
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Excited states engineering enables efficient near-infrared lasing in nanographenes†
Giuseppe M.
Paternò‡
 a,
Qiang
Chen‡§
b,
Rafael
Muñoz-Mármol‡
c,
Michele
Guizzardi
d,
Víctor
Bonal
c,
Ryota
Kabe
e,
Alexander J.
Barker
a,
Pedro G.
Boj
f,
Shreyam
Chatterjee
g,
Yutaka
Ie
g,
José M.
Villalvilla
c,
José A.
Quintana
f,
Francesco
Scotognella
d,
Klaus
Müllen
bh,
María A.
Díaz-García
*c,
Akimitsu
Narita
*bi and
Guglielmo
Lanzani
*ad
a,
Qiang
Chen‡§
b,
Rafael
Muñoz-Mármol‡
c,
Michele
Guizzardi
d,
Víctor
Bonal
c,
Ryota
Kabe
e,
Alexander J.
Barker
a,
Pedro G.
Boj
f,
Shreyam
Chatterjee
g,
Yutaka
Ie
g,
José M.
Villalvilla
c,
José A.
Quintana
f,
Francesco
Scotognella
d,
Klaus
Müllen
bh,
María A.
Díaz-García
*c,
Akimitsu
Narita
*bi and
Guglielmo
Lanzani
*ad
aCenter for Nano Science and Technology, Istituto Italiano di Tecnologia (IIT), Via Pascoli 10, 20133, Milano, Italy. E-mail: guglielmo.lanzani@iit.it
bMax Planck Institute for Polymer Research, Ackermannweg 10, 55128, Mainz, Germany. E-mail: narita@mpip-mainz.mpg.de
cDepartamento de Física Aplicada and Instituto Universitario de Materiales de Alicante, Universidad de Alicante, 03080 Alicante, Spain. E-mail: maria.diaz@ua.es
dPhysics Department, Politecnico di Milano, Piazza L. da Vinci 32, 20133 Milano, Italy
eOrganic Optoelectronics Unit, Okinawa Institute of Science and Technology Graduate University, 1919-1 Tancha, Onna-son, Okinawa 904-0495, Japan
fDepartamento de Óptica, Farmacología y Anatomía and Instituto Universitario de Materiales de Alicante, Universidad de Alicante, 03080 Alicante, Spain
gThe Institute of Scientific and Industrial Research (SANKEN), Osaka University, 8-1 Mihogaoka, Ibaraki, Osaka 567-0047, Japan
hInstitute of Physical Chemistry, Johannes Gutenberg University Mainz, Duesbergweg 10-14, 55128 Mainz, Germany
iOrganic and Carbon Nanomaterials Unit, Okinawa Institute of Science and Technology Graduate University, 1919-1 Tancha, Onna-son, Okinawa 904-0495, Japan. E-mail: akimitsu.narita@oist.jp
First published on 24th September 2021
Abstract
The spectral overlap between stimulated emission (SE) and absorption from dark states (i.e. charges and triplets) especially in the near-infrared (NIR), represents one of the most effective gain loss channels in organic semiconductors. Recently, bottom-up synthesis of atomically precise graphene nanostructures, or nanographenes (NGs), has opened a new route for the development of environmentally and chemically stable materials with optical gain properties. However, also in this case, the interplay between gain and absorption losses has hindered the attainment of efficient lasing action in the NIR. Here, we demonstrate that the introduction of two fluoranthene imide groups to the NG core leads to a more red-shifted emission than the precursor NG molecule (685 vs. 615 nm) and also with a larger Stokes shift (45 nm vs. 2 nm, 1026 cm−1vs. 53 cm−1, respectively). Photophysical results indicate that, besides the minimisation of ground state absorption losses, such substitution permits to suppress the detrimental excited state absorption in the NIR, which likely arises from a dark state with charge-transfer character and triplets. This has enabled NIR lasing (720 nm) from all-solution processed distributed feedback devices with one order of magnitude lower thresholds than those of previously reported NIR-emitting NGs. This study represents an advance in the field of NGs and, in general, organic semiconductor photonics, towards the development of cheap and stable NIR lasers.
