
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMacrophages secrete murinoglobulin-1 and galectin-3 to regulate neutrophil degranulation after myocardial infarction†
Upendra
Chalise
 ab,
Michael J.
Daseke
II
abc,
William J.
Kalusche
c,
Shelby R.
Konfrst
ab,
Jocelyn R.
Rodriguez-Paar
ab,
Elizabeth R.
Flynn
c,
Leah M.
Cook
d,
Mediha
Becirovic-Agic
ab and
Merry L.
Lindsey
*ab
ab,
Michael J.
Daseke
II
abc,
William J.
Kalusche
c,
Shelby R.
Konfrst
ab,
Jocelyn R.
Rodriguez-Paar
ab,
Elizabeth R.
Flynn
c,
Leah M.
Cook
d,
Mediha
Becirovic-Agic
ab and
Merry L.
Lindsey
*ab
aDepartment of Cellular and Integrative Physiology, Center for Heart and Vascular Research, University of Nebraska Medical Center, Omaha, NE 68198, USA. E-mail: merry.lindsey@unmc.edu
bResearch Service, Nebraska-Western Iowa Health Care System, Omaha, NE 68105, USA
cDepartment of Physiology and Biophysics, University of Mississippi Medical Center, Jackson, MS 39216, USA
dDepartment of Pathology and Microbiology, University of Nebraska Medical Center, Omaha, NE 68198, USA
First published on 1st March 2022
Abstract
Inflammation presides early after myocardial infarction (MI) as a key event in cardiac wound healing. Ischemic cardiomyocytes secrete inflammatory cues to stimulate infiltration of leukocytes, predominantly macrophages and neutrophils. Infiltrating neutrophils degranulate to release a series of proteases including matrix metalloproteinase (MMP)-9 to break down extracellular matrix and remove necrotic myocytes to create space for the infarct scar to form. While neutrophil to macrophage communication has been explored, the reverse has been understudied. We used a proteomics approach to catalogue the macrophage secretome at MI day 1. Murinoglobulin-1 (MUG1) was the highest-ranked secreted protein (4.1-fold upregulated at MI day 1 vs. day 0 pre-MI cardiac macrophages, p = 0.004). By transcriptomics evaluation, galectin-3 (Lgals3) was 2.2-fold upregulated (p = 0.008) in MI day 1 macrophages. We explored the direct roles of MUG1 and Lgals3 on neutrophil degranulation. MUG1 blunted while Lgals3 amplified neutrophil degranulation in response to phorbol 12-myristate 13-acetate or interleukin-1β, as measured by MMP-9 secretion. Lgals3 itself also stimulated MMP-9 secretion. To determine if MUG1 regulated Lgals3, we co-stimulated neutrophils with MUG1 and Lgals3. MUG1 limited degranulation stimulated by Lgals3 by 64% (p < 0.001). In vivo, MUG1 was elevated in the infarct region at MI days 1 and 3, while Lgals3 increased at MI day 7. The ratio of MUG1 to Lgals3 positively correlated with infarct wall thickness, revealing that MUG1 attenuated infarct wall thinning. In conclusion, macrophages at MI day 1 secrete MUG1 to limit and Lgals3 to accentuate neutrophil degranulation to regulate infarct wall thinning.
Introduction
In response to myocardial infarction (MI), the cardiac wound healing response involves an early phase of robust inflammation. Inflammation is a prerequisite event for later transition to resolution and repair culminating in infarct scar formation.1–5 Leukocyte infiltration is a key hallmark of the early inflammatory phase, with neutrophils and macrophages serving as the predominant cell contributions.6–9 During the inflammatory phase, neutrophils instigate tissue repair by releasing proteases such as matrix metalloproteinase (MMP)-9 that degrade extracellular matrix (ECM) to remove necrotic cardiomyocytes.10,11Neutrophils polarize to a pro-inflammatory phenotype upon activation by pro-inflammatory stimuli and release preformed granules by a process termed degranulation.11,12 Neutrophil granules contain MMP-8, MMP-9, myeloperoxidase (MPO), neutrophil gelatinase-associated lipocalin (NGAL), and neutrophil elastase (NE), all of which support infarct debris removal.12 While neutrophil degranulation is essential to create an optimum environment for later scar formation, moderation is needed to prevent excessive infarct wall thinning and dilation of the left ventricle (LV).13,14 Neutrophil numbers, MMP-9, and MPO have all been positively associated with increased risk of heart failure progression and mortality after MI.15–19
Various attempts have been made to globally target neutrophils or neutrophil activity after MI, and the majority of these studies resulted in detrimental effects on cardiac remodeling, demonstrating the importance of neutrophils in MI wound repair.20–22 MI on neutrophil depleted mice showed impaired clearance of apoptotic cardiomyocytes and reduced numbers of phagocytic macrophages, reflective of neutrophil to macrophage communication.20 Neutrophil degranulation is a crucial step for the initiation of macrophage transdifferentiation to reparative phenotype. Neutrophils secrete NGAL as a required switch for reparative macrophage polarization and negative feedback induction of phagocytosis to remove neutrophils.20 Neutrophil to macrophage cross talk, therefore, is relevant for MI remodeling.
In addition to neutrophils, macrophages are the other crucial cell type in MI wound healing.23–25 Monocytes infiltrate the infarct along with neutrophils, differentiate into macrophages in the infarct regions and initially serve as a pro-inflammatory cell.9,26 At MI day (D)1, macrophages are the second most abundant cell type in the infarct, transiting to the most abundant cell by MI D3-5.7 By MI D3, macrophages are transitioning to anti-inflammatory phenotype, with efferocytosis of apoptotic neutrophils being a key feature.4,7 While neutrophil to macrophage cellular communication has been well studied by several groups,27,28 the converse – macrophage to neutrophil cellular communication – is understudied. Because anti-inflammatory and reparative macrophages regulate other cell types (e.g., endothelial cells and cardiac fibroblasts) and both macrophages and neutrophils are present at MI D1, the pro-inflammatory macrophage may regulate neutrophil physiology as well.29,30
