
Thermal and UV light adaptive polyurethane elastomers for photolithography-transfer printing of flexible circuits†
Jiaxin
Shi
 ,
Zhiqi
Wang
,
Tianze
Zheng
,
Xueyan
Liu
,
Baohua
Guo
and
Jun
Xu
*
,
Zhiqi
Wang
,
Tianze
Zheng
,
Xueyan
Liu
,
Baohua
Guo
and
Jun
Xu
*
Advanced Materials Laboratory of Ministry of Education (MOE), Department of Chemical Engineering, Tsinghua University, Beijing, 100084, China. E-mail: jun-xu@mail.tsinghua.edu.cn
First published on 7th October 2022
Abstract
Flexible polymers are widely used in the fields of wearable devices, soft robots, sensors, and other flexible electronics. Combining high strength and elasticity, electrical conductivity, self-healability, and surface tunable properties in one material becomes a challenge for designing polymeric materials for these applications, especially in flexible electronics. Herein, we propose a “two birds with one stone” strategy to synthesize thermal and UV light adaptive polyurethane elastomers with high-strength, self-healable, surface-modifiable and patternable functions for photolithography-transfer printing flexible circuits. The “stone”, dihydroxybenzophenone, plays two roles in the synthesized polyurethanes as both a dynamic covalent bond and a UV-sensitive unit. On one hand, the phenolic group reacts with isocyanate to form a dynamic covalent phenol–carbamate bond, making the polymer self-healable, processable, and surface-embeddable with conductive fillers utilizing dynamic network rearrangement. On the other hand, the benzophenone group acts as a UV-sensitive unit to graft other functional groups to the polymer surface or self-crosslink on the surface under UV irradiation. Based on the dynamic covalent network and UV self-crosslinking properties, self-healable patterned flexible circuits can be obtained by photolithography-transfer printing. The flexible circuits prepared by loading silver nanowires on the dynamically crosslinked polyurethane substrate show little change of electric resistance when stretched up to 125% and can withstand thousands of stretching cycles.
New conceptsPrevious polymers for flexible electronics are mostly inert elastomers. In this work, we prepare a new type of elastomer for application as a substrate for photolithography and heat transfer printing of flexible electronics. The polymers with dynamic covalent bonds possess both excellent mechanical properties (high toughness and high elasticity) and other functions, such as thermal and light-triggering functions. We used dihydroxybenzophenone as a functional monomer to prepare a multifunctional polyurethane elastomer with the following advantages. Dynamic covalent bonds endow the polymer with self-healing and strong surface adhesion to conductive fibers at the heat-transfer printing temperature. The benzophenone unit acts as the UV initiator and enables the polymer to be tailor designed by UV light, e.g., chemical modification of surface hydrophilicity, self-crosslinking at selected regions to change mechanical properties and surface adhesion. Low modulus, high strength, and high resilience ensure that the polymer can withstand repeated deformation. This molecular design strategy provides a facile way to prepare multifunctional polymers applicable in the fields of flexible electronics, wearable devices, soft robots, and sensors. |
Introduction
Flexible polymers are widely used as stretchable matrices in wearable devices,1 soft robotics2,3 and artificial skin,4 especially stretchable circuits.5 Researchers have developed a variety of techniques to fabricate stretchable circuits based on flexible polymers, which fall into two main categories. One is to directly compound the conductive filler with the polymer matrix. These preparation methods are complicated, and the dispersibility of the conductive filler affects the electrical properties. The other category is surface printing or transfer printing, which is easy to operate, but the bonding between the circuit and the substrate is weak, so it easily fails when deformed. Surface modifications are often required to improve the adhesion between the polymer and circuit.6,7In addition, the self-healing ability would be an advantage for long term application of materials in flexible electronics, especially after the circuit is damaged. Although conductive particles such as metal nanowires cannot be repaired after being damaged, the conductive pathways can be rebuilt by the healing of flexible substrates. Self-healing elastomers have been extensively studied and there are many examples of their compounding with conductive particles or liquid metals.8–13 How to balance the electrical conductivity, self-healing, and mechanical properties, while being easy to pattern-print, remains a hot topic. In recent years, the concept of dynamic covalent bonds has shined in the field of flexible electronics. Polymers with a crosslinked network containing dynamic covalent bonds, termed as a covalent adaptable network (CAN),14 can not only maintain the mechanical properties and solvent resistance as traditional cross-linked polymers but also possess other functions, e.g., self-healing and reprocessing.15–18 Many types of CANs have been applied in self-healing conductive polymers such as boroxine bonds,19,20 disulphide bonds,21 imine bonds,22 oxime urethane bonds23 and hindered urea bonds.24 In addition, CAN-containing polymers can also embed conductive particles on the surface through network rearrangement, forming a very stable composite structure.24,25 Therefore, fine molecular design of polymers containing CAN would create many possibilities in the field of flexible electronics.
The surface of polymers for stretchable circuits is also important, especially for patterned circuit printing. Surface modification is of great significance for conductive inks printing,26 antibacterial property,27 adhesion to other materials,28 cell adhesion in biomedical fields,29etc. For commonly used PDMS substrates, plasma is a common means to improve the surface hydrophilicity.30,31 In addition, the hydrophilicity and hydrophobicity of PDMS can be regulated by deformation.32 However, surface modification of high-strength self-healable flexible circuit substrates has rarely been explored. Patterned printing is a key step of fabricating flexible electronic devices. In addition to traditional inkjet printing, other methods such as screen printing and imprinting have appeared in recent years. Screen printing typically requires casting and curing of flexible substrates on a patterned transfer layer, thus cannot print circuits directly on rigid substrates. Imprinting requires a conductive fluid medium such as a liquid metal to fill the indentation. Photolithography is a predominant method for integrated circuit fabrication, but it relies on small-molecule photoresists and is not widely studied in flexible circuits. It will be very promising if the substrate material of the flexible circuit can be directly used for photolithography-transfer printing.
In the present work, we synthesized a multi-functional soft polyurethane elastomer. Conductive materials such as silver nanowires (Ag NWs) could be patterned on the surface of the elastomer via a simple UV irradiation using a mask and transfer method. Cross-linked polyurethane with thermal and UV light-triggering groups was prepared (Fig. 1). On one hand, the dynamic covalent bonds make flexible circuits self-healable and the mobility of chains due to bond dissociation at higher temperatures during transfer printing can enable strong adhesion between the polymer matrix and conductive fillers. On the other hand, the UV light-initiator groups in the polymer backbone can be utilized for surface grafting of vinyl monomers with short time ultraviolet irradiation or self-crosslinking initiated by long time exposure to ultraviolet light. UV photolithography with masking led to self-crosslinking with permanent crosslinks at the exposed region, thus conductive circuits can be transfer printed at the masked, un-crosslinked region with mobile chains due to dissociation of the dynamic covalent bonds. The obtained transparent PU-DHBP elastomer showed a tensile strength of over 20 MPa and a breaking elongation of more than 1000%. The material demonstrated highly efficient self-healing at 80 °C. This molecular design strategy will expand the range of applications and processing of flexible electronic substrate materials.
