
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn the photostability and luminescence of dye-sensitized upconverting nanoparticles using modified IR820 dyes†
Mannu
Kaur
 ,
Gabrielle A.
Mandl
,
Steven L.
Maurizio
,
Gabriella
Tessitore
and
John A.
Capobianco
*
,
Gabrielle A.
Mandl
,
Steven L.
Maurizio
,
Gabriella
Tessitore
and
John A.
Capobianco
*
Department of Chemistry and Biochemistry, Centre for NanoScience Research, Concordia University, 7141 Sherbrooke Street West, Montreal, QC H4B 1R6, Canada. E-mail: john.capobianco@concordia.ca
First published on 22nd December 2021
Abstract
Dye sensitization is a promising route to enhance luminescence from lanthanide-doped upconverting nanoparticles (LnUCNPs) by improving the photon harvesting capability of LnUCNPs through the use of dye molecules, characterized by higher absorption coefficients. The literature does not fully address the poor photostability of NIR dyes, hindering solution-based applications. The improvements achieved by dye-sensitized LnUCNPs are usually obtained by comparison with non-dye sensitized LnUCNPs. This comparison results in exciting the LnUCNPs at different wavelengths with respect to the dye-sensitized LnUCNPs or at the same wavelengths, where, however, the non-dye sensitized LnUCNPs do not absorb. Both these comparisons are hardly conclusive for a quantification of the improvements achieved by dye-sensitization. Both shortcomings were addressed by studying the photodegradation via thorough spectroscopic evaluations of a 4-nitrothiophenol-modified and unmodified IR820-LnUCNP system. The modified IR820 dye system exhibits a 200% enhancement in the emission of NaGdF4:Er3+,Yb3+/NaGdF4:Yb3+ nanoparticles relative to the unmodified IR820-sensitized LnUCNPs and emits for over twice the duration, demonstrating a substantial improvement over previous dye–LnUCNP systems. Upconversion dynamics between the dyes and Er3+ establish the importance of back-transfer dynamics in modulating the dye–LnUCNP luminescence. Quantum yield measurements further illustrate the mechanism of sensitization and increased efficiency of this new nanosystem.
Introduction
Upconversion is an anti-Stokes process in which the successive absorption of two or more low-energy photons results in the production of higher energy emissions. The long-lived intermediate energy states of some lanthanides enable them to emit in the UV, visible or NIR region when excited with NIR radiation, which makes them an ideal candidate for generating upconverting nanoparticles. Lanthanide-doped upconverting nanoparticles (LnUCNPs) have narrow absorption and emission bands, high chemical stability, and are non-blinking and low toxicity nanomaterials.1–4Despite promising properties of LnUCNPs, their practical applicability is still limited due to their low upconversion efficiencies. This drawback is, in part, caused by the low absorption cross-section of lanthanide sensitizer ions (on the order of 10−20 cm2 in the NIR), usually based on the use of Yb3+ as a sensitizer.5 Furthermore, Yb3+ is excited at 976 nm, which can cause significant heating of aqueous media. In contrast, Nd3+ has been used as a sensitizer and has a narrow absorbance band at 800 nm, which does not induce heating of water, however it has a low absorption cross-section. Several structural manipulations have been studied to improve upconversion luminescence (UCL), such as controlling the crystal phase of the host lattice, tailoring the local crystal fields around lanthanide dopants or adopting core/shell structures, which can enhance UCL by 10 to 20 times.6–8 In 1942, Weissman proposed to overcome the inherent low absorption cross-section of the lanthanides by encapsulating the lanthanide ion with a suitable organic ligand to harvest the light, which allows for an indirect excitation of the lanthanide ion via energy transfer.9 In the early 1990s, lanthanide complexes were investigated as luminescent materials, and Lehn proposed that such complexes could be perceived as light conversion molecular devices and coined the term antenna effect.10 This term denotes the absorption, the energy transfer and the emission sequence involving the ligand and the emitting lanthanide ion, thus surmounting the low absorption cross-section of the lanthanide ions. These concepts proposed by Weissman and Lehn form the basis for the concept of dye-sensitized upconversion for LnUCNPs.
Since 2012, several studies have proven the advantages in the use of dye sensitization for enhancing UCL.11–15 According to this method, an organic NIR-emitting dye with a high absorption cross-section (on the order of 10−16 to 10−17 cm2)12 is coordinated to the nanoparticle surface, and the dye molecules act as an antenna to harvest the incoming radiation and transfer it to lanthanide sensitizer ions, after which upconversion luminescence is observed.11,14–16 Apart from brighter UCL, NIR dye-sensitized LnUCNPs can also be excited in the region of 780–830 nm, depending on the absorbance of the NIR dye used, in contrast to conventional Yb3+-doped LnUCNPs (excited at 976 nm), resulting in broadband absorption of LnUCNPs at different wavelengths.17,18
In spite of the attractive nature of this approach, low photostability is a major drawback associated with the use of NIR dyes.19–21 The lack of photostability makes them unsuitable for applications which require excitation of the system for prolonged times, as the benefits obtained from dye sensitization, in terms of luminescence intensity, are restricted to the short period of time prior to the degradation of the dye. Unfortunately, the photostability of NIR dyes with respect to dye-sensitized upconversion has been often overlooked in the myriad of reported dye–LnUCNP systems.22 Because the drawback of photostability is rarely acknowledged, the practical applicability of these systems is at a disadvantage since there is a lack of knowledge of the duration for which these systems are effective. Additionally, despite the myriad of demonstrated dye-sensitized systems, there is no clear convention for reporting the improvements made by new dye–UCNP systems with respect to luminescence enhancement and again, photodegradation studies are lacking. Thus, it is difficult to gauge how efficient one reported dye–LnUCNP system is versus another and whether or not the systems are useful for prolonged irradiation times. Consequently, it is of the utmost importance to both properly study the photostability of NIR dyes and thoroughly investigate their dynamics upon coordination to the LnUCNPs. These studies are necessary in order to report a meaningful enhancement relative to previously reported systems, such that practical applicability can be realized and even greater improvements can be made.
IR820 is commercially available and is commonly utilized for upconversion sensitization.12,14,15 This dye belongs to the cyanine family, well-known for having high absorption cross-sections and low photostability.23 IR820 is utilized because it exhibits good spectral overlap with Yb3+, facilitating energy transfer when excited at 800 nm.14 Additionally, IR820 has a –Cl substituted aryl ring situated at the center of the heptamethine conjugation, allowing for versatile and facile structural modifications that can be used to modulate the steric and optical properties of the dye.11,12,17
The structure of the dye also makes it attractive for use with LnUCNPs, owing to the pendant sulfonate moieties, which are crucial for obtaining strong electrostatic interactions to the positively-charged surface of unfunctionalized fluoride nanoparticles.24 It is well-established that the heptamethine conjugation of cyanine dyes can enhance the lifetime of the dye's triplet state when in the presence of a heavy atom such as Gd3+. This phenomenon is known as the heavy atom effect and has been studied as early as 1949 by McClure et al.16,25 The enhancement of the lifetime of the triplet state is also known to be deleterious to the photostability of the dyes, as it results in an increased production of singlet oxygen, which is implicated in the photodecomposition of the dyes.16,26 Fortunately, the pendant sulfonate groups of IR820 can be used to anchor the dye to the LnUCNPs, without any electronic contribution to the heptamethine conjugation, hence avoiding the deleterious enhancement of the triplet state lifetime. Thus, these functional groups help to avoid the heavy atom effect and make these dyes applicable for different LnUCNP compositions, useful in situations where heavy atoms like Gd3+ may not be an option.