The goal of this study was to explore the hypothesis that macrophages communicate to neutrophils at MI D1 to regulate degranulation during the pro-inflammatory phase. The experimental design is as detailed in Fig. 1. We catalogued the macrophage secretome and coupled the results with a previously acquired transcriptomics dataset to identify major MI D1 macrophage secreted proteins that regulated neutrophil degranulation. The top candidates were used to stimulate neutrophils in vitro and were monitored in vivo to explore the mechanisms of macrophage to neutrophil crosstalk.
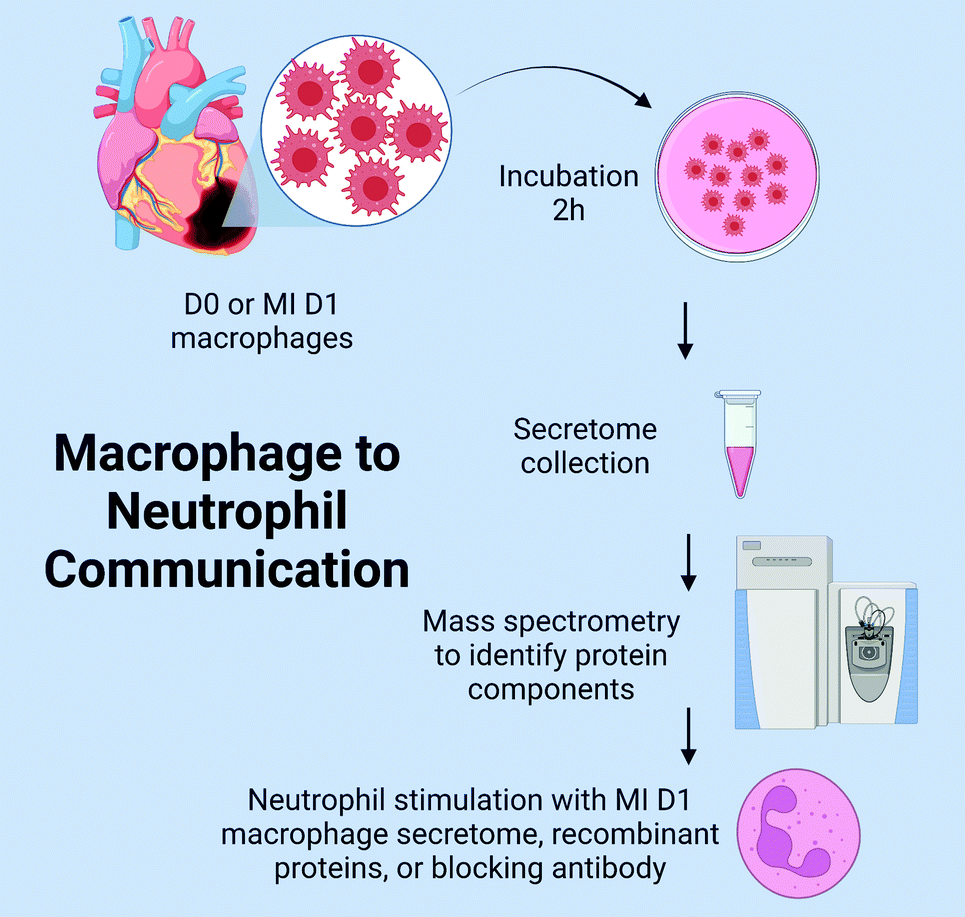 | ||
| Fig. 1 Experimental Design. D-day; MI-myocardial infarction. | ||
Results
MUG1 was the major upregulated protein in the MI D1 macrophage secretome
By mass spectrometry, 246 proteins were detected in either no MI D0 or MI D1 macrophage secretomes. Out of the 246 proteins, 39 proteins were statistically different (all p < 0.05); 22 of which were upregulated and 17 were downregulated (Fig. 2A). By volcano plot, MUG1 was the highest ranked upregulated protein, followed by β-actin like protein 2 (ACTBL2), vimentin (VIM), arf-GAP with SH3 domain, and ANK repeat and PH domain-containing protein 2 (ASAP1). Ubiquitin carboxyl-terminal hydrolase 48 (USP48) was the highest ranked downregulated protein, followed by centrosomal protein of 162 kDa (CEP162), fatty acid-binding protein heart (FABP3), myoglobin (MB), and nuclear receptor coactivator 3 (NCOA3; Fig. 2B). Partial least squares discriminant analysis (PLS-DA) scores plot showed a significant shift between D0 and D1 macrophage secretome (Fig. 2C). For the upregulated proteins, Interleukin (IL)-17 signaling was the most enriched pathway, while for the downregulated proteins, peroxisome proliferator-activated receptor (PPAR) signaling was the most enriched pathway (Fig. 2D).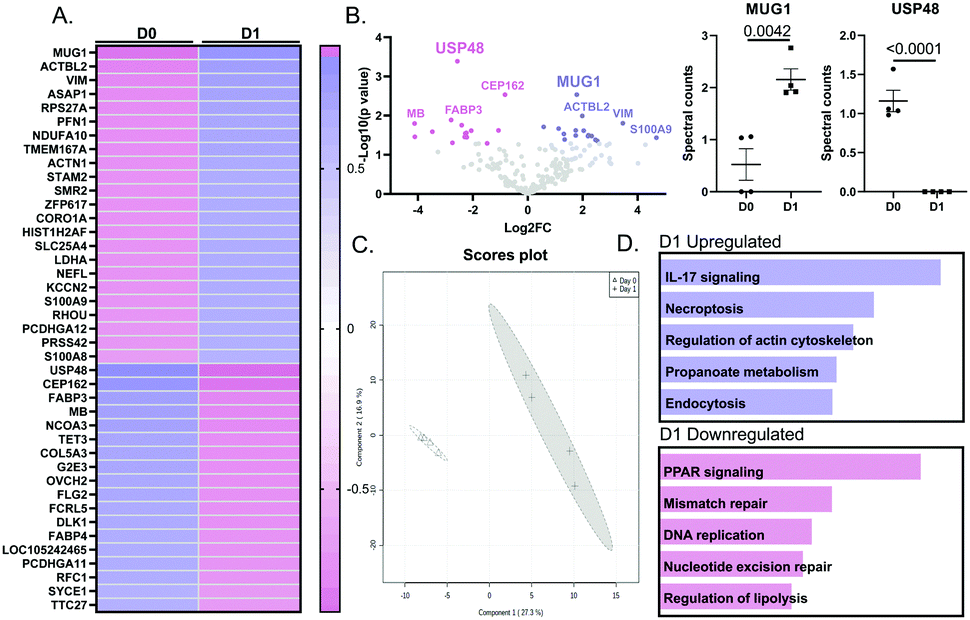 | ||
| Fig. 2 MI D1 Macrophage secretome analysis revealed MUG1 as the highest upregulated protein and USP48 as the highest downregulated protein. (A) Heat map ranking of proteins by p value and fold-change at MI D1 vs. D0. (B) By Volcano plot, MUG1 was the most upregulated and USP-48 the most downregulated protein. (C) PLS-DA 2D scores plot shows a significant shift in proteome of D0 vs. D1 macrophage secretome. (D) Enrichment analysis shows that for MI D1 upregulated proteins (top), IL-17 signaling was the most enriched pathway. Of the downregulated proteins (bottom), PPAR signaling was the most enriched pathway. Sample sizes were n = 4 pooled M sets; analysis by unpaired Students t-test and Enrichr pathway analysis. | ||
The D1 MI macrophage secretome signals through MUG1 to blunt neutrophil degranulation
The design for this experiment is detailed in Fig. 3A. Bone marrow derived neutrophils treated with PMA, a positive control for neutrophil degranulation, showed robust release of MMP-9 into the secretome (Fig. 3B). Degranulation of MMP-9 was attenuated by the addition of D1 MI macrophage secretome, an effect that was reversed by addition of a MUG1 blocking antibody. Out of 111 cytokines examined, 62 proteins were different by ANOVA, and the top 25 differentially expressed proteins are shown in Fig. 3C. For the proteins induced by PMA that were attenuated by the MI D1 macrophage secretome, cytokine-cytokine receptor interaction was the major enriched pathway (Fig. 3D top). The ability of the MI D1 macrophage secretome to reduce proteins induced by PMA stimulation was reversed by the addition of a MUG1 antibody, and cytokine-cytokine receptor interaction was the major enriched pathway affected (Fig. 3D bottom). The MI D1 macrophage secretome, therefore, acted in part through MUG1 to blunt neutrophil degranulation.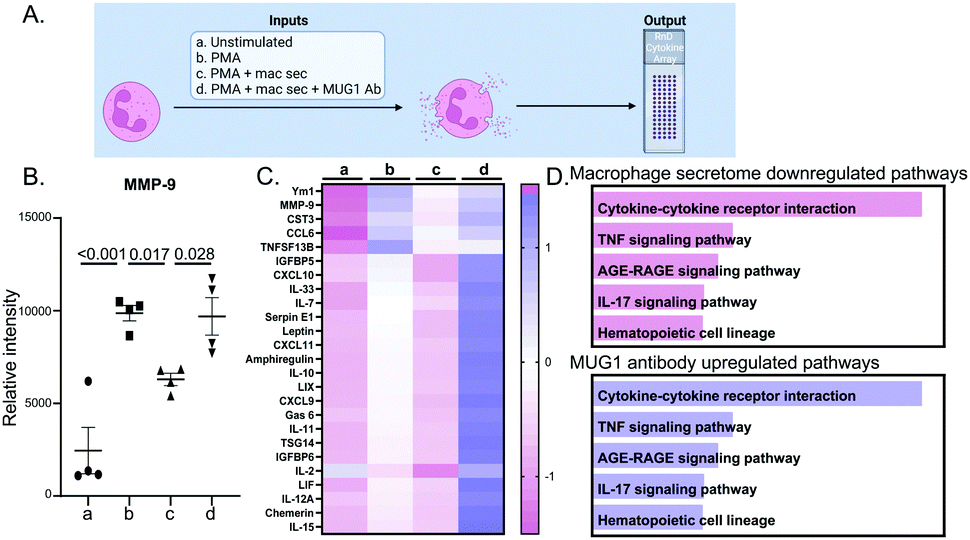 | ||
| Fig. 3 D1 MI macrophage secretome reduced neutrophil degranulation through MUG1. (A) Experimental design. (B) Neutrophils treated with phorbol 12-myristate-13-acetate (PMA) induced MMP-9 release, a surrogate marker of degranulation. Treatment with MI D1 macrophage secretome (10%, by volume) significantly reduced MMP-9 release, an effect reversed by addition of a MUG1 blocking antibody. (C) Heatmap of the top 25 proteins ranked by p value for the comparison of PMA + MI D1 macrophage secretome + MUG1 Ab vs. PMA + MI D1 macrophage secretome. (D) For the proteins downregulated by the D1 MI macrophage secretome, cytokine-cytokine receptor interaction signaling was most enriched (top). For the proteins upregulated by addition of a MUG1 blocking Ab to PMA+ MI D1 macrophage secretome, cytokine-cytokine receptor interaction was most enriched (bottom). Sample sizes were n = 4M per paired group; analysis by one way ANOVA with Student Newman–Keuls post-hoc test, paired Students t-test, and Enrichr pathway analysis. | ||
MUG1 directly inhibited IL-1β stimulated neutrophil degranulation
The design for this experiment is detailed in Fig. 4A. IL-1β upregulated 92 proteins out of 111 (all p < 0.05 for IL-1β vs. unstimulated). The top 4 were NGAL (3.9-fold, p < 0.0001), MMP-9 (5.7-fold, p < 0.0001), CCL6 (2.2-fold, p = 0.0006) and CHI3L1 (2.0-fold, p = 0.001). MUG1 attenuated IL-1β stimulated neutrophil degranulation by 60%, as measured by release of MMP-9 (Fig. 4B). Of the 92 proteins induced by IL-1β, MUG1 reduced expression for 13 of the proteins (Fig. 4C). Of these, MMP-9, MPO, and NGAL had the greatest reduction (Fig. 4D top). MUG1 by itself did not induce changes in the neutrophil secretome (all 111 proteins were p = not significant), signifying that MUG1 blunted degranulation only in the presence of a pro-inflammatory stimulus. Enrichment analysis of the 13 proteins downregulated by MUG1 treatment showed that IL-17 signaling, and chemokine and cytokine mediated signaling were the two most enriched pathways (Fig. 4D bottom). We did not observe any sex differences in the response.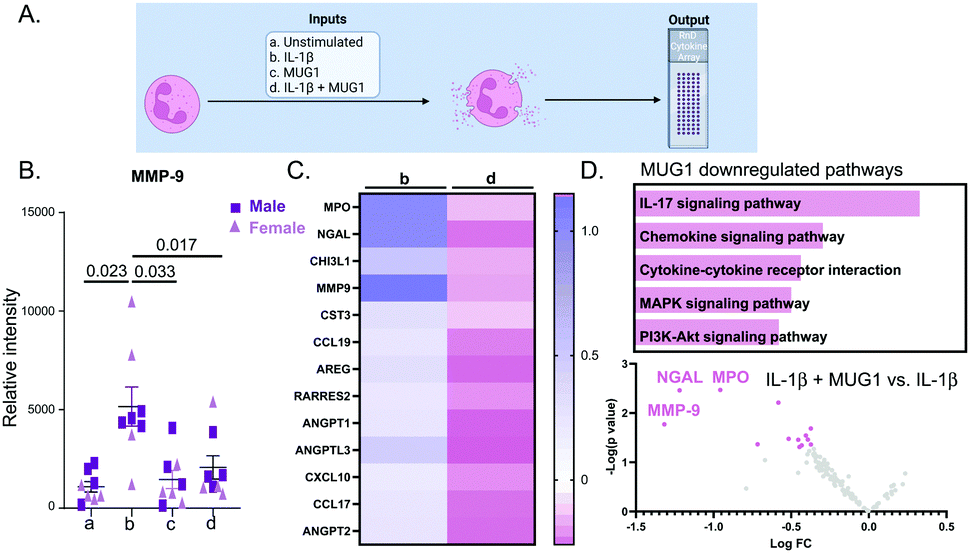 | ||
