
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceContinuous flow fabrication of green graphene oxide in aqueous hydrogen peroxide†
Kasturi
Vimalanathan
 *a,
James
Scott
b,
Xun
Pan
a,
Xuan
Luo
ac,
Soraya
Rahpeima
ad,
Qiang
Sun
ef,
Jin
Zou
ef,
Nidhi
Bansal
g,
Elisabeth
Prabawati
g,
Wei
Zhang
c,
Nadim
Darwish
d,
Mats R.
Andersson
a,
Qin
Li
b and
Colin L.
Raston
*a
*a,
James
Scott
b,
Xun
Pan
a,
Xuan
Luo
ac,
Soraya
Rahpeima
ad,
Qiang
Sun
ef,
Jin
Zou
ef,
Nidhi
Bansal
g,
Elisabeth
Prabawati
g,
Wei
Zhang
c,
Nadim
Darwish
d,
Mats R.
Andersson
a,
Qin
Li
b and
Colin L.
Raston
*a
aFlinders Institute for Nanoscale Science and Technology, College of Science and Engineering, Flinders University, Adelaide, SA 5001, Australia. E-mail: colin.raston@flinders.edu.au; kasturi.vimalanathan@flinders.edu.au
bEnvironmental Engineering and Queensland Micro and Nanotechnology Centre, Griffith University, Brisbane, QLD 4111, Australia
cCentre for Marine Bioproducts Development, College of Medicine and Public Health, Flinders University, Adelaide, SA 5042, Australia
dSchool of Molecular and Life Sciences, Curtin Institute for Functional Molecule and Interfaces, Curtin University, Bentley, Western Australia 6102, Australia
eCentre for Microscopy and Microanalysis, The University of Queensland, Brisbane, QLD 4072, Australia
fMaterials Engineering, The University of Queensland, St Lucia, QLD 4072, Australia
gSchool of Agriculture and Food Sciences, The University of Queensland, St Lucia, QLD, Australia
First published on 19th May 2022
Abstract
Highly processible graphene oxide (GO) has a diversity of applications as a material readily dispersed in aqueous media. However, methods for preparing such free-standing GO use hazardous and toxic reagents and generate significant waste streams. This is an impediment for uptake of GO in any application, for developing sustainable technologies and industries, and overcoming this remains a major challenge. We have developed a robust scalable continuous flow method for fabricating GO directly from graphite in 30% aqueous hydrogen peroxide which dramatically minimises the generation of waste. The process features the continuous flow thin film microfluidic vortex fluidic device (VFD), operating at specific conditions while irradiated sequentially by UV LED than a NIR pulsed laser. The resulting ‘green’ graphene oxide (gGO) has unique properties, possessing highly oxidized edges with large intact sp2 domains which gives rise to exceptional electrical and optical properties, including purple to deep blue emission of narrow full width at half maximum (<35 nm). Colloidally stable gGO exhibits cytotoxicity owing to the oxidised surface groups while solid-state films of gGO are biocompatible. The continuous flow method of generating gGO also provides unprecedented control of the level of oxidation and its location in the exfoliated graphene sheets by harnessing the high shear topological fluid flows in the liquid, and varying the wavelength, power and pulse frequency of the light source.
Introduction
The Anthropocene era necessitates the development of processes and products that negate adverse effects on the natural environment. Materials play a pivotal role in societal needs, and this includes various forms of nanocarbon which have wide ranging applications, from electronics to medicine and commodity products. Accordingly, it is important to incorporate green chemistry metrics into the fabrication of such materials, as ‘top down’ or ‘bottom up’ processes, by avoiding the use of chemical stabilizers and other auxiliary substances, and featuring just in time production, coupled with simplicity of the process and minimal processing time. This includes the ‘top down’ fabrication of two-dimensional (2D) graphene which has remarkably diverse properties,1–4 attracting extensive research and development for small scale niche applications through to large scale products. Graphite consists of single graphene layers of sp2 hybridised carbon atoms assembled through van der Waals forces arising from the overlap of π–π orbitals between the graphene layers. Different methods have been developed for the production of graphene from graphite involving either induced mechanical energy or chemical exfoliation.5 However, its applications are somewhat limited because graphene is a zero-band gap semiconductor making it challenging for implementation into electronic and optoelectronic applications.6 The large-scale production of high-quality graphene sheets has also remained a significant challenge which hinders industry uptake.7 In 1859, British chemist, Benjamin Brodie unbeknown discovered a method to prepare atomically thin graphene oxide (GO) sheets in large scales. They are chemically modified derivative of graphene decorated with oxygen moieties, as hydroxyl and epoxide functionalities, and carboxyl and carbonyl functionalities, on basal plane and edges, respectively.8–10 These modify the properties of graphene, which becomes an insulator with fluorescence over a broad range of wavelengths, and they facilitate solution-based processability and uptake of the material in applications.9,11 The ‘Brodie method’ of generating GO has evolved into a well-established procedure.12 Contemporary chemical methods for fabricating GO include those based on the Brodie, Staudenmaier and Hummers' methods,13–15 all typically involving but not limited to the use of strong acids and oxidants, including potassium chlorate (KClO3), nitric acid (HNO3), concentrated sulfuric acid (H2SO4) and potassium permanganate (KMnO4).There has been considerable effort towards improving the synthesis of GO while maintaining the level of surface oxidation without compromising on its structural integrity, but the use of strong acids and oxidizing agents has thus far been unavoidable.16–18 Notwithstanding the economic, environmental and sustainability issues for the abovementioned processes, the diversity in properties and applications of GO, the commercial scale production of GO has attracted considerable interest amongst the scientific community. The electronically hybrid hexagonal carbon network in GO consists of a mixture of both π states from the sp2 hybridised carbon and σ states from the sp3 carbon domains resulting from the oxygen functionalisation within the lattice.19,20 The plausibility of tuning the sp2 and sp3 domains via a subsequent reduction process offers a path towards for tuning the bandgap of GO and transforming its insulating property to semiconducting or metallic property in a controlled way. The tunability in the sp2 and sp3 domains also allows for GO to exhibit luminescence properties, a feature that is pivotal, in particular, for biomedical applications.21,22 In addition, such processing is important for materials specific applications, including in transparent conductive films, supercapacitors, fuel cells, multifunctional separation membranes, conductive fibres, lithium-ion batteries, polymer composites, aerogels and more.10,23–28 The reduction of GO can recover the disrupted sp2 conjugated graphene network within the realm of utility imparting properties similar to those of graphene prepared directly by mechanically exfoliating graphene from graphite. However, there is a significant compromise in the quality of the graphene sheets from such extensive processing thereby limiting its potential applications.29–33
Thus, an ultimate goal is to produce uniform high-quality solution processable GO sheets under continuous flow conditions ideally as a zero-waste process that is safe and scalable with the additional capabilities of being able to tune the sp2 and sp3 fractions and circumvent the subsequent reduction step in simplifying the overall process.
