
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZnO nanostructured materials and their potential applications: progress, challenges and perspectives
Sauvik
Raha
and
Md.
Ahmaruzzaman
 *
*
Department of Chemistry, National Institute of Technology Silchar, 788010, Assam, India. E-mail: mda2002@gmail.com
First published on 9th March 2022
Abstract
Extensive research in nanotechnology has been conducted to investigate new behaviours and properties of materials with nanoscale dimensions. ZnO NPs owing to their distinct physical and chemical properties have gained considerable importance and are hence investigated to a detailed degree for exploitation of these properties. This communication, at the outset, elaborates the various chemical methods of preparation of ZnO NPs, viz., the mechanochemical process, controlled precipitation, sol–gel method, vapour transport method, solvothermal and hydrothermal methods, and methods using emulsion and micro-emulsion environments. The paper further describes the green methods employing the use of plant extracts, in particular, for the synthesis of ZnO NPs. The modifications of ZnO with organic (carboxylic acid, silanes) and inorganic (metal oxides) compounds and polymer matrices have then been described. The multitudinous applications of ZnO NPs across a variety of fields such as the rubber industry, pharmaceutical industry, cosmetics, textile industry, opto-electronics and agriculture have been presented. Elaborative narratives on the photocatalytic and a variety of biomedical applications of ZnO have also been included. The ecotoxic impacts of ZnO NPs have additionally been briefly highlighted. Finally, efforts have been made to examine the current challenges and future scope of the synthetic modes and applications of ZnO NPs.
1. Introduction
Found in the Earth's crust as the mineral zincite, ZnO appears as a white powder and is nearly insoluble in water. Zinc oxide is the second most abundant metal oxide after iron. It is inexpensive and safe, and can be prepared easily. The ZnO powder has been widely used as an additive in a plethora of materials and products including ceramics, glass, cement, rubber, lubricants, paints, ointments, adhesives, plastics, sealants, pigments, foods, batteries, ferrites and fire retardants. ZnO can assume three possible polymorphs, rock salt, wurtzite and zinc blende. The rock salt polymorph exists under conditions of high pressure and is therefore very rare. At ambient pressure and temperature, ZnO has a hexagonal wurtzite crystal structure with two lattice parameters, a and c, with values of 0.3296 nm and 0.52065 nm, respectively. In hexagonal wurtzite, O2− ions occupy the hexagonal close packing (hcp) array of lattice sites while Zn2+ ions place themselves in alternate tetrahedral holes in contrast with the zinc blende structure wherein O2− ions form a cubic close packing (ccp) array with Zn2+ ions occupying tetrahedral voids. ZnO has a wide band gap of ∼3.37 eV and a large exciton binding energy of 60 meV. The physical and chemical behaviors of zinc oxide nanoparticles are simply tuned by changing the morphology using various synthesis routes and different materials.ZnO has a slew of unique chemical and physical properties, viz., high chemical stability, high electrochemical coupling coefficient, broad range of radiation absorption and high photostability, which make it among all metal oxides a key technological material and confer upon it its wide applications in varied fields. ZnO is categorized as a group II–VI semiconductor in materials science because zinc belongs to the 2nd group while oxygen belongs to the 6th group of the periodic table. Its covalence is on the borderline demarcating ionic and covalent semiconductors. Besides, it has good transparency, high electron mobility, an outsized exciton binding energy (60 meV), wide band gap (3.37 eV),1 strong room temperature luminescence, high thermal and mechanical stability at room temperature, broad range of radiation absorption and high photostability that make ZnO the most favorite multitasking material.2,3,5,6 As a result of its distinctive optical and electrical properties4 it is considered to be a possible material in electronic applications, optoelectronic applications and laser technology. ZnO among nano-sized metal oxides has also been further extensively exploited to derive possible benefits from its antimicrobial and antitumor activities.7 Because of its blocking and absorbing capabilities ZnO finds inclusion in some cosmetic lotions.8 ZnO can also be used in human medicine as an astringent (for wound healing), and to treat hemorrhoids, eczema and excoriation.9 ZnO nanoparticles have recently attracted attention owing to their unique features. There are numerous promising applications of ZnO nanoparticles in veterinary science due to their wound healing, antibacterial, antineoplastic and antigenic properties. Recently, many research studies and experimental analyses have improved the efficiency of zinc oxide (ZnO) materials by producing nano-structures where each nano-dimension is reduced to generate nanowires, thin films and other structures for plenty of applications including defense against intracellular pathogens and brain tumors.10 One-dimensional structures include nanorods,11–13 nanoneedles,14 nanohelixes, nanosprings, nanorings,1 nanoribbons,15 nanotubes,16–18 nanobelts,19 nanowires20–22 and nanocombs.23 Nanoplates/nanosheets and nanopellets24,25 are their two-dimensional forms while flowers, dandelions, snowflakes, coniferous urchin-like structures, etc.26–29 count as the three-dimensional morphologies of ZnO nanoparticles. Nevertheless, the challenges in terms of the potential toxic effects of ZnO nanoparticles do require special attention.
2. Chemical methods for synthesis of zinc oxide nanoparticles
A range of precursors and a variety of conditions such as temperature, time, concentration of reactants and so forth can be employed for the chemical synthesis of ZnO nanoparticles. These chemical techniques by virtue of adjustment of the various aforesaid parameters allow better control of morphology in terms of size and geometry. ZnO NPs can be chemically synthesized using the following techniques: the mechanochemical process, controlled precipitation, sol–gel method, vapour transport method, solvothermal and hydrothermal methods, and methods using emulsion and micro-emulsion environments (Fig. 1). The details giving the gist of the various aforementioned strategies are presented in Table 1. The detailed synthesis conditions, experimental variables, main mechanisms, properties and applications are highlighted (Table 1). The different chemical methods used for the synthesis of ZnO NPs are enumerated below.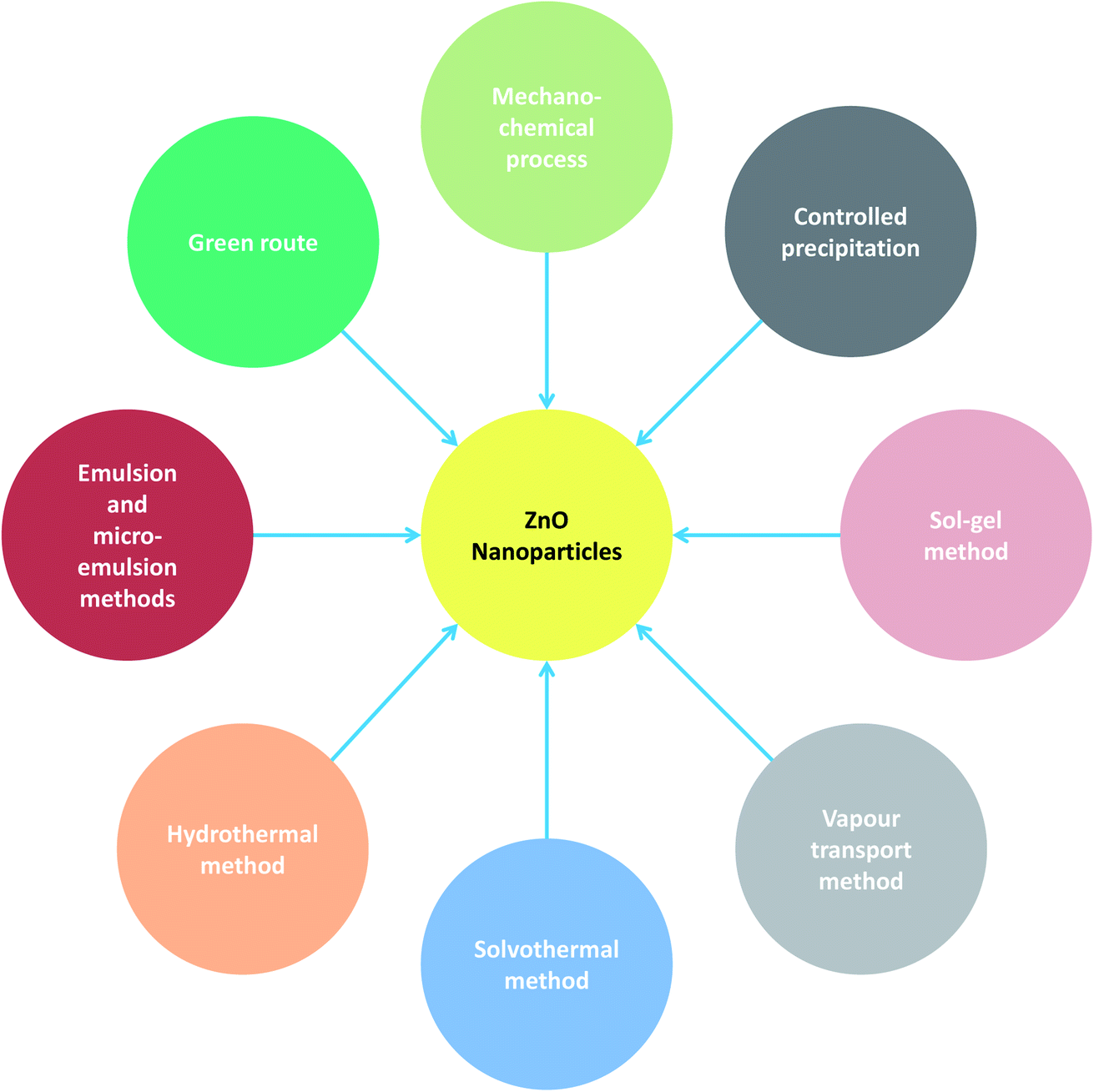 | ||
| Fig. 1 Various strategies for the fabrication of ZnO NPs. | ||
| Chemical methods of synthesis | Precursors | Synthesis conditions | Experimental variables | Main mechanisms | Properties and applications | Advantages | Ref. |
|---|---|---|---|---|---|---|---|
| Mechanochemical process | ZnCl2, Na2CO3 and NaCl | Calcination, 2 h, 600 °C, milling for 2–6 h | Milling time and heat-treatment temperature on ZnO nanocrystallite sizes | ZnO nanocrystallite growth is homogeneous, crystal nuclei were formed with decomposition of ZnCO3 and grew by emergence of the secondary formed ZnO. The driving force of the interfacial reaction came from the activation energy. Higher activation energy above 600 °C leads to a higher growth rate for the ZnO nanocrystallite | Hexagonal structure; particle diameter: 21–25 nm | Simplicity, relatively low-cost equipment, large-scale production, and applicability for a variety of materials. Operates at room temperature, which increases safety and reduces energy utilization. Induces not only morphological and structural changes of the particles but also modifies their optical and electrical properties and prevents the agglomeration of the synthesized particles | 32 |
| ZnCl2 and oxalic acid | Calcination, 1 h, 400 °C, milling for 0.5–4 h | Oxalic acid and wet-milling conditions on the ZnO average particle size and morphology | — | Hexagonal structure; particle diameter: 1 μm to 50–90 nm | 33 | ||
| ZnCl2, NaCl and Na2CO3 | Calcination, 0.5 h, 300–450 °C, milling for 9 h | Calcination temperature on particle size and structural properties of ZnO nanoparticles | — | Hexagonal structure; particle diameter: 27.7–56.3 nm | 35 | ||
| Precipitation process | ZnSO4 and NH4OH | Reaction: 50–60 °C; drying: 60 °C, 8 h | — | — | Hexagonal structure; flakes, particle diameter: 30 nm | The precipitation method is an unsophisticated method. High quality of production typifies the method. The method further has the advantage of being monetarily cheap with high production yield | 37 |
| Zn(OAc)2·2H2O and NaOH | Reaction: 30 min, 75 °C; drying: Room temperature, overnight | — | On heating the solution of zincate ions, the molecules start to rearrange into hexagonal ZnO nanorods after growing along the 〈0001〉 direction. When the molecules got saturated, the ZnO nuclei grew to give rod shaped ZnO. Over time, these freshly formed nanorods deposited on the surface of formerly formed crystalline nanorods resulting in a leaf-like structure first and a number of such leaves came together in an ordered array which appeared as flower shaped ZnNSs | Hexagonal structure; flower shape (length of each petal did not exceed 800 nm); application: antimicrobial activity | 38 | ||
| Zn(OAc)2·2H2O, (NH4)2CO3, and polyethylene glycol | Drying: 100 °C, 12 h. ZnO (A): the dried precipitate was ball-milled for 1 h followed by calcination at 450 °C for 3 h to produce ZnO powder which was further ball-milled for 3 h. ZnO (B): the precipitate was ball-milled for an hour and then a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 powder and 1-butanol mixture was prepared which was dried by heterogeneous azeotropic distillation; calcination at 450 °C for 3 h :10 powder and 1-butanol mixture was prepared which was dried by heterogeneous azeotropic distillation; calcination at 450 °C for 3 h |
Reaction temperature and time, concentration of oleic acid | Hexagonal structure; ZnO (A): particle diameter is 40 nm. ZnO (B): particle diameter is 40 nm. Photocatalytic degradation of methyl orange dye | 39 | |||
| ZnCl2, NH4OH, and CTAB | Aging: 96 h, ambient temperature, calcination: 2 h, 500 °C | Particle formation is a very complex process and involves nucleation, growth, coagulation and flocculation. Addition of surfactant CTAB affects the nucleation during the crystallization process. After nucleation, the surfactant can influence particle growth, coagulation and flocculation | Zincite structure; particle diameter: 54–60 nm, BET = ∼17 m2 g−1 | 41 | |||
| Zn(NO3)2, NaOH, SDS, and TEA (triethanolamine) | Precipitation: 50–55 min, 101 °C | Addition of sodium dodecyl sulfate (SDS) and triethanolamine (TEA) | Dissolution–reprecipitation mechanism | Wurtzite structure, rod-like shape (L: 3.6 μm, D: 400–500 nm), nut-like and rice-like shapes, size: 1.2–1.5 μm | 42 | ||
| Sol–gel | Zn(OAc)2·2H2O, polyvinyl pyrrolidone (PVP) and NaOH | Reaction: 60 °C; vigorous stirring for 1 h. Calcination: 600 °C, 1 h | Wurtzite structure; platelet-like ZnO with a grain size of 150 nm transformed into rod-shaped ZnO with a diameter of 100 nm at 3 × 10−4 M PVP | Sol–gel shows many advantages over other techniques such as its simplicity and low equipment cost | 43 | ||
| Zn(OAc)2·2H2O and oxalic acid | Reaction: refluxed at 50 °C, 1 h; drying at 80 °C for 20 h; calcination: 650 °C, 4 h | Wurtzite structure; uniform, spherically shaped ZnO nanoparticles with a crystallite size of 20 nm; BET surface area of 10 m2 g−1; 69.75% degradation of phenol and 67.98% degradation of benzoic acid in 120 min under UV light | 44 | ||||
| Zinc 2-ethylhexanoate, 2-propanol, and tetramethylammonium (TMAH) | Reaction: room temperature; aging: 30 min; drying: 60 °C | Weight ratio of 2-propanol and tetramethylammonium (TMAH) | Cylinder-shaped crystallites, diameter: 25–30 nm; height: 35–45 nm | 46 | |||
| Vapour transport method | Zn and water vapour or oxygen | Heating: 1 h, 800 °C, pressure: 0.03–0.05 MPa, cooling rate: 7 °C per minute | Influence of the atmosphere | For crystal growth, after initial nucleation, the subsequent growth stage strongly governs the final morphology of the crystal. In O2 gas, the growth of ZnO is simply along the 〈001〉 direction due to the fastest growth kinetics in this direction and absence of side or reverse reactions | With H2O: nanoflowers constructed by tens of ZnO nanosheets with random orientations. With O2: hexagonal nanorod arrays, non-uniform sized nanorods | The vapour transport method has been emphasised because of the easy control of thicknesses, morphologies and crystal structures of ZnO films and nanostructures by varying the precursor gas, substrate temperature and substrate materials | 48 |
| ZnO powder | Heating in a horizontal tube furnace: 1350 °C, 30 min; deposition: 400–500 °C under an Ar pressure of 250 Torr | Due to the small thickness of the nanobelts, spontaneous polarization normal to the nanobelt leads to the growth of helical nanostructures. The mechanism for the helical growth is attributed to the consequence of minimizing the total energy contributed by spontaneous polarization and elasticity | Wurtzite; nanobelts with widths of 10–60 nm, thickness of 5–20 nm and lengths up to several hundreds of micrometers | 49 | |||
| Zn powder and O2 | Heating in a furnace at 450 °C, 550 °C, and 650 °C at a rate of 10 °C min−1, feeding O2 into the reaction zone at a rate of 5 mL min−1 for 30, 45, and 60 min after reaching a furnace temperature of 450 °C | Growth temperature and growth time | The growth mechanism of 1D ZnO nanostructures can be divided into three stages, as follows: first, the Zn vapor and catalytic Cu form liquid alloy droplets during the heating process at a certain temperature, representing the initial stage of the nucleation process. Second, crystal nucleation occurs upon gaseous species adsorption until supersaturation is reached, and the formed sites serve as nucleation sites on the substrate. Finally, the axial growth of the nanorods begins from these sites | 450 °C: ZnO nanorods with a diameter and length of 19–27 nm and 2.8 μm, respectively. 550 °C: ZnO nanorods with a diameter and length of 85 nm and 3.8 μm, respectively. 650 °C: ZnO nanorods with a diameter and length of 190–350 nm and 3.9 μm, respectively, covered with short nanorods with a diameter of 95 nm and length of 900 nm at the tips. 30 min growth time: ZnO nanorods with a diameter of 19–27 nm and a length of 2.8 μm. 45 min growth time: ZnO nanorods with a diameter of 65–190 nm and a length of 3.2 μm. 60 min growth time: ZnO nanorods with a diameter of 80–250 nm and a length of 3.8 μm | 50 | ||
| Zn and O2 | Heating in a furnace at 750 °C, feeding O2 into the reaction zone at a rate of 50 mL min−1 for 15 min | Gas flow rate, growth temperature, position from the zinc source, and reaction time can affect the size, morphology, and density of the zinc oxide nanostructures | The growth mechanism of zinc oxide tetrapods is believed to occur by growth of four wurtzitic arms from an octahedral zinc-blende embryo, each at a 109.5° angle from the adjacent one. The tapered ends of some of the tetrapod arms indicate continued growth of zinc oxide when the oxygen flow had been turned off but residual oxygen remains in the growth chamber | ZnO nanotetrapods: arm lengths, 0.5–3.5 μm and diameters of 120–350 nm | 51 | ||
| Zn and O2 | Heating in a furnace at 700 °C, 800 °C and 900 °C, 50 sccm of oxygen flow for 2 h | Different evaporation temperatures | Wurtzite; ZnO tetrapods with an arm diameter of 22 nm and length of 90 nm. ZnO tetrapods have excellent supercapacitive performance. The maximum capacitance is 160.4 F g−1 at a current density of 1.0 A g−1. Excellent capacitance retention of 94.3% over 1000 cycles | 52 | |||
| Hydrothermal method | ZnCl2 and NaOH | pH 5–8 | Reaction temperature and template agents (organic compounds) | As temperature was increased, the ZnO particle morphologies changed | The hydrothermal technique is a promising alternative synthetic method because of the low process temperature and great ease of controlling the particle size. The hydrothermal process has several advantages over other growth processes such as use of simple equipment, catalyst-free growth, low cost, large area uniform production, environment friendliness and less hazardous nature. The low reaction temperatures make this method an attractive one for microelectronics and plastic electronics. This method has also been successfully employed to prepare nanoscale ZnO and other luminescent materials. The particle properties such as morphology and size can be controlled via the hydrothermal process by adjusting the reaction temperature, time and concentration of precursors | 53 | |
| 100 °C | 10 h | Bullet-like; 100–200 nm | |||||
| 160 °C | 6 h | Rod-like; 100–200 nm | |||||
| 180 °C | 6 h | Sheets; 50–200 nm | |||||
| 200 °C | 6 h | Polyhedra; 200–400 nm | |||||
| 220 °C | 5 h | Crushed stone-like; 50–200 nm | |||||
| Zn(OAc)2·2H2O, NaOH and methanol | 100–200 °C; 6–12 h; 0.2–0.5 M NaOH | Concentration of precursors (NaOH), reaction temperature and growth time | With 0.3 M NaOH and employing a growth time of 6 h the grain size was found to increase from 7 nm to 16 nm with temperature rise from 100 °C to 200 °C. The average grain size of ZnO synthesized at 200 °C for 12 h revealed an increase from 12 nm to 24 nm with elevation in concentration of NaOH from 0.2 M to 0.5 M | 54 | |||
| Solvothermal method | ZnSO4, NaOH, Na2CO3 and stearic acid; using the resulting ZnO nanoparticles as precursors | Reaction temperature: 60 °C; water–ethanol medium in an autoclave at 180 °C for 72–186 h | The ZnO wurtzite phase was formed. Average grain diameter of 27 nm using the Scherrer formula | 56 | |||
| 180 °C: 72 h | Precursors and time | The process appears to occur via an agglomeration/melting mechanism and leads to nanoneedles of relatively large dimensions | Nanoneedles with a diameter of 450–900 nm, length of 8–20 nm and aspect ratio of 0.05 | ||||
| 180 °C: 168 h | The formation mechanism of one-dimensional nanostructures does appear to be related more to a rolling-up/surfactant-segregation process than with the characteristic ZnO crystallite growth | Nanorods with a diameter of 40–160 nm, length of 5–8 nm and aspect ratio of 0.014 | |||||
| 180 °C: 168 h | Nanowires with a diameter of 30–50 nm, length of 0.8–1 nm and aspect ratio of 0.04. Photocatalytic activities with respect to the degradation of methylene blue | ||||||
| Zn powder, trimethylamine N-oxide and 4-picoline N-oxide in organic solvents | Reaction: 24–100 h, 180 °C | Oxidants and solvents, trace amount of water in solvent | ZnO rod-like and particle-like nanostructures with diameters ranging in between 24 and 185 nm | 57 | |||
| Emulsion or microemulsion method | Zinc oleate in decane and NaOH in water or ethanol | Stirring: 2 h, room temperature or 90 °C, and maintaining the decane/water interface during stirring | Morphologies obtained: spherical agglomerates, needle shapes, near-hexagonal shapes, near-spherical shapes and irregular agglomerates. Diameters obtained: 2–10 μm, 90–600 nm, 100–230 nm and ∼150 nm | 58 | |||
| Zinc acetate and KOH or NaOH. Cyclohexane as an organic phase, and nonylphenyl polyoxyethylene glycol ethers as a mixture of emulsifiers in emulsion formation | Stirring: 9000 rpm; destabilization: 80 °C; drying at 120 °C | Concentration of Zn(CH3COO)2 solution. Precipitating agent. Amount of zinc acetate/cyclohexane (cm3). Dosing rate of KOH (or NaOH) to Zn(CH3COO)2 (cm3 min−1) | Morphologies such as solids (Z1), ellipsoids (Z2), rods (Z3) and flakes (Z4) with modal diameters of ∼396 nm, ∼396 nm, ∼1110 nm and ∼615 nm. Values of 8 m2 g−1, 10.6 m2 g−1, 12 m2 g−1 and 23 m2 g−1 could be respectively assigned to samples Z1, Z2, Z3, and Z4 | 59 |
2.1 Mechanochemical process
In the mechanochemical process the starting materials are subjected to a high-energy dry milling which sets off a reaction by way of ball–powder impacts in a ball mill, at a low temperature. A diluent, often the by-product of the reaction (salt viz. NaCl), accompanies the starting materials during the procedure.30,31 The diluent functions as a reaction medium and effects separation of the nanoparticles thereby restricting their subsequent growth. This in turn prevents serious agglomeration. The starting materials conventionally used in this method are mainly anhydrous ZnCl2, Na2CO3 and NaCl. The following reactions take place during the procedure:| ZnCl2 + Na2CO3 → Zn CO3 + 2NaCl |
| ZnCO3 → ZnO + CO2 |
Ao et al.32 carried out a mechanochemical process of synthesizing ZnO NPs by exploiting the reaction between ZnCl2 and Na2CO3 and using NaCl as a diluent.32 The pure nanocrystalline ZnO was obtained by removing the by-product NaCl and finally drying in a vacuum. TEM images showed moderately aggregated ZnO nanoparticles of size less than 100 nm which were prepared by a 6 h milling followed by a thermal treatment at 600 °C for 2 h. The effect of milling time and annealing was carefully investigated in the study. A decrease in nanocrystallite size from 25 nm to 21.5 nm was observed as the milling time increased from 2 to 6 h after which it attained steadiness. This phenomenon was chalked up to a critical effect prevailing in the course of milling. The crystal size, however, was found to increase with temperature with the rise being steep after 600 °C. The activation energies for nanocrystallite growth in different temperature ranges were calculated using the Scott equation. The activation energy was found to be 3.99 for growth in between 400 and 600 °C while it reached 20.75 kJ mol−1 beyond 600 °C. The higher growth rate at higher temperatures was thus attributed to extensive interfacial reactions driven by greater activation energy.
A reduction in particle size on prolonging the milling time was reported by Stanković et al.33 in their work of preparing ZnO NPs using ZnCl2 and oxalic acid as starting materials. Their scheme involved milling the powders of starting materials in a Retsch PM4 planetary ball mill using a ball-to-powder weight ratio of 10:1. Different intervals of time were chosen for milling to investigate the influence of milling time on particle size. Pastes of zinc oxalate thus obtained were then air dried at 100 °C for 1 h and then subjected to thermal decomposition at 450 °C to obtain ZnO NPs.
| ZnCl2 + H2C2O4·2H2O → ZnC2O4·2H2O + 2HCl |
| ZnC2O4·2H2O + 0.5O2 → ZnO + 2H2O + 2CO2 |
While the XRD analysis substantiated a perfect long-range order and a pure wurtzite structure of the synthesized ZnO powders regardless of the milling time, Raman spectroscopy revealed that lattice defects and impurities were introduced into ZnO powders at the middle-range scale depending on milling duration. Extended milling was found to reduce crystal defects but introduce impurities. The SEM images suggested that the milling duration of the reactant mixture positively regulated the morphology of the particles irrespective of the additional thermal treatment.
ZnO NPs were also prepared through a mechanochemical method by using ZnCl2, NaCl and Na2CO3 as starting materials.34 A solid phase reaction triggered by milling the starting powders led to the isolation of ZnCO3 in the NaCl matrix. The ZnCO3 was finally subjected to a thermal treatment at 400 °C which induced its decomposition to ZnO. The anatomization of TEM results indicated a mean particle size of 26.2 nm. The mean nanocrystallite size evaluated from the XRD peak width at 2θ = 36° using the Scherrer equation was found to be 28.7 nm. Meanwhile, the surface area of the ZnO nanopowder evaluated from BET analysis was 47.3 m2 g−1 corresponding to a spherical particle size of 27 nm.
Another study on the optical properties of ZnO NPs synthesized through mechanochemical means and using ZnCl2, NaCl and Na2CO3 as raw materials was conducted by Moballegh et al.35 The XRD and TEM results revealed that particle size increased with calcination temperature. The work proposed improved optical properties as a result of the decrease in particle size owing to the enhanced ratio of surface to volume in ZnO NPs. In another study36 a mixture of starting powders (anhydrous ZnCl2, Na2CO3 and NaCl) was milled at 250 rpm and then calcined at 450 °C for 0.5 h to yield ZnO NPs with a crystallite size of 28.5 nm as estimated from subsequent XRD analysis. The particle size that emerged from TEM and SEM analysis ranged in between 20 and 30 nm. The incongruent particle size estimated from BET analysis was ascribed to an agglomeration of nanoparticles in the course of drying.
The foremost shortcoming of the procedure exists in its fundamental difficulty encountered in the homogeneous grinding of the powder and controlled minimization of the particles to the required size. Note that the particle size reduces with increasing time and intensity of milling. However, if the powder is subjected to milling for longer periods of time, the chances of contamination increase. A highly shrunk size of nanoparticles is the prime advantage that can be extracted from the method apart from the benefit of a significantly low cost of generation coupled with diminished agglomeration of particles and pronouncedly homogeneous crystallite morphology and architecture. The mechanochemical process is particularly desirable for large-scale production of ZnO NPs.
2.2 Controlled precipitation
Controlled precipitation is often resorted to when the idea is to set off a large-scale generation of nanoparticles in the powder form with reproducible properties for their use as industrial products. The method involves two stages: (a) precipitation of a precursor of ZnO from the solution, and (b) thermal treatment of this precursor with subsequent milling to induce physical disintegration of the calcined agglomerates and to get rid of impurities. Various methods may be employed for precipitation of ZnO by exercising control over a range of limiting factors that include reaction temperature, pH conditions, duration of precipitation and the medium used for washing. Using ZnSO4 and NH4OH, Sadraei37 prepared nano-ZnO flakes. Nano-sized ZnO flakes as confirmed by SEM analysis were eventually achieved. XRD analysis of the flakes revealed the hexagonal structure of ZnO flakes with high crystallinity and a diameter of 30 nm.Kumar et al.38 used zinc acetate (Zn(OAc)2·2H2O) and NaOH as reagents, and the settled white powder was separated followed by washing with deionized water thrice and dried overnight under dust-free conditions at room temperature. XRD revealed the formation of hexagonal ZnO nanostructures. SEM and TEM analyses revealed the formation of crystalline ZnO flowers in which a bunch of ZnO nanorods assembled together to form a leaf-like structure followed by flower-shaped ZnO nanostructures. The ZnO nanoflowers were each formed by the combination of 8–10 leaf-like petals as shown. The length of each petal did not exceed 800 nm. The as-synthesized ZnO nanostructures showed good antimicrobial activity towards Gram-positive bacteria Staphylococcus aureus as well as Gram-negative bacteria Escherichia coli with a MIC/MBC of 25 mg L−1. Zn(CH3COO)2·2H2O and (NH4)2CO3 were employed as reagents by Hong et al.39 in their method of synthesizing ZnO NPs. XRD and TEM tests revealed particle sizes of 40 and 30 nm. Heterogeneous azeotropic distillation thoroughly prevents agglomeration and reduces the size of ZnO NPs.
Raoufi et al.40 used the aqueous solutions of Zn(NO3)2 and (NH4)2CO3 as reagents in their experiments. The precursors were synthesized by adding Zn(NO3)2 solution in a drop-wise manner into (NH4)2CO3 solution under stirring. The molar ratio of Zn(NO3)2 to (NH4)2CO3 was maintained at 1:1.5. The precipitate obtained was filtered. It was further repeatedly washed with deionized water and ethanol. Subsequently, it was subjected to drying at 100 °C for 6 h. The precursor was annealed at temperatures ranging in between 250 and 550 °C for 4 h to obtain ZnO NPs. XRD analysis demonstrated an enhancement in crystallinity with a rise in annealing temperature. Using the Scherrer equation, the crystallite size was however found to increase from 8.34 to 27.58 nm as the annealing temperature was increased from 250 to 550 °C. The mean crystallite size obtained from BET study also confirmed an increase from 8.60 to 42.51 nm upon an increase in annealing temperature from 250 to 550 °C. The conspicuous incongruence of crystallite size caused agglomeration with a rise in temperature due to the increased growth rate.
In the precipitation method of synthesizing nanopowders, it is more or less a ritual these days to use surfactants that would enable control over the growth of particles with the simultaneous prevention of coagulation and flocculation of particles thereby preventing an appreciable reduction in the final yield. The surfactants act as chelates encapsulating the metal ions in an aqueous medium. Wang et al.41 used ZnCl2 and NH4OH and a cationic surfactant, CTAB (cetyltrimethyl-ammonium bromide), for the generation of ZnO NPs. The formation of sharply crystalline ZnO NPs with a wurtzite structure and crystallite size of 40.4 nm was confirmed by XRD data, while TEM examination of the powder bore out the formation of spherical nanoparticles of size 50 nm.
Nano-sized crystals of ZnO possessing a wide variety of shapes were generated using Zn(NO3)2·6H2O and NaOH by Li et al.42 with sodium dodecyl sulfate (SDS) and triethanolamine (TEA) as cationic surfactants. These surfactants were found to exert great influences on the shape and size of the ZnO NPs. Heating a Zn(OH)2 precursor in H2O solvent at 101 °C for 4 h gave rod-like ZnO crystals, with diameters ranging from 400 to 500 nm and an average length of about 3.6 mm (aspect ratio 8:1). When 0.01 M SDS was added into the reaction solution (keeping the other conditions identical), nut-like ZnO crystals with a twin structure were obtained. The diameter of the hexagonal cross-section ranged from 1.2 to 1.5 mm, and the aspect ratio was a little smaller than 1:1. A new well-defined rice-like morphology of ZnO appeared when 0.1 M TEA replaced 0.01 M SDS in the reaction solution. The diameters of the particles were similar to those of nut-like ZnO crystals, while the aspect ratio was slightly larger than 1:1. The precipitation method is an unsophisticated method. High quality of production typifies the method. The method further has the advantage of being monetarily cheap with high production yield.
2.3 Sol–gel method
The synthesis of ZnO NPs by the sol–gel method has recently gained widespread use. Its ease, effortlessness, soundness, repeatability and relatively mild conditions have resulted in its extensive use in the generation of metal-oxide nanoparticles. This method facilitates the surface modification of zinc oxide with selected organic substances, thereby altering its traits and attributes and expanding its realm of applications.Suwanboon et al.43 using Zn(CH3COO)2·2H2O, polyvinyl pyrrolidone (PVP) and NaOH prepared nano-structured ZnO crystallites via the sol–gel method. The XRD characterization revealed a wurtzite structure having an average crystallite size of about 45 nm. The role of PVP at its different concentrations on the morphology was checked. There occurred a shift from a platelet-like to a rod shape with an increase in PVP concentration. TEM images bore out the grain size of platelet-like ZnO to be 150 nm while the diameter of the rod-shaped ZnO was likewise determined to be 100 nm. In another sol–gel method-based synthesis by Benhebal et al.44 zinc acetate dihydrate and oxalic acid were used to generate ZnO nanopowder with ethanol as a solvent which showed a hexagonal wurtzite structure. The crystallite size obtained from the Scherrer equation was found to be 20 nm. The SEM micrograph confirmed the formation of uniform, spherically shaped ZnO nanoparticles. BET analysis revealed a surface area of 10 m2 g−1. This was characteristic of a material with low porosity, or a crystallized material.
Sharma45 obtained ZnO NPs with outstanding antibacterial properties using the sol–gel method. Zinc acetate, oxalic acid and water were employed as raw materials in this process. A white gel precipitate was first obtained. It was then thermally treated at 87 °C for 5 h, and then at 600 °C for 2 h. The ZnO NPs exhibited high crystallinity as borne out by XRD data. A diameter of 2 μm was obtained for the ZnO nano-aggregates from SEM analysis.