The photodegradation of NIR dyes occurs via in situ production of singlet oxygen, which further diffuses and reacts with the unsaturated bonds in the molecule. The use of dye-sensitized LnUCNPs embedded into a solid matrix, such as a polymer, has been proposed in order to prevent photodegradation that occurs at a greater rate in solution, owing to the increased diffusion efficiency of singlet oxygen in less-viscous media.27 Thus, currently reported systems are limited to solid-phase applications.28,29 Strategies aimed at preventing the degradation of cyanine dyes via structural modifications that sterically hinder the formation of deleterious dioxetane intermediates19,20,30 have shown great promise in enhancing their stability.
In this study, we proposed for the first time a structural modification of the IR820 dye with a sterically hindering 4-nitrothiophenol group (IR820–NO2) onto the central aryl ring, with improved photostability and greater spectral overlap with Yb3+. We also report the comparison of enhancement in emissions and quantum yields of NaGdF4:Er3+,Yb3+/NaGdF4:Yb3+ functionalized with modified and unmodified IR820, both excited at 800 nm. The functionalization of IR820 imparts resistance to photoinduced degradation, as proven by spectroscopic evaluation of the photodegradation dynamics. The enhancement of UCL and photostability by using the modified NIR dye with LnUCNPs was demonstrated by linking the dye onto the surface of oleate-free NaGdF4:Er3+,Yb3+/NaGdF4:Yb3+ core/shell nanoparticles and recording the temporal evolution of their UCL spectra. As the linking of the modified and unmodified IR820 dye is accomplished via the sulfonate pendants, we investigate the potential for the heavy atom effect in Gd3+ and Y3+-based core/shell nanoparticle systems. A comprehensive study of the luminescence lifetimes provides further evidence of back-transfer from the LnUCNPs to the coordinated dye molecules. The proposed approach is the first, to our knowledge, to address the limitations in applicability of dye sensitized LnUCNPs in the solution phase. The achieved prolonged stability and increased UCL intensity and quantum yields demonstrate significant improvements toward practical utilization of dye-sensitized upconverting nanoparticles for solution-based applications or when embedded into a matrix.
Experimental section
Materials and methods
Instrumentation
Transmission electron microscopy (TEM) was performed using at 200 kV JEOL JSM2100F microscope. TEM samples were prepared on a 300 mesh copper grid (3 mm in diameter) by depositing 10 μL of 1 mg mL−1 LnUCNPs dispersed in hexanes. The images obtained were analyzed in ImageJ, free software courtesy of the National Institutes of Health.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 v/v mixture of hexane and ethanol and was precipitated by centrifugation.
000 rpm for 20 min and the pellet was collected and stored in ethanol.
000 rpm for 15 min. The pellet was washed with ethanol 3 times and kept for drying overnight for FT-IR and UV-vis calibration studies.
:5 v/v mixture of hexane and ethanol and was precipitated by centrifugation.
000 rpm for 20 min and the pellet was collected and stored in ethanol.
000 rpm for 15 min. The pellet was washed with ethanol 3 times and kept for drying overnight for FT-IR and UV-vis calibration studies.
Results and discussion
Effects of structural modifications on the photostability of IR820 dye
Singlet oxygen is a highly reactive species which is implicated in the photo-induced degradation of many cyanine dyes.19,20,26 However, to our knowledge, this has never been conclusively established for IR820 photodegradation. The photodegradation dynamics of IR820 were studied in order to ascertain the role of singlet oxygen in the decomposition mechanism of the dye, as well as to establish a baseline from which improvements (upon structural modification) could be evaluated. The changes in photostability of IR820 in different atmospheres and solvents, as well as in the presence of sodium azide (a singlet oxygen scavenger), were measured, and confirm the role of singlet oxygen in the photodegradation of the dye. This was evidenced by a decrease in IR820 absorbance (40–95%) within 30 minutes of irradiation under the different reported conditions (Fig. S1–S3†).Investigations of the photodegradation products by mass spectrometry illustrate that the degradation of IR820 occurs primarily due to a bimolecular reaction of the heptamethine conjugation with photogenerated singlet oxygen. This is the first time, to our knowledge, that the photodegradation products of IR820 have been reported. As a result of this reaction, a strained dioxetane intermediate is formed, followed by decomposition of the dye into carbonyl compounds (Fig. S4 and Table S2†). Therefore, by introducing steric hindrance to the region around the heptamethine conjugation, the formation of the dioxetane intermediate can be inhibited, reducing the likelihood for photodegradation reactions to occur.30
Structural modifications aimed at the inhibition of the bimolecular reaction of the dye with singlet oxygen were investigated as a route to improved photostability of the NIR dye. The –Cl group of the central aryl ring was modified with a variety of para-substituted phenylthioether groups with a range of different electronegativities at the para-position (Fig. 1A, S5 and S6†). The photostability of each modified dye under 800 nm excitation was evaluated. Irradiation time, laser power density and concentration of the functionalized dyes were identical to the photostability studies for IR820 discussed above (Fig. 1B).