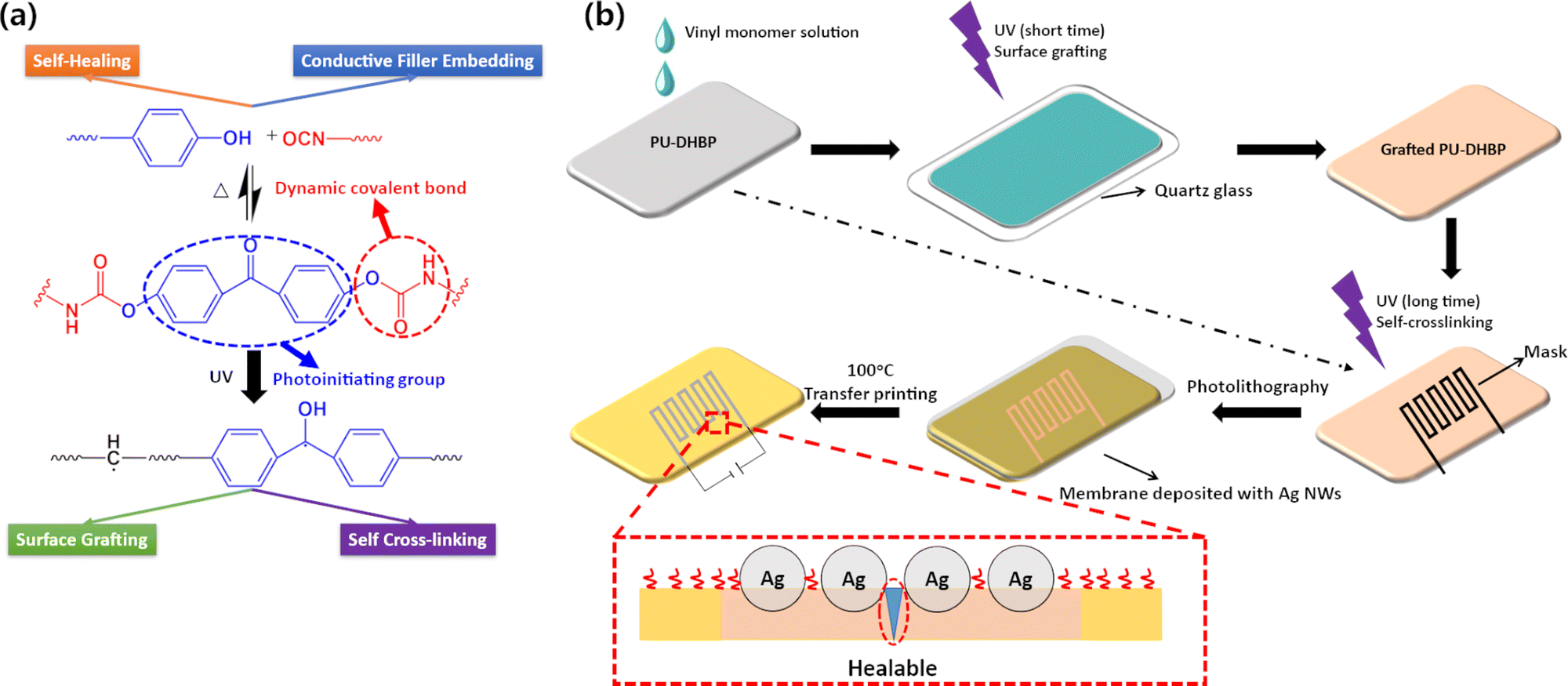 | ||
| Fig. 1 (a) Quadruple functions of PU-DHBP. (b) Schematic diagram of surface grafting and photolithography-transfer printing circuits on PU-DHBP. Based on dynamic phenol–carbamate bonds and benzophenone units, PU-DHBP can realize various functions such as self-healing, repeated processing, surface grafting, self-crosslinking, embedding conductive fillers, etc. | ||
Results and discussion
Polymeric materials for flexible electronics should meet many requirements, among which the mechanical properties are crucial. Since the materials need to have sufficient durability under repeated deformation, mechanical properties such as low modulus, high strength, and low permanent deformation are necessary. These can be controlled by the molecular structure of the hard and soft segments of the polyurethane and the crosslinking degree. In this study, we designed dynamic covalently cross-linked polyurethane as a substrate polymer for flexible electronics, in which we utilized the inherently reversible reaction of the addition of isocyanate and active hydrogen.33,34 4,4′-dihydroxybenzophenone was used as a key monomer to construct the dynamic covalent network and to incorporate the UV-initiator in the PU elastomer (Fig. 1a).In order to obtain transparent polyurethanes, we chose the flexible poly(ε-caprolactone) (PCL) diol as the soft segment. The usage of bio-degradable polyesters also enhances the environmental friendliness of the polymer. In the synthesized polyurethanes, there is a possibility of PCL to undergo crystallization, which can significantly affect the resilience and sensitivity of mechanical properties to temperature, so it should be avoided. However, due to the presence of chemical cross-linking and the hard segments containing aliphatic rings, crystallization of PCL is suppressed. For flexible electronic elastomers, amorphous PCL soft segments are advantageous. The DSC results show that there is no obvious melting and crystallization signal during the heating and cooling process, respectively (Fig. S1, ESI†). It can be confirmed that in PU-DHBPs, PCL exists in an amorphous form.
