DOI:
10.1039/D2NA00103A
(Paper)
Nanoscale Adv., 2022,
4, 2313-2320
A conjugated 2D covalent organic framework as a drug delivery vehicle towards triple negative breast cancer malignancy†
Received
14th February 2022
, Accepted 1st April 2022
First published on 27th April 2022
Abstract
Cancer, one of the deadliest diseases for both sexes, has always demanded updated treatment strategies with time. Breast cancer is responsible for the highest mortality rate among females worldwide and requires treatment with advanced regimens due to the higher probability of breast cancer cells to develop drug cytotoxicity followed by resistance. Covalent organic framework (COF) materials with ordered nanoscale porosity can serve as drug delivery vehicles due to their biocompatible nature and large internal void spaces. In this research work, we have employed a novel biocompatible COF, TRIPTA, as a drug delivery carrier towards breast cancer cells. It served as a drug delivery vehicle for cisplatin in triple negative breast cancer (TNBC) cells. We have checked the potency of TRIPTA in combating the proliferation of metastatic TNBC cells. Our results revealed that cisplatin loaded over TRIPTA-COF exhibited a greater impact on the CD44+/CD24− cancer stem cell niche of breast cancer. Retarded migration of cancer cells has also been observed with the dual treatment of TRIPTA and cisplatin compared to that of cisplatin alone. Epithelial–mesenchymal transition (EMT) has also been minimized by the combinatorial treatment of cisplatin carried by the carrier material in comparison to cisplatin alone. The epithelial marker E-cadherin is significantly increased in cells treated with cisplatin together with the carrier COF, and the expression of mesenchymal markers such as N-cadherin is lower. The transcriptional factor Snail has been observed under the same treatment. The carrier material is also internalized by the cancer cells in a time-dependent manner, suggesting that the organic carrier can serve as a specific drug delivery vehicle. Our experimental results suggested that TRIPTA-COF can serve as a potent nanocarrier for cisplatin, showing higher detrimental effects on the proliferation and migration of TNBC cells by increasing the cytotoxicity of cisplatin.
1. Introduction
Among the various disease states, cancer shows high mortality and poor survival. Breast cancer is the most common heterogeneous malignancy among females across the world and represents around 30% of all the malignancies occurring in females.1 It is categorized into three subtypes depending on the expression of hormone receptors, namely the estrogen receptor (ESR1), progesterone receptor (PGR), and HER2 epidermal growth factor receptor (ERBB2). The absence of expression of all three of these receptors is recognized as triple negative breast cancer (TNBC),2 which shows more metastasis and a more aggressive disease state. Based on the gene expression patterns, 79% of TNBC belongs to the basal-like subtype,3 one of the six stratified intrinsic molecular subtypes, the others of which are luminal subtype A, luminal subtype B, luminal subtype C, normal breast-like and ERBB2+. The absence of three receptors confines the treatment strategy towards TNBC; thus, chemotherapy is regarded as the sole standard for treating TNBC. Altered BRCA1 gene expression due to promoter methylation and p53 frameshift or nonsense mutations has been correlated with good response to chemotherapeutic regimens using cisplatin, a platinum-based drug, towards TNBC;4 however, several factors account for the development of chemoresistance in cancer cells, such as accumulation of the drug in cells either by suppression of uptake or increased efflux, detoxification of the drug by redox mechanisms, altered regulation of the apoptotic pathway or impairment of DNA repair mechanisms.5–8 Moreover, recent data by Hill et al. stated that there exist multiple mechanisms by which TNBC can acquire the property of cisplatin resistance.9
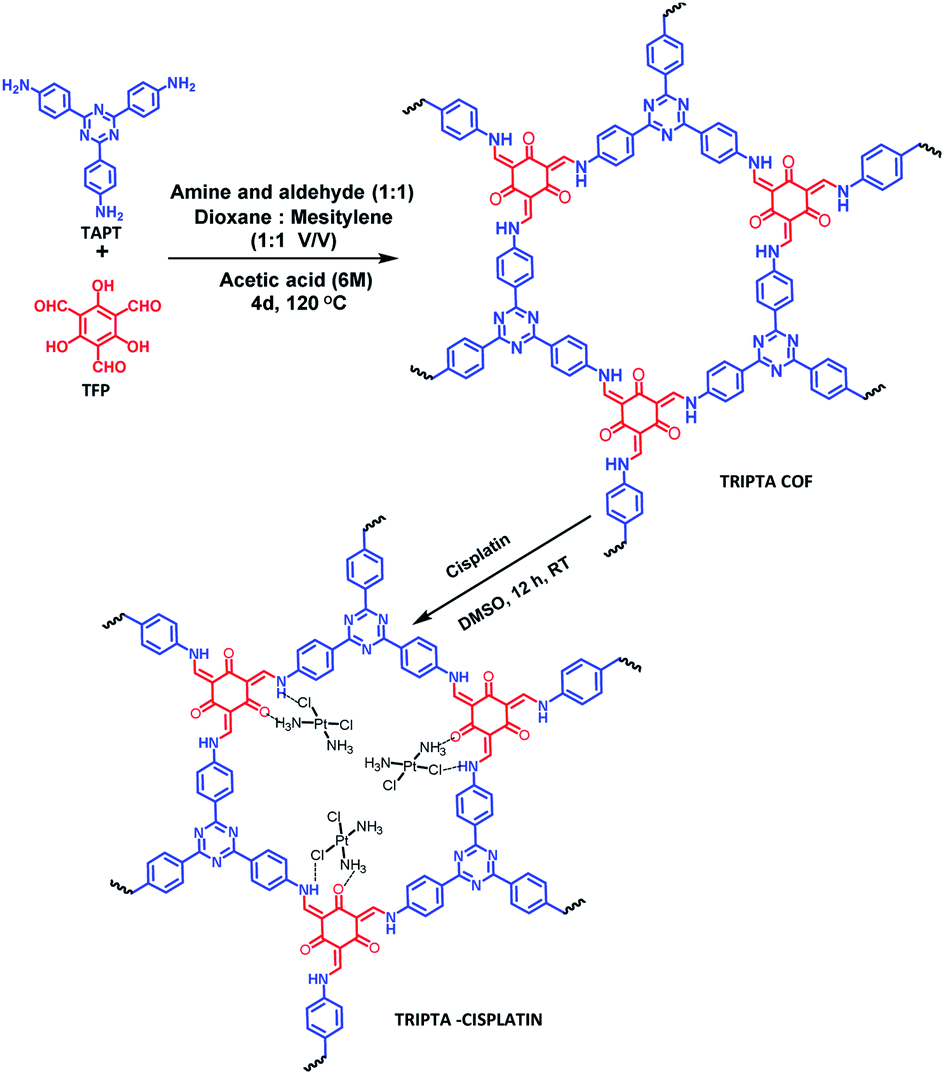
Thus, keeping this background in mind, here, we have checked the anticancer property of cisplatin in TNBC cells, MDA-MB-231, in combination with a novel nanoporous carrier material. For drug carriers, drug delivery vehicles or potential drugs, different organic, inorganic, and organic–inorganic hybrid molecules or nanomaterials are widely used.10–13 Due to the advantages of cell viability, high surface area, and nanoscale porosity together with high void space and reactive surface functional groups, porous nanomaterials play significant roles as drug delivery vehicles.14–16 Silica-based mesoporous materials, MOFs, COFs and porous polymeric materials have drawn significant attention due to their drug delivery capability.17–20 Chemical stability and metal leaching are the serious drawbacks of MOF materials for bio-medicinal applications. Covalently bonded highly crystalline COF materials with metal-free framework structures can overcome those drawbacks. Easy tailoring of the structural and functional moieties of COF materials through pre and post synthesis modification21,22 provides diverse opportunities in several potential applications, such as adsorption,23 separation,24 sensing,25 electrochemical studies,26,27 and biomedical applications.28,29 Thus, here, we have loaded cisplatin over a 2D crystalline COF material, TRIPTA,30,31 and used the resulting nanocomposite as a drug delivery vehicle towards metastatic breast cancer cells (Scheme 1). In order to determine whether this novel carrier material increases the efficacy of cisplatin, this study has been conducted; usage of TRIPTA may prove to be beneficial as a novel therapeutic approach towards the cisplatin treatment of TNBC cells.
