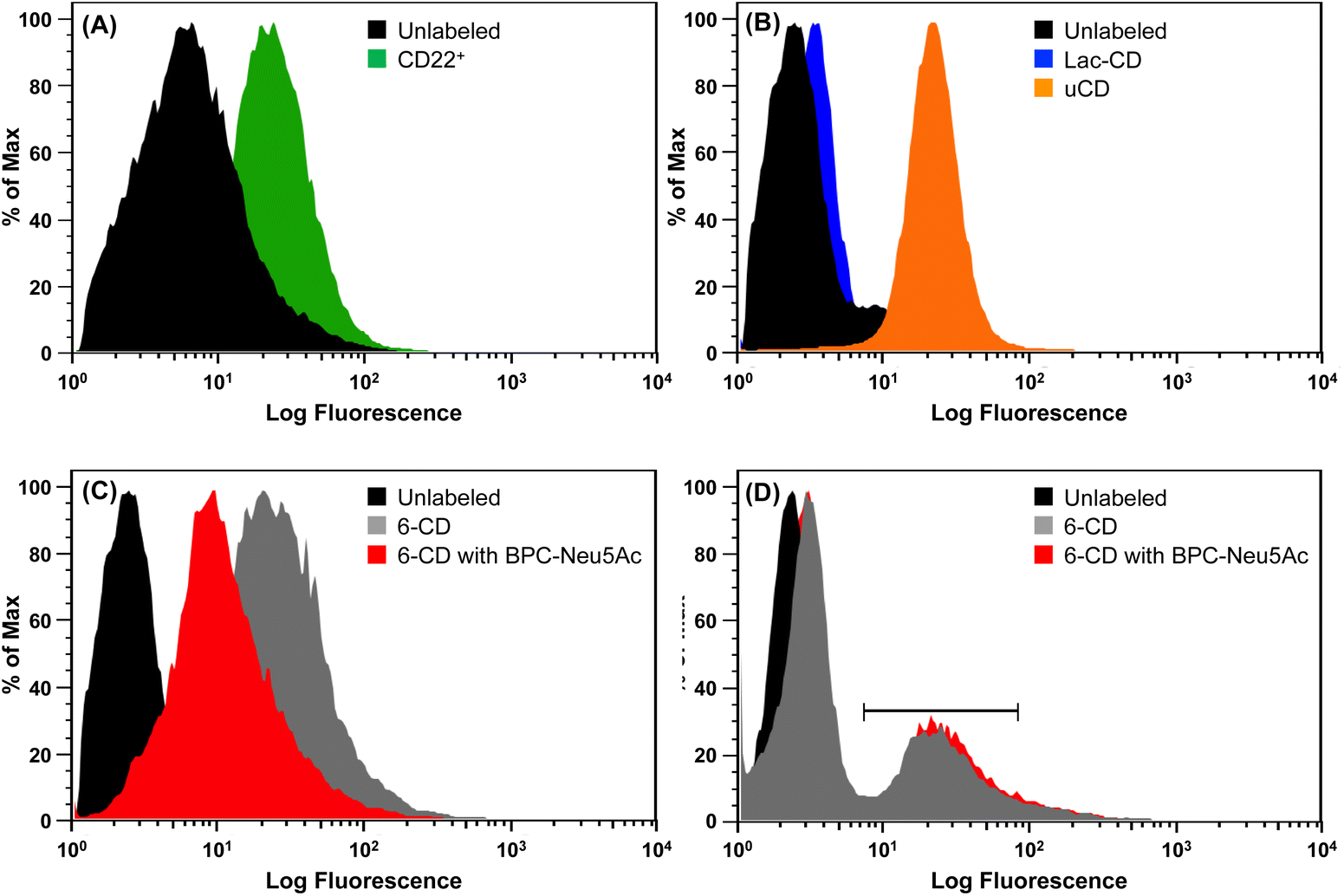 | ||
| Fig. 3 Flow cytometric analysis of various CDs interacting with CD22+ Daudi cells. (A) Unlabeled (black), median: 9.87; anti-CD22 antibody (green), median: 29.8. (B) Unlabeled (black), median: 2.65; 100 μg Lac-CD (blue), median: 3.6; 100 μg uCD (orange), median: 22.6. (C) Unlabeled (black), median: 2.65; 100 μg 6-CD (grey), median: 21.6; 100 μg 6-CD with 10 μg BPC-Neu5Ac (red), median: 10.3. (D) B cells were not pretreated with periodate. Unlabeled (black), median: 2.31; 100 μg 6-CD (grey), median 3.67, scale bar represents 30% of Daudi population, median; 23.7; 100 μg 6-CD with 10 μg BPC-Neu5Ac (red), median: 3.84, scale bar represents 35% of Daudi population, median; 27. | ||
Given the specific interaction of 6-CD to BL Daudi cell, we sought to further explore whether multivalent siglec ligands could be used to probe masked (non-periodate treated) BL Daudi cells. Fig. 3D shows that 6-CD possessed high enough avidity to compete with the cis ligands that normally mask CD22 on BL Daudi cells. Here, only a small population of BL Daudi cells (∼30%) demonstrated a 10-fold increase in interaction with 6-CD. This indicates that masked CD22 is still capable of dampening the affinity of 6-CD to the entire BL Daudi population. Importantly, this interaction could not be inhibited by monovalent BPC-Neu5Ac, that has a 2-fold better IC50 compared to 2,6-linked sialosides, suggesting that 6-CD is binding to CD22 only through trans interactions.