New conceptsThe interplay between optical gain and absorption losses in conjugated molecules, especially in the near-infrared (NIR) part of the spectrum, represents a crucial issue to tackle for building up efficient organic laser devices operating in this region. This has important practical implications, as there is a large demand for NIR organic emitters and lasers owing to their applications in biomedical imaging, night vision devices and telecommunications. Within this context, nanographenes have been introduced as promising photonic materials in the last decade, which display intense light emission in a wide spectral range with relatively high environmental and photostability. However, also these systems exhibit strong absorption losses in the NIR, which have been attributed to a dark state with charge-transfer character. This has hampered the attainment of NIR lasing emission at a low threshold in nanographenes, and might represent an impediment to their effective application as gain materials in lasers. Here, we report on a novel nanographene molecule, in which absorption losses are strongly minimised. This allows achieving lasing action in the NIR from solution-processed distributed feedback devices at a considerably lower threshold (one order of magnitude) than previously reported NIR emitting nanographenes. |
Introduction
The research of new π-conjugated organic semiconductors with optical gain properties has been an extremely popular topic in the last two decades,1–4 with the aim of building-up efficient, stable and low-cost laser devices operating from the visible to the near-infrared (NIR) spectral region. In particular, there is a large demand for NIR organic emitters and lasers,5,6 owing to their applications in biomedical imaging,7 night vision devices and telecommunications.8 Organic materials offer advantages in the development of optical gain media due to wide tunability of their optical features, notably stimulated emission (SE) cross section, by means of chemical design,9,10 and a very high processability from solution.11,12 On the other hand, progress in NIR lasing from organic semiconductors is limited by two fundamental issues: the fast internal conversion back to the ground state, accounted for by the energy gap law,13 which requires high pumping rate to reach population inversion; and the intra-gap absorption of the charge transfer state and triplets that overlaps spectrally with the SE band,14 reducing the gain bandwidth and hampering the occurrence of efficient lasing action. Lastly, photo-degradation under intense laser pumping and instability against air and moisture represent usually a weakness of organic materials.15In the last decade, graphene nanostructures have been proposed as efficient organic semiconductors that lend themselves to applications in optoelectronics and photonics.16–23 In particular, although graphene itself does not exhibit a band-gap, the quantum confinement of its wave function through the structural patterning into graphene nanostructures, as occurs in quasi-one-dimensional graphene nanoribbons24–27 and quasi-zero-dimensional graphene quantum dots, or nanographenes (NGs),28–30 allows opening of a finite band-gap in its electronic structure. Here, the greatest advantage of bottom-up synthesis over top-down fabrication methods lies in the possibility to finely tailor the size, shape, and edge structures of such graphene nanostructures, and, hence, to perform wavefunction engineering that in turn allows controlling the photophysical properties.25,31–34 For instance, it has been shown that NGs possessing zigzag edges have a small energy gap compatible with NIR emission and/or an open-shell biradical character.35,36 However, NGs with zigzag edges usually suffer from kinetic instabilities and are subject to oxidation.37–40
To this end, we have recently proposed a NG with both armchair and zigzag edges, namely dibenzo[hi,st]ovalene (DBOV), affording a small optical gap (1.93 eV), a highly intense red-emission,41,42 and amplified spontaneous emission (ASE) at 695 nm with remarkable operational stability.43 More recently, novel NGs with four zigzag edges (FZ1-3 derivatives) have shown strong ASE and lasing action from all-solution processed distributed feedback (DFB) devices in a wide spectral range.20 However DBOV,44 as well as the larger FZ compound (FZ3),45,46 exhibits a strong excited state absorption in the NIR that reduces considerably the optical gain in such a region. We have recently observed that this absorption is enhanced in the solid state,43 while it is also present in dilute solution and films.44,47 We thus attributed this to an intermolecular dark state with likely charge transfer (CT) character, which can be a precursor of free charges or triplets, similar to what has been found in dipentacene.48,49 Further detailed experimental and computational studies are needed to elucidate the nature of such a state. For instance, this can have topological origin, for instance connected to the edge-states.50 Furthermore, given that the CT would be stabilised in supramolecular aggregates (i.e. dimers and trimers47) also in dilute solution due to the strong tendency for these systems to undergo π-stacking, the minimisation of the aggregation behaviour of NGs, for instance via the judicious introduction of bulky substituents, is an important route to be explored for suppressing such an interplay. In any case, although this spectral overlap can be favourably exploited to achieve ultrafast optical switching,47 it represents a serious limitation for the attainment of efficient NIR NG-based lasers.