2. Experimental
2.1. Chemicals
Phloroglucinol was obtained from Sigma-Aldrich and used for the synthesis of 1,3,5-triformylphloroglucinol (TFP).27 4-Aminobenzonitrile and trifluoromethanesulfonic acid were purchased from Sigma-Aldrich and used for the synthesis of 1,3,5-tris-(4-aminophenyl)-triazine (TAPT).19 Anhydrous N,N-dimethylformamide, hexamine, trifluoroacetic acid and hydrochloric acid were obtained from Merck, India, and all were used without further purification.
2.2. Material characterization techniques
A Bruker AXS D-8 Advance SWAX diffractometer was used to obtain the powder X-ray diffraction pattern of TRIPTA, where the radiation source was Cu-Kα (λ = 1.5406 Å). High-resolution transmission electron microscopy (TEM) images of the COF material were collected from a HR-TEM system (JEOL JEM 2100) operated with a 200 kV electron source. For the HRTEM analysis of TRIPTA, a sonicated ethanolic dilute solution of the carrier was dropped over a carbon-coated copper grid and then dried under high vacuum for 12 h. A Quantachrome Autosorb-iQ surface area analyzer (USA) was used for surface area and porosity analysis at 77 K. From the N2 adsorption/desorption isotherms, we calculated the Brunauer–Emmett–Teller (BET) surface area and pore size distribution plot using non-local density functional theory (NLDFT), taking N2 sorption at 77 K on carbon with the slit pore model as a reference. The powder sample of TRIPTA-COF was Soxhlet extracted for 72 h using anhydrous MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :THF (1:1) followed by drying under vacuum at 160 °C for 24 h. A Mettler Toledo TGA/DTA 851e TA-SDT Q-600 instrument was used for the thermal stability analysis. The FTIR spectrum of TRIPTA was recorded using a PerkinElmer spectrum 100 spectrophotometer. The amount of cisplatin loading in the drug-loaded TRIPTA was analyzed by measuring the total Pt content with the help of ICP-OES analysis by using a PerkinElmer Optima 2100 DV optical emission spectrometer.
:THF (1:1) followed by drying under vacuum at 160 °C for 24 h. A Mettler Toledo TGA/DTA 851e TA-SDT Q-600 instrument was used for the thermal stability analysis. The FTIR spectrum of TRIPTA was recorded using a PerkinElmer spectrum 100 spectrophotometer. The amount of cisplatin loading in the drug-loaded TRIPTA was analyzed by measuring the total Pt content with the help of ICP-OES analysis by using a PerkinElmer Optima 2100 DV optical emission spectrometer.
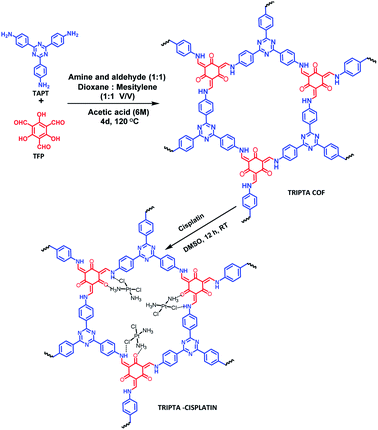 |
| | Scheme 1 Synthesis of the TRIPTA-COF and cisplatin-loaded TRIPTA-CISPLATIN. | |
2.3. Synthesis of TRIPTA
TRIPTA was synthesized through a solvothermal Schiff base polycondensation reaction using a Pyrex sealed tube.30 141.2 mg TAPT (0.4 mmol) and 84 mg TFP (0.4 mmol) were taken in the dry Pyrex tube, followed by the addition of 6 ml 1:1 mesitylene and 1,4-dioxane along with 0.5 ml 6 N acetic acid. Sonication was performed for 20 min to afford a homogeneously dispersed reaction mixture. The as-prepared mixture was degassed three times with a freeze–pump–thaw cycle. After that, the Pyrex tube was flame sealed, and the solvothermal reaction was allowed to proceed for 4 days. Finally, the deep orange-red fine powder material was filtered off and washed with ethanol followed by THF to remove unreacted starting monomers. Finally, the guest-free porous COF material was obtained by Soxhlet extraction with THF:methanol (1:1) for 48 h and dried under vacuum at 150 °C overnight. The isolated yield of the COF material was 81 wt%.
2.4. Synthesis of TRIPTA-CISPLATIN
Cisplatin was impregnated into the void spaces of the COF through overnight stirring of TRIPTA and cisplatin in a round bottom flask using DMSO as a solvent. The desired product was collected through a filtration process followed by washing with DI water several times and drying at room temperature under vacuum. After loading of the drug into the COF material, we performed the quantitative drug loading assay using gravimetric as well as ICP-OES techniques (detailed calculations are provided in the ESI, Section S1†). The obtained cisplatin loading efficiency was 31.19%. From the ICP-OES analysis, considering the amount of Pt, the calculated ratio of cisplatin and COF was found to be 2:1, i.e., two cisplatin molecules per hexagonal unit.