As shown in Table S1 and Fig. S2, S3 (ESI†), we compare PU-DHBPs with different degrees of cross-linking and find that PU with a moderate degree of cross-linking demonstrates low modulus, high strength, and high resilience, and is most suitable as an elastomer substrate for flexible electronics. The samples with a higher cross-linking degree show a larger modulus and a higher Tg, while the samples with a lower cross-linking degree exhibit higher elongation at break. Indeed, in polyurethane or polyurea, such characteristics as low modulus and high strength exhibited during deformation are common. A large number of hydrogen bonds can be formed between urethane groups to constrain the movement of molecular chains. At the beginning of stretching, the hard segments forming the continuous phase can bear a considerable amount of stress. Due to the asymmetric structure of the IPDI, the hard segments are loosely stacked and will be partially mixed with the soft segments. Therefore, as the strain increases, the soft segments will be gradually released under the action of force, accompanied by the dissociation of hydrogen bonds among hard segments. Thus, a significant decrease in the slope of the stretching stress–strain curve, the so-called “softening”, can be observed at this stage. In the final stage, the soft segments are completely released. Under the combined constraints by the hard segments and chemical cross-linkages, the finite elongation effect rapidly increases the stress to demonstrate significant strain hardening and high tensile strength.35–37
In the DMA curve (Fig. S2, ESI†), each sample shows a rapid decrease in storage modulus at high temperature, caused by the dissociation of phenol–carbamate dynamic bonds. The characteristic reaction of polyurethane, the addition of isocyanate and active hydrogen, is inherently reversible.33,34 This is well known in traditional blocked isocyanate chemistry, but it has only recently been really applied to dynamic covalently cross-linked polyurethane. In our previous reports, we have thoroughly studied the design strategy of phenol–carbamate dynamic covalent networks.43,44 The phenol–carbamate bond is a type of dissociative dynamic covalent bond, and the dissociation rate depends on the electronic effect of the phenol substituent and the chemical structure of the isocyanate.43,44 Herein, the substituent of bisphenol is a carbonyl group, which has a strong electron withdrawing effect, resulting in a low initial dissociation temperature (∼70 °C). The dissociation of the phenol–carbamate bond is evidenced via the infrared spectra during heating (Fig. S4, ESI†).
The self-healing properties under mild conditions are very important for flexible electronic polymers, which means that they can have a longer life without damaging the remaining electrical components. PU-DHBPs exhibit good self-healing properties. As shown in Fig. 2a and b, all the samples can achieve almost complete self-healing at 80 °C. The self-healing properties of PU-DHBP are attributed to the rearrangement of dynamic phenol–carbamate bonds upon heating, which has been discussed in our previous work.43–45 The preparation of polymers for flexible electronic devices has certain requirements on shape and precision, which means that polymeric materials produced in batches need to be remoulded. The samples were cut into pieces and hot pressed at 100 °C for 20 minutes. All samples could be reprocessed almost perfectly. The tensile test shows that the stress–strain curves of the three samples are very similar before and after reprocessing, indicating good re-processability (Fig. S5, ESI†).
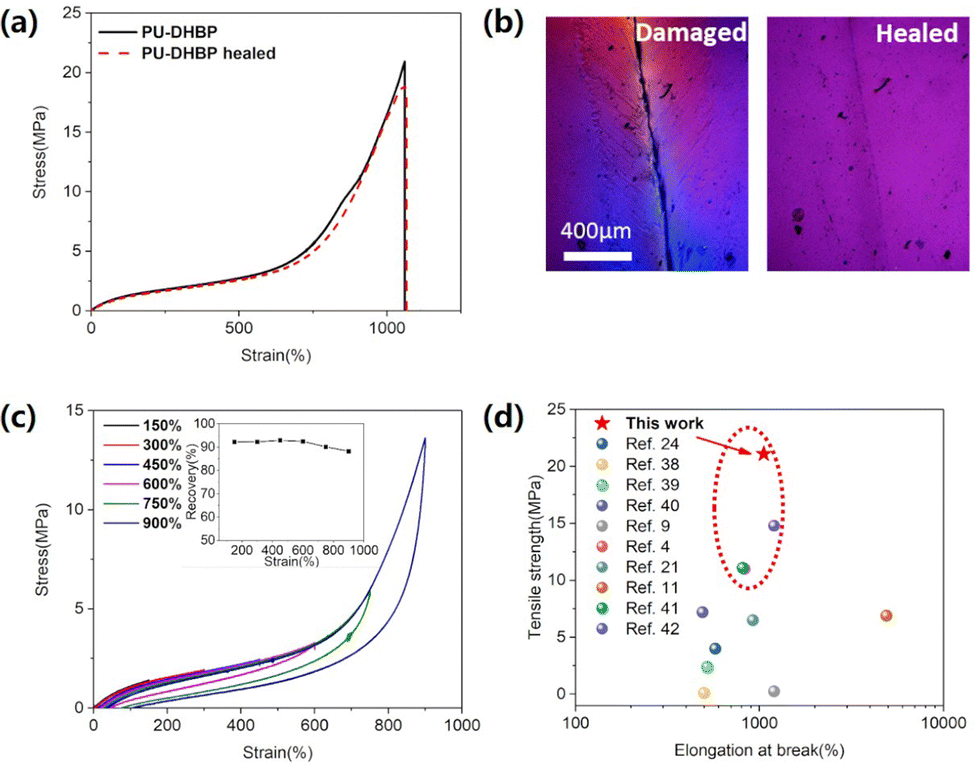 | ||
| Fig. 2 (a) The stress–strain curves of the original and self-healed PU-DHBP after cut. (b) Micrographs of scratched PU-DHBP before and after self-healing at 80 °C for 6 h. (c) Stress–strain curve of PU-DHBP in the cyclic tensile test. The inset plot shows the spring back exhibited by PU-DHBP under different cyclic tensile maximum strains, and the ordinate “recovery” is defined as the percentage of the recoverable strain to the maximum strain during the deformation process. (d) Comparison of the mechanical properties between PU-DHBP and other typical studies of self-healing flexible electronic polymers.4,9,11,21,24,38–42 | ||
Polymers for flexible electronics need to withstand repeated deformations during application, and therefore require excellent resilience after rapid deformation, which can be characterized by cyclic tensile experiments. In addition, the stress–strain curves during cyclic tensile experiments can also further reveal the structure–property relationship. The PU-DHBP elastomers were subjected to cyclic tensile experiments with stepwise increasing strain but with no waiting time between tensile tests (Fig. 2c). The energy dissipation of PU-DHBP showed a trend that the greater the maximum strain in each cycle, the faster the increase in dissipation. In polyurethane or polyurea systems, this strong dissipation often results from the dissociation of the hard segment structure.35,46–48 Between 0 and 450% strain, the amount of dissipation increases slowly with increasing strain, which approximates a linear change, indicating that only recoverable interchain interactions occur. With the further increase of the strain, the dissipation increases rapidly, which means that a strong dissipation mechanism appears under large strains. PU-DHBP has a moderate cross-linking degree and hard segment content, so it has low permanent deformation and high repeatability. As shown in Fig. 2d, the strength of PU-DHBP is the highest among the reported self-healing polymers for flexible electronics.