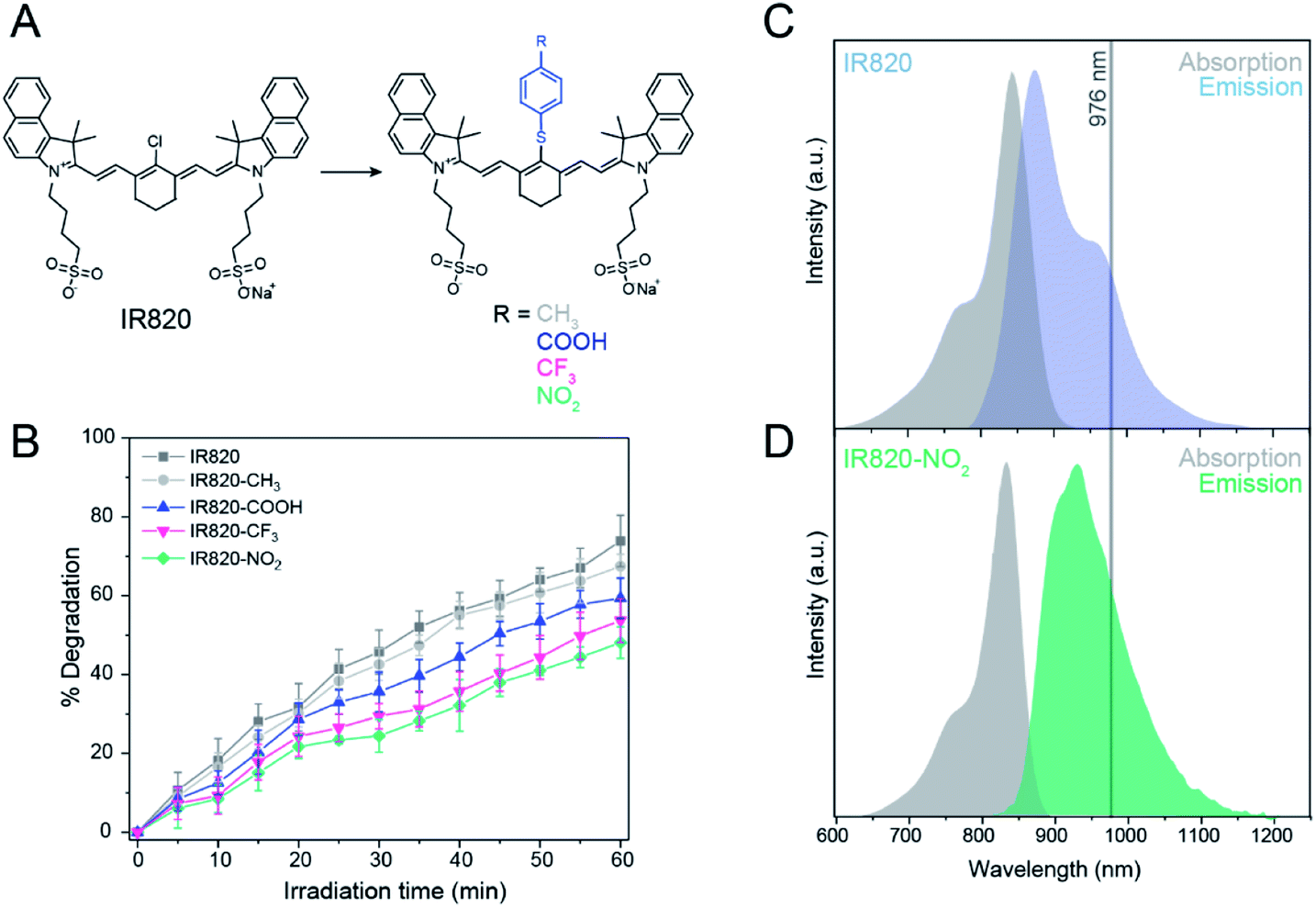 | ||
| Fig. 1 (A) Chemical structure of IR820 modified with a thiophenol group and corresponding studied groups placed at the para position. (B) Percentage photodegradation of the IR820 derivatives as a function of irradiation time (800 nm irradiation, 4.5 W cm−2). Errors established based on three repetitions. (C) Absorption (gray) and emission (blue) of IR820 dye in methanol (λmax abs 820 nm, λmax em 849 nm). (D) Absorption (gray) and emission (green) of IR820–NO2 dye in methanol (λmax abs 837 nm, λmax em 924 nm). | ||
Upon comparing the photostability of the different functionalized dyes, the degradation was observed to follow a pseudo-first order reaction, due to the self-generation of singlet oxygen that is dependent on the concentration of dye.19 IR820–NO2 was found to be the most photostable, degrading 44% under ambient conditions after 60 min of irradiation, compared to the unmodified IR820, which exhibited 74% degradation and double the rate constant under the same conditions (Fig. S3C†). In comparison, IR820–CF3, IR820–COOH, and IR820–CH3 exhibit decreasing photostability, respectively, with the –CH3 modified dye exhibiting a 67% degradation (Fig. 1B and S7†).
The increase in photostability can be attributed to two main properties of the modification. Firstly, the bulkiness of the aromatic thioether group hinders the ability of singlet oxygen to approach the heptamethine conjugation, as previously discussed.18,20 Secondly, the strength of the electron withdrawing group at the para-position of the thiophenol alters the electron density within the conjugation, increasing photostability of the dye. Since the –CH3 substituted thiophenol group is electron-donating, this results in electronic activation of the heptamethine conjugation towards electrophilic reaction with singlet oxygen by increasing the electron density.30
For the CH3-modified dye, the increased electron density of the conjugation is likely to facilitate the reaction with singlet oxygen in spite of the increased steric hindrance due to the thiophenol group, resulting in the highest degradation percentage among the studied functionalized dyes. Thus, introducing a strong electron withdrawing group at the para-position reduces the electron density within the conjugation, thereby reducing the likelihood of an electrophilic reaction with singlet oxygen.30 These results illustrate that the combination of steric hindrance and strong electron withdrawing capability of the para-substituted group are necessary to improve the photostability of the IR820 dye, with IR820–NO2 being the most photostable.
After confirming that the modified dyes were all more photostable than native IR820, shifts in the absorption and emission maxima of the modified IR820 dyes were evaluated in order to determine optimal suitability for energy transfer with Yb3+. Native IR820 has singlet-state absorption and emission maxima at 820 and 850 nm, respectively (Fig. 1C), which partially overlaps with Yb3+ absorption, between 910 and 1052 nm with a maximum at 976 nm. The functionalized dyes exhibited red shifts in absorbance and emission, which are desirable for increasing the spectral overlap of the singlet state of the dye with the Yb3+ sensitizer in the upconverting nanoparticles (Table S3†). The Yb3+ sensitizer in β-NaGdF4:Yb3+, Er3+ is known to absorb between 910 nm and 1052 nm, with maximum absorption at 976 nm.33 In particular, substitution with a 4-nitrothiophenol moiety allowed for a particularly good spectral overlap with Yb3+ as the emission maximum shifted to 950 nm (λex 800 nm) for this functionalized dye (Fig. 1D). Furthermore, it can be postulated that the red shift in the singlet emission band is also likely to mean a red shift occurs in the triplet state emission maxima, further reducing the efficiency of singlet oxygen production due to a mismatch in resonance with the energy gap of singlet oxygen, potentially also contributing to increased photostability. Thus, since IR820–NO2 had the greatest photostability, and most optimal spectral overlap with Yb3+, it was chosen as the ideal candidate to use for dye-sensitized upconversion.
The increase in photostability can be attributed to two main properties of the modification. Firstly, the bulkiness of the aromatic thioether group hinders the ability of singlet oxygen to approach the heptamethine conjugation, as previously discussed.18,20 Secondly, the strength of the electron withdrawing group at the para-position of the thiophenol alters the electron density within the conjugation, increasing photostability of the dye. Since the –CH3 substituted thiophenol group is electron-donating, this results in electronic activation of the heptamethine conjugation towards electrophilic reaction with singlet oxygen by increasing the electron density.30 For the CH3-modified dye, the increased electron density of the conjugation is likely to facilitate the reaction with singlet oxygen in spite of the increased steric hindrance due to the thiophenol group, resulting in the highest degradation percentage among the studied functionalized dyes. Thus, introducing a strong electron withdrawing group at the para-position reduces the electron density within the conjugation, thereby reducing the likelihood of an electrophilic reaction with singlet oxygen.30 These results illustrate that the combination of steric hindrance and strong electron withdrawing capability of the para-substituted group are necessary to improve the photostability of the IR820 dye, with IR820–NO2 being the most photostable.