In a study conducted by Ristic et al.46 nano-structured ZnO crystallites were obtained using the sol–gel route. From XRD examination and using the Scherrer formula, the average value of the basal diameter of the cylinder-shaped crystallites was found to be 25–30 nm, while the height of the crystallites was 35–45 nm. The sol–gel method presents a host of advantages in comparison with the previously mentioned methods. Prime amongst its merits are the low cost of the apparatus and raw materials, reproducibility and flexibility of generating nanoparticles.47
2.4 Vapour transport method
In this process, zinc and oxygen or oxygen mixture vapours are transported and are allowed to react with each other resulting in the formation of ZnO nanoparticles. The vapour transport method has been emphasized because of the easy control of thicknesses, morphologies and crystal structures of ZnO films and nanostructures by varying the precursor gas, substrate temperature and substrate materials.48 Kong and Wang49 reported free-standing ZnO nanobelts that grew along the a-axis, and their large top and bottom surfaces were the polar (0001) facets. The structurally controlled, high-purity and high-yield ZnO nanobelts were synthesized by a solid–vapor process. SEM and TEM analyses show that the product has a belt shape with widths of 10–60 nm, thickness of 5–20 nm and lengths up to several hundreds of micrometers. The dominant component of the as-synthesized sample is nanobelts with a uniform size distribution, but a significant number of nanobelts have a ring shape, which has not been found for single crystal nanobelts or nanowires of any other materials.ZnO nanoparticles were also prepared on Au-coated Si substrates by Wu et al.48 using water vapour and oxygen gas. The advantage of using a Au catalyst is the fast growth rate of the oxide material because of the catalytic effect of Au, but the main disadvantage is the remains of the Au catalyst, which may influence the purity of the ZnO nanostructures. Au covered Si substrates were prepared by depositing a 200 nm thick layer of Au film on cleaned Si(100) wafers using radio frequency sputtering (JC500-3/D) from an Au target. The synthesis procedure included the following steps: (a) flowing Ar gas through the quartz tube for 0.5 h to remove the air in it; (b) increasing the furnace temperature to 800 °C with the Ar gas flow; (c) maintaining the temperature at 800 °C for 1 h and replacing the Ar flow by a mixture of either Ar/H2O vapour or Ar/O2 gas. The pressure of the quartz tube is maintained at 0.03–0.05 MPa; (d) switching off the furnace and cooling down the sample to RT at a rate of 7 °C per minute. The reactions of metallic Zn with H2O vapour and O2 gas can be written, respectively, as follows:
| Zn + H2O → ZnO + H2 |
| 2Zn + O2 → 2ZnO |
In water vapour, ZnO nanoflowers were synthesized. The nanoflowers were constructed from tens of ZnO nanosheets with random orientations. In oxygen gas, ZnO hexagonal nanorods were obtained. The size of the nanorods was not uniform. It was argued that the size of the Au catalyst underneath might have influenced the size of the ZnO nanorods. Both the samples, however, exhibited a hexagonal wurtzite structure. Though the samples showed different morphologies and crystal structures, surprisingly, they had almost the same optical properties. The PL spectra revealed only one UV peak close to 389 nm wavelength for both samples, indicating the high quality of the synthesized ZnO samples.
Novel one-dimensional single-crystalline ZnO nanorod and nanoneedle arrays on a Cu catalyst layer-coated glass substrate were investigated by Alsultany et al.50via a simple physical vapour deposition method by thermal evaporation of Zn powder in the presence of O2 gas. The ZnO nanorods and nanoneedles were synthesized along the c-axis growth direction of the hexagonal crystal structure. The diameter and growth rate of the high-quality and well oriented one-dimensional ZnO nanostructures were achieved as a function of varying growth temperature and growth time. At 450 °C, ZnO nanorods were uniformly distributed at a high density on the entire substrate surface and quasi-aligned, and small average diameters were obtained. The diameters and lengths of the obtained nanorods were in the range of 19–27 nm and 2.8 μm, respectively. When the temperature was increased to 550 °C, ZnO nanorods grew perpendicular to the substrate, uniformly throughout their length, and with more consistent shape and dimensions, with approximately 85 nm width and 3.8 μm length. The morphological change and distribution occurred at a growth temperature of 650 °C, and ZnO nanorods with a hexagonal shape at the tips of rods of hexagonal hierarchical structures were formed. These rods possessed a typical hierarchical structure with lengths and diameters of approximately 190–350 nm and 3.9 μm, respectively, whereas short nanorods with a diameter of 95 nm and length of 900 nm were observed on the tip of each rod of hexagonal hierarchical structures. As Cu metal catalysts were used in the study, the growth mechanism of 1D ZnO nanostructures presented therein followed the VLS method. This method could be divided into three stages, as follows: first, the Zn vapor and catalytic Cu formed liquid alloy droplets during the heating process at a certain temperature, representing the initial stage of the nucleation process. Second, crystal nucleation occurred upon gaseous species adsorption until supersaturation was reached, and the formed sites served as nucleation sites on the substrate. Finally, the axial growth of the nanorods began from these sites. Based on this study of the mechanism in the presence of Cu metal catalysts at different growth temperatures and according to the nucleation theory of the VLS growth mechanism, the Cu catalyst nanoclusters formed because of capillarity, which caused beading of the Cu layer at high growth temperature. Consequently, the Cu–Zn alloy process reached a certain solubility depending on the temperature; then, the Zn vapor began to precipitate out at the interface between the surface and droplet. That in turn determined the diameter and size of the nanostructures depending on the size of the liquid alloy droplets. Notably, large-scale ZnO nanorods with a lower diameter were formed at a low growth temperature of 450 °C. The Zn metal powder (melting point of 419 °C) vapor pressure at 450 °C was sufficiently high to investigate the growth of ZnO nanorods on the glass substrate via the VLS method, and the decrease in Zn vapor as a result of the decrease in the growth temperature led to a low lateral growth rate compared with the axial growth rate of the 1D nanostructure. In contrast, the higher growth temperature could also lead to the formation of hierarchical nanostructures. In addition, at high growth temperature along with the consumption of the Zn vapor during growth, the diameter of the nanorods markedly decreased. This condition consequently caused the production of rods with a typical hierarchical structure. At a growth time of 30 min, ZnO nanorods were obtained with a diameter of 19–27 nm and a length of 2.8 μm. When the growth time increased to 45 min, nanoneedles were obtained. The needles exhibited mean diameters of 65–190 nm and length of 3.2 μm. On the other hand, nanoneedles grown at 60 min were approximately 80–250 nm in diameter and 3.8 μm in length.
Diep and Armani51 designed a flexible light-emitting nanocomposite based on ZnO nanotetrapods (NTPs) which they prepared using a vapour transport technique. The CVT synthesis of the ZnO NTPs was self-catalyzed. In the TEM images, the lattice fringes were clearly visible, indicating the single-crystalline nature of the nanostructures. The lattice spacing was found to be 2.6 Å, indicating growth in the [0001] direction. X-ray diffraction (XRD) and energy-dispersive X-ray spectroscopy (EDX) analysis were also performed to confirm the crystal structure and elemental composition of the NTPs. Based on an analysis of the TEM and SEM images, the ZnO NTP arm lengths ranged from 0.5 μm to 3.5 μm and the diameters varied from 120 nm to 350 nm.
Luo et al.52 also constructed ZnO tetrapods as potential electrode materials for low-cost and effective electrochemical supercapacitors using an oxidative-metal-vapor-transport method. The SEM images of the ZnO tetrapods collected at different temperatures showed that the products obtained were pure and uniform, and the tetrapods consist of four arms branching from one center, and the angles between the arms were nearly the same, analogous to the spatial structure of the methane molecule. As for the size variation with collected temperatures, it transpired that smaller size tetrapods were obtained with lower evaporation temperature. This demonstrated the power of the technique for controlling the size of the tetrapods. ZnO tetrapods with arms as thin as about 170 nm and shorter than 4000 nm were revealed by SEM analysis. The XRD pattern of the ZnO tetrapods showed that all the diffraction peaks could be indexed to a wurtzite 5 structure with lattice constants of a = 0.324 nm and c = 0.519 nm. The TEM and high resolution TEM (HRTEM) images of the ZnO tetrapods revealed that the arm diameter and length of the tetrapods are, on average, about 22 nm and 90 nm, respectively. The HRTEM image of a single arm revealed clear fringes perpendicular to the arm axis and these fringes were spaced by about 0.25 nm consistent with the interplanar spacing of (0002) suggesting that the nanowire growth direction was along [0001].
2.5 Hydrothermal method
The hydrothermal method is a technique often used for the easy generation of nanoparticles. This method of chemical synthesis bears an inherently favourable attribute of being harmless to the environment. Conventionally, the method involves the use of an autoclave. The substrates in the form of a mixture are placed within the autoclave and then subjected to a gradual heating up to a temperature of 100–300 °C. The heating continues over a specific period of time and cooling then follows. This causes the formation of crystal nuclei followed by nuclei growth.A 1:2 mixture of ZnCl2 and NaOH in an aqueous medium was employed by Chen et al.53 A white Zn(OH)2 precipitate was formed and hydrothermal treatment yielded ZnO NPs. XRD and TEM tests were conducted to ascertain the average size and morphology of the ZnO NPs so formed. Two factors, namely temperature and reaction time, were found to exert great effects on the size and architecture of ZnO NPs. On increasing the temperature, the particle morphology was found to change from rod-like to polyhedra. Further in the study, it was also revealed that different templates cast varying influences on the size and morphology of the nanostructured particles. Furthermore, a noticeable rise in crystallinity and lengthening of particle size with pH were recorded as a shortcoming of the procedure.
Aneesh et al.54 carried out an experiment in which they used Zn(CH3COO)2·2H2O, NaOH and methanol as reagents. The ZnO NPs thus formed had a hexagonal wurtzite structure. XRD analysis demonstrated an enhancement in average grain size with rising temperature and concentration of the substrates. The average grain size of ZnO NPs prepared from 0.3 M NaOH employing a growth time of 6 h was found to increase from 7 to 16 nm with temperature rise from 100 to 200 °C. The average grain size of ZnO synthesized at 200 °C for 12 h revealed an increase from 12 to 24 nm with elevation in concentration of NaOH from 0.2 M to 0.5 M.
This process has many advantages over other methods. Organic solvents do not find use in this process. This coupled with the omission of supplementary processes like grinding and calcination within the ambit of the method endows it with the much sought after eco-friendly character. Low operating temperatures, the diversified morphologies and sizes of the resulting nano-crystals depending on the composition of the starting mixture and the process temperature and pressure, the greatly pronounced crystallinity of the nanoparticles and their high purity are factors that surely make the process more advantageous than others.54,55
2.6 Solvothermal method
The prime objective of using the solvothermal technique of generating a wide range of nano-structured metal oxides is to achieve enormous surface area, marked crystallinity and great thermal stability. Thermal decomposition of an organometallic compound in an organic medium forms the basis of this procedure. A solvothermal method was adopted by Segovia et al.56 to fabricate ZnO NPs. The work resulted in the production of ZnO with a range of morphologies – nanoneedles, nanorods and nanowires – obtained under different solvothermal conditions. ZnSO4, NaOH, Na2CO3 and stearic acid were employed as chemical reagents while ethanol–water mixtures were used as solvents. Constant agitation of the reaction mixture consisting of ZnSO4, Na2CO3 and NaOH at 60 °C yielded the ZnO precursor. XRD data revealed the formation of the ZnO wurtzite phase. Estimation of particle size using the Scherrer formula approach indicated an average grain diameter of 27 nm. With ZnO nanoparticles as precursors and 180 °C thermal treatment carried out for 72 h, nanoneedles having a diameter of 450–900 nm, length of 8–20 nm and aspect ratio of 0.05 were formed. With ZnO nanoparticles and stearic acid as precursors and 180 °C thermal treatment carried out for 168 h, nanorods having a diameter of 40–160 nm, length of 5–8 nm and aspect ratio of 0.014 were formed. With the ZnO(stearic acid)0.38 nanocomposite as a precursor and 180 °C thermal treatment carried out for 168 h, nanowires having a diameter of 30–50 nm, length of 0.8–1 nm and aspect ratio of 0.04 were formed.Chen et al.57 also used a solvothermal route to generate ZnO NPs. They eventually prepared nano-structured ZnO crystals that were devoid of hydroxyl groups. They carried out a reaction of zinc powder with trimethylamine N-oxide (Me3N→O) and 4-picoline N-oxide (4-pic→NO). The medium for the reaction was a mixture of organic solvents (toluene, ethylenediamine (EDA) and N,N,N′,N′-tetramethylenediamine (TMEDA)) contained in an autoclave which was kept at 180 °C. It was observed that the size and morphology of the ZnO nanoparticles/nanowires were greatly influenced by the oxidants used and the ligating capacities of the solvents. The ramifications of the presence of water in the system were additionally investigated. It emerged that the presence of traces of water catalyzed the zinc/4-picN→O reaction and exerted an effect on the size of the nano-structured ZnO crystallites thus obtained. Depending on the reaction conditions, the ZnO nanostructures had diameters ranging in between 24 and 185 nm. The solvothermal synthesis method has many advantages. Foremost among them is the fact that reactions can be carried out under determined conditions. As a result, nano-structured ZnO with a range of architectures can be generated by exercising due control over the reaction conditions.
2.7 Method using an emulsion or microemulsion environment
Emulsion techniques have recently gained extensive use in preparing ZnO nanoparticles. Vorobyova et al.58 employed an emulsion to precipitate ZnO by allowing zinc oleate (mixed in decane) to react with sodium hydroxide (mixed in ethanol or water). The following reaction illustrates the entire process:| Zn(C17H33COO)2 (decane) + 2 NaOH → ZnO (water and ethanol) + H2O + 2NaC17H33COO |
SEM and XRD analysis showed that the particle size and phase location were both dependent upon the conditions (ratio of two-phase components, substrates and temperature) employed for the accomplishment of the process. Depending on the process conditions, ZnO NPs with different particle morphologies were obtained. The morphologies that formed during the process included spherical agglomerates, needle shapes, near-hexagonal shapes, near-spherical shapes and irregular agglomerates. These NPs further had a wide range of diameters. Some had diameters ranging in between 2 and 10 μm, while the diameters of others ranged from 90 to 600 nm, some others had diameters in between 100 and 230 nm and yet others were characterized by diameters hovering around 150 nm.
Kołodziejczak-Radzimska et al.59 used zinc acetate and KOH or NaOH in an emulsion system. For the generation of an emulsion, cyclohexane was utilized. Cyclohexane was held to have furnished a ready organic phase, and also essayed the role of a surfactant that wasn't ionic. In this method for emulsion formation cyclohexane was used as an organic phase, and nonylphenyl polyoxyethylene glycol ethers NP3 and NP6 were used as a mixture of emulsifiers. By tailoring the ZnO precipitation process by way of altering the precipitating agent, substrate ingredients and the tempo of substrate dosing, an amazing variety of ZnO nanostructures were designed. Four samples were obtained, labelled Z1, Z2, Z3, and Z4, composed of particles of different shapes. Morphologies such as solids (Z1), ellipsoids (Z2), rods (Z3) and flakes (Z4) with modal diameters of ∼396 nm, ∼396 nm, ∼1110 nm and ∼615 nm were obtained. They were further characterized by their considerable surface areas. Values of 8 m2 g−1, 10.6 m2 g−1, 12 m2 g−1 and 23 m2 g−1 could be respectively assigned to samples Z1, Z2, Z3, and Z4.
If a surfactant possessing balanced hydrophilic and lipophilic properties is used in the right proportion, a different oil and water system will be produced. The system remains an emulsion, but exhibits some characteristics that are different from emulsions. These new systems are “microemulsions”. The drop size in a microemulsion is significantly smaller than in an emulsion, and lies in the range 0.0015–0.15 μm.60,61 In contrast to emulsions, microemulsions form spontaneously under appropriate conditions. This synthesis method does not require any complex preparation procedure, sophisticated equipment or rigorous experimental conditions, but still provides possibilities in controlling the size and morphology of the ZnO powders in a size scale approaching nanometers. Even though the product yield is low, the narrow size distribution due to well-dispersed cage-like small reactors (5–100 nm) formed under uniform nucleation conditions is the superior aspect of the ZnO nanoparticles obtained by microemulsion routes. Such low-dimensional uniform ZnO nanostructures offering size and morphology dependent tunable electrical and optical properties are of particular technological interest for applications such as quantum dots, UV-emission optoelectronic and lasing devices, and transparent conducting thin films.
Li et al.62 prepared ZnO NPs by a facile process based on microemulsion. The synthesized ZnO NPs were marked by a wide range of morphologies and further exhibited significant optical properties. The microemulsion (ME) was generated by using a solution of heptane and hexanol in the mole ratio 3:1 as the oil phase and polyoxyethylene tert-octylphenyl ether (Triton X-100) as nonionic surfactant. A definite amount of Triton X-100 was put in the oil phase, and the mixture was subjected to stirring for a while up until the mixture became transparent. Thus, a microemulsion (ME) consisting of 0.20 M Triton X-100 in the oil phase was prepared. The next step entailed the preparation of two microemulsion solutions, namely ME-1 and ME-2, containing different reactants. 3 mL of a 0.25 M Zn(NO3)2 aqueous solution having various concentrations of PEG400 additive was added to 30 mL ME. This microemulsion solution was named ME-1. Likewise, ME-2 was obtained by adding 3 mL of a 0.5 M NaOH aqueous solution to 30 mL ME. Next, ME-1 was first added to ME-2 in a slow manner with constant stirring. This in turn resulted in a mixture that was subjected to hydrothermal treatment at 140 °C for 15 h in a 100 mL Teflon-lined autoclave. The autoclave was then left to cool down to room temperature by itself. Collection of precipitates was done by way of their centrifugation at 5000 rpm. This was followed by washing and then drying in air at 60 °C. XRD peaks confirmed the hexagonal wurtzite structure of the prepared ZnO NPs. The marked crystallinity of the nanoparticles was figured out from the sharpness of the diffraction peaks. The crystallite size was found to decrease from 40 to 27 nm with a rise in the concentration of PEG400 additive from zero to 50.0% in Zn(NO3)2 aqueous solution exhibiting the regulatory influence of microemulsions as well as the PEG400 additive on the nucleation and growth of ZnO NPs and their end architecture. TEM analysis further revealed that the morphology of the ZnO NPs underwent an alteration from needle-like to columnar and finally to spherical as the PEG400 concentration rose from zero to 50.0%.
Yildirim and Durucan63 also synthesized ZnO NPs through the use of microemulsions. They made an endeavour to reshape the microemulsion modus operandi with an eye to generate monodisperse ZnO nanostructures. They subjected the zinc complex precipitate obtained in the course of the microemulsion method to thermal decomposition. Subsequent calcination was adopted. The use of glycerol as the internal phase of a reverse microemulsion imparted the intended modification. The synthesized ZnO NPs had spherical shapes. They were monodisperse and their diameter measured in between 15 and 24 nm.
All the procedures involving chemical synthesis of ZnO NPs generate a few toxic chemicals and their adsorption on the surface increases the likelihood of harmful effects being wielded in medical applications. Further, these approaches include reactions requiring high temperature and intense pressure for their commencement while some reactions require operations in an inert atmosphere or under inert conditions. Toxic materials such as metallic precursors, toxic templates and capping agents and even H2S find application in quite a few chemical routes.64 Very often toxic substances are employed for the generation of nano-structured particles and for their stabilization as well. This in turn produces secondary products and residues that are detrimental to the ecosystem.65,66
3. Green methods for the synthesis of ZnO nanoparticles
The usual procedures adopting the chemical routes are costly. They often involve the use of chemical substances as reducing agents and these substances more often than not turn out to be toxic.67 Green routes of synthesis of nanoparticles cut down the risk of pollution at the originary level. These procedures lay greater focus on prevention of generation of wastes than their treatment or removal after they are formed. The principle emphasizes upon the choice of reagents which are eco-friendly. Thus, eco-friendly chemicals and safe solvents such as water are used alongside gentle natural extracts in such procedures. Although chemical methods are quick and easier for the generation of nanoparticles the biogenic techniques are superior and eco-friendly.68–70 Biogenic routes involving the use of extracts generated from microorganisms and plants have thus been preferred over chemical procedures of fabricating ZnO NPs. Besides, ZnO NPs obtained by way of green methods demonstrate pronounced biocidal activity against a host of pathogens in comparison with chemically generated ZnO NPs.71 Bio-inspired procedures of nanoparticle generation have of late been observed to employ systems including bacteria, yeast and other types of fungi.72 Nevertheless, a greatly detailed procedure of maintaining cell cultures and intracellular synthesis causes inevitable complications in the use of microorganisms in the generation of nanoparticles while numerous purification measures additionally increase the difficulty. Table 2 is a brief presentation of the various methods adopted by researchers to fabricate ZnO nanostructures using green routes. The detailed synthesis conditions, experimental variables, surface morphology, mechanisms and applications are discussed (Table 2).| Source | Synthesis conditions | Experimental variables | Shape/morphology | Mechanism and applications | Size | Ref. |
|---|---|---|---|---|---|---|
| Carom-Trachyspermum ammi seed extract | 2 mL of the extract was slowly added dropwise to a 25 mL solution of 0.05 M ZnNO3. Magnetic stirring for 2 h at 50 °C. Centrifugation and drying at room temperature at 35 °C | — | Uniform hexagonal plates, irregular and highly aggregated nanoparticles with a rough surface | Anti-bacterial activities on both Gram-positive (Staphylococcus aureus) and Gram-negative (Pseudomonas aeruginosa) bacteria | ∼41 nm | 70 |
| Nyctanthes arbor-tristis flower extract | 0.01 M solution of zinc acetate and flower extract were added at a pH of 12 and the solution was stirred for 2 h. A white precipitate was obtained and dried at 60 °C overnight | Concentration of zinc acetate, pH, and temperature | Aggregate of nanoparticles | Nanoparticles were tested for their antifungal potential and were found to be active against all five tested phytopathogens with the lowest MIC value recorded being 16 μg mL−1 | ∼12–32 nm | 72 |
| Ulva lactuca seaweed extract | U. lactuca extract was added into 1 mM zinc acetate and kept under magnetic stirring at 70 °C for 3–4 h. The mixture was centrifuged at 4000 rpm for 10 min and the solid product was collected and heated at 450 °C for 4 h | — | Agglomeration of asymmetrically shaped nanoparticles | Excellent photocatalytic activity on methylene blue. High antibiofilm activity on 4 species of Gram-positive and -negative bacteria | ∼15 nm | 73 |
| Muraya koenigii seed extract | 20 mL of Murraya koenigii seed extract was mixed with 80 mL of zinc nitrate (ZnNO2) and 2.0 M NaOH solution was added with vigorous stirring for 3–5 h, and incubated overnight at room temperature. Zinc oxide nanoparticles (white precipitate) were washed with distilled water and ethanol and dried at room temperature | — | Spherical, triangle, radial, hexagonal, rod and rectangle shaped | ZnO nanoparticles used for antimicrobial activities using human pathogenic bacterial and fungal species | ∼100 nm | 74 |
| Calotropis procera leaf extract | Leaf extract was added to 50 mL distilled water and heated up to 70 °C and 6 g of Zn(NO3)2·6H2O was added and evaporated. Calcined at 400 °C for 3 h | — | Spherical | When Zn(NO3)2·6H2O is mixed with C. procera leaf extract, the Zn2+ ions dispense consistently and form a complex with active sites of hydroxyl groups. Polyphenolic molecules that interact with divalent Zn2+ cations forming a bridge between two hydroxyl groups from two different chains come from the polyphenolic groups in close contact with Zn2+. The divalent cations keep the molecules together and form various structures of zinc complex. Photocatalytic degradation of methyl orange with an efficiency of 81% within 100 min under UV light | ∼15–25 nm | 75 |
| Artocarpus heterophyllus leaf extract | 5 g of zinc nitrate hexahydrate was added to 150 mL leaf extract and heated at 80 °C and calcined in a muffle furnace at 400, 600 and 800 °C for 1 h | Calcination temperature | Spherical | Photo-degradation of Congo red dye | ∼10–15 nm at 400 °C, ∼15–25 nm at 600 °C and ∼25–30 at 800 °C | 76 |
| Moringa oleifera | 2.97 g of zinc nitrate hexahydrate was dissolved in Moringa oleifera natural extract and heated on a hot plate with a stirrer to form a gel kind of product and kept in a muffle furnace maintained at 400 °C | Concentration of Moringa oleifera natural extract and time | Clusters of spherical nanoparticles | ZnO nanoparticles with smaller size show better H2 evolution rates up to 360 μmol h−1 g−1. It is noteworthy that ZnO nanoparticles prepared via novel green synthesis exhibit oxygen vacancies and register enhanced photocatalytic activity as well as good photostability | 100–200 nm | 77 |
| Carica papaya leaf extract | To zinc acetate dihydrate (5 mmol) papaya leaf extract was added and the mixture heated at a temperature of 60 °C for 2 h under stirring at a pH of 8. Finally, it was washed with a water and ethanol mixture and dried at 80 °C for 12 h | — | Spherical | Photocatalyst for methylene blue dye degradation (complete degradation within 180 min in the presence of UV) and photo-anode with an energy conversion efficiency of 1.6% with a current density of 8.1 mA cm−2 in dye sensitized solar cells | ∼50 nm | 78 |
| Nephelium lappaceum L. fruit peel extract | A volume of 50 mL was prepared and then 10 mL of rambutan peel extract was added to 0.1 M Zn(NO3)2·6H2O with heating at a temperature of 80 °C for 2 h and then incubated at room temperature for 1 day to form zinc-ellagate and dried in an oven at 40 °C for 8 h. ZnO nanoparticles were obtained on direct decomposition of the zinc-ellagate complexes in a muffle furnace at 450 °C | — | Multidimensional chain-like structures in which spherical nanoparticles were intertwined with each other | — | ∼20–50 nm | 79 |
| Moringa oleifera leaf extract | 50 mL of Moringa oleifera extract was added to Zn(NO3)2·6H2O at room temperature with a pH of 5 and subjected to heat treatment in air at 500 °C for 1 h | Concentration of zinc salt and calcination temperature | Drying at 100 °C: agglomerates of spherical particles; annealing at 500 °C: nanorods in addition to the clusters of spherical nanostructures | Three chemical reactions of the solvated Zn2+ ions are considered with the phytochemicals of Moringa oleifera, i.e. with a phenolic acid, a flavonoid and vitamin based compounds. An altered chemical behavior of L-ascorbic acid and zinc nitrate, probable oxidation of biological compound i.e.L-ascorbic acid to L-dehydroascorbic acid via free radicals, followed by electrostatic attraction between the free radical and cation of the precursors. Electrochemical investigations by cyclic and square wave voltammetry | ∼12.27–30.51 nm | 80 |
| Catharanthus roseus leaf extract | An aqueous leaf extract of C. roseus was added to 0.025 M aqueous zinc acetate and pH adjusted to 12 and the solution was dried in a vacuum | — | Spherical | Antibacterial activity was evaluated. Among the four bacterial species tested, Pseudomonas aeruginosa is more susceptible when compared with the other three species and may be used for the preparation of antibacterial formulations against Pseudomonas aeruginosa | ∼23–57 nm | 81 |
| Camellia sinensis leaf extract | ZnO NPs using the aqueous extract of green tea leaves. In the prepared extract zinc acetate was dissolved by way of magnetic stirring. Intense stirring was eventually applied on this solution for 5–6 h; a temperature of about 150 °C was maintained during this time. The solid mass thus obtained subsequently underwent a 4500 rpm centrifugation for 15 min; this act was repeated again. Finally washing and drying at 80 °C for 7 to 8 h yielded agglomerates of irregularly shaped ZnO NPs. UV spectroscopy analysis showed maximum absorption at about 330 nm. The size of the particles was determined using a particles size analyzer. The average diameter of the particles was found to be 853 nm | — | Agglomerates of irregularly shaped nanoparticles | These nano-sized ZnO demonstrated remarkable antimicrobial properties against Gram-positive and Gram-negative bacteria as well as against a fungal strain | ∼853 nm | 82 |
| Citrus aurantifolia fruit extract | 50 mL of aqueous Citrus aurantifolia extract was boiled to 60–80 °C.83 It was followed by the addition of a specific amount (5 g) of Zn(NO3)2 to the solution as its temperature rose to 60 °C. The reaction mixture so prepared was then boiled until a deep yellow coloured paste was left. This paste was then collected and heated in a furnace in the presence of air at 400 °C for 2 h to eventually yield a powder. This powder bearing a faint white colour was further ground in a mortar-pestle. The synthesized nanoparticles were characterized by moderate stability. They had near spherical shapes with the most probable particle-size in the range of 9–10 nm | — | Near spherical shaped nanoparticles | — | ∼9–10 nm | 83 |
| Oryza sativa rice extract | ZnO NPs were prepared by the hydrothermal method. The method involved the use of zinc acetate, sodium hydroxide, and uncooked rice flour at several ratios at 120 °C for 18 h. The rice bio-template was found to exert considerable influences upon the size and morphology of ZnO NPs | — | Flake-, flower-, star-, toothed-edge flake-like, rose- and rod-like structures for 0.25 g, 0.50 g, 1.0 g, 2.0 g, 4.0 g and 8.0 g uncooked rice, respectively | — | ∼200–800 nm, ∼800–2000 nm, ∼200–1000 nm, ∼250–700 nm, ∼200–700 nm, ∼150–700 nm and ∼40–100 nm for 0.25 g, 0.50 g, 1.0 g, 2.0 g, 4.0 g and 8.0 g uncooked rice, respectively | 84 |
| Passiflora caerulea. L. leaf extract | The leaf extract was prepared by maintaining a temperature of 70 °C for 8 min. 50 mL of aqueous 1 mM zinc acetate [Zn(O2CCH3)2·(H2O)2] was prepared and subjected to stirring for 1 h. Subsequently, to this solution, a 20 mL of NaOH solution was slowly added. This was followed by a slow addition of 25 mL of plant extract. As a consequence, the color of the reaction mixture was found to change after incubation for an hour. This solution was again subjected to stirring for 3 h. The subsequent appearance of a yellow color confirmed the generation of ZnO NPs. The precipitate so obtained was centrifuged at 8000 rpm at 60 °C for 15 min. Thereafter, the pellets that resulted were dried in a hot air oven at a temperature of 80 °C for 2 h | — | Spherical | — | ∼30–50 nm | 85 |
| Sucrose (as a capping agent) | Zinc acetate (Zn(CH3COO)2·2H2O) and sucrose (C12H22O11) served as the precursor and capping agent, respectively. The precursor was prepared by dissolving 4.3900 g of zinc acetate in 50 mL of double distilled water and stirring for 30 min at 60 °C. During the process, 3.4229 g of sucrose solution was slowly added. The resultant solution was stirred for 2 h at the same temperature. The solution was then bone dried at 80 °C and was calcined in an atmosphere of air at 400 °C for an hour. The end product was finely ground using an agate mortar to obtain the required ZnO/C nanocomposite. Similarly, without sucrose we synthesized pure ZnO nanoparticles | — | Granular | Carbon coated ZnO nanoparticles are used for symmetric supercapacitor device fabrication. The symmetric device yields a specific cell capacitance of 92 F g−1 at a specific current of 2.5 A g−1 | ∼10–100 nm | 86 |
| Whey (as a chelating agent) | Firstly, zinc citrate was obtained by mixing Zn(NO3)2·6H2O with citric acid (CA), previously dissolved in distilled water (0.1 g mL−1), at a molar ratio of 1:3. Whey (W) was subsequently added to these solutions at a mass ratio of CA:W = 60:40 to promote citrate polymerization. The resultant mixture was homogenized and maintained under magnetic stirring for 1 h at 80 °C to complete gelatinization. The obtained brown colored and transparent stable resin (xerogel) aspect was heated to 100 °C in order to eliminate excess water. Then, the xerogel was calcined at temperatures ranging from 200 to 1000 °C for one hour to obtain the resultant ZnO nanoparticles |
Calcination temperature | Spherical | — | With an increase in calcination temperature from 400 to 1000 °C, the size of nanoparticles increased from 18.3 to 88.6 nm | 87 |
| Citrus sinensis fruit peel extract | An aqueous extract of orange peel was used as the biological reducing agent for the synthesis of ZnO NPs from zinc acetate dihydrate. The ZnO NPs were synthesized by mixing 2 g of zinc nitrate with 42.5 mL of the extracts. These mixtures were then stirred for 60 minutes and then placed in a water bath at 60 °C for 60 minutes. Subsequently, the mixtures were dried at 150 °C and then heat-treated at 400 °C for 1 hour | Annealing temperature and synthesis pH | Spherical | Ligation takes place between the functional components of the orange peel and the zinc precursor. The organic substances (flavonoids, limonoids, and carotenoids) in orange peel extract act as ligand agents. These hydroxyl aromatic ring groups, one of the extract components, form complex ligands with zinc ions. Through the process of nucleation and shaping, nanoparticles are stabilized and formed. The mixture of the organic solution is then decomposed directly upon calcination at 400 °C resulting in the release of ZnO nanoparticles. Antibacterial activities toward E. coli and S. aureus: without UV light, the bactericidal rate towards E. coli was over 99.9%, while the bactericidal rate towards S. aureus varied in the relatively wide range of 89–98% | 400 °C, 700 °C and 900 °C: 35–60 nm, 70–100 nm and 200–230 nm, respectively. pH values of 6.0 and 8.0: 10–20 nm and 400 nm. pH value of 10.0: Agglomerates of blocks with lengths of ∼370 nm and widths of ∼160 nm | 88 |
An extract prepared from Ajwain (Carom-Trachyspermum ammi) seeds has also been used to synthesize ZnO NPs.70 The work boasts of its operation under ambient temperature conditions. The ZnO NPs were found to have a wurtzite structure. The synthesized ZnO nanostructures were morphologically characterized by FE-SEM images. The ZnO nanostructure showed uniform hexagonal plates, as well as irregular and highly aggregated nanoparticles with a rough surface. The average diameter of the nano-sized ZnO clusters has been observed to be ∼41 nm. XRD results showed an increase in interplanar spacing with an increase in the extract volume from 0.2474 nm to 0.2765 nm with a simultaneous decrease in crystallite size from 39.51 nm to 28.112 nm. The band gap also fell from 3.592 eV to 3.383 eV as the amount of extract increased. Phytoconstituents in the extract thus evidently played a key role of reductants and furthermore acted as capping agents in the generation and stabilization of ZnO NPs.
Jamdagni et al.72 used an aqueous flower extract of Nyctanthes arbortristis for making ZnO NPs. The starting materials consisted of zinc acetate dihydrate and sodium hydroxide. XRD results showed an average crystallite size of 16.58 nm while TEM analysis revealed that the individual particle size ranged within 12–32 nm and the nanoparticles were obtained in the form of aggregates. In a very recent study,73Ulva lactuca seaweed extract was used to prepare ZnO nanoparticles. XRD analysis revealed strong characteristic peaks of ZnO suggesting high crystallinity of the synthesized material. Further, the average crystallite size thus calculated was found to range in between 5 and 15 nm. TEM micrographs revealed an agglomeration of asymmetrically shaped NPs bearing an average crystallite size of 15 nm.
Muraya koenigii seed extract was also recently reported to have been used as a stabilizer as well as a reductant in the preparation of ZnO NPs.74 Sharp diffraction peaks in XRD results indicated remarkable crystallinity of the NPs whose average crystallite size was calculated to be 70–100 nm. Both SEM and TEM micrographs revealed nanoparticles with an average size of about 100 nm and bearing a wide range of morphologies – spherical, triangular, radial, hexagonal, rod-like and rectangle-shaped.