Discussion
The challenge to design targeted siglec sialosides has been explored in great detail,63–69 however the use of sialosides on multivalent nanoparticles is limited.28,70–72 Based on our findings, we demonstrate that CDs are suitable scaffolds for biologically relevant multivalent display of sialosides to target siglecs as well Sia-recognizing receptors. In light of the varied cellular responses seen across BL Daudi cells treated with CDs, our work highlights unique biological responses from exposure to multivalent glycan scaffolds. Additionally, further research into the effect that the CD itself has on cellular interactions would provide useful insight into how these architectures can be modified to promote targeted endocytosis and uptake. Herein, we have sought to provide the framework for further design of intelligent multivalent sialylated CDs that can be used as targeted drug delivery systems for BL Daudi cells.Thanks to the inherent fluorescence of these next-generation sialosides we were able to explore through flow cytometric analysis that cytotoxicity of 6-CD in BL Daudi cells was driven through trans interactions with CD22 (see Fig. 3D). We propose, that 6-CD interaction with CD22 allows phosphorylation of ITIMS by the kinase Lyn and subsequent recruitment of SHP-1 to negatively regulate BCR signal. Studies have demonstrated that the concentration of α(2,6)-linked Sia present on the B cell surface is estimated to be in the region of 25–30 mM,25,73 around 100-fold higher than the KD of CD22 for α2,6-linked Sia (0.1–0.3 mM (ref. 74–76)). These findings indicate CD22 is capable of cis interaction with glycoprotein ligands on the same cell. Our hapten inhibition assays revealed only a 2-fold increase in affinity for 6-CD to CD22 compared to α2,6-linked Sia, therefore it is challenging to suggest that 6-CD is capable of cis interactions with CD22.
Flow cytometry experiments revealed that ‘unmasking’ of CD22 by mild periodate treatment and removal of active Sia moieties, enables strong 6-CD binding. Binding of 6-CD to CD22 was limited in non-periodate treated BL Daudi cells, suggesting that 6-CD interact with CD22 on the B-cell surface through trans interactions. Our work supports previous findings77 that cis ligands downregulate but do not prohibit binding of ligands in trans. In vivo, B cells are known to exploit trans interactions to allow recirculating back to the bone marrow by binding to ligands that are expressed on sinusoidal endothelium.78 Additionally, it has been shown that binding of higher affinity multivalent probes to CD22 (ref. 77) or redistribution of CD22 (ref. 75) does not require unmasking. These studies highlighted a dynamic equilibrium where CD22 can switch between cis and trans interactions, depending on their relative affinity/avidity.
Further, our 6-CD findings are consistent with work performed by Collins et al., where it was concluded that cis ligands regulate siglec binding to biologically relevant trans ligands. This is achieved through thresholds set by cis ligands to control binding of CD22 by trans interactions.25,79,80 Importantly, studies into subpopulations of B cells have shown that cells can exist in both an unmasked state and that CD22 unmasking can occur after B cell activation.81,82 Taking this into consideration, as 6-CD binding to CD22 in masked BL Daudi cells could not be inhibited by the addition of BPC-Neu5Ac, this indicates that CD22 interacts with 6-CD in trans and this is favoured over the binding of ligands in cis.