2.5. Cell culture
MDA-MB-231 triple negative breast cancer cells were grown in DMEM:F12 media supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 1% antibiotic (PSN) at 37 °C in a humidified atmosphere under 5% CO2. After 75–80% confluency, the cells were harvested with 0.025% trypsin and 0.52 mM EDTA in phosphate buffered saline (PBS) and seeded at the desired density to allow them to re-equilibrate a day before the start of the experiment.
2.6. Cell viability assays
To define the cell viability, an MTT assay was performed. Cells were plated at a count of 5 × 103 cells in a 96-well flat-bottom transparent plate and allowed to seed overnight at 37 °C in 5% CO2.32 After their adhesion, the cells were treated under different conditions, with only cisplatin, only carrier, and a combination of cisplatin and carrier. Treatments were given at specific time intervals of 0, 24, 48 and 72 h. Following treatment with the drug and carrier, 20 μl 3-(4,5-dimethylthiazol-2-yl)-2,5 diphenyltetrazolium bromide (MTT) tetrazolium dissolved in PBS were added to each well and incubated for 3 h at 37 °C. After incubation, 150 μl dimethyl sulphoxide (DMSO) was added to each well. The plates were analyzed in a microplate plate reader at 595 nm.33
2.7. Scratch wound assay
For the scratch assay, MDA-MB-231 cells were seeded in three 35 mm plates (n = 6) with a cell population density of 0.025 × 106 cells per well followed by incubation for 24 h for their growth. After obtaining confluent monolayers of cells, a scratch wound was created as a straight line with a 200 μl sterile pipette tip, and the time was designated as 0 h. One plate was the control, and the other two plates were treated with only 100 μl cisplatin and 10 μl TRIPTA-COF of interest. Images were captured at 0 h and after 48 h. The percent wound confluence was calculated using [(A − B) × 100/A], where A is the width of the scratch at 0 h and B is the width upon achieving confluency after 48 h.34
2.8. Cellular uptake
After 12 h of incubation, the cellular uptake study revealed that TRIPTA-COF was internalized by the cell membrane, leading to a cytosolic portion of cells with a maximum uptake of more than 60%.35 To check the uptake of carrier molecules within the cells, the carrier molecule was administered in a six-well plate seeded with MDA-MB-231 cells. In the control wells, no carrier molecule was added, while carrier molecules were added to the other wells at 0, 0.5, 1, 3 and 12 h intervals in order to check their uptake at specific time intervals. Data were collected using a spectrophotometer at 510 nm.36
2.9. Stemness assay
For the flow cytometry analysis, MDA-MB-231 cells in the logarithmic growth phase were digested with 0.25% trypsin and washed with PBS three times, followed by being re-suspended in 100 μl PBS; then, they were stained with anti-CD44-FITC and anti-CD24-PE or stained with their isotype control at room temperature for 40 min. The samples were then washed with PBS three times and finally re-suspended in 200 μl PBS. Flow cytometry analysis was performed on a BD FACSverse Flow Cytometer (BD Bioscience). The expression ratio of CD44 and CD24 (CD44+/CD24−) in the breast cancer cell line was calculated from the percentage of CD44-positive and CD24-negative sub-populations in the flow cytometry analysis.
2.10. Analysis of protein expression
After treatment with cisplatin, the resulting TRIPTA-CISPLATIN fixation in cells was conducted in 4% paraformaldehyde in PBS (pH 7.4) for 20 min at room temperature. 0.1% TritonX-100 in PBS was used for permeabilization with 0.1% FBS for 5 min. After washing twice with PBS with 3% FBS, the permeabilized cells were incubated with the primary antibody on ice for 2 h and washed with PBS. Then, the cells were incubated with FITC-conjugated goat anti-rabbit IgG as the secondary antibody for 2 h on ice and washed twice with PBS. The stained cells were acquired and analyzed using a BD FACSverse Flow Cytometer (San Jose, CA, USA) with the FlowJo software.
2.11. Statistical analysis
All values are expressed as mean ± SD. Statistical significance was compared between various treatment groups and controls using one-way analysis of variance (ANOVA). Data were considered statistically significant when P values were <0.01.
3. Results and discussion
3.1. PXRD and spectroscopic analysis
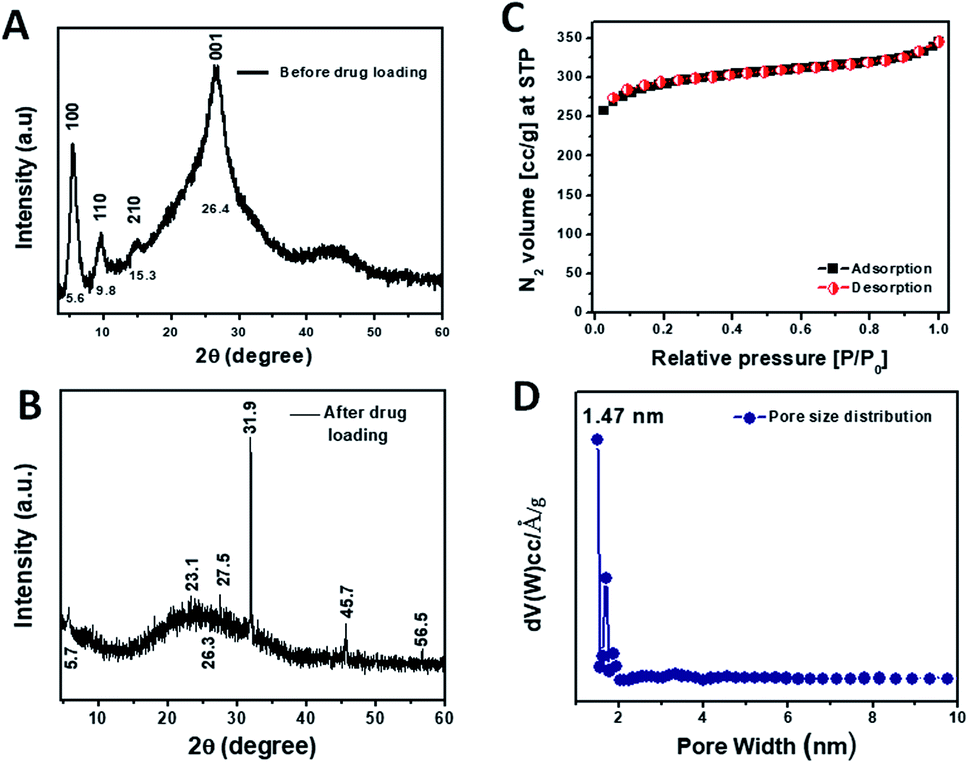
The formation of the crystalline organic framework of TRIPTA was confirmed from the PXRD analysis. The PXRD pattern of the COF in the 2θ value range from 4 to 60° is shown in Fig. 1A, where the peaks at 5.6°, 9.8°, 15.2° and 26.4° can be assigned to the crystal planes of 100, 110, 210 and 001, respectively. The PXRD pattern is very similar to that of TRIPTA previously synthesized by our group.30 To investigate the incorporation of cisplatin into the COF, a PXRD study was performed with a powder sample of cisplatin-loaded TRIPTA-COF (Fig. 1B). The PXRD peaks at 5.74° (100 plane) and 26.3° (001 plane) remain almost identical, which indicates the structural integrity of TRIPTA-COF. Very slight shifting of the PXRD peaks may have arisen from the loading of cisplatin into the porous network. Additional peaks that appeared at 23.1°, 27.6°, 31.9°, 45.7° and 56.5° indicate the incorporation of cisplatin in the COF matrix.37 FTIR data (Fig. S1†) were collected using a powder sample of TRIPTA-COF in the range of 400 to 4000 cm−1, where the characteristic peaks appeared at 1623, 1575 and 1256 cm−1. These peaks are assigned to the ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) C
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (α,β-unsaturation), CC
O (α,β-unsaturation), CC![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) and C–NH– (keto-enamine) groups, respectively.30 Thus, the FTIR analysis confirms the formation of TRIPTA as well as the presence of different bonding connectivities in the porous polymer framework.