One recent experiment used Calotropis procera leaf extract and Zn(NO3)2·6H2O to synthesize ZnO NPs.75 An XRD test confirmed a hexagonal wurtzite structure of the nanoparticles with marked crystallinity. The average crystallite size was calculated using the Scherrer equation and found to be 24 nm. Diffuse Reflectance Spectroscopy (DRS) revealed a band gap of 3.1 eV for the synthesized nanoparticles. In the FT-IR analysis of the synthesized ZnO NPs, a peak attributed to the metal–oxygen bond of ZnO appeared in between 500 and 700 cm−1. Further, a conspicuous shift and broadening of peaks corresponding to functional groups like hydroxyl, aldehyde, amine, ketone, and carboxylic acid suggests their participation in the stabilization of ZnO by the extract. Surface attachment of groups like aldehyde, amine, phenol and terpenoid enhances stabilization additionally allowing the extract to function as a bio-template thereby preventing aggregation of ZnO NPs. TEM images revealed an average particle size of 15–25 nm, while SAED and HR-TEM further confirmed the high crystallinity of the material prepared.
The effects of Artocarpus heterophyllus leaf extract and varying temperatures on the morphology and properties of the ZnO NPs thus prepared were studied by Vidya C. et al.76 XRD results show an increase in crystallinity and average crystallite size with temperature, the diffraction peaks being increasingly sharper and narrower with temperature. The particles were all spherical and a grain size of 50 nm was obtained from SEM images. SEM analysis also shows similar trends of size and morphology upon temperature variation. TEM analysis revealed a particle size of ∼10–15 nm at 400 °C, ∼15–25 nm at 600 °C and ∼25–30 nm at 800 °C. This further corroborated the results of XRD and SEM tests. Diffuse Reflectance Spectroscopy (DRS) showed a decrease in the calculated band gaps with increasing calcination temperatures.
Archana et al.77 used Moringa oleifera natural extract and Zn(NO3)2·6H2O for the preparation of ZnO NPs. They took different volumes of the extract, viz. 2, 6, 10 and 14 mL, to prepare ZnO NPs which were accordingly labeled ZnO-2, ZnO-6, ZnO-10 and ZnO-14. The PXRD results of all the samples showed great crystallinity. They had a hexagonal wurtzite structure. And the average crystallite size was found to be 21.6 nm. Field Emission Scanning Electron Microscopy (FE-SEM) analysis showed highly crystalline ZnO-10 and ZnO-14 having a spherical shape and average crystallite size of 20–150 nm. HR-TEM micrographs revealed d-spacing of 0.28 and 0.19 nm for the (001) and (101) planes of wurtzite ZnO. The band gaps calculated using the results from Diffuse Reflectance Spectroscopy (DRS) had values of 2.92 eV for ZnO-2, 3.05 eV for ZnO-6, 3.12 eV for ZnO-10 and 3.10 eV for ZnO-14. The increase in band gap with the amount of fuel was attributed to quantum size effects.
In their research work, Rajeswari Rathnasamy et al.78 used papaya leaf extract for the synthesis of ZnO NPs. Both FESEM and TEM data revealed an average size of ∼50 nm for the individual nanoparticles. The extract of Nephelium lappaceum L. (rambutan) peels (a natural ligation agent) was put into use for the preparation of ZnO NPs in another investigation.79 The bio-mediated ZnO NPs were found to be spherical in shape. They were characterized by diameters between 20 and 50 nm. Some of the particles were found in agglomerated form. After a day, multi-dimensional chain-like structures formed. In these chains spherical nanoparticles were found intertwined to each other.
An investigation conducted by Matinise et al.80 used Moringa oleifera extract as a remarkably operative chelating agent to prepare ZnO nanoparticles. The ZnO NPs eventually obtained were characterized by a particle size in between 12.27 and 30.51 nm. The sample obtained just after drying at 100 °C consisted of agglomerates of spherical particles while that obtained after annealing at 500 °C also had nanorods in addition to the clusters of spherical nanostructures.
The biocomponents of leaves of Catharanthus roseus have also been utilized to prepare ZnO NPs with zinc acetate and sodium hydroxide as reagents.81 SEM micrographs revealed that in addition to the individual ZnO-NPs, aggregates were also formed and they were spherical with diameter ranging from 23 to 57 nm. Sharp and clear XRD peaks confirmed high purity and excellent crystallinity. Shah et al.82 generated ZnO NPs using the aqueous extract of green tea (Camellia sinensis) leaves. The size of the particles was determined using a particle size analyzer. The average diameter of the particles was found to be 853 nm. These nano-sized ZnO particles demonstrated remarkable antimicrobial properties against Gram-positive and Gram-negative bacteria as well as against a fungal strain.
In another experiment, 50 mL of aqueous Citrus aurantifolia extract was boiled to 60–80 °C.83 It was followed by the addition of a specific amount (5 g) of Zn(NO3)2 to the solution as its temperature rose to 60 °C. The synthesized nanoparticles were characterized by moderate stability. They had near-spherical shapes with the most probable particle size in the range of 9–10 nm. The extract of Oryza sativa rice84 was also used to generate ZnO NPs. The extract has been considered a renewable bio-resource. Its abundance adds to its list of merits. The extract has also been cited as a source of bio-template that typically assists the generation of a variety of multifunctional nano-structured materials. ZnO NPs were prepared using the hydrothermal method. The method involved the use of zinc acetate, sodium hydroxide, and uncooked rice flour at several ratios at 120 °C for 18 h. The rice bio-template was found to exert considerable influences upon the size and morphology of ZnO NPs. Fig. 2 shows field emission scanning electron microscopy (FESEM) images of the samples synthesized at different concentrations of uncooked rice (UR). To investigate the effects of raw rice on the resulting ZnO morphology, FESEM was conducted on ZnO synthesized without UR (Fig. 2a and b). As seen in Fig. 2c and d, the ZnO structures were mostly flake-like structures assembling together. They were much more ordered in contrast to the one synthesized without UR (as a control) (Fig. 2a and b). The diameter of ZnO flakes dramatically decreased after adding 0.25 g UR. This was proposed to have occurred due to the inhibition of lateral growth of ZnO crystals. It was further proposed that the accessibility of the zinc ions to the ZnO crystal seeds was controlled by a bio-template. However, the size of particles seemed to increase when the synthesis was done using 0.25 g UR. Different morphologies of the as-synthesized ZnO were observed with increasing the amount of uncooked rice to 0.5 g. Particles with a very small flower-like shape could be observed (Fig. 2e and f). A lower magnification FESEM image indicated that the mentioned structure showed denticulated petals aggregated and form larger flowers of particles. Notably the size of the ZnO particles had been obviously decreased for the sample prepared using 0.5 g UR. In addition, the tooth-like flakes were more dominant for the ZnO sample prepared using 0.5 g UR compared to the one synthesized using 0.25 g UR. Fig. 1g and h indicate the FESEM images of the ZnO sample synthesized using 1 g UR. A very unique star-like structure could be clearly observed at low to high magnification. The star-like structure contained small flakes with denticulated edges which attach to other similar flakes in the center. A closer look showed that the lateral flake acted as a substrate for other flakes to grow on the surface and form a star-like structure. It was therefore argued that the branched pattern for soft templates of starch revealed that the semicrystalline granules of starch were made from concentric rings in which the amylose and amylopectin basic components were aligned perpendicularly to the growth rings and to the granule surface. Fig. 2g and h show that the size of the star-like ZnO particles decreased in comparison with the previous lower amount of uncooked rice. In the case of ZnO crystals synthesized at 2 g UR, increasing the amount of bio-template resulted in different morphologies of ZnO particles being produced. It formed lots of agglomerated toothed-edge flakes which became a secondary unit for larger particles. The star-like shape of the particles could be perceived in some areas but aggregation seemed to be dominant and prevented clearer observation of the particles as they really are. Fig. 2k and l show the FESEM images of the as-synthesized ZnO particles synthesized using 4 g UR. The ZnO morphology changed to flower-like structures, mostly rose-like shapes. A detailed view of the flower-like particles revealed that their flakes had the largest diameter compared to other samples. In the case of ZnO synthesized using 8 g UR, a new morphology, different from other and control samples, was observed. The ZnO crystals appear mostly as rods with around 100 nm size. Moreover, agglomerated without any specific shape, particles coexisted with nanorods in the structure of ZnO synthesized using 8 g UR. Fig. 3 shows the particle size distribution of the ZnO samples synthesized using 0.25, 0.5, 1, 2, 4, and 8 g UR. The particle size distribution of ZnO synthesized without rice is also given for comparison. As shown in Fig. 3, the range of particle size for ZnO synthesized without UR lies between 200 and 800 nm. When 0.25 g UR was used in the synthesis, the size of particles increased dramatically to 800–2000 nm. Notably the size of ZnO synthesized using 0.5 g UR considerably decreased to the 200–1000 nm range. The decreasing trend continued for the sample synthesized using 1 g UR and with a size range of 250–700 nm. Although this distribution was quite similar to that of ZnO synthesized without a bio-template, it was slightly narrower. On the basis of the particle size distribution for the samples synthesized using 2 and 4 g UR, it could be clearly observed that the size of particles decreased to 200–700 nm and 150–700 nm, respectively. In the case of the ZnO sample synthesized using 8 g UR, the size of particles was within the nano regime, between 40 and 100 nm. As mentioned in the growth mechanism suggested by the study, adding a bio-template, which presumably acts as a flocculant, forces aggregation. Therefore, the surface-active sites of the template might influence the size and state of aggregation during the particle growth process and ultimately the resulting ZnO particle size distribution. Another procedure used the aqueous leaf extract of Passiflora caerulea. L. (Passifloraceae).85 The SEM analysis revealed that the ZnO NPs had diameters ranging in between 30 and 50 nm.
 | ||
| Fig. 2 FESEM images of ZnO prepared using different concentrations of uncooked rice (g): 0 (a and b), 0.25 (c and d), 0.5 (e and f), 1 (g and h), 2 (i and j), 4 (k and l), and 8 (m and n) (reproduced from ref. 84 with permission from Springer). | ||
 | ||
| Fig. 3 Particle size distribution of ZnO samples synthesized using various concentrations of UR (g); 0, 0.25, 0.5, 1, 2, 4, and 8 (w/w%) (reproduced from ref. 84 with permission from Springer). | ||
Sucrose was used in a study as the capping agent to synthesize a ZnO/C nanocomposite adapting the sol–gel method.86 The presence of carbon in the prepared ZnO/C was confirmed through EDAX. SEM images of the ZnO/C samples indicate a wide distribution of particles ranging from 10 to 100 nm and exhibit only an irregular granular feature. This kind of surface morphology was argued to be more suitable for supercapacitor electrode materials. Electrochemical investigations of the ZnO/C electrode were carried out using cyclic voltammetry, galvanostatic charge–discharge, and electrochemical impedance spectroscopy. The ZnO/C electrode exhibits a maximum specific capacitance of 820 F g−1 at a constant specific current of 1 A g−1. The symmetric aqueous supercapacitor device exhibits a specific cell capacitance of 92 F g−1 at a specific current of 2.5 A g−1. The aqueous symmetric supercapacitor device achieved an energy density of 32.61 W h kg−1 and a power density of approximately 1 kW kg−1 at a discharge current of 1.0 A g−1. It has been found that the cells have an excellent electrochemical reversibility (92% after 400 continuous cycles) and capacitive characteristics in 1 M Na2SO4 electrolyte.
Zinc oxide (ZnO) nanoparticles were successfully synthesized using a whey-assisted sol–gel method.87 X-ray diffraction (XRD) and Raman spectroscopy analysis revealed a wurtzite crystalline structure for ZnO nanoparticles with no impurities present. Transmission electron microscopy (TEM), XRD observations, and UV-vis absorption spectroscopy results showed that with an increase in calcination temperature from 400 to 1000 °C, the size of the spherical nanoparticles increased from 18.3 to 88.6 nm, while their optical band gap energy decreased to ∼3.25 eV. The whey-assisted sol–gel method proved to be highly efficient for the synthesis of crystalline ZnO nanoparticles whose applications are of great interest in materials science technology. Eryngium foetidum L. leaf extract was also used for the nontoxic, cost-effective biosynthesis of ZnO nanoparticles (NPs) following the hydrothermal route.88 The biosynthesized ZnO NPs served as an excellent antibacterial agent against pathogenic bacteria like Escherichia coli, Pseudomonas aeruginosa, Staphylococcus aureus susp. aureus and Streptococcus pneumoniae. The maximum zone of inhibition in ZnO NPs is 32.23 ± 0.62 and 28.77 ± 1.30 mm for P. aeruginosa and E. coli, respectively.
Another report presented an efficient, environmentally friendly, and simple approach for the green synthesis of ZnO nanoparticles (ZnO NPs) using orange fruit peel extract.89 The approach aimed to both minimize the use of toxic chemicals in nanoparticle fabrication and enhance the antibacterial activity and biomedical applications of ZnO nanoparticles. The sample obtained without annealing exhibited relatively small spherical particles (10–20 nm) which were coagulated in large clusters on a matrix of residual organic material from the reducing agents. In the samples annealed at 400 °C and 700 °C, the particle sizes were randomly distributed and ranged from 35 to 60 nm and 70 to 100 nm, respectively. For an annealing temperature of 900 °C, the particle size increased intensively in the range of 200–230 nm. It was thus found that the morphology and size of the ZnO NPs depended on the annealing temperature. Specifically, with increasing annealing temperature, the particle size tended to increase and shape larger particles due to crystal growth. For pH values of 4.0 and 6.0, the particles were sphere-like in shape, and were distorted with distinct grain boundaries and low coagulation. At pH = 6, the particle size was in the 10–20 nm range and exhibited relative separation. Meanwhile, for a pH of 8.0, the particles had a variable shape and were coagulated in large clusters around 400 nm in size with indistinct grain boundaries. For a pH of 10.0, the particles were coagulated into large blocks with lengths of ∼370 nm and widths of ∼160 nm. The ZnO NPs exhibited strong antibacterial activity toward Escherichia coli (E. coli) and Staphylococcus aureus (S. aureus) without UV illumination at an NP concentration of 0.025 mg mL−1 after 8 h of incubation. In particular, the bactericidal activity towards S. aureus varied extensively with the synthesis parameters. This study presents an efficient green synthesis route for ZnO NPs with a wide range of potential applications, especially in the biomedical field.
4. Modification of zinc oxide nanoparticles
The ultimate motive behind surface modification of ZnO nanoparticles is to harness their high affinity for adhesion and agglomeration through reduction of surface energy thereby controlling their growth at the nano level. Modification of ZnO has been carried out with different classes of modifiers, namely inorganic compounds, organic compounds and polymer matrices. Inorganic compounds like SiO2, Al2O3, LiCoO27, metal ions, etc. when used as modifiers brought about a change of surface area or particle size, diminished the photocatalytic efficiency of the metal oxide and improved the dispersion degree of ZnO particles. Organic modifiers like silanes and carboxylic acids made the following modifications: introduction of characteristic groups on the surface of ZnO and alteration of its physicochemical properties, increase in the compatibility of ZnO with an organic matrix, reduced aggregation of particles and enhanced long-term stability in an organic matrix and improved ZnO dispersion in rubber mixtures. Modifiers such as polyethylene glycol, polystyrene, poly(methyl methacrylate), chitin, etc. improved the unique electrical, thermal and optical properties of ZnO/polymer composites.Cao et al.90 used silica and trimethyl siloxane (TMS) for modifying ZnO in order to achieve a two-fold benefit: enhancing the compatibility of ZnO and cutting down on its agglomeration in the organic phase. A chemical precipitation method using zinc sulfate heptahydrate (ZnSO4·7H2O), ammonium solution (NH4OH) and ammonium bicarbonate (NH4HCO3) was adopted to first obtain the precursor, zinc carbonate hydroxide (ZCH). The surface of the ZCH was then successively modified by an in situ method using TEOS and hexamethyldisilazane (HMDS) in water. The functionalized ZHC was subjected to calcination, to yield extremely fine nanoparticles of ZnO. Reduced agglomeration was thereby effected through such functionalization of the surfaces of ZnO NPs although a lowered photocatalytic activity of the oxide was observed. Nevertheless, a marked increase in the compatibility of ZnO with the organic matrix lent credence to the method. Further, the greater shielding capacity of UV radiation renders the synthesized nanomaterial an excellent candidate for use in cosmetics. Below is a schematic representation (Fig. 4) of the synthesis of surface-modified ZnO ultrafine particles using an in situ modification method.
 | ||
| Fig. 4 Schematic representation of the synthesis of surface-modified ZnO ultrafine particles using an in situ modification method (reproduced from ref. 89 with permission from Elsevier). | ||
Xia and Tang91 also carried out modification of ZnO using silica. They had a silica surface coated with ZnO either in the form of a thin layer or as nanoparticles depending on the reaction parameters. ZnO clusters were generated using triethanolamine (TEOH) on a silica surface having silanol (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–OH) and siloxane (Si–O–Si) groups via a procedure of controlled precipitation. TEOH was observed to play a dual role in the synthesis. It first induced surface modification of silica by getting adsorbed on the silica surface via a mechanism involving the splitting of the siloxane and silanol networks and then subsequently reacting with Zn(OAc)2 to form ZnO clusters whose further growth on the silica surface is sustained via ripening and aggregation. The attachment of ZnO on a larger silica surface was argued to enable rapid collision with other ZnO clusters leading to the significant growth of ZnO on the silica surface.
Si–OH) and siloxane (Si–O–Si) groups via a procedure of controlled precipitation. TEOH was observed to play a dual role in the synthesis. It first induced surface modification of silica by getting adsorbed on the silica surface via a mechanism involving the splitting of the siloxane and silanol networks and then subsequently reacting with Zn(OAc)2 to form ZnO clusters whose further growth on the silica surface is sustained via ripening and aggregation. The attachment of ZnO on a larger silica surface was argued to enable rapid collision with other ZnO clusters leading to the significant growth of ZnO on the silica surface.
Silica was also used by Hong et al.92 for the modification of ZnO NPs. In a separate experiment they also used oleic acid as the capping agent. A reaction between Zn(OAC)2·2H2O and (NH4)2CO3 produced the precursor that was then subjected to a procedure culminating in calcination to get ZnO. Oleic acid was then coated on one sample of ZnO while through a separate pH controlled procedure silica was coated on another ZnO sample using sodium silicate. The oleic acid coated ZnO exhibited remarkable compatibility with the organic matrix. The FTIR spectra revealed a chemical bond between the –OH groups on the ZnO surface and the –COOH groups of the oleic acid chains. The corresponding reaction is given below:
ZnO(OH)x + yOHC(CH2)7CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH(CH2)7CH3 → ZnO(OH)x−y[OOC(CH2)7CH3]y + yH2O CH(CH2)7CH3 → ZnO(OH)x−y[OOC(CH2)7CH3]y + yH2O |
The FTIR spectra for the SiO2-modified ZnO revealed interphase bonds between ZnO and SiO2. A thin film coating of SiO2 on the ZnO surface resulted in enhanced dispersion and reduced agglomeration of nanoparticles, a fact fairly well corroborated by HR-TEM data. The photocatalytic activity of SiO2-modified ZnO however suffered a setback in comparison with that demonstrated by uncoated ZnO. The work further demonstrated that the thorough reduction of the crystallinity of ZnO achieved through heterogeneous azeotropic distillation of the zinc oxide precursor not only precludes aggregation but also brings about a decline in the average particle size.
Yuan et al.93 modified ZnO using Al2O3. A basic carbonate of zinc was obtained from the reaction between zinc sulfate and ammonium bicarbonate followed by precipitating aluminum hydroxide over it. The resulting compound-precipitate was then calcined at 400–600 °C to obtain ZnO NPs coated with Al2O3. It was discovered from TEM analysis that as the Al2O3-coating content rose from 3 to 5%, agglomeration decreased significantly and correspondingly the particle size decreased from an average value of 100 nm to 30–80 nm. The coating thus designed was 5 nm thick and was highly uniform. The coating-core interphase possibly had the structure of ZnAl2O4. Zeta potential data clearly confirm modifications on the ZnO surface by Al2O3 deposition. The change in pH at the isoelectric point for ZnO NPs upon coating with Al2O3 from around 10 to a value of 6 might have assisted a greater degree of dispersion of ZnO NPs.
In a study by Hu et al.,94 nano-sized ZnO rods doped with transition metals such as Mn, Ni, Cu, and Co were designed by a plasma enhanced chemical vapor deposition method. The ZnO thus modified had a greater amount of crystal defects within its structure. This led to its greater sensitivity towards formaldehyde. When the 1.0 mol% Mn doped ZnO nanorods were activated by 10 mol% CdO, a maximum sensing of ∼25 ppm was obtained and the corresponding response and recovery time were found to be appreciably short.
Wysokowski et al.95 developed a β-chitin/ZnO nanocomposite material. The β-chitin used in the synthesis was derived from Sepia officinalis, a cephalopod mollusk. This nanocomposite was found to exhibit remarkable anti-bacterial activity and was touted as an excellent ingredient for the making of wound-dressing materials.
Ong et al.96 in their work synthesized a heterogeneous photocatalytic material by loading ZnO on solvent exfoliated graphene sheets. For anchoring ZnO onto the graphene sheet, they used poly(vinyl pyrrolidone) as an inter-linker which was also found to enhance the functionalization of the acid treated graphene sheets. The thermal stability of the decorated ZnO was found to be higher than that of the undecorated oxide. The modified ZnO proved to be an outstanding photocatalyst being able to cause 97% degradation of Reactive Black 5 under visible light. This improvement was attributed to a host of favourable parameters achieved through the modification, namely, an enhancement of light absorption intensity, widening of the light absorption range, suppression of charge carrier recombination, improvement of surface active sites and rise in the chemical stability of the designed photocatalyst.
Tang et al.97 demonstrated a way to tackle the agglomeration tendency of ZnO NPs. They prepared ZnO/polystyrene nanocomposites via a mini-emulsion polymerization method. For this, a silane coupling agent, namely γ-glycidoxypropyl trimethoxysilane (KH-560, AR), was first allowed to cling to ZnO NPs via reaction between its Si–OCH3 groups and the hydroxyl groups on the surface of the nanoparticles followed by anchoring of 4,4′-azobis(4-cyanovaleric acid) (ACVA) onto their surface through reaction of its carboxyl groups with the terminal epoxy groups of the aforementioned coupling agent. Subsequently, polymerization of the styrene monomer was initiated using the azo group of ACVA for designing the final nanocomposites. The monomer droplet of the mini-emulsion polymerization system thus obtained contained well dispersed ZnO/polystyrene nanocomposites with a high grafting efficiency of 85% as calculated from TGA. It was evident from scanning electron microscopy (SEM) that while pure ZnO NPs suffered considerable agglomeration in poly(vinyl chloride) (PVC) film, the ZnO/polystyrene nanocomposite particles underwent homogeneous dispersion in the PVC matrix. The scheme depicted in Fig. 5 explains the mechanism of the mini-emulsion polymerization method to construct ZnO/polystyrene nanocomposites adopted by Tang and his research group. From SEM micrographs (Fig. 5), it was observed that functionalized ZnO (f-ZnO) nanoparticles had been well dispersed in the polymer matrix because the f-ZnO nanofiller had outstanding adhesion and strong interfacial bonding to PEA. As was observed, f-ZnO nanoparticles were homogeneously dispersed in the polymer matrix and their sizes were estimated to be between 20 and 50 nm.
 | ||
| Fig. 5 A schematic diagram showing the synthesis of ZnO/polystyrene nanocomposites by anchoring 4,4′-azobis(4-cyanovaleric acid) (ACVA) onto the surface of ZnO nanoparticles to initiate styrene polymerization (reproduced from ref. 97 with permission from Elsevier). | ||
Cyclodextrins (CDs) make up a class of cyclic torus-shaped oligosaccharides. CD has a hydrophilic external surface and a hydrophobic internal cavity. CDs have been extensively used as eco-friendly coupling agents.98,99 Among the derivatives of CDs, monochlorotriazinyl-β-cyclodextrin (MCT-β-CD) with a monochlorotriazinyl group as a reactive anchor was found to possess the ability to form covalent bonds with substituents of the nucleophilic type, viz., –OH or –NH2 groups.100–103 Therefore, MCT-β-CD provides an interesting way of surface modification for inorganic nanomaterials. Abdolmaleki et al.104 accomplished surface modification of ZnO NPs by covalently grafting MCT-β-CD onto the surfaces of ZnO NPs through a facile and single-step procedure. In the next step, f-ZnO nanoparticles were employed for construction of a new series of poly(ester-amide)/ZnO bionanocomposites (PEA/ZnO BNCs) whose TEM image is shown in Fig. 6. MCT-β-CD has monochloro-triazinyl groups that react with –OH groups on the surfaces of ZnO NPs through nucleophilic reaction (Fig. 7). After the incorporation of MCT-β-CD on the surfaces of ZnO NPs, polymer/ZnO bionanocomposites (BNCs) were designed using a biodegradable amino acid containing poly(ester-amide) (PEA). ZnO NPs with β-CD functional groups incorporated on their surfaces exhibited a near-complete suppression of their tendencies towards agglomeration while simultaneously displaying enhanced compatibility with the polymer matrix. Scores of functional groups on the surfaces of ZnO NPs enable possible interactions with PEA chains that lead to excellent dispersion and compatibility with the polymer matrix. FE-SEM and TEM results bore out a reduction of agglomeration that can be safely attributed to the steric hindrance induced by the organic chains of MCT-β-CD between the inorganic nanoparticles. The dispersibility, surface morphology and particle dimensions of functionalized ZnO (f-ZnO) with β-CD are shown in Fig. 8.
 | ||
| Fig. 6 FESEM of pure ZnO NPs (a) and grafted ZnO/polystyrene nanocomposite particles (b) dispersed in PVC matrices (reproduced from ref. 104 with permission from Elsevier). | ||
 | ||
| Fig. 7 Modification of ZnO nanoparticles with MCT-β-CD (reproduced from ref. 104 with permission from Elsevier). | ||
 | ||
| Fig. 8 (A) Photograph of aqueous dispersions of pure ZnO (left) and f-ZnO (right), and (B) FESEM and (C) TEM micrographs of f-ZnO (reproduced from ref. 104 with permission from Elsevier). | ||
Farzi et al.105 carried out surface modifications of ZnO with oleic acid, trimethoxyvinyl silane and their different mixtures as capping agents. The compatibility of ZnO with different organic matrices was found to increase considerably. A greater degree of dispersion of the inorganic nanoparticle was simultaneously achieved. Modifications with trimethoxyvinyl silane were however found to be more effective than those with oleic acid clearly evident from the greater extent of compatibility with the organic phase achieved using the former. Thus, different combinations of these coupling agents were used to get a grip on the hydrophobicity and the dispersion capacity of the inorganic oxide in solvents that are polar to varied extents. Modifications with a 1:3 (by weight) mixture of trimethoxyvinyl silane and oleic acid exhibited more efficient dispersion in the highly hydrophobic styrene. Meanwhile, ZnO NPs modified with a 1:1 (by weight) mixture of trimethoxyvinyl silane and oleic acid dispersed more efficiently in the moderately hydrophobic butylacrylate. Modifications attained by a 3:1 (by weight) mixture of trimethoxyvinyl silane and oleic acid endowed the nanoparticles with greater dispersion in the weakly hydrophobic methyl methacrylate. The magnitudes of grafting yield and grafting density of both the coupling agents were also found to be within an acceptable range.
5. Potential applications
Recent years have witnessed a sharp rise in focus on ZnO NPs in the realm of research courtesy of their unique properties that have been exploited to achieve a wide range of applications in transparent electronics, ultraviolet (UV) light emitters, piezoelectric devices, chemical sensors, opto-electronics, solar cells and spin electronics.106,107 Being nontoxic, ZnO has of late found extensive use as an excellent photocatalyst for the degradation of a great many emerging organic pollutants. ZnO has also attracted great interest as a raw material in the pharmaceutical and cosmetic industries, as a pigment in the paint, concrete and rubber industries, as a UV filter in products, the textile industry, and medical and biological fields, and so on (Fig. 9). A few of these applications of ZnO NPs have been presented below. | ||
| Fig. 9 Applications of ZnO NPs. | ||
5.1 Concrete and rubber industries
In the rubber industry, ZnO is extensively used as an additive. ZnO like a host of other metal oxides and inorganic substances raises the thermal conductivity of silicone rubber when added even in low proportions. This it does while retaining the high electrical resistance of the silicone rubber. However, poor dispersion of the nanoparticles in the polymer matrix owing to feeble interactions between the surfaces of nanoparticles and the polymer is one area that requires improvement and therefore has been extensively investigated. Agglomeration of ZnO NPs bearing high surface energy has been reported.108 This necessitates surface modification of the filler ZnO NPs in order to achieve simultaneous reduction in their agglomeration and an improved degree of dispersion in the polymer matrix.109 Furthermore, ZnO is widely known to activate sulphur vulcanization via enhancement of the degree of cross-linking. Therefore, the mechanical properties of silicone rubber are found to improve as well when ZnO is incorporated into it as a filler.In their attempt to enhance the interactions between the nano-sized ZnO particles and the polymer, Yuan et al.110 by incorporating vinyl silane groups on the surfaces of ZnO NPs using vinyl triethoxysilane through a procedure premised on the hydrosilylation reaction during curing carried out their surface modification. The vinyl silane groups on the ZnO surface enabled improved cross-linking with the rubber matrix. In order to solve this problem, surface modification techniques are applied to improve the interaction between the nanoparticles prepared by the sol–gel method and the polymer. In comparison with the nanocomposites of silicone rubber with ZnO, the nanocomposites of silicone rubber with vinyl triethoxysilane modified ZnO possessing extensive cross-linking and a higher degree of dispersion with the rubber matrix exhibited superior mechanical properties and enhanced thermal conductivity.
ZnO NPs have been widely used as an efficient material for the enrichment of cross-linking in elastic polymers.111,112 The cured polymer produced through incorporation of ZnO NPs exhibited high ultimate tensile strength, tear strength, toughness and hysteresis. The slippage of polymer chains on the surfaces of ionic clusters and the renewal of ionic bonds when the sample gets externally deformed give rise to enhanced capacity of the ionic elastic polymer for stress relaxation which in turn results in its upgraded mechanical properties. Furthermore, the thermoplastic properties of such polymers enable their processing in a fused state in a manner akin to a thermoplastic polymer.113 Nevertheless, carboxylic elastic polymers with ZnO as a cross-linker suffer from a few drawbacks prominent among which are their tendency to get scorched, feeble flex properties and high value of compression set. The tendency to get scorched is gotten rid of by the incorporation of either zinc peroxide (ZnO2) or ZnO2/ZnO cross-linkers. ZnO2 serves to not only create ionic cross-links but also generate covalent cross-links as a result of peroxide action. However, prolonged curing is needed to obtain elastomers with an ultimate strength and cross-link density comparable to that of ZnO-cross-linked elastomers. The three vital processes that amount to the curing of XNBR by ZnO2/ZnO cross-linkers are rapid creation of ionic crosslinks due to the initial ZnO present, covalent links resulting from peroxide cross-links and further ionic cross-linking due to the generation of ZnO from the decomposition of ZnO2. Leaving aside the problem of scorching, ZnO NPs make good and therefore widely used cross-linkers in carboxylated nitrile rubbers.
The prime factors affecting the involvement of ZnO in the formation of ionic cross-links with the carboxylic groups of the elastic polymers are its particle size, surface area and morphology. They are also found to govern the dimensions of the interphase between the cross-linkers and elastomer chains.114 With a view to ascertain the correlation between the characteristics of ZnO NPs and their roles in the curing of elastic polymers, Przybyszewska et al.115 employed a variety of ZnO NPs with different morphological characteristics (spheres, whiskers, and snowflakes) as cross-linkers in a carboxylated nitrile elastomer. It emerged from their investigation that ZnO NPs as cross-linkers imparted improved mechanical properties to vulcanizates than commercially used ZnO micro-particles. The ultimate tensile strength of vulcanizates with ZnO NPs was found to be four times higher than that of ZnO micro-particles containing vulcanizates. As a result, there is a 40% reduction of the quantity of ZnO that is put to such use. Since ZnO is known to have deleterious effects on aquatic life, an approach that reduces its usage is highly commendable from the point of view of eco-friendliness. However, ZnO cross-linked XNBR undergoes shrinkage on prolonged exposure to heat.
Among all the aforesaid morphologies, it was observed that ZnO snowflakes with a surface of approximately 24 m2 g−1 had the highest activity. However, surface area and particle size exerted little influence on the activity of ZnO cross-linkers. It was also observed that the ZnO NPs exhibited a minimum tendency to agglomerate in the rubber matrix. There gathered smaller agglomerates with ZnO NPs as cross-linkers upon sample deformation as compared to the large agglomerates observed with ZnO microparticles.
The usage of ZnO as a cross-linker in rubber has an adverse impact on the environment, particularly when it is discharged into the surroundings upon degradation of rubber.116 Zinc is known to cause great harm to aquatic species117 and efforts to cut down on the content of ZnO in rubber are hence being made.118 Bringing down the ZnO level in rubber, therefore, may follow any of the following three fundamental procedures:
(i) substituting the commonly used micro-dimensional ZnO of surface area 4–10 m2 g−1 with nano-structured ZnO with surface area of up to 40 m2 g;
(ii) carrying out surface modifications of ZnO with carboxylic acids (viz., stearic acid, maleic acid and the like);
(iii) using additional activators.119
In order to get over the eco-toxicity associated with the usage of ZnO in large quantities, Thomas et al.120 designed a few unique accelerators, namely, N-benzylimine aminothioformamide (BIAT)-capped-stearic acid-coated nano-ZnO (ZOBS), BIAT-capped ZnO (ZOB), and stearic acid-coated nano-zinc phosphate (ZPS), to probe their effects on the curing of natural rubber (NR) and thereby its mechanical properties. ZnO NPs prepared by the sol–gel route were surface-decorated using accelerators such as BIAT and fatty acids such as stearic acid. The capping agents functioned to reduce the size of agglomerates leading to an improvement of vulcanization and physicochemical properties of NR. Capping of ZnO further ensured a decline in the time and energy required for dispersion in the rubber matrix. As a result, there happened a further enhancement of the acceleration of vulcanization and a remarkable upgrade of the mechanical properties of the emerging vulcanizates. The rubber vulcanized with an optimal dose of BIAT-capped-stearic acid-coated zinc oxide (ZOBS) was found to possess superlative curing and mechanical properties in comparison with other countertypes and the reference polymer containing pristine ZnO NPs. The rigidity of vulcanizates containing ZPS was found to increase as a result of an enhanced cross-link density. The vulcanizates exhibited reduced tendency to get scorched as a result of incorporation of capped ZnO NPs and this was attributed to the delayed release of BIAT from the capped ZnO into the rubber matrix for interaction with CBS (conventional accelerator). Sabura et al.121 adopted a solid-phase pyrolytic procedure to synthesize ZnO NPs of particle size in between 15 and 30 nm and surface area in the range 12–30 m2 g−1 for use in neoprene rubber as cross-linkers. Two findings emerged from this study. One, the optimal content of ZnO required was found to be low in comparison with commercially used ZnO. Two, the cure characteristic and mechanical properties of the rubber showed a marked improvement when compared with those containing conventional ZnO.