After confirming that the modified dyes were all more photostable than native IR820, shifts in the absorption and emission maxima of the modified IR820 dyes were evaluated in order to determine optimal suitability for energy transfer with Yb3+. Native IR820 has singlet-state absorption and emission maxima at 820 and 850 nm, respectively (Fig. 1C), which partially overlaps with Yb3+ absorption, between 910 and 1052 nm with a maximum at 976 nm. The functionalized dyes exhibited red shifts in absorbance and emission, which are desirable for increasing the spectral overlap of the singlet state of the dye with the Yb3+ sensitizer in the upconverting nanoparticles (Table S3†). The Yb3+ sensitizer in β-NaGdF4:Yb3+, Er3+ is known to absorb between 910 nm and 1052 nm, with maximum absorption at 976 nm.33 In particular, substitution with a 4-nitrothiophenol moiety allowed for a particularly good spectral overlap with Yb3+ as the emission maximum shifted to 950 nm (λex 800 nm) for this functionalized dye (Fig. 1D). Furthermore, it can be postulated that the red shift in the singlet emission band is also likely to mean a red shift occurs in the triplet state emission maxima, further reducing the efficiency of singlet oxygen production due to a mismatch in resonance with the energy gap of singlet oxygen, potentially also contributing to increased photostability. Thus, since IR820–NO2 had the greatest photostability, and most optimal spectral overlap with Yb3+, it was chosen as the ideal candidate to use for dye-sensitized upconversion.
Spectroscopic characterization of dye–LnUCNP nanosystems
To evaluate the efficacy of the dyes for upconversion sensitization, β-NaGdF4:Er3+,Yb3+/NaGdF4:Yb3+ core/shell upconverting nanoparticles were selected. A core/shell architecture was chosen because of the increased intensity of the Er3+ emissions in solution, as compared to the core-only particles, which often suffer from quenching due to interactions of solvent phonons with the nanoparticle surface.1,6,7 An “active” shell, containing Yb3+ as a sensitizer, was used to allow for non-radiative energy transfer between the dye molecules and the Yb3+ ions, which requires close proximity of the involved species to interact. These core/shell nanoparticles were synthesized by a double injection thermal decomposition method.7 The as-synthesized nanoparticles were physically characterized by XRD and TEM (Fig. S8†), confirming a hexagonal crystal phase, uniform morphology, and an average nanoparticle size of 35 ± 2 nm.IR820 and IR820–NO2 were each coordinated to the nanoparticle surface via electrostatic interactions. To ensure a close contact between the dyes and nanoparticle surface to facilitate efficient energy transfer, the oleate capping ligand was removed from the nanoparticle surface.32 Removal of the oleate ligand was confirmed by FT-IR (Fig. S9A†) and zeta potential (+29.5 mV for oleate-free LnUCNPs, +4.5 mV for dye-coated LnUCNPs). TEM was used to confirm there was no aggregation post-oleate removal and dye linking (Fig. S10A and B†). Confirmation of the dye linking was obtained using FT-IR (Fig. S9B†) and subsequent spectroscopy experiments.
Functionalization of the LnUCNP surface with different concentrations of IR820 and IR820–NO2 was performed in order to determine the optimal number of dyes per nanoparticle for luminescence enhancement with respect to the green Er3+ emissions. It is well-established that too high of a concentration of dye molecules can result in self-quenching, whereas too low of a dye concentration will not provide sufficient enhancement of the absorbed NIR light. Consequently, in both cases, the UCL would be negatively affected.11,12,14–17 Therefore, upconversion emission spectra of the dye–LnUCNPs were recorded for varied amounts of dye molecules per nanoparticle (Fig. 2A, Table S1, Fig. S11 and 12†). Additionally, the dye-coating was stable for a minimum of 72 hours, with no observed leakage (Fig. S13†). Upconversion emission spectra of dye-functionalized LnUCNPs excited at 800 nm exhibit the characteristic Er3+ green and red emissions, as shown in Fig. 2B. Green emissions were observed between 510–570 nm and ascribed to the 2H11/2 → 4I15/2 and 4S3/2 → 4I15/2 transitions (centered at 525 and 550 nm, respectively). Red emissions were observed between 630–680 nm from the 4F9/2 → 4I15/2 transition. To evaluate the difference in intensity of Er3+ emissions from IR820 and IR820–NO2 LnUCNPs with varying amounts of dye/LnUCNP, the Er3+ emissions sensitized by IR820 were normalized with respect to the most intense emission of IR820–NO2 sensitized LnUCNPs (Fig. S12†). The emission spectra of the dye-functionalized LnUCNPs at different concentrations of dyes shows that approximately 94 IR820–NO2 dye molecules/LnUCNP and approximately 99 IR820 dye molecules/LnUCNP constituted the optimum concentrations in each case (Fig. S12†). Furthermore, the photostability of the dye–LnUCNP constructs were evaluated using NaYF4 as a host to evaluate the role of the heavy atom effect in dye degradation. Stabilization of the triplet state through the “heavy atom” effect should influence the emission from the dye-sensitized LnUCNPs if the triplet states overlap with the Yb3+ sensitizer ions. No contribution from the heavy atom effect was observed, as Y3+ is not considered a “heavy atom” and no change in UCL was observed as a function of a change in host composition (Fig. S14†). This confirms the energy transfer occurs from the singlet state of the dye to Yb3+, as there is no spectral overlap of Yb3+ with the triplet state of the dye. As previously discussed, this is due to the alkyl chain length of the pendant sulfonate moieties on the dye, which are used to conjugate the IR820–NO2 to the LnUCNP surface. This is important, as it allows for versatile utilization of this photostable NIR dye with different LnUCNP compositions.