and C–NH– (keto-enamine) groups, respectively.30 Thus, the FTIR analysis confirms the formation of TRIPTA as well as the presence of different bonding connectivities in the porous polymer framework.
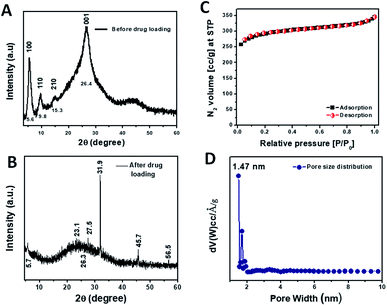 |
| | Fig. 1 PXRD patterns of TRIFTA-COF (A) and TRIPTA-CISPLATIN (B); N2 adsorption/desorption isotherms (C) along with the pore size distribution plot (D). | |
3.2. Surface area and porosity analysis
Specific surface area and pore size distribution analyses were carried out using the N2 adsorption/desorption isotherms (Fig. 1C) taken at 77 K. The N2 adsorption/desorption isotherms of TRIPTA could be classified as typical Type I, indicating the microporous nature of the material.38 The specific surface area calculated from the isotherms was 1074 m2 g−1. The pore size distribution plot (Fig. 1D) was obtained from the N2 adsorption/desorption isotherms by employing the NLDFT model; the pore sizes appeared in the microporous region, with a peak pore size distribution at ca. 1.47 nm.
3.3. Morphological analysis
HRTEM analysis was carried out to understand the morphology of the COF carrier TRIPTA and cisplatin-loaded COF, which are shown in Fig. 2. HRTEM images confirm the flower-like particle morphology of the COF (Fig. 2A and B). Further, the images of samples treated with different acids, 7.4 pH buffer and neutral pH are shown in Fig. S2.† It can be seen from these images that the morphology of the COF remained unchanged under different conditions. This result suggests the high structural integrity of TRIPTA. TRIPTA-COF has been successfully utilized as a drug delivery vehicle to deliver cisplatin towards triple negative breast cancer malignancy. TRIPTA-CISPLATIN showed enhancement of the in vitro anticancer activity of cisplatin against breast cancer cells. HRTEM images of the cisplatin-loaded COF are shown in Fig. 2C, D and S3,† where the actual sizes of the COF and cisplatin are electronically visualized at different magnifications (100, 20, 10 and 5 nm) with an average particle size of less than 10 nm. Materials or nanoparticles with particle sizes less than 50 nm are highly efficient for cellular uptake. Zhao et al. summarized the intercellular uptake of different kinds of materials with wide particle size ranges through endocytosis.39
 |
| | Fig. 2 HRTEM images of TRIPTA (A and B) and TRIPTA-CISPLATIN (C and D) at different resolutions. | |
3.4. The COF increases the cytotoxicity of cisplatin while decreasing the viability of MDA-MB-231 cells
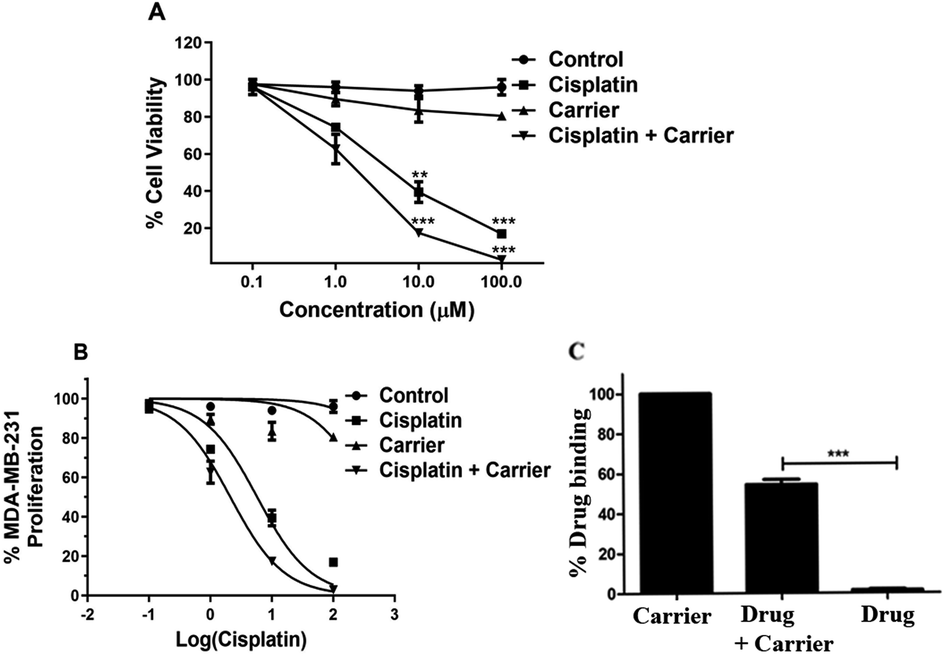
When checking the percentage viability of breast cancer cells, MTT analysis revealed that the percentage viability decreased gradually with increasing concentration of cisplatin and carrier. Decreased viability was observed under individual treatment of cisplatin and carrier; however, the rate of decreased viability with increasing concentration was considerably lower than that of cisplatin and TRIPTA-CISPLATIN. The cell viability under cisplatin and carrier treatment decreased to zero at 100 μM concentration and was recorded to be maximum in the control, followed by only carrier treatment and only cisplatin treatment, as per Fig. 3A and C. The IC50 values of cisplatin and TRIPTA-CISPLATIN were calculated to be 20 μM and 2.5 μM, respectively.