Since an improvement of the processing time and resistance of concrete against water can be made by incorporation of ZnO NPs in concrete, ZnO NPs have also been used for manufacturing concrete. Nivethitha et al.122 have reported positively about the aforesaid fact in their work. They had the cement partially replaced by ZnO of 0, 1, 3 and 5% by weight of cement. They prepared the blended material by using a cement–sand ratio of 1:1 and 1:2 by weight while the water–binder ratio was fixed at 0.35. The compressive and split tensile strength was evaluated after 7 and 28 days. Studies were also carried out on durability parameters that include water absorption, impact strength, sorptivity and microstructure of the mortar. The results show that in general an improvement in mechanical and impact strength of the mortar was observed for ZnO incorporation up to 3% by weight of cement. However, a slight decline of these parameters was observed for 5% by weight of cement incorporation of ZnO. SEM data revealed that the nanoparticles filled cement pores, promoted the hydration process and given their homogeneous dispersion greatly enhanced the microstructure of the cement mortar.
5.2 Opto-electronic industry
Because of its large exciton binding energy (60 meV), ZnO is considered as a potentially attractive material for light-emitting devices in the ultraviolet (UV) region. Due to its direct, wide bandgap of 3.37 eV, ZnO can be used as a light-emitting diode and photodetector, in sensors, and in solar cells and devices emitting a surface acoustic wave.123,124 The optical and electrical properties of ZnO have been greatly exploited in recent years to generate a large photocurrent in ZnO based dye-sensitized solar cells (DSSCs). Since the performance efficiency of solar cells is considered to be substantially influenced by the varying geometries of ZnO nanostructures, a plethora of investigative studies on the effects of morphology of ZnO have unfolded in recent years. With ZnO nanosheets having inadequate surface area, a conversion efficiency of 1.55% was obtained for a DSSC.125 However, the ZnO nanosheets/spheres synthesized with oxalic acid as surfactant led to an expansion of the surface area thereby enhancing the conversion efficiency of the DSSC to 2.61%.126 An electrodeposited ZnO nanobelt film was used in a DSSC which was found to exhibit 2.6% photovoltaic efficiency.127 DSSCs with films comprising ZnO tetrapods had conversion efficiencies in the range 1.20–3.27%.128,129 Additionally, it came to the fore that incorporation of ZnO NPs into such films increased their surface area which in turn boosted their efficiency.128 One-dimensional nano-sized structures have been known to reduce recombination by increasing the electron diffusion length via direct electron transport. A number of one-dimensional ZnO nanostructures have thus been put to use in DSSCs for enhancement of their efficiency. Notably, a DSSC with ZnO nanotube film exhibited a conversion efficiency of 2.3%.130 Several dopants have also been tested for their effects on photovoltaic efficiency of ZnO based DSSCs. A dye-sensitized device with ZnO nanoporous nanosheets doped with boron was probed131 and a conversion efficiency of 6.75% was obtained. A DSSC with fluorine-doped ZnO produced a conversion efficiency of 3.43%.132 With a lithium-doped ZnO photoanode, the DSSC generated an efficiency of 6.1%.133The last decade has seen an upsurge in the fabrication of ZnO-based perovskite solar cells (PSCs). Although the conventional choice for an electron transport layer has been TiO2, ZnO with higher electron mobility is increasingly replacing it as an efficient and low-cost material for electron transport in PSCs. Additionally, the power conversion efficiency of PSCs at large has exceeded 20% of late giving the necessary impetus to delve deep into the fabrication of ZnO electron transport layers (ETLs) for yet more brilliant perovskite solar devices. Bi et al.134 fabricated a PSC with ZnO nanorods aligned vertically over the substrate. With the length of nanorods, the Jsc (short-circuit current density), FF (fill factor) and PCE of solar cells were found to increase. They however reported a decrease in Voc with nanorod length. They reasoned that nanorod length has a bearing on the electron transport time and lifetime that in turn influence the performance of the solar cell. They achieved a maximum overall cell efficiency of 5%. Son et al.135 substituted the single-step method used by Bi et al. by a two-step coating procedure. Such a treatment generated a fully filled perovskite film that covered all ZnO nanorods of varying lengths without voids and formed an overlayer on the surface of nanorods. As a further consideration, the two-step coating treatment induced optimization of the cuboid size of MAPbI3 and reduced the series resistance of the solar cell.136 As a result, a maximum PCE of 11.13% was obtained. Tang et al. designed ZnO nanowall ETLs.137 The best performance PSC based on ZnO nanowalls produced a Jsc of 18.9 mA cm−2, Voc of 1.0 V, FF of 72.1%, and PCE of 13.6%. Meanwhile, the control device shows a Jsc of 18.6 mA cm−2, Voc of 0.98 V, FF of 62%, and PCE of 11.3%. The introduction of ZnO nanowalls led to an evident boost in the FF and PCE of the PSCs and this can be ascribed to the greater contact area between ZnO and perovskite offered by the ZnO nanowalls in comparison with the planar ZnO film which improves not only the electron collection but also transportation efficiency at the interface of the ZnO nanowalls and perovskite. Moreover, the decomposition of ZnO by perovskite triggered by the alkaline nature of the ZnO surface leads to the formation of PbI2 on the perovskite/ZnO interface. The presence of PbI2 can suppress the surface recombination and improve the FF.138
5.3 Gas-sensing
ZnO nanostructures have exhibited excellent gas sensing properties largely driven by the change in modifications in sensor resistance when gas molecules interact with their surface. The overriding feature of ZnO nanostructures when it comes to gas sensing ability is the large surface-to-volume ratio. Under ambient conditions, oxygen molecules get adsorbed on the surface through a phenomenon termed as ionosorption. In the course of this phenomenon, they collect electrons from the conduction band of ZnO and assume various forms (O−, O2− and O2−) depending on the temperature.139,140 This creates a surface depletion layer that leads to an increase of resistance in ZnO. Upon approach of reductive gases in the vicinity of the ZnO surface, the adsorbed negatively charged oxygen species interact with the gas molecules and the trapped electrons are released back to the conduction band of ZnO thereby lowering the sensor resistance. On the other hand, oxidative gases undergo interaction that raises resistance in the material.Hydrothermally obtained ZnO nanorods reported in a study by Cho et al.141 exhibited remarkable sensitivity of NO2 gas in the presence of CO. The sensor resistance elicited a change by 1.8 times its pristine value upon exposure to 1 ppm NO2 while an imperceptible change was noticed for a 50 ppm CO. It also emerged from the study that ZnO nanorods with larger crystallite size turned out to be more sensitive than ones with smaller crystallite diameters. Sensitivity towards NO2 was found to be more pronounced under conditions of low temperature. Vanalakar et al.142 also prepared ZnO nanorods with preferential orientation along the (002) direction through the wet chemical route and studied their gas sensing ability towards NO2 and the parameters affecting it, such as the aspect ratio of the rod lengths, time, operating temperature and gas concentrations. The sample of ZnO nanorods with a (103)/(002) ratio of 0.036 and intermediate spacing between them produced remarkable sensing performance at a temperature of 175 °C for detection of NO2 at a concentration of 100 ppm with a high sensitivity of 3100%. The mechanism of gas sensing outlined in the work is as follows:
| NO2 (gas) + e− (CB) → NO2− (adsorption) |
| NO2 (gas) + O2− (adsorption) + 2e− → NO2− (adsorption) + 2O− (adsorption) |
Zhang et al.143 constructed a Ag loaded ZnO–MoS2 gas sensing film through a LBL (layer-by-layer) self-assembly route and investigated its response upon exposure to a wide range of gases such as H2, CH4, CO, C2H2, C2H6 and C3H8. It was found that the nanocomposite film showed remarkable selectivity towards CO (Fig. 10). Upon contact with CO gas, the Ag NPs of the nanocomposite sensing film catalyze the oxidation of CO triggering the release of electrons trapped by anionic oxygen species which in turn causes a sharp fall of sensor resistance to manifest a brilliant sensing response. The Ag–ZnO–MoS2 nanocomposite's gas sensing performance towards CO was found to be higher than that of Pt–ZnO–MoS2 or ZnO–MoS2 or ZnO film in terms of response/recovery capability, repeatability, stability and selectivity (Fig. 11). Notably, under the same conditions of the experiment, the Ag–ZnO–MoS2 sensing film functioned way better than the Pt–ZnO–MoS2 sensor. This was elucidated invoking the Schottky barrier (SB). Fig. 12a and b show how a noble metal's SB would tamper with the electrical properties of the ZnO–MoS2 film. The equation ΦSB = Φ − ψ allows the calculation of the SB height ΦSB, where Φ is the work function of the noble metal and ψ is the electron affinity of ZnO–MoS2. Metals with a lowered work function Φ would therefore produce a SB of smaller magnitude. Hence, a metal with a small work function is desirable for a reduced SB and therefore an enhanced sensitivity of the resultant nanocomposite film. Since Ag has a lower work function (4.26 eV) than that of Pt (5.65 eV), the Ag loaded ZnO–MoS2 gas sensing nanocomposite film elicited greatly elevated sensing responses towards CO. Using an improvised sol–gel procedure, Hjiri et al.144 prepared an Al-doped ZnO gas sensor showing excellent selectivity towards CO. Samples coded A0ZO, A1ZO, A3ZO and A5ZO had [Al/Zn] ratios of 0, 0.01, 0.03 and 0.05, respectively. The response behaviour of all these samples towards 50 ppm CO at various operating temperatures was monitored. The response (R0/R) of all samples initially increased with temperature, reached a maximum and then exhibited a decline (Fig. 13). At lower temperatures of 200 to 300 °C, only A1ZO showed a better response than the other samples that responded almost equally. However, in the temperature range 300–400 °C, all Al-loaded samples showed a greater response than the unloaded ZnO. With different concentrations of CO at 300 °C (Fig. 14), the A3ZO sensor showed the best response. Thus, impregnating ZnO with a low content of Al in the range of 1–3 wt% resulted in an enhanced response towards CO sensing. Besides, a significant response could be observed at a very low CO concentration of 5 ppm. Also, since exposure of the Al-loaded ZnO samples to CO produced a decrease in the resistance of the samples, it was concluded that Al-doping sustained the n-type semiconducting properties of the pristine ZnO. Exposure to CO caused a great variation in the resistance of the sensing film (Fig. 15). The signal was further found to go back to its initial baseline value after each pulse indicating that the CO adsorption on the surface layer was reversible. The following mechanism was furnished to explain the decrease of resistance of Al-loaded ZnO upon exposure to CO:
| CO(gas) + O−(adsorbed) → CO2 + e− |
 | ||
| Fig. 10 Sensing responses of pure ZnO, ZnO–MoS2, Pt–ZnO–MoS2 and Ag–ZnO–MoS2 film sensors towards 100 ppm CO gas (reproduced from ref. 143 with permission from Elsevier). | ||
 | ||
| Fig. 11 Selectivity of the Ag–ZnO–MoS2 nanocomposite sensor towards 100 ppm gas species of H2, CH4, CO, C2H2, C2H6 and C3H8 (reproduced from ref. 143 with permission from Elsevier). | ||
 | ||
| Fig. 12 (a) Schematic diagram of the Ag–ZnO–MoS2 nanocomposite sensor towards CO gas; (b) energy band diagram of the Ag–ZnO–MoS2 nanocomposite (reproduced from ref. 143 with permission from Elsevier). | ||
 | ||
| Fig. 13 Response of the Al-loaded and unloaded ZnO samples towards 50 ppm CO at different operating temperatures (reproduced from ref. 144 with permission from Elsevier). | ||
 | ||
| Fig. 14 Response of AZO nanoparticles as a function of CO concentration at a temperature of 300 °C (reproduced from ref. 144 with permission from Elsevier). | ||
 | ||
| Fig. 15 Response of the A3ZO sensor as a function of CO concentration at 300 °C (reproduced from ref. 144 with permission from Elsevier). | ||
The electrons injected into the conduction band lower the resistance of the Al-doped ZnO gas sensors. The response and recovery times observed for all Al-loaded ZnO samples were 6–8 s sand 16–30 s, respectively. The unloaded ZnO sample was marked by longer response and recovery times of 30 s and 70 s, respectively. The sensing films exhibited excellent thermo-mechanical and electrical stability.
Chen et al.145 synthesized mesoporous ZnO sensors for selectively detecting ethanol under UV light at operating temperatures lower than 150 °C. With the assistance of a low-powered UV LED (2 mW), outstanding response and selectivity towards varying concentrations (10 to 1000 ppm) of ethanol were demonstrated by the sensing layer at 80 °C. UV illumination induced electron–hole pairs that activated adsorbed oxygen leading to their extensive interaction with ethanol thereby eliciting a remarkable sensing response from the synthesized ZnO sensors. The response and recovery times were 6 s and 94 s, respectively, and the gas sensor displayed repeatability and brilliant stability. Another study146 reported a facile three-stage synthesis of ZnO–SnO2 composite hollow spheres for the selective sensing of ethanol by exploiting the synergic benefits of the n–n heterojunction. The basic properties of ZnO played a pivotal role in the sensing mechanism via dehydrogenation of ethanol as elaborated in the work.
| C2H5OH (g) → CH3CHO (g) + H2 (g) (basic oxide) |
The CH3CHO intermediate is subsequently oxidized to form CO2 and H2O, as depicted in the following equation:
| CH3CHO (ad) + 5O− → 2CO2 + 2H2O + 5e− |
Therefore, the ZnO/SnO2 nanocomposite gas sensor demonstrated a sharper response to ethanol gas than the pristine SnO2 sensor. Moreover, a possible increase in the effective barrier height of the n–n heterojunction enabled better engagement with adsorbed oxygen causing greater depletion of electrons from the conduction band eventually leading to an enhanced gas sensing response by the system. Additionally, remarkable detection at a lower (ppb) limit was shown by the heterostructured sensor.
5.4 Cosmetic industry
Due to its antibacterial and good antifungal activity, zinc oxide is widely used in production of various raw materials used in medicine such as disinfectant agents and for dermatological applications.147 ZnO NPs absorb UVA and UVB radiations and can be used in sun protective creams.148In a study by Reinosa et al.,149 it was brought to light that a nano/micro-composite comprising nanosized TiO2 dispersed on ZnO micro-particles showed a higher sun protection factor (SPF) than individual TiO2 and ZnO particles. The SPF of the synthesized nano-sized TiO2 was found to be higher than that of its micro-sized counterpart with the former showing maximum absorption at 319 nm while the latter showed maximum absorption at 360 nm. The synthesized micro-sized ZnO had a higher SPF than its nano equivalent. Both exhibited maximum absorption at 368 nm. These data suggested that ZnO has a higher critical wavelength because it covers the entire UV range and has a higher UVA/UVB ratio since the maximum of the SPF curve lies in the UVA region (Fig. 16a). Additionally, it was observed that TiO2, with a lower UVA/UVB ratio owing to the presence of the SPF maximum in the lower wavelength region, has a lower critical wavelength (Fig. 16b). Therefore, to boost the SPF output, a suitable combination of the two oxides was thought out. A dry dispersion procedure was adopted to prepare the composite consisting of 15 wt% TiO2 NPs and 85 wt% ZnO micro-structured particles. The results obtained from this composite were compared with those obtained by the standard procedure. Raman spectroscopy revealed a superior dispersion of the NPs and their anchoring with higher quantum confinement resulting from dry dispersion by using ZnO micro-structures as host particles. The SPF output was found to be higher for the sunscreen with the filter prepared by the dry dispersion method than the one with the filter synthesized following the standard method (Fig. 17). This observation was chiefly attributed by the authors to the correct dispersion of TiO2 NPs over the host ZnO micro-sized particles.
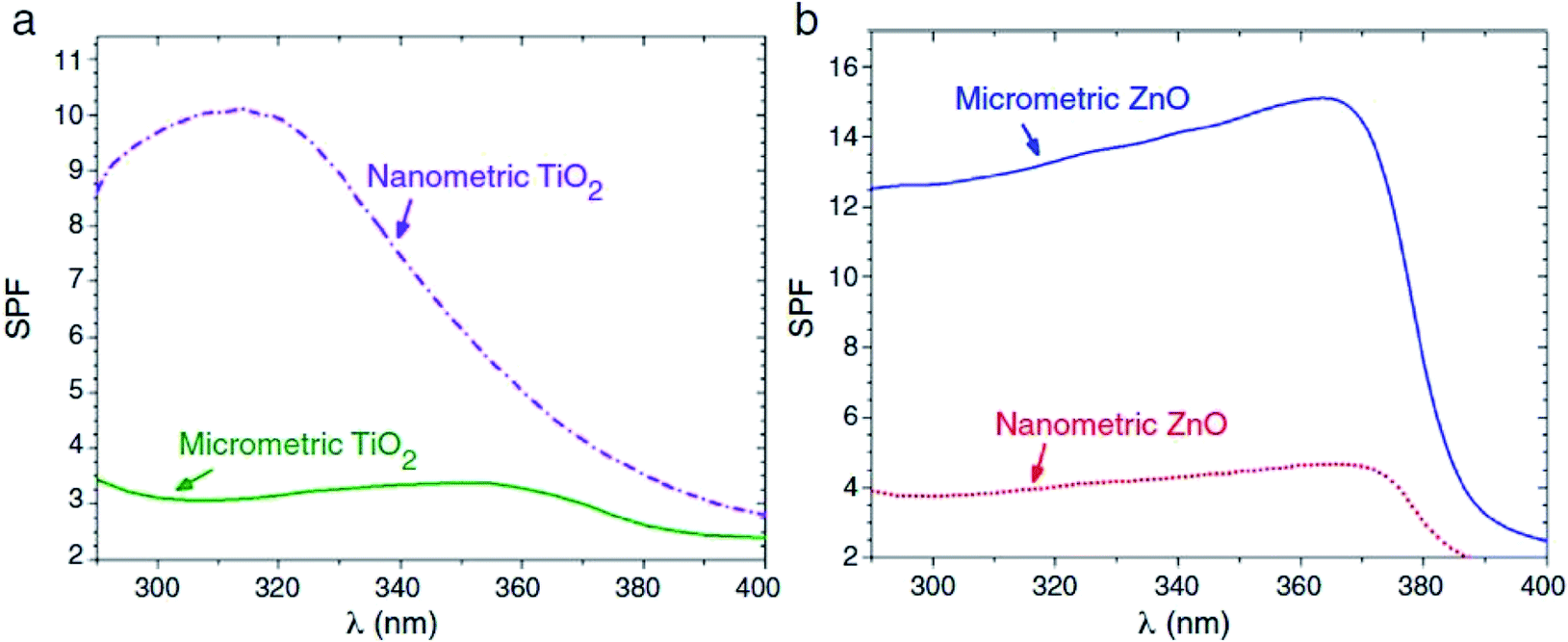 | ||
| Fig. 16 SPF curves of COLIPA sunscreen incorporating an inorganic UV filter: nanometric (dashed lines) and micrometric (solid lines) (a) TiO2 and (b) ZnO particles (reproduced from ref. 149 with permission from Elsevier). | ||
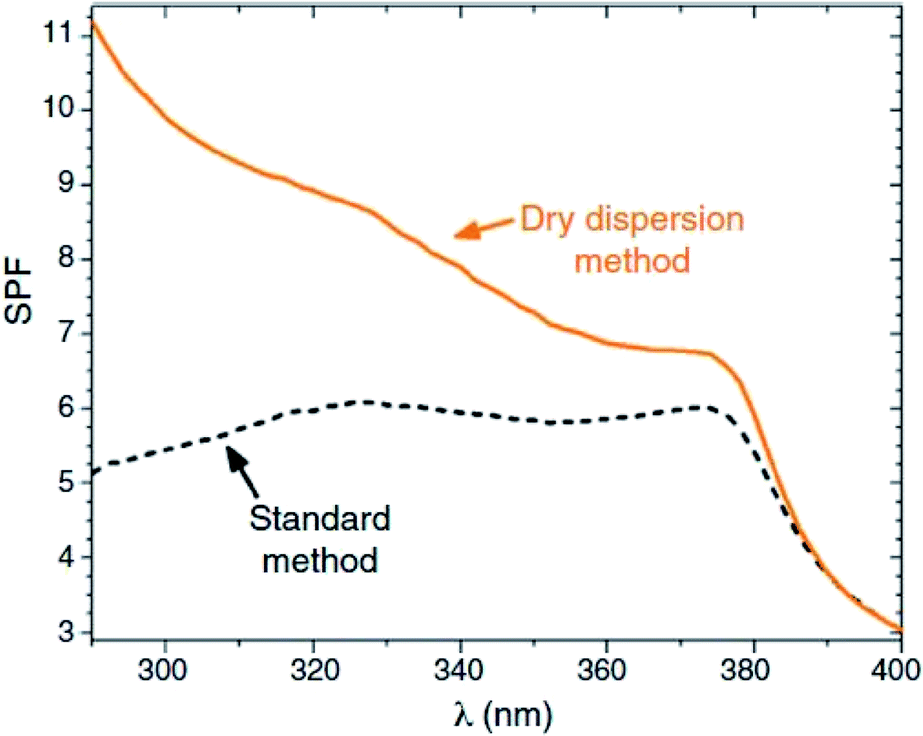 | ||
| Fig. 17 SPF curves of sunscreens with micro–nanocomposite filters. The solid line represents the SPF curve of the new micro–nano composite obtained by a nano-dispersion method and the dashed line represents the curve obtained by a standard method (reproduced from ref. 149 with permission from Elsevier). | ||
5.5 Textile industry
Water-repellent and self-cleaning fibers come handy in the military world where laundering is difficult and inconvenient with regard to both time and scope. On the other hand, in the business sectors, water-repellant and self-cleaning fabrics become useful in preventing undesirable stains. Furthermore, blocking the UV portion of sunlight is another aspect that has long concerned researchers. All these facts eventually underline the massive potential offered by the textile industry for the commercialization of nanotechnological products. Research seeking to manufacture fabrics with water-repellant, self-cleaning and UV-blocking properties has, as a result, surfaced in recent years.150–156 The extended surface area and the correspondingly high surface energy associated with nanostructured materials enhance their affinity for fabrics and induce an improvement in their textile properties. This, therefore, warrants their increasingly widespread applications in the textile sector.157It has been shown in many research investigations that the use of ZnO in the processing of fabrics promotes their anti-bacterial and self-cleaning properties apart from upgrading their UV absorption capacity.158 Moreover, in textile applications, coatings of ZnO in the nano-dimensions aside from being bio-compatible are found to exhibit air-permeability and UV-blocking ability far greater than their bulk equivalents.159 Therefore, ZnO nanostructures have become very attractive as UV-protective textile coatings. Different methods have been reported for the production of UV-protective textiles utilizing ZnO nanostructures. For instance, hydrothermally grown ZnO nanoparticles in SiO2-coated cotton fabric showed excellent UV-blocking properties.160 Synthesis of ZnO nanoparticles elsewhere through a homogeneous phase reaction at high temperatures followed by their deposition on cotton and wool fabrics resulted in a significant improvement in UV-absorbing activity.161 Similarly, ZnO nanorod arrays that were grown onto a fibrous substrate by a low-temperature growth technique provided excellent UV protection.162
Zinc oxide nanowires were grown on cotton fabric by Ates et al.163 to impart self-cleaning, superhydrophobicity and ultraviolet (UV) blocking properties. The ZnO nanowires were grown by a microwave-assisted hydrothermal method and subsequently functionalized with stearic acid to obtain a water contact angle of 150°, demonstrating their superhydrophobic nature, which is found to be stable for up to four washings. The UV protection offered by the resulting cotton fabric was also examined, and a significant decrease in transmission of radiation in the UV range was observed. The self-cleaning activity of the ZnO nanowire-coated cotton fabric was also studied, and this showed considerable degradation of methylene blue under UV irradiation. These results suggest that ZnO nanowires could serve as ideal multifunctional coatings for textiles.
Research on the use of zinc oxide in polyester fibres has also been carried out at Poznan University of Technology and the Textile Institute in Lodz.164 Zinc oxide was obtained by an emulsion method, with particles measuring approximately 350 nm and with a surface area of 8.6 m2 g−1. These results indicate the product's favourable dispersive/morphological and adsorption properties. Analysis of the microstructure and properties of unmodified textile products and those modified with zinc oxide showed that the modified product could be classed as providing protection against UV radiation and bacteria.
5.6 Antibacterial activity
The remarkable antibacterial activity of ZnO has recently received wide interest and efforts are being continuously made to tune its size, concentration, morphology and defects to achieve enhanced properties of ZnO with regards to its powerful nanobiological toxicity.165,166 Currently, Gram-negative Escherichia coli (E. coli) and Gram-positive Staphylococcus aureus (S. aureus) are largely chosen as prototype bacteria to investigate the antibacterial activity of ZnO NPs.167,168 Some other Gram-negative bacteria such as Vibrio cholerae (V. cholerae),169Pseudomonas aeruginosa (P. aeruginosa),170,171Proteus vulgaris (P. vulgaris),73 and other Gram-positive bacteria such as Bacillus subtilis (B. subtilis)172 and Enterococcus faecalis (E. faecalis)173 are also subjected to investigations. It has been observed that ZnO NPs synthesized via green routes responded with greater capacities of growth inhibition of Staphylococcus aureus, Escherichia coli and the like than chemically synthesized ZnO NPs and other commonly used antimicrobials. Due to their small size and higher surface area to volume ratio, there is a marked increase in the bioactivity of ZnO NPs. Although the mechanism by which ZnO NPs exhibit antibacterial activity is still being investigated in the world of research, the latest probes into this topic have revealed a few probable pathways: (a) ZnO NPs are found to directly interact with the cell surfaces causing a disruption of cell membrane permeability,174–176 (b) generation of ROS on the surface of the ZnO NPs, and (c) particle dissolution and subsequent release of free Zn2+ ions in the cytoplasm (and particularly cellular organelles such as lysosomes which have lower pH values) causing a disruption of cellular zinc ion homeostasis which in turn leads to an elevation of the ROS level inside cells, thereby destroying the integrity of the plasma membrane and mitochondrial functions, and ultimately inducing cell necrosis.177–180 It is also possible that the antibacterial effects of ZnO NPs are a cumulative consequence of the aforesaid pathways.Epidemic disease cholera mainly affects populations in developing countries.169,180 It is a serious diarrheal disease caused by the intestinal infection of Gram-negative bacterium V. cholerae. The effective antibacterial activity of ZnO NPs and their mechanism of toxicity were explored against Vibrio cholerae (two biotypes of cholera bacteria (classical and El Tor)) by Sarwar et al.176 Strong arguments and detailed justifications of the toxicity mechanism emerged as a result of this rigorous investigation. The bacterial membrane bears an overall negative charge that can be ascribed to the acidic phospholipids and lipopolysaccharides in it while ZnO NPs possess a positive charge in water suspension. An initial NP–membrane interaction via electrostatic attraction may result from this charge difference following which membrane disruption occurs. As the membrane plays an essential role by maintaining the vital function of the cell, such damage induces depolarization of the membrane, increased membrane permeabilization – loss in membrane potential and protein leakage and denaturation upon subsequent contact with ZnO NPs. Besides, ZnO NPs also have the ability of interacting with DNA as well as forming abrasions on it. Significant oxidative stress was also noticed inside the bacteria cells. They thus arrived at a conclusion that binding of ZnO NPs with the bacterial cell surface induces membrane damage followed by internalization of NPs into the cells, leakage of cytoplasmic content, DNA damage and cell death. Disruption of the membrane by ZnO NPs would additionally give easy access of antibiotics into the cell. Their findings further corroborated a synergic effect produced by the actions of ZnO NPs and antibiotics. They also encountered the antibacterial activity of the ZnO NPs in cholera toxin (CT) mouse models. It emerged that ZnO NPs could induce the CT secondary structure collapse gradually and interact with CT by interrupting CT binding with the GM1 ganglioside receptor.181
In bacteria treated with NPs of ZnO, it was observed that the damage to cell membranes was an inevitable phenomenon. The pathways of the antibacterial activity of ZnO NPs were investigated using Escherichia coli (E. coli) as a prototype organism.182 As was evident from the SEM images of E. coli obtained after treatment with ZnO NPs, a greater number of cell damage sites were noted at higher doses of ZnO NPs. This cell damage has been ascribed to pathways involving both the presence and absence of ROS. In the absence of ROS, the interaction of ZnO NPs with bacterial membranes would lead to damage to the molecular structure of phospholipids culminating in cell membrane damage.
Jiang et al.183 studied the potential antibacterial mechanisms of ZnO NPs against E. coli. They reported that ZnO NPs with an average size of about 30 nm caused cell death by coming into direct contact with the phospholipid bilayer of the membrane and destroying the membrane integrity. The significant role of ROS production in the antibacterial properties of ZnO NPs surfaced when it emerged that the addition of radical scavengers such as mannitol, vitamin E, and glutathione could block the bactericidal action of ZnO NPs. However, the antibacterial effect triggered by Zn2+ released from ZnO NP suspensions was not apparent. Reddy synthesized ZnO NPs with sizes of ∼13 nm and investigated their antibacterial (E. coli and S. aureus) activities.168 It was discovered that ZnO NPs effected complete cessation of the growth of E. coli at concentrations of about 3.4 mM but induced growth inhibition of S. aureus at much lower concentrations (≥1 mM). Besides, Ohira and Yamamoto184 also discovered that the antibacterial (E. coli and S. aureus) activity of ZnO NPs with small crystallite sizes was far more pronounced than for those with large crystallite sizes. From ICP-AES measurement, it emerged that the amount of Zn2+ released from the small ZnO NPs was much higher than from the large ZnO powder sample and E. coli was more sensitive to Zn2+ than S. aureus. This is a further confirmation that eluted Zn2+ ions from ZnO NPs also play a key role in antibacterial action.
Iswarya et al.,185 having extracted crustacean immune molecule β-1,3-glucan binding protein (Phβ-GBP) from the haemolymph of Paratelphusa hydrodromus, successfully designed Phβ-GBP-coated ZnO NPs. The Phβ-GBP-ZnO NPs were spherical shaped having a particle size of 20–50 nm and halted the growth of S. aureus and P. vulgaris. S. aureus was found to be more prone to the bactericidal action of Phβ-GBP-ZnO NPs than P. vulgaris. In addition, Phβ-GBP-ZnO NPs could induce drastic modification in cell membrane permeability and set off outrageous levels of ROS formation both in S. aureus and P. vulgaris. This work was thus pivotal in bringing to the forefront the immensely great antibacterial hallmark of Phβ-GBP-ZnO NPs.
The mechanism of breaking into bacterial cells by membrane disruption and then inducing oxidative stress in bacterial cells, thereby stalling cell growth and eventually causing cell death has been reported in many recent research studies.186–191 Important bacterial biomolecules can also adsorb on ZnO NPs. Bacterial toxicity, in the recent past, has been heavily reported to have resulted from structural changes in proteins and molecular damage to phospholipids.192 The antibacterial activity of ZnO NPs thus finds its apt application in the discipline of food preservation. As a formidable sanitizing agent, it can be used for disinfecting and sterilizing food industry equipment and containers against attack and contamination by food-borne pathogenic bacteria. ZnO NPs showed both toxicity on pathogenic bacteria (e.g., Escherichia coli and Staphylococcus aureus) and beneficial effects on microbes, such as Pseudomonas putida, which has bioremediation potential and is a strong root colonizer.193
Investigations into the antibacterial activities of ZnO micro-sized particles, ZnO NPs, and ZnO NPs capped with oxalic acid against S. aureus were carried out in the presence and absence of light.194 It was observed that the efficiency of ZnO NPs was just 17% in the dark. However, their antibacterial properties saw a surge up to 80% upon application of light. The antibacterial behaviour was greatest for ZnO NPs while it was minimum for ZnO micro-sized particles, suggesting a higher release of Zn2+ ions from ZnO NPs than ZnO micro-sized particles. The examination revealed that surface defects of the ZnO NPs boosted ROS production in the presence as well as absence of light. Additionally, it was also found that capping lowers the amount of superoxide radicals generated because capping blocks the oxygen vacancies that are chiefly accountable for the generation of superoxide radicals. In another investigation, the influence of NP size on bacterial growth inhibition by ZnO NPs and the mechanistic routes of their action were demonstrated.195 ZnO NPs with diameters ranging from 12 nm to 307 nm were first generated. Thereafter, they were administered to Gram-positive and Gram-negative microorganisms (Fig. 18). The results clearly illustrated the greater bactericidal efficacy of smaller ZnO NPs under dark conditions. The use of UV light resulted in an enhanced antibacterial behaviour of ZnO NPs owing to the enhanced formation of ROS from them. The antibacterial properties were rooted in the generation of ROS and the build-up of ZnO nano-sized particles in the cytoplasm and on the external membranes.
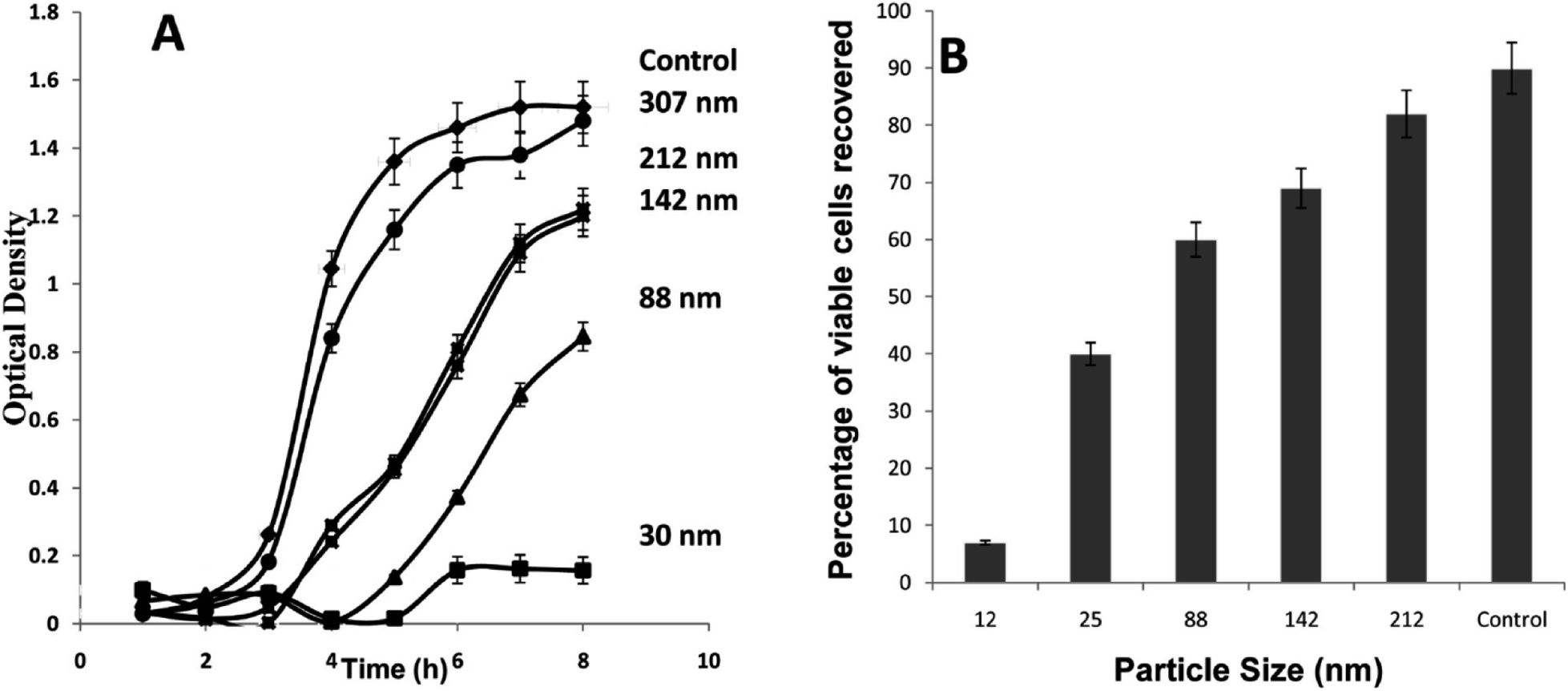 | ||
| Fig. 18 The influence of different sizes of ZnO NPs on the growth of a methicillin sensitive S. aureus strain. (A) Growth analysis curves obtained by tracking the optical density at 600 nm. (B) Percentage of viable S. aureus recovered after treatment with ZnO NPs of different sizes (reproduced from ref. 195 with permission from the American Chemical Society). | ||
In another intriguing investigation, the toxicity induced in antibiotic resistant nosocomial pathogens such as Acinetobacter baumannii (A. baumannii) and Klebsiella pneumoniae (K. pneumoniae) by photocatalytic ZnO NPs was studied.196 It was seen that A. baumannii and K. pneumoniae were significantly destroyed by 0.1 mg mL−1 of ZnO nano-structures with 10.8 J cm−2 of blue light. Further, the mechanistic pathway of the antibacterial activity of photocatalytic ZnO NPs against antibiotic defiant A. baumannii was investigated. While cytoplasm leakage and membrane disruption of A. baumannii were evident after treatment with ZnO NPs under blue light exposure, there was no sign of plasmid DNA fragmentation. Therefore, membrane disruption could be associated with the mechanistic route via which the photocatalytic ZnO NPs demonstrated antibacterial activity. The possibility of the role of DNA damage therein was categorically ruled out.
