
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembled dipeptide based fluorescent nanoparticles as a platform for developing cellular imaging probes and targeted drug delivery chaperones†
Subramaniyam
Sivagnanam‡
 a,
Kiran
Das‡
b,
Madhuri
Basak
b,
Tarun
Mahata
b,
Adele
Stewart
c,
Biswanath
Maity
*b and
Priyadip
Das
*a
a,
Kiran
Das‡
b,
Madhuri
Basak
b,
Tarun
Mahata
b,
Adele
Stewart
c,
Biswanath
Maity
*b and
Priyadip
Das
*a
aDepartment of Chemistry, SRM Institute of Science and Technology, SRM Nagar, Potheri, Kattankulathur, Tamil Nadu 603203, India. E-mail: priyadipcsmcri@gmail.com; priyadip@srmist.edu.in
bCentre of Biomedical Research, Sanjay Gandhi Post Graduate Institute of Medical Sciences (SGPGI) Campus, Raebareli Road, Lucknow, Uttar Pradesh 226014, India. E-mail: bmaity28@gmail.com; bmaity@cbmr.res.in
cDepartment of Biomedical Science, Charles E. Schmidt College of Medicine, Florida Atlantic University, Jupiter, FL 33458, USA
First published on 15th February 2022
Abstract
Self-assembled peptide-based nanostructures, comprised of naturally occurring amino acids, display excellent biocompatibility, biodegradability, flexible responsiveness, and synthetic feasibility and can be customized for various biomedical applications. However, the lack of inherent optical properties of peptide-based nanoparticles is a limitation on their use as imaging probes or drug delivery vehicles. To overcome this impediment, we generated Boc protected tyrosine–tryptophan dipeptide-based nanoparticles (DPNPs) with structure rigidification by Zn(II), which shifted the peptide's intrinsic fluorescent properties from the ultraviolet to the visible range. These DPNPs are photostable, biocompatible and have visible fluorescence signals that allow for real-time monitoring of their entry into cells. We further show that two DPNPs (PS1-Zn and PS2-Zn) can encapsulate the chemotherapeutic drug doxorubicin (Dox) and facilitate intracellular drug delivery resulting in cancer cell killing actions comparable to the unencapsulated drug. Finally, we chemically modified our DPNPs with an aptamer directed toward the epithelial cell surface marker EPCAM, which improved Dox delivery to the lung cancer epithelial cell line A549. In contrast, the aptamer conjugated DPNPs failed to deliver Dox into the cardiomyocyte cell line AC16. Theoretically, this strategy could be employed in vivo to specifically deliver Dox to cancer cells while sparing the myocardium, a major source of dose-limiting adverse events in the clinic. Our work represents an important proof-of-concept exercise demonstrating that ultra-short peptide-based fluorescent nanostructures have great promise for the development of new imaging probes and targeted drug delivery vehicles.
Introduction
Nanodrug delivery systems (NDDs) hold great promise in the development of novel molecules for the diagnosis or treatment of several diseases, such as cancer, inflammation, neurological diseases and cardiovascular diseases, which represent a large burden on the global healthcare system.1–5 The augmentation of existing cancer chemotherapeutics with drug-chaperone systems capable of directing delivery of the drug to cancer cells while sparing healthy tissue is in demand given the high incidence of dose-limiting adverse events associated with many chemotherapeutic agents. When compared to low molecular weight substrates, NDDs often exhibit extended blood half-life, enhanced penetration into tumor tissues, and the ability to release drugs into the cytoplasm in a controllable manner.6–8To date, liposomes, nanoparticles and micelles have been identified as potential transporters for drug delivery9,10 and used to develop clinical nanomedicines, such as Doxil/Caelyx and Abraxane.11,12 Doxil, a pegylated, liposomal formulation of the anthracycline chemotherapeutic drug doxorubicin, was developed with the goal of mitigating the dose-limiting, often irreversible, and occasionally life-threatening cardiotoxicity associated with doxorubicin exposure. Liposomes fail to penetrate the vasculature in the heart due to the presence of tight junctions limiting cardiac accumulation. While Doxil™ displays improved cardiac safety and less myelosuppression, alopecia, and nausea/vomiting when compared to unencapsulated Dox regimens,13 the polyethylene glycol coating results in preferential accumulation of Dox in the skin where the drug can leak from the capillaries resulting in redness, tenderness, and peeling of the skin, a phenomenon known as palmar plantar erythrodysesthesia (PPE), in 50% of patients.14 Thus, there is a demand for improved drug delivery vehicles that allow for increased doxorubicin dosing without corresponding adverse events.
Monomeric units composed of peptides or peptide derivatives are able to self-assemble into thermodynamically stable supramolecular nanostructures with various morphologies.15–18 These peptide-based, self-assembled nanostructures can function as drug delivery systems with excellent biocompatibility and biodegradability, flexible responsiveness, specific biological recognition ability, and synthetic feasibility.19 Furthermore, the inherent low toxicity of peptide-based superstructures has facilitated growing interest in their use in biological as well as biomedical applications.15,16 Researchers have taken advantage of self-assembling, peptide-based NDDs to achieve site-specific drug delivery.20,21 Peptide nanoparticles with luminescent properties are also of interest owing to their biodegradability and potential as imaging and sensing probes.22 However, the limited inherent optical properties of peptides restrict their use in imaging applications. The introduction of organic fluorophores23–25 or quantum dots (QDs)26 has been proposed as a way to modify peptides for fluorescent based bio-imaging. Unfortunately, fluorescent organic dyes are highly susceptible to photo-bleaching and lack site-specificity during bio-conjugation.27,28 The cytotoxicity of QDs remains a major issue for biomedical applications.29,30 Though several studies have sought to direct peptide self-assembly into desired nanostructures,31,32 few investigators have investigated the impact of the self-assembly process on the intrinsic optical properties of peptide nanostructures composed of short peptides. Inspired by the bathochromic shift in emission maxima in yellow fluorescent protein (YFP) compared to green fluorescent protein (GFP),33 which has been attributed to π–π stacking interactions between tyrosine (Tyr) and the phenolate anion of the chromophore unit,34 and the impact of Zn(II) on the emission intensity, quantum yield, and fluorophore mobility of the GFP derivative BFPms1, Fan et al. demonstrated that the formation of tryptophan–phenylalanine dipeptide nanoparticles could shift the peptide's intrinsic fluorescent signal from the ultraviolet to the visible range.20 Aptamer conjugation allowed for functionalization of the resultant fluorescent dipeptide nanoparticles (DPNPs) targeting doxorubicin to cancer cells expressing the MUC1 protein and allowing for cancer cell visualization.20 Another group utilized tripeptides, which also self-assemble and, in the presence of Zn(II) conjugation display visible intrinsic fluorescence.35
Here, we describe the development and characterization of novel, self-assembled DPNPs from a tyrosine (Tyr)/tryptophan (Trp) backbone. Tryptophan was chosen because of its long emission wavelength and high quantum yield compared to other aromatic amino acids.36 As was previously shown,20 Zn(II) coordination shifted the optical properties of both Boc-Tyr-Trp-OMe (PS1) and its C-termini deprotected analogue Boc-Tyr-Trp-OH (PS2) from the ultraviolet to the visible spectrum. These DPNPs are photostable, biocompatible and have visible luminescence properties. Furthermore, the DPNPs obtained by the biomolecular self-assembly of PS1 and PS2 with Zn(II) coordination are nontoxic and cell penetrating in nature with the ability to carry and deliver drug into cells to facilitate cancer cell death. Notably, conjugation with an EPCAM aptamer allowed for the selective targeting of cancer cells, but not cardiomyocytes.