Cross-linking studies have indicated that multivalency is important to achieve functional interactions with CD22.74 In our study, the multivalent nature of sialoglyco-CDs permits binding of more than one copy of CD22 receptor molecules. This is compounded especially given that CD22 forms homo-oligomers in resting B cells which would greatly increase the avidity of CD22–ligand interactions.83–85 A study by Gasparrini et al., (2016)86 using super-resolution imagining techniques demonstrated that CD22 is organised as nanoscale clusters in resting B cells. Transgenic mice expressing CD22 mutants showed that the Sia binding domain of CD22 has a major contribution in defining both lateral mobility and nanocluster organisation.86 Given this nanoscale clustering of CD22, it is likely that initial trans-interactions promote further binding of adjacent 2,6-SL moieties on our multivalent 6-CD. This in line with previous observations that suggest that low affinity binding events might be important in order to ensure that CD22 association, and allow the signalling event to be rapidly reversible.87 Further, as CD22 redistributes to the site of cell contact,80 it follows that the avidity of this interaction would also be increased by this process. This is supported by the cell viability results (Fig. 2, bar graph insert) where monovalent 2,6-SL decreased the cytotoxic effects of 6-CD.
Conclusions
The significance of the interaction between Sia and siglecs in immune regulation, cancer and infectious disease is well documented, however its role in physiological and pathological processes remains poorly understood. Here, we report for the first time the use of our SAGM approach to generate sialoglyco-CDs as a scaffold for simple targeting of siglecs. Of note, hapten inhibition assays with 3-CD demonstrated an IC50 of 15 μM against siglec-1, equating to a 15-fold increase in affinity compared to 2,3-SL. Additionally, for 6-CD we show that the preference of α(2,3)-linked Sia for siglec-1 can be altered as a result of the multivalent display of Sia ligands present on the CD. Here, the high surface/volume ratios, and abundant surface functional groups of the CDs, provide a locally high glycan concentration that has a direct effect on linkage specificity. Finally, trans interactions of 6-CD with CD22 was confirmed as a driver of cytotoxicity in BL Daudi cells. Taken together, our work highlights the potential of glycoengineered CDs as novel fluorescent molecular probes for widespread applications in sialoglycoscience, pharmaceutical and medical applications. This versatile functional behaviour of sialoglyco-CDs generated using our simple SAGM approach provides non-invasive diagnostic imaging and therapeutic opportunities especially for drug delivery applications.Author contributions
The manuscript was written through contributions of all authors. OC wrote the initial draft of the paper, performed all the experiments, data analysis and generated the figures. QL provided the carbon dots. TH and OC performed the NMR characterization. MW and SK devised the hapten inhibition assays which were performed by OC. JT and TH conceived, designed, coordinated and revised the paper. JT planned together with OC all experiments. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
TH and SK thank the Australia–Germany Joint Research Co-operation Scheme, Universities Australia for a research exchange programme. The authors thank Prof. Paul Crocker for the siglec-1 clone; Dr Judith Weber, Dr Andrea Maggioni and Mrs Stephanie Holt for expression of siglec-1; Prof. Mark von Itzstein, Dr Paul Madge and Dr Robin Thomson for supplying BPC-Neu5Ac; and James Scott for acquiring the photoluminescence spectra of 6-CD and 3-CD.Notes and references
- A. Varki and T. Angata, Glycobiology, 2006, 16, 1R–27R CrossRef PubMed.
- P. R. Crocker, J. C. Paulson and A. Varki, Nat. Rev. Immunol., 2007, 7, 255–266 CrossRef PubMed.
- S. Sato, A. S. Miller, M. Inaoki, C. B. Bock, P. J. Jansen, M. L. K. Tang and T. F. Tedder, Immunity, 1996, 5, 551–562 CrossRef CAS PubMed.
- L. Nitschke, R. Carsetti, B. Ocker, G. Köhler and M. C. Lamers, Curr. Biol., 1997, 7, 133–143 CrossRef CAS.