| Fig. 4 MUG1 downregulated neutrophil degranulation and pro-inflammatory cytokine release. (A) Experimental design. (B) Neutrophils treated with IL-1β induced MMP-9 release, a surrogate marker of neutrophil degranulation. IL-1β stimulated release of MMP-9 was attenuated by supplementation of MUG1. (C) Heatmap ranking of the 13 IL-1β stimulated neutrophil proteins decreased by MUG1. (D) By enrichment analysis, IL-17 signaling was the most enriched pathway for the MUG1 downregulated proteins (top). By volcano plotting, MMP-9, MPO, and NGAL were the IL-1β induced neutrophil proteins most downregulated by MUG1 (bottom). Sample sizes were n = 8, 4M/4F per paired group; analysis done by one way ANOVA with Student Newman–Keuls post-hoc test, paired Students t-test, and Enrichr pathway analysis. | ||
Lgals3 expressed by MI D1 macrophages promotes neutrophil degranulation
Lgals3 (Mac2) is a pro-inflammatory protein highly expressed by MI D1 macrophages (Fig. 5A).7 Lgals3 is a macrophage secreted protein with roles in neutrophil chemotaxis, neutrophil apoptosis, and phagocytosis of apoptotic neutrophils by macrophages.31–33 Since, Lgals3 interacts with soluble fibrinogen, a neutrophil protein increased in MI D1 that correlates with infarct wall thinning, we hypothesized Lgals3 could be another macrophage secreted protein that affected neutrophil physiology.34,35 The design for this experiment is detailed in Fig. 5B. IL-1β alone robustly increased neutrophil degranulation. Lgals3 alone also showed strong pro-degranulatory activity (Fig. 5C). By two-way ANOVA, there was no difference between sex and treatment groups (p = 0.34). The p value for sex alone was 0.23. The only differences observed were between treatments (p = 0.0025). Lgals3 stimulation did not amplify the IL-1β effect, indicating that Lgals3 and IL-1β signaled through the same pathway. Compared to the unstimulated control, Lgals3 directly increased expression of 9 proteins (Fig. 5D); the top of which were MPO, MMP-9, Cystatin C (CST3), NGAL and CCL6. All 9 proteins were also upregulated by IL-1β, further evidence that the two stimuli share similar signaling pathways. Enrichment analysis of the proteins upregulated by Lgals3 revealed leukocyte transendothelial migration as the most upregulated pathway (Fig. 5E top), showing that Lgals3 may also serve a role in stimulating neutrophil influx to the infarct. Volcano plot visualization showed that the highest ranked proteins were MPO followed by MMP-9 and NGAL, all of which are components of neutrophil degranulation (Fig. 5E bottom). This experiment demonstrated that Lgals3 directly stimulated neutrophil degranulation.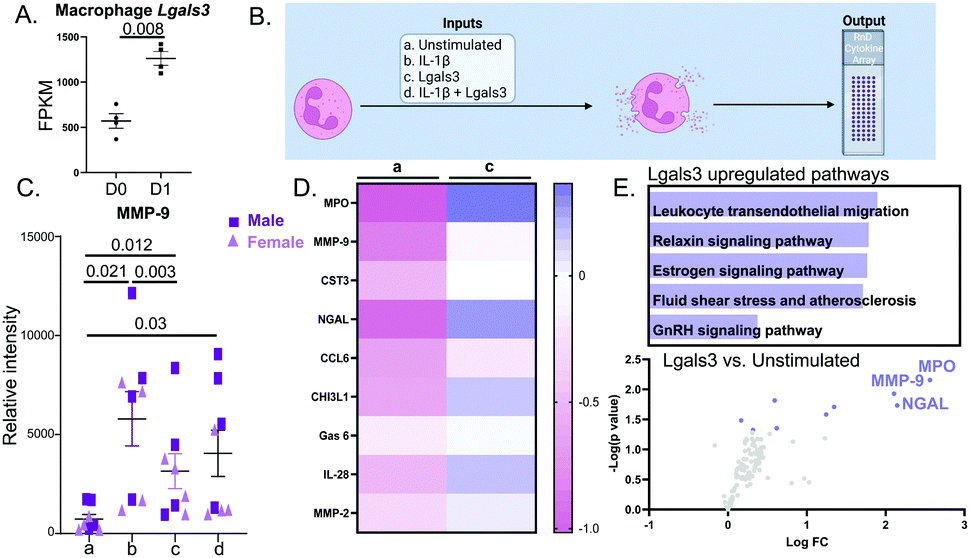 | ||
| Fig. 5 Lgals3 was upregulated in the D1 MI macrophage and induced MMP-9 release from neutrophils. (A) Lgals3 mRNA expression increased 2.2-fold in MI D1 macrophages compared to D0 no MI controls. (B) Experimental design. (C) Neutrophils treated with IL-1β induced MMP-9 release, a surrogate marker of neutrophil degranulation. Lgals3 independently induced MMP-9 release. Co-stimulating Lgals3 with IL-1β did not have an additive effect (p = 0.885 for IL-1β vs. IL-1β + Lgals3). (D) Heatmap showing the top 9 proteins increased by IL-1β compared to unstimulated controls, ranked by p value. (E) Enrichment analysis of proteins upregulated by Lgals3 revealed leukocyte transendothelial migration as the most upregulated pathway (top). By volcano plot, MPO was the most upregulated neutrophil protein stimulated by Lgals3. Sample sizes were n = 8, 4M/4F each paired group; analysis done by one way ANOVA with Student Newman–Keuls post-hoc test, paired Students t-test, and Enrichr pathway analysis. | ||
MUG1 prevented Lgals3 induced degranulation
As Lgals3 and MUG1 were both upregulated in D1 macrophages and both proteins actively regulated neutrophil degranulation, we examined the combined effect of MUG1 and Lgals3 on neutrophil degranulation. We know that a homologue of MUG1, α2-macroglobulin, binds Lgals3; therefore, we hypothesized that MUG1 would bind Lgals3 to inhibit its action on neutrophil degranulation.36 Neutrophils stimulated with a combination of MUG1 and Lgals3 showed a 64% reduction in Lgals3-induced release of MMP-9 (Fig. 6). MUG1 and Lgals3 therefore, could be endogenous mechanisms employed by macrophages to regulate neutrophil degranulation.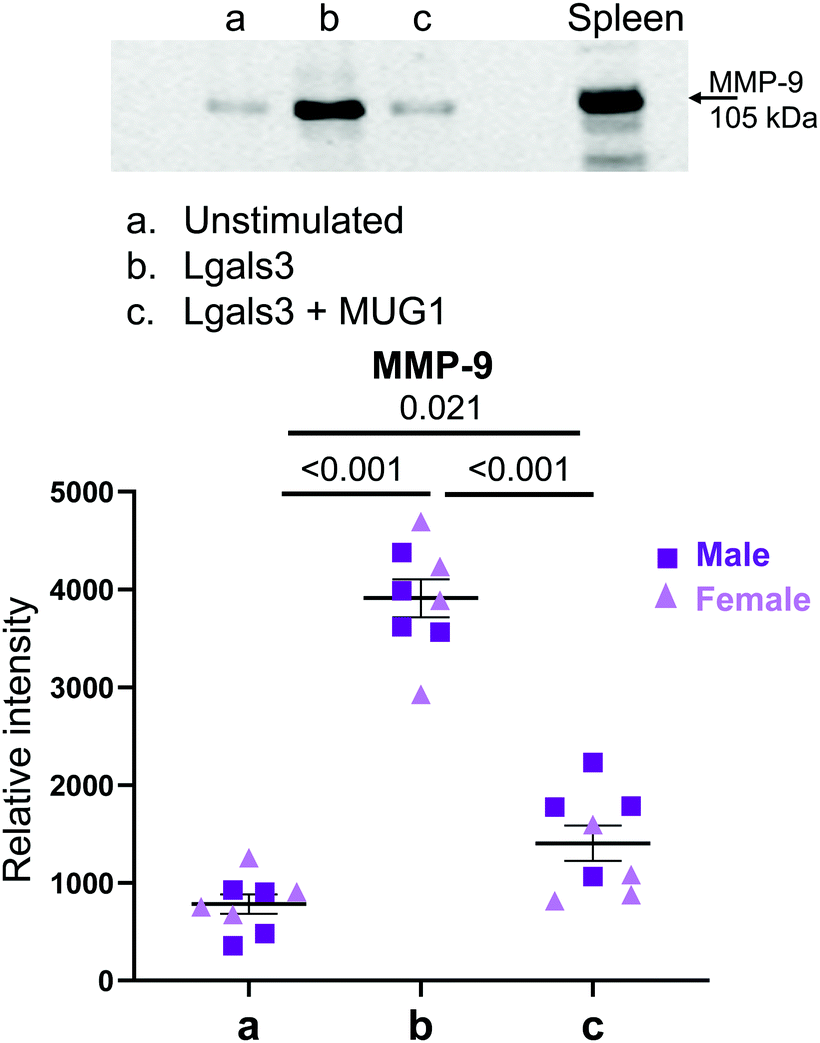 | ||
| Fig. 6 MUG1 inhibited LGALS3 induced neutrophil degranulation. By immunoblotting for MMP-9 release into the neutrophil conditioned media, MUG1 inhibited neutrophil degranulation induced by Lgals3 by 64%. Sample sizes were n = 8, 4M/4F each paired group; analysis done by one-way ANOVA with Student Newman–Keuls post-hoc test. | ||
In vivo, the MUG1 to Lgals3 ratio peaked at MI D1 and positively correlated with infarct wall thickness