Results and discussion
The generalized scheme for the bio-inspired synthesis of the fluorescent DPNPs is illustrated in Scheme 1. As shown in Schemes S1–S4,† we have synthesized two dipeptides with the Tyr–Trp backbone, PS1 (Boc-Tyr-Trp-OMe) and its C-termini deprotected analogue PS2 (Boc-Tyr-Trp-OH). The Zn(II) coordination of these dipeptides was achieved by dissolving them in ethanol with subsequent addition of aqueous solution of ZnCl2. Field emission scanning electron microscopy (FE-SEM) analysis was performed to determine the morphology of the self-assembled structures with or without Zn(II) coordination. PS1 generated well-ordered spherical nanostructures (Fig. 1A). Dynamic light scattering (DLS) analysis revealed that the average diameter of these spherical nanostructures is approximately 1744 ± 201.04 nm (Fig. S9A†). PS2, conversely, self-assembled into a 3D network of aggregated spheres (Fig. 1B). The average diameter of the spheres obtained from DLS measurement is 1794 ± 206.86 nm (Fig. S9B†), though the spheres present in the aggregated network differed in size. Metal–ligand coordination can modify the self-assembly process of short peptides to yield more well-ordered and morphologically homogeneous nanostructures.37,38 In the presence of Zn(II), PS1 self-assembled into well-ordered nano-capsules like nanostructures (Fig. 1C) and PS2 self-assembled into nano-flower like nanostructures (Fig. 1D). The coordination of Zn(II) in the self-assembled state was further confirmed by energy-dispersive X-ray spectroscopy (EDS) analysis (Fig. 1E and S10†). To gain insight into the molecular configuration of the self-assembled nanostructures (DPNPs), Fourier transform infrared (FTIR) and powder X-ray diffraction (XRD) analysis were performed to identify the nature of non-covalent interactions involved within the DPNPs. FTIR characteristic peaks were identified at ∼1615 cm−1 and ∼1655 cm−1 for DPNPs formed by the self-assembly of both PS1 and PS2 through Zn(II) coordination. These peaks in the amide I region correspond to the amide group and indicate strong intramolecular H-bonding39 (Fig. 1F and G). The peak at ∼1595 cm−1 in the amide II region can be attributed to the bending mode of ‘–NHamide’ (Fig. 1F and G). In general, the characteristic peak at 1530 ± 30 cm−1 is mainly due to N–H bending trans to the carbonyl oxygen. In a self-assembled state, this absorption band often exhibited a prominent shift due to the intermolecular H bonding characteristic of well-organized nanostructures.40,41 The XRD pattern of the DPNPs formed by both PS1 and PS2 through Zn(II) coordination exhibited a prominent crystalline nature (Fig. 1H and I) compared to the self-assembled structures obtained from the self-assembly of PS1 and PS2 in the absence of Zn(II) (Fig. S11†). At higher degrees (2θ; 25°–45°), the DPNPs exhibited a diffraction pattern typical of crystalline materials with distinct sharp peaks of high intensity. These results clearly demonstrate that the driving forces of self-assembly in the presence or absence of Zn(II) coordination are quite distinct. | ||
| Scheme 1 Design and development of the self-assembled fluorescent DPNPs by bio-molecular self-assembly of PS1 (BOC-Tyr-Trp-OMe) and PS2 (BOC-Tyr-Trp-OH) with the Tyr–Trp dipeptide as the basic unit. Through hydrogen bonding, π–π stacking interactions between the aromatic residues in the peptide backbone, and Zn(II) coordination the dipeptides self-assemble to form DPNPs. | ||
 | ||
| Fig. 1 FE-SEM micrographs of the self-assembled structures formed by (A) BOC-Tyr-Trp-OMe (PS1) and (B) BOC-Tyr-Trp-OH (PS2) without Zn(II), and FE-SEM micrographs of the self-assembled structures (DPNPs) formed by (C) BOC-Tyr-Trp-OMe and (PS1) (D) BOC-Tyr-Trp-OH (PS2) in the presence of Zn(II). (E) EDS analysis of PS1-Zn based DPNPs using SEM. FTIR absorption spectrum of the self-assembled nanostructures (DPNPs) in the amide I & II region formed by (F) PS1 and (G) PS2 with Zn(II) coordination. Powder X-ray diffraction spectra of DPNPs formed by PS1 (H) and PS2 (I) with Zn(II) coordination. The diffraction pattern indicates the formation of crystalline nanostructures. | ||
Fluorescence microscopy analysis of the DPNPs obtained from PS1 and PS2 after Zn(II) coordination yielded a bright green fluorescence signal (Fig. 2A–D). This red shifted visible green fluorescence of PS1 and PS2 in the presence of Zn(II) may result from quantum confinement.42–45 It was previously reported that the intrinsic luminescence properties of peptide-based nanostructures are not influenced by the presence of aromatic side chain residues in the polypeptide backbone.46,47 Therefore, hydrogen bonding in the self-assembled nanostructures responsible for electron delocalization may permit low energy transitions. The red shifted visible emission maxima of the DPNPs obtained from PS1 and PS2 with Zn(II) coordination likely results from π–π stacking interactions and metal–ligand interaction. However, it is possible that electron delocalization caused by the extensive H-bonding within the DPNPs could contribute to the shift of emission maxima from the ultra violet to the visible range.
 | ||
| Fig. 2 Bright field and fluorescence images of the PS1/PS2-Zn based DPNPs on a glass slide. Bright field images of (A) PS1-Zn and (C) PS2-Zn; fluorescence images of (B) PS1-Zn and (D) PS2-Zn. The green colour represents the fluorescence signal of DPNPs. Effects of metal ions: emission spectral scan of the (E) PS1 and (F) PS2 dipeptide self-assembly with various metal ions (K(I), Mg(II), Ca(II), Fe(II), Cu(II) and Zn(II)). The maximum and significant enhancement occurred with Zn(II), which possesses the highest nuclear charge and polarizability among all the tested metal ions. Emission spectra of (G) BOC-Tyr, Trp-OMe, the BOC-Tyr-Trp-OMe (PS1) monomer, and the BOC-Tyr-Trp-OMe (PS1) self-assembly with or without Zn(II) coordination and (H) BOC-Tyr, Trp-OH, the BOC-Tyr-Trp-OH (PS2) monomer, and the BOC-Tyr-Trp-OH (PS2) self-assembly with or without Zn(II) coordination. (I) Emission spectra of the BOC-Tyr-Trp-OMe (PS1) self-assembly at different concentrations (excitation, 280 nm) without Zn(II) coordination. The emission peak at 356 nm originated from the PS1 monomer unit. (J) Emission spectra of the BOC-Tyr-Trp-OH (PS2) self-assembly at different concentrations (excitation, 280 nm) without Zn(II) coordination. The emission peak at 355 nm originated from the PS2 monomer unit. (K) Emission spectra of PS1 self-assemblies with Zn(II) coordination at several concentrations (excitation, 365 nm). The emission peak at 428 nm is originated from the self-assembled nanostructures (DPNPs) with Zn(II) coordination. (L) Emission spectra of PS2 self-assemblies with Zn(II) coordination at several concentrations (excitation, 365 nm). The emission peak at 433 nm is originated from the self-assembled nanostructure (DPNPs) with Zn(II) coordination. | ||
To confirm that Zn(II) was optimal for metal–ligand coordination, we applied various metal ions (K(I), Mg(II), Ca(II), Cu(II), and Fe(II) along with Zn(II)) to trigger the self-assembly of PS1 and PS2 and evaluated the optical properties of the resulting DPNPs. No significant increase in emission intensity was observed in the presence of K(I), Mg(II), Ca(II), Fe(II) and Cu(II). Maximal enhancement was observed in the presence of Zn(II), which possesses the highest nuclear charge and polarizability among all the scanned metal ions (Fig. 2E and F). By exposing PS1 and PS2 to increasing concentrations of Zn(II) we determined that the optimal enhancement of emission intensity was achieved at 10 mM and 12 mM Zn(II), respectively (Fig. S12†).
Next, we recorded the UV-Vis absorption spectra of PS1 and PS2 with or without Zn(II) coordination. The UV-vis spectra of both self-assembled PS1 and PS2 without Zn(II) exhibited absorption maxima at 280 nm with a shoulder peak at 290 nm (Fig. S13A and B†). Generally, a weaker n–π* transition was submerged under a strong π–π* transition and appeared as a shoulder peak at ∼290 nm. The UV-Vis spectra of the self-assembled PS1 and PS2 in the presence of Zn(II) showed two absorption peaks at ∼280 nm and ∼365 nm (Fig. S13C and D†). The red shifted absorption band at ∼365 nm is responsible for the spin allowed charge transfer transition after Zn(II) coordination. For PS1 self-assemblies without Zn(II) coordination the emission maxima appeared at ∼386 nm and with Zn(II) coordination the red shifted emission maxima appeared at ∼428 nm, which is longer than that of the PS1 monomer (357 nm), BOC-Tyr (305 nm) and Trp-OMe (353 nm) (Fig. 2G). A similar spectral pattern was observed for PS2 self-assemblies with Zn(II) coordination. The red shifted emission maxima appeared at ∼433 nm with Zn(II) coordination, which is longer than that of PS2 self-assemblies without Zn(II) coordination (394 nm) or for the PS2 monomer (358 nm), BOC-Tyr (305 nm) and Trp-OH (354 nm) (Fig. 2H).