- K. L. Otipoby, K. B. Andersson, K. E. Draves, S. J. Klaus, A. G. Farr, J. D. Kerner, R. M. Perlmutter, C. L. Law and E. A. Clark, Nature, 1996, 384, 634–637 CrossRef CAS PubMed.
- T. L. O'Keefe, G. T. Williams, S. L. Davies and M. S. Neuberger, Science, 1996, 274, 798–801 CrossRef.
- S. Kelm, A. Pelz, R. Schauer, M. T. Filbin, S. Tang, M. E. de Bellard, R. L. Schnaar, J. A. Mahoney, A. Hartnell and P. Bradfield, et al. , Curr. Biol., 1994, 4, 965–972 CrossRef CAS.
- S. Kelm, R. Brossmer, R. Isecke, H. J. Gross, K. Strenge and R. Schauer, Eur. J. Biochem., 1998, 255, 663–672 CrossRef CAS PubMed.
- L. D. Powell and A. Varki, J. Biol. Chem., 1994, 269, 10628–10636 CrossRef CAS PubMed.
- N. Razi and A. Varki, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 7469–7474 CrossRef CAS.
- H. Floyd, L. Nitschke and P. R. Crocker, Immunology, 2000, 101, 342–347 CrossRef CAS.
- N. Kawasaki, J. L. Vela, C. M. Nycholat, C. Rademacher, A. Khurana, N. van Rooijen, P. R. Crocker, M. Kronenberg and J. C. Paulson, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7826–7831 CrossRef.
- M. De Schryver, A. Leemans, I. Pintelon, D. Cappoen, L. Maes, G. Caljon, P. Cos and P. L. Delputte, Immunobiology, 2017, 222, 797–806 CrossRef.
- T. K. Bera, M. Onda, R. J. Kreitman and I. Pastan, Leuk. Res., 2014, 38, 1224–1229 CrossRef.
- P. M. Chaudhary, S. Toraskar, R. Yadav, A. Hande, R.-A. Yellin and R. Kikkeri, Chem.–Asian J., 2019, 14, 1344–1355 CrossRef PubMed.
- A. H. Courtney, E. B. Puffer, J. K. Pontrello, Z.-Q. Yang and L. L. Kiessling, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2500–2505 CrossRef CAS PubMed.
- L. Nitschke, Immunol. Rev., 2009, 230, 128–143 CrossRef CAS PubMed.
- C. M. Nycholat, C. Rademacher, N. Kawasaki and J. C. Paulson, J. Am. Chem. Soc., 2012, 134, 15696–15699 CrossRef CAS.
- H. Prescher, A. Schweizer, E. Kuhfeldt, L. Nitschke and R. Brossmer, ACS Chem. Biol., 2014, 9, 1444–1450 CrossRef CAS PubMed.
- S. Mesch, K. Lemme, M. Wittwer, H. Koliwer-Brandl, O. Schwardt, S. Kelm and B. Ernst, ChemMedChem, 2012, 7, 134–143 CrossRef CAS PubMed.
- P. D. Madge, A. Maggioni, M. Pascolutti, M. Amin, M. Waespy, B. Bellette, R. J. Thomson, S. Kelm, M. von Itzstein and T. Haselhorst, Sci. Rep., 2016, 6, 36012 CrossRef CAS.
- S. V. Shelke, G. P. Gao, S. Mesch, H. Gathje, S. Kelm, O. Schwardt and B. Ernst, Bioorg. Med. Chem., 2007, 15, 4951–4965 CrossRef CAS PubMed.
- C. D. Rillahan, E. Schwartz, R. McBride, V. V. Fokin and J. C. Paulson, Angew. Chem., Int. Ed. Engl., 2012, 51, 11014–11018 CrossRef.
- C. D. Rillahan, E. Schwartz, C. Rademacher, R. McBride, J. Rangarajan, V. V. Fokin and J. C. Paulson, ACS Chem. Biol., 2013, 8, 1417–1422 CrossRef.
- B. E. Collins, O. Blixt, S. Han, B. Duong, H. Li, J. K. Nathan, N. Bovin and J. C. Paulson, J. Immunol., 2006, 177, 2994–3003 CrossRef.
- J. Paulson, Nat. Preced., 2009 DOI:10.1038/npre.2009.3549.1.