UV irradiation is a common method for the synthesis and functionalization of polymeric materials. There are some research studies using photosensitive groups to construct functional polymers. For example, shape memory materials49 and self-healing materials50–52 were prepared by using the structure of reversible 2+2 cyclization by different wavelengths of UV irradiation, with molecular topoisomerization triggered by UV-induced release of transesterification catalysts.53 In this work, we achieve two important functions by UV irradiation of PU-DHBP: surface modification and patterned cross-linking (Fig. 3a). Surface modification can regulate the surface hydrophilicity, antibacterial properties, adhesion, etc., enabling polymers to play an important role in flexible electronics, biomedical implants, smart wearables, and other fields. Patterned cross-linking can prefabricate a non-adhesive area on the polymer surface, and flexible electronic circuits can be easily prepared by the method of transfer-printing. Due to the poor penetrating ability of UV, the irradiation can only work in the thin surface layer, so these functional materials are all in the form of thin films or coatings. But from another point of view, the poor penetration ability means that it is suitable for surface treatment.
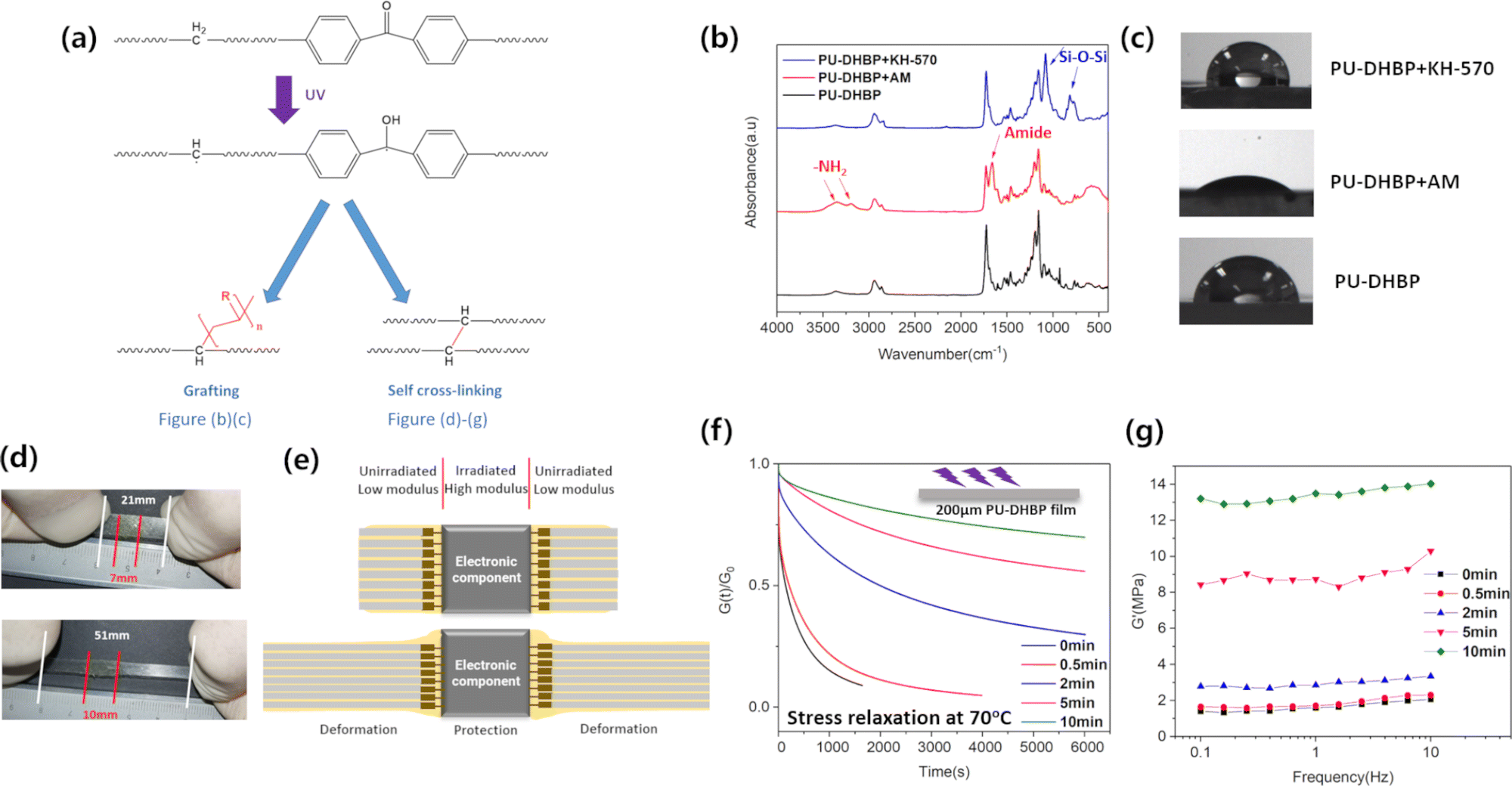 | ||
| Fig. 3 (a) Possible reactions of BP units upon UV irradiation. (b) ATR-IR spectra of ungrafted, grafted acrylamide and KH-570 of PU-DHBP. (c) Water contact angle photographs of ungrafted, grafted acrylamide and KH-570 of PU-DHBP. (d) Photographs of UV irradiated PU-DHBP films in the middle during stretching. (e) Manipulating local modulus to protect electronic components from deformation (conceptual image). (f) Stress relaxation curves of PU-DHBP 200 μm thin films after UV double-sided irradiation for different times. (g) Modulus of PU-DHBP 200 μm thin films at room temperature after UV double-sided irradiation for different times. | ||
Benzophenone (BP) can be used as a type II photoinitiator,54–56 which can subtract hydrogen from polymer chains to form hydrocarbon radicals and semi-aryl pinacol radicals under UV irradiation. Although semi-aryl pinacol radicals can be coupled with other radicals, or connected with polymerizable monomers such as vinyl monomers, the resulting chemical bonds are not stable and are easy to break again to form semi-aryl pinacol radicals. So semi-aryl pinacol radicals are generally not considered to be sufficiently reactive. In contrast, hydrocarbon radicals can stably initiate radical polymerization, and can also couple with each other to form irreversible C–C bonds, forming additional permanent chemical crosslinks on the surface (Fig. 3a). Therefore, the introduction of a benzophenone structure into the thermally dissociated dynamic covalently cross-linked polymer can achieve two functions simultaneously. One is coating the surface with monomers, which can be initiated for radical polymerization under UV irradiation to obtain surface grafting. The other is UV exposure under a mask to form irreversible C–C bonds at the irradiation site, which further self-crosslink so that a simple transfer-printing can be used to selectively adhere the conductive material at the unirradiated position to construct a patterned flexible electric circuit.