Next, we recorded the concentration dependent emission spectra of the DPNPs (PS1 and PS2 with Zn(II) coordination) at two different excitation wavelengths: 365 nm and 280 nm. Typically, the emission intensity is directly proportional to the concentration regardless of the excitation wavelength. However, with excitation at 280 nm, we observed a steady decrease in the emission intensity (the emission originated from PS1/PS2 monomers) with increasing concentrations of the dipeptides (PS1 or PS2) (Fig. 2I and J) likely due to the self-assembly of more dipeptides into DPNPs in the presence Zn(II). An opposite trend was observed with excitation at 365 nm. More specifically, we observed a steady increase in emission intensity with increasing concentrations of dipeptides (Fig. 2K and L). The emission peak at ∼430 nm corresponds to self-assembled DPNPs in the presence of Zn(II) (PS1-Zn and PS2-Zn). We hypothesize that, with an increasing concentration of the dipeptides, more dipeptides start to self-assemble into DPNPs resulting in a steady decrease or increase in emission intensity originating from dipeptide monomers or self-assembled DPNPs. When the concentration of the dipeptide was higher than 1.5 mg mL−1, concentration-dependent changes in emission intensity became saturated indicating that the majority of dipeptides (PS1 and PS2) have self-assembled into DPNPs.
In the presence of Zn(II), the emission maxima for PS1- and PS2-based DPNPs were shifted from ∼390 nm to 430 nm indicating the formation of fluorescent nanostructures. With excess Zn(II), the sequential addition of dipeptides (PS1 and PS2) resulted in a steady increase in the emission intensity with an increasing dipeptide concentration (Fig. S14†) indicating that the amount of metal coordinated nanostructures generated depends on the dipeptide concentration. The calculated quantum yield (Φ) of PS1 and PS2 self-assembled nanostructures in the presence of Zn(II) was found to be 14.49 and 15.14% respectively. Both PS1 and PS2 self-assembled nanostructures without Zn(II) coordination retained their spherical morphology with a comparatively much larger average diameter (∼1.75 μM) (Fig. S9†).
The emission intensity of the self-assembled PS1- and PS2-based nanostructures remains stable after continuous irradiation for 120 seconds (Fig. 3A). This reflects better photostability compared to the organic fluorescent dye rhodamine 6G (Rh6G). Additionally, we measured the emission intensity in media with varying pH (pH = 2 to 12). Under moderately acidic (pH = 2) and basic conditions (pH = 10), the emission intensities were minimal. At a pH between 4 and 8, the emission intensities were moderately high with a maximum fluorescence signal observed at physiological pH of 7 (Fig. 3B and C), ideal for biomedical applications. Ideally, the fluorescence of the donor and acceptor moieties should remain consistent with a change in pH. Nevertheless, almost all the GFP mutants are pH sensitive. For example, 50% emission intensity of an enhanced blue fluorescence protein is quenched at pH 6.48 Our experimental results indicate that the fluorescence of the DPNPs obtained from PS1 and PS2 with Zn(II) coordination were comparatively stable in the pH range 6–8 (Fig. 3B and C). GFP also denatures at high temperatures (78 °C) with a loss of 50% emission intensity compared to its room temperature (RT) emission. In our case the emission intensity of these DPNPs showed no substantial change at 50 °C and 75 °C compared to the RT emission spectra (Fig. 3D and S15†).
 | ||
| Fig. 3 (A) Photostability assessment of DPNPs formed by PS1 and PS2 with Zn(II) coordination and the organic dye Rh6G. The fluorescence intensity of DPNPs remained stable after continuous irradiation for 2 minutes, which indicates better photostability compared with that of the organic dye Rh6G. Emission intensity of the (B) PS1 (λmon = 428 nm) and (C) PS2 (λmon = 433 nm) dipeptide self-assembly with Zn(II) coordination at various aqueous pH. (D) Emission spectra of DPNPs formed by the self-assembly of PS1 with Zn(II) coordination at RT, 50 °C and 75 °C. The emission intensity of the DPNPs remained stable from RT to 75 °C. | ||
We tested the biocompatibility of PS1 (PS1-Zn) and PS2 (PS2-Zn) based self-assembled nanostructures with Zn(II) coordination in the human embryonic kidney cell line HEK 293 and human cardiomyocyte cell line AC16 in vitro (Fig. 4A and B). No significant loss in cell viability was observed in either cell line for concentrations of PS1-Zn or PS2-Zn up to 100 micromolar (Fig. 4A and B). The cellular incorporation of PS1-Zn and PS2-Zn was also measured using these cells as their propensity to form a single adherent layer in culture allows for facile visualization of fluorescence in imaging studies. Within 24 h, intracellular accumulation of the compounds was confirmed by prominent green fluorescence (Fig. 5A). Fluorometric analysis showed statistically significant uptake in a concentration-dependent manner for both test compounds (Fig. 4C and D).
 | ||
| Fig. 4 Cellular incorporation and cytotoxicity of PS1-Zn and PS2-Zn. Survival of normal human kidney HEK293 cells and AC16 human cardiomyocytes at 40 h following incubation with PS1-Zn (A) and PS2-Zn (B) at the mentioned concentrations as measured by MTT assays. Spectrofluorometric analysis of cellular incorporation of PS1-Zn (C) and PS2-Zn (D) post 24 h incubation in both cells. The results are means ± S.E. of multiple experiments (n = 5, and *p < 0.05; **p < 0.01, and **p < 0.001 compared with the control at the 0 h time point). | ||
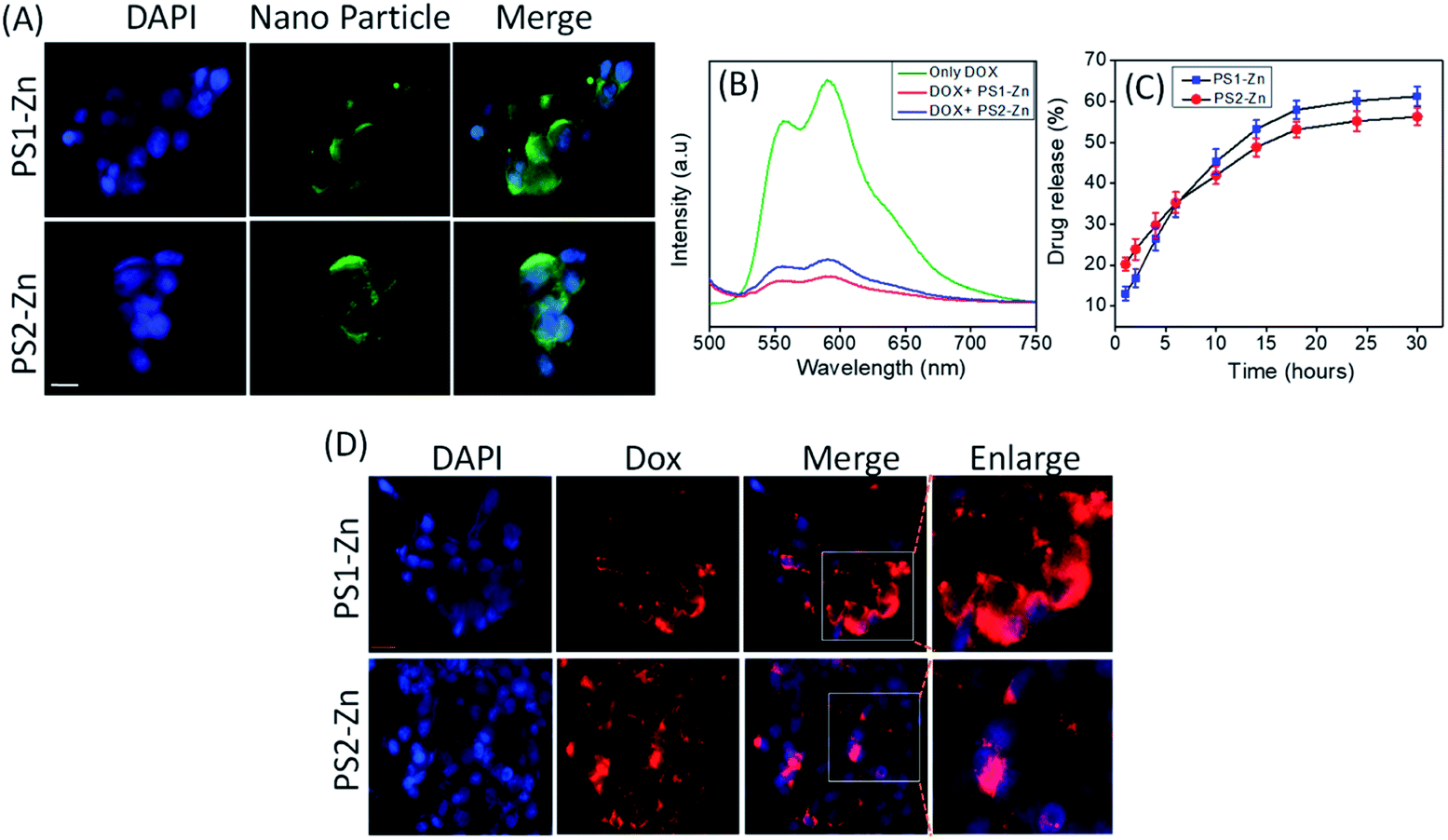 | ||
| Fig. 5 (A) Incorporation of fluorescent DPNPs (PS1-Zn and PS2-Zn) in A549 and visualized with a fluorescence microscope. (B) Emission spectra of Dox in the absence and presence of DPNPs formed by PS1 and PS2 with Zn(II) coordination. A decrease in the fluorescence intensity of Dox was observed after conjugation with the DPNPs. (C) The release profile of Dox from the DPNPs-Dox conjugates. The release behavior of Dox from the DPNPs-Dox conjugates in PBS buffer (pH 7.2) at RT was examined and plotted by varying the emission intensity at 590 nm (λmon = 490 nm). The % of drug release is given as a mean value. The error bars represent the standard deviation (n = 3). PS1-Zn and PS2-Zn encapsulated Dox enters cells and promotes oxidative stress and apoptosis. (D) Incorporation of Dox in A549 cells post encapsulation in PS1-Zn and PS2-Zn and visualized with a fluorescence microscope. | ||