To evaluate MUG1 and Lgals3 in the LV infarct, we measured the expression time course by immunoblotting for samples in a previously published data and tissue bank. Cardiac physiology for the MI groups is shown in Fig. 7A. Representative immunoblots are shown in Fig. 7B. MUG1 protein expression was elevated at MI D1 and peaked at MI D3 (6-fold increased vs. D0), returning to D0 values by MI D7 (Fig. 7C left). Lgals3 protein expression peaked at MI D7 (634-fold increase vs. D0). MUG1 to Lgals3 ratio peaked at MI D1 (Fig. 7C middle), and infarct wall thickness at all MI times correlated positively with MUG1 to Lgals3 ratio (r = 0.60, p = 0.02; Fig. 7C right). This indicates that macrophage MUG1 expression is an endogenous protective mechanism to limit neutrophil degranulation and excessive infarct wall thinning early in MI.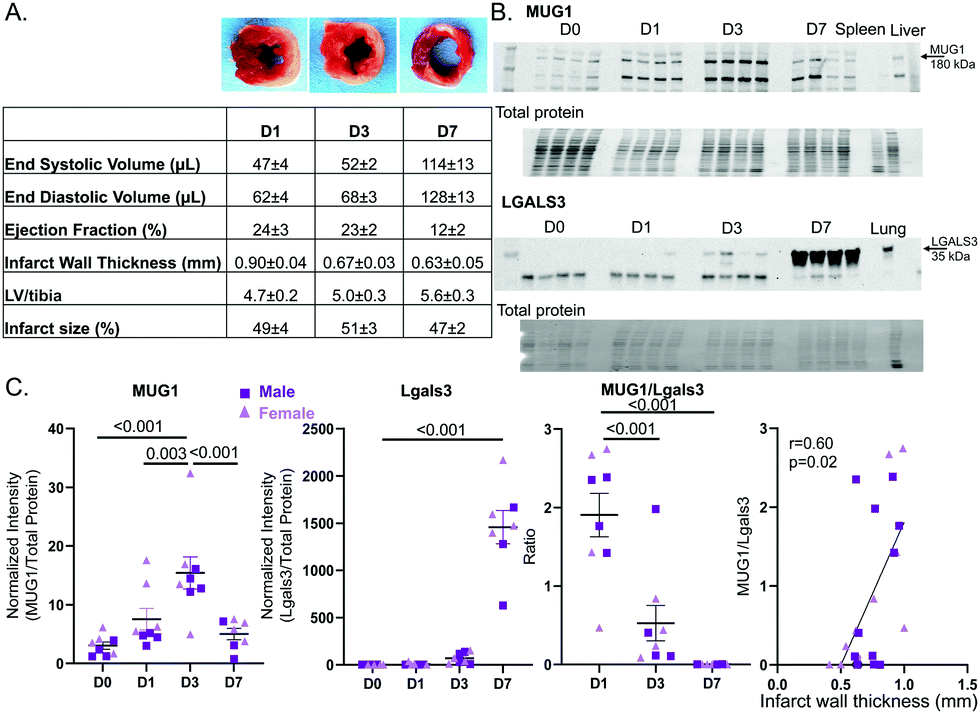 | ||
| Fig. 7 In the LV infarct region, MUG1 increased at MI D1-D3 and tracked with preserved LV wall thickness, while LGALS3 peaked at MI D7. (A) Cardiac physiology after MI at days 1, 3, and 7, including representative infarct size images for each day. (B) Representative MUG1 immunoblot of LV infarct samples (top). Representative Lgals3 immunoblot of LV infarct samples (bottom). (C) MUG1 increased after MI, peaking at MI D3 (6-fold increase vs. D0) and returned to D0 levels by MI D7. Lgals3 increased from MI D3 and peaked at MI D7 of MI (634-fold compared to D0). The MUG1 to Lgals3 ratio peaked at MI D1, and the MUG1 to Lglas3 ratio positively correlated with infarct wall thickness. Sample sizes were n = 7–8, 3–4M/4F per group; analysis done by one way ANOVA with Student Newman–Keuls post-hoc test. | ||
Discussion
The goal of this study was to explore the hypothesis that macrophages communicate to neutrophils at MI D1 to regulate degranulation during the pro-inflammatory phase. The striking findings from this study were: (1) MUG1 was the highest ranked protein secreted by the MI D1 macrophage; (2) The MI D1 macrophage secretome attenuated neutrophil degranulation induced by PMA, an effect reversed by addition of a MUG1 blocking antibody; (3) MUG1 attenuated IL-1β induced degranulation, while Lgals3 induced degranulation, and the effects of both IL-1β and Lgasl3 were blocked by MUG1; and (4) the MUG1 to Lgals3 ratio in vivo linked to infarct wall thickness. Combined, our results revealed that D1 MI macrophages regulate neutrophil degranulation by secreting MUG1 as an anti-degranulatory protein and Lgals3 as a pro-degranulatory protein. At MI D1, the balance favors MUG1 activity to contain and prevent excessive neutrophil degranulation early in MI when inflammation is high.Excessive neutrophil activation and degranulation can exacerbate inflammation after MI and worsen cardiac dysfunction; at the same time, neutrophil degranulation early in MI is essential for initiation of infarct wound healing.37,38 Increased neutrophil activity can induce wall thinning after MI and lead to LV aneurysms and death due to LV rupture.35,39 LV wall thinning after MI peaks at D1, which occurs due to the loss of cardiomyocytes resulting from ischemia. Neutrophils remove the necrotic debris and lay ground for ECM deposition to form the scar by releasing ECM degrading enzymes from its granules.12 Our study sheds light on an endogenous mechanism to regulate excessive neutrophil activity through macrophage to neutrophil communication. Macrophages releasing MUG1 to limit neutrophil degranulation could potentially be a mechanism to initiate resolution of inflammation and tissue repair.
MUG1 was the highest ranked upregulated protein secreted by D1 macrophages and USP48 was the highest ranked downregulated protein. MUG1 is a 190 kDa protein characterized as a protease inhibitor belonging to the α2-macroglobulin family.40 USP48 is a 119 kDa deubiquitinating protease which is a part of ubiquitin carboxyl-terminal hydroxylase family.41 The D1 macrophage secretome reflected upregulated IL-17 signaling. Similarly, necroptosis and endocytosis enrichment suggests a crucial role of macrophages in early wound healing phase and direct regulation of necrotic debris removal.42 Regulation of the actin-cytoskeleton by macrophages indicates its role in neutrophil degranulation.43 Upregulated proteins are mainly associated with inflammatory processes as per the enrichment analysis. PPAR signaling pathway being the most enriched pathway that was downregulated further established a central role of macrophages in MI wound healing, as activation of PPAR signaling has been implicated in ischemia reperfusion repair and reduction of infarct size.44–46 PPARγ activation in neutrophils turns off NF-κB activation and stimulates LXR/RXR signaling to initiate resolution.47 Downregulated proteins are mainly associated with cellular repair processes according to the enrichment analysis. USP48 downregulation is associated with histone 2A downregulation. USP48 repression induces cell survival.41
As we focused on macrophage to neutrophil communication through its major upregulated protein in the secretome, we observed that neutrophil degranulation was one of the components modulated by D1 MI macrophages. Macrophage secretome treatment downregulated most of the cytokines released, and cytokine-cytokine receptor interaction was the major enriched downregulated pathway in neutrophils stimulated by the MI D1 macrophage secretome, followed by TNF signaling and AGE-RAGE signaling. This showed that D1 macrophages secrete factors to limit major inflammatory pathways in neutrophils. The macrophage secretome could be the negative feedback required to dwindle neutrophil numbers after D1.6 With addition of MUG1 blocking antibody to the macrophage secretome or PMA, we see upregulation of cytokine release indicating MUG1 actively regulated neutrophil activity. This was further highlighted by the upregulation of cytokine-cytokine receptor interaction, TNF signaling, and AGE-RAGE signaling pathways with MUG1 blocking antibody treatment.