Since the method used for grafting is solution coating and irradiation, UV can fully initiate free radical polymerization on the surface in a short time. It should be noted that if the UV irradiation time is too long, additional self-crosslinking will be induced and the surface will lose its dynamic covalent bond character. We used UV to graft acrylamide or silane coupling agent KH-570 to the surface of the PU-DHBP to achieve hydrophilic or hydrophobic modification. The ATR-IR spectrum and contact angle results (Fig. 3b and c) show that the surface of the grafted acrylamide PU-DHBP has characteristic absorption peaks of polyacrylamide, and the water contact angle is reduced from ∼80° to ∼40°. The PU-DHBP grafted with KH-570 was subjected to hydrolysis-drying treatment after being irradiated with UV, and a Si–O–Si network was formed on the surface. The results of ATR-IR prove the existence of this chemical structure. Meanwhile, the water contact angle increases to ∼105° due to the enhanced hydrophobicity after hydrolysis. In addition, surface elemental analysis also demonstrates the successful grafting of silicon-containing molecules (Fig. S7, ESI†). In order to prove that the surface grafting is indeed initiated by the benzophenone unit, the control sample PU-AF (Scheme S2, ESI†) was prepared with bisphenol AF instead of dihydroxybenzophenone. Under the same processing conditions, there is no change in the ATR-IR spectra and water contact angle of PU-AF.
Since excessive UV irradiation can cause irreversible cross-linking, and a small amount of other chemical components are introduced into the surface after surface grafting polymerization, it is necessary to verify whether the grafting affects the self-healing of the surface. Fig. S8 (ESI†) shows that the grafted sample can still achieve obvious self-healing after deep scratching. In fact, the UV irradiation time is short and does not cause a lot of irreversible crosslinking. Due to the poor penetration ability of UV, a 200 μm PU-DHBP film was exposed to verify the self-cross-linking ability. Fig. 3f and g show that as the UV exposure time increases, more irreversible cross-linking occurs, the stress relaxation rate decreases, and the modulus increases. However, irradiation for a longer period was required to generate sufficient self-crosslinking. Fig. 3d shows the stretching process of the exposed film splines in the middle. Under the same stress, the deformation of the self-cross-linked position is significantly lower than that of the unexposed position. Fig. 3g demonstrates that the modulus of the material can be easily controlled by the exposure time to UV irradiation. Some previous studies suggest that the improvement of the local modulus of the elastomer helps to protect the electronic components in the corresponding position under large deformation.57 In PU-DHBP, the UV-irradiated part cannot embed Ag NWs, but can serve as the embedment site for electronic components. The high-modulus self-crosslinked part after long time UV irradiation deforms much less than the unirradiated part when stretched, thus protecting fragile electronic components (Fig. 3d and e).
Dissociative dynamic covalent bonds have special advantages in surface-printed conductive networks. The mobile polymer chains during network rearrangement can be used to make conductive fillers adhere to the polymer surface, thereby forming a stable surface conductive network.24
Embedding of Ag NWs into PU-DHBP (1 mg cm−2) were realized by thermal transfer printing. First, Ag NWs were deposited on the PTFE membrane by suction filtration of the Ag NW dispersion, and then the PTFE membrane was tightly covered on the surface of PU-DHBP and heated at 100 °C for an hour. After sufficient annealing to rebuild the phenol-carbamate bond, the filter membrane was removed to obtain PU-DHBP with surface conductivity. At 100 °C, the dynamic covalent bond of PU-DHBP has rapid dissociation–reconstruction, like slow flow, so the polymer can form a strong embedded structure with Ag NWs (Fig. 4a and Fig. S11, ESI†), which makes the stretchable conductor more stable. However, heated at lower temperature, e.g., 80 °C for 1 hour could not obtain an embedded conductive network, and Ag NWs could only sit on the surface of the polymer (Fig. S12, ESI†). Benefiting from the embedded conductive network and the large aspect ratio of Ag NWs, the PU-DHBP loaded with Ag NWs exhibits excellent electrical conductivity (∼4.9 Ω sq−1). There is still no dramatic increase in resistance when stretched to 100% deformation (Fig. 4b and Fig. S13, ESI†). When the strain is too large (e.g., larger than 125%), the overlap between the Ag NWs decreases and the resistance increases sharply. In addition, the stretchable conductor exhibits good cycling performance (Fig. 4c and Fig. S14, ESI†), even after hundreds of cycles. The resistance remains nearly constant, which can be attributed to the high elasticity of PU-DHBP and the stability of the embedded conductive network. Due to the self-healing properties of PU-DUBP, the scissored Ag NW conductive network can be rebuilt by dynamic covalent network rearrangement at 80 °C in 6 h, and its conductivity is almost the same as that of the original sample (Fig. 4d and e). In conclusion, PU-DHBP loaded with Ag NWs is suitable as a flexible electronic material.