We then investigated the feasibility of PS1- and PS2-based DPNPs as drug delivery vehicles by real time monitoring of drug release. Doxorubicin (Dox), an efficient anticancer chemotherapy drug with a strong fluorescence (λems = 590 nm, λext = 490 nm), has a propensity to stack with aromatic moieties through π–π stacking interactions. We self-assembled PS1 and PS2 in the presence of Zn(II) and Dox resulting in Dox-DPNPs conjugates in which the fluorescent drug was incorporated into the metal coordinated dipeptide self-assemblies. The calculated encapsulation efficiency (EE) was 57.34% for the nano-capsules (PS1-Zn DPNPs) and 61.28% for nano-flowers (PS2-Zn DPNPs), while the loading capacity (LC) was 15.5% (nano-capsules) and 16.6% (nano-flowers). The conjugation of Dox with DPNPs was characterized by observing the fluorescence spectra of Dox in the presence and absence of our DPNPs (Fig. 5B). The noticeable quenching in emission intensity (λems = 590 nm) was ascribed to the electrostatic interaction between the DPNPs and Dox.49,50
We evaluated the release of encapsulated Dox by monitoring the steady state luminescence of Dox-DPNPs conjugates dispersed in PBS buffer and relocated into a dialysis bag (MWCO 3 kDa) in PBS buffer at RT. The emission intensity of the aliquots was measured at several time intervals for 36 hours revealing a steady increase in the emission intensity with time (Fig. 5C) corresponding to an increase in the concentration of the drug molecule (Dox) in the buffer solution outside the dialysis bag. After 30 hours, we did not notice any significant increase in the emission intensity and it is likely that the drug release process was completed and reached a plateau at this stage (Fig. 5C).
Here, we move to a more clinically relevant cellular system. A549 lung cancer cells were utilized because Dox-containing chemo-therapeutic regimens are used to treat lung cancer. First, we sought to confirm whether Dox, which displays autofluorescence enabling easy visualization, could be delivered into cells when encapsulated with either PS1-Zn or PS2-Zn. Fig. 5D demonstrates clear accumulation of Dox within A549 cells following exposure to either PS1-Zn-Dox or PS2-Zn-Dox. Dox inhibits topoisomerase 2β inducing DNA damage, activating the pro-apoptotic DNA damage response (DDR), triggering phosphorylation of ATM, p53, and the ATM substrate γH2AX, decreasing anti-apoptotic Bcl-2, increasing pro-apoptotic Bax, and culminating in mitochondrial-dependent oxidative stress and cell death (Fig. 7B).51 Indeed, the ability of PS1-Zn-Dox or PS2-Zn-Dox to compromise cell viability (Fig. 6A), trigger chromatin fragmentation (Fig. 6B), and damage DNA (Fig. 6C and D) did not differ from the unencapsulated compound. PS1-Zn & PS2-Zn alone had little to no impact on the DDR (Fig. 7A and S16A†). However, Dox, PS1-Zn-Dox, and PS2-Zn-Dox elevated pATM, p53, γH2AX, and the Bax/Bcl-2 ratio to a similar extent. Similar results were obtained for the colon cancer cell line HCT116 (Fig. S19A–C†). Though canonically believed to function as G protein coupled receptor modulators, RGS6 and its co-stabilizing partner Gβ5 have recently emerged as novel tumor suppressors downregulated in breast and bladder cancer.52,53
 | ||
| Fig. 6 (A) Survival of cancer cells (A549 human lung cancer) at 36 h following incubation with Dox, PS1-Zn-Dox and PS2-Zn-Dox (1 μM) as measured by MTT assays. The results are the means ± S.E. of multiple experiments (n = 6 and *p < 0.01 compared with the control, which received only 0.1% DMSO). (B) Apoptosis was measured by the formation of cytoplasmic histone-associated DNA fragments (enrichment factor) as described under “Experimental procedures”. Dox encapsulation with PS1-Zn and PS2-Zn promotes DNA damage in A549 cancer cells. Self-assembly with PS1-Zn and PS2-Zn induced DNA damage (DSBs) in A549 cancer cells as represented by (C) DNA laddering and (D) comet assay. Representative images of single cells treated with or without Dox, PS1-Zn-Dox and PS2-Zn-Dox with quantification of comet tail length (bar diagram in D). The results represent the means ± S.E. of independent experiments (n = 10 and *p < 0.01). | ||
 | ||
| Fig. 7 Dox encapsulation with PS1-Zn and PS2-Zn promotes DNA damage in A549 cancer cells. (A) Expression of different proteins in A549 cells exposed to Dox or Dox encapsulated with PS1-Zn and PS2-Zn. No changes were observed in control cells with PS1-Zn and PS2-Zn treatment. β-Actin is shown as a loading control. (B) Schematic representation showing mitochondrial regulation in normal and apoptotic cells. Cell death signals induce oxidative stress and DNA damage in the cells that has the potential to shift the Bax/Bcl-2 ratio, trigger the mitochondrial permeability transition, activate caspases, and induce cell death. (C) Effects of Dox-PS1-Zn/PS2-Zn on mitochondrial membrane potential (Δψm) in cancer cells. Cells were harvested 24 h following treatment, and mitochondrial membrane potential was measured as described under “Experimental procedures”. The results are expressed as means ± S.E. (n = 6 and *p < 0.01 compared with control cells). (D) PS1-Zn-Dox and PS2-Zn-Dox promote the generation of ROS in A549 cells. ROS was measured by CM-H2DCFDA fluorescence 24 h later. The results are expressed as means ± S.E. (n = 6 and *p < 0.01 compared with control cells with vehicles only). | ||
Importantly, the ability of Dox to promote reactive oxygen species (ROS) generation and apoptosis in breast cancer cells requires RGS6/Gβ5 complexes.54 Indeed, Dox, PS1-Zn-Dox, and PS2-Zn-Dox failed to significantly impact RGS6/Gβ5 expression in A549 cells, which could result from cancer-cell specific mutation of these proteins as has been proposed.55 Finally, we noted that Dox, PS1-Zn-Dox, and PS2-Zn-Dox decreased the mitochondrial membrane potential (ΔψM) (Fig. 7C) and increased oxidative stress (Fig. 7D) to a similar degree. Together, these data represent an important proof of concept showing that both PS1-Zn and PS2-Zn can effectively shuttle the chemotherapeutic drug Dox into cells while maintaining therapeutic efficacy.