Results and discussion
Herein we report a ‘top down’ fabrication process, for oxidising graphene sheets from graphite in a controlled way, incorporating the principles of green chemistry, in dramatically reducing the waste stream. The continuous flow process allows large scale production of graphene oxide, which we designate as such as green graphene oxide. The ability to control levels of oxidation is envisaged as an avenue towards balancing between achieving sufficient oxygen functionalities to allow for dispersibility in aqueous media while limiting disruption to the sp2 conjugated graphene network. This effectively eliminates the need for any subsequent reduction step for product specific applications. The benign process herein involves oxidation of graphite using 30% aqueous H2O2 in the continuous flow vortex fluidic device (VFD) which has been shown to be efficient for oxidation of small molecules compared to traditional processing platforms.34 Aqueous H2O2 was considered an ideal oxidant of choice based on its safety and environmentally credentials, affording innocuous by-products as such, as well as its higher oxidation potential (2.80 V) relative to that of ozone (2.07 V).35The VFD is a thin film microfluidic platform with a diverse range of applications, including in materials processing, organic chemistry, biocatalysis, and probing the structure of self-organised systems.36–41 It features a glass tube (usually quartz) rotating at high speed (2000–9000 rpm) at 45° tilt angle which is optimal for any process in the device (Fig. 1). Liquid is continuously delivered to the bottom of the glass tube in the VFD (or at points along the tube), which then forms a thin film as it moves up the tube, exiting at the top. The VFD has high shear topological fluid flow at submicron dimensions, as spinning top flow generated by the Coriolis force generated at the hemispherical base of the tube, double helical flow as eddies from Faraday waves twisted by the Coriolis force associated with curvature along the tube, and a special case where these fluid flows have the same diameter.42 The VFD also has high heat transfer, for example in controlling highly exothermic reactions34 and the thin film in the device allows uniform processing when irradiated with external light sources.43 We hypothesised that these unique effects in the VFD can accelerate and control the in situ oxidation and exfoliation of graphene from graphite, forming gGO.
 | ||
| Fig. 1 Synthesis and characterization of oxidized graphene sheets. (a) Schematic of the sequential continuous flow processing in the vortex fluidic device (VFD) while (1) irradiated with a UV lamp for photo-dissociation of aqueous H2O2, step 1, and (2) then in a separate VFD set up irradiated using a pulsed NIR laser operating at 1064 nm at different laser powers, step 2. (b) Resulting powder sample of the gGO after washing 5 times in water and then freeze dried (laser power 650 mJ); the freeze-dried sample was then dispersed in water at a concentration of 1 mg mL−1. (c) SEM and (d and e) AFM height images of gGO sheets (generated at 260 mJ power) which are mostly between 2 to 5 nm thick, corresponding to 2 to 5 sheets of gGO, the higher areas, >5 nm in thickness, corresponding to areas of high corrugation and wrinkling. (f and h) TEM and (g and i) HRTEM images of gGO sheets. Note: The height analysis and associated average distribution plot is presented in the ESI as Fig. S1.† | ||
Graphite ore flakes were supplied by First Graphene Ltd, with a purity of ≥99% and particle dimensions ranging between 200 μm to 1 mm. The graphite flakes were dispersed in 30% aqueous H2O2 and allowed to stand overnight, whereupon a sequential two-step continuous flow process involving the VFD was developed, involving systematically exploring the processing parameters (flow rates, concentration of the graphite, tilt angle and rotational speed of the 20 mm OD quartz tube) (20 mm OD, 17.5 mm ID, 18.5 cm long), for maximising the level of oxidation and exfoliation of the graphite, as follows: (1) UV irradiation (20 W, λ = 235 nm) of a suspension of graphite in 30% aqueous hydrogen, and (2) irradiating the resulting suspension from (1) with a pulsed Nd:YAG laser (λ = 1064 nm) at a laser power of 260 mJ. The resulting material was then centrifuged to remove any contaminants and any bulk graphite, followed by high-speed centrifugation to collect the gGO material. This was then washed three times with Milli-Q water with the final material freeze dried to afford a dry black powder. Characterisation of the materials used X-ray photoelectron spectroscopy (XPS) and Raman spectroscopy, as primarily techniques for determining the percent of oxidation and nature of the functional groups, scanning electron microscopy (SEM), high resolution transmission electron microscopy (TEM), atomic force microscopy (AFM), electrochemistry, UV-vis spectroscopy, photoluminescence and 4-point probe conductivity measurements.
The overall operation of the processing of the graphite ore is summarised in Fig. 1a. We chose aqueous hydrogen peroxide (H2O2) as the oxidising agent, as the ultimate reagent in improving the green chemistry metrics, after air and pure oxygen as oxidants, with a concentration of 30% as a trade-off between maximising potential for oxidation and safety. After dispersing the graphite overnight in 30% H2O2, the mixture was passed through the VFD while UV irradiated, noting that UV irradiation is a highly efficient method (and low costing) to dissociate H2O2 into OH˙.44,45 While OH˙ is short lived, it rapidly undergoes non-selective reactions with organic compounds within its vicinity.46 This accounts for ≥20% oxidisation predominantly on the edges of the graphene sheets in the graphite flakes during this first step (Fig. 2) (see below). The VFD process was optimised by systematically varying the rotational speed (4000 rpm to 9000 rpm), the inclination angle (0° to 90°) and the flow rate under continuous flow conditions. Based on this, we established the optimised conditions were a rotational speed of 8000 rpm, inclination angle of θ 45° and a flow rate of 0.5 mL min−1.
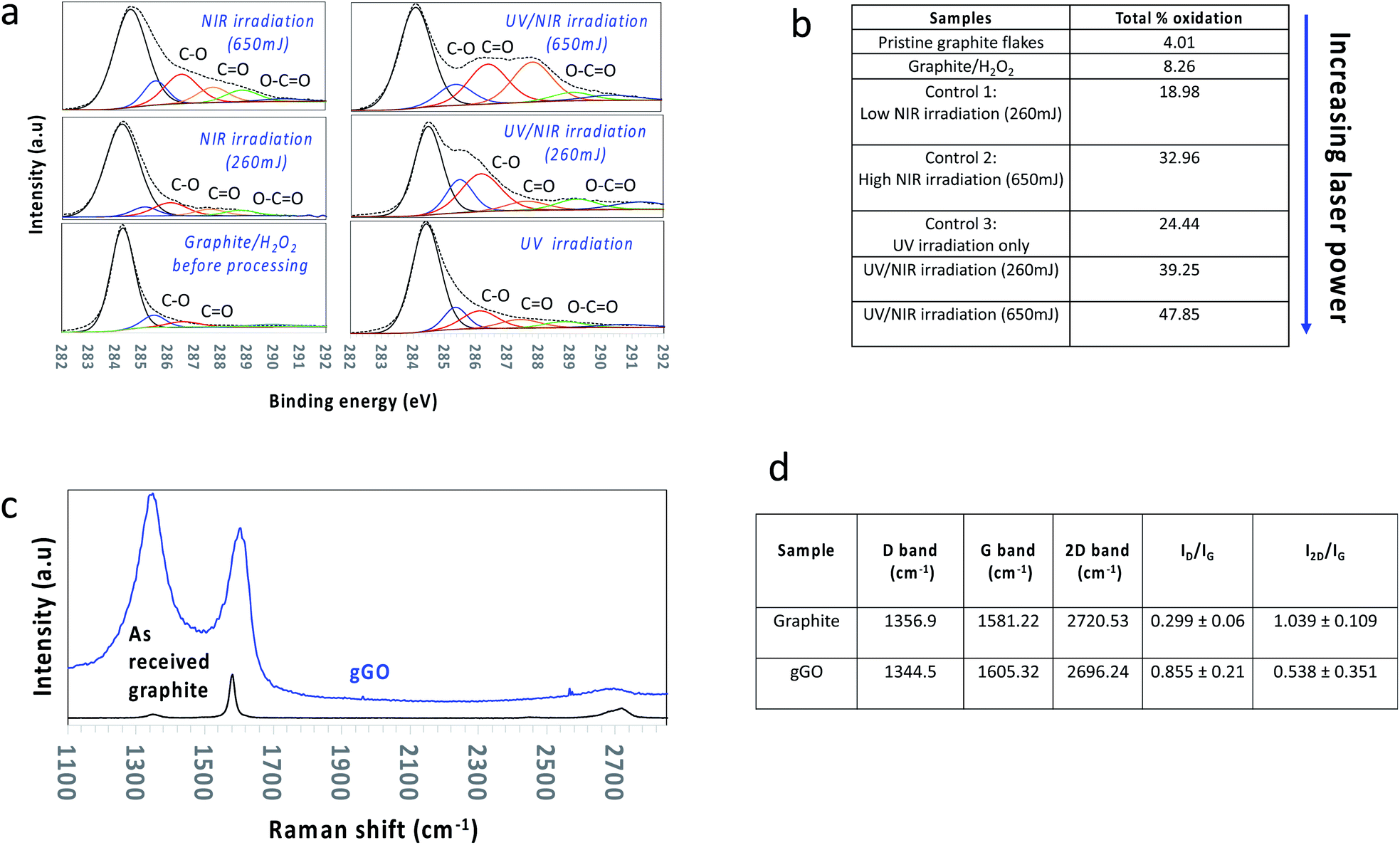 | ||
| Fig. 2 Surface characteristics of oxidised graphene. (a and b) XPS analysis based on the deconvolution of the C1s peak of the oxidized graphene generated under the different operating conditions and described in (c and d) Raman spectra, comparing peak shifts, widening of the peaks (FWHM) and increase in band intensity relative to as received graphite flakes (black line), and the oxidised graphene sheets (blue line). | ||