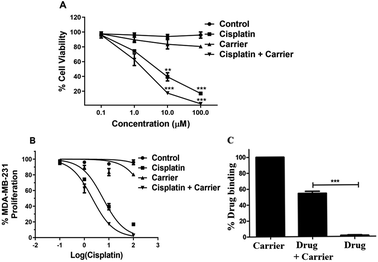 |
| | Fig. 3 Cells were treated with TRIPTA-COF along with cisplatin in the concentration range of 0–100 μM of cisplatin and TRIPTA-COF for 48 h. A significant result was found with TRIPTA-COF and cisplatin as compared to cisplatin alone, where (A) cell viability, (B) % MDA-MB-231 proliferation and (C) % drug binding were assayed by flow cytometric analyses with cisplatin, carrier and cisplatin + carrier. The data are represented as mean ± standard deviation (SD) from triplicate independent experiments (*P ≤ 0.05, ***P ≤ 0.001). | |
3.5. The carrier molecule enhances the activity of cisplatin in decreasing the population of CD44+/CD24− stemness in MDA-MB-231 cells
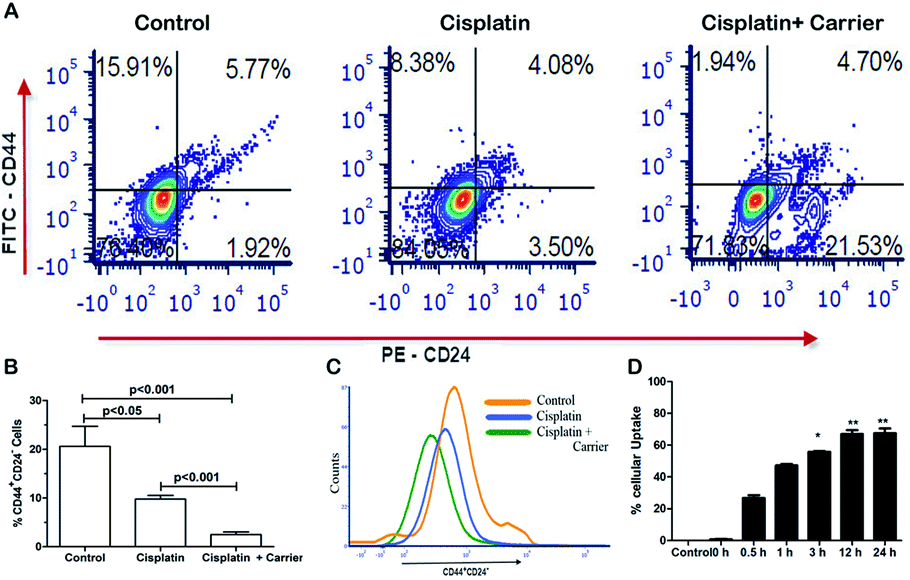
Cancer stem cells are a highly tumorigenic population of cells that are endowed with self-renewal ability and are often associated with drugs.40 We compared the expression of CD44+/CD24− in cisplatin and cisplatin with carrier-treated cells, as shown in Fig. 4A. We found significant changes in the CD44+/CD24− stem cell population among the control, cisplatin and cisplatin with carrier groups. As shown in Fig. 4B and C, the CD44+/CD24− stem cell population decreased in the cisplatin with carrier molecule group compared with the only cisplatin group and control group. The control group has the highest population of CD44+/CD24− stem cells, which eventually decreased with the treatment of cisplatin and cisplatin with the carrier molecule. We found a significant difference in the CD44+/CD24− stem cell population of the control and cisplatin groups as well as in the cisplatin and cisplatin and carrier molecule groups.
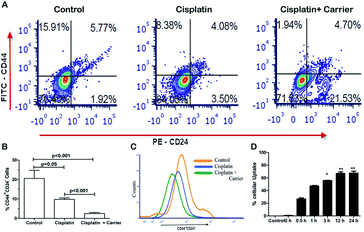 |
| | Fig. 4 Cells were pretreated with TGF-β to induce EMT and treated with TRIPTA-COF along with cisplatin. After 12 h of treatment, the cells stained with anti-FITC-CD44 and anti-PE-CD24 were compensated and analyzed with flow cytometry. (A) Flow cytometry data (control, cisplatin, control + cisplatin as TRIPTA-CISPLATIN) with anti-CD44 and anti-CD24, (B) percentages of CD44+/CD24− cells, (C) histogram positive events (CD44+/CD24− cells), and (D) percentages of cellular uptake of TRIPTA-COF in a time-dependent manner by spectrometry. The data are represented as mean ± standard deviation (SD) from triplicate independent experiments. | |
The uptake of a carrier molecule increases with incubation time. In order to check the potency and efficacy of the carrier molecule, a cellular uptake assay was performed. The uptake of the carrier molecules was observed to be maximum under 12 h of treatment, where the uptake percentage was above 60% and decreased gradually with decreasing treatment time; the lowest percentage was obtained at 0 h, as per Fig. 4D. The uptake at 12 h and 24 h was significantly high compared to that of the control. As shown in Fig. S4,† we performed a drug binding assay where we checked the drug (cisplatin), carrier (TRIPTA-COF) and covalent organic framework (TRIPTA-CISPLATIN) by flow cytometry analysis and drug release (Fig. S5†).
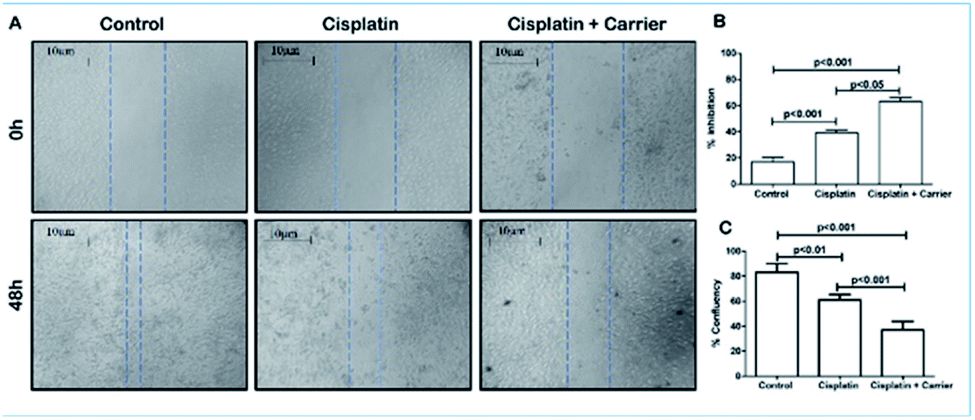
3.6. TRIPTA-CISPLATIN is more potent in inhibiting the migration of breast cancer cells
The control group did not receive any dosage of treatment, and after 48 h, the scratch created was filled with migrating breast cancer cells with 80% confluency. However, it was noted that treatment of MDA-MB-231 cells with cisplatin (2.5 μM) had a retarding effect on the migration of the cells and had approximately 60% confluency in the scratch. This migration retarding effect on the cancer cells was further enhanced in the cell plate treated with cisplatin and the carrier of interest, where the confluency was reduced to below 40% (Fig. 5A–C). Thus, it could be concluded that although cisplatin has an inhibiting effect on cancer cells, its inhibitory activity can be increased when administered in combination with the carrier.