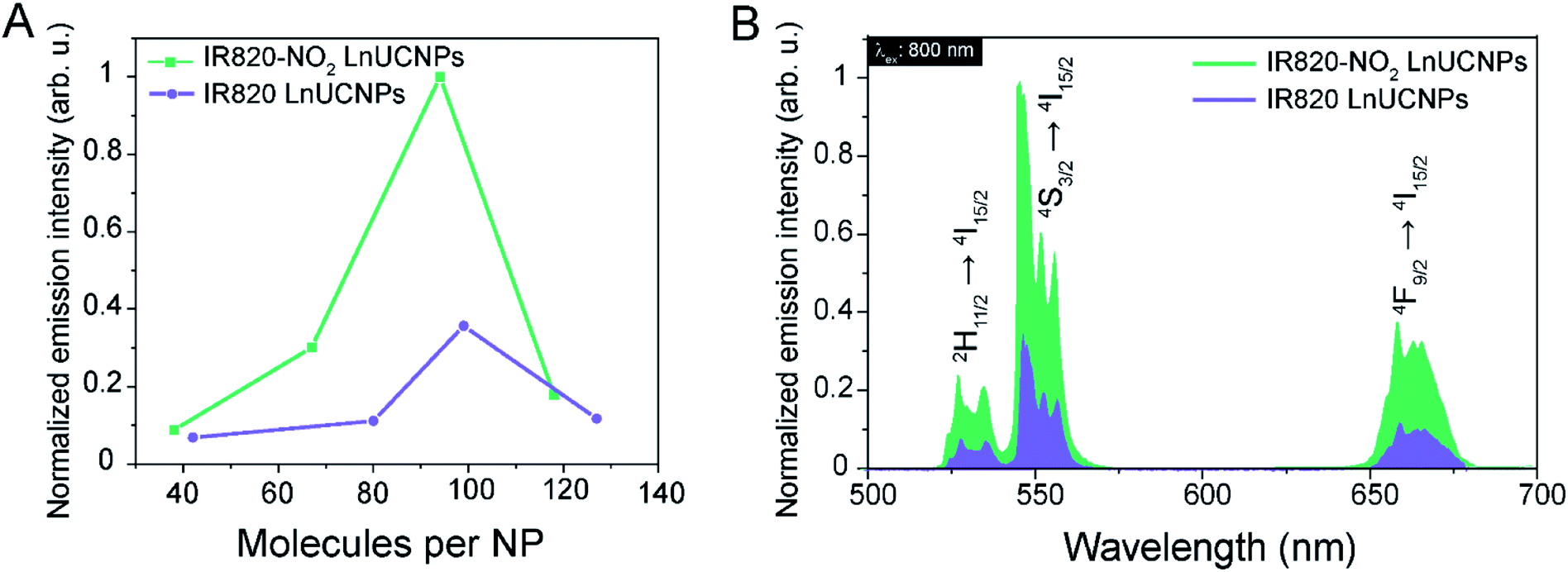 | ||
| Fig. 2 (A) Integrated emission intensities of the 4S3/2 → 4I15/2 transition of colloidal dispersions of IR820–NO2-NPs (green) and IR820-NPs (blue) as a function of the number of dye molecules per nanoparticle. (B) Upconversion emission spectra of IR820–NO2-NPs (green) and IR820-NPs (blue) with the optimum number of dye molecules per nanoparticle. | ||
In order to assess the improvement imparted by the structural modification of IR820 upon 800 nm excitation, the enhancement of the UCL had to be carefully analyzed, as the literature on dye-sensitized upconversion exhibits no standard methods of reporting enhancement values or a standard material/circumstance for which the enhancement values are compared to. It is especially difficult to compare improvements in newly reported dye-sensitized LnUCNP systems because many of them are evaluated with respect to the emissions of the non-sensitized LnUCNPs either upon 800 nm or 976 nm excitation.14,15,28 In the case of comparisons to LnUCNP emissions upon 800 nm, large enhancement values are reported since the native nanoparticles do not absorb (and therefore do not emit) at this excitation wavelength, unless Nd3+ is used as a sensitizer. Calculation of enhancement factors based on this method were carried out using the nanoparticles from this work, and a 3100-times enhancement was demonstrated (Fig. S15A†). This value is strongly influenced by the experimental setup, making it impossible to compare to other obtained values and easily manipulated by changing the recording parameters such as the integration time of the luminescence signal, underscoring the difficulty in comparing to other systems. Comparison of bare LnUCNPs excited at 976 nm and dye–LnUCNPs excited at 800 nm are also commonly made in the literature, however, the reported improvements are even more difficult to contextualize under these conditions since, even under the same power densities of the different excitation wavelengths, the photon flux differs and the wavelengths interact with solvents differently in terms of transmittance and refraction, and signal are often not corrected for the relative absorbance at each wavelength. As a result of the differing photon flux, the quantum yields of the system will vary drastically, making it virtually impossible to make a meaningful comparison between the two scenarios in terms of enhancement. More importantly, the population dynamics during upconversion when exciting at these two different wavelengths will differ. Upon 976 nm excitation of bare LnUCNPs, direct excitation of Yb3+ and energy transfer to Er3+ occurs, while upon 800 nm excitation of the dye-coated LnUCNPs, an indirect, two-step excitation must occur through the dye, then Yb3+ before exciting Er3+. Despite this, the comparison was made to illustrate the “practical” difference between the dye-sensitized emissions upon 800 nm excitation, and Yb3+-sensitized emissions upon 976 nm excitation, at the same power densities, from which a 1.7-times increase in the luminescence was observed for the IR820–NO2 LnUCNPs (Fig. S15B†).
For the above-mentioned reasons, comparison between a previously established dye–LnUCNP system and the new dye–LnUCNP system reported herein allows for a clear evaluation of the improvements made to the field of dye-sensitized LnUCNPs since excitation dynamics are identical in both scenarios. The emission spectra of IR820-functionalized LnUCNPs and IR820–NO2-functionalized LnUCNPs at their optimum loadings were compared by evaluating the integrated emission intensities over the entire visible region (400–750 nm). As shown in Fig. 2B, the IR820–NO2-functionalized LnUCNPs exhibit 200% enhancement in Er3+ emissions relative to the intensity of the IR820-functionalized LnUCNPs. Therefore, this result represents an enhancement over the baseline dye-sensitized upconversion system (IR820-LnUCNPs), indicating that significant and measurable progress has been demonstrated here in improving the luminescence of dye sensitized upconversion nanosystems with respect to the commonly used IR820 dye.
In addition to observing enhanced luminescence from the IR820–NO2 LnUCNPs upon 800 nm excitation, we also evaluated the dynamics of the dye-coated nanoparticles upon 976 nm excitation to establish the potential for deleterious back-transfer from Er3+ to the dye, resulting in luminescence quenching. The broad nature of the absorbance bands of organic dyes makes it likely that most currently employed dye–LnUCNP systems involving Er3+ have the potential for deleterious back-transfer processes. While this is likely the case for the majority of current dye–LnUCNP systems, to our knowledge, it has been seldomly explored or reported. By exciting the particles at 976 nm, the dye molecules are not expected to contribute to the population of the emitting states of Er3+. Contrarily, it provides insights into the dynamics of the depopulation of Er3+ in presence of the dye molecules, as both dyes have absorption bands that overlap with the Er3+ 4F9/2 → 4I15/2 transition at 655 nm. As previously shown in Fig. 1, IR820 has an absorption coefficient of 8860 M−1 cm−1, while IR820–NO2 has an absorption coefficient of 3340 M−1 cm−1 at 655 nm (Fig. S16†). Thus, both dyes have the potential for back-transfer from the Er3+ to the dye (Fig. 3A). As shown in Fig. 3B, evaluation of the upconversion emission spectra of both IR820 LnUCNPs and IR820–NO2 LnUCNPs upon 976 nm excitation, there is a significant reduction in the overall red and green emission intensities from IR820 