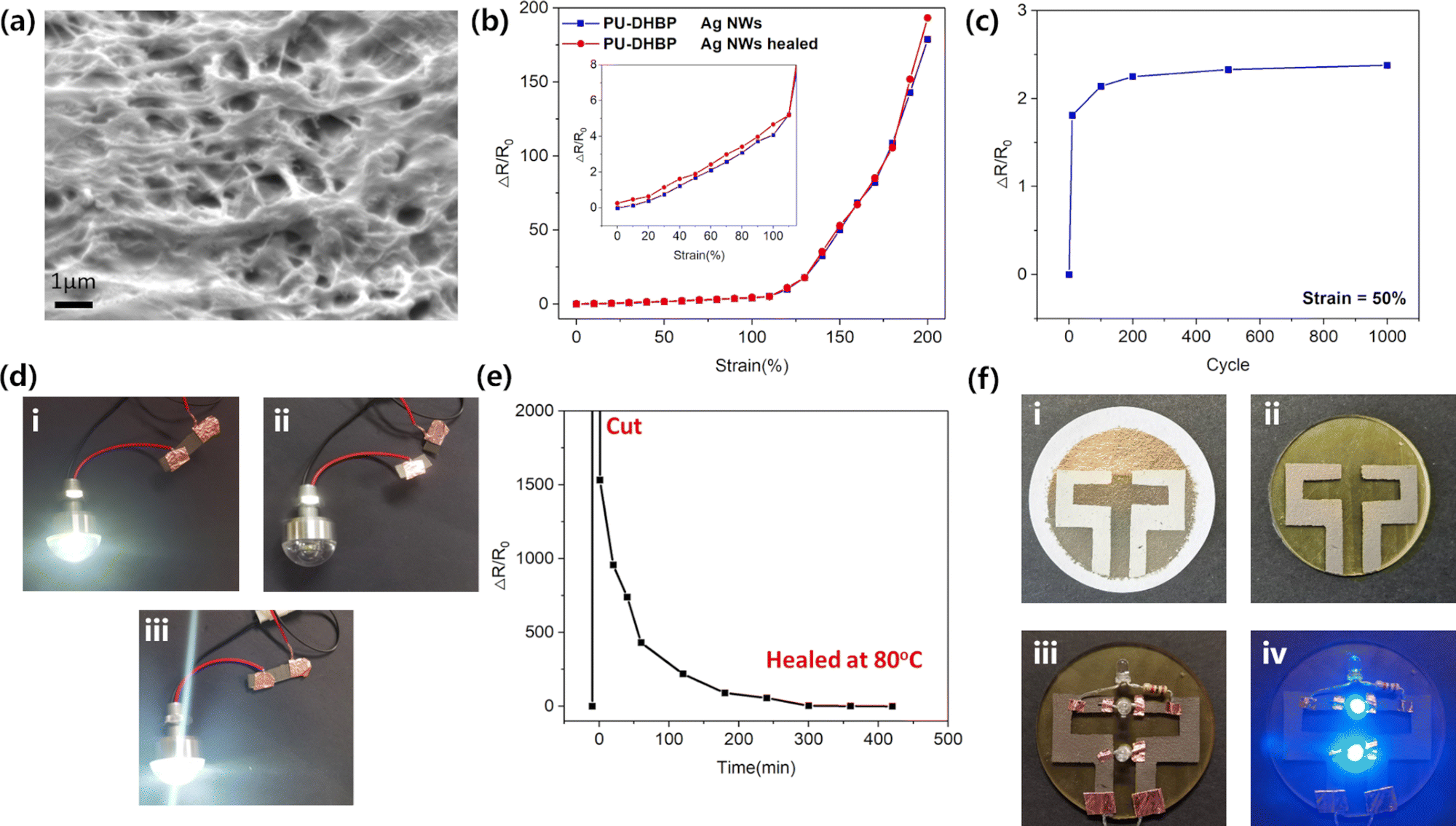 | ||
| Fig. 4 (a) SEM photograph of PU-DHBP embedded with Ag NWs. (b) The relationship between conductivity and tensile deformation of pristine and healed PU-DHBP-2 embedded with Ag NWs. (c) Conductivity changes due to 50% stretching cycle of PU-DHBP embedded with Ag NWs. (d) The pristine PU-DHBP loaded with Ag NWs (i), cut off (ii) and after self-healing (iii) as a conductor to light up the LED. (e) Resistance changes of PU-DHBP embedded with Ag NWs during self-healing at 80 °C (f) PTFE membrane (i) and PU-DHBP (ii) after patterned adhering of Ag NWs. Power-off (iii) and power-on (iv) photos of the PU-DHBP-based LED circuit. | ||
The patterned self-cross-linking function of PU-DHBP can be applied in flexible electronics. Using the UV self-crosslinking properties on the surface of PU-DHBP, it is possible to adhere conductive fillers such as silver nanowires simply and selectively at masked regions without self-crosslinking to achieve surface printing of electric circuits. Since the long-time UV-irradiated region of surface loses its dynamic nature, Ag NWs cannot be firmly adhered to the surface even under heating. However, the masked regions without UV exposure still retain the characteristics of dynamic covalent cross-linking. After heat treatment, the covalent dynamic network dissociates, so the polymer can be firmly combined with Ag NWs (Fig. S15, ESI†). After cooling, the phenol–carbamate bonds were reformed, and the selectively patterned Ag NWs could be peeled off from the PTFE membrane. Therefore, after mask illumination and thermal transfer printing, the desired electric circuit pattern can be transferred from the PTFE membrane onto the masked region. As shown in Fig. 4f, a simple flexible LED parallel circuit was fabricated via the above method. To demonstrate the possible accuracy with this approach, we performed a simple evaluation using a 360 mesh screen as a mask. The mesh-like structure was largely preserved after fabrication (Fig. S16, ESI†), proving that this process can reach a precision of tens of microns. In principle, any conductive filler can be embedded into the polymer surface through network rearrangement. To demonstrate the versatility of the photolithography-transfer printing strategy, we printed multi-walled carbon nanotubes onto the UV exposed half of a rectangular PU-DHBP sample and demonstrated the conductivity of the printed circuit by lighting up a LED (Fig. S17, ESI†).
In this work, through delicate molecular design, we prepared PU-DHBP with multiple functions and excellent properties. First, compared with commonly used inert flexible electronic polymers, PU-DHBP with dynamic covalent bonds and UV-initiator groups can respond to heat and light for controllable processing, healing, surface modification, and heat transfer printing. The dynamic feature of the polymer surface enables embedding and strong adhesion of the conductive fillers. Second, PU-DHBP can be used to prepare flexible circuits by the photolithography-transfer printing method, which has high precision and is easy for large-scale application. Third, material modulus at selected regions can be tailor designed via UV irradiation with a mask and the modulus at un-masked region will increase with prolonged irradiation time. Fourth, self-healable PU-DHBP has high strength, high toughness, and high resilience, which guarantee a long life of flexible circuits.