- S. Spence, M. K. Greene, F. Fay, E. Hams, S. P. Saunders, U. Hamid, M. Fitzgerald, J. Beck, B. K. Bains, P. Smyth, E. Themistou, D. M. Small, D. Schmid, C. M. O'Kane, D. C. Fitzgerald, S. M. Abdelghany, J. A. Johnston, P. G. Fallon, J. F. Burrows, D. F. McAuley, A. Kissenpfennig and C. J. Scott, Sci. Transl. Med., 2015, 7, 303ra140 Search PubMed.
- C. L. Schofield, M. J. Marin, M. Rejzek, P. R. Crocker, R. A. Field and D. A. Russell, Analyst, 2016, 141, 5799–5809 RSC.
- M. Ohmae, M. Kojima, K. Mihara, Y. Yamazaki, I. Hara, E. Hara and S. Kimura, Bioorg. Med. Chem. Lett., 2016, 26, 4976–4982 CrossRef CAS.
- R. Yadav, P. Madhukar Chaudhary, B. Subramani, S. Toraskar, H. Bavireddi, R. V. Murthy, S. Sangabathuni and R. Kikkeri, ACS Appl. Mater. Interfaces, 2018, 10, 28322–28330 CrossRef CAS.
- H. Zhang, J. Zhao, X. Gu and Y. Wen, J. Nanopart. Res., 2019, 21, 154 CrossRef.
- W. Peng and J. C. Paulson, J. Am. Chem. Soc., 2017, 139, 12450–12458 CrossRef CAS PubMed.
- M. K. O'Reilly and J. C. Paulson, Trends Pharmacol. Sci., 2009, 30, 240–248 CrossRef.
- P. R. Crocker and P. Redelinghuys, Biochem. Soc. Trans., 2008, 36, 1467–1471 CrossRef.
- W. C. Chen, G. C. Completo, D. S. Sigal, P. R. Crocker, A. Saven and J. C. Paulson, Blood, 2010, 115, 4778–4786 CrossRef PubMed.
- C. D. Rillahan, M. S. Macauley, E. Schwartz, Y. He, R. McBride, B. M. Arlian, J. Rangarajan, V. V. Fokin and J. C. Paulson, Chem. Sci., 2014, 5, 2398–2406 RSC.
- W. C. Chen, D. S. Sigal, A. Saven and J. C. Paulson, Leuk. Lymphoma, 2012, 53, 208–210 CrossRef CAS.
- L. Otten, D. Vlachou, S. J. Richards and M. I. Gibson, Analyst, 2016, 141, 4305–4312 RSC.
- D. Benito-Alifonso, S. Tremel, B. Hou, H. Lockyear, J. Mantell, D. J. Fermin, P. Verkade, M. Berry and M. C. Galan, Angew. Chem., Int. Ed. Engl., 2014, 53, 810–814 CrossRef CAS.
- M. D. Oliveira, C. A. Andrade, M. T. Correia, L. C. Coelho, P. R. Singh and X. Zeng, J. Colloid Interface Sci., 2011, 362, 194–201 CrossRef CAS PubMed.
- T. D. Farr, C. H. Lai, D. Grunstein, G. Orts-Gil, C. C. Wang, P. Boehm-Sturm, P. H. Seeberger and C. Harms, Nano Lett., 2014, 14, 2130–2134 CrossRef CAS.
- D. Benito-Alifonso, S. Tremel, B. Hou, H. Lockyear, J. Mantell, D. J. Fermin, P. Verkade, M. Berry and M. C. Galan, Angew. Chem., Int. Ed. Engl., 2014, 53, 810–814 CrossRef CAS PubMed.
- C. Dalal and N. R. Jana, J. Phys. Chem. C, 2018, 122, 25651–25660 CrossRef CAS.
- E. Han, L. Ding, S. Jin and H. Ju, Biosens. Bioelectron., 2011, 26, 2500–2505 CrossRef CAS.
- Y. Guo, I. Nehlmeier, E. Poole, C. Sakonsinsiri, N. Hondow, A. Brown, Q. Li, S. Li, J. Whitworth, Z. Li, A. Yu, R. Brydson, W. B. Turnbull, S. Pohlmann and D. Zhou, J. Am. Chem. Soc., 2017, 139, 11833–11844 CrossRef.