A novel approach comprising a combined application of ultrasonication and light irradiation to ZnO NPs has been developed to boost their antibacterial properties.197 The sono-photocatalytic activity of ZnO nanofluids against E. coli was tested. The results revealed a 20% rise in the antibacterial efficacy of ZnO nanofluids. Further, ROS generation by ZnO nanofluids played a crucial role in bacterial elimination. The sono-photocatalysis of ZnO nanofluids also enhanced the permeability of bacterial membranes, inducing more efficacious penetration of ZnO NPs into the bacteria.
Although ZnO NPs make a promising antibacterial agent owing to their wide-ranging activities against Gram-positive as well as Gram-negative bacteria, the exact antibacterial pathway of ZnO NPs has not been adequately established. Hence, deep investigations into it hold a lot of important theoretical and practical value. In the future, ZnO NPs can be explored as antibacterial agents, such as ointments, lotions, and mouthwashes. Additionally, they can be overlayed on various substrates to prevent bacteria from adhering, spreading, and breeding in medical devices.
5.7 Drug delivery
Nanotechnology has enabled the exploration of better and more efficient avenues concerning drug delivery vehicles. Various research studies suggest the potency of ZnO NPs for drug delivery in a number of diseases. ZnO NPs encapsulated with chitosan, bearing enhanced stability, were found to be effective for importing doxorubicin (DOX) into cancer cells in a recent study.195 In various other studies, ZnO NPs were successfully used as vehicles for gene delivery and gene silencing (Fig. 17 and 18).198–200 The use of this apparatus for gene delivery had advantages. For example, the expression of plasmid containing genes on the surface of nanoparticles could ensure safe and efficient gene targeting to the recipient tissues201,202 enabling the nanoparticles to be used as suitable tools for targeting genes to various cells such as tumor cells. Nie et al.199 in their study discovered that synthesized ZnO tetrapod-like nanostructures could be used as pioneering vehicles for gene delivery. They showed that silica-coated amino-modified tetrapod-like ZnO nanostructures could, via electrostatic interactions, effectively bind to DNA which in turn improved the transfection efficiency of melanoma cells. In another study conducted by Zhang et al., it was unveiled that polycation-capped ZnO quantum dots had the potential to deliver DNA into COS-7 cells.203 Further, the utilization of this apparatus additionally enabled real-time imaging of gene delivery.Reports bearing evidence of the applications of ZnO NPs in the delivery of chemotherapeutic agents to treat cancers have emerged prolifically in the last few years. For instance, a porous ZnO nanorod based DDS (ZnO-FA-DOX), enclosing folic acid (FA) as a targeting agent and doxorubicin (DOX) as a chemotherapeutic drug, was fabricated by Mitra et al.204 The ZnO-FA-DOX nano-apparatus was found to exhibit pH-triggered release of DOX and potent cytotoxicity in MDA-MD-231 breast cancer cells. The biocompatible nature of the ZnO-FA material, as observed from the acute toxicity study in a murine model also emerged from the investigation. In another research study by Zeng et al.,205 a lymphatic-targeted DDS with lipid-coated ZnO-NPs (L-ZnO-NPs) enclosing 6-mercaptopurine (6-MP) as an anticancer agent was designed. The L-ZnO-NP apparatus demonstrated pH-susceptive drug release and remarkable cytotoxicity to cancer cells as a result of the generation of intracellular reactive oxygen species (ROS). Liu et al.206 also reported the fabrication of DOX-loaded ZnO-NPs. The researchers encountered a pH-susceptive drug release from the DDS and diminished drug efflux with enhanced cytotoxicity in drug defiant breast cancer cells (MCF-7R). Likewise, Li et al.207 fabricated a novel DDS enclosing hollow silica nanoparticles (HSNPs) embedded with ZnO quantum dots to co-deliver DOX and camptothecin. The nano-apparatus evinced pH-susceptive drug release and cytotoxicity to drug defiant cancer cells. A research study used ZnO NPs as caps to cover the pores of mesoporous silica NPs (MSNs), and when the designed drug delivery apparatus came into contact with acids, there took place a decomposition of ZnO NPs followed by a release of doxorubicin (DOX) molecules from the MSN nanostructures.208 One major drawback of such an apparatus was that it had difficulty in degradation thereby resulting in an incomplete release of drugs.209,210 Another scheme employed the technique of loading drugs onto the ZnO NPs directly.211 Upon contact with acids, the drug molecules are released following the complete decomposition of ZnO NPs. In another investigation, a liposome-incorporated ZnO-NP based DDS (ZNPs-liposome-DNR) enclosing anticancer drug daunorubicin (DNR) was designed by Tripathy et al.212 The incorporation of ZnO-NPs in the DDS was observed to prevent the premature release of DNR, which could be prompted only in acidic medium, thereby efficiently exerting an anticancer effect on A549 cells. The study of intracellular release in cancer cells with confocal laser scanning microscopy (CLSM) revealed that treatment with ZNPs-liposome-DNR induced a marked DNR release, causing greater cytotoxicity to cancer cells, compared to pure DNR and DNR-conjugated liposomes (liposome-DNR), as evidenced by the green fluorescence intensity. For the treatment of lung cancer, Cai et al. accomplished the construction of ZnO quantum dot-based drug delivery apparatus that was conjugated with a targeting agent (hyaluronic acid) and an anticancer agent (DOX).213 The nano-apparatus demonstrated CD44 receptor-specific uptake and pH-driven drug release in lysosomal compartments of the cancer cells. Kumar et al.214 also designed sub-micron sized self-assembled spherical capsules of ZnO nanorods that successfully effected the delivery of anticancer agent DOX to K562 cancer cells. Furthermore, Han et al.215 also synthesized ZnO NPs conjugated with an aptamer as a functionalization agent and DOX as an anticancer agent and demonstrated the effect of combined chemo- and radiation therapy in MCF-7 breast cancer cells employing the nano-apparatus. Recently, Zhang et al.216 as a part of their investigation devised a new scheme to restrain the proliferation of human hepatocarcinoma cells (SMMC-7721) via a combined application of ZnO nanorod based DNR in photodynamic therapy (PDT), where ROS generation had the possibility to play a key role in the net anticancer behaviour of the hybrid nano-apparatus (Fig. 19). The researchers further discovered that ZnO NPs were able to transport a larger quantity of DNR via internalization into SMMC-7721 cells, thereby inducing outstanding restraint on the multiplication of these cancerous cells. Besides, UV irradiation on this drug delivery nano-apparatus further reinforced the arrest of cell proliferation through photocatalysis of ZnO nanorods. To look into the signaling pathway of anticancer activity of the DDS in PDT, the researchers monitored the caspase-3 activity, which is a hallmark of apoptosis. The results of immunocytochemistry study confirmed that upon treatment with a DNR–ZnO nanocomposite under UV irradiation, the cells demonstrated far more pronounced activation of caspase-3 molecules in cancer-afflicted cells. It was consequently proposed that ZnO nanorods could raise the drug's targeting efficiency and minimize the associated toxicity. Therefore, the DNR–ZnO hybrid nano-apparatus with UV irradiation was claimed to have the potential of a fruitful scheme for the treatment of cancers (Fig. 20 and 21). In another study, Ye et al.217 using a copolymerization process also prepared water soluble ZnO–polymer core–shell quantum dots, and designed a drug delivery apparatus based on these quantum dots containing Gd3+ ions and anticancer drug DOX. The ZnO-Gd-DOX nano-system was found to be biocompatible, pH-responsive and led to a marked release of DOX into the acidic environment of cancer-afflicted cells and tumors. When administered to human pancreatic cancer (BxPC-3) tumor containing nude mice, this polymer-modified drug delivery nano-apparatus was found to display higher therapeutic efficiency compared to the FDA-approved liposomal DOX formulation DOXIL at 2 mg kg−1 DOX concentration. The histopathology study and ICP-AES analysis of the vital organs further confirmed that this ZnO-Gd-DOX nano-apparatus could substantially bring about growth-inhibition of tumors without exerting any toxic effects 36 days post administration. Additionally, the histopathology study of tumor sections also demonstrated severe damage to the tumor cells caused by the administration of the DDS, compared to the control, DOX and DOXIL groups.
 | ||
| Fig. 19 Schematic illustration of possible processes of ZnO nanorods encapsulating chemotherapeutic agents for anticancer therapy (reproduced from ref. 216 with permission from Elsevier). | ||
 | ||
| Fig. 20 Possible mechanism of ROS production by ZnO nanorods under UV irradiation (reproduced from ref. 216 with permission from Elsevier). | ||
 | ||
| Fig. 21 Cytotoxicity of DNR or the DNR–ZnO nanocomposite in the absence or presence of UV irradiation against SMMC-7721 cells. The inset graph shows the IC50 of DNR and the DNR–ZnO nanocomposite in the absence or presence of UV irradiation for SMMC-7721 cells (reproduced from ref. 216 with permission from Elsevier). | ||
5.8 Anti-cancer activity
Cancer, a condition of uncontrolled malignant cell proliferation, has typically been subjected to treatment by chemotherapy, radiotherapy, and surgery during the past several decades. Although in theory all these therapies appear to be exquisitely effective in killing cancer cells, these non-specific therapy methods also introduce a lot of serious side effects.218 Recently, nanomaterial-based nanomedicine possessing high biocompatibility, easy surface functionalization, cancer targeting, and drug delivery capacity has displayed the much coveted flair to rise above these side effects. Substantial research progress has been made in biocompatible nanosized zinc oxide (ZnO) because of its unique physical properties. ZnO nanomaterials, inspired by the fact that Zn2+ is an essential nutrient for adults, are considered to be safe in vivo. Considering these leverages, ZnO NPs can be aptly chosen as biocompatible and biodegradable nano-platforms and can also be investigated for cancer treatment.219,220Several studies have thus suggested the cytotoxic effects of ZnO NPs on cancer cells. The cancer cell viability percentage on the MCF7 cell line, A549 cell line, HL60 cell line and VERO cell line has been studied at various concentrations of ZnO. Results show that the cell viability of the above cell lines exhibits a marked decrease with a rise in ZnO concentration221,222 with minimal damage to healthy cells.
The mitochondrial electron transport chain is known to be closely linked to intracellular ROS generation, and anticancer agents accessing cancer cells could impair the electron transport chain and release huge amounts of ROS.223,224 However, an inordinate amount of ROS brings about mitochondrial damage thereby resulting in the loss of protein activity balance that eventually induces cell apoptosis.225 ZnO NPs introduce certain cytotoxicity in cancer cells chiefly by a mechanism that involves a higher intracellular release of dissolved Zn2+ ions, followed by enhanced ROS induction and induced cancer cell death by way of the apoptosis signaling pathway. The effects of ZnO NPs on human liver cancer HepG2 cells and their possible pharmacological mechanism were investigated by Sharma et al.226 They observed that ZnO NP-exposed HepG2 cells exhibited higher cytotoxicity and genotoxicity, which were related to cell apoptosis conciliated by the ROS triggered mitochondrial route. The loss of the mitochondrial membrane potential led to the opening of outer membrane pores following which some related apoptotic proteins including cytochrome c were released into the cytosol thereby activating the caspase in due course. Mechanistic studies had proved that the loss of mitochondrial membrane potential-mediated HepG2 cell apoptosis was mainly due to the decrease in mitochondrial membrane potential and Bcl-2/Bax ratios as well as accompanying the activation of caspase-9. Besides, ZnO NPs could noticeably activate p38 and JNK and induce and attract p53ser15 phosphorylation but this was not dependent on JNK and p38 pathways (Fig. 21). These results afforded valuable insights into the mechanism of ZnO NP-induced apoptosis in human liver HepG2 cells. Moghaddam et al.227 took recourse to biogenic synthesis and successfully generated ZnO NPs using a new strain of yeast (Pichia kudriavzevii GY1) and examined their anticancer activity in breast cancer MCF-7 cells. ZnO NPs have been observed to exhibit powerful cytotoxicity against MCF-7 cells. This cytotoxicity is affected more likely via apoptosis than cell cycle arrest. The apoptosis induced by ZnO NPs was largely by way of both extrinsic and intrinsic apoptotic pathways. A few antiapoptotic genes of Bcl-2, AKT1, and JERK/2 were subjected to downregulation, while upregulation of some proapoptotic genes of p21, p53, JNK, and Bax was prompted. ZnO NPs have been widely employed in cancer therapy and reported to promote a selective cytotoxic effect on cancer cell proliferation. Chandrasekaran and Pandurangan evaluated the cytotoxicity of ZnO nanoparticles against cocultured C2C12 myoblastoma cancer cells and 3T3-L1 adipocytes. The study revealed that ZnO NPs could be more cytotoxic to C2C12 myoblastoma cancer cells than 3T3-L1 cells. Compared to 3T3-L1 cells, it emerged that ZnO NPs stalled C2C12 cell proliferation and brought about a more pronounced apoptosis by way of a ROS-conciliated mitochondrial intrinsic apoptotic route, an upregulation of p53, tempered Bax/Bcl-2 ratio, and caspase-3 routes.228
In a study, biogenic zinc oxide nanoparticles (ZnO NPs) were developed from aqueous Pandanus odorifer leaf extract (POLE) with spherical morphology and approximately 90 nm size.229 The anticancer activity of the ZnO NPs was evaluated by MTT assay and neutral red uptake (NRU) assays in MCF-7, HepG2 and A-549 cells at different doses (1, 2, 5, 10, 25, 50, and 100 μg mL−1). Moreover, the morphology of the treated cancer cells was examined by phase contrast microscopy. The results suggest that the synthesized ZnO NPs inhibited the growth of the cells when applying a concentration from 50–100 μg mL−1. Overall, the study demonstrated that POLE derived biogenic ZnO NPs could serve as a significant anticancer agent. Phytomediated synthesis of metal oxide nanoparticles have become a key research area in nanotechnology due to its wide applicability in various biomedical fields. The work by Kanagamani et al.230 explored the biosynthesis of zinc oxide nanoparticles (ZnO-NPs) using Leucaena leucocephala leaf extract. Biosynthesized ZnO-NPs were found to have a wurtzite hexagonal structure with particles distributed in the range of 50–200 nm as confirmed by TEM studies. The anticancer activity of ZnO-NPs against MCF-7 (breast cancer) and PC-3 (human prostate cancer) cell lines was evaluated using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. From the assay, biosynthesized ZnO-NPs were found to have better cytotoxic activity on PC-3 cell lines than MCF-7 cell lines. The in vitro cytotoxicity studies of biosynthesized ZnO-NPs against Dalton lymphoma ascites (DLA) cells revealed better antitumor activity with 92% inhibition at a ZnO-NP concentration of 200 μg mL−1, and as the concentration increased, the anticancer efficiency also increased. These results suggested that ZnO NPs could selectively induce cancer cell apoptosis making them a bright candidate for cancer therapy.
Photodynamic therapy requires the administration of a photosensitizing agent that is subjected to activation by light of a specific wavelength thereby generating ROS. The application of ZnO NPs as effective photosensitizers can be ascribed to their capability to generate ROS in response to visible light or UV light. Recent studies exhibited that photo-triggered toxicity of ZnO NPs renders them aptly suitable for targeted PDT in a spatiotemporal manner, providing a surer way to selectively terminate cancerous cells.231–234 An attempt was made to utilize the synergic effects of anticancer drugs with ZnO NPs in PDT to induce cell-death in cancer cells.231 The cytotoxic effects of daunorubicin (DNR), an anti-cancer drug, on drug defiant leukemia K562/A02 cancer cells were put to the test in combination with ZnO NPs. The combination of DNR and ZnO NPs under UV irradiation could appreciably check the proliferation of drug-defiant cancer cells in a dose-dependent manner. Additionally, ZnO NPs were found to induce an enhanced cellular uptake of DNR.
An anticancer treatment using DNR-conjugated ZnO nanorods in PDT was investigated with human hepatocarcinoma cells (SMMC-7721) (Fig. 19).216 The fabrication of photo-excited ZnO nanorods with DNR displayed an outstanding boost in the anticancer properties of the ZnO nanorods (Fig. 20). The ZnO nanorods raised the intracellular concentration of DNR and augmented the anticancer efficiency. This is further evidence of the drug carrying capacity of ZnO nanorods into target cancer cells. UV irradiation additionally reinforced the growth inhibition of cancerous cells via photocatalytic activity of ZnO nanorods. In this study, the promoted mortality of cancer cells indicates that ZnO nanorods under UV irradiation could efficiently induce the formation of ROS and further attack the cell membrane (mainly by lipid peroxidation), nucleic acids, and proteins (such as enzyme deactivation). The mechanism of ROS generation of ZnO nanorods under UV irradiation is displayed in Fig. 21. ZnO is a direct band gap semiconductor with a band gap energy of 3.36 eV at room temperature, high exciton binding energy of 60 meV and high dielectric constant, which under UV irradiation will produce a hole (h+) in the valence band and an electron (e−) in the conduction band, namely electron/hole pairs. These electron/hole pairs will induce a series of photochemical reactions in an aqueous suspension of colloidal ZnO nanorods, generating ROS. Generally, at the surface of the excited ZnO nanorods, the valence band holes abstract electrons from water and/or hydroxyl ions, generating hydroxyl radicals (˙OH). Electrons reduce O2 to produce the superoxide anion O2−˙. ZnO nanorods can be one of the promising nanomaterials for PDT in cancer.
The size of ZnO NPs has been reported to have a strong association with their anticancer activities. The UV light-activated anti-cancer effects of various ZnO NPs with different sizes have been examined against human hepatocarcinoma cells (SMMC-7721).232 To achieve synergetic cytotoxicity, a combination of ZnO NPs and an anticancer agent, DNR, was subjected to investigation. A schematic illustration of the anticancer effect of DNR-conjugated ZnO NPs under UV irradiation is shown in Fig. 22. The outcome showed higher cytotoxicity of smaller NPs. UV irradiation greatly boosted the cytotoxic effect on SMMC-7721 cells treated with ZnO NPs via generation of ROS and a consequent cell apoptosis. Additionally, when the ZnO NPs were conjugated with DNR, their cytotoxicity against the cancer cells further increased by leaps and bounds.
 | ||
| Fig. 22 The schematic image of ZnO nanoparticle cytotoxicity and the PDT process in cooperation with daunorubicin in vitro (reproduced from ref. 232 with permission from SpringerOpen). | ||
To secure concomitant intracellular drug delivery and PDT for cancer treatment, poly(ethylene glycol) (PEG)-capped ZnO NPs enclosing DOX were fabricated.233 It was found that DOX-loaded PEG-ZnO NPs on exposure to UV irradiation achieved significantly enhanced cell cytotoxicity through light-driven ROS production from the NPs. The synergistic anticancer activity of a combined treatment with PEG-ZnO NPs and DOX under UV irradiation came to the fore as a result of this investigation.
Likewise, poly(vinylpyrrolidone) (PVP)-capped ZnO nanorods (PVP-ZnO nanorods) were designed as a drug carrying nano-apparatus for the delivery of daunorubicin (DNR), as well as a photosensitizer for PDT.234 The DNR-loaded PVP-ZnO nanorods (DNR-PVP-ZnO) encouraged an exceptional upswing in the anticancer activity of DNR due to elevated cellular uptake of the DNR delivered by the nanorods. The DNR-PVP-ZnO nanorods also demonstrated efficient PDT under UV light irradiation. It has been demonstrated that NPs can furnish solutions to confront the acute demerits of conventional photosensitizers.235 By a dramatic enhancement of the solubility of photosensitizers, NPs can facilitate their increased cellular internalization. They also upgrade the target-specificity of photosensitizers by way of passive targeting to tumor tissues through the enhanced permeability and retention (EPR) effect. Further, cell-specificity of photosensitizers can be remarkably increased by surface modification of the NPs to bind active targeting components. Complexation of ZnO NPs with other photosensitizers has been widely researched to increase the efficacy of ZnO NPs in PDT by synergistically enhancing the ROS generation.234,235Meso-tetra(o-aminophenyl)porphyrin (MTAP)-conjugated ZnO nanocomposites were fabricated and examined for synergistic PDT against ovarian cancer cells.236 The MTAP-ZnO NPs induced generation of ROS upon UV irradiation, the controlling parameters being concentration and light intensity. It emerged that 30 μM MTAP-ZnO NPs wielded high light-induced toxic effects in cancer-afflicted ovarian cells under UV illumination, while they remained inactive in the dark. The cytotoxic activity of MTAP-ZnO NPs under UV illumination was markedly boosted weighed against that of porphyrin alone.235 This study elucidated the targeted and synergistic PDT by nanoparticles of ZnO loaded with photosensitizing substances. ZnO NPs were combined with protoporphyrin IX (PpIX) as a drug delivery nano-apparatus for photosensitizers.237 Simple ZnO NPs and PEG-capped ZnO NPs were synthesized and examined for their cancer-eliminating effect against human muscle carcinoma cells. In the absence of laser light, ZnO NPs at 1 mM concentration were found to exert very low cytotoxicity (98% viability). In the presence of 630 nm laser light, PEG-capped ZnO NPs loaded with PpIX exhibited outstanding cytotoxicity owing to the increased ROS generation. Additionally, a high build-up of PpIX in the tumor area was observed when it was delivered by ZnO NPs, exhibiting the potency of ZnO NPs as a tumor-selective drug delivery system for photosensitizers.
5.9 Anti-diabetic activity
Diabetes mellitus has been deemed to be among the most serious public health scares, and according to the WHO 2014 estimation, there are more than 400 million adults with diabetes all over the world.238 It has been known to mankind for more than 2000 years.239 Diabetes mellitus is a cluster of lifelong progressive metabolic diseases caused by the body's incapacity to produce insulin or by the ineffective use of the insulin produced which in turn induces a high blood glucose level (hyperglycemia).240–244 Insulin therapy often forms a critical part of the treatment for people with diabetes. The goal of insulin therapy is to maintain the optimum blood glucose level. Nevertheless, the use of insulin gives rise to several complications, viz., insulin resistance, fatty liver, brain atrophy, etc.247 This has prompted a number of scientific investigations in recent years to discover new alternative therapeutic schemes for checking diabetes via a nanomedicine approach. Zinc is a trace element and a mineral found in abundance in all human tissues and tissue fluids. Zinc has a distinct reputation for maintaining the structural integrity of insulin. It plays a vital role in the secretion of insulin from pancreatic cells. It also participates in the synthesis, storage, and secretion of insulin.245 The deficiency of zinc has further been observed to be related to the progress of diabetes and associated complications. All these reasons make zinc-based therapy an attractive strategy for scientists to combat diabetes. Accordingly, the anti-diabetic potential of ZnO has been put under scrutiny by a large number of researchers of late.A natural extract of red sandalwood (RSW) as an effective anti-diabetic agent in conjugation with ZnO NPs has been tested by Kitture et al.246 The anti-diabetic activity was evaluated with the help of α-amylase and α-glucosidase inhibition assay with murine pancreatic and small intestinal extracts. Results revealed that the ZnO–RSW conjugate effected a moderately higher percentage of inhibition (20%) against porcine pancreatic α-amylase and proved more effective against the crude murine pancreatic glucosidase than either of the two components alone (RSW and ZnO NPs). The conjugated ZnO–RSW induced 61.93% inhibition in glucosidase while the bare ZnO NPs and RSW exhibited 21.48 and 5.90% inhibition, respectively.
In an investigation conducted to compare the anti-diabetic activity and oxidative stress of ZnO NPs and ZnSO4 in diabetic rats it was observed that ZnO NPs with small dimensions at higher doses (3 and 10 mg kg−1) had a much greater antidiabetic effect compared to ZnSO4 (30 mg kg−1). The observation was backed up by a marvelous decline in the blood glucose level, a steep rise in the insulin level and a refinement of the serum zinc status in a time- and dose-dependent manner. However, it was finally inferred in the study that ZnO nanoparticles severely elicited oxidative stress particularly at higher doses corroborated by the altered erythrocyte antioxidant enzyme activity, enhancement in malondialdehyde (MDA) production, and remarkable drop in serum total antioxidant capacity.240 Hyperglycemia can squarely trigger off an inflammatory state via activation of C-reactive protein (CRP) and cytokines, such as interleukins, eventually resulting in the development of cardiovascular diseases. Hussein et al. designed ZnO NPs using hydroxyl ethyl cellulose as a stabilizing agent to alleviate diabetic complications.247 The study demonstrated that ZnO NPs could significantly decrease malondialdehyde (MDA), fast blood sugar and asymmetric dimethylarginine (ADMA) levels. The inflammatory markers, interleukin-1 (IL-1α) and CRP, were also notably lowered after ZnO NP treatment, concomitant with a rise in nitric oxide (NO) and serum antioxidant enzyme (PON-1) levels in diabetic rats.
An investigation was conducted in 2014 into the anti-diabetic potential of ZnO NPs in streptozotocin-induced diabetic albino (Sprague-Dawley) rats.243 The researchers inferred that the administration of ZnO NPs in diabetic rats brought about a marked lowering of the blood glucose level, boosted the serum insulin level, and elicited the expression of insulin receptor and GLUT-2 proteins, suggesting the inherent capacity of ZnO NPs for diabetic remedy. The anti-diabetic activity of ZnO NPs in streptozotocin-induced diabetic (types 1 and 2) Wistar rats was also demonstrated by Umrani et al. in their research work.248 The research revealed that ZnO NPs raised the levels of parameters like glucose, insulin, and lipid in rats attesting to the efficient anti-diabetic activity of ZnO NPs. The same research group recently undertook an enquiry into the mechanistic pathway behind the anti-diabetic properties of ZnO NPs in vitro.249 They demonstrated that ZnO NPs led to protein kinase B (PKB) activation, enhanced glucose transporter 4 (GLUT-4) translocation and uptake of glucose, reduced glucose 6 phosphatase expression, proliferation of pancreatic beta cells, etc., which were critically responsible for the anti-diabetic behaviour of ZnO NPs. The antidiabetic effectiveness of ZnO nanoparticles prepared using U. diocia leaf extract for treating alloxan-caused diabetic rats was evaluated.250 From the characterization of the samples, the envelopment of extract over the ZnO-extract sample resulted in individual particles with enhanced properties compared to bulk ZnO. The occurrence of the nettle phytochemicals linked to the ZnO-extract sample was verified by various techniques, especially using TGA, FT-IR, and GC-MS analyses. Among all the employed treatments, the ZnO-extract performed the best for controlling the common complications accompanying diabetes. This biologically produced sample significantly lowered the levels of Fasting Blood Sugar (FBS), Total Triglycerides (TG), and Total Cholesterol (TC) and enhanced the high-density lipoprotein cholesterol (HDLC) and insulin levels in the diabetic rats when compared to the rest of the remedies. The results confirmed the synergistic relationship between ZnO and U. diocia leaf extract where ZnO-extract performed the best compared with the only extract and ZnO. From the results, the as-prepared ZnO-extract sample can be introduced as a non-toxic, applicable, and active phyto-nanotherapeutic agent for controlling diabetes complications.
ZnO nanoparticles were synthesized using a microwave-assisted method in the presence of Vaccinium arctostaphylos L. fruit extract.251 A decrease in crystallite size was observed for the biologically synthesized ZnO compared to the chemically synthesized sample. Furthermore, the existence of organic moieties over the biologically synthesized ZnO NPs was approved using characterizing methods. Then, the alloxan-induced diabetic rats were divided into an untreated diabetic control group and a normal healthy control group, and the groups received insulin, chemically synthesized ZnO, plant extract, and biologically synthesized ZnO. After treatment, fasting blood glucose (FBS), high-density lipoprotein (HDL), total triglyceride (TG), total cholesterol (TC) and insulin were measured. Analysis showed a significant decrease in FBS and increase in HDL levels in all groups under treatment. However, the results for cholesterol reduction were only significant for the group treated with biologically synthesized ZnO. Despite the changes in the triglyceride and insulin levels, the results were not significant. For all the studied parameters, bio-mediated ZnO NPs were found to be the most effective in treating the alloxan-diabetic rats compared to the other studied treatment agents. All reports of ZnO NPs for diabetes treatment indicated that ZnO NPs could be employed as a promising agent in treating diabetes as well as attenuating its complications.