To further demonstrate the suitability for bioimaging, the DPNPs were next processed to bind with EPCAM aptamers forming DPNPs/aptamer conjugates for recognition of the overexpressed EPCAM proteins located on the membrane of epithelial cells such as A549 cells (Fig. 8A). The covalent conjugation of the DPNPs with the EPCAM aptamers was verified by an electrophoretic mobility shift assay (Fig. S17A†). Epithelial marker-based aptamer conjugation with the DPNPs significantly increased the uptake into A549 cancer cells (Fig. 8B and S17B†). To demonstrate the selectivity of the DPNPs/aptamer conjugates, AC16 human cardiomyocyte cells (EPCAM-negative) and A549 human carcinoma epithelial cells (EPCAM-positive) were treated with aptamer conjugated PS2-Zn-Dox. Fluorescent imaging revealed cancer cell-specific aptamer-DPNPs (green) and Dox (red) uptake into A549 cells, but not AC16 cells (Fig. 8C). Consistent with a lack of intracellular Dox delivery into the AC16 cells, the ability of PS2-Zn-Apt-Dox to promote ATM & γH2AX phosphorylation, p53 induction, and increase in the Bax/Bcl-2 ratio was lost in AC16 (Fig. 8E and S16†), but not in A549 cells (Fig. 8D and S17C†). Overall, our findings clearly demonstrate that the DPNPs/aptamer exhibit the capability to function as fluorescent nanoprobes for cancer cell targeting and sensing.
 | ||
| Fig. 8 Effect of DPNPs (PS2-Zn) modified with the EPCAM aptamer in cells. (A) Schematic representation showing the DPNPs functionalized by the EPCAM aptamer (DPNPs/aptamer). DPNPs modified with the EPCAM aptamer (wavy lines) bind to the EPCAM protein (pointed arrow structure) found on the cell membrane of A549 cells (yellow, cytoplasm; blue, nucleus). A carboxyl-terminated DPNP was conjugated with the EPCAM aptamer using the reagents EDC and NHS. (B) In vitro fluorescence imaging of A549 cells incubated with PS2-Zn alone. The blue colour represents the nucleus of the cells and green fluorescence signal of the DPNPs (scale bar = 100 μm). (C) A549 and AC16 cells were incubated with 100 nM DPNPs/aptamer for 1 h at 37 °C. Fluorescence cellular imaging has been performed. Expression of different proteins with exposure to PS2-Zn, Dox only (n = 3) and PS2-Zn-Apt-Dox (n = 6, 100 nM DPNPs/aptamer for 12 h) was documented in A549 (D) and AC16 (E) cells. Quantification of the proteins is shown in the ESI Fig. S17C (A549) and S18 (AC16).† | ||
Indeed, histochemical approaches are used to evaluate EPCAM levels in cancer diagnosis particularly for tumours of gastrointestinal origin.56 A fluorescent peptide-based approach has the potential to streamline EPCAM detection in malignant tumours as has been proposed recently for detection of amyloid-beta aggregation in patients with Alzheimer's disease with cyclic peptide nanoparticles (c-PNPs).57 Similarly, Song et al., have demonstrated that biometallohydrogels based on the self-assembly and local mineralization of Ag+-coordinated Fmoc-amino acids can aid in the localized delivery of antimicrobial agents58 emphasizing the range of potential biomedical applications for small, peptide-based nanoparticles. Notably, PS2-Zn-Apt may have utility as both a probe and a drug delivery platform.
Conclusion
We have designed and developed BOC-Tyr-Trp dipeptide-based DPNPs with structural rigidification by Zn(II), which shifts the peptides’ inherent fluorescence properties from the ultraviolet to the visible region. Compared with common organic fluorophores (prone to photobleaching), QDs (lack of biocompatibility, ease of agglutination and solubility) and GFPs (slow folding rate and pH and temperature sensitive), the DPNPs exhibit photostability, biocompatibility and have visible fluorescence properties. Additionally, the DPNPs chemically modified with the EPCAM aptamer and encapsulated with Dox target the A549 cancer cells without impacting cardiomyocytes providing evidence that these DPNPs/aptamer conjugates represent a novel platform for the development of highly selective drug chaperones for various biomedical applications. Future work will also focus on employing DPNPs for real-time monitoring of drug release and cellular uptake.Materials and methods
All the chemicals and solvents are commercially available and were used as received. L-Tyrosine, L-tryptophan, N,N-dicyclohexylcarbodiimide (DCC), 1-hydroxybenzotriazole (HOBt), BOC anhydride, trimethylchlorosilane (TMSCl), and triethyl amine were purchased from Sisco Research Laboratories Pvt. Ltd (SRL, India). Potassium hydrogen sulphate, HCl, sodium chloride, sodium sulfate, sodium hydroxide, and sodium carbonate were purchased from Finar Chemicals Pvt. Ltd (India). 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), N-hydroxysuccinimide (NHS), 1,1,1,3,3,3-hexafluoro-2-propanol (HFP), doxorubicin hydrochloride (98.0–102.0% (HPLC)) and rhodamine-B dye were purchased from Sigma Aldrich (St. Louis, MO, USA). Aprotinin, PMSF, leupeptin, sodium orthovanadate, 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), 2′,7′-dichlorodihydrofluorescein diacetate, N-acetyl cysteine, JC-1, carbonyl cyanide 3-chlorophenylhydrazone, L-N6-nitroarginine methyl ester hydrochloride, and antibodies for β-actin were obtained from Sigma (St. Louis, MO, USA). The Caspases-3 activity kit was obtained from Biovision (San Francisco, CA, USA), and the 3,3′-diaminobenzidine HRP substrate kit, antibodies for RGS6, and H2AX were obtained from Abcam (Cambridge, UK). Antibodies for p53, Bcl2 and Bax were from Cell Signalling Technology, Inc. (Danvers, MA, USA). DMEM and FBS were obtained from Himedia Laboratories (Mumbai, MH, India). The Cell Death Detection kit was purchased from Roche Applied Science (San Francisco, CA, USA). Antibodies for Gβ5, ATM, pATM are purchased from Invitrogen (Carlsbad, CA, USA). The Gβ5 antibody was obtained from Millipore (Burlington, MA, USA) and nitrocellulose membrane was purchased from Bio-Rad. Other regular laboratory chemicals were purchased from Sisco Research Laboratory and Loba Chemicals (Mumbai, MH, India). The 5′-amine-anti-EpCAM DNA aptamer (sequence of 5′-CACTACAGAGGTTGCGTCTGTCCCACGTTGTCATGGGGGGTTGGCCTG-3′) was purchased from IDT.Synthesis of PS1, PS2, PS1-Zn and PS2-Zn
The detailed protocol utilized to synthesize PS1, PS2 and PS1-Zn(II) and PS2-Zn(II) with proper characterization is available in the ESI Experimental section (Schemes S1–S4 and S1–S8†).Self-assembly of PS1, PS2, PS1-Zn and PS2-Zn
A fresh stock solution of the peptide was prepared by dissolving the lyophilized forms of PS1, PS2, PS1-Zn and PS2-Zn in HFP to a concentration of 100 mg mL−1. Then, we blended this peptide solution in several different proportions and diluted them with aqueous ethanol (50%) to get the desired concentrations of the peptide for self-assembly. The polarized solvent allowed the molecules to self-assemble.Field emission scanning electron microscopy
A 10 μL drop of a self-assembled solution of PS1, PS2, PS1-Zn and PS2-Zn was placed on a glass cover slip and allowed to dry at RT. The substrates were then coated with gold using a Leica EM ACE200 2–3 nm gold coater. SEM analysis was performed using a field emission scanning electron microscope (FE-SEM, JEOL JSM-7100F) operating at 18 kV.Microanalysis
C, H, and N analysis was performed using a Vario Micro Cube (Elementar) instrument.Fourier transform infrared spectroscopy (FT-IR)
Fourier transform infrared spectra were recorded using an IRTracer-100 FT-IR spectrometer (Shimadzu) with a deuterated lanthanum α-alanine doped triglycine sulphate (DLaTGS) detector. The peptide self-assembled solutions were deposited on a CaF2 window and dried under vacuum. The peptide deposits were suspended with D2O and subsequently dried to form thin films. The re-suspension procedure was repeated twice to ensure maximal hydrogen-to-deuterium exchange. The measurements were taken using 4 cm−1 resolution and an average of 2000 scans. The transmittance minimal values were determined using the Lab solutions IR analysis program (IR Tracer).UV-vis spectroscopy