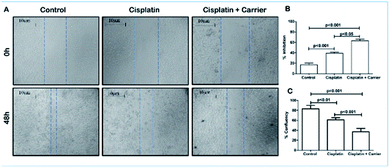 |
| | Fig. 5 Effect of cisplatin + carrier as TRIPTA-CISPLATIN on cell migration in MDA-MB-231 cells. (A) Analysis of MDA-MB-231 cell migration by scratch assay. (B) Percentages of inhibition and quantitative representation of the percentages of cell migration into the wound scratch after TRIPTA-CISPLATIN treatment for 24 h. (C) Percentages of confluency-quantitative representation of the percentages of cell migration into the wound scratch after TRIPTA-CISPLATIN treatment for 24 h. Representative images of wound healing at 0 and 24 h following scratch induction and TRIPTA-CISPLATIN treatment. The data are represented as mean ± standard deviation (SD) from triplicate independent experiments. | |
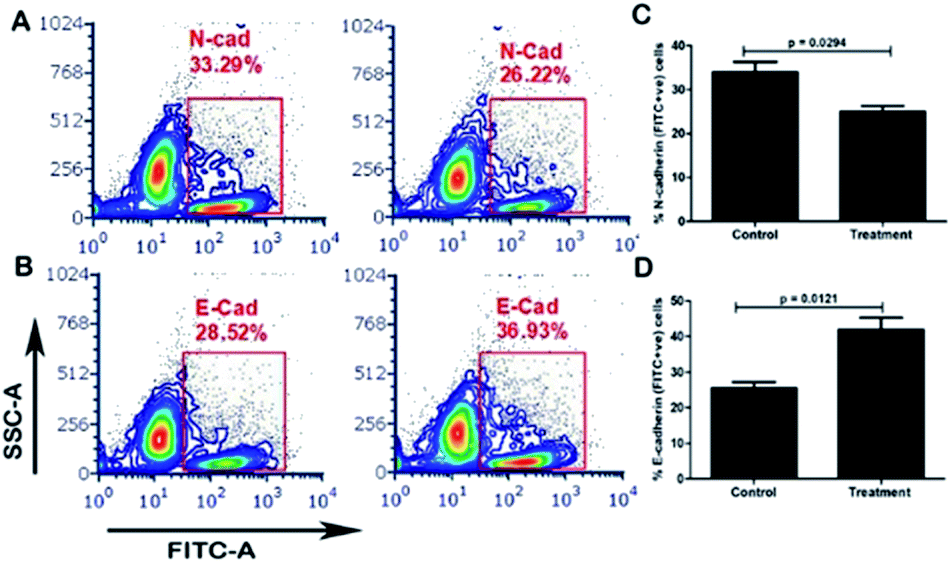
The carrier COF with cisplatin attenuates EMT (epithelial–mesenchymal transition) in MDA-MB-231 cells. EMT, in which cancer cells lose their epithelial property and adopt mesenchymal features, is one of the important mechanisms involved in stemness, metastasis and the recurrence of breast cancer. We investigated the role of the carrier molecule in the EMT of TGF-β-induced MDA-MB-231 cells, as shown in Fig. 6. We observed that the carrier material itself does not attenuate EMT; however, together with cisplatin, it decreases mesenchymal markers and increases the expression of epithelial markers (Fig. S6†). The carrier material enhances the cellular uptake of cisplatin and thus enhances its functional activity. We found significant changes in the expression of E-cadherin and N-cadherin between the carrier and the carrier with cisplatin groups, as shown in Fig. 6A–D. The expression of the epithelial marker E-cadherin (E-Cad) increased significantly in the carrier with cisplatin group compared with that in the carrier group, and the expression of the mesenchymal markers N-cadherin (N-Cad) and Snail decreased significantly.
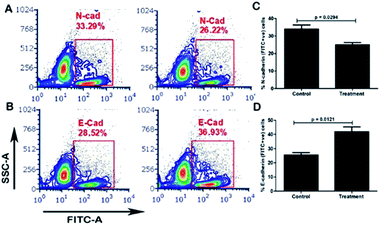 |
| | Fig. 6 Evaluation of the expression of E-cadherin and N-cadherin by flow cytometry. After treatment with cisplatin + carrier as TRIPTA-CISPLATIN along with only cisplatin and followed by TGF-β induction, we evaluated the (A) N-cadherin (N-Cad) expression; (C) quantitative analysis of the % N-Cad positive cells; (B) E-cadherin (E-Cad) expression; and (D) quantitative analysis of the % E-Cad positive cells. The data are represented as mean ± standard deviation (SD) from triplicate independent experiments. | |
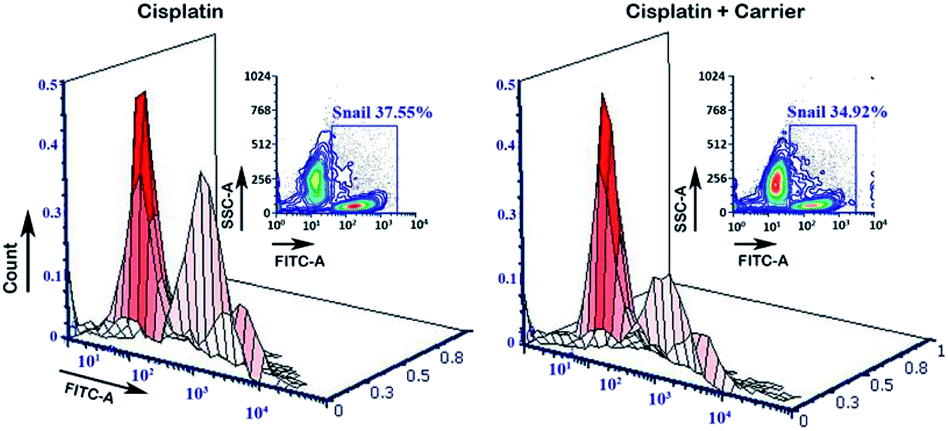
The transcriptional factor Snail promotes and regulates the expression of EMT proteins such as E-Cad and N-Cad. Under treatment with the carrier molecule along with cisplatin, we found reduced expression of Snail in the flow cytometry analysis (Fig. 7), which in turn may be the underlying reason for the greater expression of E-Cad as compared to N-Cad.