5.10 Anti-inflammatory activity
Inflammation is a part of the complex biological response of body tissues to harmful stimuli, viz., pathogens, damaged cells, or irritants.252 It functions to localize and eliminate the injurious agent and to remove damaged tissue components so that the body can begin to heal. The response consists of changes in blood flow, an increase in permeability of blood vessels and the migration of fluid proteins and white blood cells (leukocytes) from the circulation to the site of tissue damage. Inflammation is thus a defense mechanism that evolved in higher organisms to protect them from infection and injury. In the wake of several bio-medical applications of ZnO NPs, the anti-inflammatory effects of ZnO NPs have garnered wide interest among researchers. Atopic dermatitis (AD) is a chronic inflammatory skin disease. It is characterized by a significant disruption of the skin-barrier which is associated with complex interaction between genetic and environmental factors.253,254 Textiles have the longest and most intense contact with the human skin. Wiegand investigated the role of ZnO-functionalized textile fibers in the control of oxidative stress in AD in vitro and in vivo.255 It emerged from the study that there was an obvious improvement in AD pruritus and subjective sleep quality when AD patients wore the ZnO textiles overnight on 3 consecutive days. This is likely owing to the high anti-oxidative and intense antibacterial potential of the ZnO textile. Ilves et al.256 explored whether ZnO NPs of different sizes would be able to penetrate injured skin and injured allergic skin in the mouse AD model. Their experiments clearly offered vivid evidence of the fact that only nano-sized ZnO was able to reach into the deep layers of the allergic skin, while bulk-sized ZnO stayed in the upper layers of both damaged and allergic skin. Compared with bulk-sized ZnO, nano-sized ZnO exerted higher anti-inflammatory effects by drastically cutting down on pro-inflammatory cytokines (IL-10, IL-13, IFN-γ, and Th2 cytokines) in the mouse model of AD. These results confirmed that ZnO NPs with a small size had remarkable effects on reducing skin inflammation in AD models. The anti-inflammatory activity of ZnO NPs is not just restricted to the treatment of atopic dermatitis but has also produced great results in combatting other inflammatory diseases. Nagajyothi et al. made ZnO NPs using the root extract of P. tenuifolia and the anti-inflammatory activity was investigated in LPS-stimulated RAW 264.7 macrophages.257 ZnO NPs exhibited outstanding anti-inflammatory activity via dose-dependently suppressing NO production as well as the related protein expressions of iNOS, COX-2, IL-1β, IL-6, and TNF-α. The ZnO NPs synthesized by Thatoi et al. under photoconditions using the aqueous extracts of two mangrove plants, Heritiera fomes and Sonneratia apetala, were found to possess a higher potential for anti-inflammatory activity (79%) compared to silver nanoparticles (69.1%).258 Thus, ZnO NPs also have the potential to be employed for anti-inflammatory treatment.5.11 Immunotherapy
In the past few decades, immunotherapy, via the delivery of immune molecules (antigens, adjuvants, and antibodies), has become a leading-edge strategy to battle against different diseases.259 However, delivery of these molecules in a targeted manner to achieve therapeutic benefits has remained a major challenge. In this context, nanotechnology plays a key role in the development of schemes for delivering profitably different types of immune molecules for vaccination, adjuvant therapy, and immunotherapy. Recently, ZnO NPs garnered ample interest in the field of immunotherapy. For instance, Roy et al.260 brought the adjuvant effect of ZnO NPs when intraperitoneally administered with ovalbumin (OVA) to the forefront. They illustrated that the serum levels of IgG1 and IgE were significantly increased as a result of treatment with ZnO NPs. In another study,259 researchers fabricated iron oxide–zinc oxide core–shell nanoparticles and demonstrated their cell imaging feature and delivery of carcinoembryonic antigen (CEA) to dendritic cells (DC). The study reported that after immunizing the mice with DC loaded with zinc oxide/iron oxide nanocomposites impregnated with ZBP (ZnO binding peptide) and CEA, enhanced tumor regression was observed with an improved survival rate, compared to other treatment equivalents, including ZBP-CEA, nanoparticle/CEA, CEA, nanoparticles and only DC (Fig. 23).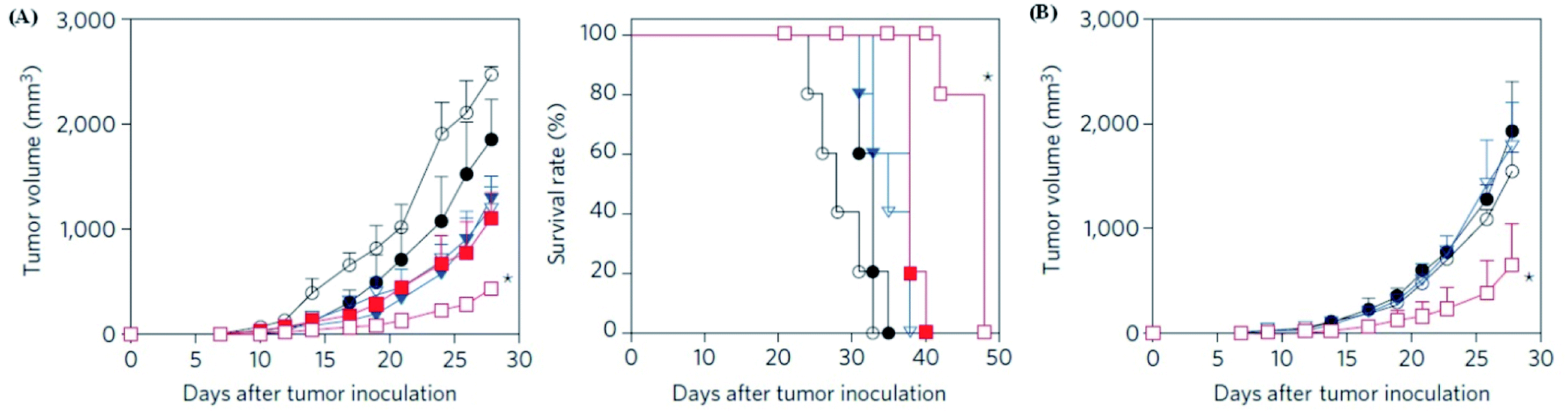 | ||
| Fig. 23 Tumor growth and survival of immunized mice. (A) Tumor volume (left) and survival rate (right) of mice (five mice per group) injected with MC38/CEA cells. (B) Tumor growth in human CEA-transgenic mice (five mice per group) inoculated with MC38/CEA cells (reproduced from ref. 259 with permission from Nature Publishing Group). | ||
5.12 Wound healing
Wound healing is a complex and remarkable biological course of action a body resorts to in order to replace devitalized and missing cellular structures and tissue layers. The process usually consists of four phases: coagulation of blood, inflammation, proliferation, and tissue remodeling.261,262 It is widely known that the healing process is subjected to a setback under several pathological circumstances such as major blood loss, diabetes, antibacterial infection, etc.,263 which warrants the treatment of wounds urgently with utmost care.264 The use of several wound dressings made of natural (alginate, chitosan, gelatin, etc.)265 and synthetic (polyvinyl alcohol, polyurethane, polylactic acid, etc.)266–268 polymers to heal wounds is quite common. However, synthetic polymer-based wound dressings present some disbenefits such as toxicity of the degraded product and non-hemostatic properties. Notwithstanding that natural polymers are biocompatible, non-immunogenic and biodegradable, wound dressings containing these polymers too suffer from some disadvantages. For instance, the ionic compounds of alginate, especially calcium salts, might induce swelling and degradation while chitosan (CS) is greatly amenable to degradation by lysozyme.261 In the light of these complications, researchers seek incorporation of nano-materials into wound dressing materials to prevail over these drawbacks. Among different metal-based nanomaterials, ZnO NPs often find use in wound-healing applications. In an investigation by Diez-Pascual et al.269 castor oil (CO)-based biocompatible polymeric films containing CS-decorated ZnO-NPs (≤5 wt%) were developed and their wound-healing capacity in Sprague-Dawley rats was put under scrutiny. They demonstrated that the hybrid nano-wound-healing-apparatus was able to effect wound-healing in rats at a greater pace than effected by the administration of only castor oil-based polymer or gauze (control experiment), as clearly evident from the snapshots of wounds at different points of time (Fig. 24). | ||
| Fig. 24 Photographs of the wounds treated with (1) CO/CS-ZnO (5.0 wt%), (2) CO, and (3) gauze as a control at (A) day 0, (B) day 5, and (C) day 14 (reproduced from ref. 269 with permission from the American Chemical Society). | ||
The ensuing results revealed that, two weeks after administration, the synthesized nanocomposite induced a 90% reduction of wound area, while mere 70% wound repair was noted in the control experiment thereby bearing evidence of its commanding wound-fixing capacity. Augustine et al.270 also fabricated ZnO NP decorated polycaprolactone (PCL) scaffolds and demonstrated that their implantation was able to boost faster wound-fixing by elevating the proliferation and migration of fibroblasts in an in vivo model (wound-healing model of American satin guinea pigs), without showing any marked signs of inflammation. Similarly, Bellare et al.271 designed biocompatible ZnO NP based scaffolds of gelatin and poly(methyl vinyl ether)/maleic anhydride (PMVE/MA) with remarkable antibacterial effects. Their report threw light on the ability of the scaffolds for endothelial progenitor cell (EPC) adhesion and proliferation. Further, the topical application of the scaffolds on wounds of Swiss/alb mice displayed the potential to expedite the process of wound-fixing. Modern wound care materials suffer from several serious shortcomings that include inadequate porosity, inferior mechanical strength, lessened flexibility, lack of antibacterial properties, etc. Given this backdrop, a CS hydrogel/nanoparticulate ZnO-based bandage which exerted antibacterial effects against both Gram-negative (E. coli) and Gram-positive (S. aureus) bacteria was fabricated by Kumar et al.272 The nanocomposite bandage characterized by biodegradability, microporosity and biocompatibility produced elevated wound healing in Sprague-Dawley rats and boosted re-epithelialization and collagen deposition at a remarkable pace. Taking into account the crucial factors of biocompatibility, antibacterial effects, and wound-fixing capacity, the researchers held that the hybrid nanomaterial-based bandage could be valuable for the healing of chronic wounds, burn wounds, diabetic foot ulcers, etc. Likewise, a porous bandage consisting of ZnO NPs conjugated with alginate hydrogel and exhibiting blood clotting capacity and bactericidal effects against E. coli, S. aureus, Candida albicans, and methicillin resistant S. aureus was fabricated by Mohandas et al.261 The bandage made from the nanocomposite was observed to exhibit biocompatibility at a lower concentration of ZnO-NPs. Further, an ex vivo re-epithelialization investigation with porcine ear skin demonstrated that faster wound-fixing was effected by the hybrid nanomaterial-based bandage than only alginate control bandage. This was ascribed to the release of zinc ions that would enhance the proliferation and migration of keratinocyte cells to the wound area. Nair et al.273 also developed a bandage consisting of a biocompatible nanocomposite of ZnO NPs conjugated with β-chitin hydrogel. The bandage showed efficient antibacterial activity (against S. aureus and E. coli) and had the ability of blood clotting and activation of platelets. It was elucidated that the application of the bandage on wounds of Sprague Dawley rats led to faster healing, with enhanced collagen deposition and a reduced number of bacterial colonies than in the control experiment, indicating the remarkable wound repairing capacity of the hybrid nanomaterial-based bandage. A novel, biocompatible ZnO QDs@GO-CS hydrogel was constructed by Liang et al.274 through the simple assembly of ZnO quantum dots (QDs) with GO sheets and via a simple electrostatic interaction with the loaded CS hydrogel. The antibacterial efficacy could reach 98.90% and 99.50% against S. aureus and E. coli bacteria, respectively, with a low-cost, rapid, and effective treatment. ZnO QDs in antibacterial nanoplatforms could immediately produce ROS and Zn2+ under acidic intracellular conditions. In parallel, when exposed to 808 nm laser irradiation, hyperthermia from GO sheets could simultaneously kill bacteria. Thus, the excellent performance of the material stems from the combined effects of hyperthermia produced under the near-infrared irradiation of GO sheets, reactive oxygen species, the release of Zn2+ from ZnO QDs under an acidic environment, and the antibacterial activity of the hydrogel. This work demonstrated that the synergy of antibacterial nanoplatforms could be used for wound anti-inflammatory activity in vivo indicated by the wound healing results. The hybrid hydrogel caused no evident side effects on major organs in mice during wound healing. Therefore, the biocompatible multimodal therapeutic nanoplatforms were proposed to possess great potential for antibacterial activity and wound healing. In a study by Dodero et al.,275 the possibility of using for biomedical purposes alginate-based membranes embedding ZnO nanoparticles that were prepared via an electrospinning technique was extensively evaluated. The morphological investigation showed that the prepared mats were characterized by thin and homogeneous nanofibers (diameter of 100 ± 30 nm), creating a highly porous structure; moreover, EDX spectroscopy proved ZnO-NPs to be well dispersed within the samples, confirming the efficiency of the electrospinning technique to prepare nanocomposite membranes. Mouse fibroblast and human keratinocyte cell lines were used to assess the biological response of the prepared mats; cytotoxicity tests evidenced the safety of all the samples, which overall showed very promising outcomes in terms of keratinocyte adhesion and proliferation. In particular, the strontium- and barium-cross-linked mats were characterized by similar cell viability results to those obtained with a commercial porcine collagen membrane used as a control; moreover, except for the calcium-cross-linked sample, the prepared mats exhibited a good stability over a period of 10 days under physiological conditions. Antibacterial assays confirmed the proficiency of using ZnO nanoparticles against E. coli without compromising the biocompatibility of the membranes. The mechanical properties of the strontium cross-linked mats were similar to those of human skin (i.e., Young's modulus and tensile strength in the range 280–470 MPa and 15–21 MPa for the samples with and without nanoparticles, respectively), as well as the water vapor permeability (i.e., 3.8–4.7 × 10−12 g m−1 Pa−1 s−1), which was held to be extremely important in order to ensure gas exchange and exudate removal; furthermore, due to the low moisture content (i.e., 11%), the prepared mats could be easily and safely stored for quite a long period without any negative effect on their properties. Consequently, the achieved results demonstrated that the prepared mats could be successfully employed for the preparation of surgical patches and wound healing products by using alginate as an economic and safer alternative to the commonly employed commercial animal collagen-derived membranes.
Ahmed et al.276 fabricated chitosan/PVA/ZnO nanofiber membranes by using the electrospinning technique. The samples of chitosan/PVA and chitosan/PVA/ZnO tested for antibacterial efficacy and antioxidant potential demonstrated very encouraging results in diabetic wound healing. The nanofiber mats displayed outstanding antibacterial properties against various strains of bacteria. The samples of chitosan/PVA and chitosan/PVA/ZnO nanofiber membranes also manifested higher antioxidant properties which made them promising candidates for applications in diabetic wounds. In experiments involving diabetic rabbits, chitosan/PVA and chitosan/PVA/ZnO nanofiber mats exhibited increased performance of wound contractions in a time interval of 12 days. It was thus concluded in the study that the chitosan/PVA/ZnO nanofibrous membranes could serve as useful dressing materials for diabetic wounds, a major problem for type-2 diabetic patients worldwide.
5.13 Agriculture
ZnO NPs have been observed to increase the growth and yield of food crops. In peanuts, ZnO induced considerable stimulation of seed germination, seedling vigor, and stem and root growth.277 A colloidal solution of ZnO is used as a nanofertilizer. Apart from providing the plants with nutrients, the nanofertilizer also revives the soil to an organic state without the harmful impacts resulting from the use of a chemical fertilizer. What gives a nanofertilizer a cutting edge is that it can be used in very low quantities. In a study by Mahajan et al.,278 it was observed that there was stimulated growth in the roots and shoots of mung seedlings till the concentration of ZnO reached 20 ppm from 1 ppm and beyond this significant retardation was noted. In the same study an optimum ZnO concentration of 1 ppm was found to excite growth in gram seedlings. The effective growth at a specific optimum concentration and a retarded growth beyond this concentration were ascribed to the accumulation and uptake of ZnO NPs by the roots. The key role played by the concentration of nano-sized ZnO NPs was also reported by Prasad et al.277 in their work on the benefits in the usage of ZnO NPs in agriculture. An optimum concentration of 1000 ppm of ZnO NPs led to an improvement of germination, root growth, shoot growth, dry weight and pod yield of the treated peanut seeds. Likewise, at 1.5 ppm concentration of ZnO NPs, Burman et al.279 noted an improved shoot dry weight and antioxidant activity in chickpea seedlings. Raliya et al.280 also reported a boost in plant growth and nutrient content in cluster bean seedlings upon treatment with 10 ppm ZnO NPs. In another study, enhanced plant height and dry weight were observed in maize plants.2815.14 Photodegradation
Studies involving fabrication of metal oxide semiconductors for the photocatalytic degradation of Persistent Organic Pollutants (POPs) in water have been conducted on a regular basis of late with an eye to construct a low-cost, non-toxic and highly efficient photocatalyst. ZnO with its low cost, non-toxicity and high absorption efficiency across a large range of the solar spectrum seems to fit the bill.282,283 This resulted in various enthusiastic investigations that have been carried out in recent years to design ZnO NPs that can successfully exploit the Advanced Oxidation Process (AOP) mechanism thereby causing enhanced photodegradation of otherwise recalcitrant POPs via very pronounced generation of hydroxyl (˙OH) radicals.Ishwarya et al.73 reported the degradation of methylene blue dye in the presence of ZnO NPs prepared using Ulva lactuca seaweed extract and solar irradiation in their study. With an optimum initial dye concentration of 25 ppm and an optimum catalyst loading of 200 mg, the dye present in 100 mL of water got degraded to 90.4% in 120 min.
Gawade et al.75 carried out photocatalytic degradation of methyl rrange dye using green fabricated ZnO NPs. 81% of the dye (20 ppm) was degraded after 100 min exposure to UV light. This they carried out after the dye solution was stirred with the catalyst for 30 min in the dark for complete equilibrium of the adsorption–desorption phenomenon when 2% of the dye was found to be adsorbed. The optimum catalyst dose was observed to be 1.5 g dm−3 after the dose had been varied in between 0.1 and 2.0 g dm−3. The increase in degradation efficiency is ascribed to two favourable factors: (a) an increase in the number of active sites and (b) an increase in the number of photons absorbed by the catalyst. Beyond the optimal quantity of the catalyst, aggregation of the catalyst results in the active sites on the catalyst surface becoming unavailable for light absorption. The turbidity of the suspension leading to the inhibition of photon absorption on the catalytic surface of ZnO NPs because of the scattering effect was cited as an additional cause for the lowered degradation efficiency after the optimal catalyst dose.
Vidya et al.76 performed the photodegradation of Congo red dye using ZnO NPs. With an optimal dose of 60 mg ZnO NPs in 250 mL of dye solution in which the concentration had been optimized at 30 ppm, they achieved a 90% degradation in an hour's time while maintaining an optimum pH of 9. They reported that at very low pH, the ZnO NPs display a marked tendency to agglomerate and the photon absorption on to the metal oxide surface reduces and in Congo red, the azo linkage (–NN–) is prone to electrophilic attack by the hydroxyl radicals. At low pH, despite excess H+ concentration, the H+ ion interacts with azo linkage decreasing the electron densities at the azo groups thereby discouraging the onslaught of hydroxyl radicals via the electrophilic mechanism. At high pH, electrostatic repulsion between the negatively charged ZnO catalyst and the negatively charged anionic Congo red dye hinders efficient photocatalysis. Using ZnO NPs synthesized by the sol–gel route, Senthilkumaar et al.284 in their brief study, achieved total mineralization of Crystal Violet (CV) dye at pH 10.0 for 15 min of UV irradiation. The degradation was found to follow first order kinetics. The adsorption of the dye onto the ZnO surface in the absence of light was found to follow the Langmuir isotherm. In their study Daniel E. León et al.285 performed photodegradation of an antibiotic cefotaxime under simulated solar irradiation. The ZnO dose was optimized at 1.5 g L−1. An optimum pH of 7.5 was maintained during the experiment. The photoreaction showed 60% efficiency and attained equilibrium after 60 min. It was found to follow pseudo-first order kinetics and the rate constant was evaluated to be 0.466 min−1.
Enhanced photocatalytic activity of the Mg doped ZnO/reduced graphene oxide nanocomposite has been recently reported by Nithiyadevi et al.286 They investigated photodegradation of cationic dyes Methylene Blue (MB) and Malachite Green (MG) under visible light irradiation. They achieved a 94.41% degradation of MB and a 99.56% degradation of MG after exposure to visible light for 75 min in each case. Both the photocatalytic degradations showed a marked increase in efficiency in comparison with that effected by bare ZnO NPs. They obeyed pseudo-first order kinetics and the rate constant assumed values of 0.0391 and 0.0493 min−1 respectively in the case of MB and MG. They cited the following reasons for the enhanced photocatalytic ability of the nanocomposite: (a) the introduction of reduced graphene oxide (RGO) enabled better adsorption of dye molecules through π–π conjugation between the dye and aromatic compounds of RGO, (b) the ability of RGO to facilitate the growth of the ZnO particles on RGO sheets, (c) the availability of a large reactive surface area, (d) the greater interfacial contact between ZnO and RGO, (e) increase in the lifetime of charge carriers most probably attributed to RGO, (f) narrowing of the energy bands of ZnO due to Mg2+ substitution, (g) presence of oxygen vacancies and (h) the reduction of particle size.
Photodegradation of Methylene Blue (MB) was performed by Debasmita Sardar et al.287 with an Ag-doped-ZnO nanocatalyst. On increasing the percentage of loaded Ag the rate of photocatalytic decomposition gradually increased and reached the maximum for 20% Ag loading on ZnO. The rate constant was found to be 0.0087 min−1 with a fairly high degradation efficiency of 55.87%. However, a sharp fall in the value of rate constant was observed for 25% Ag loading which remained almost the same on further increasing the Ag content, i.e. for 30 and 35% loading. It was also observed from TEM analysis that too much loading of Ag led to agglomeration and thus covered up the surface of ZnO preventing light absorption. Moreover, there were large numbers of unattached Ag nanoparticles which could be oxidized in the presence of reactive oxygen species. Oxidized silver would not initiate any charge separation in the system. It was thus assumed that silver up to this optimum amount could act as an electron–hole separation centre. Beyond the optimum amount, it could help in charge carrier recombination. In fact, a large number of negatively charged Ag particles (which had already accumulated electrons) on ZnO could capture holes and thus would start acting as a recombination site itself essentially by forming a bridge between an electron and a hole. Thus, the efficiency of charge separation and hence the photocatalytic capability declined to an appreciably large extent.
Very recently, Vaianoa et al.288 too tried photo-catalytically favourable modification of ZnO by Ag. They too achieved similar results with regard to removal of phenol from water. The loading of Ag responded favourably in the range of 0.14–0.88 wt% but backfired beyond 1.28 wt%. Similar reasons as mentioned above were cited for the trends observed. A photocatalytic test was thus performed by using 0.15 g of the optimized catalyst (1% Ag/ZnO) to treat drinking water containing phenol with an optimized initial concentration of 50 mg L−1 in 100 mL aqueous solution. Near-complete mineralization was accomplished within 180 min of exposure to UV irradiation. Photoreaction was found to fit in the pseudo-first order kinetic model. Another investigation289 reported a facile microwave assisted synthesis of two-dimensional ZnO nano-triangles with a band gap of around 3.33 eV. The as-synthesized ZnO nano-triangles were applied for the reduction of noxious p-nitroaniline within 50 min. They were further used for the effective elimination of Rose Bengal dye within 150 min.
Likewise, ZnO-nanorods were synthesized290 by adopting a facile microwave assisted green route of synthesis for the complete reduction of nitro compounds. Lauric acid was used as a complexing and capping agent in the ethanol phase. The nanorods had an average diameter of 5.5–10.0 nm with a hexagonal crystal structure and further demonstrated unusual luminescence properties wherein high intensity UV and yellow emission bands were observed along with negligible blue and green emission bands. Toxic nitro-compounds p-nitrophenol, p-nitroaniline and 2,4,6-trinitrophenol were completely reduced into amino derivatives by NaBH4 in the presence of these nanorods within 120, 45, and 18 min, respectively.
Chidambaram et al.291 effectively constructed a ZnO/g-C3N4 heterojunction using a facile, economically viable pyrolysis synthetic route for the photodegradation of methylene blue under visible light illumination. The nanocomposites prepared using 0.1, 0.2 and 0.3 molar ratios of zinc nitrate precursor are labeled 0.1ZnO/GCN, 0.2ZnO/GCN and 0.3ZnO/GCN, respectively. The nanocomposites are found to exhibit a fall in charge recombination corroborated by their photoluminescence spectra that showed a fall in the intensity of the concerned emission peak (Fig. 25). A maximum photodegradation of 86% was achieved with 0.2ZnO/GCN in 60 min following a pseudo-first order kinetic rate constant of 0.032 min−1 while graphitic carbon nitride, 0.1ZnO/GCN and 0.3ZnO/GCN attained 44%, 73% and 76% degradation of methylene blue dye in the same time with lower rate constants. The loading of ZnO over g-C3N4 sheets created a heterojunction (Fig. 26). The excitation of electrons by visible light occurs from the valence band to the conduction band of g-C3N4. The excited electrons are transferred to the conduction band of ZnO while there occurs a simultaneous movement of holes from the valence band of ZnO to the valence band of g-C3N4via the smooth interface of the heterostructure. This enabled the generation of the superoxide anion radical and hydroxyl radicals that effected improved mineralization of the dye. An excess of Zn was deemed to cause recombination of photo-induced charges that led to decreased photocatalytic efficiency. In a recent investigation by the authors of the current work,292 a destructive photocatalyst made up of ZnO nanorods/Fe3O4 nanoparticles anchored onto g-C3N4 sheets was synthesized using hydrothermal synthesis and ultrasonication techniques. HRTEM micrographs shed light on the coupling of Fe3O4 nanoparticles with ZnO nanorods and the successful formation of the intended ternary heterojunction. The g-C3N4 sheets fostered close contact between ZnO nanorods and Fe3O4 nanoparticles thereby inducing a mellowed agglomeration of nanostructured ZnO/Fe3O4 particles. The Tauc plot derived from UV-visible absorbance data showed that the ZnO/Fe3O4/g-C3N4 nano-hybrid had a band gap of 2.48 eV. PL studies further confirmed the successful development of a staggered type II heterojunction with wide separation between light-induced charge carriers (Fig. 27). The hybrid catalyst showed remarkable photocatalytic activity under visible light, as evident from the efficient degradation of pantoprazole, a pharmaceutical drug widely known as a recalcitrant organic water pollutant. This could be attributed to the synergistic interactions between ZnO, Fe3O4 and g-C3N4. A degradation efficiency of 97.09% was achieved within 90 min with a remarkable pseudo-first order rate constant of 0.0433 min−1. The incorporation of Fe3O4 expectedly facilitated the ready recovery of the catalyst and the degradation efficiency displayed fair consistency up to 4 cycles. The work thus offered a cost-efficient strategy for tackling organic water pollutants.
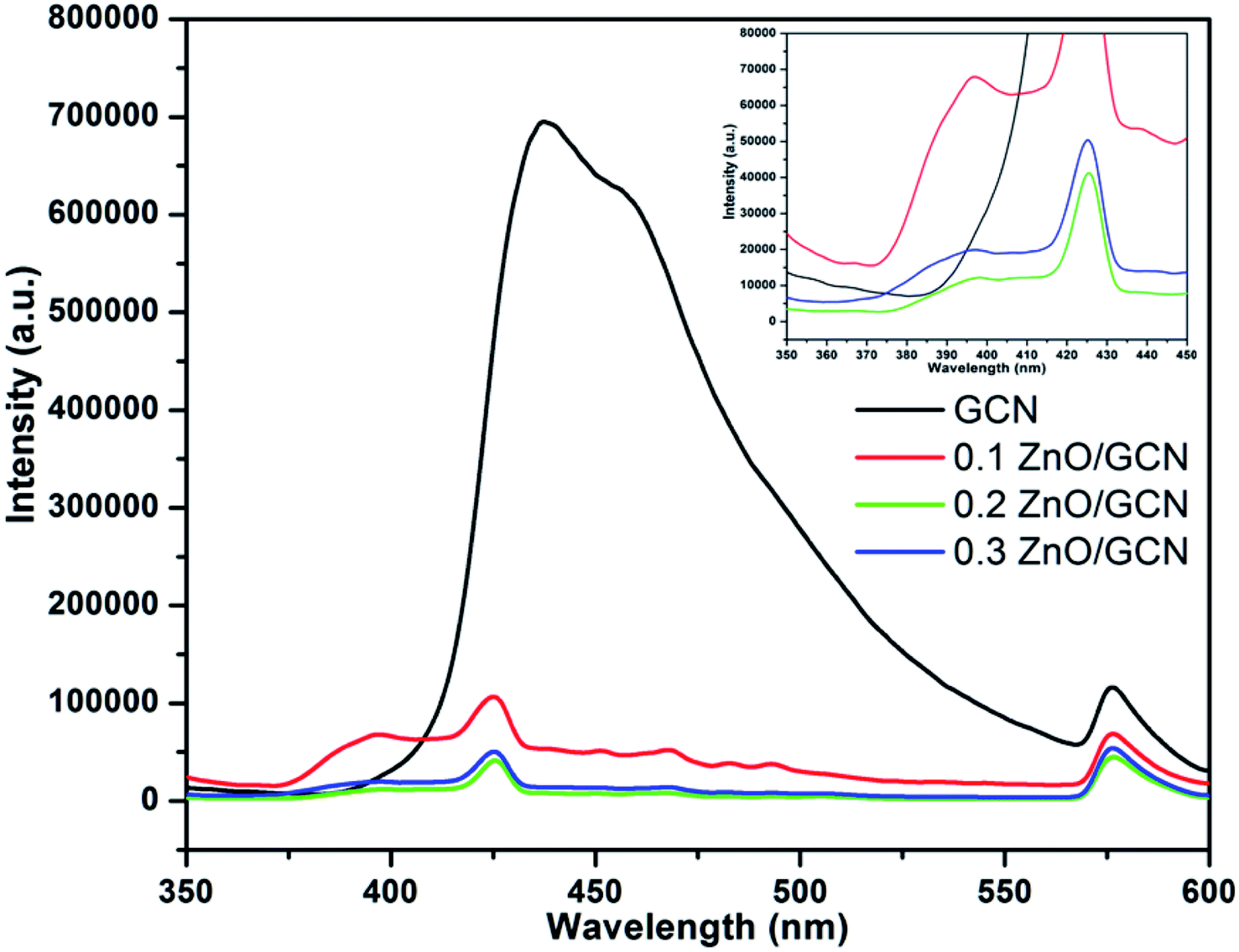 | ||
| Fig. 25 Photoluminescence spectra of GCN and ZnO/GCN nanocomposites (the inset shows the enlarged PL spectra in the wavelength region of 350–450 nm) (reproduced from ref. 291 with permission from IOP Publishing). | ||
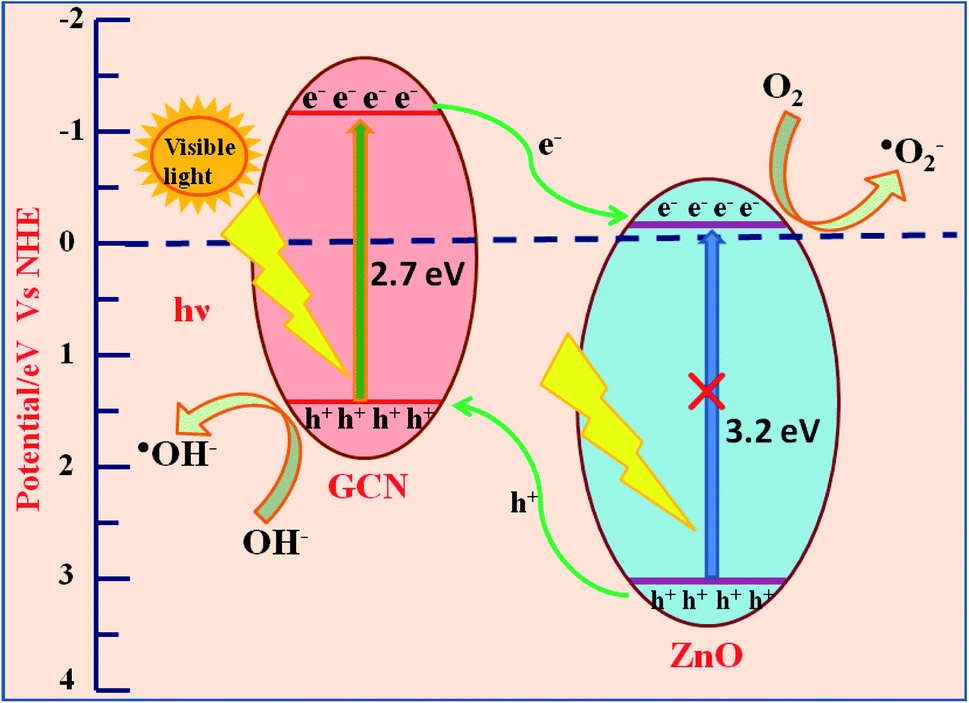 | ||
| Fig. 26 Schematic depiction of the photocatalytic degradation mechanism of the ZnO/GCN heterojunction (reproduced from ref. 291 with permission from IOP Publishing). | ||
 | ||
| Fig. 27 Schematic depiction of the photocatalytic degradation mechanism of the g-C3N4/ZnO/Fe3O4 heterojunction (reproduced from ref. 292 with permission from Elsevier). | ||
In another study,293 a facile generation of a quaternary nano-structured hybrid photocatalyst, g-C3N4/NiO/ZnO/Fe3O4, was proposed for photodegradation of an ecotoxic pharmaceutical drug, esomeprazole, in aqueous solution. The photocatalytic annihilation of esomeprazole as a prototypical organic contaminant was executed under LED irradiation. By itself the designed ternary heterojunction accomplished a maximum 95.05% photodegradation of esomeprazole and a TOC removal of 81.66% and COD reduction up to 70.68% under optimum conditions of catalyst dose, esomeprazole concentration and pH within 70 min at a superior pseudo-first order kinetic rate constant of 0.06616 min−1. This actually implied an improvement of degradation over NiO/ZnO, g-C3N4/NiO and g-C3N4/ZnO up to ∼74, ∼57, and ∼42%, respectively. The specific reaction rate also went up remarkably by almost ∼3.8, ∼3.18, and ∼2.85 times in comparison with the values obtained for NiO/ZnO, g-C3N4/NiO and g-C3N4/ZnO, respectively. The remarkable photocatalytic potential of the heterostructured photocatalyst in practical applications was evident from its reconcilable performances under varying initial concentrations of esomeprazole and initial pH of the solution. The effect of the addition of H2O2 was also put under scrutiny and it was found that the photocatalytic degradation, TOC removal and COD reduction increased to 98.43, 84.72, and 73.86%, respectively, upon addition of an optimum quantity of H2O2 over the same time span. The impacts made by inorganic and organic species on photodegradation and the associated reaction kinetics were investigated and the results were reported. The inhibiting influence of water matrices on esomeprazole degradation was also evaluated for better assessment of the performance of the designed photocatalyst in a real aqueous environment.
CdS/ZnO photocatalysts were prepared by two steps via hydrothermal and photochemical methods for the photodegradation of rhodamine B (RhB) dye.294 The UV/Vis absorption spectra revealed that the absorption performance of the heterostructure is extended toward the visible light region. The photocatalytic activities of both ZnO nanorod and CdS/ZnO heterostructures were investigated for the photodegradation of RhB dye. It was found that the CdS/ZnO heterostructure prepared with 30 min light illumination shows the best photocatalytic efficiency compared to the one at 15 min and pure ZnO nanorods. The better and enhanced photocatalytic efficiency of the CdS/ZnO heterostructure was ascribed to the high charge separation efficiency. The maximum photocatalytic efficiency of 85% was achieved within 8 h with the CdS/ZnO-30 min photocatalyst.
The photocatalytic degradation of rhodamine B (RhB) over chlorophyll-Cu co-modified ZnO catalysts (Chl-Cu/ZnO) was studied under visible-light irradiation by Worathitanon et al.295 It was found that chlorophyll as an electron donor and copper in Cu2+ form help inhibit the recombination of electron–hole pairs and improve the photoactivity of the catalyst. The synergistic effect between chlorophyll and Cu was found to improve the visible-light response of ZnO nanoparticles, resulting in excellent performance in photodegradation of RhB. The appropriate ratio of chlorophyll and Cu loadings over ZnO was 0.5Chl-0.10Cu/ZnO. At this ratio, under visible-light irradiation for 2 h, the degradation efficiency was approximately 99% (60 mg L−1 of RhB solution), of which 18% of RhB adsorption occurred under dark conditions. Moreover, outstanding reusability of Chl-Cu/ZnO, for up to six cycles, was found, with more than 80% degradation efficiency.
In yet another investigation,296 ZnO nanowires (NWs) were successfully synthesized onto commercially available civil engineering materials using a hydrothermal synthesis method. This easy and low-cost method allowed obtaining an almost homogeneous repartition of nanostructures on the entirety of the surface of the substrates. The measured gap values were similar to those of the ZnO NWs grown on typical substrates, i.e., ∼3.18 eV and 3.20 eV for concrete and tiling, respectively. The excellent photocatalytic efficiency of our samples was demonstrated on three commonly used dyes, namely, Methyl Orange (MO), Methylene Blue (MB) and Acid Red 14 (AR 14). All of the dyes were fully degraded in less than 2 h for MB and AR 14, and less than 3 h for the more difficult to degrade MO. Investigating the durability of the samples so prepared, very promising results were found, as they showed no loss of efficiency after four experiment cycles. The ability of implementing ZnO NWs on civil engineering materials, their good photocatalytic properties, and the possibility to re-use samples with minimal efficiency losses, even after several months, were found very promising for the use of the nanostructures as road surfaces for air or water depollution.
6. Toxic impacts and mechanisms of ZnO NPs
Several toxicological studies have been carried out on zinc oxide nanoparticles in recent years. These studies revealed the potential adverse effects of ZnO NPs on health and the environment. ZnO NPs have been revealed to be seriously toxic to bacteria, Daphnia magna, freshwater microalgae, mice, and even human cells.297–301 Although the exact mechanistic pathways of the interaction of ZnO NPs with living cells are yet to be known, studies have demonstrated their engagement with cell membranes, DNA and proteins and the key role they play in producing oxidative stress. Animals in certain regions face significant exposure to ZnO NPs that can have toxic effects on them. Rats on being in contact with ZnO NPs were found to demonstrate biochemical abnormalities in blood and pathological lesions in the stomach, liver and kidney.302 Another study on male mice showed drastic physiological changes in them; an increase in the weight of the brain, pancreas and lungs accompanied by altered blood enzyme behaviour and mRNA expressions was observed.303 The study conducted on male Sprague Dawley rats by Tang et al.304 reported adverse effects on cytokines, enzyme activity, oxidative stress, histological and hematological parameters of liver and mRNA expression. Renal functions were also found to be disrupted by exposure to ZnO NPs. Tweaked oxidative stress and subdued catalase and superoxidase activity were observed in affected rats.305 Bone marrow affected by oxidative stress and liver-DNA degradation were also reported in yet another investigation.306 Exposure to ZnO NPs also leaves a host of pathological effects on aquatic organisms as has emerged from various investigations carried out on fish as a representative of an aquatic animal. Altered gene expression and subdued embryo development have been reported in zebrafish.307 Similar abnormalities in the growth of zebrafish embryos were also noted by Du et al.308 Exposure to ZnO NPs could also upset immunity and the serum protein level.309 The toxic effects of ZnO NPs on plants have also been recorded in several studies. One investigation on Arabidopsis noted an 80% reduction in plant growth and 50% decline in chlorophyll.310 Studies have also revealed that ZnO NPs reduced the number of roots and stunted the length of rice seedlings,311 induced drastic inhibition of chlorophyll-synthesis and reduced the efficiency of photosynthesis in Arabidopsis.312 Yet another investigation showed their consequences on germination as well as shoot-growth of Brassica nigra.313 Toxicological studies of ZnO NPs on ryegrass show that ZnO NPs cause a considerable reduction in the biomass of ryegrass and shrinking of the root tip. Further, in the presence of ZnO NPs, root epidermal as well as cortical cells of ryegrass became highly vacuolated and collapsed.314 These days ZnO NPs are regularly used in sunscreen creams due to their intrinsic ability to block UVA and UVB radiations effectively. However, these nanoparticles can easily penetrate the skin to reach the viable cells thereby imposing potential toxicity.The toxicity mechanism of ZnO-NPs in zebrafish was investigated by Yu et al.315 The toxicity caused by ZnO is primarily because of the release of Zn2+ ions and through mechanical damage in zebrafish. ZnO-NPs induced elevation of intracellular Zn2+ concentration, leading to over-generation of intracellular reactive oxygen species, leakage of plasma membrane, dysfunction of mitochondria, and ultimately cell death.316 Therefore, it is demonstrated that cell uptake, intracellular dissolution and release of Zn2+ are the inherent causes for high toxicity of ZnO-NPs. However, there are some disagreements regarding the role of dissolved Zn2+ in the toxicity mechanisms of ZnO-NPs. Several researchers suggested that dissolved Zn2+ from ZnO-NPs played a minor role in the toxicity of ZnO-NPs,317,318 while other investigations indicated that most of the toxicity of ZnO-NPs is due to the dissolved Zn2+.315,316 This discrepancy may be ascribed to the sensitivities of different organisms to dissolved Zn2+, such as single tissue cells, bacteria, zebrafish and so on. In the study of Stella et al.,319 dissolved Zn2+ from nZnO was considered to play the vital role in the toxicological mechanisms, which was inferred from the levels of the biomarkers of metallothionein (MT) and heat shock protein 70 (HSP70) in the body of O. melastigma larvae, but this dissolved Zn2+ was obtained by filtering the ZnO-NP suspensions with a 0.1 μm sterile syringe filter and it might include ZnO-NPs whose diameters were smaller than 100 nm.