Here, we show that the attachment of fluoranthene imide (FAI) side moieties to the DBOV core makes it possible to shift the emission towards the NIR and, interestingly, to limit the occurrence of such detrimental absorption. These advantageous features allow achieving ASE and lasing action at 712–720 nm from solution-processed DFB devices at a considerably lower threshold (one order of magnitude) than previously reported NIR emitting NGs (down to 50 μJ cm−2). We preliminary attribute such an effect to the minimisation of the aggregation tendency in this molecule owing to the bulky substituents, and to wavefunction delocalization into the FAI side groups, which would permit synergistically to circumvent the formation of CT states linked to the NG core and/or to edge states.50
Results and discussion
Synthesis of DBOV-FAI
As shown in Fig. 1a, DBOV-FAI was synthesized by one-step Suzuki–Miyaura coupling of 3,11-dibromo-6,14-dimesityldibenzo[hi,st]ovalene (1)51 with fluoranthene diimide (FAI)-pinacol borate ester 2 in 50% yield. The 1H NMR spectrum of DBOV-FAI showed sharp and well-resolved peaks, which could be assigned with the assistance of 2D NMR (Fig. 1b). The high-resolution MALDI-TOF MS spectrum exhibited one intense peak with m/z of 1442.6043, in good agreement with the calculated value of 1442.5956 (Fig. 1c). Furthermore, the experimental isotopic distribution pattern (black) was consistent with the calculated spectrum based on the chemical composition of C106H78N2O4 (red), indicating the successful preparation of DBOV-FAI. Additional 1H and 13C NMR spectra are reported in Fig. S1 and S2 (ESI†).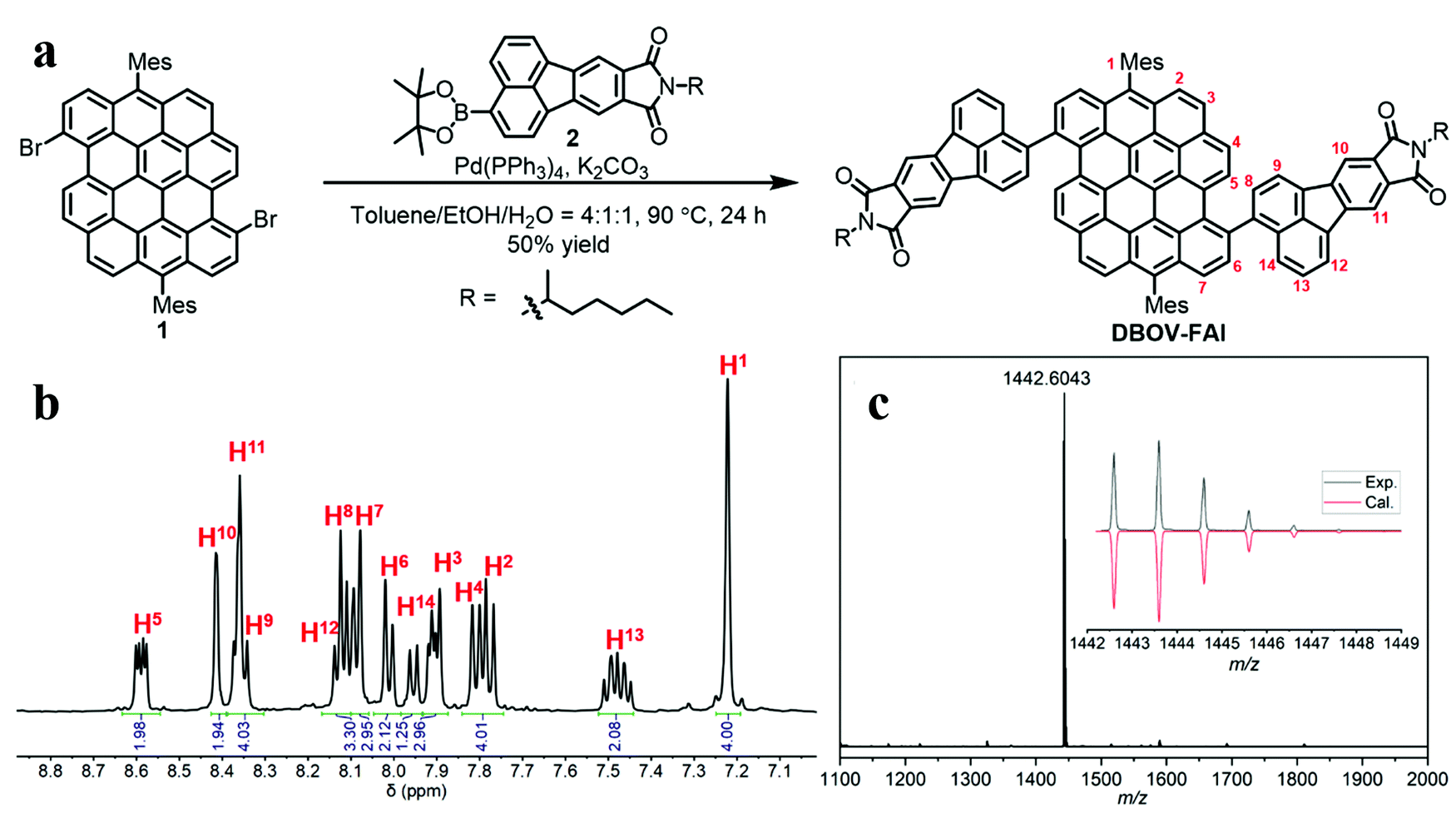 | ||
Fig. 1 (a) Synthetic route towards DBOV-FAI; (b) aromatic region of the 1H NMR spectrum of DBOV-FAI in C2D2Cl4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CS2 = 2:1 (500 MHz, 303 K); (c) high-resolution MALDI-TOF MS spectrum of DBOV-FAI (inset shows comparison of the experimental isotopic distribution pattern with the simulated pattern for C106H78N2O4). :CS2 = 2:1 (500 MHz, 303 K); (c) high-resolution MALDI-TOF MS spectrum of DBOV-FAI (inset shows comparison of the experimental isotopic distribution pattern with the simulated pattern for C106H78N2O4). | ||