However, the material from the first step was colloidally unstable (≤24 hours), and this was overcome by inducing oxidation in the basal plane of the graphene, which was achieved in the second step. This involved in situ irradiating a suspension from the first process with a pulsed near infrared (NIR) Nd:YAG laser operating at 1064 nm. Absorbing energy from the NIR laser after UV irradiation facilitated oxidation via nucleophilic addition on either side of the basal plane but without disregarding the potential for further edge oxidation, as shown in Fig. 2b, which enlists the quantities of the carbon–oxygen bonding states under different processing conditions. This depended on the choice of laser power, which was investigated at 260, 450, and 650 mJ. Absorption of the NIR radiation will effectively heat the graphitic material in the VFD, increasing for increasing power,35,47 with additional heating at localised regions on the surface of the tube where the spinning top and topological fluid flows strike its surface (Fig. 1a).42 At 650 mJ, the oxidation levels in the gGO were close to that achieved using the conventional Hummers' method and the material is colloidally stable (4–5 months). Nevertheless, even at the lowest power, 260 mJ, stable colloidal suspensions were generated (2–3 months), despite the lower levels of resulting oxidation (see below) (Fig. 2a). Our previous study on in situ VFD processing of carbon nanotubes while irradiated with a pulsed NIR laser resulted in controlled slicing of the material at lengths depending on the laser power40,48,49 which is now understood as a combined effect of bending from the topological fluid flows and heating from the laser. We also note that carrying this reaction out in 30% H2O2 resulted in breakdown of multiwalled carbon nanotubes in forming carbon dots, as a controlled oxidation process.41 Overall, in the present study the level of exfoliation and surface oxidation of graphene is similar to that obtained using Hummers' method, but in contrast to Hummers' method, the level of oxidation can now be controlled (see below) without compromising on dispersibility in aqueous media and maintaining high quality and crystallinity of the material. We note the ability to achieve a yield of 40–50% of the oxidized material for all the oxidation levels with then the unoxidized graphite separated, collected and fed back into the system with additional graphite starting material, such that there is ultimately no unused graphite.
Raman spectroscopy and XPS (Fig. 2) were used to determine the level of defects/oxygen functionalities present in the lattice of material post VFD processing, for both processing steps. Graphitic material in general has a number of Raman active bands with their positions strongly dependent on the purity of the material. They include the D band (∼1355 cm−1) corresponding to the disorder from the graphite edge, the G band (∼1575 cm−1) originating from the activation of the doubly degenerate zone centre E2g phonon mode, and the 2D band (∼2678 cm−1) arising from a double resonance process which relates to the electronic band structure and the phonon dispersion.50 Surface oxidation with a high level of defects of graphene sheets herein is supported by (a) a significant increase in the averaged Raman ID/IG ratio (corresponding to the D-band and G-band intensities), (b) broadening of the G band associated with a blue shift to higher wavenumbers, (c) an increase in full width of half maximum (FWHM) of both the D and G band, and (d) shifting of the 2D band to a lower wavenumber (red shift) with a decrease in intensity (Fig. 2b). All Raman spectra were conducted as a bulk measurement using the same laser excitation frequency (λ = 532 nm), such that the data is directly comparable from one sample to another.
XPS was used to quantify the elemental composition with respect to the C/O ratio, along with determining the chemical environments of the atoms in the gGO. Deconvolution of the peak area of the C1s peak gave assignable peaks corresponding to the asymmetric sp2 carbon (binding energy (BE) at 284.3 eV), sp3 carbon (285.6 eV), C–O (286.7 eV), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (287.6 eV) and –O–CO (288.6 eV). Deconvolution of the C1s peak for increasing laser power (260 mJ, 450 mJ, 650 mJ) established (i) a decrease in intensity of the sp2 carbon peak with a gradual increase in the sp3 carbon peak, (ii) an increase in peaks corresponding to hydroxyl and epoxide species (weak oxidation), and (iii) introduction of carboxyl groups (heavy oxidation) (Fig. 2b and S2†). The maximum laser power output, 650 mJ afforded graphene sheets with the highest surface oxidation, ∼45–48%. Control experiments showed that laser power < 650 mJ afforded lower surface oxidation than the above with the lower power limit being ∼260 mJ which resulted in ∼33–40% surface oxidation. Laser power ≤ 260 mJ gave minimal to no increase in surface oxidation relative to the material generated in step 1, with the resulting material colloidally unstable in aqueous medium, unlike all other gGO prepared at higher laser power. The zeta potential of the colloidally stable suspensions of the gGO was measured at different pH values corresponding to moderate colloidal stability (Fig. S3, ESI†). The negative zeta potential values (−44 mV) are due to the electrostatic repulsion between the negatively charged functional groups on the edges and surface of the oxidised sheets.51
O (287.6 eV) and –O–CO (288.6 eV). Deconvolution of the C1s peak for increasing laser power (260 mJ, 450 mJ, 650 mJ) established (i) a decrease in intensity of the sp2 carbon peak with a gradual increase in the sp3 carbon peak, (ii) an increase in peaks corresponding to hydroxyl and epoxide species (weak oxidation), and (iii) introduction of carboxyl groups (heavy oxidation) (Fig. 2b and S2†). The maximum laser power output, 650 mJ afforded graphene sheets with the highest surface oxidation, ∼45–48%. Control experiments showed that laser power < 650 mJ afforded lower surface oxidation than the above with the lower power limit being ∼260 mJ which resulted in ∼33–40% surface oxidation. Laser power ≤ 260 mJ gave minimal to no increase in surface oxidation relative to the material generated in step 1, with the resulting material colloidally unstable in aqueous medium, unlike all other gGO prepared at higher laser power. The zeta potential of the colloidally stable suspensions of the gGO was measured at different pH values corresponding to moderate colloidal stability (Fig. S3, ESI†). The negative zeta potential values (−44 mV) are due to the electrostatic repulsion between the negatively charged functional groups on the edges and surface of the oxidised sheets.51
The first processing step, involving UV irradiation, is likely to result in OH˙ reacting predominantly with the edges of the graphite lattice.46 XPS data is consistent with an increase in CO and O–CO groups after such UV treatment. NIR laser irradiation presumably promotes the reaction of OH˙ with CC moieties in the basal plane of graphene. Theoretical studies support that pulsed NIR laser processing provides sufficient energy to cleave CC bonds in single walled carbon nanotubes;40 increasing the vibrational energy necessary for this is likely to facilitate reaction with H2O2 and also OH˙ for graphene in the present study. XPS data for material post VFD processing in the second step using NIR laser irradiation is consistent with an increase in the content of hydroxyl and epoxide moieties, with an increase in laser power from 260 mJ to 650 mJ associated with an increase in surface oxidation, from 33% to 48%. We note that all applications of gGO presented below were for an overall surface oxidation of gGO at ∼35%, i.e. gGO generated using 260 mJ laser power.