LnUCNPs. Considering the higher absorption coefficient of IR820 at 655 nm, the red-to-green emission ratio is significantly lower for the IR820 dye upconversion spectrum than for the modified IR–NO2 dye (0.625 for IR820, 0.833 for IR820–NO2). However, the spectral overlap in the red region would not justify the decrease in the green emission intensities, thus upconversion lifetimes upon 976 nm excitation were performed to assess the interactions of the dyes and Er3+ (Fig. 3C). When comparing unfunctionalized LnUCNPs to IR820 functionalized LnUCNPs, the dye coating shortens the decay time by about 50% for both the green and red emission bands. This is compared to IR820–NO2 functionalized LnUCNPs, which only shortens the decay times by 36% for both the green and red emissions. The shortening of the red emission decay time is justified by the spectral overlap between both dyes and the Er3+ 4F9/2 → 4I15/2 transition, resulting in a non-radiative energy transfer from the Er3+ to the dye. While this is consistent with their respective absorption coefficients, it does not explain the shortening of the green emission decay time, which follows the same trend. This is postulated to be, in part, a result of a known cross-relaxation mechanism occurring between the green and red emitting states of Er3+ (2H11/2 + 4I11/2 → 4F9/2 + 4F9/2).34 The non-radiative depopulation of the red-emitting 4F9/2 state from the dye interaction results in improved efficiency of this mechanism, consequently reducing the green emission intensity and decay time, relative to the unfunctionalized LnUCNPs. Moreover, the reported depopulation of the 4I9/2 NIR intermediate state through back transfer from Er3+ to the dye molecules can further justify the observed trend for the green emitting states.35 Quenching of any of the NIR intermediate states involved in the population of the upper states would further justify the observed trend. Considering that back transfer to Yb3+ ions has been known to often occur both from NIR transitions involving the 4I11/2 and 4S3/2 states, these transitions would also be resonant with the absorption of the dye, providing possible additional routes for the depopulation of the Er3+ excited states.36,37
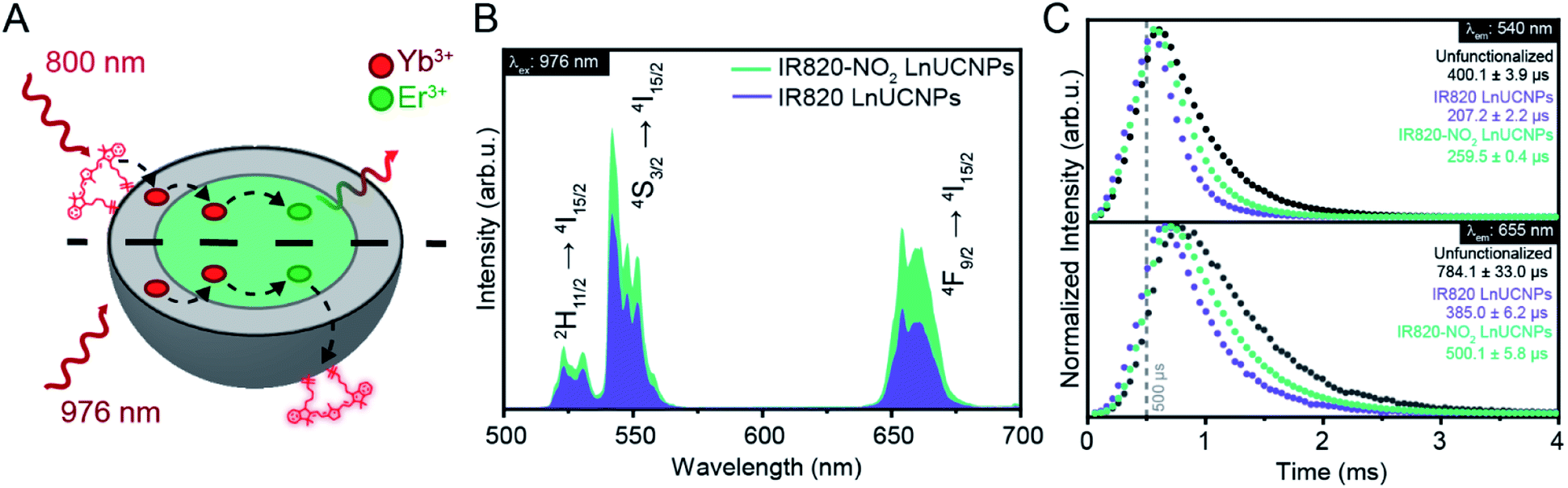 | ||
| Fig. 3 (A) Schematic of the interactions between the dye, sensitizer, and activator ions upon 800 nm excitation and 976 nm excitation. (B) Upconversion emission spectra of IR820–NO2-LnUCNPs and IR820 LnUCNPs upon 976 nm excitation. (C) Upconversion lifetimes of the green (top) and red (bottom) emissions of Er3+ in unfunctionalized (black), IR820-functionalized (blue) and IR820–NO2-functionalized LnUCNPs upon 976 nm excitation. | ||
To summarize, due to the increased spectral overlap between IR820 and the Er3+ red emission, there is a slight quenching of the overall upconversion luminescence from Er3+. This process is less efficient for IR820–NO2, due to its red-shifted absorbance band. As observed in Fig. 3B, the IR820–NO2 functionalized LnUCNPs exhibits a 60% increase in intensity relative to IR820 functionalized LnUCNPs, under 976 nm excitation. Back transfer occurs upon 800 nm excitation as well, since the Er3+ energy transfer to the dye is still expected, thus implying that up to 60% of the observed enhancement under 800 nm excitation is due to a decrease in back-transfer from the activator to the dye. This is an especially important finding, as it illustrates the importance of considering the back-transfer dynamics between the activator and dye when engineering dye-sensitized LnUCNPs.
As previously discussed, IR820–NO2 dye exhibited increased photostability relative to IR820. Thus, kinetic studies were performed on the IR820 and IR820–NO2 LnUCNPs to evaluate the photostability of the dye upon coordination to the nanoparticle surface. In particular, the luminescence intensity of the 4S3/2 → 4I15/2 transition of Er3+ as a function of irradiation time was evaluated. As observed in Fig. 4A, the green emissions of the IR820–NO2-LnUCNPs are substantially more intense than the IR820-LnUCNPs, even after 30 minutes of irradiation, illustrating the vast improvement of the modified dye with respect to facilitating dye-sensitized UCL. To quantify the results observed in Fig. 4A, the emissions from IR820–NO2 functionalized NPs were observable for more than double the duration of the IR820 functionalized LnUCNPs. The emissions from the IR820–NO2 functionalized LnUCNPs could be observed for over 1.5 hours, in comparison to only 30 minutes for IR820-LnUCNP emissions (Fig. 4B). Interestingly, the dye-functionalized LnUCNPs follow the same pseudo-first order rate of degradation when observing the upconversion emissions. Since both the dye degradation kinetics, as discussed above, and the UCL follow the same trend, it can be determined that the concentration of dye remaining on the surface of the nanoparticles (that is not degraded during irradiation) is related to the UCL enhancement.
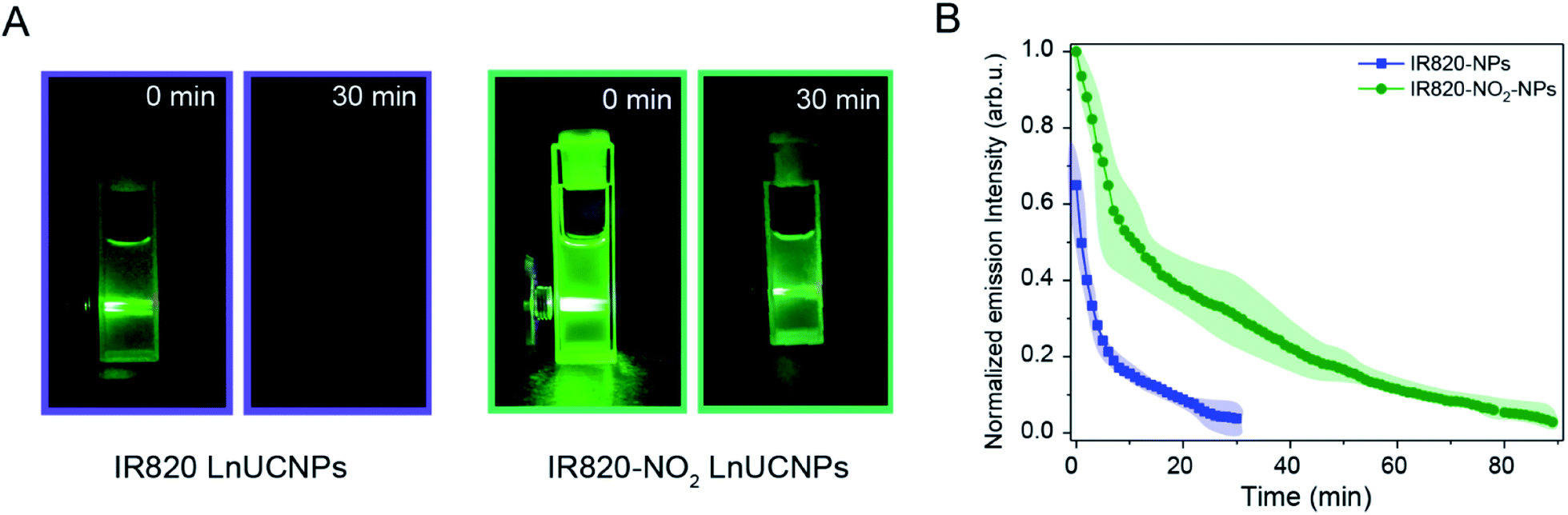 | ||
| Fig. 4 (A) Photographs of colloidal dispersions of IR820 LnUCNPs (top) and IR820–NO2 LnUCNPs (bottom) at 0 and 30 minutes of irradiation with 800 nm excitation. (B) Overall emission intensities of IR820–NO2-NPs (green) and IR820-NPs (blue) as a function of irradiation time upon 800 nm excitation (2.1 W cm−2). Light green and light blue regions around the curves represent the error margins. Errors are based on a minimum of two repetitions. | ||
The combination of the 200% enhancement in the luminescence and the more than doubled luminescence duration of this new nanosystem over the previous dye–LnUCNP system shows strong promise for practical use in a variety of applications, especially in solution.