UV-Vis absorption spectra of the synthesized peptides in the absence and presence of Zn(II) were recorded using a UV/Vis spectrophotometer (Agilent, Cary 5000, Double beam UV-Vis absorption spectrometer).Fluorescence spectroscopy
Fluorescence measurements were performed at RT using a fluorescence spectrometer (Fluorolog, HORIBA). The emission spectra were collected for PS1 and PS2 from 290 to 600 nm with an excitation wavelength of 280 nm and for PS1-Zn and PS2-Zn the emission spectra were collected from 375 nm to 650 nm with an excitation wavelength of 365 nm. For the optical in vitro drug release assay, fluorescence measurements were performed and the emission spectra were collected from 500 to 750 nm with an excitation wavelength of 490 nm. The fluorescence intensity at 590 nm was used for the quantitative analysis. Both the excitation and emission slit widths were set to 2.0 nm.Dynamic light scattering (DLS) analysis and zeta potential measurements
Dynamic light scattering (DLS) analysis of the spherical assemblies formed by PS1 and PS2 without Zn coordination was performed using a Nano-zeta sizer (Horiba Sz-100) and these measurements were performed at RT, 25 °C.Drug loading and drug release of PS1-Zn and PS2-Zn self-assembled DPNPs
The incorporation of doxorubicin was conducted during the self-assembly of PS1-Zn and PS2-Zn. Dox at a concentration of 10−1 mol L−1 (dissolved in water) was added to PS1-Zn and PS2-Zn (dissolved in HFP at 100 mg mL−1 concentration) to achieve the desired final concentration (2.0 mg mL−1; effective concentration). Then the mixture was left overnight. This mixture underwent spontaneous accommodation of the drug molecule within the PS1-Zn and PS2-Zn based DPNPs. Following conjugation, samples were prepared by drop-casting 25 μL of the conjugate mixture on the glass coverslip and dried in air. The remaining solvent was left to dry overnight at RT under vacuum. Then, the assemblies were washed carefully with ultrapure water several times to eliminate the residual free Dox and then dried properly at RT. The drug incorporated DPNPs were prepared by the above-mentioned protocol and dispersed in PBS (10 mM NaCl pH = 7.4, 150 mM). After that, this suspension (2 mL) was transferred into a dialysis bag (MWCO 3 kDa), and the bag was dipped in 40 mL of PBS at RT. The emission intensity of the buffer solution outside the dialysis bag was measured at different time intervals for 30 hours. The volume of the solution was kept constant by adding 1 mL of the original PBS solution after each measurement. The emission intensities were measured at RT using a fluorescence spectrophotometer. The emission spectra were recorded from 500 nm to 750 nm for the % drug release vs. time plot (λext = 490 nm and λmon = 590 nm).Drug-encapsulation efficiency (EE) and loading capacity (LC) calculations
PS1-Zn-Dox and PS2-Zn-Dox conjugates were prepared as reported above, and left to precipitate overnight. The aqueous medium was decanted and the emission intensity at the desired wavelength was measured. The drug-encapsulation efficiency (EE), which is correlated with the concentration of the drug not incorporated or the free unentrapped drug molecule, can be expressed using eqn (1)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 59
59 | (1) |
 | (2) |
 | (3) |
As the concentration of the drug is directly proportional to the emission intensity (eqn (2)) the emission of the drug incorporated in nanoparticles is equal to the total emission subtracted by the emission intensity of the drug not incorporated; EE can be calculated using eqn (3).
The loading capacity (w/w LC%) can be calculated using the following expression:

The molecular weight of Dox = 543.52 g mol−1. For the drug encapsulation study, the final volume of the resultant solution is 1 and the final effective concentration of PS1-Zn and PS2-Zn is 1.5 mg mL−1. The concentration of the fluorescent drug (Dox) actually loaded is 10−3 mol L−1. The drug encapsulation efficiency calculated for the PS1-Zn based DPNPs and PS2-Zn based DPNPs is 57.36% and 61.28%, respectively.
The amount of the entrapped drug by the PS1-Zn based DPNPs is
| 0.543 × EE = 0.543 × 57.34% = 0.311 mg. |
| LC = (0.311/2.0) × 100% = 15.5% |
The amount of the entrapped dye by the PS2-Zn based DPNPs is
| 0.543 × EE = 0.543 × 61.28% = 0.332 mg. |
| LC = (0.332/2.0) × 100% = 16.6% |
Cell culture
The human lung carcinoma (A549), colon carcinoma (HCT116) and normal kidney (HEK293) cell lines were procured from the National Centre for Cell Science (NCCS), Pune, India. The human cardiac myocyte cell line (AC16, SCC109) was procured from Merck, Millipore, USA. These cells were cultured in DMEM media, supplemented with 10% FBS, 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin. The cells were grown at 37 °C with 5% CO2.MTT assay
The growth and proliferation of the control cells (HEK293 and AC16) and those treated with various concentrations of PS1-Zn and PS2-Zn were determined by the MTT reduction assay. First, the cells were seeded with a density of 5 × 104 per well in 48 well plates and further incubated with the DMEM medium supplemented with 10% FCS (Gibco). The culture medium was replaced after 24 h and the cells were then incubated with the test compounds for 40 h. The stock solution of MTT was prepared at a concentration of 1 mg mL−1 in the culture medium without phenol red and 200 μL of MTT solution was added into each well after removing the old medium. Then, the cells were kept at 37 °C with 5% CO2 for at least 2 h and 200 μL of DMSO was added into each well after 2 h for the solubilization of the formed formazan crystals. The optical density was then recorded using a plate reader (Biotek Instrument) at a wavelength of 550 nm.60Fluorometric analysis
HEK293, A549 and AC16 cells were seeded at a density of 3 × 105 in a 35 mm cell culture dish and grown in DMEM supplemented with 10% FCS. After 30 h, the medium was replaced with the one containing PS2-Zn and PS2-Zn-Apt respectively at different concentrations, and the cultures were incubated for the next 24 h. Then, the cells were harvested, spun down, washed with chilled PBS and then lysed in PBS using 1% Tween 20. The cell lysates were next centrifuged, the supernatant was collected and the fluorescence level was determined as the ratio of excitation at 560 nm to emission at 633 nm.61DNA fragmentation assay
Briefly, the A549 and HCT cells (1 × 106) were seeded and treated with Dox, PS1-Zn-Dox, or PS2-Zn-Dox for 30 h. The cells were then harvested, thoroughly rinsed with chilled PBS, and lysed using an appropriate buffer containing 0.5% Triton X-100, 20 mM Tris HCl buffer and 15 mM EDTA at RT. The lysate solution was further incubated with RNase (0.1 mg mL−1) and proteinase K (1 mg mL−1) for a minimum of 1 h, extracted using phenol/chloroform/isoamyl alcohol (25:24:1), and the DNA was precipitated by incubating the upper aqueous phase with 0.1 volume of 3 M sodium acetate (pH 5.2) and 3 volumes of absolute alcohol overnight at −20 °C. The final pellet obtained post centrifugation was further washed with 70% ethanol, air dried, and dissolved using 50 μL TE buffer (10 mM Tris, 1 mM EDTA, pH 8.0). Then the extracted DNA was resolved by electrophoresis on a 2% agarose gel and visualized after staining with ethidium bromide.62
Immunofluorescence study