Colloidal aqueous suspensions of gGO were used to prepare uniform films to measure electrical conductivity and resistance, using a 4-point probe system, noting that the electrical conductivity in such films depends on the conjugated sp2 network in the graphene sheets and the number of effective contacts established between sheets within the film (Fig. S2, ESI†). All conductivity measurements were in triplicate and are based on three different oxidised graphene films to ensure reliability and reproducibility of the results. The gGO sheets fabricated using our described method showed a high sheet resistivity of 3.25 ± 0.79 MΩ, which is consistent with the disruption of the conjugated framework of the graphene lattice.
Applications
Photoluminescence properties of the gGO
gGO was subjected to membrane dialysis (Spectrapor™ MWCO of 3.5 kDa), yielding a transparent dialysate, with purple to deep blue photoluminescence under 365 nm illumination. The emissions present a high spectral purity rarely seen in carbon-based fluorescent materials, with a FWHM of 35 nm, which is on par with some of the best narrow bandwidth materials. Of the recently reported materials with narrow emissions bandwidth,52–55 gGO possesses the shortest emission wavelength, with the main peak at 379 nm. There are also secondary and tertiary peaks visible in the main photoluminescence spectrum, at 398 and 419 nm respectively (Fig. 3b). These peaks correspond to energy differences on the order of 100 meV which may be attributed to resolved vibrational modes of the photoluminescent material, as seen in rigid molecules such as polycyclic hydrocarbons. The emission–excitation matrix (EEM) and photoluminescence excitation spectra (PLE) (Fig. 3) show that the photoluminescence may be induced by two electronic transitions at 281 and 375 nm with additional resolved vibronic peaks at 270, 336 and 353 nm. The emission spectra at these frequencies remain essentially unchanged, only varying in relative intensity (Fig. S3, ESI†). We can therefore attribute all the fluorescent signals in this series to a single fluorescent species. Given the short wavelength, narrow FWHM and resolved vibrational structure, the photoluminescence is most likely attributed to π–π* transitions in small uniform sp2 hybridised domains as a sub-population of the gGO.56 This is in-line with previously reported carbon nanomaterials with narrow bandwidth emissions.49,51 It should be noted that there is another weaker photoluminescent signal emitting at 330 nm centred at an excitation frequency λex = 273 nm. HRTEM analysis of the transparent gGO dialysate shows gGO nanofilms (Fig. 3c and S4, ESI†). Layered lattices can be seen at the edge of the film, exhibiting a lattice distance of ∼0.34 nm, which corresponds to the (002) facet of graphite. Dispersive crystalline nanoparticles size < 5 nm in diameter were also observed on the gGO nanofilms. A lattice distance of ∼0.28 nm was observed in these crystalline nanoparticles which is slightly larger than the lattice spacing of the typical graphene quantum dots (GQDs). This is consistent with the presence of oxygen atoms on the edges of the nanoparticles.57 The presence of the gGO nanofilms in the 3.5 kDa dialysate is somewhat peculiar. They may be single layers sheared off from the edge of the graphite, therefore inheriting the (002) facet. However, they are likely to be rolled up in the as synthesized gGO mixture, thereby able to pass through the 3.5 kDa dialysis membrane, and the open up on subsequent dilution. In this context, we note previous studies on the formation of graphene scrolls in the VFD directly from graphite.39 The perfect crystalline structure, uniform size (<5 nm) but non-uniform shape of the GQDs also suggests that the GQDs are likely the corner fragments sheared off the gGO sheets under the double helical flow force field in the thin film, Fig. 1 and 3.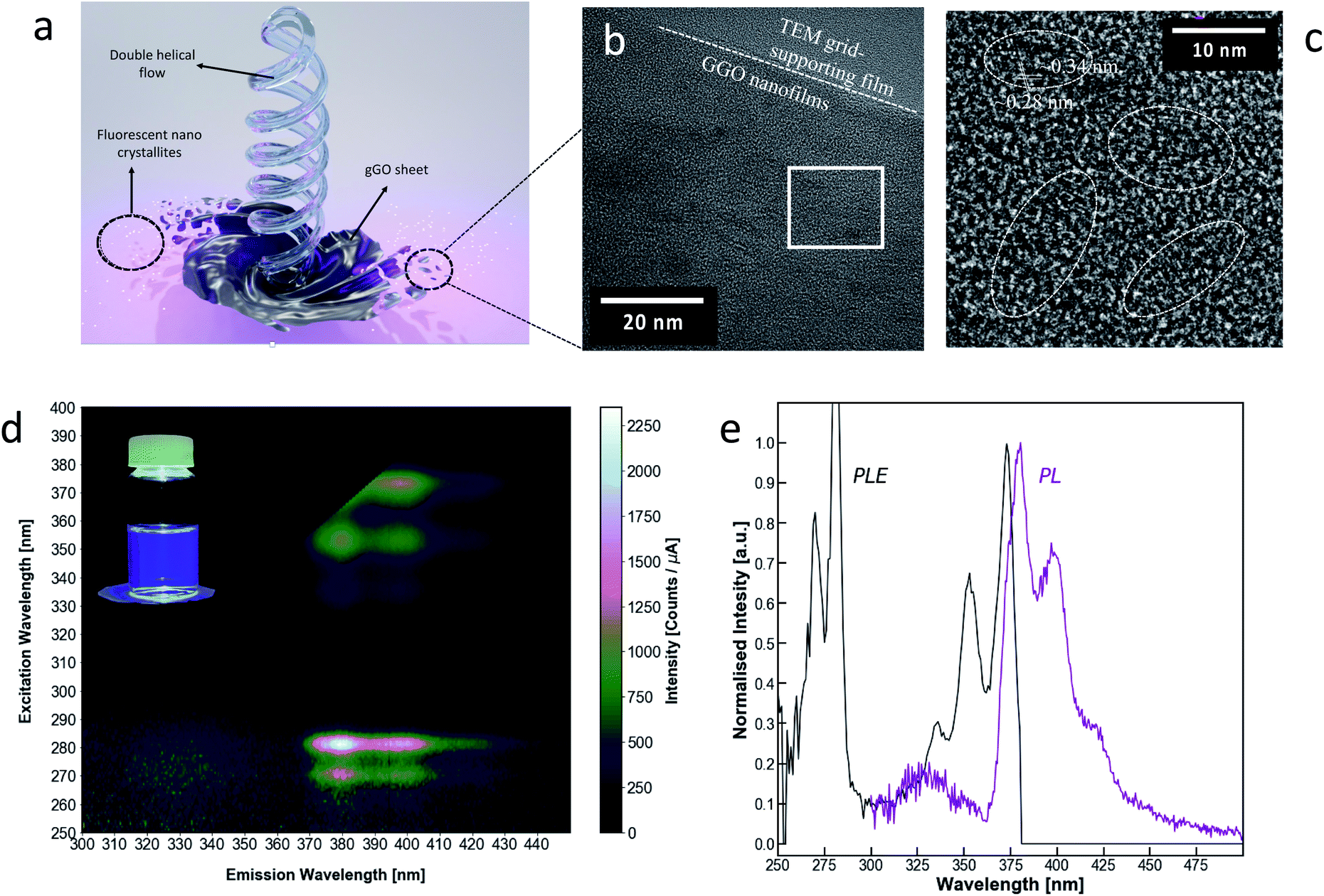 | ||
| Fig. 3 Photoluminescent properties of oxidised graphene quantum dots. (a) Schematic describing the mechanism of formation of the fluorescent nano crystallites as a result of the double helical flow effect. (b) Photoluminescence excitation-emission matrix for gGO dialysate (<3.5 kDa); inset: gGO dialysate under 365 nm excitation. (c) Photoluminescence spectra at λex = 282 nm and photoluminescence excitation spectra at λem = 378 nm of gGO dialysate. (d) TEM image showing the crystalline nanoparticles observed on the gGO nanofilm, and (e) a zoomed-in HRTEM showing small graphitic nanoparticles with a lattice spacing of ∼0.28 nm and the underlying gGO nanofilm showing (002) interplanar fringes with a lattice spacing of ∼0.34 nm corresponding to single layer graphene sheets. | ||