The dissolution of Zn2+ ions from ZnO was also suggested to be the main mechanism for the toxicity of ZnO-NPs as claimed recently.320,321 Li et al.322 also reported the same mechanism for the toxicity of ZnO-NPs. They have studied the toxicity of ZnO-NPs with various initial concentrations to E. coli in ultrapure water, NaCl and PBS solutions. For higher concentrations of ZnO-NPs, although a few ZnO particles may attach to the bacterial cells, it was difficult to determine the contribution of nano-ZnO itself considering the high toxicity of co-existing Zn2+ ions. In addition, bacteria could also release the solutes in response to osmotic down-shock in ultrapure water, resulting in damage to the normal physiological functions and the decrease of tolerance of bacteria to toxicants.323 Therefore, the toxicity of nano-ZnO at 1 mg L−1 in ultrapure water was much higher than that in 0.85% NaCl solution. To confirm the toxicity mechanism of ZnO-NPs, the ultrastructural characteristics of normal E. coli cells and those treated with ultrapure water, ZnO-NPs, and Zn2+ ions were investigated with TEM by Li and his research group. The morphologies of E. coli cells treated with ZnO-NPs or Zn2+ ions were significantly different from those of normal E. coli cells. The cytoplasmic membranes were deformed, wherein some cells swelled and the intracellular substances leaked out under both Zn stress and osmotic stress. Combined with the toxicity results of nano-ZnO, bulk-ZnO, and Zn2+ ions in ultrapure water, Li and co-workers concluded that the toxicity of nano-ZnO to E. coli was mainly attributed to the released Zn2+ ions.
7. Challenges and prospects
Although a large variety of morphologies, particle sizes and structures have been designed for engineered ZnO nano-sized structures, no accurate mechanisms have been put forward elucidating their formation during the reported procedures. Although investigations concentrating on the influence of the conditions of synthesis on the structure and morphology have made inroads, there is still a long way to go until a comprehensive understanding of the effects of the synthesis parameters on structure, particle size and morphology has been achieved.With higher electron diffusivity than TiO2, high electron mobility, exceptionally large exciton binding energy, low cost and considerable stability against photo-corrosion, ZnO has been widely considered a perfect substitute for TiO2 as the electron transport material in DSSCs and PSCs. However, ineffective surface passivation, interfacial charge recombination and long-term stability have collectively yielded poor electron injection efficiency and thereby low current density and efficiency of the ZnO based photovoltaic device. Probable remedies involve incorporation of organic and inorganic dopants for effective surface passivation and effecting surface modification for marked electronic contact. Poor control of the properties of individual building blocks and low device-to-device reproducibility are further areas that require investigative attention. As a yet further consideration, adequate studies devoted to the impact of facet selectivity, structure and morphology of ZnO nano-structures on the overall efficiency of solar cells and the associated mechanism have to be conducted.
ZnO nanostructured particles have revolutionized the field of photocatalysis. And their efficacy in water splitting and degradation of recalcitrant organic water pollutants has been widely investigated and taken advantage of. However, a few concerning aspects about their photocatalytic activity still need to be dealt with through possible corrective measures. First, the photodegrading ability of a prepared ZnO nano-catalyst needs to be checked by taking the pollutant of interest in lieu of a representative substance which in usual cases is a dye. This is because dye-degradation is relatively plain sailing while removal of pharmaceutical wastes, pesticides, insecticides or other endocrine disruptors offers greater challenges and complicacies. Moreover, the archives of scientific literature are brimming with thorough investigative reports concerning degradation of dyes. Furthermore, waste water contains a mix of different contaminants with varying ranges of pH and ionic strength. Few photodegradation studies have been conducted on organic pollutants in such a simulated sample of water while taking into account the effect of the presence of other contaminants, varying pH and ionic strength on the degradation kinetics. Second, a detailed insight into the mechanistic routes of the degradation of these compounds and their interaction with ZnO based nano-catalysts is elusive as of now and its development remains imperative and will unfold approaches to tackle other emerging contaminants. Third, many improvements in the very architecture of ZnO nanostructures are due specifically in areas such as surface area, particle size, separation and lifespan of charge carriers and so forth. Fourth, since band positions and band gaps are dependent on particle size, it becomes difficult to create heterojunctions able enough to achieve effective charge separation and thereby efficient photocatalytic activity. Systemic studies with a focus on discovering specific synthesis protocols for the achievement of ZnO based nanostructures with desired band positions and band gaps have to be embarked on. Also, there are a few difficulties associated with the operating procedures, such as loss and recovery of nano-structured photocatalysts in the course of post-synthesis treatment and photocatalytic activity. Furthermore, more sweeping research investigations are required to develop and verify the mathematical models for photocatalytic operations/systems for water/wastewater treatment in order to predict the quantum yield, kinetics and optimum conditions of the process.
ZnO nanomaterials may be outstanding candidates as biocompatible and biodegradable nanoplatforms for cancer targeted imaging and therapy. For in vivo imaging and therapy applications, the future of nanomedicine lies in multifunctional nanoplatforms combining both therapeutic components and multimodality imaging. Biocompatibility is also a concern for the applications of nanomaterials in biomedicine. Surface modification of nanomaterials plays a vital role in this context. Biocompatibility of ZnO nanomaterials might be enhanced by slowing down the dissolution rate through Fe doping324 or surface capping.325 Therefore, surface coating of ZnO NPs with biocompatible macromolecules, such as poly(lactic) acid, PEG, PEI and chitosan, was attempted to increase their suitability for further clinical usage. Another idea is the synthesis of ZnO nanoplatforms using the biodegradable and biocompatible materials already proven clinically. Some biocompatible polymers, such as liposomes and dendrimers, have been clinically approved for various pharmaceutical applications. Hence, the modification or conjugation of already approved therapeutic formulations or materials with functional ligands which will improve their diagnostic index could be essential. Much effort is needed for long-term in vivo toxicology studies to pave the way for future biomedical applications of these intriguing nanomaterials. Facile conjugation of various biocompatible polymers, imaging labels, and drugs to ZnO nanomaterials can be achieved because of the versatile surface chemistry.
Some other issues of ZnO NPs concerning their biomedical application and their impact on biological systems still need further meticulous inspection. Following are a few such concerns: (a) lack of comparative analysis of the biological advantages of ZnO NPs to other metal nanoparticles, (b) the limitations imposed by the toxicity of ZnO NPs toward biological systems continue to remain a hot potato in recent research, (c) limitations of biocompatible/biodegradable ZnO nanoplatforms for tumor targeted drug/gene delivery, (d) lack of evidence-based research carrying out as its focal point a thorough survey of the therapeutic roles of ZnO NPs in improving anticancer, antibacterial, anti-inflammatory, and anti-diabetic activities, and (e) lack of extensive in vivo investigations into the anticancer, antibacterial, anti-inflammatory, and anti-diabetic activities of ZnO. Fresh studies focused on the abovementioned issues would bring forth further elucidation and comprehension of the potential use of ZnO nanoparticles in biomedical diagnostic and therapeutic fields.
8. Conclusion
This overview on ZnO is an attempt to conduct a survey of the various strategies that have been adopted to fabricate ZnO nanostructures and their applications across a wide range of fields. One of the merits of the work lies in putting together the work done in biological and pure materials science fields. The other merit lies in the inclusion of a discussion on studies carried out on the toxicity of ZnO nanostructures. Additionally, a holistic picture of the future prospects of nano-structured ZnO vis-à-vis its application in the fields discussed has been presented. This overview demonstrates ZnO as a multitasking material displaying diverse properties, functionality and application in the emerging field of nanotechnology. The high chemical stability, high photostability, wide range of radiation absorption, biodegradability and biocompatibility enable ZnO NPs to be used in new materials and potential applications in a wide range of fields of technology. Moreover, the emergence of various eco-friendly bio-mediated syntheses of ZnO particles with enhanced properties which can be exploited for their better applications in various fields of technology has attracted greater interest in them in the world of research. The opto-electronic properties of ZnO NPs are largely exploited in the fabrication of ZnO based ETLs in DSSCs and PSCs. Modifications based on doping the ZnO ETLs with organic and inorganic substances and surface treatment with functionalizing agents for efficient electron transfer will surely lead to noteworthy advancements in the field of photovoltaics. The biomedical applications of ZnO have drawn great attention from researchers in recent years. Efforts are underway in designing more effective applications of ZnO NPs in regard to drug delivery systems, antimicrobial activity, anti-cancer activity, anti-inflammatory activity, and anti-diabetic activity as well as bio-imaging activity. Owing to their inherent toxicity, ZnO NPs exert profound inhibition effects upon cancerous cells and bacteria. This they achieve by inducing intracellular ROS generation and activating apoptotic signaling pathways, which make ZnO NPs potential anticancer and antibacterial agents. Additionally, ZnO NPs have also been widely known to promote the bioavailability of therapeutic drugs or biomolecules when functioning as drug carriers to achieve enhanced therapy efficiency. Besides, the ability to lower blood glucose and raise insulin levels does attest to the promising potential of ZnO NPs in treating diabetes and abating its complications, which can be subjected to further evaluation. ZnO NPs are listed as a safe substance by the FDA. We believe that nanomaterials would dramatically promote the development of medicine, and ZnO nanoparticles are expected to make more exciting contributions in these fields. The role of ZnO nanofertilizer in inducing growth and yield of crops finds stress in recent research studies. Food demand has been mounting at an alarming rate with every passing day and regrettably the yield of staple food crops has not been able to catch up to the pace. It, therefore, is the need of the hour to design and commercialize metal oxide nanoparticles with favourable properties for sustainable agriculture. ZnO NPs have thus evinced a wide range of areas of applications in science and industry and with investigation on them still on, they offer yet newer possibilities of potential applications.Author contributions
S. R. wrote the main manuscript, text and figures. M. A. discussed the results and commented on, revised and corrected the whole manuscript.Conflicts of interest
The authors declare no competing financial or non-financial interests.Acknowledgements
The authors deeply appreciate the Director, NIT Silchar and TEQIP for pecuniary assistance.References
- X. Wang, Y. Ding, C. J. Summers and Z. L. Wang, J. Phys. Chem. B, 2004, 108(26), 8773–8777 CrossRef CAS.
- D. Segets, J. Gradl, R. K. Taylor, V. Vassilev and W. Peukert, ACS Nano, 2009, 3(7), 1703–1710 CrossRef CAS PubMed.
- X. Lou, H. Shen and Y. Shen, J. Sens. Trans. Technol., 1991, 3(1), 1–34 Search PubMed.
- L. Vayssieres, K. Keis, A. Hagfeldt and S. E. Lindquist, Chem. Mater., 2001, 13(12), 4395–4398 CrossRef CAS.
- Z. Deng, M. Chen, G. Gu and L. Wu, J. Phys. Chem. B, 2008, 112(1), 16–22 CrossRef CAS PubMed.
- S. J. Yang and C. R. Park, Nanotechnology, 2007, 19(3), 035609 CrossRef PubMed.
- C. Petchthanasombat, T. Tiensing and P. Sunintaboon, J. Colloid Interface Sci., 2012, 369(1), 52–57 CrossRef CAS PubMed.
- A. Becheri, M. Dürr, P. L. Nostro and P. Baglioni, J. Nanopart. Res., 2008, 10(4), 679–689 CrossRef CAS.
- U. Ozgur, D. Hofstetter and H. Morkoc, Proc. IEEE, 2010, 98(7), 1255–1268 CAS.
- Y. Muktar, T. Bikila and M. Keffale, World Appl. Sci. J., 2015, 33(10), 1588–1596 Search PubMed.
- D. Banerjee, J. Y. Lao, D. Z. Wang, J. Y. Huang, Z. F. Ren, D. Steeves and M. Sennett, Appl. Phys. Lett., 2003, 83(10), 2061–2063 CrossRef CAS.
- Y. B. Hahn, Korean J. Chem. Eng., 2011, 28(9), 1797–1813 CrossRef CAS.
- T. Frade, M. M. Jorge and A. Gomes, Mater. Lett., 2012, 82, 13–15 CrossRef CAS.
- R. Wahab, S. G. Ansari, Y. S. Kim, H. K. Seo and H. S. Shin, Appl. Surf. Sci., 2007, 253(18), 7622–7626 CrossRef CAS.
- Z. W. Pan, Z. R. Dai and Z. L. Wang, Science, 2001, 291(5510), 1947–1949 CrossRef CAS PubMed.
- J. J. Wu, S. C. Liu, C. T. Wu, K. H. Chen and L. C. Chen, Appl. Phys. Lett., 2002, 81(7), 1312–1314 CrossRef CAS.
- W. J. Chen, W. L. Liu, S. H. Hsieh and T. K. Tsai, Appl. Surf. Sci., 2007, 253(16), 6749–6753 CrossRef CAS.
- J. Liu, X. Huang, J. Duan, H. Ai and P. Tu, Mater. Lett., 2005, 59(28), 3710–3714 CrossRef CAS.
- Y. Huang, J. He, Y. Zhang, Y. Dai, Y. Gu, S. Wang and C. Zhou, J. Mater. Sci., 2006, 41(10), 3057–3062 CrossRef CAS.
- B. Nikoobakht, X. Wang, A. Herzing and J. Shi, Chem. Soc. Rev., 2013, 42(1), 342–365 RSC.
- L. C. Tien, S. J. Pearton, D. P. Norton and F. Ren, J. Mater. Sci., 2008, 43(21), 6925–6932 CrossRef CAS.
- J. Cui, Mater. Charact., 2012, 64, 43–52 CrossRef CAS.
- T. Xu, P. Ji, M. He and J. Li, J. Nanomater., 2012, 2012, 12 Search PubMed.
- W. S. Chiu, P. S. Khiew, M. Cloke, D. Isa, T. K. Tan, S. Radiman and C. H. Chia, Chem. Eng. J., 2010, 158(2), 345–352 CrossRef CAS.
- M. Jose-Yacaman, C. Gutierrez-Wing, M. Miki, D. Q. Yang, K. N. Piyakis and E. Sacher, J. Phys. Chem. B, 2005, 109(19), 9703–9711 CrossRef CAS PubMed.
- V. Polshettiwar, B. Baruwati and R. S. Varma, ACS Nano, 2009, 3(3), 728–736 CrossRef CAS PubMed.
- Q. Xie, Z. Dai, J. Liang, L. Xu, W. Yu and Y. Qian, Solid State Commun., 2005, 136(5), 304–307 CrossRef CAS.
- J. Liu, X. Huang, Y. Li, K. M. Sulieman, F. Sun and X. He, Scr. Mater., 2006, 55(9), 795–798 CrossRef CAS.
- M. Bitenc and Z. C. Orel, Mater. Res. Bull., 2009, 44(2), 381–387 CrossRef CAS.
- K. Galatsis, L. Cukrov, W. Wlodarski, P. McCormick, K. Kalantar-Zadeh, E. Comini and G. Sberveglieri, Sens. Actuators, B, 2003, 93(1–3), 562–565 CrossRef CAS.
- K. S. Venkataraman and K. S. Narayanan, Powder Technol., 1998, 96(3), 190–201 CrossRef CAS.
- W. Ao, J. Li, H. Yang, X. Zeng and X. Ma, Powder Technol., 2006, 168(3), 148–151 CrossRef CAS.
- A. Stanković, Lj. Veselinović, S. D. Škapin, S. Marković and D. Uskokovic, J. Mater. Sci., 2011, 46, 3716–3724 CrossRef.
- T. Tsuzuki and P. G. McCormick, Scr. Mater., 2001, 44(8–9), 1731–1734 CrossRef CAS.
- A. Moballegh, H. R. Shahverdi, R. Aghababazadeh and A. R. Mirhabibi, Surf. Sci., 2007, 601(13), 2850–2854 CrossRef CAS.
- R. Aghababazadeh, B. Mazinani, A. Mirhabibi and M. Tamizifar, J. Phys.: Conf. Ser., 2006, 26(1), 312 CrossRef.
- R. Sadraei, Res. Rev.: J. Chem., 2016, 2319, 9849 Search PubMed.
- K. M. Kumar, B. K. Mandal, E. A. Naidu, M. Sinha, K. S. Kumar and P. S. Reddy, Spectrochim. Acta, Part A, 2013, 104, 171–174 CrossRef PubMed.
- R. Hong, T. Pan, J. Qian and H. Li, Chem. Eng. J., 2006, 119(2–3), 71–81 CrossRef CAS.
- D. Raoufi, Renewable Energy, 2013, 50, 932–937 CrossRef CAS.
- Y. D. Wang, C. L. Ma, X. D. Sun and H. D. Li, Inorg. Chem. Commun., 2002, 5(10), 751–755 CrossRef CAS.
- P. Li, Y. Wei, H. Liu and X. K. Wang, J. Solid State Chem., 2005, 178(3), 855–860 CrossRef CAS.
- S. Suwanboon, ScienceAsia, 2008, 34, 031–034 CrossRef CAS.
- H. Benhebal, M. Chaib, T. Salmon, J. Geens, A. Leonard, S. D. Lambert, M. Crine and B. Heinrichs, Alexandria Eng. J., 2013, 52(3), 517–523 CrossRef.
- V. Sharma, J. Appl. Chem., 2012, 2, 52–55 Search PubMed.
- M. Ristić, S. Musić, M. Ivanda and S. Popović, J. Alloys Compd., 2005, 397(1–2), L1–L4 CrossRef.
- N. Shakti and P. S. Gupta, Appl. Phys. Res., 2010, 2(1), 19–28 CAS.
- Q. H. Wu, J. Exp. Nanosci., 2015, 10(3), 161–166 CrossRef CAS.
- X. Y. Kong and Z. L. Wang, Nano Lett., 2003, 3(12), 1625–1631 CrossRef CAS.
- F. H. Alsultany, Z. Hassan, N. M. Ahmed and M. A. Almessiere, Ceram. Int., 2017, 43(1), 610–616 CrossRef CAS.
- V. M. Diep and A. M. Armani, Nano Lett., 2016, 16(12), 7389–7393 CrossRef CAS PubMed.
- Q. Luo, P. Xu, Y. Qiu, Z. Cheng, X. Chang and H. Fan, Mater. Lett., 2017, 198, 192–195 CrossRef CAS.
- D. Chen, X. Jiao and G. Cheng, Solid State Commun., 1999, 113(6), 363–366 CrossRef CAS.
- A. N. P. Madathil, K. A. Vanaja and M. K. Jayaraj, in Nanophotonic Materials IV, International Society for Optics and Photonics, 2007, vol. 6639, p. 66390J Search PubMed.
- D. Polsongkram, P. Chamninok, S. Pukird, L. Chow, O. Lupan, G. Chai, H. Khallaf, S. Park and A. Schulte, Phys. B, 2008, 403, 3713–3717 CrossRef CAS.
- M. Segovia, C. Sotomayor, G. Gonzalez and E. Benavente, Mol. Cryst. Liq. Cryst., 2012, 555(1), 40–50 CrossRef CAS.
- S. J. Chen, L. H. Li, X. T. Chen, Z. Xue, J. M. Hong and X. Z. You, J. Cryst. Growth, 2003, 252(1–3), 184–189 CrossRef CAS.
- S. A. Vorobyova, A. I. Lesnikovich and V. V. Mushinskii, Mater. Lett., 2004, 58(6), 863–866 CrossRef CAS.
- A. Kołodziejczak-Radzimska, E. Markiewicz and T. Jesionowski, J. Nanomater., 2012, 2012, 15 Search PubMed.
- P. Couvreur, M. J. Blanco-Prieto, F. Puisieux, B. Roques and E. Fattal, Adv. Drug Delivery Rev., 1997, 28(1), 85–96 CrossRef CAS PubMed.
- R. Kumar, M. S. Kumar and N. Mahadevan, Int. J. Recent Adv. Pharm. Res., 2012, 2(1), 9–19 Search PubMed.
- X. Li, G. He, G. Xiao, H. Liu and M. Wang, J. Colloid Interface Sci., 2009, 333(2), 465–473 CrossRef CAS PubMed.
- Ö. A. Yıldırım and C. Durucan, J. Alloys Compd., 2010, 506(2), 944–949 CrossRef.
- M. Hudlikar, S. Joglekar, M. Dhaygude and K. Kodam, J. Nanopart. Res., 2012, 14(5), 865 CrossRef.
- G. Singhal, R. Bhavesh, K. Kasariya, A. R. Sharma and R. P. Singh, J. Nanopart. Res., 2011, 13(7), 2981–2988 CrossRef CAS.
- G. Alagumuthu and R. Kirubha, Int. J. Nanomater. Biostructures, 2012, 2(3), 30–33 Search PubMed.
- C. Mason, S. Vivekanandhan, M. Misra and A. K. Mohanty, World J. Nano Sci. Eng., 2012, 2(02), 47 CrossRef CAS.
- Green Chemistry: Challenging Perspectives, ed. P. Tundo and P. T. Anastas, Oxford University Press, USA, 2000 Search PubMed.
- S. M. Reed and J. E. Hutchison, J. Chem. Educ., 2000, 77(12), 1627 CrossRef CAS.
- D. Saravanak Kumar, S. Sivaranjani, M. Umamaheswari, S. Pandiarajan and B. Ravikumar, Der Pharma Chem., 2016, 8(7), 173–180 CAS.
- S. Gunalan, R. Sivaraj and V. Rajendran, Prog. Nat. Sci.: Mater. Int., 2012, 22(6), 693–700 CrossRef.
- P. Jamdagni, P. Khatri and J. S. Rana, J. King Saud Univ., Sci., 2018, 30(2), 168–175 CrossRef.
- R. Ishwarya, B. Vaseeharan, S. Kalyani, B. Banumathi, M. Govindarajan, N. S. Alharbi, S. Kadaikunnan, M. N. Al-anbr, J. M. Khaled and G. Benelli, J. Photochem. Photobiol., B, 2018, 178, 249–258 CrossRef CAS PubMed.
- G. Sundaraselvan and S. D. Quine, J. Nanosci. Nanotechnol., 2017, 3(4), 289–292 Search PubMed.
- V. V. Gawade, N. L. Gavade, H. M. Shinde, S. B. Babar, A. N. Kadam and K. M. Garadkar, J. Mater. Sci.: Mater. Electron., 2017, 28(18), 14033–14039 CrossRef CAS.
- C. Vidya, C. Manjunatha, M. N. Chandraprabha, M. Rajshekar and A. R. MAL, J. Environ. Chem. Eng., 2017, 5(4), 3172–3180 CrossRef CAS.
- B. Archana, K. Manjunath, G. Nagaraju, K. C. Sekhar and N. Kottam, Int. J. Hydrogen Energy, 2017, 42(8), 5125–5131 CrossRef CAS.
- R. Rathnasamy, P. Thangasamy, R. Thangamuthu, S. Sampath and V. Alagan, J. Mater. Sci.: Mater. Electron., 2017, 28(14), 10374–10381 CrossRef CAS.
- R. Yuvakkumar, J. Suresh and S. I. Hong, Adv. Mater. Res., 2014, 952, 137–140 CAS.
- N. Matinise, X. G. Fuku, K. Kaviyarasu, N. Mayedwa and M. Maaza, Appl. Surf. Sci., 2017, 406, 339–347 CrossRef CAS.
- G. Bhumi and N. Savithramma, Int. J. Drug Dev. Res., 2014, 6(1), 208–214 Search PubMed.
- R. K. Shah, F. Boruah and N. Parween, Int. J. Curr. Microbiol. Appl. Sci., 2015, 4(8), 444–450 CAS.
- P. Ramesh, A. Rajendran and A. Subramanian, Asian J. Phytomed. Clin. Res., 2014, 2(4), 189–195 CAS.
- D. Ramimoghadam, M. Z. B. Hussein and Y. H. Taufiq-Yap, Chem. Cent. J., 2013, 7(1), 136 CrossRef PubMed.
- J. Santhoshkumar, S. V. Kumar and S. Rajeshkumar, Resour.-Effic. Technol., 2017, 3(4), 459–465 Search PubMed.
- C. Sasirekha, S. Arumugam and G. Muralidharan, Appl. Surf. Sci., 2018, 449, 521–527 CrossRef.
- V. A. Soares, M. J. S. Xavier, E. S. Rodrigues, C. A. de Oliveira, P. M. A. Farias, A. Stingl, N. S. Ferreira and M. S. Silva, Mater. Lett., 2020, 259, 126853 CrossRef CAS.
- S. Begum, M. Ahmaruzzaman and P. P. Adhikari, Mater. Lett., 2018, 228, 37–41 CrossRef CAS.
- T. U. D. Thi, T. T. Nguyen, Y. D. Thi, K. H. T. Thi, B. T. Phan and K. N. Pham, RSC Adv., 2020, 10(40), 23899–23907 RSC.
- Z. Cao, Z. Zhang, F. Wang and G. Wang, Colloids Surf., A, 2009, 340(1–3), 161–167 CrossRef CAS.
- H. L. Xia and F. Q. Tang, J. Phys. Chem. B, 2003, 107(35), 9175–9178 CrossRef CAS.
- R. Hong, T. Pan, J. Qian and H. Li, Chem. Eng. J., 2006, 119(2–3), 71–81 CrossRef CAS.
- F. Yuan, H. Peng, Y. Yin, Y. Chunlei and H. Ryu, Mater. Sci. Eng. B, 2005, 122(1), 55–60 CrossRef.
- P. Hu, N. Han, D. Zhang, J. C. Ho and Y. Chen, Sens. Actuators, B, 2012, 169, 74–80 CrossRef CAS.
- M. Wysokowski, M. Motylenko, H. Stöcker, V. V. Bazhenov, E. Langer, A. Dobrowolska, K. Czaczyk, R. Galli, A. L. Stelling, T. Behm and Ł. Klapiszewski, J. Mater. Chem. B, 2013, 1(46), 6469–6476 RSC.
- W. J. Ong, S. Y. Voon, L. L. Tan, B. T. Goh, S. T. Yong and S. P. Chai, Ind. Eng. Chem. Res., 2014, 53(44), 17333–17344 CrossRef CAS.
- E. Tang, C. Fu, S. Wang, S. Dong, F. Zhao and D. Zhao, Powder Technol., 2012, 218, 5–10 CrossRef CAS.
- S. V. Kurkov and T. Loftsson, Int. J. Pharm., 2013, 453(1), 167–180 CrossRef CAS PubMed.
- E. M. Del Valle, Process Biochem., 2004, 39(9), 1033–1046 CrossRef CAS.
- E. S. Abdel-Halim, F. A. Abdel-Mohdy, S. S. Al-Deyab and M. H. El-Newehy, Carbohydr. Polym., 2010, 82(1), 202–208 CrossRef CAS.
- N. A. Ibrahim, W. R. E-Zairy and B. M. Eid, Carbohydr. Polym., 2010, 79(4), 839–846 CrossRef CAS.
- N. A. Ibrahim and E. M. R. El-Zairy, Carbohydr. Polym., 2009, 76(2), 244–249 CrossRef CAS.
- L. Rehmann, H. Yoshii and T. Furuta, Starch/Staerke, 2003, 55(7), 313–318 CrossRef CAS.
- A. Abdolmaleki, S. Mallakpour and S. Borandeh, Carbohydr. Polym., 2014, 103, 32–37 CrossRef CAS PubMed.
- G. A. Farzi, R. Tayebee and S. Naghibinasa, Int. J. Nano Dimens., 2015, 6, 67–75 CAS.
- K. Nomura, H. Ohta, K. Ueda, T. Kamiya, M. Hirano and H. Hosono, Science, 2003, 300(5623), 1269–1272 CrossRef CAS PubMed.
- T. Nakada, Y. Hirabayashi, T. Tokado, D. Ohmori and T. Mise, Sol. Energy, 2004, 77(6), 739–747 CrossRef CAS.
- G. Momen and M. Farzaneh, Rev. Adv. Mater. Sci., 2011, 27(1), 1–13 CrossRef CAS.
- A. Kołodziejczak-Radzimska, T. Jesionowski, M. Maciejewska and M. Zaborski, Pol. J. Chem. Technol., 2014, 16(3), 63–68 CrossRef.
- Z. Yuan, W. Zhou, T. Hu, Y. Chen, F. Li, Z. Xu and X. Wang, Surf. Rev. Lett., 2011, 18, 33–38 CrossRef CAS.
- U. K. Mandal, D. K. Tripathy and S. K. De, Polymer, 1996, 37(25), 5739–5742 CrossRef CAS.
- L. Ibarra, A. Marcos-Fernandez and M. Alzorriz, Polymer, 2002, 43(5), 1649–1655 CrossRef CAS.
- K. Chatterjee and K. Naskar, eXPRESS Polym. Lett., 2007, 1, 527–534 CrossRef CAS.
- G. R. Hamed and K. C. Hua, Rubber Chem. Technol., 2004, 77(2), 214–226 CrossRef CAS.
- M. Przybyszewska and M. Zaborski, eXPRESS Polym. Lett., 2009, 3, 542–552 CrossRef CAS.
- A. V. Chapman, M. Porter and A. D. Roberts, Nat. Sci. Technol., 1988, 21(4), 362 Search PubMed.
- G. J. Fosmire, Zinc toxicity, Am. J. Clin. Nutr., 1990, 51(2), 225–227 CrossRef CAS PubMed.
- G. Heideman, R. N. Datta, J. W. Noordermeer and B. V. Baarle, J. Appl. Polym. Sci., 2005, 95(6), 1388–1404 CrossRef CAS.
- L. Pyskło, W. Parasiewicz, P. Pawłowski and K. Niciński, Zinc Oxide in Rubber Compounds, Instytut Przemyslu Gumowego, Piastow, Poland, 2007 Search PubMed.
- S. P. Thomas, E. J. Mathew and C. V. Marykutty, J. Appl. Polym. Sci., 2012, 124(4), 3099–3107 CrossRef CAS.
- P. M. Sabura Begum, K. K. Mohammed Yusuff and R. Joseph, Int. J. Polym. Mater., 2008, 57(12), 1083–1094 CrossRef CAS.
- D. Nivethitha and S. Dharmar, Int. J. Earth Sci. Eng., 2016, 09(03), 175–181 Search PubMed.
- T. K. Kundu, N. Karak, P. Barik and S. Saha, Int. J. Soft Comput. Eng., 2011, 1, 19–24 Search PubMed.
- P. Verma, S. Pandey and A. C. Pandey, Proc. Of ASID, New Delhi, 2006, pp. 256–258 Search PubMed.
- A. E. Suliman, Y. Tang and L. Xu, Sol. Energy Mater. Sol. Cells, 2007, 91(18), 1658–1662 CrossRef.
- M. S. Akhtar, M. A. Khan, M. S. Jeon and O. B. Yang, Electrochim. Acta, 2008, 53(27), 7869–7874 CrossRef CAS.
- C. Lin, H. Lin, J. Li and X. Li, J. Alloys Compd., 2008, 462(1–2), 175–180 CrossRef CAS.
- Y. F. Hsu, Y. Y. Xi, C. T. Yip, A. B. Djurišić and W. K. Chan, J. Appl. Phys., 2008, 103(8), 083114 CrossRef.
- W. Chen, H. Zhang, I. M. Hsing and S. Yang, Electrochem. Commun., 2009, 11(5), 1057–1060 CrossRef CAS.
- M. Guo, P. Diao and S. M. Cai, Chin. Chem. Lett., 2004, 15(9), 1113–1116 CAS.
- K. Mahmood and S. B. Park, J. Mater. Chem. A, 2013, 1(15), 4826–4835 RSC.
- L. Luo, W. Tao, X. Hu, T. Xiao, B. Heng, W. Huang and Y. Tang, J. Power Sources, 2011, 196(23), 10518–10525 CrossRef CAS.
- Q. Zhang, C. S. Dandeneau, S. Candelaria, D. Liu, B. B. Garcia, X. Zhou and G. Cao, Chem. Mater., 2010, 22(8), 2427–2433 CrossRef CAS.
- D. Bi, G. Boschloo, S. Schwarzmüller, L. Yang, E. M. Johansson and A. Hagfeldt, Nanoscale, 2013, 5(23), 11686–11691 RSC.
- D. Y. Son, J. H. Im, H. S. Kim and N. G. Park, J. Phys. Chem. C, 2014, 118(30), 16567–16573 CrossRef CAS.
- J. H. Im, I. H. Jang, N. Pellet, M. Grätzel and N. G. Park, Nat. Nanotechnol., 2014, 9(11), 927–932 CrossRef CAS PubMed.
- J. F. Tang, Z. L. Tseng, L. C. Chen and S. Y. Chu, Sol. Energy Mater. Sol. Cells, 2016, 154, 18–22 CrossRef CAS.
- D. Bi, W. Tress, M. I. Dar, P. Gao, J. Luo, C. Renevier, K. Schenk, A. Abate, F. Giordano, J. P. C. Baena and J. Decoppet, Sci. Adv., 2016, 2(1), e1501170 CrossRef PubMed.
- A. Kolmakov and M. Moskovits, Annu. Rev. Mater. Res., 2004, 34, 151–180 CrossRef CAS.
- N. Barsan and U. Weimar, J. Electroceram., 2001, 7(3), 143–167 CrossRef CAS.
- P. S. Cho, K. W. Kim and J. H. Lee, J. Electroceram., 2006, 17(2–4), 975–978 CrossRef CAS.