Our study is the first to our knowledge to demonstrate MUG1 release from macrophages, and the literature available on MUG1 is meager. Murine MUG1 is a homologue of human α2-macroglobulin, another protease inhibitor and a major component of human plasma.48 Both mouse MUG1 and mouse α2-macroglobulin inhibit thrombin, plasmin, and pancreatic elastase. In addition, mouse α2-macroglobulin inhibits clotting factor Xa, plasma kallikrein, submaxillary gland trypsin-like proteinase, and neutrophil elastase.49 Before this study, MUG1 was thought to act primarily in the plasma as a protease inhibitor. Interestingly, MUG1 deficient mice also show elevated plasma levels of TNFα and Interferon gamma (IFNγ) indicating its role in inflammation.50,51 Here, we show that a major action of MUG1 secreted by macrophages was to blunt neutrophil degranulation, indicated by reduced secretion of MPO, MMP-9, and NGAL. Cytokines downregulated by MUG1 matched similar pathway enrichment as shown by those downregulated by the macrophage secretome, with chemokine signaling and cytokine-cytokine receptor interaction as most enriched pathways. Our study shows enrichment for downregulation of MAPK and PI3K-AKT signaling pathway, and further work on the signaling paradigm affected by MUG1 is warranted.
Lgals3 was the second protein we focused on in this study as it was one of the pro-inflammatory genes upregulated by macrophages at MI D1 and is a secreted protein. Lgals3 alone stimulated release of degranulation components (i.e., MPO, MMP-9 and NGAL). The most enriched pathway was leukocyte transendothelial migration. Lgals3 induces β1 integrin mediated cellular adhesion and migration by increasing binding to fibronectin, laminin, and collagen 1.52 Lgals3 is a lectin of approximately 35 kDa identified as a biomarker of inflammation and fibrosis.53–56 In the 2013 Guidelines from the American College of Cardiology Foundation/American Heart Association, Lgals3 was recognized as a predictive biomarker for hospitalization and death in heart failure patients.57,58 Activated macrophages are the major source of Lgals3,54 and Lgals3 is highly expressed when monocytes differentiate to macrophages.59 We have previously shown that neutrophils also secrete Lgals3, and MI neutrophils show their peak Lgals3 expression at MI D7- a time consistent with our immunoblotting results of the infarct tissue.60 Most of the literature available has focused on the role of Lgals3 in fibrosis, fibroblast activation, and proliferation.61,62 Our study uniquely identifies the role of Lgals3 in neutrophil degranulation.
Time course analysis of MUG1 and Lgals3 in the LV infarct showed that both proteins increase after MI, peaking at different time points in MI. MUG1 was early in MI and Lgals3 later. The highest ratio of MUG1 to Lgals3 occurred at MI D1, suggesting MUG1 activity culminates as pro-inflammation peaks in the LV. This is in line with our findings that MUG1 activity required pro-inflammatory stimuli, while Lgals3 action does not require pro-inflammation (and Lgals3 peaked when inflammation was subsiding in the infarct region). While macrophage production of Lgals3 mRNA was elevated at MI D1, protein expression in the entire LV infarct region was not increased until MI D3. This could mean macrophages are the major contributor to Lgals3 levels at D1, while neutrophils and fibroblasts are major contributors at D7.60,63 As both MUG1 and Lgals3 were macrophage secreted proteins, we evaluated the possibility that the two shared signaling pathways as a built-in mechanism to regulate neutrophil degranulation. The decrease in MMP-9 release seen with combined stimulation of bone marrow derived neutrophils with MUG1 and Lgals3 revealed that MUG1 was an upstream regulator of Lgals3. Lgals3 binds to various protease inhibitors, including α2-macroglobulin.36 Therefore, there is a potential for MUG1 inhibition of Lgals3 induced degranulation to occur through Lgals3 binding to MUG1.36
Lgals3 is proteolyzed by both MMP-2 and MMP-9 to activate it.64 We have previously revealed a similar mechanism for MMP-9 regulation of fibronectin.60 Lgals3 degradation by MMP-9 further increases MMP-9 release indicating a positive feedback response. This is also consistent with the higher MMP-9 concentration at MI D1 and D3 when Lgals3 degradation is also higher compared to D7.60,65 Lgals3 null mice show reduced macrophage infiltration and early polarization to M2 like phenotype worsening cardiac physiology after MI. Lgals3 null mice also show increased neutrophil infiltration and reduced apoptosis.66,67 As full length Lgals3 is downstream of MMP-9 and Lgals3 activated by MMP-9, this indicates that Lgals3 role(s) in MI wound healing are dependent on quality (degradation status) as much as quantity. Careful tapping of the MUG1-Lgals3 signaling axis for optimization of neutrophil degranulation after MI could be of potential means to optimize the MI response.
Since direct targeting of neutrophils numbers and activity has negative impacts in MI resolution, an optimal regulation of neutrophil degranulation may be achieved by focusing on neutrophil kinetics instead. Neutrophil specific proteins such as S100A8/A9 are potential targets, and this study shows a new and unique way to modulate neutrophils i.e. through macrophages.35 While rupture is not as common in humans with the advent of reperfusion, excessive wall thinning can lead to LV aneurysms and dilation.68,69 Hence, treatments that limit excessive wall thinning could potentially prove useful in preventing poor outcomes. Exogenous MUG1 treatment following MI could mitigate excessive wall thinning after MI (Fig. 8).
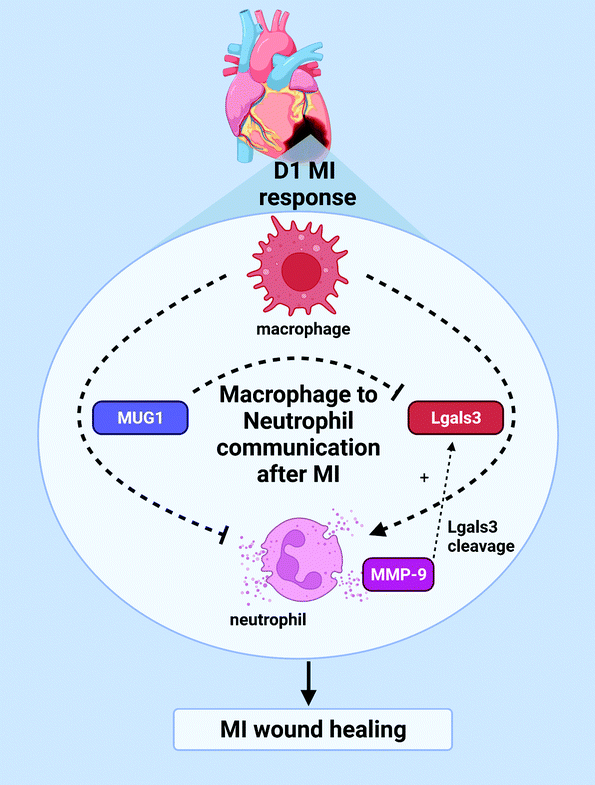 | ||
| Fig. 8 Macrophage coordination of neutrophil physiology. Macrophages inhibit neutrophil degranulation by secretion of MUG1 and induce neutrophil degranulation by secretion of Lgals3 to coordinate LV wall thinning and early MI wound healing. Release of MMP-9 provides an additional feedback to proteolyze and activate Lgals3. | ||
Materials and methods
Animal use
All animal procedures were performed according to the Guide for the Care and Use of Laboratory Animals, and all protocols were pre-approved by the Institutional Animal Care and Use Committee at the University of Mississippi Medical Center or the University of Nebraska Medical Center.70 C57BL/6J wild-type mice of both sexes (3–6 months old) were either purchased from Jackson Laboratory or in house bred and housed in the animal facility. All mice were maintained together in the same room, under a 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12 h light–dark cycle with ad libitum access to standard mouse chow and water. For the time course of MI samples, the tissue was obtained from the mouse heart attack research tool (mHART) tissue bank.35,71 All MI samples selected had undergone cardiac ligation surgery and cardiac physiology measurements by echocardiography as per the published guidelines.72–74 Infarct size was calculated using adobe photoshop as the percentage of the area infarcted over the total area from the images of MI hearts stained by 2,3,5-triphenyl tetrazolium chloride (TTC).
:12 h light–dark cycle with ad libitum access to standard mouse chow and water. For the time course of MI samples, the tissue was obtained from the mouse heart attack research tool (mHART) tissue bank.35,71 All MI samples selected had undergone cardiac ligation surgery and cardiac physiology measurements by echocardiography as per the published guidelines.72–74 Infarct size was calculated using adobe photoshop as the percentage of the area infarcted over the total area from the images of MI hearts stained by 2,3,5-triphenyl tetrazolium chloride (TTC).