Biocompatibility and cytotoxicity of the gGO
We have explored the therapeutic potential of gGO, noting that oxidised graphene in general has recognised utility not only in device technology but also in biological and medical applications.58,59 For this purpose, we conducted in vitro toxicity studies using the MTT cell viability assay against MCF-7 breast cancer cells. The results from MTT assay are consistent with the toxicity of GO against various cell lines.60,61 The MTT for gGO also showed dose-dependent cytotoxic effects on MCF-7 cells (Fig. 4a), which is consistent with that for GO, as reported by Chaudhari et al.62 Three separately prepared samples of gGO (generated at 260 mJ) showed similar toxic effects to MCF-7, and this is consistent with similar functional groups in gGO and the GO prepared using the Hummers' method. A concentration of gGO at 50 μg mL−1 resulted in a decrease in viability of MCF-7 by 60%, in contrast to 90% cell viability established by Chaudhari et al. for the same concentration of oxidized graphene.64 The higher inhibition of cell viability using gGO (zeta potential −44 mV) correlates with an increase in colloidal stability of gGO (Fig. S5, ESI†) relative to the GO used by Chaudhari et al. (zeta potential −16.3 mV). Above 50 μg mL−1, the solubility of gGO in DMEM medium was saturated and the amount of gGO penetrating the cells reached a maximum, as evident by a plateaued cell viability (∼60%) above this concentration. At a low concentration of 3.125 μg mL−1, more than 80% cell viability was maintained, and the cellular morphology appeared intact after 24 h treatment. gGO showed good solubility and exerts sufficient toxicity for reducing cancer cell viability when applied at low concentrations, likely due to the abundant oxygen-containing groups. This is a desirable property for the use of gGO as drug delivery vehicle, and in biomedical applications in general.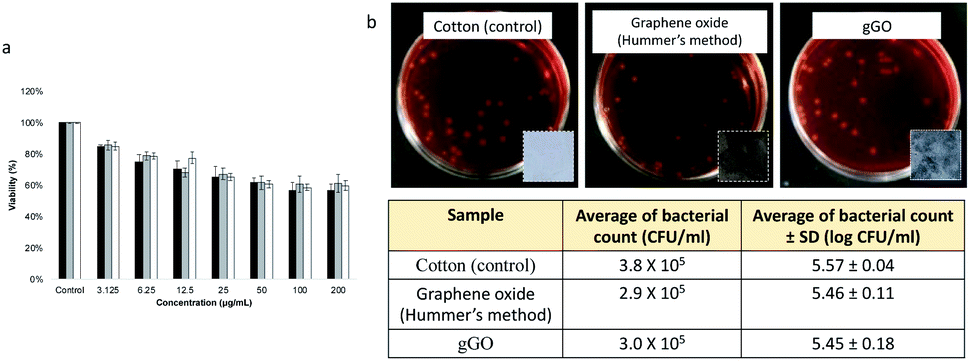 | ||
| Fig. 4 Cytotoxicity evaluation of gGO against MCF-7 breast cancer cells and antimicrobial activity. (a) Cell viability of MCF-7 cells treated with different concentrations of gGO (3.125, 6.25, 12.5, 25, 50, 100 and 200 μg mL−1), as evaluated by the MTT assay after a 24 h exposure time, noting that cells in the control group were not treated with gGO. (b) Escherichia coli JM109 colonies on MacConkey agar plates (photos of the cotton fabric, GO-coated and gGO-coated fabrics shown as insets) with a countable dilution (10−3) treated on cotton, cotton coated with Hummers' GO and cotton coated with gGO and incubated at 37 °C for 4 hours. | ||
In view of the potential opto- and electronic device applications, it is important to understand the biocompatibility and environmental impact of the gGO in the solid state. This could be first assessed by its interactions with microbes such as bacteria. For this purpose, gGO was coated on sterilised cotton fabrics (1.8 × 1.8 cm) by soaking the cotton pieces in gGO suspension (2 g L−1) followed by drying in oven 150 °C for 30 min. GO synthesized by Hummers' method (2 g L−1) was used for comparison. Untreated cotton fabric was sterilised as a control. Photographs of the coated cotton fabrics are displayed as insets in Fig. 2b. The metallic black colour of gGO coating in comparison to the dark brown GO coating confirms the presence of high quality of sp2 domains on the gGO. As shown in Fig. 3b, the coated GO and gGO does not exhibit an antibacterial effect on E. coli JM109. In other words, upon forming films or other solid-state structures, the oxygen-containing moieties on gGO appear to be passivated and are unable to interact with biological systems. Also noteworthy is that GO and gGO coatings on the cotton pieces remained intact after the addition of a saline solution and shaking, suggesting strong adhesion between the GO and gGO sheets and good bonding with the cotton fabrics. Overall, this study forms a preliminary assessment on the biocompatibility of gGO in the solid state.