Because the modified IR820–NO2 dye is characterized by a lower absorption cross-section than IR820 (Fig. S11†), quantum yield analyses on the 2H11/2, 4S3/2 → 4I15/2 transitions were carried out to evaluate the efficiencies of each system, as this method yields results which take into consideration the differences in the absorption cross-sections. Quantum yield measurements can also provide insight on the energy transfer mechanisms between the dyes and the Yb3+, and on the dynamics of the system. The quantum yield of IR820–NO2-functionalized LnUCNPs immediately after excitation was found to be over four times as efficient as IR820-functionalized LnUCNPs (Fig. S17†). This increase in efficiency can be credited to the improved energy transfer between IR820–NO2 and Yb3+ compared to the unmodified IR820 dye, since IR820–NO2 has improved spectral overlap with Yb3+ despite its slightly lower absorption coefficient (Fig. S11†).
The quantum yields were measured as a function of irradiation time and both systems were found to be nearly constant (Fig. S17†). The quantum yields for both systems remained constant as a function of irradiation time, despite the occurrence of photodegradation. This implies that a decrease in absorbance due to the decomposition of the dye corresponds to a proportional decrease in emission; in other words, the UCL is dependent only on the dye concentration. This excludes any direct population of Er3+ or energy transfer from Yb3+ ions when exciting at 800 nm, which would otherwise result in a non-constant trend in the quantum yields. Simply put, in the event of having direct population of Er3+ or through absorption from the Yb3+ ions, there would be a certain value of the quantum yield directly dependent on these phenomena and, more importantly, independent of the irradiation time, since Yb3+ and Er3+ absorption and Er3+ emission would not be affected by the exposure time to the exciting source. Consequently, with decreasing of the absorption and emission due to the dye-sensitization process, an asymptotic trend would be expected, converging to the quantum yield value obtained for the non-dye LnUCNPs, i.e. the value of the quantum yield due to direct population of Er3+. The constant trend of the quantum yield, contrarily, confirms the assumption that the Er3+ emissions achieved in the dye-sensitized LnUCNPs upon 800 nm excitation only arise from the dye energy transfer to the LnUCNPs.
Conclusions
For nearly a decade, NIR dyes have been used for upconversion sensitization. To our knowledge, studies on dye sensitized LnUCNPs do not address the poor photostability of NIR dyes, and no significant measures have been taken to improve it. Furthermore, there is a lack of information on the dynamics between the dye, sensitizer and activator ions in these dye–LnUCNP systems. This calls for urgent attention towards this aspect, as the available dye sensitized LnUCNPs systems are unsuitable for applications that require prolonged irradiation times, especially in solution, and there is little information on the degradation dynamics, and therefore a lack of knowledge on how to improve it.In this study, for the first time, we report on the photostability of IR820 and the role of singlet oxygen in the photodegradation of this dye. To address the photostability of NIR dyes for upconversion sensitization, we introduced structural modifications to IR820 which were aimed at preventing the reaction of singlet oxygen with the heptamethine conjugation of the dye through steric and electronic effects. Of the modifications studied, IR820 functionalized with 4-nitrothiophenol group at the para-position was found to exhibit enhanced photostability relative to IR820 and possessed the greatest spectral overlap with Yb3+.
IR820 and IR820–NO2 were coordinated to NaGdF4:Er3+,Yb3+/NaGdF4:Yb3+ upconverting nanoparticles by electrostatic interaction and the emission enhancement, dynamics and photostability were studied. Significant attention was paid to providing a meaningful comparison between the dye-functionalized LnUCNP systems, as there is a lack of consistency in how these values are reported in the literature. The emission spectra of the IR820–NO2-LnUCNPs exhibit 200% more intense emissions as compared to the IR820-sensitized LnUCNPs. The brighter emissions can be attributed to better spectral overlap between IR820–NO2 and Yb3+, which ensures a more efficient energy transfer, as well as a reduction in back-transfer from Er3+ to the dye, as proven by lifetime and quantum yield measurements. Additionally, kinetic studies of the Er3+ 2H11/2 → 4I15/2 and 4S3/2 → 4I15/2 transitions illustrate that the IR820–NO2-functionalized LnUCNPs exhibit more intense and observable emissions for more than twice the duration of IR820-sensitized LnUCNPs. The increased intensity, luminescence efficiency and duration of the emissions from IR820–NO2-functionalized LnUCNPs represents a significant improvement in dye-sensitized upconverting nanoparticle properties.
These studies pave the way for dye-sensitized upconversion applications that require brighter emissions and prolonged irradiation times, especially with regard to solution-based and broadband absorbance applications. Future dye-sensitized upconversion systems should benefit from the knowledge gained from this work.
Author contributions
Mannu Kaur was responsible for all synthesis of dyes and nanoparticles and their characterization. Gabrielle A. Mandl was responsible for aiding in experimental design. Steven L. Maurizio performed the quantum yield and lifetime measurements and analyses. Gabriella Tessitore guided the spectroscopy experiments. John A. Capobianco supervised the project. All authors discussed the final results and contributed to the final manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
M. K. is grateful to Concordia University for support through Concordia International Tuition Award of Excellence. G. A. M. is grateful to the National Science and Engineering Research Council of Canada for support through the Alexander Graham Bell CGS-D fellowship. S. L. M. is grateful to the National Science and Engineering Research Council for support through the Alexander Graham Bell CGS-D fellowship and to Concordia University for support through the Doctoral Incentive fellowship. G. T. is grateful to Concordia University for financial support from the Horizon Post-Doctoral Program. J. A. C. is a Concordia University Research Chair in Nanoscience and is grateful to Concordia University for sustained support of his research. J. A. C. is grateful to the National Science and Engineering Research Council of Canada for continued financial support.References
- B. Chen and F. Wang, Trends Chem., 2020, 2, 427–439 CrossRef CAS.