- S. A. Vanalakar, V. L. Patil, N. S. Harale, S. A. Vhanalakar, M. G. Gang, J. Y. Kim, P. S. Patil and J. H. Kim, Sens. Actuators, B, 2015, 221, 1195–1201 CrossRef CAS.
- D. Zhang, C. Jiang, Y. Yao, D. Wang and Y. Zhang, Sens. Actuators, B, 2017, 253, 1120–1128 CrossRef CAS.
- M. Hjiri, L. El Mir, S. G. Leonardi, A. Pistone, L. Mavilia and G. Neri, Sens. Actuators, B, 2014, 196, 413–420 CrossRef CAS.
- Y. Chen, X. Li, X. Li, J. Wang and Z. Tang, Sens. Actuators, B, 2016, 232, 158–164 CrossRef CAS.
- J. Liu, T. Wang, B. Wang, P. Sun, Q. Yang, X. Liang, H. Song and G. Lu, Sens. Actuators, B, 2017, 245, 551–559 CrossRef CAS.
- E. M. El-Diasty, M. A. Ahmed, N. A. G. W. A. Okasha, S. F. Mansour, S. I. El-Dek, H. M. El-Khalek and M. H. YOUSSIF, Rom. J. Biophys., 2013, 23(3), 191–202 CAS.
- P. Singh and A. Nanda, J. Chem. Pharm. Res., 2013, 5, 457–463 Search PubMed.
- J. J. Reinosa, P. Leret, C. M. Álvarez-Docio, A. del Campo and J. F. Fernández, Bol. Soc. Esp. Ceram. Vidrio, 2016, 55(2), 55–62 CrossRef.
- M. J. Uddin, F. Cesano, S. Bertarione, F. Bonino, S. Bordiga, D. Scarano and A. Zecchina, J. Photochem. Photobiol., A, 2008, 196(2–3), 165–173 CrossRef CAS.
- Q. Gao, Q. Zhu, Y. Guo and C. Q. Yang, Ind. Eng. Chem. Res., 2009, 48(22), 9797–9803 CrossRef CAS.
- P. Atienzar, T. Ishwara, B. N. Illy, M. P. Ryan, B. C. O'Regan, J. R. Durrant and J. Nelson, J. Phys. Chem. Lett., 2010, 1(4), 708–713 CrossRef CAS.
- Z. H. Lim, Z. X. Chia, M. Kevin, A. S. W. Wong and G. W. Ho, Sens. Actuators, B, 2010, 151(1), 121–126 CrossRef CAS.
- J. L. Gomez and O. Tigli, J. Mater. Sci., 2013, 48(2), 612–624 CrossRef CAS.
- D. Tanasa, N. Vrinceanu, A. Nistor, C. M. Hristodor, E. Popovici, I. L. Bistricianu, F. Brinza, D. L. Chicet, D. Coman, A. Pui and A. M. Grigoriu, Text. Res. J., 2012, 82(8), 832–844 CrossRef CAS.
- N. Vigneshwaran, S. Kumar, A. A. Kathe, P. V. Varadarajan and V. Prasad, Nanotechnology, 2006, 17(20), 5087 CrossRef CAS.
- F. Zhang and J. Yang, Int. J. Chem., 2009, 1(1), 18 CAS.
- S. Kathirvelu, L. D'souza and B. Dhurai, Indian J. Fibre Text. Res., 2009, 34(3), 267–273 CAS.
- A. Yadav, V. Prasad, A. A. Kathe, S. Raj, D. Yadav, C. Sundaramoorthy and N. Vigneshwaran, Bull. Mater. Sci., 2006, 29(6), 641–645 CrossRef CAS.
- Z. Mao, Q. Shi, L. Zhang and H. Cao, Thin Solid Films, 2009, 517(8), 2681–2686 CrossRef CAS.
- A. Becheri, M. Dürr, P. L. Nostro and P. Baglioni, J. Nanopart. Res., 2008, 10(4), 679–689 CrossRef CAS.
- R. Wang, J. H. Xin, X. M. Tao and W. A. Daoud, Chem. Phys. Lett., 2004, 398(1–3), 250–255 CrossRef CAS.
- E. S. Ates and H. E. Unalan, Thin Solid Films, 2012, 520(14), 4658–4661 CrossRef CAS.
- T. Jesionowski, A. Kołodziejczak-Radzimska, F. Ciesielczyk, J. Sójka-Ledakowicz, J. Olczyk and J. Sielski, Fibres Text. East. Eur., 2011, 2(85), 70–75 Search PubMed.
- S. B. Ghaffari, M. H. Sarrafzadeh, Z. Fakhroueian, S. Shahriari and M. R. Khorramizadeh, Mater. Sci. Eng. C, 2017, 79, 465–472 CrossRef CAS PubMed.
- Y. Li, C. Zhang, L. Liu, Y. Gong, Y. Xie and Y. Cao, Toxicol. Mech. Methods, 2018, 28(3), 167–176 CrossRef CAS PubMed.
- R. K. Dutta, B. P. Nenavathu, M. K. Gangishetty and A. V. R. Reddy, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2013, 48(8), 871–878 CrossRef CAS PubMed.
- K. M. Reddy, K. Feris, J. Bell, D. G. Wingett, C. Hanley and A. Punnoose, Appl. Phys. Lett., 2007, 90(21), 213902 CrossRef CAS PubMed.
- T. Chatterjee, S. Chakraborti, P. Joshi, S. P. Singh, V. Gupta and P. Chakrabarti, FEBS J., 2010, 277(20), 4184–4194 CrossRef CAS PubMed.
- C. Mahendra, M. Murali, G. Manasa, P. Ponnamma, M. R. Abhilash, T. R. Lakshmeesha, A. Satish, K. N. Amruthesh and M. S. Sudarshana, Microb. Pathog., 2017, 110, 620–629 CrossRef CAS PubMed.
- B. N. Singh, A. K. S. Rawat, W. Khan, A. H. Naqvi and B. R. Singh, PLoS One, 2014, 9(9), e106937 CrossRef PubMed.
- Y. H. Hsueh, W. J. Ke, C. T. Hsieh, K. S. Lin, D. Y. Tzou and C. L. Chiang, PLoS One, 2015, 10(6), e0128457 CrossRef PubMed.
- M. Divya, B. Vaseeharan, M. Abinaya, S. Vijayakumar, M. Govindarajan, N. S. Alharbi, S. Kadaikunnan, J. M. Khaled and G. Benelli, J. Photochem. Photobiol., B, 2018, 178, 211–218 CrossRef CAS PubMed.
- R. Brayner, R. Ferrari-Iliou, N. Brivois, S. Djediat, M. F. Benedetti and F. Fiévet, Nano Lett., 2006, 6(4), 866–870 CrossRef CAS PubMed.
- L. Zhang, Y. Jiang, Y. Ding, M. Povey and D. York, J. Nanopart. Res., 2007, 9(3), 479–489 CrossRef CAS.
- S. Sarwar, S. Chakraborti, S. Bera, I. A. Sheikh, K. M. Hoque and P. Chakrabarti, Nanomed.: Nanotechnol. Biol. Med., 2016, 12(6), 1499–1509 CrossRef CAS PubMed.
- B. Wang, Y. Zhang, Z. Mao, D. Yu and C. Gao, J. Nanosci. Nanotechnol., 2014, 14(8), 5688–5696 CrossRef CAS PubMed.
- N. Li, T. Xia and A. E. Nel, Free Radical Biol. Med., 2008, 44(9), 1689–1699 CrossRef CAS PubMed.
- K. Kasemets, A. Ivask, H. C. Dubourguier and A. Kahru, Toxicol. In Vitro, 2009, 23(6), 1116–1122 CrossRef CAS PubMed.
- W. Salem, D. R. Leitner, F. G. Zingl, G. Schratter, R. Prassl, W. Goessler and S. Schild, Int. J. Med. Microbiol., 2015, 305(1), 85–95 CrossRef CAS PubMed.
- S. Sarwar, A. Ali, M. Pal and P. Chakrabarti, J. Biol. Chem., 2017, 292(44), 18303–18311 CrossRef CAS PubMed.
- Y. H. Leung, X. Xu, A. P. Ma, F. Liu, A. M. Ng, Z. Shen, L. A. Gethings, M. Y. Guo, A. B. Djurišić, P. K. Lee and H. K. Lee, Sci. Rep., 2016, 6, 35243 CrossRef CAS PubMed.
- Y. Jiang, L. Zhang, D. Wen and Y. Ding, Mater. Sci. Eng. C, 2016, 69, 1361–1366 CrossRef CAS PubMed.
- T. Ohira and O. Yamamoto, Chem. Eng. Sci., 2012, 68(1), 355–361 CrossRef CAS.
- A. Iswarya, B. Vaseeharan, M. Anjugam, B. Ashokkumar, M. Govindarajan, N. S. Alharbi, S. Kadaikunnan, J. M. Khaled and G. Benelli, Colloids Surf., B, 2017, 158, 257–269 CrossRef CAS PubMed.
- M. Heinlaan, A. Ivask, I. Blinova, H. C. Dubourguier and A. Kahru, Chemosphere, 2008, 71(7), 1308–1316 CrossRef CAS PubMed.
- S. Shafaei, M. Lackner, R. Voloshchuk, I. Voloshchuk, P. Voloshchuk, J. Guggenbichler and C. Zollfrank, Recent Pat. Mater. Sci., 2014, 7(1), 26–36 CrossRef.
- S. D. Sarker, L. Nahar and Y. Kumarasamy, Methods, 2007, 42(4), 321–324 CrossRef CAS PubMed.
- Y. Xie, Y. He, P. L. Irwin, T. Jin and X. Shi, Appl. Environ. Microbiol., 2011, 77(7), 2325–2331 CrossRef CAS PubMed.
- O. Yamamoto, M. Komatsu, J. Sawai and Z. E. Nakagawa, J. Mater. Sci.: Mater. Med., 2004, 15(8), 847–851 CrossRef CAS PubMed.
- A. Nel, T. Xia, L. Mädler and N. Li, Science, 2006, 311(5761), 622–627 CrossRef CAS PubMed.
- U. Manzoor, S. Siddique, R. Ahmed, Z. Noreen, H. Bokhari and I. Ahmad, PLoS One, 2016, 11(5), e0154704 CrossRef PubMed.
- M. A. Molina, J. L. Ramos and M. Espinosa-Urgel, Environ. Microbiol., 2006, 8(4), 639–647 CrossRef CAS PubMed.
- V. L. Prasanna and R. Vijayaraghavan, Langmuir, 2015, 31, 9155–9162 CrossRef PubMed.
- K. R. Raghupathi, R. T. Koodali and A. C. Manna, Langmuir, 2011, 27(7), 4020–4028 CrossRef CAS PubMed.
- M. Y. Yang, K. C. Chang, L. Y. Chen, P. C. Wang, C. C. Chou, Z. B. Wu and A. Hu, J. Photochem. Photobiol., B, 2018, 180, 235–242 CrossRef CAS PubMed.
- L. Zhang, H. Qi, Z. Yan, Y. Gu, W. Sun and A. A. Zewde, Ultrason. Sonochem., 2017, 34, 232–238 CrossRef CAS PubMed.
- Q. Yuan, S. Hein and R. D. K. Misra, Acta Biomater., 2010, 6(7), 2732–2739 CrossRef CAS PubMed.
- L. Nie, L. Gao, P. Feng, J. Zhang, X. Fu, Y. Liu, X. Yan and T. Wang, Small, 2006, 2(5), 621–625 CrossRef CAS PubMed.
- P. Zhang and W. Liu, Biomaterials, 2010, 31(11), 3087–3094 CrossRef CAS PubMed.
- E. Taylor and T. J. Webster, Int. J. Nanomed., 2011, 6, 1463 CrossRef CAS PubMed.
- P. V. Asharani, Y. L. Wu, Z. Gong and S. Valiyaveettil, Nanotechnology, 2008, 19(25), 255102 CrossRef CAS PubMed.
- P. Zhang and W. Liu, Biomaterials, 2010, 31(11), 3087–3094 CrossRef CAS PubMed.
- S. Mitra, B. Subia, P. Patra, S. Chandra, N. Debnath, S. Das, R. Banerjee, S. C. Kundu, P. Pramanik and A. Goswami, J. Mater. Chem., 2012, 22(45), 24145–24154 RSC.
- K. Zeng, J. Li, Z. Zhang, M. Yan, Y. Liao, X. Zhang and C. Zhao, J. Mater. Chem. B, 2015, 3(26), 5249–5260 RSC.
- J. Liu, X. Ma, S. Jin, X. Xue, C. Zhang, T. Wei, W. Guo and X. J. Liang, Mol. Pharm., 2016, 13(5), 1723–1730 CrossRef CAS PubMed.
- Z. Li, H. Li, L. Liu, X. You, C. Zhang and Y. Wang, RSC Adv., 2015, 5(94), 77097–77105 RSC.
- Y. Wang, S. Song, J. Liu, D. Liu and H. Zhang, Angew. Chem., Int. Ed., 2015, 54(2), 536–540 CAS.
- Y. S. Lin, K. R. Hurley and C. L. Haynes, J. Phys. Chem. Lett., 2012, 3(3), 364–374 CrossRef CAS PubMed.
- J. Zhu, L. Liao, X. Bian, J. Kong, P. Yang and B. Liu, Small, 2012, 8(17), 2715–2720 CrossRef CAS PubMed.
- Z. Y. Zhang, Y. D. Xu, Y. Y. Ma, L. L. Qiu, Y. Wang, J. L. Kong and H. M. Xiong, Angew. Chem., Int. Ed., 2013, 52(15), 4127–4131 CrossRef CAS PubMed.
- N. Tripathy, R. Ahmad, H. A. Ko, G. Khang and Y. B. Hahn, Nanoscale, 2015, 7(9), 4088–4096 RSC.
- X. Cai, Y. Luo, W. Zhang, D. Du and Y. Lin, ACS Appl. Mater. Interfaces, 2016, 8(34), 22442–22450 CrossRef CAS PubMed.
- V. B. Kumar, K. Kumar, A. Gedanken and P. Paik, J. Mater. Chem. B, 2014, 2(25), 3956–3964 RSC.
- Z. Han, X. Wang, C. Heng, Q. Han, S. Cai, J. Li, C. Qi, W. Liang, R. Yang and C. Wang, Phys. Chem. Chem. Phys., 2015, 17(33), 21576–21582 RSC.
- H. Zhang, B. Chen, H. Jiang, C. Wang, H. Wang and X. Wang, Biomaterials, 2011, 32(7), 1906–1914 CrossRef CAS PubMed.
- D. X. Ye, Y. Y. Ma, W. Zhao, H. M. Cao, J. L. Kong, H. M. Xiong and H. Möhwald, ACS Nano, 2016, 10(4), 4294–4300 CrossRef CAS PubMed.
- H. Sharma, K. Kumar, C. Choudhary, P. K. Mishra and B. Vaidya, Artif. Cells, Nanomed., Biotechnol., 2016, 44(2), 672–679 CrossRef CAS PubMed.
- Y. Zhang, T. R. Nayak, H. Hong and W. Cai, Curr. Mol. Med., 2013, 13(10), 1633–1645 CrossRef CAS PubMed.
- M. Martínez-Carmona, Y. Gun'ko and M. Vallet-Regí, Nanomaterials, 2018, 8(4), 268 CrossRef PubMed.
- M. Premanathan, K. Karthikeyan, K. Jeyasubramanian and G. Manivannan, Nanomed.: Nanotechnol. Biol. Med., 2011, 7(2), 184–192 CrossRef CAS PubMed.
- D. Selvakumari, R. Deepa, V. Mahalakshmi, P. Subhashini and N. Lakshminarayan, J. Eng. Appl. Sci., 2015, 10, 5418–5421 Search PubMed.
- D. F. Stowe and A. K. Camara, Antioxid. Redox Signaling, 2009, 11(6), 1373–1414 CrossRef CAS PubMed.
- E. Moghimipour, M. Rezaei, Z. Ramezani, M. Kouchak, M. Amini, K. A. Angali and S. Handali, Life Sci., 2018, 194, 104–110 CrossRef CAS PubMed.
- C. Guo, L. Sun, X. Chen and D. Zhang, Neural Regener. Res., 2013, 8(21), 2003 CAS.
- V. Sharma, D. Anderson and A. Dhawan, Apoptosis, 2012, 17(8), 852–870 CrossRef CAS PubMed.
- A. Boroumand Moghaddam, M. Moniri, S. Azizi, R. Abdul Rahim, A. Bin Ariff, M. Navaderi and R. Mohamad, Genes, 2017, 8(10), 281 CrossRef PubMed.
- M. Chandrasekaran and M. Pandurangan, Biol. Trace Elem. Res., 2016, 172(1), 148–154 CrossRef CAS PubMed.
- A. Hussain, M. Oves, M. F. Alajmi, I. Hussain, S. Amir, J. Ahmed, M. T. Rehman, H. R. El-Seedi and I. Ali, RSC Adv., 2019, 9(27), 15357–15369 RSC.
- K. Kanagamani, P. Muthukrishnan, K. Saravanakumar, K. Shankar and A. Kathiresan, Rare Met., 2019, 38(4), 277–286 CrossRef CAS.
- D. Guo, C. Wu, H. Jiang, Q. Li, X. Wang and B. Chen, J. Photochem. Photobiol., B, 2008, 93(3), 119–126 CrossRef CAS PubMed.
- J. Li, D. Guo, X. Wang, H. Wang, H. Jiang and B. Chen, Nanoscale Res. Lett., 2010, 5(6), 1063 CrossRef CAS PubMed.
- R. Hariharan, S. Senthilkumar, A. Suganthi and M. Rajarajan, J. Photochem. Photobiol., B, 2012, 116, 56–65 CrossRef CAS PubMed.
- R. Hariharan, S. Senthilkumar, A. Suganthi and M. Rajarajan, J. Photochem. Photobiol., A, 2013, 252, 107–115 CrossRef CAS.
- C. K. Lim, J. Heo, S. Shin, K. Jeong, Y. H. Seo, W. D. Jang, C. R. Park, S. Y. Park, S. Kim and I. C. Kwon, Cancer Lett., 2013, 334(2), 176–187 CrossRef CAS PubMed.
- Y. Liu, Y. Zhang, S. Wang, C. Pope and W. Chen, Appl. Phys. Lett., 2008, 92(14), 143901 CrossRef.
- M. Fakhar-e-Alam, S. Rahim, M. Atif, M. H. Aziz, M. I. Malick, S. S. Z. Zaidi, R. Suleman and A. Majid, Laser Phys. Lett., 2014, 11(3), 039501 CrossRef.
- S. N. Seclen, M. E. Rosas, A. J. Arias and C. A. Medina, BMJ Open Diabetes Res. Care, 2017, 5(1), e000401 CrossRef PubMed.
- M. A. Woldu and J. L. Lenjisa, Int. J. Basic Clin. Pharmacol., 2017, 3(2), 277–284 CrossRef.
- A. Nazarizadeh and S. Asri-Rezaie, AAPS PharmSciTech, 2016, 17(4), 834–843 CrossRef CAS PubMed.
- R. D. Umrani and K. M. Paknikar, Nanomed, 2014, 9(1), 89–104 CrossRef CAS PubMed.
- A. Alkaladi, A. M. Abdelazim and M. Afifi, Int. J. Mol. Sci., 2014, 15(2), 2015–2023 CrossRef PubMed.
- S. Rahiman and B. A. Tantry, J. Nanomed. Nanotechnol., 2012, 3(5), 1–7 Search PubMed.
- O. Veiseh, B. C. Tang, K. A. Whitehead, D. G. Anderson and R. Langer, Nat. Rev. Drug Discovery, 2015, 14(1), 45 CrossRef CAS PubMed.
- R. Malizia, A. Scorsone, P. D'Angelo, C. P. Lo, L. Pitrolo and C. Giordano, J. Pediatr. Endocrinol. Metab., 1998, 11, 981–984 Search PubMed.
- R. Kitture, K. Chordiya, S. Gaware, S. Ghosh, P. A. More, P. Kulkarni, B. A. Chopade and S. N. Kale, J. Nanosci. Nanotechnol., 2015, 15(6), 4046–4051 CrossRef CAS PubMed.
- J. Hussein, M. El-Banna, T. A. Razik and M. E. El-Naggar, Int. J. Biol. Macromol., 2018, 107, 748–754 CrossRef CAS PubMed.
- R. D. Umrani and K. M. Paknikar, Nanomed, 2014, 9(1), 89–104 CrossRef CAS PubMed.
- S. C. Asani, R. D. Umrani and K. M. Paknikar, Nanomed, 2016, 11(13), 1671–1687 CrossRef CAS PubMed.
- A. Bayrami, S. Haghgooie, S. R. Pouran, F. Mohammadi and A. Habibi-Yangjeh, Adv. Powder Technol., 2020, 31(5), 2110–2118 CrossRef CAS.
- A. Bayrami, S. Parvinroo, A. Habibi-Yangjeh and S. Rahim Pouran, Artif. Cells, Nanomed., Biotechnol., 2018, 46(4), 730–739 CrossRef CAS PubMed.
- L. Ferrero-Miliani, O. H. Nielsen, P. S. Andersen and S. E. Girardin, Clin. Exp. Immunol., 2007, 147(2), 227–235 CrossRef CAS PubMed.
- M. Boguniewicz and D. Y. Leung, Immunol. Rev., 2011, 242(1), 233–246 CrossRef CAS PubMed.
- R. Jurakić Tončić and B. Marinović, Acta Dermatovenerol. Croat., 2016, 24(2), 95 Search PubMed.
- C. Wiegand, U. C. Hipler, S. Boldt, J. Strehle and U. Wollina, Clin., Cosmet. Invest. Dermatol., 2013, 6, 115 CAS.
- M. Ilves, J. Palomäki, M. Vippola, M. Lehto, K. Savolainen, T. Savinko and H. Alenius, Part. Fibre Toxicol., 2014, 11(1), 38 CrossRef PubMed.
- P. C. Nagajyothi, S. J. Cha, I. J. Yang, T. V. M. Sreekanth, K. J. Kim and H. M. Shin, J. Photochem. Photobiol., B, 2015, 146, 10–17 CrossRef CAS PubMed.
- P. Thatoi, R. G. Kerry, S. Gouda, G. Das, K. Pramanik, H. Thatoi and J. K. Patra, J. Photochem. Photobiol., B, 2016, 163, 311–318 CrossRef CAS PubMed.
- N. H. Cho, T. C. Cheong, J. H. Min, J. H. Wu, S. J. Lee, D. Kim, J. S. Yang, S. Kim, Y. K. Kim and S. Y. Seong, Nat. Nanotechnol., 2011, 6(10), 675–682 CrossRef CAS PubMed.
- R. Roy, S. Kumar, A. K. Verma, A. Sharma, B. P. Chaudhari, A. Tripathi, M. Das and P. D. Dwivedi, Int. Immunol., 2013, 26(3), 159–172 CrossRef PubMed.
- A. Mohandas, P. T. Sudheesh Kumar, B. Raja, V. K. Lakshmanan and R. Jayakumar, Int. J. Nanomed., 2015, 10(s1), 53 CAS.
- N. Ninan, M. Muthiah, I. K. Park, A. Elain, T. W. Wong, S. Thomas and Y. Grohens, ACS Appl. Mater. Interfaces, 2013, 5(21), 11194–11206 CrossRef CAS PubMed.
- G. Daeschlein, Int. Wound J., 2013, 10(s1), 9–14 CrossRef PubMed.
- Y. Dong, W. U. Hassan, R. Kennedy, U. Greiser, A. Pandit, Y. Garcia and W. Wang, Acta Biomater., 2014, 10(5), 2076–2085 CrossRef CAS PubMed.
- T. Wang, X. K. Zhu, X. T. Xue and D. Y. Wu, Carbohydr. Polym., 2012, 88(1), 75–83 CrossRef CAS.
- S. Aoki, M. Kinoshita, H. Miyazaki, A. Saito, T. Fujie, K. Iwaya, S. Takeoka and D. Saitoh, Plast. Reconstr. Surg., 2013, 131(2), 236–240 CrossRef CAS PubMed.
- A. R. Unnithan, N. A. Barakat, P. T. Pichiah, G. Gnanasekaran, R. Nirmala, Y. S. Cha, C. H. Jung, M. El-Newehy and H. Y. Kim, Carbohydr. Polym., 2012, 90(4), 1786–1793 CrossRef CAS PubMed.
- L. Varshney, Nucl. Instrum. Methods Phys. Res., Sect. B, 2007, 255(2), 343–349 CrossRef CAS.
- A. M. Díez-Pascual and A. L. Díez-Vicente, Biomacromol, 2015, 16(9), 2631–2644 CrossRef PubMed.
- R. Augustine, E. A. Dominic, I. Reju, B. Kaimal, N. Kalarikkal and S. Thomas, RSC Adv., 2014, 4(93), 51528–51536 RSC.
- H. Chhabra, R. Deshpande, M. Kanitkar, A. Jaiswal, V. P. Kale and J. R. Bellare, RSC Adv., 2016, 6(2), 1428–1439 RSC.
- P. T. Sudheesh Kumar, V. K. Lakshmanan, T. V. Anilkumar, C. Ramya, P. Reshmi, A. G. Unnikrishnan, S. V. Nair and R. Jayakumar, ACS Appl. Mater. Interfaces, 2012, 4(5), 2618–2629 CrossRef CAS PubMed.
- S. Kumar, V. K. Lakshmanan, M. Raj, R. Biswas, T. Hiroshi, S. V. Nair and R. Jayakumar, Pharm. Res., 2013, 30(2), 523–537 CrossRef PubMed.
- Y. Liang, M. Wang, Z. Zhang, G. Ren, Y. Liu, S. Wu and J. Shen, Chem. Eng. J., 2019, 378, 122043 CrossRef CAS.
- A. Dodero, S. Scarfi, M. Pozzolini, S. Vicini, M. Alloisio and M. Castellano, ACS Appl. Mater. Interfaces, 2019, 12(3), 3371–3381 CrossRef PubMed.
- R. Ahmed, M. Tariq, I. Ali, R. Asghar, P. N. Khanam, R. Augustine and A. Hasan, Int. J. Biol. Macromol., 2018, 120, 385–393 CrossRef CAS PubMed.
- T. N. V. K. V. Prasad, P. Sudhakar, Y. Sreenivasulu, P. Latha, V. Munaswamy, K. R. Reddy, T. S. Sreeprasad, P. R. Sajanlal and T. Pradeep, J. Plant Nutr., 2012, 35(6), 905–927 CrossRef CAS.
- P. Mahajan, S. K. Dhoke and A. S. Khanna, J. Nanotechnol., 2011, 2011, 696535 Search PubMed.
- U. Burman, M. Saini and P. Kumar, Toxicol. Environ. Chem., 2013, 95(4), 605–612 CrossRef CAS.
- R. Raliya and J. C. Tarafdar, Agric. Res., 2013, 2(1), 48–57 CrossRef CAS.
- T. Adhikari, S. Kundu, A. K. Biswas, J. C. Tarafdar and A. Subba Rao, J. Plant Nutr., 2015, 38(10), 1505–1515 CrossRef CAS.
- R. Qiu, D. Zhang, Y. Mo, L. Song, E. Brewer, X. Huang and Y. Xiong, J. Hazard. Mater., 2008, 156(1–3), 80–85 CrossRef CAS PubMed.
- N. Madhusudhana, K. Yogendra and K. M. Mahadevan, Int. J. Eng. Res. Appl., 2012, 2(5), 1300–1307 Search PubMed.
- S. Senthilkumaar and R. Thamizselvi, Int. J. Res. Eng. Technol. Sci., 2017, 4(11), 898–905 Search PubMed.
- D. E. León, H. Zúñiga-Benítez, G. A. Peñuela and H. D. Mansilla, Water, Air, and Soil Pollut., 2017, 228(9), 361 CrossRef.
- K. Nithiyadevi and K. Ravichandran, J. Mater. Sci.: Mater. Electron., 2017, 28(15), 10929–10939 CrossRef CAS.
- D. Sardar, J. Maity, M. K. Ghosalya, C. S. Gopinath and T. Bala, Mater. Res. Express, 2017, 4(5), 055011 CrossRef CAS.
- V. Vaiano, M. Matarangolo, J. J. Murcia, H. Rojas, J. A. Navío and M. C. Hidalgo, Appl. Catal., B, 2018, 225, 197–206 CrossRef CAS.
- A. Bhattacharjee and M. Ahmaruzzaman, J. Environ. Chem. Eng., 2018, 6(4), 4970–4979 CrossRef CAS.
- A. Bhattacharjee and M. Ahmaruzzaman, RSC Adv., 2016, 6(1), 527–533 RSC.
- N. Chidhambaram and K. Ravichandran, Mater. Res. Express, 2017, 4(7), 075037 CrossRef CAS.
- S. Raha and M. Ahmaruzzaman, Chem. Eng. J., 2020, 387, 123766 CrossRef CAS.
- S. Raha and M. Ahmaruzzaman, Chem. Eng. J., 2020, 395, 124969 CrossRef CAS.
- K. A. Adegoke, M. Iqbal, H. Louis and O. S. Bello, Mater. Sci. Energy Technol., 2019, 2(2), 329–336 Search PubMed.
- C. Worathitanon, K. Jangyubol, P. Ruengrung, W. Donphai, W. Klysubun, N. Chanlek, P. Prasitchoke and M. Chareonpanich, Appl. Catal., B, 2019, 241, 359–366 CrossRef CAS.
- M. Le Pivert, R. Poupart, M. Capochichi-Gnambodoe, N. Martin and Y. Leprince-Wang, Microsyst. Nanoeng., 2019, 5(1), 1–7 CrossRef PubMed.
- V. Sharma, R. K. Shukla, N. Saxena, D. Parmar, M. Das and A. Dhawan, Toxicol. Lett., 2009, 185(3), 211–218 CrossRef CAS PubMed.
- R. Brayner, R. Ferrari-Iliou, N. Brivois, S. Djediat, M. F. Benedetti and F. Fiévet, Nano Lett., 2006, 6(4), 866–870 CrossRef CAS PubMed.
- N. M. Franklin, N. J. Rogers, S. C. Apte, G. E. Batley, G. E. Gadd and P. S. Casey, Environ. Sci. Technol., 2007, 41(24), 8484–8490 CrossRef CAS PubMed.
- M. Heinlaan, A. Ivask, I. Blinova, H. C. Dubourguier and A. Kahru, Chemosphere, 2008, 71(7), 1308–1316 CrossRef CAS PubMed.
- B. Wang, W. Feng, M. Wang, T. Wang, Y. Gu, M. Zhu, H. Ouyang, J. Shi, F. Zhang, Y. Zhao and Z. Chai, J. Nanopart. Res., 2008, 10(2), 263–276 CrossRef CAS.
- A. K. Srivastav, M. Kumar, N. G. Ansari, A. K. Jain, J. Shankar, N. Arjaria, P. Jagdale and D. Singh, Hum. Exp. Toxicol., 2016, 35(12), 1286–1304 CrossRef CAS PubMed.
- C. Wang, J. Lu, L. Zhou, J. Li, J. Xu, W. Li, L. Zhang, X. Zhong and T. Wang, PLoS One, 2016, 11(10), e0164434 CrossRef PubMed.
- H. Q. Tang, M. Xu, Q. Rong, R. W. Jin, Q. J. Liu and Y. L. Li, Int. J. Nanomed., 2016, 11, 4275 CrossRef CAS PubMed.
- L. Xiao, C. Liu, X. Chen and Z. Yang, Food Chem. Toxicol., 2016, 90, 76–83 CrossRef CAS PubMed.
- M. Ghosh, A. Jana, S. Sinha, M. Jothiramajayam, A. Nag, A. Chakraborty, A. Mukherjee and A. Mukherjee, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2016, 807, 25–32 CrossRef CAS PubMed.
- J. S. Choi, R. O. Kim, S. Yoon and W. K. Kim, PLoS One, 2016, 11(8), e0160763 CrossRef PubMed.
- J. Du, S. Wang, H. You and Z. Liu, J. Environ. Sci., 2016, 47, 153–164 CrossRef CAS PubMed.
- L. Chupani, E. Zusková, H. Niksirat, A. Panáček, V. Lünsmann, S. B. Haange, M. von Bergen and N. Jehmlich, Sci. Total Environ., 2017, 579, 1504–1511 CrossRef CAS PubMed.
- X. Wang, X. Yang, S. Chen, Q. Li, W. Wang, C. Hou, X. Gao, L. Wang and S. Wang, Front. Plant Sci., 2016, 6, 1243 Search PubMed.
- P. Boonyanitipong, B. Kositsup, P. Kumar, S. Baruah and J. Dutta, Int. J. Biosci., Biochem. Bioinf., 2011, 1(4), 282 Search PubMed.
- S. V. Raskar and S. L. Laware, Int. J. Curr. Microbiol. Appl. Sci., 2014, 3(2), 467–473 CAS.
- H. Zafar, A. Ali, J. S. Ali, I. U. Haq and M. Zia, Front. Plant Sci., 2016, 7, 535 Search PubMed.
- D. Lin and B. Xing, Environ. Sci. Technol., 2008, 42(15), 5580–5585 CrossRef CAS PubMed.
- L. Yu, T. Fang, D. Xiong, W. Zhu, X. Sima, L. Yu, T. Fang and D. Xiong, J. Environ. Monit., 2011, 13, 1975–1982 RSC.
- B. Wang, Y. Zhang, Z. Mao, D. Yu and C. Gao, J. Nanosci. Nanotechnol., 2014, 4(8), 5688–5696 CrossRef PubMed.
- J. M. Guerreiro-Tanomaru, K. F. Pereira, C. A. Nascimento, M. I. Bernardi and M. Tanomaru-Filho, Acta Odontol. Latinoam., 2013, 26(3), 144–148 Search PubMed.
- M. B. Smeets, J. Fontijn, A. Kavelaars, G. Pasterkamp and D. P. V. De Kleijn, Int. J. Exp. Pathol., 2003, 84(2), 69–74 CrossRef CAS PubMed.
- L. He, Y. Liu, A. Mustapha and M. Lin, Microbiol. Res., 2011, 166, 2007–2015 CrossRef PubMed.
- N. M. Franklin, N. J. Rogers, S. C. Apte, G. E. Batley, G. E. Gadd and P. S. Casey, Environ. Sci. Technol., 2007, 41, 8484–8490 CrossRef CAS PubMed.
- A. Kahru and H. C. Dubourguier, Toxicol, 2010, 269, 105–119 CrossRef CAS PubMed.
- M. Li, L. Zhu and D. Lin, Environ. Sci. Technol., 2011, 45, 1977–1983 CrossRef CAS PubMed.
- L. W. Hall and R. D. Anderson, Crit. Rev. Toxicol., 1995, 25, 281–346 CrossRef CAS PubMed.
- S. George, S. Pokhrel, T. Xia, B. Gilbert, Z. Ji, M. Schowalter, A. Rosenauer, R. Damoiseaux, K. A. Bradley, L. Mädler and A. E. Nel, ACS Nano, 2010, 4, 15–29 CrossRef CAS.
- S. Nair, A. Sasidharan, V. V. Divya Rani, D. Menon, S. Nair, K. Manzoor and S. Raina, J. Mater. Sci.: Mater. Med., 2009, 20(S1), S235–S411 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |