
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of SERS-active core–satellite nanoparticles using heterobifunctional PEG linkers†
Angela Michelle T.
San Juan
 ab,
Suhash Reddy
Chavva
ab,
Dandan
Tu
ab,
Melanie
Tircuit
a,
Gerard
Coté
ab and
Samuel
Mabbott
*ab
ab,
Suhash Reddy
Chavva
ab,
Dandan
Tu
ab,
Melanie
Tircuit
a,
Gerard
Coté
ab and
Samuel
Mabbott
*ab
aTexas A&M University, Health Technologies and Innovations Building, 3006 TAMU, College Station, Texas 77843, USA. E-mail: smabbott@tamu.edu
bDepartment of Biomedical Engineering, Emerging Technologies Building, 3120 TAMU, College Station, Texas 77843, USA
First published on 15th November 2021
Abstract
Surface-enhanced Raman scattering (SERS) is a sensitive analytical technique capable of magnifying the vibrational intensity of molecules adsorbed onto the surface of metallic nanostructures. Various solution-based SERS-active metallic nanostructures have been designed to generate substantial SERS signal enhancements. However, most of these SERS substrates rely on the chemical aggregation of metallic nanostructures to create strong signals. While this can induce high SERS intensities through plasmonic coupling, most chemically aggregated assemblies suffer from poor signal reproducibility and reduced long-term stability. To overcome these issues, here we report for the first time the synthesis of gold core–satellite nanoparticles (CSNPs) for robust SERS signal generation. The novel CSNP assemblies consist of a 30 nm spherical gold core linked to 18 nm satellite particles via linear heterobifunctional thiol–amine terminated PEG chains. We explore the effects that the varying chain lengths have on SERS hot-spot generation, signal reproducibility and long-term activity. The chain length was varied by using PEGs with different molecular weights (1000 Da, 2000 Da, and 3500 Da). The CSNPs were characterized via UV-Vis spectrophotometry, transmission electron microscopy (TEM), ζ-potential measurements, and lastly SERS measurements. The versatility of the synthesized SERS-active CSNPs was revealed through characterization of optical stability and SERS enhancement at 0, 1, 3, 5, 7 and 14 days.
Introduction
Surface-Enhanced Raman Spectroscopy (SERS) is a versatile analytical technique commonly recognized for its superior sensitivity and ability to generate molecular fingerprints of chemicals that can allow them to be uniquely identified.1–3 SERS causes significant signal amplification of target molecules once adsorbed onto a noble metal surface resulting in intensified Raman scattering. The intensity of SERS spectra is several orders of magnitude larger than that of conventional Raman scattering.1–3 Over the course of nearly 4 decades since the discovery of SERS, the technique has been applied to a multitude of applications including diagnostic assays,4,5 identification of environmental contaminants,6,7 and food monitoring applications.7,8SERS enhancement relies on the unique optical and plasmonic properties associated with metallic nanostructures. Conventionally, plasmonic nanostructures are often designed to offer intense localized surface plasmon resonance (LSPR) caused by the collective oscillation of electrons when excited by light of a resonant frequency. Areas of high electric field found in the interstitial gaps between metal surfaces are often referred to as SERS hot-spots.1,2,9 The SERS signal generated from molecules that get trapped in the hot spots is significantly amplified. Hot spots can also be generated at the tips or edges of a noble metal surface.1,2,9 Ideal SERS-active nanostructures for sensing applications exploit this phenomenon by drastically enhancing Raman scattering in a reproducible and uniform manner.3 However, many of the tuneable SERS-active nanostructures involve complex synthetic processes and suffer from poor structural reproducibility often resulting in widely distributed enhancement factors (EFs) limiting clinical application by hindering the ability to quantify biomarkers in a reliable manner.2,7,9,10
As mentioned previously, reliability and robustness have been among the key challenges for SERS measurements especially under aqueous conditions. A number of approaches have been employed by researchers to create high performance SERS-active NPs,11–14 including the variation of physical properties associated with the particles such as size,15–17 shape/morphology,18–20 material composition,21–23 and gap distance/interstitial junction.24–26 Specific examples include work by Yang et al. that introduces the synthesis of bimetallic Ag@Au nanocubes by deposition of Au atoms on the surface of the Ag nanocubes.27 A similar study by Gopalakrishnan et al. demonstrates the synthesis of bimetallic 3-D nanostar dimers in a ring cavity to further enhance the overall SERS performance.28 In both studies, SERS-active bimetallic NPs with unique morphologies have been demonstrated to exhibit greater SERS enhancement of associated analytes when compared to bare unmodified NPs.29 However, although these SERS-active NPs offer large EFs, the poor reproducibility and instability resulting from the desorption of Raman reporters through competitive binding of other molecules cannot be ignored. Additionally, many SERS-active NPs require further subsequent surface functionalization to enhance stability. To be applied in a real-world application, integration of these SERS-active NPs in diagnostic assays requires further addition of targeting moieties to adhere to the surface such as aptamers,30,31 antibodies,32–34 oligonucleotide probes35,36 and Raman reporter molecules (RRMs).37 This mixed monolayer often results in competitive binding on the exposed NP surface, causing inequivalent surface deposition. Loss of RRMs due to surface desorption limits the SERS sensitivity once applied to a diagnostic assay.
To overcome these challenges, we have devised a synthesis method for the development of novel core–satellite nanostructures whereby a gap is created between the core and the satellite using a heterobifunctional PEG linker. The effect of changing the length of the PEG linker on the overall SERS performance was explored through assessing the two most important key parameters of signal intensity and reproducibility. To evaluate the SERS performance, a commonly employed RRM, malachite green isothiocyanate (MGITC), was utilized. In particular, a core–satellite NP assembly was chosen due to hot-spot generation through core–satellite plasmonic coupling and satellite–satellite plasmonic coupling. This synergistic combination leads to a strong SERS enhancement due to the high electromagnetic coupling between the two moieties.38 Based on a theoretical prediction by Singamaneni et al., it was illustrated that the electric field intensity generated at the interstitial space between the core and satellite NPs is about 660× higher than that at the surfaces of the cores. Therefore, in comparison to conventionally aggregated NP systems, core–satellite assemblies have better repeatability due to the high affinity for core–satellite interaction.39 To synthesize these nanoparticle systems, bottom-up approaches are predominantly utilized using either organic40 or inorganic templates41 as core nanostructures, while smaller inorganic nanoparticles such as gold, silver, or semiconductor NPs are explored as satellites. The nanospacer linkers that are commonly employed include (1) protein-based assemblies,42 (2) polymer-based assemblies,43–45 (3) DNA-based assemblies,37,46–50 and (4) molecular linkers that directly bridge the plasmonic units using either covalent or electrostatic interactions of the functional groups.38,51–53 For example, Chen et al. demonstrated the synthesis of a streptavidin-coated polymeric bead core–Au satellite assembly via DNA linkers.54 Two different types of DNA linker were immobilized on the surface of the nanoparticles to enhance stability, and a pre-treatment step for both linkers and gold nanoparticles (AuNPs) was required to initiate the functionalization. However, the use of DNA is often laborious and expensive. Typically, DNA mediated core–satellite assemblies require at least ∼17 hours to complete the reaction with many subsequent centrifugation steps.37,46–50 Moderate enhancing capabilities are observed within the DNA mediated assembly due to the intrinsic DNA length required to ensure stable hybridization precluding tunability of the interparticle gap distances between the core and satellite nanostructures.54,55 Similar limitations are seen utilizing bulky macromolecules such as protein-mediated assembly or branched polymer-mediated assembly. On the other hand, efficient distribution of molecular linkers on the nanoparticle surface is hampered due to the irreversible colloidal instability they introduce to the NP system.56 To overcome this challenge, there is a need to develop a more cost-effective and scalable synthesis of these CSNPs with tunable SERS hotspot generation.