Mapping the MI D1 macrophage secretome
We evaluated the MI D1 macrophage secretome from male mice (C57BL/6J, ages 3–6 months) subjected to coronary artery ligation surgery as described previously.7,72 Macrophages from no MI D0 were used as the negative control, and sample sizes were n = 4 pooled sets per group. Transcriptomics was performed in the cell pellets from the same paired cell sets, and this dataset has previously been evaluated and published.7 Macrophage secretomes were collected by incubating freshly isolated cells from the infarct region for 2 h at 37 °C in RPMI 1640 media (Gibco #11-875-093) supplemented with 1% penicillin/streptomycin (Thermo, #15140-122). The secretome was evaluated by mass spectrometry using the Q Exactive (ThermoFisher, Waltham, MA) as previously described.75 The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD011780 and presented in Table S1 (ESI†).76 Volcano plotting was used to rank individual proteins based on p value and fold change expression.Bone marrow derived neutrophil isolation and stimulation
Bone marrow derived neutrophils were isolated from male and female mice using the autoMACS Pro Separator (Miltenyi Biotec) with anti-Ly6G microbeads (Miltenyi Biotech; #130-120-337) as previously described.77 The bone marrow from the femur and tibia was flushed using a 26-guage needle and 10 ml syringe filled with RPMI 1640 media (Gibco) supplemented with 1% penicillin/streptomycin (Thermo) and 2 mM EDTA. Single cell suspension was obtained by passing the cells over 30 μm pre-separation filters (Miltenyi Biotec, #130-041-407). Red blood cell lysis buffer (Miltenyi Biotec, #130-094-183) was added to the suspension to lyse erythrocytes. The single cell suspension was centrifuged at 400 x g for 10 min, and the cell pellet was resuspended in PEB buffer (PBS containing 2 mM EDTA and 0.5% BSA). Neutrophils were labeled using magnetic anti-Ly-6G MicroBeads (Miltenyi Biotec, #130-120-337) by incubation for 10 min at 4 °C and sorted using a AutoMACS Pro Separator. Cells were counted using a hemocytometer and plated at 1 × 106 cells ml−1. Neutrophils were stimulated for 15 min at 37 °C in RPMI 1640 media (Gibco) with 1% antibiotics (Thermo).6,78For the first set of experiments, the groups were: (a) unstimulated, (b) phorbol myristate acetate (PMA, 2 nM), (c) PMA + MI D1 macrophage secretome (10% by volume), (d) PMA + MI D1 macrophage secretome + murinoglobulin-1 blocking antibody (200 ng ml−1; My BioSource, #MBS1496515, sodium azide free). This data is presented in Table S2 (ESI†). For the second set, the groups were: (a) unstimulated, (b) IL-1β (Rnd Systems, #401-ML, 200 ng ml−1) as an MI relevant positive control stimulus, (c) MUG1 (MyBioSource, #MBS1035048, 500 ng ml−1), and d) IL-1β + MUG1. Data is presented in Table S3 (ESI†). For the third set, the groups were: (a) unstimulated, (b) IL-1β (200 ng ml−1), (c) Lgals3 (RnD Systems, #1197-GA, 200 ng ml−1), and (d) IL-1β + Lgals3. Data is presented in Table S4 (ESI†). For the fourth set, the groups were: (1) unstimulated, (2) Lgals3, and (3) Lgals3 + MUG1. Doses were selected as being <50% of the recommended maximum limit dose, to avoid cellular toxicity.79 Following stimulation, the cells were centrifuged at 800 g for 8 min to separate secretome (supernatant) and cell pellet, which were separately snap frozen at −80 °C and stored until use.
Cytokine array
The Proteome Profiler Mouse XL Cytokine Array Kit (ARY028; RnD Systems) containing 111 mouse proteins was used to analyze the secretome according to manufacturer instructions.80 Images were captured using the iBright FL1000 imaging system (Thermo Fisher) and chemiluminescence intensity was quantified using HLimage++ (Western vision) software. Duplicate technical replicates were averaged for each sample and reported as relative intensity.Immunoblotting
Immunoblotting was performed according to the published guidelines.81 For immunoblotting of the neutrophil secretome, samples were volume loaded (10 μL) onto 4–12% Criterion XT Bis-Tris precast gel (Bio-Rad) and transferred to nitrocellulose membranes using the Trans-Blot Turbo Transfer Pack (Bio-Rad). MMP-9 antibody (EMD Millipore, #Ab19016 dilution:1:1000) was used as the primary antibody, and goat anti-rabbit IgG (Vector Laboratories, #PI1000 dilution:1:1000) was used as secondary antibody. Because each group had the same number of cells in the same total volume of media, the secretome samples were volume loaded. The data were plotted as relative chemiluminescence intensity.
For immunoblotting of LV infarct for the MI time-course of MUG1 and Lgals3, total protein (10 μg) was run on 4–12% Criterion XT Bis-Tris precast gels (Bio-Rad) and transferred to nitrocellulose membranes using the Trans-Blot Turbo Transfer Pack (Bio-Rad). Membranes were stained with Peirce Reversible Protein Stain Kit (Thermo Fisher) and images were taken for total membrane protein quantification. Rabbit anti-MUG1 (MyBiosource, #MBS1496515, dilution:1:1000) and rat anti-mouse Lgals3 (Invitrogen, #MAI-940, dilution:1:1000) were used as primary antibodies. The corresponding secondary antibodies used were goat anti-rabbit IgG (Vector Laboratories, #PI1000 dilution:1:1000) and horse anti-mouse (Vector Laboratories, #PI2000; dilution:1:1000). Liver was used a positive control for MUG1, and lung was used as a positive control for Lgals3. Chemiluminescent images were captured using the iBright FL1000 imaging system (Thermo Fisher) and quantified using iBright analysis software 4.0.0. The blots were normalized to the total membrane protein, and the data plotted as normalized relative intensity.
Statistics and bioinformatics analysis
Statistical analyses were performed using GraphPad Prism 9 according to the guidelines outlined in Statistical Considerations in Reporting Cardiovascular Research.82 Data are reported as mean ± SEM for group comparisons. One-way ANOVA was used to compare more than 2 groups with a Newman-Keuls post-hoc test for multiple comparisons. A paired Students t-test was used to compare two groups from the paired stimulus experiments. Two-way ANOVA was used to compare sex differences and treatment groups. For data visualization by volcano plotting, the data were autoscaled and run in Metaboanalyst 5.0 (https://www.metaboanalyst.ca/).83 Partial least squares discriminant analysis (PLS-DA) was performed in Metaboanalyst to show the shift in proteome from D0 to MI D1. Enrichment analysis was done using Enrichr, a bioinformatics tool developed by Ma’ayan Laboratory for a comprehensive multi-omics evaluation of data.84–86 GO and KEGG pathways were used for enriched processes and pathways. A p value <0.05 was considered statistically significant.Conclusions
In conclusion, this study revealed that MI D1 macrophages secrete various factors to positively and negatively regulate neutrophil degranulation. MUG1 secreted by macrophages inhibited, while Lgals3 secreted by macrophages induced, neutrophil degranulation. The key to understanding their overall in vivo balance lies in the temporal expression of these two proteins in the LV infarct. MUG1 was elevated at MI D1 and D3 as an endogenous mechanism to temper neutrophil degranulation. Therefore, effective optimization of MUG1 to limit excessive neutrophil degranulation could prevent adverse LV remodeling after MI.Data availability
All data is available in Tables S1–S4 (ESI†). Data from Table S1 (ESI†) has previously been uploaded to PRIDE with identifier PXD011780.Author contributions
Conceptualization: U. C. and M. L. L. data curation: U. C., M. J. D., W. J. K., S. R. K., J. R. R. P., and E. F. formal analysis: U. C. funding acquisition: M. L. L. resources: M. L. L. supervision: M. L. L. Visualization: U. C. and M. L. L. Writing – original draft: U. C., M. J. D., and M. L. L. Writing – review & editing: U. C., M. J. D., W. J. K., S. R. K, J. R. R. P, E. F., L. M. C., M. B. A., and M. L. L.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr Lindsey is a Stokes-Shackleford Professor at UNMC. We acknowledge funding from National Institutes of Health under Award Numbers HL137319, U54GM115458, and F31HL156315, and from the Biomedical Laboratory Research and Development Service of the Veterans Affairs Office of Research and Development under Award Numbers 5I01BX000505; the American Cancer Society Research Scholar Grant under award number RSG-19-127-01-CSM; and the Swedish Society for Medical Research under award number P19-0144. The content is solely the responsibility of the authors and does not necessarily represent the official views of any of the funding agencies. All authors have reviewed and approved the article.Notes and references
- M. A. Tenkorang, U. Chalise, I. Daseke, J. Michael, S. R. Konfrst and M. L. Lindsey, Biochem. Soc. Trans., 2019, 47, 1679–1687 CrossRef CAS PubMed.
- N. G. Frangogiannis, C. W. Smith and M. L. Entman, Cardiovasc. Res., 2002, 53, 31–47 CrossRef CAS PubMed.
- S. D. Prabhu and N. G. Frangogiannis, Circ. Res., 2016, 119, 91–112 CrossRef CAS PubMed.
- M. J. Daseke, 2nd, M. A. A. Tenkorang-Impraim, Y. Ma, U. Chalise, S. R. Konfrst, M. R. Garrett, K. Y. DeLeon-Pennell and M. L. Lindsey, J. Mol. Cell. Cardiol., 2020, 145, 112–121 CrossRef PubMed.
- M. Becirovic-Agic, U. Chalise, M. Jung, J. R. Rodriguez-Paar, S. R. Konfrst, E. R. Flynn, J. D. Salomon, M. E. Hall and M. L. Lindsey, Am. J. Physiol.: Heart Circ. Physiol., 2022 DOI:10.1152/ajpheart.00612.2021.