Optical properties and DFT calculations
The absorption and photoluminescence (PL) spectra of DBOV-FAI are shown in Fig. 2a. To properly analyse the effect of the FAI substituents, spectra for FAI and for the mesityl-substituted DBOV molecule (DBOV-Mes) have also been included in the figure. DBOV-Mes exhibits mirroring absorption/PL spectra peaked at 613–615 nm with minimal Stokes shift (2 nm, 53 cm−1) owing to its rigid structure. In the new DBOV-FAI molecule the introduction of the FAI substituent, featuring absorption in the UV range and a large Stokes-shifted PL, leads to new absorption bands at 388 nm and 638 nm and to PL lying at 685 nm with the absolute PL quantum yield (PLQY) of 75% and fluorescence lifetime of 6.1 ns (Fig. S5, ESI†). Both absorption and PL lineshapes are less resolved in DBOV-FAI, due to spectral broadening. The energy levels and the frontier molecular orbital profile computed via a DFT method are displayed in Fig. 2b and c (see the ESI† for full details on DFT calculations and Fig. S6 for the calculated spectra). In DBOV-FAI, we see that while the HOMO orbital is totally localized in the DBOV unit, the LUMO partially extends to the FAI substituent. Conversely, the higher lying LUMO+1 and LUMO+2 states are localized in the FAI unit only. This suggests a substantial modification of the low-lying electronic structure compared to the DBOV core alone. Finally, in LUMO+3 the electron density lies in the DBOV unit, leading to a strong oscillator strength for the HOMO–LUMO+3 transition (Fig. 2a and Fig. S7, ESI†). Functionalizing the DBOV core with the side units offers a handle to carry out dynamic wavefunction engineering. The excited state wavefunctions in DBOV-FAI are indeed spread on the full molecular structure, but the delocalization is governed by the dihedral angle between the core and the side-groups. These introduce a distortion of the geometry and a consequent broad distribution of conformers. The former can be linked to the enlarged Stokes shift, while the latter is responsible for the observed inhomogeneous broadening in the DBOV-FAI spectrum. However, we will show that transition to the excited state changes dramatically the electron delocalization and affects the molecular geometry through electron-torsional coupling. In this way, we managed to further extend the emission to the NIR (35% of the total PL above 700 nm) and increase the Stokes shift to 45 nm (1026 cm−1) for the NG system.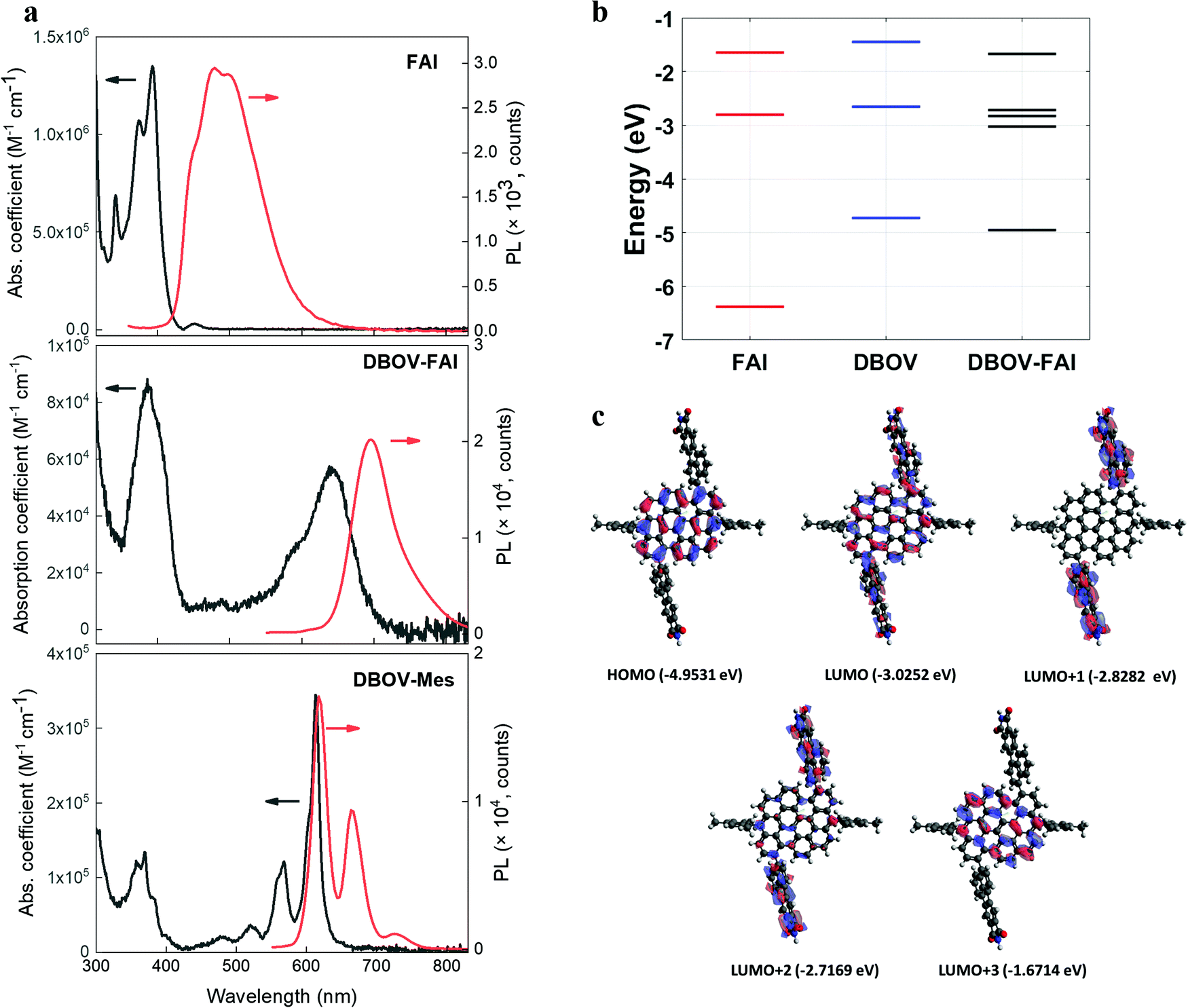 | ||