In this study, we report a facile synthesis method of gold core–satellite nanoparticles that exhibit high SERS intensity with great reproducibility. We explore these SERS hotspot generations by immobilizing linear heterobifunctional polyethylene glycol (PEG) that is composed of an amine and a thiol group. This linear PEG chain serves as a tunable nanospacer for the core–satellite structure. By altering the distance between the core and satellite nanostructures, we can induce controllable SERS hotspot generation. The SERS hotspot generation can be tuned by altering the distance between the nanostructures by using various PEG polymer thicknesses. The core–satellite nanoparticles were generated through immobilization of the satellite nanoparticles through electrostatic interactions of the positively charged amine groups of the PEG chain and the negatively charged citrate stabilized seed nanoparticles. It is shown that these core–satellite gold nanoparticles are synthesized in an easy and low-cost manner and generate strong and reproducible SERS signals, thus addressing the need for a SERS-active nanostructure with high sensitivity and stability.
Materials and methods
Chemicals
Gold(III) chloride trihydrate (HAuCl4·3H2O, >99.9%), trisodium citrate dihydrate (≥99.0%), and malachite green isothiocyanate (MGITC) were all purchased from Sigma-Aldrich. Linear heterobifunctional polyethylene glycol (SH-PEG-NH2) linkers with a molecular weight (MW) of 1000 Da were obtained from Nanosoft Polymers (North Carolina, USA), and linkers weighing 2000 Da and 3500 Da were obtained from JenKem Technology (Texas, USA). Ultrapure water (Milli-Q) was used for the preparation of all solutions.Methods
Results and discussion
Synthesis of the gold core–satellite NPs
Fig. 1 depicts the synthesis and assembly process of the CSNPs. The core and satellite AuNPs were synthesized via the Turkevich method57 (verified by TEM in Fig. 3A). In this assembly, highly stabilized CSNPs were synthesized by anchoring linear heterobifunctional polyethylene glycol (SH-PEGx-NH2, where x denotes the various MWs of PEG (1000, 2000, and 3500)) on the surface of the core AuNPs. The SH-PEGx-NH2 (referred to as PEGx in this paper) serves as a tunable nanospacer altering the distance between the core and surface immobilized satellite nanoparticles. Additionally, Raman reporter molecules (MGITC) were simultaneously anchored on the surface of the core particles. Here, competition for surface attachment on the AuNP core occurs between PEGx and MGITC to form a mixed monolayer. Due to the comparatively weak isothiocyanate binding of the MGITC, PEGx adsorbs more readily allowing more active groups for binding of the satellite NPs. The use of the PEGx linker allows MGITC to become trapped at the interface of the core and satellite particles, which is an area of high electric field density amenable to large increases in SERS enhancement. Exploration of different PEG lengths offers a facile, cost-effective method to tune the distance between the core and satellite nanostructures, thereby modulating the SERS signal through hotspot generation.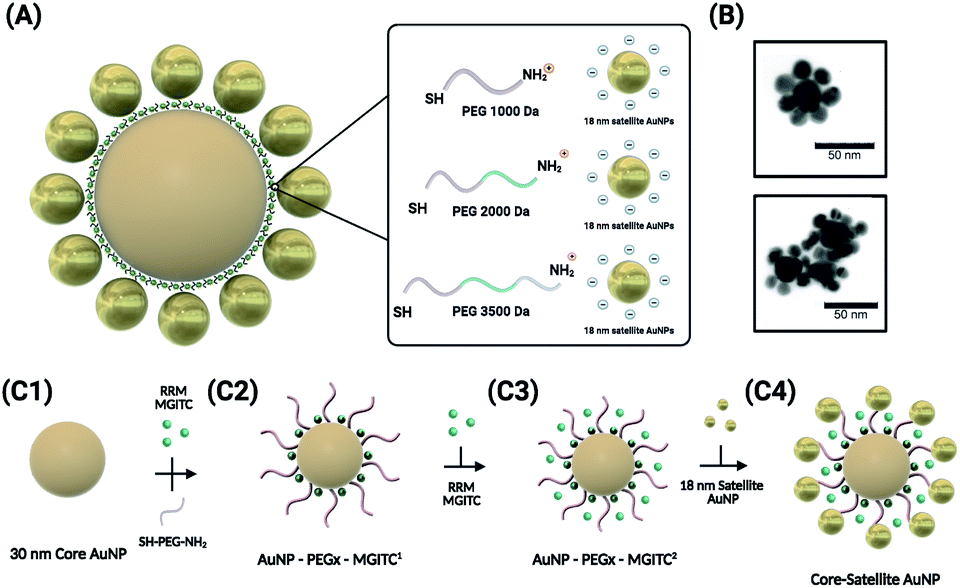 | ||
| Fig. 1 Schematic illustration of the synthesis of the core–satellite AuNPs. (A) The overall morphology of the CSNPs with MGITC entrapped between the core and satellite nanostructures. A tunable hot-spot formation can be achieved by modulating PEG lengths. (B) TEM images of CSNPs–PEG1000–MGITC2 forming a discrete core–satellite nanostructure. (C) Illustration of the nanoparticle assembly forming the distinct core–satellite NPs through functionalization of heterobifunctional PEG and MGITC. | ||
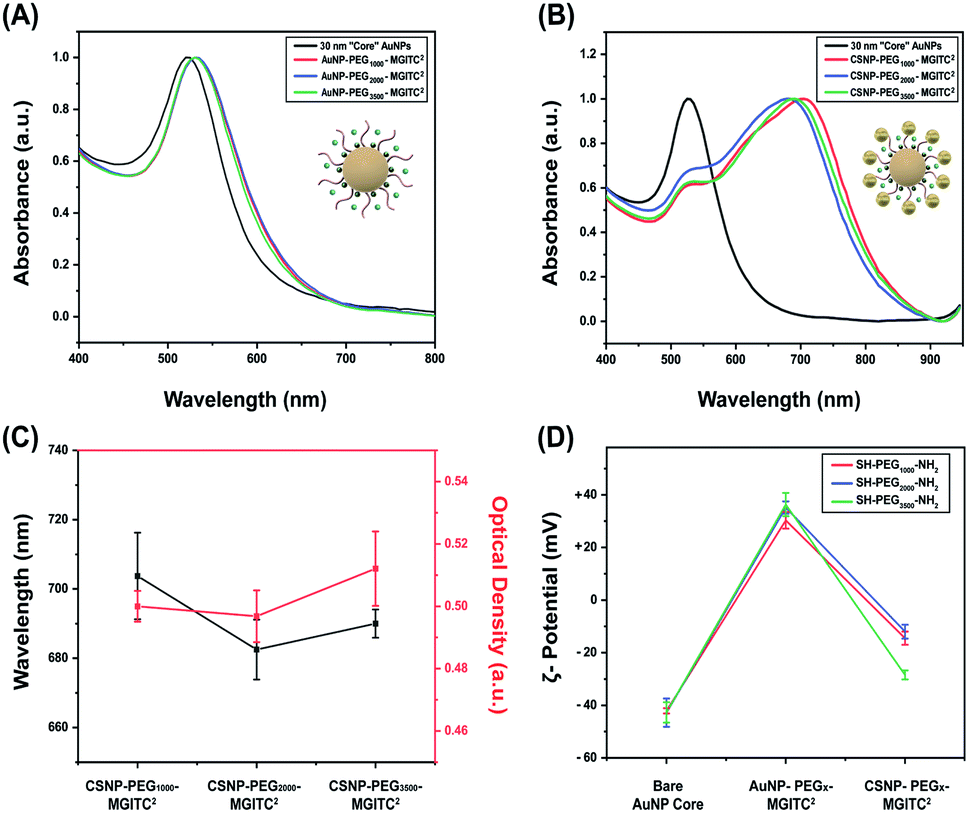 | ||
| Fig. 2 (A) UV-Vis spectra to verify the optical stability of the 30 nm AuNP core nanoparticles and PEGx–MGITC2 modified core nanoparticles at various polymer thicknesses (MW: 1000 Da, 2000 Da, and 3500 Da), (B) UV-Vis spectra illustrating the spectral shift after formation of CSNPs modified by PEG with different polymer thicknesses, (C) CSNP wavelength shift for each PEG polymer thickness and optical density after synthesis, and (D) ζ-potential measurements illustrating successful surface modification at each functionalization step through surface charge differences for each synthesized CSNP. | ||
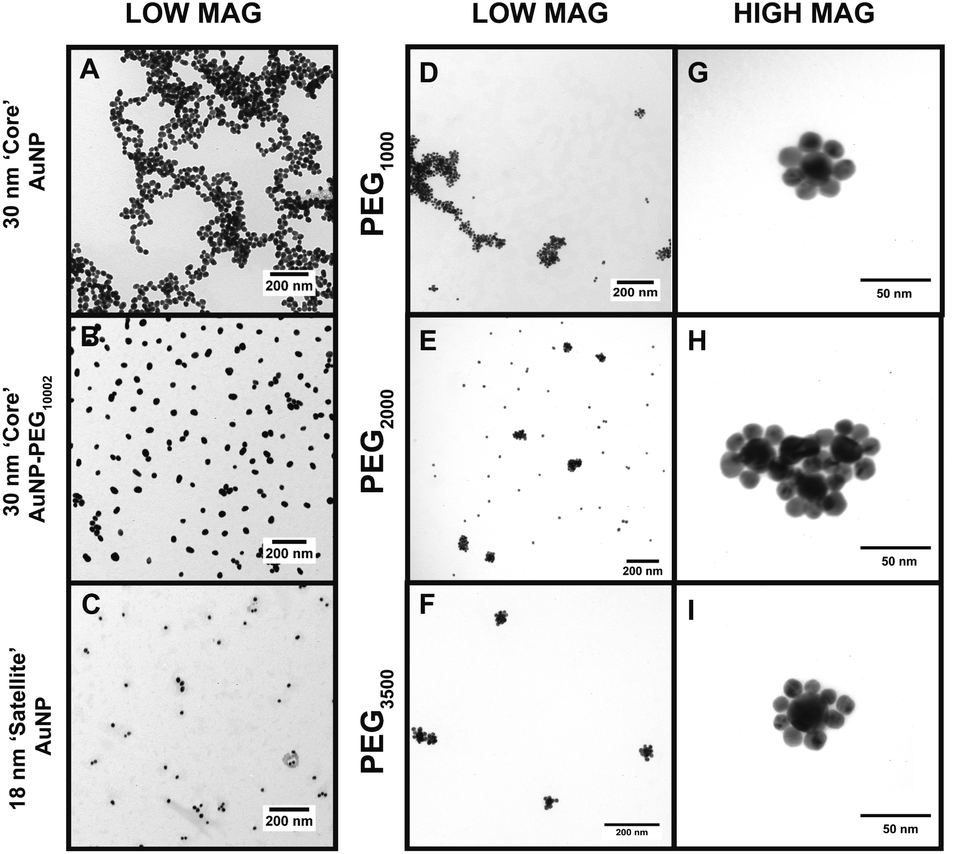 | ||
| Fig. 3 TEM images of the CSNP assemblies with (A) 30 nm core AuNPs at low magnification, (B) 30 nm core AuNP–PEG1000–MGITC2, (C) 18 nm satellite AuNPs, (D–F) CSNP–PEGx–MGITC2 with MWs of 1000 Da, 2000 Da, and 3500 Da, respectively, all at low magnification, and (G–I) CSNP–PEGx–MGITC2 with MWs of 1000 Da, 2000 Da, and 3500 Da, respectively, at high magnification. | ||
The simultaneous attachment of PEGx and MGITC results in modified AuNP–PEGx–MGITC1 as shown in Fig. 1(C2), where MGITC1 denotes one loading cycle of MGITC. An additional loading of MGITC after PEGx modification was made to ensure optimal RRM entrapment between the core and satellite nanostructures, which results in AuNP–PEGx–MGITC2 as shown in Fig. 1(C3). Following this reaction, 18 nm AuNP seeds were then deposited onto the AuNP–PEGx–MGITC2 as shown in Fig. 1(C4). CSNP formation and morphology were characterized using UV-Vis spectrophotometry (UV-Vis), ζ-potential measurements, and transmission electron microscopy (TEM).
Characterization of the CSNP assembly
UV-Vis measurements obtained for the 30 nm core AuNPs exhibited a LSPR peak at 525 nm shown in Fig. 2A. Successful functionalization of the core AuNPs with the PEGx–MGITC2 was confirmed upon observation of a 5 nm redshift to 530 nm caused by a change in the refractive index. Experimental observations revealed that the addition of too much MGITC onto the AuNP surface in one go would lead to immediate particle instability due to a reduction in charge repulsion between particles causing AuNP aggregation. Therefore, PEGx and MGITC were added simultaneously to enhance the stability of the nanoparticles as well as to serve as an anchor site for satellite AuNP attachment. Due to the simultaneous attachment of MGITC and PEGx, competitive binding between the two moieties arises. ESI Fig. S1(A–C)† report the hydrodynamic size of 36.67 nm and SERS enhancement of CSNPs–PEG1000–MGITC1 and CSNPs–PEG1000–MGITC2. Fig. S1(B and C)† illustrate minimal enhancement when MGITC was only deposited once due to the competitive binding between the two moieties.To control the simultaneous loading of MGITC and PEGx, various amounts of PEGx deposited on the surface of the nanoparticles were explored. The deposition of MGITC and different PEG lengths were modulated by fixing one parameter. In this case, MGITC was fixed at a concentration of 10−5 M and different concentration ranges of PEG1000 were explored ranging from 1.53 mM (0.5 mg PEG) to 153 μM (0.05 mg PEG) and 15.3 μM (0.005 mg PEG) attached to a fixed concentration of AuNPs (OD of 0.6 A.U.) shown in Fig. S2.† Fig S2A† illustrates the UV-Vis measurement for the 30 nm AuNP core exhibiting a LSPR peak at 525 nm, and a shift to 530 nm confirmed the successful attachment of PEG1000 at all concentration ranges. Through ζ-potential measurements, a minimum threshold amount of PEGx was identified for each MW of PEGx. Fig. S2B† reports the minimum saturation point for PEG1000 attachment at a fixed 10−5 M concentration of MGITC, which is 15.3 μM PEG1000. SERS measurements were also conducted to evaluate whether enhancement of MGITC1 and various concentration ranges of PEG1000 are observed shown in Fig. S2(C and D).† The simultaneous addition of MGITC and PEGx leads to competitive binding of the two moieties on the surface of the nanoparticle leading to minimal SERS enhancement due to poor attachment of MGITC. As mentioned previously, an additional MGITC loading was added to the surface after PEGx functionalization to ensure effective deposition of MGITC in the nanoparticle assembly.
Attachment of the satellite nanoparticles to the CSNPs was achieved by adding a fixed concentration of 18 nm satellite nanoparticles to the solution containing PEGx modified core AuNPs. In this experiment, a volumetric ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 Au core–Au satellite seed was used for CSNPs–PEG1000–MGITC2. Meanwhile, a volume ratio of 1:3 Au core–Au satellite seed ratio was used for both CSNPs–PEG2000–MGITC2 and CSNPs–PEG3500–MGITC2. It was noticed that if we increased the ratiometric amount of Au satellites above 3 the particles became destabilized causing sedimentation. This may be attributed to the reduction of coulombic repulsion between assemblies due to excess satellite NP inclusion in the synthesis step. The initial wine-red color of the core turns light blue due to plasmonic coupling between the core and satellite particles which indicates successful gold satellite anchoring. UV-Vis was used to monitor the color change (Fig. 2B and C), and a red shift from the λmax at 530 nm to higher wavelengths of 704 nm ± 12.5 nm, 683 ± 8.7 nm, and 690 ± 4.08 nm was observed for CSNPs modified with PEGx–MGITC2 with MWs of 1000 Da, 2000 Da, and 3500 Da, respectively. The change in extinction profile morphology is observed after the addition of the 18 nm satellite AuNPs and is believed to be caused by the newly created plasmon coupling between the tightly packed core and satellite nanostructures. ζ-Potential measurements further confirmed the successful assembly of CSNPs by assessing the change of surface charge on the nanoparticle surface illustrated in Fig. 2D. The bare AuNP displayed an average ζ-potential of −42.1 ± 0.38 mV. During the assembly of the core–satellite nanoparticles, the ζ-potential increased to +30.29 ± 1.04 mV, +35.16 ± 0.78 mV, and +36.3 ± 1.48 mV after the addition of the PEGx–MGITC with MWs of 1000 Da, 2000 Da, and 3500 Da, respectively. Due to the heterobifunctional nature of the PEGx chain, the thiol groups (SH) favourably bind to the AuNP surface through Au–S interactions while the amine (NH2) groups are left exposed on the surface resulting in an overall positive charge on the nanoparticle surface. Previous studies have reported that the gold–sulphur (Au–S) covalent interaction is exceptionally strong. Meanwhile, amine groups have been shown to bind through electrostatic interaction with the negatively charged AuNP surface.58,59 Specifically, it has been noted that thiol groups have shown the strongest binding affinity towards the surface of noble gold nanoparticles due to the nature of the covalent interaction or “chemisorption binding”.59,60 In comparison, amine groups are noted to have weak covalent bonding with the negatively charged surface of the gold nanoparticles.58,59,61 Specifically, a study reported by Ftouni et al. evaluated the effects of citrate capped AuNP binding on two linkers, (3-aminopropyl)triethoxysilane (APTES) and (3-mercaptopropyl)triethoxysilane (MPTES).61 MPTES was utilized for its thiol functionality, and APTES linker was chosen due to the weak covalent affinity of the amino group to the gold nanoparticles reported previously as well by Genzer et al.61,62 In this work, they evaluated the catalytic activity and utilized scanning electron microscopy and electron probe microanalysis to confirm the presence of the gold nanoparticles.61 The author reported that using MPTES linkers with thiol modification exhibited better stability due to stronger bonding to the gold nanoparticles.61 Additionally, a study by Tao et al. evaluated the effects of anchoring groups such as amine, thiol, and carboxylic acid functional groups on the single-molecule conductance with gold.59 Binding strength information was obtained by measuring the average length over which one can stretch each molecular junction until it breaks, which varies in the order of Au–S > Au–NH2 > Au–COOH, which is consistent with the binding strengths of the three anchoring groups to gold.59 Additionally, a study by Cortie et al. confirmed the weak adsorption of amine compounds with only significant binding in the under-coordinated atom site of the gold structure.60 The authors confirmed these findings using a density functional theory study of the adsorption energetics of various amine compounds on the gold surface.60 In our work, it is believed that preferential binding of the thiol groups occurs on the NP surface due to the resulting overall positive ζ-potential measurement after functionalization with each respective PEG length chain. The surface charge switch from a negatively charged nanoparticle surface to a positively charged surface further confirmed the successful functionalization of the PEGx–MGITC. The amine groups serve as active binding sites for satellite particle attachment via AuNP–amine electrostatic interaction.63 Through this electrostatic interaction between the amine active groups and negatively charged citrate stabilized surface of the satellite particles, the well-defined CSNPs display a mean ζ-potential of −14.50 ± 0.83 mV, −12.0 ± 0.878 mV, and −28.5 ± 0.56 mV, respectively, for PEGx–MGITC with MWs of 1000 Da, 2000 Da, and 3500 Da.
:4 Au core–Au satellite seed was used for CSNPs–PEG1000–MGITC2. Meanwhile, a volume ratio of 1:3 Au core–Au satellite seed ratio was used for both CSNPs–PEG2000–MGITC2 and CSNPs–PEG3500–MGITC2. It was noticed that if we increased the ratiometric amount of Au satellites above 3 the particles became destabilized causing sedimentation. This may be attributed to the reduction of coulombic repulsion between assemblies due to excess satellite NP inclusion in the synthesis step. The initial wine-red color of the core turns light blue due to plasmonic coupling between the core and satellite particles which indicates successful gold satellite anchoring. UV-Vis was used to monitor the color change (Fig. 2B and C), and a red shift from the λmax at 530 nm to higher wavelengths of 704 nm ± 12.5 nm, 683 ± 8.7 nm, and 690 ± 4.08 nm was observed for CSNPs modified with PEGx–MGITC2 with MWs of 1000 Da, 2000 Da, and 3500 Da, respectively. The change in extinction profile morphology is observed after the addition of the 18 nm satellite AuNPs and is believed to be caused by the newly created plasmon coupling between the tightly packed core and satellite nanostructures. ζ-Potential measurements further confirmed the successful assembly of CSNPs by assessing the change of surface charge on the nanoparticle surface illustrated in Fig. 2D. The bare AuNP displayed an average ζ-potential of −42.1 ± 0.38 mV. During the assembly of the core–satellite nanoparticles, the ζ-potential increased to +30.29 ± 1.04 mV, +35.16 ± 0.78 mV, and +36.3 ± 1.48 mV after the addition of the PEGx–MGITC with MWs of 1000 Da, 2000 Da, and 3500 Da, respectively. Due to the heterobifunctional nature of the PEGx chain, the thiol groups (SH) favourably bind to the AuNP surface through Au–S interactions while the amine (NH2) groups are left exposed on the surface resulting in an overall positive charge on the nanoparticle surface. Previous studies have reported that the gold–sulphur (Au–S) covalent interaction is exceptionally strong. Meanwhile, amine groups have been shown to bind through electrostatic interaction with the negatively charged AuNP surface.58,59 Specifically, it has been noted that thiol groups have shown the strongest binding affinity towards the surface of noble gold nanoparticles due to the nature of the covalent interaction or “chemisorption binding”.59,60 In comparison, amine groups are noted to have weak covalent bonding with the negatively charged surface of the gold nanoparticles.58,59,61 Specifically, a study reported by Ftouni et al. evaluated the effects of citrate capped AuNP binding on two linkers, (3-aminopropyl)triethoxysilane (APTES) and (3-mercaptopropyl)triethoxysilane (MPTES).61 MPTES was utilized for its thiol functionality, and APTES linker was chosen due to the weak covalent affinity of the amino group to the gold nanoparticles reported previously as well by Genzer et al.61,62 In this work, they evaluated the catalytic activity and utilized scanning electron microscopy and electron probe microanalysis to confirm the presence of the gold nanoparticles.61 The author reported that using MPTES linkers with thiol modification exhibited better stability due to stronger bonding to the gold nanoparticles.61 Additionally, a study by Tao et al. evaluated the effects of anchoring groups such as amine, thiol, and carboxylic acid functional groups on the single-molecule conductance with gold.59 Binding strength information was obtained by measuring the average length over which one can stretch each molecular junction until it breaks, which varies in the order of Au–S > Au–NH2 > Au–COOH, which is consistent with the binding strengths of the three anchoring groups to gold.59 Additionally, a study by Cortie et al. confirmed the weak adsorption of amine compounds with only significant binding in the under-coordinated atom site of the gold structure.60 The authors confirmed these findings using a density functional theory study of the adsorption energetics of various amine compounds on the gold surface.60 In our work, it is believed that preferential binding of the thiol groups occurs on the NP surface due to the resulting overall positive ζ-potential measurement after functionalization with each respective PEG length chain. The surface charge switch from a negatively charged nanoparticle surface to a positively charged surface further confirmed the successful functionalization of the PEGx–MGITC. The amine groups serve as active binding sites for satellite particle attachment via AuNP–amine electrostatic interaction.63 Through this electrostatic interaction between the amine active groups and negatively charged citrate stabilized surface of the satellite particles, the well-defined CSNPs display a mean ζ-potential of −14.50 ± 0.83 mV, −12.0 ± 0.878 mV, and −28.5 ± 0.56 mV, respectively, for PEGx–MGITC with MWs of 1000 Da, 2000 Da, and 3500 Da.