HEK293 and A549 cells were plated on cover slips in 6-well plates with DMEM containing 10% FCS. After 30 hours, the medium was removed and Dox, PS1-Zn-Dox and PS2-Zn-Dox containing DMEM medium were added to the cells. After 8 h, the cells were washed with chilled PBS solution twice, further fixed in 4% paraformaldehyde at RT for 10 min and mounted with Vectashield mounting media (Invitrogen). The PS2-Zn-aptamer was also used for microscopic studies in a separate set of experiments. Then cells were visualized by fluorescence microscopy (Optika B-100FL HBO, Italy) for labelled fluorescence.63Comet assay
Comet assay using A549 cells was performed with Dox (0.5 mmol L−1, 8 h), PS1-Zn-Dox and PS2-Zn-Dox or with the corresponding test compound without Dox. Comet assay (alkaline single cell gel assay, pH > 13) was performed following a published protocol to determine the extent of DNA damage. The measurement of comet tail length was further assessed with Comet Assay IV software (Perceptive Instruments Ltd).54ROS generation study
Intracellular ROS levels were measured with the cell-permeable oxidation-sensitive probe, CM-H2DCFDA. A549 cells were first treated with Dox, PS1-Zn-Dox and PS2-Zn-Dox for 24 h. Cells were then scraped, centrifuged, pelleted down, washed (twice) with chilled PBS and further resuspended in PBS with CM-H2DCFDA (5 μM) at 37 °C for at least 20 min in the dark. Next, the cells were washed again with chilled PBS and lysed using PBS with 1% Tween 20. The generation of intracellular ROS level in lysates was determined by the measurement of the fluorescence of dichlorofluorescein (DCF) (excitation at 480 nm and emission at 530 nm).64Measurement of mitochondrial membrane potential
Mitochondrial membrane potential (MMP ψM) was measured by the incorporation of the cationic fluorescent dye JC-1 (5,5′,6,6′-tetracholoro-1,1′,3,3′-tetraethyl benzimidazolylcarbo cyanine iodide) directly into the mitochondria. A549 cells were grown and treated with PS1-Zn-Dox and PS2-Zn-Dox for 30 h. Cells were next harvested as mentioned in the earlier methodology section for ROS studies and incubated with shaking for 30 min at 37 °C in a PBS solution containing JC-1 (2.5 μg mL−1). Cells were then washed thrice with chilled PBS and resuspended in PBS again, and mitochondrial membrane potential was quantified by measuring the ratio of fluorescence at 590/530 nm.64Apoptosis assay
The Roche cell death detection kit was used for the quantification of the extent of apoptosis in A549 cancer cells treated with Dox (positive control), PS1-Zn-Dox and PS2-Zn-Dox. This ELISA kit quantifies the formation of cytoplasmic histone-associated DNA fragments (both mono- and oligosomes) associated with apoptotic cell death. The results are expressed as a fold increase in the enrichment factor (cytoplasmic nucleosomes).65Immunoblotting
Cells post treatment were flash frozen in liquid nitrogen and lysates were prepared using RIPA buffer with protease (p8340) and phosphatase (#3) inhibitor cocktails (Abcam), quantified and then probed. Twenty micrograms of protein from each sample was subjected to SDS-PAGE and immunoblotting using standard techniques and immunoblots were developed using the chemiluminescence method with horseradish peroxidase-labelled secondary antibodies. Quantification of western blots by densitometry was done using Image J software (NIH). For each experiment protein expression was normalized with loading controls and expressed relative to control conditions.65Conjugation of peptides to an aptamer
The DPNPs were conjugated covalently with an EPCAM aptamer using the EDC catalyst (Sigma Aldrich) and NHS (Sigma Aldrich) reaction. To activate the carboxyl groups, DPNPs were treated with EDC and NHS for 25 minutes. The mixture was centrifuged and dissolved in distilled water. After activation of the DPNPs, the EPCAM aptamer was added into the activated DPNPs solution at RT overnight. After the conjugation, the samples were centrifuged to remove any unreacted aptamers.Electromobility shift assay
The covalent binding between the EPCAM aptamer and DPNPs was assessed by an electromobility shift assay according to a previously published protocol.20 First, the DPNPs/aptamer were disassembled through slow heating to 95 °C for at least 15 minutes. Then, 5 μL of 50 ng μL−1 aptamer-labelled DPNPs (after disassembly) were processed for electrophoresis on an agarose gel with ETBr. Equivalent amounts of dipeptides and aptamers were used as the controls for this experiment, and a 1 kb DNA ladder as the marker. The gel was then visualized and imaged by using the chemiluminescence documentation system (Analytic Jena).20Statistical analysis
Data were analyzed by Student's t-test or one- or two-way ANOVA. Statistical analyses were performed using Origin 6.1 software. The results were considered significantly different at p < 0.05. Values are expressed as means ± SEM.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. D. acknowledges Council of Scientific and Industrial Research (CSIR), India, for research funding (File no. 01(3077)/21/EMR-II) and the support of the Interdisciplinary Institute of Indian System of Medicine, Nano Research Centre (NRC), Department of Biotechnology of SRM IST for several characterization studies. B. M. acknowledges the financial support from Department of Biotechnology, India (BT/PR21156/MED/30/1753/2016 and BT/PR28635/MED/30/2145/2019) and DST-SERB (EMR/2016/006873) for conducting the research.Notes and references
- H. Chen, W. Zhang, G. Zhu, J. Xie and X. Chen, Nat. Rev. Chem., 2017, 2, 17024 CAS.
- L. L. Li and H. Wang, Nat. Biomed. Eng., 2018, 2, 56–57 CrossRef PubMed.
- F. Huang, J. Wang, A. Qu, L. Shen, J. Liu, J. Liu, Z. Zhang, Y. An and L. Shi, Angew. Chem., 2014, 126, 9131–9136 CrossRef.
- M. Mahmoudi, M. Yu, V. Serpooshan, J. C. Wu, R. Langer, R. T. Lee, J. M. Karp and O. C. Farokhzad, Nat. Nanotechnol., 2017, 12, 845–855 CrossRef CAS PubMed.
- N. Kandoth, S. Barman, A. Chatterjee, S. Sarkar, A. K. Dey, S. K. Pramanik and A. Das, Adv. Funct. Mater., 2021, 31, 2104480 CrossRef CAS.
- Y.-F. Wang, L. Liu, X. Xue and X.-J. Liang, F1000Research, 2017, 6, 681 Search PubMed.
- R. Tiwari, S. Banerjee, D. Tyde, K. Das Saha, A. Ethirajan, N. Mukherjee, S. Chattopadhy, S. K. Pramanik and A. Das, Bioconjugate Chem., 2021, 32, 245–253 CrossRef CAS PubMed.
- H. Singh, K. Tiwari, R. Tiwari, S. K. Pramanik and A. Das, Chem. Rev., 2019, 119, 11718–11760 CrossRef CAS PubMed.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2005, 4, 145–160 CrossRef CAS PubMed.
- E. Villemin, Y. C. Ong, C. M. Thomas and G. Gasser, Nat. Rev. Chem., 2019, 3, 261–282 CrossRef CAS.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- M. E. Davis, Z. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS PubMed.
- S. M. Rafiyath, M. Rasul, B. Lee, G. Wei, G. Lamba and D. Liu, Exp. Hematol. Oncol., 2012, 1, 1–9 CrossRef PubMed.
- J. J. M. Kwakman, Y. S. Elshot, C. J. A. Punt and M. Koopman, Oncol. Rev., 2020, 14, 57–63 Search PubMed.
- R. V. Ulijn and A. M. Smith, Chem. Soc. Rev., 2008, 37, 664–675 RSC.
- C. A. E. Hauser and S. Zhang, Nature, 2010, 468, 516–517 CrossRef CAS PubMed.
- E. Gazit, Chem. Soc. Rev., 2007, 36, 1263–1269 RSC.
- L. Adler-Abramovich and E. Gazit, Chem. Soc. Rev., 2014, 43, 6881–6893 RSC.
- J. Zhou, J. Li, X. Du and B. Xu, Biomaterials, 2017, 129, 1–27 CrossRef CAS PubMed.
- Z. Fan, L. Sun, Y. Huang, Y. Wang and M. Zhang, Nat. Nanotechnol., 2016, 11, 388–394 CrossRef CAS PubMed.
- Q. Wang, N. Jiang, B. Fu, F. Huang and J. Liu, Biomater. Sci., 2019, 7, 4888–4911 RSC.
- Z. Fan, Y. Chang, C. Cui, L. Sun, D. H. Wang, Z. Pan and M. Zhang, Nat. Commun., 2018, 9, 2605 CrossRef PubMed.
- M. Sameiro and T. Gonçalves, Chem. Rev., 2009, 109, 190–212 CrossRef PubMed.
- S. K. Pramanik and A. Das, Chem. Commun., 2021, 57, 12058–12073 RSC.
- R. Tiwari, P. S. Shinde, S. Sreedharan, A. K. Dey, K. A. Vallis, S. B. Mhaske, S. K. Pramanik and A. Das, Chem. Sci., 2021, 12, 2667–2673 RSC.
- S. Ranjbarvaziri, S. Kiani, A. Akhlaghi, A. Vosough, H. Baharvand and N. Aghdami, Biomaterials, 2011, 32, 5195–5205 CrossRef CAS PubMed.
- X. Wu and W. Zhu, Chem. Soc. Rev., 2015, 44, 4179–4184 RSC.
- E. M. S. Stennett, M. A. Ciuba and M. Levitus, Chem. Soc. Rev., 2014, 43, 1057–1075 RSC.
- H. Soo Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. Itty Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165–1170 CrossRef PubMed.
- T. S. Hauck, R. E. Anderson, H. C. Fischer, S. Newbigging and W. C. W. Chan, Small, 2010, 6, 138–144 CrossRef CAS PubMed.
- S. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS PubMed.
- N. S. De Groot, T. Parella, F. X. Aviles, J. Vendrell and S. Ventura, Biophys. J., 2007, 92, 1732–1741 CrossRef PubMed.
- R. Y. Tsien, Annu. Rev. Biochem., 1998, 67, 509–544 CrossRef CAS PubMed.
- R. M. Wachter, M. A. Elsliger, K. Kallio, G. T. Hanson and S. J. Remington, Structure, 1998, 6, 1267–1277 CrossRef CAS PubMed.
- D. Fu, D. Liu, L. Zhang and L. Sun, Chin. Chem. Lett., 2020, 31, 3195–3199 CrossRef CAS.
- F. W. Teale and G. Weber, Biochem. J., 1957, 65, 476–482 CrossRef CAS PubMed.
- R. Zou, Q. Wang, J. Wu, J. Wu, C. Schmuck and H. Tian, Chem. Soc. Rev., 2015, 44, 5200–5219 RSC.
- P. Das, I. Pan, E. Cohen and M. Reches, J. Mater. Chem. B, 2018, 6, 8228–8237 RSC.
- P. W. J. M. Frederix, G. G. Scott, Y. M. Abul-Haija, D. Kalafatovic, C. G. Pappas, N. Javid, N. T. Hunt, R. V. Ulijn and T. Tuttle, Nat. Chem., 2015, 7, 30–37 CrossRef CAS PubMed.
- A. Barth and C. Zscherp, Q. Rev. Biophys., 2002, 35, 369–430 CrossRef CAS PubMed.
- S. Fleming, P. W. J. M. Frederix, I. Ramos Sasselli, N. T. Hunt, R. V. Ulijn and T. Tuttle, Langmuir, 2013, 29, 9510–9515 CrossRef CAS PubMed.
- G. Rosenman, N. Amdursky, M. Molotskii, D. Aronov, L. Adler-Abramovich and E. Gazit, Nano Lett., 2009, 9, 3111–3115 CrossRef PubMed.
- P. Mahato, A. Ghosh, S. K. Mishra, A. Shrivastav, S. Mishra and A. Das, Chem. Commun., 2010, 46, 9134 RSC.
- F. Ali, H. A. Anila, N. Taye, D. G. Mogare, S. Chattopadhyay and A. Das, Chem. Commun., 2016, 52, 6166–6169 RSC.
- K. H. Min, Y.-H. Kim, Z. Wang, J. Kim, J. S. Kim, S. H. Kim, K. Kim, I. C. Kwon, D. O. Kiesewetter and X. Chen, Theranostics, 2017, 7, 4240–4254 CrossRef CAS PubMed.
- F. T. S. Chan, G. S. Kaminski Schierle, J. R. Kumita, C. W. Bertoncini, C. M. Dobson and C. F. Kaminski, Analyst, 2013, 138, 2156–2162 RSC.
- D. Pinotsi, A. K. Buell, C. M. Dobson, G. S. Kaminski Schierle and C. F. Kaminski, ChemBioChem, 2013, 14, 846–850 CrossRef CAS PubMed.
- G. H. Patterson, S. M. Knobel, W. D. Sharif, S. R. Kain and D. W. Piston, Biophys. J., 1997, 73, 2782–2790 CrossRef CAS.
- M. K. Yu, Y. Y. Jeong, J. Park, S. Park, J. W. Kim, J. J. Min, K. Kim and S. Jon, Angew. Chem., Int. Ed., 2008, 47, 5362–5365 CrossRef CAS PubMed.
- S. Sivagnanam, M. Basak, A. Kumar, K. Das, T. Mahata, P. Rana, A. S. Sengar, S. Ghosh, M. Subramanian, A. Stewart, B. Maity and P. Das, ACS Appl. Bio Mater., 2021, 4, 6807–6820 CrossRef CAS PubMed.
- S. Sritharan and N. Sivalingam, Life Sci., 2021, 278, 119527 CrossRef CAS PubMed.
- B. Maity, A. Stewart, Y. O'Malley, R. W. Askeland, S. L. Sugg and R. A. Fisher, Carcinogenesis, 2013, 34, 1747–1755 CrossRef CAS PubMed.
- J. Yang, L. T. Platt, B. Maity, K. E. Ahlers, Z. Luo, Z. Lin, B. Chakravarti, S.-R. Ibeawuchi, R. W. Askeland, J. Bondaruk, B. A. Czerniak and R. A. Fisher, Oncotarget, 2016, 7, 69159–69172 CrossRef PubMed.
- J. Huang, J. Yang, B. Maity, D. Mayuzumi and R. A. Fisher, Cancer Res., 2011, 71, 6310–6319 CrossRef CAS PubMed.
- V. DiGiacomo, M. Maziarz, A. Luebbers, J. M. Norris, P. Laksono and M. Garcia-Marcos, Sci. Signaling, 2020, 13, 1–18 CrossRef PubMed.
- G. Spizzo, D. Fong, M. Wurm, C. Ensinger, P. Obrist, C. Hofer, G. Mazzoleni, G. Gastl and P. Went, J. Clin. Pathol., 2011, 64, 415–420 CrossRef PubMed.
- L. Sun, D. Liu, D. Fu, T. Yue, D. Scharre and L. Zhang, Chem. Eng. J., 2021, 405, 126733 CrossRef CAS.
- J. Song, C. Yuan, T. Jiao, R. Xing, M. Yang, D. J. Adams and X. Yan, Small, 2020, 16, 1907309 CrossRef CAS PubMed.
- Z. Zhang and S. S. Feng, Biomaterials, 2006, 27, 4025–4033 CrossRef CAS PubMed.
- M. Basak, T. Mahata, S. Chakraborti, P. Kumar, B. Bhattacharya, S. K. Bandyopadhyay, M. Das, A. Stewart, S. Saha and B. Maity, Antioxid. Redox Signaling, 2020, 32, 766–784 CrossRef CAS PubMed.
- A. Bonetti, S. Pellegrino, P. Das, S. Yuran, R. Bucci, N. Ferri, F. Meneghetti, C. Castellano, M. Reches and M. L. Gelmi, Org. Lett., 2015, 17, 4468–4471 CrossRef CAS PubMed.
- M. Tyagi, B. Maity, B. Saha, A. K. Bauri, M. Subramanian, S. Chattopadhyay and B. S. Patro, Food Funct., 2018, 9, 5715–5727 RSC.
- S. Chakraborti, A. Pramanick, S. Saha, S. Sarkar, L. P. Singh, A. Stewart and B. Maity, Free Radical Biol. Med., 2020, 160, 125–140 CrossRef CAS PubMed.
- B. Maity, J. Yang, J. Huang, R. W. Askeland, S. Bera and R. A. Fisher, J. Biol. Chem., 2011, 286, 1409–1419 CrossRef CAS PubMed.
- S. Chakraborti, A. Pramanick, S. Saha, S. S. Roy, A. R. Chaudhuri, M. Das, S. Ghosh, A. Stewart and B. Maity, Cancer Res., 2018, 78, 428–441 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details including synthesis and characterization, DLS measurement, EDS analysis, concentration and temperature dependent fluorescence emission spectrum, UV-Vis analysis and XRD analysis, effects of PS1-Zn and PS2-Zn and encapsulation of Dox, effect of PS2-Zn-EPCAM aptamer on A549 cells and AC16 cells, and effects of the test compounds on HCT116 cells. See DOI: 10.1039/d1na00885d |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2022 |