Conclusions
In summary, this study proposes a strategy to produce flexible electronic substrates for producing high-toughness, self-healable, surface-modifiable, and patternable printed circuits. Besides the dynamically cross-linked substrate in previous studies, the materials reported in our work is adaptive to UV light. On exposure to UV light for a short period of time (around 30 s), the flexible polymer substrates can be easily surface-modified to change the surface hydrophilicity, which has not been previously reported. In addition, UV exposure with masking for a longer period (minutes) can be used to tune the adhesion and modulus of selected surface regions. In this work, a new photolithography-transfer printing method is established to fabricate flexible circuits.We used dihydroxybenzophenone as a monomer, through a simple synthesis method, to prepare the cross-linked polyurethane elastomer (PU-DHBP) with both dynamic covalent bonds and UV-sensitive units. The obtained materials possess excellent and controllable mechanical properties. For instance, the tensile strength can reach 20 MPa and the elongation at break can exceed 1000%. It is revealed that dynamic covalently cross-linked polymers can achieve multi-functionality through specially designed monomers, e.g., dihydroxybenzophenone in this work. On one hand, the carbonyl substituted bisphenol makes the phenol–carbamate bond dynamic at moderate temperatures (e.g. 50–80 °C), giving PU-DHBP excellent self-healing and reprocessing capabilities verified by the experiments. On the other hand, the benzophenone unit acts as a UV-sensitive group, so that the material exposed to short time UV irradiation can be surface grafted with vinyl monomers to change the surface properties. In contrast, long time UV irradiation of the pure PU samples, e.g., around 10 min, leads to self-crosslinking. The self-crosslinking function is further utilized to selectively transfer Ag NWs to the masked region of the polyurethane samples where UV irradiation could not penetrate and remains dynamic and adhesive to silver nanowires at 100 °C. The application of the multi-functional polyurethane materials as a soft substrate for heat transfer printing of flexile electronics was demonstrated. The embedded structure of Ag NWs formed by dynamic network rearrangement endows flexible electrodes with cycling stability, low electric resistance, and high stretchability. The mask exposure process also enables the material to have the possibility of fabricating fine circuits. This work proposes a facile and low-cost strategy to produce both thermal and UV adaptive multifunctional elastomers and demonstrates their application in the field of flexible electronics. Thus, the molecular design of dynamic polymers from functional monomers can be utilized to prepare various functional polymer materials for applications in different fields.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are indebted to the National Natural Science Foundation of China (Grant No. U1862205, 51673110) and the Tsinghua University Scientific Research Project (Grant No. 20194180048) for financial support.References
- J. Cao, Z. Zhou, Q. Song, K. Chen, G. Su, T. Zhou, Z. Zheng, C. Lu and X. Zhang, ACS Nano, 2020, 14, 7055–7065 CrossRef CAS PubMed.
- M. Baumgartner, F. Hartmann, M. Drack, D. Preninger, D. Wirthl, R. Gerstmayr, L. Lehner, G. Mao, R. Pruckner, S. Demchyshyn, L. Reiter, M. Strobel, T. Stockinger, D. Schiller, S. Kimeswenger, F. Greibich, G. Buchberger, E. Bradt, S. Hild, S. Bauer and M. Kaltenbrunner, Nat. Mater., 2020, 19, 1102–1109 CrossRef CAS PubMed.
- E. J. Markvicka, M. D. Bartlett, X. Huang and C. Majidi, Nat. Mater., 2018, 17, 618–624 CrossRef CAS PubMed.
- X. Xun, Z. Zhang, X. Zhao, B. Zhao, F. Gao, Z. Kang, Q. Liao and Y. Zhang, ACS Nano, 2020, 14, 9066–9072 CrossRef CAS PubMed.
- Y. Kim, O. Y. Kweon, Y. Won and J. H. Oh, Macromol. Res., 2019, 27, 625–639 CrossRef CAS.
- M. A. Rahim, F. Centurion, J. Han, R. Abbasi, M. Mayyas, J. Sun, M. J. Christoe, D. Esrafilzadeh, F. M. Allioux, M. B. Ghasemian, J. Yang, J. Tang, T. Daeneke, S. Mettu, J. Zhang, M. H. Uddin, R. Jalili and K. Kalantar-Zadeh, Adv. Funct. Mater., 2021, 31, 2007336 CrossRef CAS.
- Y. Liu, X. Xu, Y. Wei, Y. Chen, M. Gao, Z. Zhang, C. Si, H. Li, X. Ji and J. Liang, Nano Lett., 2022, 22, 3784–3792 CrossRef CAS PubMed.
- Y. Jia, Q. Guan, L. Zhang, R. E. Neisiany, N. Yan, Y. Li and Z. You, Sci. China Mater., 2022, 65, 2553–2564 CrossRef.
- F. Sun, J. Xu, T. Liu, F. Li, Y. Poo, Y. Zhang, R. Xiong, C. Huang and J. Fu, Mater. Horiz., 2021, 8, 3356–3367 RSC.
- G. Zhu, Y. Hou, J. Xiang, J. Xu and N. Zhao, ACS Appl. Mater. Interfaces, 2021, 13, 34954–34961 CrossRef CAS PubMed.
- K. Parida, G. Thangavel, G. Cai, X. Zhou, S. Park, J. Xiong and P. S. Lee, Nat. Commun., 2019, 10, 2158 CrossRef PubMed.
- X. Wu, J. Wang, J. Huang and S. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 7387–7396 CrossRef CAS PubMed.
- S. H. Kim, H. Seo, J. Kang, J. Hong, D. Seong, H. J. Kim, J. Kim, J. Mun, I. Youn, J. Kim, Y. C. Kim, H. K. Seok, C. Lee, J. B. H. Tok, Z. Bao and D. Son, ACS Nano, 2019, 13, 6531–6539 CrossRef CAS PubMed.
- G. M. Scheutz, J. J. Lessard, M. B. Sims and B. S. Sumerlin, J. Am. Chem. Soc., 2019, 141, 16181–16196 CrossRef CAS PubMed.
- J. M. Winne, L. Leibler and F. E. Du Prez, Polym. Chem., 2019, 10, 6091–6108 RSC.
- F. Ling, Z. Liu, M. Chen, H. Wang, Y. Zhu, C. Ma, J. Wu and G. Huang, J. Mater. Chem. A, 2019, 7, 25324–25332 RSC.
- H. Wei, Y. Yang, X. Huang, Y. Zhu, H. Wang, G. Huang and J. Wu, J. Mater. Chem. A, 2020, 8, 9013–9020 RSC.
- H. Wu, B. Jin, H. Wang, W. Wu, Z. Cao, J. Wu and G. Huang, Front. Chem., 2020, 8, 585569 CrossRef CAS PubMed.
- J. C. Lai, J. F. Mei, X. Y. Jia, C. H. Li, X. Z. You and Z. Bao, Adv. Mater., 2016, 28, 8277–8282 CrossRef CAS PubMed.
- Q. Huang, Z. Tang, D. Wang, S. Wu and B. Guo, ACS Macro Lett., 2021, 10, 231–236 CrossRef CAS PubMed.
- S. M. Kim, H. Jeon, S. H. Shin, S. A. Park, J. Jegal, S. Y. Hwang, D. X. Oh and J. Park, Adv. Mater., 2018, 30, 1705145 CrossRef PubMed.
- X. Dai, L. Huang, Z. Sun, Y. Du, B. Xue, M. Wong, J. Han, Q. Liang, Y. Wu, B. Dong, J. Kong and J. Hao, Mater. Horiz., 2022, 9, 1921–1934 RSC.
- L. Zhang, Z. Liu, X. Wu, Q. Guan, S. Chen, L. Sun, Y. Guo, S. Wang, J. Song, E. M. Jeffries, C. He, F. L. Qing, X. Bao and Z. You, Adv. Mater., 2019, 31, 1901402 CrossRef PubMed.