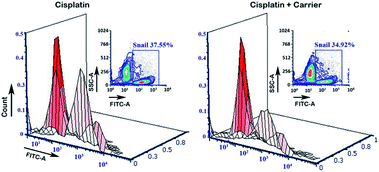 |
| | Fig. 7 Estimation of the expression of Snail by flow cytometry analysis. After treatment with cisplatin + carrier as TRIPTA-CISPLATIN along with only cisplatin and followed by TGF-β induction, the Snail expression with 3D analysis was evaluated. The data are represented as triplicate experiments. | |
3.7. Discussion
More metastatic and aggressive breast cancer cells, MDA-MB-231, are more prone to developing cisplatin resistance, with lower response to this platinum-derived agent compared to other mammary carcinoma cells. In this study, we have investigated the cytotoxicity of cisplatin in MDA-MB-231 in the presence of a novel porous carrier material, TRIPTA-COF, in order to check whether there is any increment in the efficiency of the drug to decrease the proliferation of cells and increase their sensitivity to the drug via regulating the stemness (Fig. S6†).
The proliferation and migration of cancer cells are intrinsic properties that depict the growth and metastasizing capability of cancer cells. Targeting cell proliferation can minimize the probability of metastasis. In this study, when the carrier molecule was administered with cisplatin, positive results with minimal migration were observed as compared to that with cisplatin alone. This suggests that the carrier molecules and cisplatin together have a detrimental effect on cell division, thereby minimizing the mobility of cancer cells. Furthermore, this novel carrier molecule has a high tendency to be internalized by cancer cells in a time-dependent manner, indicating that it can efficiently transfer cisplatin to cancer cells, which may increase the cytotoxicity of cisplatin. We have investigated the role of the carrier COF material in CD44+/CD24− stem cell populations and the EMT of the MDA-MB-231 cell line. It has been observed that the carrier molecule together with cisplatin significantly decreases the population of CD44+/CD24− in MDA-MB-231 cells, indicating that the stemness of the cancer cells in turn is a probable reason for retarded migration.
MDA-MB-231 cells are prone to undergo EMT associated with breast cancer metastasis by transforming themselves from the polarized epithelial state to the non-polarized mesenchymal state to initiate the process of cancer progression.41 Recently, Wang et al. reported that cisplatin in combination with paclitaxel is capable of retarding TGF-β-induced EMT in breast cancer cells.42 To enhance the cytotoxicity of cisplatin towards TNBC cells, we attempted to administer cisplatin via the TRIPTA-COF carrier material (Fig. S7†). We have studied the role of the carrier molecule in enhancing the functional activity of cisplatin on EMT. The carrier COF was unable to change the expression of EMT markers; however, together with cisplatin, it was able to significantly decrease the expression of the mesenchymal marker N-cadherin and the transcription factor Snail and increase the expression of the epithelial marker E-cadherin. The carrier material also plays a role in enhancing the activity of cisplatin in EMT; however, TRIPTA-COF alone has not shown any effect on the EMT process.
4. Conclusion
We have immobilized cisplatin on the surface of TRIPTA-COF, and the resulting nanocomposite material has been utilized for triple negative breast cancer cell destruction. The anticancer activity of cisplatin itself is significantly lower than that of cisplatin-loaded TRIPTA-COF. As the COF material alone did not play any significant role in the EMT process, it may act by precisely delivering cisplatin towards breast cancer cells for enhancement of the anticancer activity of cisplatin. Thus, the efficacy of cisplatin can be enhanced when it is encapsulated in the novel porous drug delivery carrier material, TRIPTA. Hence, our experimental results of utilizing TRIPTA-COF as a transporter of an anticancer drug reported herein will open up new opportunities in biomedical research in the future.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
SKD would like to thank IACS Kolkata and UGC, New Delhi, for Senior Research Fellowships. SR would like to thank University Grant Commission: for a Junior Research Fellowship. AB would like to thank IGSTC, New Delhi, for the Indo-German project research grant (Project no. IGSTC/Call 2018/CO2BioFeed/15/2019-20/). Cells and other biological samples used in this work were gifted by Dr A. K. Srivastava, Scientist, CSIR-Indian Institute of Chemical Biology.
Notes and references
- S. D. Cosimo and J. Baselga, Nat. Rev. Clin. Oncol., 2010, 7, 139–147 CrossRef PubMed.
- J. R. Jhan and E. R. Andrechek, Pharmacogenomics, 2017, 18, 1595–1609 CrossRef CAS PubMed.
- A. Prat, B. Adamo, M. C. U. Cheang, C. K. Anders, L. A. Carey and C. M. Perou, Oncologist, 2013, 18, 123–133 CrossRef CAS PubMed.
- D. P. Silver, A. L. Richardson, A. C. Eklund, Z. C. Wang, Z. Szallasi, Q. Li, N. Juul, C. O. Leong, D. Calogrias, A. Buraimoh, A. Fatima, R. S. Gelman, P. D. Ryan, N. M. Tung, A. D. Nicolo, S. Ganesan, A. Miron, C. Colin, D. C. Sgroi, L. W. Ellisen, E. P. Winer and J. E. Garber, J. Clin. Oncol., 2010, 28, 1145–1153 CrossRef CAS PubMed.
- C. A. Rabik, E. B. Maryon, K. Kasza, J. T. Shafer, C. M. Bartnik and M. E. D. Cara, Cancer Chemother. Pharmacol., 2009, 64, 133–142 CrossRef CAS PubMed.
- Z. Zhu, S. Du, Y. Du, J. Ren, G. Ying and Z. Yan, J. Neurochem., 2018, 144, 93–104 CrossRef CAS PubMed.
- L. Galluzzi, L. Senovilla, I. Vitale, J. Michels, I. Martins, O. Kepp, M. Castedo and G. Kroemer, Oncogene, 2012, 31, 1869–1883 CrossRef CAS PubMed.
- M. Parhizkar, P. J. T. Reardon, A. H. Harker, R. J. Browning, E. Stride, R. B. Pedley, J. C. Knowles and M. Edirisinghe, Nanoscale Adv., 2020, 2, 1177–1186 RSC.
- D. P. Hill, A. Harper, J. Malcolm, M. S. McAndrews, S. M. Mockus, S. E. Patterson, T. Reynolds, E. J. Baker, C. J. Bult, E. J. Chesler and J. A. Blake, BMC Cancer, 2019, 19, 1039 CrossRef PubMed.