- Y. Ma, A. Yabluchanskiy, R. P. Iyer, P. L. Cannon, E. R. Flynn, M. Jung, J. Henry, C. A. Cates, K. Y. Deleon-Pennell and M. L. Lindsey, Cardiovasc. Res., 2016, 110, 51–61 CrossRef CAS PubMed.
- A. J. Mouton, K. Y. DeLeon-Pennell, O. J. R. Gonzalez, E. R. Flynn, T. C. Freeman, J. J. Saucerman, M. R. Garrett, Y. Ma, R. Harmancey and M. L. Lindsey, Basic Res. Cardiol., 2018, 113, 1–18 CrossRef CAS PubMed.
- D. M. Calcagno, C. Zhang, A. Toomu, K. Huang, V. K. Ninh, S. Miyamoto, A. D. Aguirre, Z. Fu, J. Heller Brown and K. R. King, J. Am. Heart Assoc., 2021, 10, e019019 CAS.
- E. Vafadarnejad, G. Rizzo, L. Krampert, P. Arampatzi, A.-P. Arias-Loza, Y. Nazzal, A. Rizakou, T. Knochenhauer, S. R. Bandi and V. A. Nugroho, Circ. Res., 2020, 127, e232–e249 CrossRef CAS PubMed.
- N. G. Frangogiannis, Nat. Rev. Cardiol., 2014, 11, 255–265 CrossRef CAS PubMed.
- M. Becirovic-Agic, U. Chalise, M. J. Daseke, S. Konfrst, J. D. Salomon, P. K. Mishra and M. L. Lindsey, Biomolecules, 2021, 11, 491 CrossRef CAS PubMed.
- M. J. Daseke II, U. Chalise, M. Becirovic-Agic, J. D. Salomon, L. M. Cook, A. J. Case and M. L. Lindsey, Cell. Signalling, 2021, 77, 109816 CrossRef PubMed.
- B. U. Goldmann, V. Rudolph, T. K. Rudolph, A.-K. Holle, M. Hillebrandt, T. Meinertz and S. Baldus, Free Radicals Biol. Med., 2009, 47, 79–83 CrossRef CAS PubMed.
- M. Mauler, N. Herr, C. Schoenichen, T. Witsch, T. Marchini, C. Härdtner, C. Koentges, K. Kienle, V. Ollivier and M. Schell, Circulation, 2019, 139, 918–931 CrossRef CAS PubMed.
- A. M. Arruda-Olson, G. S. Reeder, M. R. Bell, S. A. Weston and V. L. Roger, Circulation, 2009, 2, 656–662 Search PubMed.
- G. V. Halade, Y.-F. Jin and M. L. Lindsey, Pharmacol. Ther., 2013, 139, 32–40 CrossRef CAS PubMed.
- J. Hansson, R. S. Vasan, J. Ärnlöv, E. Ingelsson, L. Lind, A. Larsson, K. Michaëlsson and J. Sundström, PLoS One, 2011, 6, e16185 CrossRef CAS PubMed.
- T. J. Mocatta, A. P. Pilbrow, V. A. Cameron, R. Senthilmohan, C. M. Frampton, A. M. Richards and C. C. Winterbourn, J. Am. Coll. Cardiol., 2007, 49, 1993–2000 CrossRef CAS PubMed.
- A. D. Shah, S. Denaxas, O. Nicholas, A. D. Hingorani and H. Hemingway, J. Am. Coll. Cardiol., 2017, 69, 1160–1169 CrossRef PubMed.
- M. Horckmans, L. Ring, J. Duchene, D. Santovito, M. J. Schloss, M. Drechsler, C. Weber, O. Soehnlein and S. Steffens, Eur. Heart J., 2017, 38, 187–197 CAS.
- H. Hammerman, R. A. Kloner, S. Hale, F. J. Schoen and E. Braunwald, Circulation, 1983, 68, 446–452 CrossRef CAS PubMed.
- S. Liu, D. T. Ngo, C. R. Chong, A. T. Amarasekera, N. E. Procter, G. Licari, R. F. Dautov, S. Stewart, Y. Y. Chirkov and J. D. Horowitz, Eur. J. Heart Failure, 2015, 17, 475–483 CrossRef CAS PubMed.
- A. L. Peterson, G. Siddiqui, E. K. Sloan and D. J. Creek, Mol. Omics, 2021, 17(4), 583–595 RSC.
- A. Hanna, A. V. Shinde and N. G. Frangogiannis, Am. J. Physiol.: Heart Circ. Physiol., 2020, 319, H948–H964 CrossRef CAS PubMed.
- S. Frantz, U. Hofmann, D. Fraccarollo, A. Schäfer, S. Kranepuhl, I. Hagedorn, B. Nieswandt, M. Nahrendorf, H. Wagner and B. Bayer, FASEB J., 2013, 27, 871–881 CrossRef CAS PubMed.
- C. Peet, A. Ivetic, D. I. Bromage and A. M. Shah, Cardiovasc. Res., 2020, 116, 1101–1112 CrossRef CAS PubMed.
- V. Frodermann and M. Nahrendorf, Eur. Heart J., 2017, 38, 198–200 CAS.
- S.-L. Puhl and S. Steffens, Front. Cardiovasc. Med., 2019, 6, 25 CrossRef CAS PubMed.
- J. M. Lambert, E. F. Lopez and M. L. Lindsey, Int. J. Cardiol., 2008, 130, 147–158 CrossRef PubMed.
- M. B.-A. Upendra Chalise and M. L. Lindsey, Curr. Opin. Physiol., 2022, 100485 Search PubMed.
- S. Sato, N. Ouellet, I. Pelletier, M. Simard, A. Rancourt and M. G. Bergeron, J. Immunol., 2002, 168, 1813–1822 CrossRef CAS PubMed.
- A. Karlsson, K. Christenson, M. Matlak, Å. Björstad, K. L. Brown, E. Telemo, E. Salomonsson, H. Leffler and J. Bylund, Glycobiology, 2009, 19, 16–20 CrossRef CAS PubMed.
- N. C. Henderson and T. Sethi, Immunol. Rev., 2009, 230, 160–171 CrossRef CAS PubMed.
- G. C. Fernández, J. M. Ilarregui, C. J. Rubel, M. A. Toscano, S. A. Gómez, M. Beigier Bompadre, M. A. Isturiz, G. A. Rabinovich and M. S. Palermo, Glycobiology, 2005, 15, 519–527 CrossRef PubMed.
- U. Chalise, M. Becirovic-Agic, M. J. Daseke II, S. R. Konfrst, J. R. Rodriguez-Paar, D. Feng, J. D. Salomon, D. R. Anderson, L. M. Cook and M. L. Lindsey, Am. J. Physiol.: Heart Circ. Physiol., 2021, 322(2), H145–H155 CrossRef PubMed.
- C. Cederfur, E. Salomonsson, J. Nilsson, A. Halim, C. T. Öberg, G. Larson, U. J. Nilsson and H. Leffler, Glycobiology, 2008, 18, 384–394 CrossRef CAS PubMed.
- V. Kain and G. V. Halade, Pharmacol. Ther., 2020, 205, 107424 CrossRef CAS PubMed.
- O. Soehnlein, S. Steffens, A. Hidalgo and C. Weber, Nat. Rev. Immunol., 2017, 17, 248–261 CrossRef CAS PubMed.
- Y. Amano, M. Takayama and S. Kumita, J. Comput. Assist. Tomogr., 2008, 32, 259–264 CrossRef PubMed.
- L. Overbergh, S. Torrekens, F. Van Leuven and H. Van den Berghe, J. Biol. Chem., 1991, 266, 16903–16910 CrossRef CAS.
- M. Uckelmann, R. M. Densham, R. Baas, H. H. Winterwerp, A. Fish, T. K. Sixma and J. R. Morris, Nat. Commun., 2018, 9, 1–16 CrossRef CAS PubMed.
- C. Piamsiri, C. Maneechote, N. Siri-Angkul, S. C. Chattipakorn and N. Chattipakorn, J. Biomed. Sci., 2021, 28, 1–13 CrossRef PubMed.
- P. Lacy, Allergy, Asthma, Clin. Immunol., 2006, 2, 1–11 Search PubMed.
- S. Wang, E. J. Dougherty and R. L. Danner, Pharmacol. Res., 2016, 111, 76–85 CrossRef CAS PubMed.
- Y.-B. Zhao, J. Zhao, L.-J. Zhang, R.-G. Shan, Z.-Z. Sun, K. Wang, J.-Q. Chen and J.-X. Mu, Biomed. Pharmacother., 2019, 113, 108697 CrossRef PubMed.
- C. Lotz, M. Lazariotto, A. Redel, T. M. Smul, J. Stumpner, C. Blomeyer, T. Tischer-Zeitz, J. Schmidt, J. Pociej and N. Roewer, Exp. Biol. Med., 2011, 236, 113–122 CrossRef CAS PubMed.
- K. Y. DeLeon-Pennell, A. J. Mouton, O. K. Ero, Y. Ma, R. P. Iyer, E. R. Flynn, I. Espinoza, S. K. Musani, R. S. Vasan and M. E. Hall, Basic Res. Cardiol., 2018, 113, 1–19 CrossRef CAS PubMed.
- W. Borth, FASEB J., 1992, 6, 3345–3353 CrossRef CAS PubMed.