gGO as a work function modification layer in organic photovoltaics (OPV)
There have been significant efforts towards the use of graphene oxide (GO), fabricated based on the conventional Hummers' method, as an interface layer or work function (WF) modification layer at the interface in organic photovoltaics (OPV).63–66 However, to ensure the addition of GO lowers the work function which then results in a high efficiency cell, there has been a need for further functionalisation of GO prior to use in OPVs.67 However, such further functionalisation complicates the OPV fabrication process which, as a result, increases the cost of production, an outcome not ideal for the potential up-scaling of the technology.67,68 Thus, we report the use of the gGO fabricated in the VFD as a work function modification layer when added to the cathode interface in OPV devices. The gGO powder was firstly dispersed in ethanol to afford a stable suspension devoid of any aggregation/sedimentation which was then spin coated on the cathode interface to form a homogenous uniform film. The power conversion efficiency (PCE) of the devices was then tested, and to ensure batch to batch reproducibility and to obtain confidence of the reproducibility of the data set, we carried out a series of replicates with different batches of gGO with an overall surface oxidation of ∼35%. We observed that the OPV devices with gGO added achieved a power conversion efficiency (PCE) of 9.99%, compared to the OPV without the gGO (PCE = 9.31%) (Fig. 5b). PM6 and IT-2Br are used as the electron donor and acceptor material respectively (Fig. S7, ESI†), in the OPV devices.69,70Fig. 5a also provides an illustration of the OPV device structure applied in this study.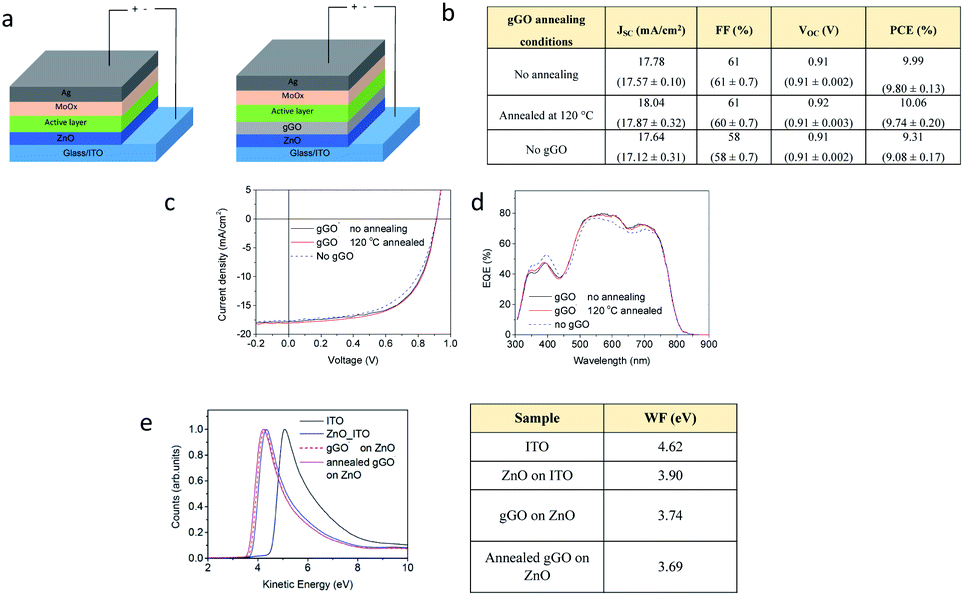 | ||
| Fig. 5 gGO as a work function modification layer in OPV. (a) Schematic of the inverted structure of the OPV device with and without the addition of the gGO. (b) The OPV device characteristics of the best devices for varied gGO annealing conditions, based on an average ± standard deviations over 6 devices in parentheses. (c and d) J–V curve and the EQE curve respectively of the OPVs with and without the addition of gGO as a work function modification layer, and (e) UPS spectra and the associated table illustrating the work function of ITO, ZnO on ITO and gGO with and without annealing. | ||
The OPV device results are summarized in Fig. 5b, and the representative J–V curves and EQE results are presented in Fig. 5c and d. The OPV devices without gGO as the control cells achieved a maximum PCE value of 9.31%, with the Voc of 0.91 V, fill factor (FF) of 58% and Jsc of 17.64 mA cm−2. The addition of gGO layer in between the ZnO and BHJ layer improved the efficiency to 9.99%. The improvement in the PCE is a result of higher Jsc (17.78 mA cm−2) and FF (61%) compared to the control cells. We then thermally annealed the gGO at 120 °C to test if the reduction step would recover the conductivity and thus increase the efficiencies of the cells. We also used Raman analysis to characterize the gGO samples before and after thermal treatment. We observed a significant reduction in the D band and an increase in intensity of the 2D band providing preliminary evidence of the recovery of the sp2 network, reduction of surface defects and oxygen functionalities (Fig. S8, ESI†). We then tested the thermally annealed gGO in the OPV cells and observed a slight increase in PCE of 10.06%. In order to understand the role of gGO in the OPV devices and to determine the origin of the improved OPV performance with the addition of the gGO layer, we characterised the work function of the samples using ultraviolet photoelectron spectroscopy (UPS). The results then provided information on the work function of the cathode interface in OPV devices before and after the addition of the gGO (Fig. 5e). The work function of ZnO coated on ITO was measured to be 3.90 eV, which is comparable to the literature.71,74 After coating with a layer of gGO on the surface of the ZnO film, the work function was observed to slightly reduce to 3.74 eV. Upon thermal treatment at 120 °C for 10 minutes, the work function was observed to further decrease to 3.69 eV. The reduced work function of gGO coated ZnO improved the electron collection at the cathode interface which is consistent with the increase in efficiency of device performance.
Electrochemical reduction of gGO versus GO (Hummers' method)
Commercial GO obtained using the Hummers' method has been recently demonstrated to spontaneously react with H– terminated silicon (Si–H) surfaces via Si–O–C covalent bonding.72 The graphene oxide modified electrode (Si–GO) can then be electrochemically reduced to reduced graphene oxide (Si–rGO).72,73 Here, we compare the electrochemical reduction of gGO and Hummers' method GO on Si electrodes. Key observations are (a) a similarity in the reduction wave position occurring at ∼950 mV vs. Ag/AgCl (Fig. 6) which indicates a similar apparent reduction potential for both systems, and (b) the reduction waves are less pronounced in the gGO modified Si, indicating less total oxide content in the gGO compared to the conventional GO. These findings further demonstrate that gGO made in the VFD can be an alternative to the GO fabricated by the Hummers' method and requires similar reduction potential with the only difference being less total oxide content in gGO – a property that can be utilized to reduce the energy cost associated with the number of potential cycles required to electrochemically reduce graphene oxide.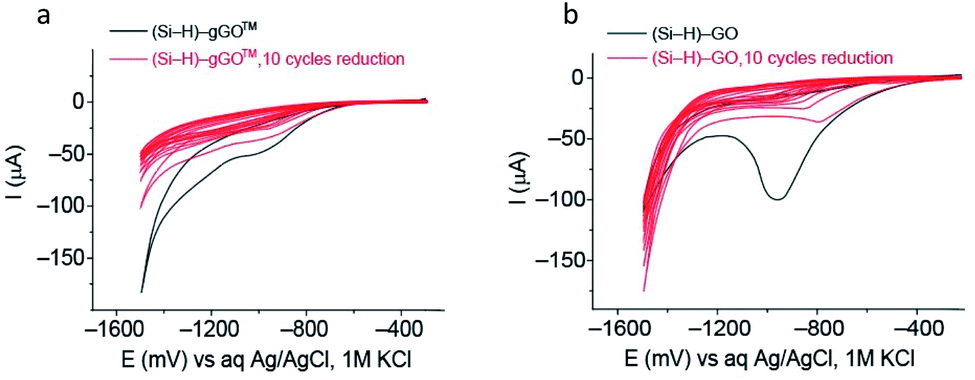 | ||
| Fig. 6 Electrochemical reduction of gGO and GO (Hummers' method). Ten consecutives cyclic voltammograms for the electrochemical reduction of (a) Si–gGO and (b) Si–GO (Hummers' method) in 0.1 M phosphate buffer solution at pH 7.4, and a scan rate of 50 mV s−1. The potential is presented in mV vs. Ag/AgCl, 1 M KCl. The reduction wave is centred around – 950 mV for both carbon-2D materials. In both systems, the charge decreases by 80–90% after 10 cycles. | ||
Conclusions
We have established a sequential two step continuous flow process for generating an oxidised form of graphene, designated green graphene oxide (gGO), with ca. ∼50% oxygen content, with unreacted graphite recycled back into the first step. It has comparable properties relative to conventional GO prepared using Hummers' method, but now with a dramatic reduction in the generation of waste. Indeed, the waste stream can be regarded as minimal, given that hydrogen peroxide can rapidly decompose in the environment to oxygen and water. The light-irradiation VFD processing of a graphite feedstock in the presence of 30% hydrogen peroxide allows: (1) The ability to control the oxidation of the graphene in the first step under UV radiation and control of the degree of oxidation in the second step by varying the power of the NIR laser power, and (2) harness the unique high mass transfer in the VFD associated with the double helical topological fluid flow, along with high heat transfer for rapid dissipation of heat. In addition, the resulting gGO has unique properties, with less oxidation in the basal plane of the sheets, being more strongly oxidized on the edges of the sheets with large intact sp2 domains, resulting in good conductivity. The lower oxide content of gGO in the basal plane is consistent with less electrochemical reduction charge requirement compared to that obtained using Hummers' method GO. The predominantly oxidized edges of gGO imparts colloidal stability and well-defined sp2 domains exhibiting remarkably purple to deep blue emission with less than 30 nm narrow full width at half maximum. Small fragments of gGO of nanometre lateral dimensions are also formed during the processing, and they are high quality graphene quantum dots (GQDs). Overall, the new tuneable process for generating graphene oxide provides a path forward for generating such material in an environmentally responsible way, potentially opening new applications and opportunities, ranging from medicine to optoelectronic device technologies and beyond.Author contributions
K. V. and C. L. R. designed the experiments and wrote the manuscript. K. V. optimized the experimental conditions and carried out the SEM, AFM, Raman, XPS, zeta potential analysis and conductivity measurements. J. S. and Q. L. performed the photoluminescent studies, X. P. and M. R. A. designed and performed organic photovoltaic studies, X. L. performed the cytotoxicity studies, N. B. and E. P. performed biocompatibility studies, S. R. and N. D. performed the electrochemical measurements, Q. S. and J. Z. carried out the TEM and HRTEM measurements. All authors contributed towards editing of the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
Support of this work by First Graphene Pty Ltd, the Australian Research Council (DP200101105) and the Government of South Australia is greatly acknowledged. The authors also acknowledge the expertise, equipment and support provided by Microscopy Australia (MA) and the Australian National Fabrication Facility (ANFF) and the South Australian Nodes under the National Collaborative Research Infrastructure Strategy.References
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS PubMed.