Examining the morphological structure of the CSNP assembly
The TEM images in Fig. 3 offer a qualitative image quantification of each characterization step of the CSNP assembly at different MWs. Herein, TEM-based quantification of core sizes was measured to evaluate morphological changes after each functionalization step. Fig. 3A shows a TEM image of discrete unmodified core AuNPs with an average diameter of 30.07 ± 6.68 nm. Fig. 3B illustrates a TEM image captured for core AuNPs modified with PEG1000–MGITC2, and Fig. 3C shows the satellite AuNPs with an average diameter of 18.39 ± 3.719 nm. Fig. 3D–F report low magnification images of the synthesized CSNPs with PEGx of various MWs (1000 Da, 2000 Da, and 3500 Da), respectively. It is common that the nanoparticles tend to cluster together as part of the drying process, as the solvent evaporates.64,65 Due to the decrease in volume as the solvent evaporates, electrostatic imbalances occur between the nanoparticles causing them to cluster together.64,65Fig. 3G–I show high magnification images for each of the CSNPs synthesized. Fig. 3G–I illustrate the clear attachment of the 18 nm AuNP satellite seed on the 30 nm core AuNP surface forming a satellite-like morphology for all MWs of PEGx–MGITC2. Through ImageJ analysis, the number of satellites detected around each nanoparticle core was 6 ± 1, 8 ± 2, and 8 ± 2, respectively, for PEGx (MW: 1000 Da, 2000 Da, and 3500 Da). ESI Fig. S3† further shows all high magnification images captured for CSNPs synthesized with different PEG length chains. These TEM images illustrate the assembly process which demonstrates the attachment of the 18 nm Au satellite seeds forming discrete core–satellite nanostructures. Herein, controlling the amount of PEGx and MGITC is essential as an ideal amount of NH2 active sites are required for proper ∼18 nm satellite attachment while also ensuring that MGITC is entrapped on the nanoparticle surface.Investigating SERS enhancement as a factor of PEG length
The overall goal of this study was to evaluate the effects of the PEGx-polymer length between the core and satellite nanostructures on overall SERS performance. Previous studies have shown that core size had only a minor effect on the overall SERS enhancement and that the majority of SERS originated from satellite–satellite interactions.66 Therefore, to observe the enhancements based on PEG lengths, a fixed concentration and volume of satellite nanoparticles were deposited onto the heterobifunctional linkers as various MWs of PEG have different surface coverage densities and exposed NH2 active sites. Additionally, equimolar concentrations of CSNPs–PEGx–MGITC2 and AuNP–PEGx–MGITC2 were analysed at an excitation of 785 nm for SERS analysis. As seen in Fig. 4, the AuNP–PEGx–MGITC2 exhibits limited SERS enhancement despite the loading of the MGITC. As seen in ESI Fig. 2C,† the addition of the linear bifunctional PEG interfered with the SERS signal enhancement of the RRM. However, after the addition of the satellite nanoparticles, an intense signal amplification of the RRM was observed as shown in Fig. 4A–D. MGITC Raman spectral bands were observed and tentatively assigned at ∼1170 cm−1 (corresponding to the in-plane aromatic C–H bending vibration), ∼1368 cm−1 (N–C stretching), ∼1398 cm−1 (C–C and C–H in-plane motion), and ∼1620 cm−1 (N–C bond and C–C stretching). For further studies, we evaluated the characteristic intensities of MGITC peaks at ∼1175 cm−1 and ∼1620 cm−1 post baseline correction. CSNPs modified with PEG1000–MGITC2 that were excited at 785 nm realized the highest signals: 17826 and 12686 for 1175 cm−1 and 1620 cm−1, respectively. There is a clear decrease in SERS intensity for CSNPs modified with PEG2000–MGITC2 and CSNPs modified with PEG3500–MGITC2 with reported SERS intensities of around 8772 and 6128 at ∼1175 cm−1 as well as 6765 and 3552 at ∼1620 cm−1, respectively. Herein, it is reported that SERS enhancement increased in the order of CSNPs modified with PEG1000 > PEG2000 > PEG3500. This is most likely due to the formation of smaller nanogaps when using linkers containing PEG1000 compared to the larger gaps created when using PEG3500. The smaller gap of the core–satellite interface is ideal for plasmon coupling/hot-spot formation to occur, resulting in a larger SERS signal being generated from trapped MGITC RRMs. In addition, it is important to keep in mind that the satellite nanoparticle size is crucial in obtaining appropriate SERS enhancement. As shown in ESI Fig. S4,† we report the use of 2 nm satellite AuNPs in the same nanoparticle assembly where minimal SERS enhancement was observed when 2 nm satellite AuNPs are used.
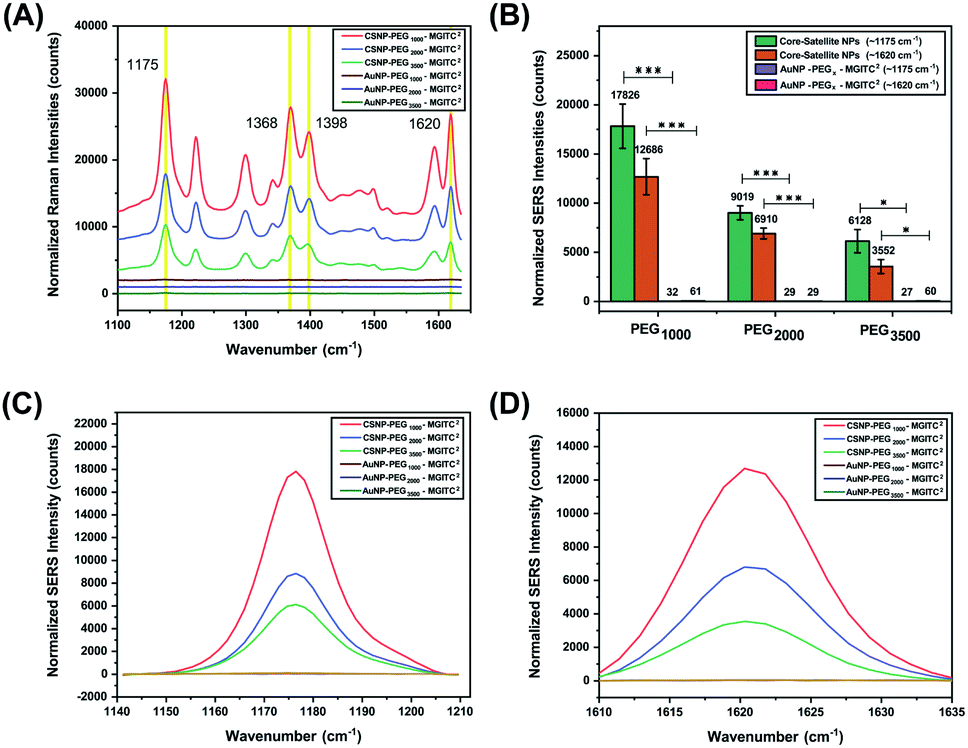 | ||
| Fig. 4 (A) The SERS spectra generated from MGITC adsorbed onto the surface of CSNPs functionalized with PEGx (MW: 1000 Da, 2000 Da, and 3500 Da) and AuNP cores modified with PEGx (MW: 1000 Da, 2000 Da, and 3500 Da). (B) The difference in SERS peak intensity observed for the CSNPs and respective AuNP ‘core’ controls of characteristic MGITC peaks positioned at ∼1175 cm−1 and ∼1620 cm−1. (C and D) SERS spectra of individual MGITC peaks (1175 cm−1 (C) and 1620 cm−1 (D)) for all CSNPs and respective AuNP controls. Notice that without attachment of the satellites hardly any SERS peak is observed. | ||
Long-term optical stability of the CSNP assembly
To characterize the long-term stability of CSNP nanostructures in an aqueous solution sensing application, UV-Vis stability measurements were obtained at various time points ranging from 0 to 14 days after synthesis. Fig. 5A and ESI Fig. S5† report a slight blue shift in wavelength, and a decrease in optical density is observed for CSNPs synthesized from the bifunctional linkers containing different PEG lengths.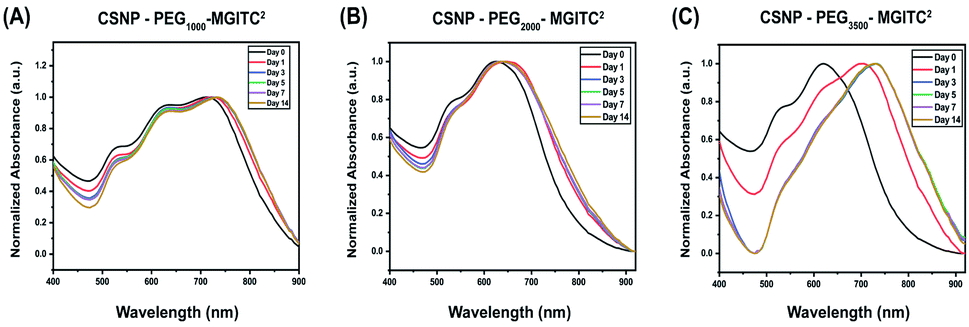 | ||
| Fig. 5 (A–C) Optical stability of CSNPs synthesized with various polymer thicknesses at 0, 1, 3, 5, 7, and 14 days. (A) CSNPs with PEG1000–MGITC2 at a volume ratio of 1:4 Au satellite to Au core NPs, (B) CSNPs with PEG2000–MGITC2 and (C) CSNPs with PEG3500–MGITC2 at a volume ratio of 1:3 Au satellite to Au core NPs. | ||
Specifically, Fig. 5A demonstrates a blue shift of CSNPs–PEG1000–MGITC2 from an average of 725 to 730 nm in the span of 14 days. Additionally, a calculated 19.48% loss of particles was observed after 14 days based on the decrease in absorbance (optical density) illustrated in ESI Fig. S5A.† In both CSNPs modified with PEGs with MWs of 2000 Da and 3500 Da, a spectral red shift was observed. CSNPs–PEG2000–MGITC2 exhibited a spectral red shift from 625 nm to 640 nm after 14 days, and the loss of NPs based on the decrease of absorbance was calculated to be around 30.24% after 14 days as shown in Fig. 5B and ESI Fig. S5B.† CSNPs–PEG3500–MGITC2 reported the largest spectral red shift from 620 nm to 730 nm stabilizing after 5 days. Similarly, CSNPs–PEG3500–MGITC2 reported a 47.68% decrease in absorbance after 14 days. Herein, it was observed that CSNPs–PEG1000–MGITC2 < CSNPs–PEG2000–MGITC2 < CSNPs–PEG3500–MGITC2 is the order of lowest to greatest loss of particles after 14 days.