- F. Auzel, Chem. Rev., 2004, 104, 139–173 CrossRef CAS PubMed.
- X. Liu, C.-H. Yan and J. A. Capobianco, Chem. Soc. Rev., 2015, 44, 1299–1301 RSC.
- G. Chen, H. Qiu, P. N. Prasad and X. Chen, Chem. Rev., 2014, 114, 5161–5214 CrossRef CAS PubMed.
- S. Banerjee, J. Koerner, M. Siebold, Q. Yang, K. Ertel, P. D. Mason, P. J. Phillips, M. Loeser, H. Zhang, S. Lu, J. Hein, U. Schramm, M. C. Kaluza and J. L. Collier, Opt. Express, 2013, 21, A726–A734 CrossRef CAS.
- G. Chen, H. Ågren, T. Y. Ohulchanskyy and P. N. Prasad, Chem. Soc. Rev., 2015, 44, 1680–1713 RSC.
- F. Vetrone, R. Naccache, V. Mahalingam, C. G. Morgan and J. A. Capobianco, Adv. Funct. Mater., 2009, 19, 2924–2929 CrossRef CAS.
- E. M. Chan, E. S. Levy and B. E. Cohen, Adv. Mater., 2015, 27, 5753–5761 CrossRef CAS PubMed.
- S. I. Weissman, J. Chem. Phys., 1942, 10, 214–217 CrossRef CAS.
- J. M. Lehn, Pure Appl. Chem., 1994, 66, 1961–1966 CAS.
- W. Zou, C. Visser, J. A. Maduro, M. S. Pshenichnikov and J. C. Hummelen, Nat. Photonics, 2012, 6, 560–564 CrossRef CAS.
- G. Chen, J. Damasco, H. Qiu, W. Shao, T. Y. Ohulchanskyy, R. R. Valiev, X. Wu, G. Han, Y. Wang, C. Yang, H. Ågren and P. N. Prasad, Nano Lett., 2015, 15, 7400–7407 CrossRef CAS.
- J. Xu, M. Sun, Y. Kuang, H. Bi, B. Liu, D. Yang, R. Lv, S. Gai, F. He and P. Yang, Dalton Trans., 2017, 46, 1495–1501 RSC.
- X. Wu, H. Lee, O. Bilsel, Y. Zhang, Z. Li, T. Chen, Y. Liu, C. Duan, J. Shen, A. Punjabi and G. Han, Nanoscale, 2015, 7, 18424–18428 RSC.
- D. Yin, Y. Liu, J. Tang, F. Zhao, Z. Chen, T. Zhang, X. Zhang, N. Chang, C. Wu, D. Chen and M. Wu, Dalton Trans., 2016, 45, 13392–13398 RSC.
- D. J. Garfield, N. J. Borys, S. M. Hamed, N. A. Torquato, C. A. Tajon, B. Tian, B. Shevitski, E. S. Barnard, Y. D. Suh, S. Aloni, J. B. Neaton, E. M. Chan, B. E. Cohen and P. J. Schuck, Nat. Photonics, 2018, 12, 402–407 CrossRef CAS.
- X. Wang, R. R. Valiev, T. Y. Ohulchanskyy, H. Ågren, C. Yang and G. Chen, Chem. Soc. Rev., 2017, 46, 4150–4167 RSC.
- M. D. Wisser, S. Fischer, C. Siefe, A. P. Alivisatos, A. Salleo and J. A. Dionne, Nano Lett., 2018, 18, 2689–2695 CrossRef CAS.
- A. Samanta, M. Vendrell, R. Das and Y. T. Chang, Chem. Commun., 2010, 46, 7406–7408 RSC.
- E. Engel, R. Schraml, T. Maisch, K. Kobuch, B. Konig, R. M. Szeimies, J. Hillenkamp, W. Baümler and R. Vasold, Invest. Ophthalmol. Visual Sci., 2008, 49, 1777–1783 CrossRef.
- G. Tessitore, G. A. Mandl, M. G. Brik, W. Park and J. A. Capobianco, Nanoscale, 2019, 11, 12015–12029 RSC.
- S. Wilhelm, ACS Nano, 2017, 11, 10644–10653 CrossRef CAS.
- C. Shi, J. B. Wu and D. Pan, J. Biomed. Opt., 2016, 21, 050901–050913 CrossRef PubMed.
- H. T. T. Duong, Y. Chen, S. A. Tawfik, S. Wen, M. Parviz, O. Shimoni and D. J. Ab, RSC Adv., 2018, 8, 4842–4849 RSC.
- D. S. McClure, J. Chem. Phys., 1949, 17, 905–913 CrossRef CAS.
- A. P. Gorka and M. J. Schnermann, Curr. Opin. Chem. Biol., 2016, 33, 117–125 CrossRef CAS PubMed.
- I. Zebger, L. Poulsen, Z. Gao, L. K. Andersen and P. R. Ogilby, Langmuir, 2003, 19, 8927–8933 CrossRef CAS.
- X. Wu, Y. Zhang, K. Takle, O. Bilsel, Z. Li, H. Lee, Z. Zhang, D. Li, W. Fan, C. Duan, E. M. Chan, C. Lois, Y. Xiang and G. Han, ACS Nano, 2016, 10, 1060–1066 CrossRef CAS PubMed.
- X. Zou, M. Xu, W. Yuan, Q. Wang, Y. Shi, W. Feng and F. Li, Chem. Commun., 2016, 52, 13389–13392 RSC.
- D. H. Li, C. L. Schreiber and B. D. Smith, Angew. Chem., Int. Ed. Engl., 2020, 59, 12154–12161 CrossRef CAS.
- L. Strekowski, C. J. Mason, H. Lee, R. Gupta, J. Sowell and G. Patonay, J. Heterocycl. Chem., 2003, 40, 913–916 CrossRef CAS.
- N. Bogdan, F. Vetrone, G. A. Ozin and J. A. Capobianco, Nano Lett., 2011, 11, 835–840 CrossRef CAS.
- A. Aebischer, S. Heer, D. Biner, K. Krämer, M. Haase and H. U. Güdel, Chem. Phys. Lett., 2005, 407, 124–128 CrossRef CAS.
- F. Vetrone, J. C. Boyer, J. A. Capobianco, A. Speghini and M. Bettinelli, Chem. Mater., 2003, 15, 2737–2743 CrossRef CAS.
- B. Xue, D. Wang, L. Tu, D. Sun, P. Jing, Y. Chang, Y. Zhang, X. Liu, J. Zuo, J. Song, J. Qu, E. J. Meijer, H. Zhang and X. Kong, J. Phys. Chem. Lett., 2018, 9, 4625–4631 CrossRef CAS PubMed.
- R. H. Page, K. I. Schaffers, P. A. Waide, J. B. Tassano, S. A. Payne, W. F. Krupke and W. K. Bischel, J. Opt. Soc. Am. B, 1998, 15, 996–1008 CrossRef CAS.
- F. T. Rabouw, P. T. Prins, P. Villanueva-Delgado, M. Castelijns, R. G. Geitenbeek and A. Meijerink, ACS Nano, 2018, 12, 4812–4823 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00710f |
| This journal is © The Royal Society of Chemistry 2022 |