- K. I. Bolotin, K. J. Sikes, Z. Jiang, M. Klima, G. Fudenberg, J. Hone, P. Kim and H. L. Stormer, Solid State Commun., 2008, 146, 351–355 CrossRef CAS.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385 CrossRef CAS PubMed.
- P. P. Brisebois and M. Siaj, J. Mater. Chem. A, 2020, 8, 1517–1547 CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS PubMed.
- L. Lin, H. Peng and Z. Liu, Nat. Mater., 2019, 18, 520–524 CrossRef CAS PubMed.
- H. He, J. Klinowski, M. Forster and A. Lerf, Chem. Phys. Lett., 1998, 287, 53–56 CrossRef CAS.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- D. R. Dreyer, A. D. Todd and C. W. Bielawski, Chem. Soc. Rev., 2014, 43, 5288–5301 RSC.
- C. Hontoria-Lucas, A. J. Lopez-Peinado, J. de D. Lopez-Gonzalez, M. L. Rojas-Cervantes and R. M. Martin-Aranda, Carbon, 1995, 33, 1585–1592 CrossRef CAS.
- B. C. Brodie, Philos. Trans. R. Soc. London, 1859, 149, 249–259 CrossRef.
- U. Hofmann and E. König, Angew. Chem., Int. Ed., 1937, 234, 311–336 CAS.
- L. Staudenmaier, Eur. J. Inorg. Chem., 1898, 31, 1481–1487 CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- H. Yu, B. Zhang, C. Bulin, R. Li and R. Xing, Sci. Rep., 2016, 6, 36143 CrossRef CAS PubMed.
- S. Pei, Q. Wei, K. Huang, H.-M. Cheng and W. Ren, Nat. Commun., 2018, 9, 145 CrossRef PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- K. Krishnamoorthy, M. Veerapandian, K. Yun and S.-J. Kim, Carbon, 2013, 53, 38–49 CrossRef CAS.
- J. W. Suk, R. D. Piner, J. An and R. S. Ruoff, ACS Nano, 2010, 4, 6557–6564 CrossRef CAS PubMed.
- X. Sun, Z. Liu, K. Welsher, J. T. Robinson, A. Goodwin, S. Zaric and H. Dai, Nano Res., 2008, 1, 203–212 CrossRef CAS PubMed.
- Z. Liu, J. T. Robinson, X. Sun and H. Dai, J. Am. Chem. Soc., 2008, 130, 10876–10877 CrossRef CAS PubMed.
- J. Zhao, S. Pei, W. Ren, L. Gao and H.-M. Cheng, ACS Nano, 2010, 4, 5245–5252 CrossRef CAS PubMed.
- Y. Ma and L. Zhi, Small Methods, 2019, 3, 1800199 CrossRef.
- K. P. Loh, Q. Bao, G. Eda and M. Chhowalla, Nat. Chem., 2010, 2, 1015–1024 CrossRef CAS PubMed.
- D. Chen, H. Feng and J. Li, Chem. Rev., 2012, 112, 6027–6053 CrossRef CAS PubMed.
- Y. Zhu, D. K. James and J. M. Tour, Adv. Mater., 2012, 24, 4924–4955 CrossRef CAS PubMed.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- L. G. Guex, B. Sacchi, K. F. Peuvot, R. L. Andersson, A. M. Pourrahimi, V. Strom, S. Farris and R. T. Olsson, Nanoscale, 2017, 9, 9562–9571 RSC.
- C. Gómez-Navarro, J. C. Meyer, R. S. Sundaram, A. Chuvilin, S. Kurasch, M. Burghard, K. Kern and U. Kaiser, Nano Lett., 2010, 10, 1144–1148 CrossRef PubMed.
- O. C. Compton and S. T. Nguyen, Small, 2010, 6, 711–723 CrossRef CAS PubMed.
- K. A. Mkhoyan, A. W. Contryman, J. Silcox, D. A. Stewart, G. Eda, C. Mattevi, S. Miller and M. Chhowalla, Nano Lett., 2009, 9, 1058–1063 CrossRef CAS PubMed.
- C. Mattevi, G. Eda, S. Agnoli, S. Miller, K. A. Mkhoyan, O. Celik, D. Mastrogiovanni, G. Granozzi, E. Garfunkel and M. Chhowalla, Adv. Funct. Mater., 2009, 19, 2577–2583 CrossRef CAS.
- S. J. Pye, S. J. Dalgarno, J. M. Chalker and C. L. Raston, Green Chem., 2018, 20, 118–124 RSC.
- R. J. G. De Moor, J. Verheyen, A. Diachuk, P. Verheyen, M. A. Meire, P. J. De Coster, F. Keulemans, M. De Bruyne and L. J. Walsh, Sci. World J., 2015, 835405 Search PubMed.
- J. Britton, K. A. Stubbs, G. A. Weiss and C. L. Raston, Chem.–Eur. J., 2017, 23, 13270–13278 CrossRef CAS PubMed.
- C. A. Totoiu, J. M. Phillips, A. T. Reese, S. Majumdar, P. R. Girguis, C. L. Raston and G. A. Weiss, PLoS One, 2020, 15, 2695 CrossRef PubMed.
- J. M. Phillips, M. Ahamed, X. Duan, R. N. Lamb, X. Qu, K. Zheng, J. Zou, J. M. Chalker and C. L. Raston, ACS Appl. Bio Mater., 2019, 2, 488–494 CrossRef CAS PubMed.
- K. Vimalanathan, I. Suarez-Martinez, M. C. R. Peiris, J. Antonio, C. de Tomas, Y. Zou, J. Zou, X. Duan, R. N. Lamb, D. P. Harvey, T. M. D. Alharbi, C. T. Gibson, N. A. Marks, N. Darwish and C. L. Raston, Nanoscale Adv., 2019, 1, 2495–2501 RSC.
- K. Vimalanathan, J. R. Gascooke, I. Suarez-Martinez, N. A. Marks, H. Kumari, C. J. Garvey, J. L. Atwood, W. D. Lawrance and C. L. Raston, Sci. Rep., 2016, 6, 22865 CrossRef CAS PubMed.