Measuring the SERS signal generated from the CSNPs for two weeks
The long-term SERS activity of CSNPs composed of different PEG lengths in aqueous solution was characterized by taking SERS measurement of the CSNPs at 0, 7, and 14 days shown in Fig. 6. The CSNPs–PEGx–MGITC2 were characterized with a laser excitation of 785 nm at 1000 ms integration time. Based on the results, a retention of 41.16% of the SERS signal after 14 days is observed for CSNPs synthesized with PEG1000–MGITC2 at ∼1170 cm−1 corresponding to the in-plane aromatic C–H bending vibration. Additionally, a similar trend is observed for ∼1620 cm−1 corresponding to the N–C (ϕ bond) and C–C stretching with a signal retention of 32.3% after Day 14. CSNPs–PEG3500–MGITC2 exhibited a similar trend after 14 days with a SERS signal retention of around 44.7% and 40.32% at ∼1170 cm−1 and 1620 cm−1, respectively. Meanwhile, CSNPs–PEG2000–MGITC2 exhibited a retention of around 100.2% and 72.3% at ∼1170 cm−1 and 1620 cm−1. This could be attributed to the aggregation of the particles on Day 14 which could possibly cause a slight increase of signal.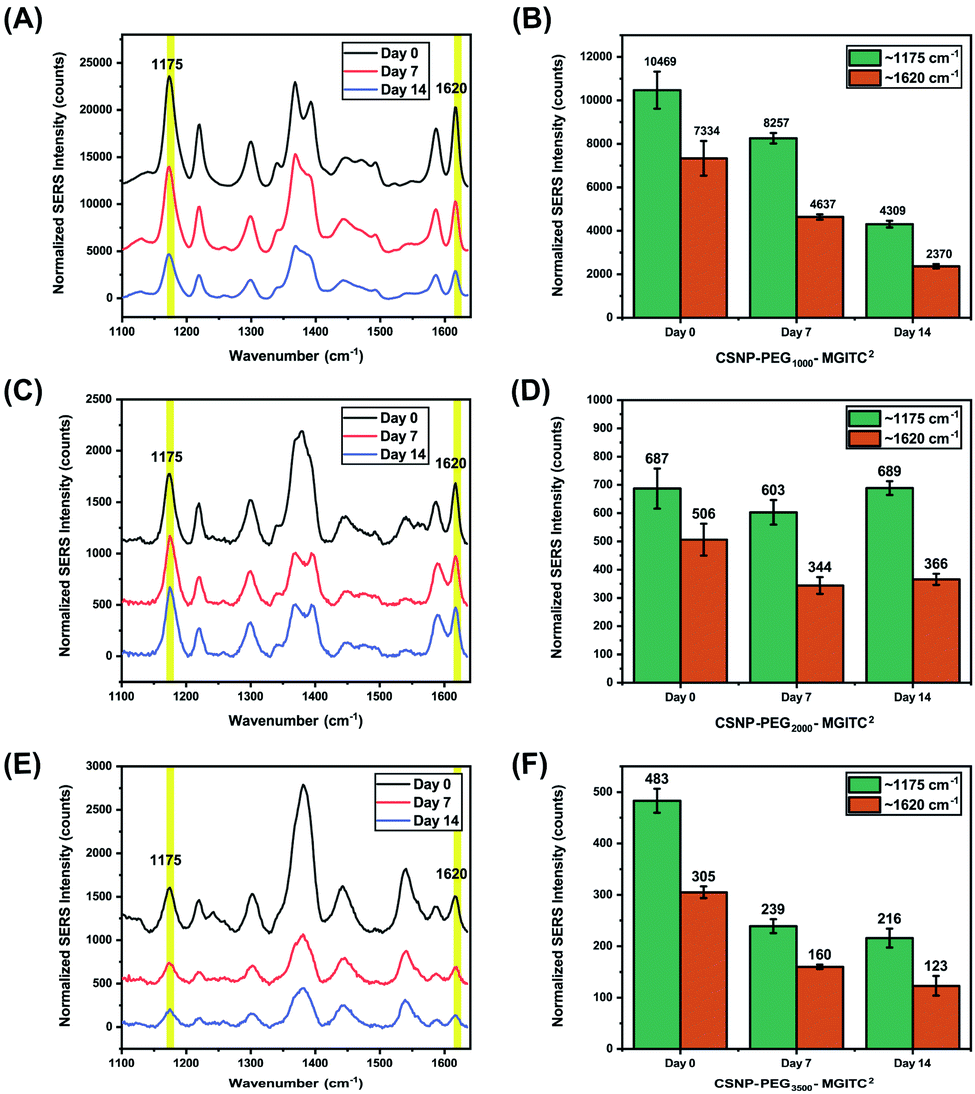 | ||
| Fig. 6 SERS measurements conducted at 0, 7, and 14 days for CSNPs with various gap distances: SERS retention of (A and B) CSNPs with PEG1000–MGITC2, (C and D) CSNPs with PEG2000–MGITC2 and (E and F) CSNPs with PEG3500–MGITC2, confirming the distinct MGITC characteristic peaks at 1175 cm−1 and 1620 cm−1. | ||
Conclusions
In this work, CSNPs modified with PEG with various MWs (1000 Da, 2000 Da, and 3500 Da) were successfully achieved using heterobifunctional PEG containing thiol (SH) and amine (NH2) functional groups. Successful formation of CSNPs was verified through UV-Vis measurements, confirming successful functionalization through spectral shift, and ζ-potential measurements by quantitatively measuring the change in surface charge. Successful modulation of PEGx and MGITC was achieved resulting in discrete CSNP formation. TEM images further confirmed the morphological changes. A tunable SERS hot spot was generated through the exploration of PEG with different length chains. Herein, it was observed that CSNPs–PEG1000–MGITC2 offered the highest signal enhancement when compared to longer PEGx polymer chains. Herein, it was observed that CSNPs–PEG1000–MGITC2, CSNPs–PEG2000–MGITC2, and CSNPs–PEG3500–MGITC2 have the lowest to highest loss of particles after 14 days as well as having the highest to lowest SERS signal enhancement due to nanogap distances. As for SERS signal retention, it was observed that CSNPs–PEG2000–MGITC2 retained most of the SERS signal at 14 days as opposed to the other PEG length chains. This nanoparticle system has a free surface monolayer that can further stabilize the NP system while also allowing application in a diagnostic assay by functionalization with target moieties such as antibodies, aptamers, and DNA oligonucleotide probes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AMSJ, SRC, DT, MT, GC and SM would like to thank the NSF-ERC (PATHS-UP) – Award Number 1648451, NSF-CBET (preeclampsia grant) – 2022805 and Mabbott's start-up funders, the Department of Biomedical Engineering and TEES for sponsoring this research.Notes and references
- J. Langer, et al., Present and Future of Surface-Enhanced Raman Scattering, ACS Nano, 2020, 14, 28–117 CrossRef CAS PubMed.