- S. A. Hill, D. Benito-Alifonso, D. J. Morgan, S. A. Davis, M. Berry and M. C. Galan, Nanoscale, 2016, 8, 18630–18634 RSC.
- O. Cooper, E. Eftekhari, J. Carter, B. Mallard, J. Kaur, M. J. Kiefel, T. Haselhorst, Q. Li and J. Tiralongo, ACS Appl. Nano Mater., 2020, 3, 7804–7817 CrossRef.
- O. Cooper and J. Tiralongo, in Comprehensive Glycoscience, Elsevier, 2021, pp. 538–565, DOI:10.1016/b978-0-12-819475-1.00012-2.
- E. Eftekhari, W. Wang, X. Li, A. Nikhil, Z. Wu, R. Klein, I. S. Cole and Q. Li, Sens. Actuators, B, 2017, 240, 204–211 CrossRef.
- D. Vetter and M. A. Gallop, Bioconjugate Chem., 1995, 6, 316–318 CrossRef CAS PubMed.
- V. E. Turula Jr, T. Gore, S. Singh and R. G. Arumugham, Anal. Chem., 2010, 82, 1786–1792 CrossRef.
- P. R. Crocker and S. Kelm, in Weir’s Handbook of Experimental Immunology, ed. L. A. Herzenberg, D. M. Weir and C. Blackwell, Blackwell Science, Cambridge, 1996, pp. 166.1–166.11 Search PubMed.
- S. Kelm, P. Madge, T. Islam, R. Bennett, H. Koliwer-Brandl, M. Waespy, M. von Itzstein and T. Haselhorst, Angew. Chem., Int. Ed., 2013, 52, 3616–3620 CrossRef CAS.
- N. Bock and S. Kelm, Methods Mol. Biol., 2006, 347, 359–375 CAS.
- E. Eftekhari, W. Wang, X. Li, A. Nikhil, Z. Wu, R. Klein, I. S. Cole and Q. Li, Sens. Actuators, B, 2017, 240, 204–211 CrossRef CAS.
- V. E. Turula, T. Gore, S. Singh and R. G. Arumugham, Anal. Chem., 2010, 82, 1786–1792 CrossRef CAS PubMed.
- E. C. Brinkman-Van der Linden and A. Varki, J. Biol. Chem., 2000, 275, 8625–8632 CrossRef.
- B. E. Collins, M. Kiso, A. Hasegawa, M. B. Tropak, J. C. Roder, P. R. Crocker and R. L. Schnaar, J. Biol. Chem., 1997, 272, 16889–16895 CrossRef.
- K. Strenge, R. Schauer, N. Bovin, A. Hasegawa, H. Ishida, M. Kiso and S. Kelm, Eur. J. Biochem., 1998, 258, 677–685 CrossRef PubMed.
- K. Hanasaki, L. D. Powell and A. Varki, J. Biol. Chem., 1995, 270, 7543–7550 CrossRef.
- S. Kelm, J. Gerlach, R. Brossmer, C.-P. Danzer and L. Nitschke, J. Exp. Med., 2002, 195, 1207–1213 CrossRef CAS.
- E. Frohlich, Int. J. Nanomed., 2012, 7, 5577–5591 CrossRef PubMed.
- C. M. Nycholat, C. Rademacher, N. Kawasaki and J. C. Paulson, J. Am. Chem. Soc., 2012, 134, 15696–15699 CrossRef CAS PubMed.
- H. Prescher, A. Schweizer, E. Kuhfeldt, L. Nitschke and R. Brossmer, ACS Chem. Biol., 2014, 9, 1444–1450 CrossRef CAS PubMed.
- S. Mesch, K. Lemme, M. Wittwer, H. Koliwer-Brandl, O. Schwardt, S. Kelm and B. Ernst, ChemMedChem, 2012, 7, 134–143 CrossRef CAS.
- P. D. Madge, A. Maggioni, M. Pascolutti, M. Amin, M. Waespy, B. Bellette, R. J. Thomson, S. Kelm, M. von Itzstein and T. Haselhorst, Sci. Rep., 2016, 6, 36012 CrossRef CAS PubMed.
- S. V. Shelke, G. P. Gao, S. Mesch, H. Gathje, S. Kelm, O. Schwardt and B. Ernst, Bioorg. Med. Chem., 2007, 15, 4951–4965 CrossRef CAS PubMed.