- J. K. Patra, G. Das, L. F. Fraceto, E. V. R. Campos, M. P. R. Torres, L. S. A. Torres, L. A. D. Torres, R. Grillo, M. K. Swamy, S. Sharma, S. Habtemariam and H.-S. Shin, J. Nanobiotechnol., 2018, 16, 71 CrossRef PubMed.
- L. Fu, H. Q. Gao, M. Yan, S. Z. Li, X. Y. Li, Z. F. Dai and S. Q. Liu, Small, 2015, 11, 2938–2945 CrossRef CAS PubMed.
- L. Bai, S. Z. F. Phua, W. Q. Lim, A. Jana, Z. Luo, H. P. Tham, L. Zhao, Q. Gao and Y. Zhao, Chem. Commun., 2016, 52, 4128–4131 RSC.
- Z. Shi, Y. Zhou, T. Fan, Y. Lin, H. Zhang and L. Mei, Smart Mater. Med., 2020, 1, 32–47 CrossRef.
- C. Chen, W. Tang, D. W. Jiang, G. L. Yang, X. L. Wang, L. N. Zhou, W. A. Zhang and P. Wang, Nanoscale, 2019, 11, 11012–11024 RSC.
- P. Kumar, K. H. Kim, A. Saneja, B. Wang and M. Kukkar, J. Porous Mater., 2019, 26, 655–675 CrossRef CAS.
- L. C. C. Fonseca, M. de Sousa, D. L. S. Maia, V. L. de Luna and O. L. L. Alves, Nanoscale Adv., 2020, 2, 1290–1300 RSC.
- C. Bharti, U. Nagaich, A. K. Pal and N. Gulati, J. Pharm. Invest., 2015, 5, 124–133 CrossRef CAS PubMed.
- M. Liu, L. Wang, X. H. Zheng, S. Liu and Z. Xie, ACS Appl. Mater. Interfaces, 2018, 10, 24638–24647 CrossRef CAS PubMed.
- S. K. Das, S. Mishra, K. Manna, U. Kayal, S. Mahapatra, K. Das Saha, S. Dalapati, G. P. Das, A. A. Mostafa and A. Bhaumik, Chem. Commun., 2018, 54, 11475–11478 RSC.
- L. L. Feng, C. Qian and Y. L. Zhao, ACS Mater. Lett., 2020, 2, 1074–1092 CrossRef CAS.
- V. Valtchev, G. Majano, S. Mintova and J. Perez-Ramirez, Chem. Soc. Rev., 2013, 42, 263–290 RSC.
- S. Das, P. Heasman, T. Ben and S. Qiu, Chem. Rev., 2017, 117, 1515–1563 CrossRef CAS PubMed.
- H. Wei, S. Chai, N. Hu, Z. Yang, L. Wei and L. Wang, Chem. Commun., 2015, 51, 12178–12181 RSC.
- B. P. Biswal, H. P. Chaudhari, R. Banerjee and U. K. Kharul, Chem. – Eur. J., 2016, 22, 4695–4699 CrossRef CAS PubMed.
- S. H. Zhang, Q. Yang, X. T. Xu, X. H. Liu, Q. Li, J. R. Guo, N. L. Torad, S. M. Alshehri, T. Ahamad, M. S. A. Hossain, Y. V. Kaneti and Y. Yamauchi, Nanoscale, 2020, 12, 15611–15619 RSC.
- X. Zhao, P. Pachfule and A. Thomas, Chem. Soc. Rev., 2021, 50, 6871–6913 RSC.
- S. K. Das, K. Bhunia, A. Mallick, A. Pradhan, D. Pradhan and A. Bhaumik, Microporous Mesoporous Mater., 2018, 266, 109–116 CrossRef CAS.
- Q. Guan, G. B. Wang, L. L. Zhou, W. Y. Li and Y. B. Dong, Nanoscale Adv., 2020, 2, 3656–3733 RSC.
- S. Bhunia, K. A. Deo and A. K. Gaharwar, Adv. Funct. Mater., 2020, 30, 2002046 CrossRef CAS.
- R. Gomes and A. Bhaumik, RSC Adv., 2016, 6, 28047–28054 RSC.
- S. K. Das, B. K. Chandra, R. A. Molla, M. Sengupta, S. M. Islam, A. Majee and A. Bhaumik, Mol. Catal., 2020, 480, 110650 CrossRef CAS.
- C. Germain, N. Niknejad, L. Ma, K. Garbui, T. Hai and J. Dimitroulakos, Neoplasia, 2010, 12, 527–538 CrossRef PubMed.
- N. Chatterjee, S. Das, D. Bose, S. Banerjee, T. Jha and K. Das Saha, PLoS One, 2015, 10, e0120509 CrossRef PubMed.
- P. Buachan, L. Chularojmontri and S. K. Wattanapitayakul, Nutrients, 2014, 6, 1618–1634 CrossRef CAS PubMed.
- F. Zhao, Y. Zhao, Y. Liu, X. Chang, C. Chen and Y. Zhao, Small, 2011, 7, 1322–1337 CrossRef CAS PubMed.
- M. Pramanik, N. Chatterjee, S. Das, K. Das Saha and A. Bhaumik, Chem. Commun., 2013, 49, 9461–9463 RSC.
- A. C. Jayasuriya and A. J. Darr, J. Biomed. Sci. Eng., 2013, 6, 586–592 CrossRef.
- S. K. Das, S. Chatterjee, S. Mondal and A. Bhaumik, Mol. Catal., 2019, 475, 110483 CrossRef CAS.
- F. Zhao, Y. Zhao, Y. Liu, X. Chang, C. Chen and Y. Zhao, Small, 2011, 7, 1322–1337 CrossRef CAS PubMed.
- W. Li, H. Ma, J. Zhang, L. Zhu, C. Wang and Y. L. Yang, Sci. Rep., 2017, 7, 13856 CrossRef PubMed.
- Z. Huang, P. Yu and J. Tang, OncoTargets Ther., 2020, 13, 5395–5405 CrossRef CAS PubMed.
- H. Wang, S. Guo, S. J. Kim, F. Shao, J. W. Kei Ho, K. U. Wong, Z. Miao, D. Hao, M. Zhao, J. Xu, J. Zeng, K. H. Wong, L. Di, A. H. H. Wong, X. Xu and C. X. Deng, Theranostics, 2021, 11, 2442–2459 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: FTIR, HRTEM, EMT with the involvement of TRIPTA COF cisplatin, drug binding assay, Cytotoxicity study. See https://doi.org/10.1039/d2na00103a |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Nabanita
Chatterjee
*b and
Asim
Bhaumik
a,
Nabanita
Chatterjee
*b and
Asim
Bhaumik