- X. Luo, A. H. M. Al-Antaki, K. Vimalanathan, J. Moffatt, K. Zheng, Y. Zou, J. Zou, X. Duan, R. N. Lamb, S. Wang, Q. Li, W. Zhang and C. L. Raston, React. Chem. Eng., 2018, 3, 164–170 RSC.
- T. M. D. Alharbi, M. Jellicoe, X. Luo, K. Vimalanathan, I. K. Alsulami, B. S. Al Harbi, A. Igder, F. A. J. Alrashaidi, X. Chen, K. A. Stubbs, J. M. Chalker, W. Zhang, R. A. Boulos, D. B. Jones, J. S. Quinton and C. L. Raston, Nanoscale Adv., 2021, 3, 3064–3075 RSC.
- T. M. D. Alharbi, A. R. M. Alghamdi and K. Vimalanathan, Chem. Commun., 2019, 55, 11438–11441 RSC.
- R. J. G. De Moor, J. Verheyen, A. Diachuk, P. Verheyen, M. A. Meire, P. J. De Coster, F. Keulemans, M. De Bruyne and L. J. Walsh, Sci. World J., 2015, 650492 Search PubMed.
- W. H. Glaze, J. W. Kang and D. H. Chapin, Ozone: Sci. Eng., 1987, 9, 335–352 CrossRef CAS.
- A. Vittore, M. R. Acocella and G. Guerra, Langmuir, 2019, 35, 2244–2250 CrossRef PubMed.
- W. Buchalla and T. Attin, Dent. Mater., 2007, 23, 586–596 CrossRef CAS PubMed.
- T. M. D. Alharbi, K. Vimalanathan, W. D. Lawrance and C. L. Raston, Carbon, 2018, 140, 428–432 CrossRef CAS.
- T. M. D. Alharbi, K. Vimalanathan, I. K. Alsulami and C. L. Raston, Nanoscale, 2019, 11, 21394–21403 RSC.
- A. C. Ferrari and D. M. Basko, Nat. Nanotechnol., 2013, 8, 235–246 CrossRef CAS PubMed.
- D. Li, M. B. Muller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- F. Yuan, Y.-K. Wang, G. Sharma, Y. Dong, X. Zheng, P. Li, A. Johnston, G. Bappi, J. Z. Fan, H. Kung, B. Chen, M. I. Saidaminov, K. Singh, O. Voznyy, O. M. Bakr, Z.-H. Lu and E. H. Sargent, Nat. Photonics, 2020, 14, 171–176 CrossRef CAS.
- F. Yuan, T. Yuan, L. Sui, Z. Wang, Z. Xi, Y. Li, X. Li, L. Fan, Z. Tan, A. Chen, M. Jin and S. Yang, Nat. Commun., 2018, 9, 2249 CrossRef PubMed.
- J. Liu, Y. Geng, D. Li, H. Yao, Z. Huo, Y. Li, K. Zhang, S. Zhu, H. Wei, W. Xu, J. Jinlan and B. Yang, Adv. Mater., 2020, 32, 1906641 CrossRef CAS PubMed.
- S. Zhao, J. Lavie, L. Rondin, L. Orcin-Chaix, C. Diederichs, P. Roussignol, Y. Chassagneux, C. Voisin, K. Mullen, A. Narita, S. Campidelli and J.-S. Lauret, Nat. Commun., 2018, 9, 3470 CrossRef PubMed.
- G. Eda, Y.-Y. Lin, C. Mattevi, H. Yamaguchi, H.-A. Chen, I.-S. Chen, C.-W. Chen and M. Chhowalia, Adv. Mater., 2010, 22, 505–509 CrossRef CAS PubMed.
- X.-T. Tian and X.-B. Yin, Small, 2019, 15, 1901803 CrossRef CAS PubMed.
- C. Liao, Y. Li and S. C. Tjong, Int. J. Mol. Sci., 2018, 19, 3564–3599 CrossRef PubMed.
- K.-H. Liao, Y.-S. Lin, C. W. Macosco and C. L. Haynes, ACS Appl. Mater. Interfaces, 2011, 3, 2607–2615 CrossRef CAS PubMed.
- J. Szczepaniak, B. Strojny, E. S. Chwalibog, S. Jaworski, J. Jagiello, M. Winkowska, M. Szmidt, M. Wierzbicki, M. Sosnowska, J. Balaban, A. Winnicka, L. Lipinska, O. W. Pilaszewicz and M. Grodzik, Int. J. Mol. Sci., 2018, 19, 3939–3965 CrossRef PubMed.
- A. Jarosz, M. Skoda, I. Dudek and D. Szukiewicz, Oxid. Med. Cell. Longevity, 2016, 5851035 Search PubMed.
- N. S. Chaudhari, A. P. Pandey, P. O. Patil, A. R. Tekade, S. B. Bari and P. K. Deshmukh, Mater. Sci. Eng., C, 2014, 37, 278–285 CrossRef CAS PubMed.
- J.-M. Yun, J.-S. Yeo, J. Kim, H.-G. Jeong, D.-Y. Kim, Y.-J. Noh, S.-S. Kim, B.-C. Ku and S.-In. Na, Adv. Mater., 2011, 23, 4923–4928 CrossRef CAS PubMed.
- S.-S. Li, K.-H. Tu, C.-C. Lin, C.-W. Chen and M. Chhowalla, ACS Nano, 2010, 4, 3169–3174 CrossRef CAS PubMed.
- C.-C. Chueh, C.-Z. Li and A. K.-Y. Jen, Energy Environ. Sci., 2015, 8, 1160–1189 RSC.
- E. Stratakis, K. Savva, D. Konios, C. Petridis and E. Kymakis, Nanoscale, 2014, 6, 6925–6931 RSC.
- D. Konios, G. Kakavelakis, C. Petridis, K. Savva, E. Stratakis and E. Kymakis, J. Mater. Chem. A, 2016, 4, 1612–1623 RSC.
- G. Kakavelakis, D. Konios, E. Stratakis and E. Kymakis, Chem. Mater., 2014, 26, 5988–5993 CrossRef CAS.
- Y. Wang, Q. Fan, X. Guo, W. Li, B. Guo, W. Su, X. Ou and M. Zhang, J. Mater. Chem. A, 2017, 5, 22180–22185 RSC.
- S. Lu, F. Li, K. Zhang, J. Zhu, W. Cui, R. Yang, L. Yu and M. Sun, Sol. Energy, 2020, 195, 429–435 CrossRef CAS.
- A. Sharma, R. Kroon, D. A. Lewis, G. G. Andersson and M. R. Andersson, ACS Appl. Mater. Interfaces, 2017, 9, 10929–10936 CrossRef CAS PubMed.
- S. Rahpeima, E. M. Dief, C. R. Peiris, S. Ferrie, A. Duan, S. Ciampi, C. L. Raston and N. Darwish, Chem. Commun., 2020, 56, 6209–6212 RSC.
- A. G. Marrani, R. Zanoni, R. Schrebler and E. A. Dalchiele, J. Phys. Chem. C, 2017, 121, 5675–5683 CrossRef CAS.
- X. Pan, A. Sharma, D. Gedefaw, R. Kroon, A. D. de Zerio, N. P. Holmes, A. L. D. Kilcoyne, M. G. Barr, A. Fahy, M. Marks, X. Zhou, W. Belcher, P. C. Dastoor and M. R. Andersson, Org. Electron., 2018, 59, 432–440 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2na00310d |
| This journal is © The Royal Society of Chemistry 2022 |