- R. Pilot, et al., A Review on Surface-Enhanced Raman Scattering, Biosensors, 2019, 9, 57 CrossRef CAS.
- C. Zong, et al., Surface-Enhanced Raman Spectroscopy for Bioanalysis: Reliability and Challenges, Chem. Rev., 2018, 118, 4946–4980 CrossRef CAS PubMed.
- T. J. Moore, et al., In Vitro and In Vivo SERS Biosensing for Disease Diagnosis, Biosensors, 2018, 8, 46 CrossRef.
- H. Marks, M. Schechinger, J. Garza, A. Locke and G. Coté, Surface enhanced Raman spectroscopy (SERS) for in vitro diagnostic testing at the point of care, Nanophotonics, 2017, 6, 681–701 CAS.
- J. Yang, G. L. Rorrer and A. X. Wang, Bioenabled SERS Substrates for Food Safety and Drinking Water Monitoring, Proc. SPIE-Int. Soc. Opt. Eng., 2015, 9488, 948808 CrossRef.
- H. Tang, C. Zhu, G. Meng and N. Wu, Review—Surface-Enhanced Raman Scattering Sensors for Food Safety and Environmental Monitoring, J. Electrochem. Soc., 2018, 165, B3098–B3118 CrossRef CAS.
- R. Pilot, SERS detection of food contaminants by means of portable Raman instruments, J. Raman Spectrosc., 2018, 49, 954–981 CrossRef CAS.
- D. Radziuk and H. Moehwald, Prospects for plasmonic hot spots in single molecule SERS towards the chemical imaging of live cells, Phys. Chem. Chem. Phys., 2015, 17, 21072–21093 RSC.
- K. A. Willets, Super-resolution imaging of SERS hot spots, Chem. Soc. Rev., 2014, 43, 3854–3864 RSC.
- Z. Zhou, et al., Silver nanocubes monolayers as a SERS substrate for quantitative analysis, Chin. Chem. Lett., 2021, 32, 1497–1501 CrossRef CAS.
- A. K. Nair, et al., In Situ Synthesis of Silver Nanospheres, Nanocubes, and Nanowires over Boron-Doped Graphene Sheets for Surface-Enhanced Raman Scattering Application and Enzyme-Free Detection of Hydrogen Peroxide, Langmuir, 2018, 34, 13603–13614 CrossRef CAS.
- A. Garcia-Leis, J. V. Garcia-Ramos and S. Sanchez-Cortes, Silver Nanostars with High SERS Performance, J. Phys. Chem. C, 2013, 117, 7791–7795 CrossRef CAS.
- T. Jiang, et al., Seed-mediated synthesis and SERS performance of graphene oxide-wrapped Ag nanomushroom, Sci. Rep., 2017, 7, 9795 CrossRef PubMed.
- C. P. Shaw, et al., Statistical Correlation Between SERS Intensity and Nanoparticle Cluster Size, J. Phys. Chem. C, 2013, 117, 16596–16605 CrossRef CAS.
- S. Hong and X. Li, Optimal Size of Gold Nanoparticles for Surface-Enhanced Raman Spectroscopy under Different Conditions, J. Nanomater., 2013, 2013, 790323 Search PubMed.
- K. G. Stamplecoskie, J. C. Scaiano, V. S. Tiwari and H. Anis, Optimal Size of Silver Nanoparticles for Surface-Enhanced Raman Spectroscopy, J. Phys. Chem. C, 2011, 115, 1403–1409 CrossRef CAS.
- N. D. Israelsen, C. Hanson and E. Vargis, Nanoparticle Properties and Synthesis Effects on Surface-Enhanced Raman Scattering Enhancement Factor: An Introduction, Sci. World J., 2015, 2015, 124582 Search PubMed.
- E.-T. Lee, H.-W. Cheng, J.-Y. Yang and Y. Li, Shape Effect of Surface-Enhanced Raman Scattering-Active-Substrate-Based Nanoparticles on Local Electric Field for Biochemical Sensing Application, J. Nanosci. Nanotechnol., 2017, 17, 871–877 CrossRef CAS.
- F. Benz, et al., SERS of Individual Nanoparticles on a Mirror: Size Does Matter, but so Does Shape, J. Phys. Chem. Lett., 2016, 7, 2264–2269 CrossRef CAS.
- B. Sharma, R. R. Frontiera, A.-I. Henry, E. Ringe and R. P. Van Duyne, SERS: Materials, applications, and the future, Mater. Today, 2012, 15, 16–25 CrossRef CAS.
- Y. Cao, D. Li, F. Jiang, Y. Yang and Z. Huang, Engineering Metal Nanostructure for SERS Application, J. Nanomater., 2013, 2013, 123812 Search PubMed.
- B. Yang, et al., Recent Development of SERS Technology: Semiconductor-Based Study, ACS Omega, 2019, 4, 20101–20108 CrossRef CAS PubMed.
- C. Mun, et al., Highly Sensitive and Selective Nanogap-Enhanced SERS Sensing Platform, Nanomaterials, 2019, 9, 619 CrossRef CAS.
- Y. Yokota, K. Ueno and H. Misawa, Essential nanogap effects on surface-enhanced Raman scattering signals from closely spaced gold nanoparticles, Chem. Commun., 2011, 47, 3505–3507 RSC.
- D. Natelson, Y. Li and J. B. Herzog, Nanogap structures: combining enhanced Raman spectroscopy and electronic transport, Phys. Chem. Chem. Phys., 2013, 15, 5262–5275 RSC.
- Y. Yang, J. Liu, Z. Fu and D. Qin, Galvanic Replacement-Free Deposition of Au on Ag for Core–Shell Nanocubes with Enhanced Chemical Stability and SERS Activity, J. Am. Chem. Soc., 2014, 136, 23 Search PubMed.
- A. Gopalakrishnan, et al., Bimetallic 3D Nanostar Dimers in Ring Cavities: Recyclable and Robust Surface-Enhanced Raman Scattering Substrates for Signal Detection from Few Molecules, ACS Nano, 2014, 8, 7986–7994 CrossRef CAS.
- Y. Yang, et al., Growth of Spherical Gold Satellites on the Surface of Au@Ag@SiO(2) Core–Shell Nanostructures Used for an Ultrasensitive SERS Immunoassay of Alpha-Fetoprotein, ACS Appl. Mater. Interfaces, 2019, 11, 3617–3626 CrossRef CAS PubMed.
- H.-X. Wang, Y.-W. Zhao, Z. Li, B.-S. Liu and D. Zhang, Development and Application of Aptamer-Based Surface-Enhanced Raman Spectroscopy Sensors in Quantitative Analysis and Biotherapy, Sensors, 2019, 19, 3806 CrossRef CAS.
- M. Muhammad and Q. Huang, A review of aptamer-based SERS biosensors: Design strategies and applications, Talanta, 2021, 227, 122188 CrossRef CAS PubMed.
- J. Smolsky, S. Kaur, C. Hayashi, S. K. Batra and A. V. Krasnoslobodtsev, Surface-Enhanced Raman Scattering-Based Immunoassay Technologies for Detection of Disease Biomarkers, Biosensors, 2017, 7, 7 CrossRef.
- A. Kamińska, et al., SERS-based Immunoassay in a Microfluidic System for the Multiplexed Recognition of Interleukins from Blood Plasma: Towards Picogram Detection, Sci. Rep., 2017, 7, 10656 CrossRef.
- Z. Kunushpayeva, et al., Sandwich SERS immunoassay of human immunoglobulin on silicon wafer compared to traditional SERS substrate, gold film, Sens. Bio-Sens. Res., 2020, 29, 100355 CrossRef.
- M. Schechinger, H. Marks, S. Mabbott, M. Choudhury and G. Cote’, A SERS approach for rapid detection of microRNA-17 in the picomolar range, Analyst, 2019, 144, 4033–4044 RSC.