- C. D. Rillahan, E. Schwartz, R. McBride, V. V. Fokin and J. C. Paulson, Angew. Chem., Int. Ed. Engl., 2012, 51, 11014–11018 CrossRef.
- C. D. Rillahan, E. Schwartz, C. Rademacher, R. McBride, J. Rangarajan, V. V. Fokin and J. C. Paulson, ACS Chem. Biol., 2013, 8, 1417–1422 CrossRef.
- S. Spence, M. K. Greene, F. Fay, E. Hams, S. P. Saunders, U. Hamid, M. Fitzgerald, J. Beck, B. K. Bains, P. Smyth, E. Themistou, D. M. Small, D. Schmid, C. M. O'Kane, D. C. Fitzgerald, S. M. Abdelghany, J. A. Johnston, P. G. Fallon, J. F. Burrows, D. F. McAuley, A. Kissenpfennig and C. J. Scott, Sci. Transl. Med., 2015, 7, 303ra140 Search PubMed.
- M. Ohmae, M. Kojima, K. Mihara, Y. Yamazaki, I. Hara, E. Hara and S. Kimura, Bioorg. Med. Chem. Lett., 2016, 26, 4976–4982 CrossRef.
- R. Yadav, P. Madhukar Chaudhary, B. Subramani, S. Toraskar, H. Bavireddi, R. V. Murthy, S. Sangabathuni and R. Kikkeri, ACS Appl. Mater. Interfaces, 2018, 10, 28322–28330 CrossRef CAS PubMed.
- B. E. Collins, O. Blixt, A. R. DeSieno, N. Bovin, J. D. Marth and J. C. Paulson, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 6104–6109 CrossRef CAS PubMed.
- L. D. Powell, R. K. Jain, K. L. Matta, S. Sabesan and A. Varki, J. Biol. Chem., 1995, 270, 7523–7532 CrossRef CAS.
- T. R. Bakker, C. Piperi, E. A. Davies and P. A. Merwe, Eur. J. Immunol., 2002, 32, 1924–1932 CrossRef CAS.
- Y. Suzuki, T. Ito, T. Suzuki, R. E. Holland Jr, T. M. Chambers, M. Kiso, H. Ishida and Y. Kawaoka, J. Virol., 2000, 74, 11825–11831 CrossRef CAS.
- O. Blixt and J. C. Paulson, Adv. Synth. Catal., 2003, 345, 687–690 CrossRef CAS.
- L. Nitschke, H. Floyd, D. J. Ferguson and P. R. Crocker, J. Exp. Med., 1999, 189, 1513–1518 CrossRef CAS.
- B. E. Collins, O. Blixt, N. V. Bovin, C. P. Danzer, D. Chui, J. D. Marth, L. Nitschke and J. C. Paulson, Glycobiology, 2002, 12, 563–571 CrossRef CAS PubMed.
- B. E. Collins, O. Blixt, A. R. DeSieno, N. Bovin, J. D. Marth and J. C. Paulson, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 6104–6109 CrossRef.
- N. Razi and A. Varki, Glycobiology, 1999, 9, 1225–1234 CrossRef.
- C. P. Danzer, B. E. Collins, O. Blixt, J. C. Paulson and L. Nitschke, Int. Immunol., 2003, 15, 1137–1147 CrossRef PubMed.
- S. Han, B. E. Collins, P. Bengtson and J. C. Paulson, Nat. Chem. Biol., 2005, 1, 93–97 CrossRef PubMed.
- C. L. Law, A. Aruffo, K. A. Chandran, R. T. Doty and E. A. Clark, J. Immunol., 1995, 155, 3368–3376 CAS.
- S. Han, B. E. Collins, P. Bengtson and J. C. Paulson, Nat. Chem. Biol., 2005, 1, 93–97 CrossRef CAS.
- F. Gasparrini, C. Feest, A. Bruckbauer, P. K. Mattila, J. Muller, L. Nitschke, D. Bray and F. D. Batista, EMBO J., 2016, 35, 258–280 CrossRef CAS.
- T. F. Tedder, J. Tuscano, S. Sato and J. H. Kehrl, Annu. Rev. Immunol., 1997, 15, 481–504 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2na00364c |
| This journal is © The Royal Society of Chemistry 2022 |