- S. Liu, S. Chen, W. Shi, Z. Peng, K. Luo, S. Xing, J. Li, Z. Liu and L. Liu, Adv. Funct. Mater., 2021, 31, 2102225 CrossRef CAS.
- X. Lu, L. Zhang, J. Zhang, C. Wang and A. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 41421–41432 CrossRef CAS PubMed.
- D. Tian, Y. Song and L. Jiang, Chem. Soc. Rev., 2013, 42, 5184–5209 RSC.
- P. Bi, X. Liu, Y. Yang, Z. Wang, J. Shi, G. Liu, F. Kong, B. Zhu and R. Xiong, Adv. Mater. Technol., 2019, 4, 1900426 CrossRef CAS.
- K. Wei, T. Wang, Z. Wang, Y. Zhang and H. Xu, Sci. Sin.: Chim., 2018, 48, 1131–1140 Search PubMed.
- J. Liu, X. Zhang, Y. Liu, M. Rodrigo, P. D. Loftus, J. Aparicio-Valenzuela, J. Zheng, T. Pong, K. J. Cyr, M. Babakhanian, J. Hasi, J. Li, Y. Jiang, C. J. Kenney, P. J. Wang, A. M. Lee and Z. Bao, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 14769–14778 CrossRef CAS PubMed.
- M. Morra, E. Occhiello, R. Marola, F. Garbassi, P. Humphrey and D. Johnson, J. Colloid Interface Sci., 1990, 137, 11–24 CrossRef CAS.
- V. Roucoules, A. Ponche, A. Geissler, F. Siffer, L. Vidal, S. Ollivier, M. F. Vallat, P. Marie, J. C. Voegel, P. Schaaf and J. Hemmerlé, Langmuir, 2007, 23, 13136–13145 CrossRef CAS PubMed.
- A. J. Mazaltarim, J. M. Taylor, A. Konda, M. A. Stoller and S. A. Morin, ACS Appl. Mater. Interfaces, 2019, 11, 33452–33457 CrossRef CAS PubMed.
- D. A. Wicks and Z. W. Wicks, Prog. Org. Coat., 2001, 41, 1–83 CrossRef CAS.
- D. A. Wicks and Z. W. Wicks, Prog. Org. Coat., 1999, 36, 148–172 CrossRef CAS.
- R. G. Rinaldi, M. C. Boyce, S. J. Weigand, D. J. Londono and M. W. Guise, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 1660–1671 CrossRef CAS.
- S. S. Sarva, S. Deschanel, M. C. Boyce and W. Chen, Polymer, 2007, 48, 2208–2213 CrossRef CAS.
- T. Choi, D. Fragiadakis, C. M. Roland and J. Runt, Macromolecules, 2012, 45, 3581–3589 CrossRef CAS.
- X. Chen, P. Sun, H. Tian, X. Li, C. Wang, J. Duan, Y. Luo, S. Li, X. Chen and J. Shao, J. Mater. Chem. C, 2022, 10, 1039–1047 RSC.
- D. Zhao, J. Yang, Y. Wang and H. Li, Dyes Pigm., 2021, 197, 109864 CrossRef.
- L. Zhang, X. Zhang, H. Zhang, L. Xu, D. Wang, X. Lu and A. Zhang, Chem. Eng. J., 2022, 434, 134751 CrossRef CAS.
- Y. Guo, L. Yang, L. Zhang, S. Chen, L. Sun, S. Gu and Z. You, Adv. Funct. Mater., 2021, 31, 2106281 CrossRef CAS.
- L. Zhang, Z. Liu, X. Wu, Q. Guan, S. Chen, L. Sun, Y. Guo, S. Wang, J. Song, E. M. Jeffries, C. He, F. L. Qing, X. Bao and Z. You, Adv. Mater., 2019, 31, 32–34 Search PubMed.
- J. Shi, T. Zheng, Y. Zhang, B. Guo and J. Xu, ACS Sustainable Chem. Eng., 2020, 8, 1207–1218 CrossRef CAS.
- J. Shi, T. Zheng, Y. Zhang, B. Guo and J. Xu, Polym. Chem., 2021, 12, 2421–2432 RSC.
- J. Shi, T. Zheng, B. Guo and J. Xu, Polymer, 2019, 181, 121788 CrossRef CAS.
- T. Zheng, Y. Zhang, J. Shi, J. Xu and B. Guo, Mol. Simul., 2021, 47, 1258–1272 CrossRef CAS.
- T. Li, C. Zhang, Z. Xie, J. Xu and B. H. Guo, Polymer, 2018, 145, 261–271 CrossRef CAS.
- T. Zheng, T. Li, J. Shi, T. Wu, Z. Zhuang, J. Xu and B. Guo, Macromolecules, 2022, 55, 3020–3029 CrossRef CAS.
- B. Jin, H. Song, R. Jiang, J. Song, Q. Zhao and T. Xie, Sci. Adv., 2018, 4, eaao3865 CrossRef PubMed.
- J. Ling, M. Z. Rong and M. Q. Zhang, Chin. J. Polym. Sci., 2014, 32, 1286–1297 CrossRef CAS.
- S. Banerjee, R. Tripathy, D. Cozzens, T. Nagy, S. Keki, M. Zsuga and R. Faust, ACS Appl. Mater. Interfaces, 2015, 7, 2064–2072 CrossRef CAS PubMed.
- M. Abdallh, M. T. W. Hearn, G. P. Simon and K. Saito, Polym. Chem., 2017, 8, 5875–5883 RSC.
- W. Miao, W. Zou, B. Jin, C. Ni, N. Zheng, Q. Zhao and T. Xie, Nat. Commun., 2020, 11, 4257 CrossRef CAS PubMed.
- J. Wei, H. Wang, X. Jiang and J. Yin, Macromol. Chem. Phys., 2006, 207, 2321–2328 CrossRef CAS.
- J. Wei, X. S. Jiang and H. Y. Wang, Macromol. Chem. Phys., 2007, 208, 2303–2311 CrossRef CAS.
- T. Yin, S. R. Lavoie, S. Qu and Z. Suo, Cell Rep. Phys. Sci., 2021, 2, 100463 CrossRef CAS.
- L. Miao, H. Guo, J. Wan, H. Wang, Y. Song, H. Chen, X. Chen and H. Zhang, J. Micromech. Microeng., 2020, 30, 045001 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional information on the DSC, IR, stress relaxation, tensile test, swelling test, conductivity test, synthesis, and experiments of PU-DHBP and PU-AF. See DOI: https://doi.org/10.1039/d2mh01005d |
| This journal is © The Royal Society of Chemistry 2022 |