- J.-A. Huang, et al., SERS discrimination of single DNA bases in single oligonucleotides by electro-plasmonic trapping, Nat. Commun., 2019, 10, 5321 CrossRef PubMed.
- D. D. Li, J. Wang, G. C. Zheng, J. H. Liu and W. H. Xu, A highly active SAB – Regiospecific core–satellite assembly of gold nanoplates (AuNPs)/gold nanorods (AuNRs) can be fabricated via ss-DNA hybridization. SERS behavior of the DNA driven assembly has been explored from inducing transition between para-ATP a, Nanotechnology, 2013, 24, 235502 CrossRef CAS PubMed.
- Y. Yang, et al., Growth of Spherical Gold Satellites on the Surface of Au@Ag@SiO2 Core–Shell Nanostructures Used for an Ultrasensitive SERS Immunoassay of Alpha-Fetoprotein, ACS Appl. Mater. Interfaces, 2019, 11, 3617–3626 CrossRef CAS.
- N. Gandra, A. Abbas, L. Tian and S. Singamaneni, Plasmonic Planet–Satellite Analogues: Hierarchical Self-Assembly of Gold Nanostructures, Nano Lett., 2012, 12, 2645–2651 CrossRef CAS.
- Z. Li, et al., Core–satellite metal–organic framework@upconversion nanoparticle superstructures via electrostatic self-assembly for efficient photodynamic theranostics, Nano Res., 2020, 13, 3377–3386 CrossRef.
- C. Rossner and A. Fery, Planet–satellite nanostructures from inorganic nanoparticles: from synthesis to emerging applications, MRS Commun., 2020, 10, 112–122 CrossRef CAS.
- R. P. M. Höller, M. Dulle, S. Thoma, M. Mayer, A. M. Steiner, S. Förster, A. Fery, C. Kuttner and M. Chanana, Protein-Assisted Assembly of Modular 3D Plasmonic Raspberry-like Core/Satellite Nanoclusters: Correlation of Structure and Optical Properties, ACS Nano, 2016, 10, 5740–5750 CrossRef PubMed.
- J. Tian, B. Huang and W. Zhang, Precise Self-Assembly and Controlled Catalysis of Thermoresponsive Core–Satellite Multicomponent Hybrid Nanoparticles, Langmuir, 2019, 35, 266–275 CrossRef CAS PubMed.
- F. Han, S. R. C. Vivekchand, A. H. Soeriyadi, Y. Zheng and J. J. Gooding, Thermoresponsive plasmonic core-satellite nanostructures with reversible, temperature sensitive optical properties, Nanoscale, 2018, 10, 4284–4290 RSC.
- C. Wang, et al., Polyethylenimine-interlayered core–shell–satellite 3D magnetic microspheres as versatile SERS substrates, Nanoscale, 2015, 7, 18694–18707 RSC.
- Y.-B. Liu, T.-T. Zhai, Y.-Y. Liang, Y.-B. Wang and X.-H. Xia, Gold core–satellite nanostructure linked by oligonucleotides for detection of glutathione with LSPR scattering spectrum, Talanta, 2019, 193, 123–127 CrossRef CAS.
- L. Tang, et al., Highly Stabilized Core-Satellite Gold Nanoassemblies in Vivo: DNA-Directed Self-Assembly, PEG Modification and Cell Imaging, Sci. Rep., 2017, 7, 8553 CrossRef PubMed.
- X. Zhao, et al., Gold-Quantum Dot Core–Satellite Assemblies for Lighting Up MicroRNA In Vitro and In Vivo, Small, 2016, 12, 4662–4668 CrossRef CAS PubMed.
- C. Hu, et al., Highly narrow nanogap-containing Au@Au core–shell SERS nanoparticles: size-dependent Raman enhancement and applications in cancer cell imaging, Nanoscale, 2016, 8, 2090–2096 RSC.
- D. Meng, W. Ma, X. Wu, C. Xu and H. Kuang, DNA-Driven Two-Layer Core–Satellite Gold Nanostructures for Ultrasensitive MicroRNA Detection in Living Cells, Small, 2020, 16, 2000003 CrossRef CAS.
- A. S. D. S. Indrasekara, R. Thomas and L. Fabris, Plasmonic properties of regiospecific core–satellite assemblies of gold nanostars and nanospheres, Phys. Chem. Chem. Phys., 2015, 17, 21133–21142 RSC.
- J. H. Yoon, J. Lim and S. Yoon, Controlled Assembly and Plasmonic Properties of Asymmetric Core–Satellite Nanoassemblies, ACS Nano, 2012, 6, 7199–7208 CrossRef CAS PubMed.
- Y. Yang, J. Zhao, G. Weng, J. Li, J. Zhu and J. Zhao, et al., Fine-tunable fluorescence quenching properties of core-satellite assemblies of gold nanorod–nanosphere: Application in sensitive detection of Hg2+, Spectrochim. Acta, Part A, 2020, 228, 117776 CrossRef CAS.
- L.-A. Wu, W.-E. Li, D.-Z. Lin and Y.-F. Chen, Three-Dimensional SERS Substrates Formed with Plasmonic Core–Satellite Nanostructures, Sci. Rep., 2017, 7, 13066 CrossRef PubMed.
- Y. Zheng, et al., DNA-Directed Self-Assembly of Core–Satellite Plasmonic Nanostructures: A Highly Sensitive and Reproducible Near-IR SERS Sensor, Adv. Funct. Mater., 2013, 23, 1519–1526 CrossRef CAS.
- N. Pazos-Perez, J. M. Fitzgerald, V. Giannini, L. Guerrini and R. A. Alvarez-Puebla, Modular assembly of plasmonic core–satellite structures as highly brilliant SERS-encoded nanoparticles, Nanoscale Adv., 2019, 1, 122–131 RSC.
- J. Kimling, et al., Turkevich Method for Gold Nanoparticle Synthesis Revisited, J. Phys. Chem. B, 2006, 110, 15700–15707 CrossRef CAS.
- R. C. Hoft, M. J. Ford, A. M. McDonagh and M. B. Cortie, Adsorption of Amine Compounds on the Au(111) Surface: A Density Functional Study, J. Phys. Chem. C, 2007, 111, 13886–13891 CrossRef CAS.
- F. Chen, X. Li, J. Hihath, Z. Huang and N. Tao, Effect of Anchoring Groups on Single-Molecule Conductance:Comparative Study of Thiol-,Amine-,and Carboxylic-Acid-Terminated Molecules, J. Am. Chem. Soc., 2006, 128, 15874–15881 CrossRef CAS PubMed.
- M. J. Ford, C. Masens and M. B. Cortie, The application of gold surfaces and particles in nanotechnology, Surf. Rev. Lett., 2006, 13, 297–307 CrossRef CAS.
- J. Ftouni, M. Penhoat, J. Girardon, A. Addad, E. Payen and C. Rolando, et al., Immobilization of gold nanoparticles on fused silica capillary surface for the development of catalytic microreactors, Chem. Eng. J., 2013, 227, 103–110 CrossRef CAS.
- R. R. Bhat and J. Genzer, Tuning the number density of nanoparticles by multivariant tailoring of attachment points on flat substrates, Nanotechnology, 2006, 18, 25301 CrossRef.
- M. Ben Haddada, et al., Optimizing the immobilization of gold nanoparticles on functionalized silicon surfaces: amine- vs thiol-terminated silane, Gold Bull., 2013, 46, 335–341 CrossRef.
- B. Michen, et al., Avoiding drying-artifacts in transmission electron microscopy: Characterizing the size and colloidal state of nanoparticles, Sci. Rep., 2015, 5, 9793 CrossRef CAS PubMed.
- A. Kaliyaraj Selva Kumar, Y. Zhang, D. Li and R. G. Compton, A mini-review: How reliable is the drop casting technique?, Electrochem. Commun., 2020, 121, 106867 CrossRef CAS.
- R. P. M. Höller, et al., Biomacromolecular-Assembled Nanoclusters: Key Aspects for Robust Colloidal SERS Sensing, ACS Appl. Mater. Interfaces, 2020, 12, 57302–57313 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00676b |
| This journal is © The Royal Society of Chemistry 2022 |