- K. Abe, K. Yamamoto and H. Sinohara, J. Biochem., 1989, 106, 564–568 CrossRef CAS PubMed.
- L. Umans, L. Serneels, L. Overbergh, L. Stas and F. Van Leuven, Am. J. Pathol., 1999, 155, 983–993 CrossRef CAS.
- K. Krause, F. Azouz, E. Nakano, V. R. Nerurkar and M. Kumar, Front. Microbiol., 2019, 10, 259 CrossRef PubMed.
- C. Margadant, I. van den Bout, A. L. van Boxtel, V. L. Thijssen and A. Sonnenberg, J. Biol. Chem., 2012, 287, 44684–44693 CrossRef CAS PubMed.
- M. Li, Y. Yuan, K. Guo, Y. Lao, X. Huang and L. Feng, Am. J. Cardiovasc. Drugs, 2020, 20, 333–342 CrossRef PubMed.
- O. Frunza, I. Russo, A. Saxena, A. V. Shinde, C. Humeres, W. Hanif, V. Rai, Y. Su and N. G. Frangogiannis, Am. J. Pathol., 2016, 186, 1114–1127 CrossRef CAS PubMed.
- C. Besler, D. Lang, D. Urban, K.-P. Rommel, M. von Roeder, K. Fengler, S. Blazek, R. Kandolf, K. Klingel and H. Thiele, Circ.: Heart Failure, 2017, 10, e003804 CAS.
- R. I. Lala, M. Puschita, D. Darabantiu and L. Pilat, Acta Cardiol., 2015, 70, 323–331 CrossRef PubMed.
- C. W. Yancy, M. Jessup, B. Bozkurt, J. Butler, D. E. Casey, M. H. Drazner, G. C. Fonarow, S. A. Geraci, T. Horwich and J. L. Januzzi, J. Am. Coll. Cardiol., 2013, 62, e147–e239 CrossRef PubMed.
- R. Asleh, M. Enriquez-Sarano, A. S. Jaffe, S. M. Manemann, S. A. Weston, R. Jiang and V. L. Roger, J. Am. Coll. Cardiol., 2019, 73, 2286–2295 CrossRef CAS PubMed.
- F.-T. Liu, D. K. Hsu, R. I. Zuberi, I. Kuwabara, E. Y. Chi and W. R. Henderson Jr, Am. J. Pathol., 1995, 147, 1016 CAS.
- M. J. Daseke, 2nd, F. M. Valerio, W. J. Kalusche, Y. Ma, K. Y. DeLeon-Pennell and M. L. Lindsey, Basic Res. Cardiol., 2019, 114, 37 CrossRef PubMed.
- N. C. Henderson, A. C. Mackinnon, S. L. Farnworth, T. Kipari, C. Haslett, J. P. Iredale, F.-T. Liu, J. Hughes and T. Sethi, Am. J. Pathol., 2008, 172, 288–298 CrossRef CAS PubMed.
- N. C. Henderson, A. C. Mackinnon, S. L. Farnworth, F. Poirier, F. P. Russo, J. P. Iredale, C. Haslett, K. J. Simpson and T. Sethi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5060–5065 CrossRef CAS PubMed.
- A. Filer, M. Bik, G. N. Parsonage, J. Fitton, E. Trebilcock, K. Howlett, M. Cook, K. Raza, D. L. Simmons and A. M. Thomas, Arthritis Rheum., 2009, 60, 1604–1614 CrossRef CAS PubMed.
- J. Ochieng, R. Fridman, P. Nangia-Makker, D. E. Kleiner, L. A. Liotta, W. G. Stetler-Stevenson and A. Raz, Biochemistry, 1994, 33, 14109–14114 CrossRef CAS PubMed.
- N. Ortega, D. J. Behonick, C. Colnot, D. N. Cooper and Z. Werb, Mol. Biol. Cell, 2005, 16, 3028–3039 CrossRef CAS PubMed.
- R. D. Wright, P. R. Souza, M. B. Flak, P. Thedchanamoorthy, L. V. Norling and D. Cooper, J. Leukocyte Biol., 2017, 101, 717–726 CrossRef CAS PubMed.
- P. Cassaglia, F. Penas, C. Betazza, F. F. Estevez, V. Miksztowicz, N. M. Naya, M. C. Llamosas, S. N. Truant, L. Wilensky and V. Volberg, Am. J. Pathol., 2020, 190, 1789–1800 CrossRef CAS PubMed.
- M. L. Lindsey, L. E. d. C. Brás, K. Y. DeLeon-Pennell, N. G. Frangogiannis, G. V. Halade, C. C. O’Meara, F. G. Spinale, Z. Kassiri, J. A. Kirk, P. Kleinbongard, C. M. Ripplinger and K. R. Brunt, Am. J. Physiol.: Heart Circ. Physiol., 2021, 321, H208–H213 CrossRef CAS PubMed.
- V. L. Roger, Circ. Res., 2013, 113, 646–659 CrossRef CAS PubMed.
- U. Albus, Lab. Anim., 2012, 46(3), 267–268 CrossRef CAS.
- K. Y. DeLeon-Pennell, R. P. Iyer, Y. Ma, A. Yabluchanskiy, R. Zamilpa, Y. A. Chiao, P. L. Cannon, A. Kaplan, C. A. Cates and E. R. Flynn, Am. J. Physiol.: Heart Circ. Physiol., 2018, 315, H522–H530 CrossRef CAS PubMed.
- M. L. Lindsey, K. R. Brunt, J. A. Kirk, P. Kleinbongard, J. W. Calvert, L. E. de Castro Brás, K. Y. DeLeon-Pennell, D. P. Del Re, N. G. Frangogiannis, S. Frantz, R. J. Gumina, G. V. Halade, S. P. Jones, R. H. Ritchie, F. G. Spinale, E. B. Thorp, C. M. Ripplinger and Z. Kassiri, Am. J. Physiol.: Heart Circ. Physiol., 2021, 321, H1056–H1073 CrossRef CAS PubMed.
- M. L. Lindsey, Z. Kassiri, J. A. Virag, L. E. de Castro Brás and M. Scherrer-Crosbie, Am. J. Physiol.: Heart Circ. Physiol., 2018, 314, H733–H752 CrossRef PubMed.
- M. L. Lindsey, R. Bolli, J. M. Canty Jr, X.-J. Du, N. G. Frangogiannis, S. Frantz, R. G. Gourdie, J. W. Holmes, S. P. Jones and R. A. Kloner, Am. J. Physiol.: Heart Circ. Physiol., 2018, 314, H812–H838 CrossRef CAS PubMed.
- A. J. Mouton, Y. Ma, O. J. Rivera Gonzalez, M. J. Daseke, 2nd, E. R. Flynn, T. C. Freeman, M. R. Garrett, K. Y. DeLeon-Pennell and M. L. Lindsey, Basic Res. Cardiol., 2019, 114, 6 CrossRef PubMed.
- J. A. Vizcaíno, A. Csordas, N. Del-Toro, J. A. Dianes, J. Griss, I. Lavidas, G. Mayer, Y. Perez-Riverol, F. Reisinger and T. Ternent, Nucleic Acids Res., 2016, 44, D447–D456 CrossRef PubMed.
- M. J. Daseke II, M. A. Tenkorang-Impraim, Y. Ma, U. Chalise, S. R. Konfrst, M. R. Garrett, K. Y. DeLeon-Pennell and M. L. Lindsey, J. Mol. Cell. Cardiol., 2020, 145, 112–121 CrossRef PubMed.
- R. P. Iyer, N. L. Patterson, F. A. Zouein, Y. Ma, V. Dive, L. E. de Castro Brás and M. L. Lindsey, Int. J. Cardiol., 2015, 185, 198–208 CrossRef PubMed.
- I. H. T. Guideline, 2009.
- K. Lim, T.-h. Kim, A. Trzeciak, A. M. Amitrano, E. C. Reilly, H. Prizant, D. J. Fowell, D. J. Topham and M. Kim, Nat. Immunol., 2020, 21, 1046–1057 CrossRef CAS PubMed.
- H. L. Brooks and M. L. Lindsey, Am. J. Physiol.: Heart Circ. Physiol., 2018, 314, H724–H732 CrossRef CAS PubMed.
- M. L. Lindsey, G. A. Gray, S. K. Wood and D. Curran-Everett, Am. J. Physiol.: Heart Circ. Physiol., 2018, 315, H303–H313 CrossRef PubMed.
- Z. Pang, J. Chong, G. Zhou, D. A. de Lima Morais, L. Chang, M. Barrett, C. Gauthier, P.-É. Jacques, S. Li and J. Xia, Nucleic Acids Res., 2021, 49(W1), W388–W396 CrossRef CAS PubMed.
- Z. Xie, A. Bailey, M. V. Kuleshov, D. J. Clarke, J. E. Evangelista, S. L. Jenkins, A. Lachmann, M. L. Wojciechowicz, E. Kropiwnicki and K. M. Jagodnik, Curr. Protoc., 2021, 1, e90 CAS.
- S. Graw, K. Chappell, C. L. Washam, A. Gies, J. Bird, M. S. Robeson and S. D. Byrum, Mol. Omics., 2021, 17(2), 170–185 RSC.
- M. Kim and I. Tagkopoulos, Mol. Omics, 2018, 14, 8–25 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1mo00519g |
| This journal is © The Royal Society of Chemistry 2022 |