| Fig. 2 (a) Absorption (expressed as molar absorption coefficient) and photoluminescence spectra of FAI (compound 2 in Fig. 1a), DBOV-FAI and DBOV-Mes recorded in toluene at 10−5 M (b). For obtaining the photoluminescence spectra, the molecules were excited at 321 nm, 525 nm and 519 nm for FAI, DBOV-FAI and DBOV-Mes, respectively. (b) Energy levels of the three molecules calculated by means of the DFT method. (c) HOMO–LUMO distribution of DBOV-FAI based on DFT calculations. | ||
Ultrafast transient absorption spectroscopy
To gain insights into the photophysics of the novel DBOV-FAI system, we carried out ultrafast transient absorption (TA) spectroscopy in the ps–ns time regime on both DBOV-FAI and on the NG unit alone (DBOV-Mes) in diluted solution (0.02 mg mL−1 in toluene). To highlight the difference in the excited states absorption of DBOV-Mes and DBOV-FAI, we pumped the higher lying states (390 nm) that usually lead to stronger NIR absorption in zigzag-edged NGs47 than pumping in resonance.44 In a transient absorption experiment, an energetic pump pulse is absorbed by the sample permitting to populate the excited states, while a delayed, weaker and broadband probe pulse monitors the excited state absorption spectra and dynamics. At wavelengths coinciding with ground state absorption, the transmission of the probe will be enhanced due to the pump-induced ground state depletion (ΔT/T > 0), and the corresponding positive signal is called photobleaching (PB). Conversely, the probe will be attenuated by the excited state absorption events (ΔT/T < 0), and the relative negative signal is called excited state absorption (ESA). Finally, when the probe stimulates the emission of photons from the excited state this is also perceived as an increase of transmission (ΔT/T > 0), and the positive signal is called stimulated emission (SE).In Fig. 3a, we present the TA spectral evolution for DBOV-Mes in the 0.3 ps–1 ns range. At short pump–probe time delays, gain in the spectral region (615–700 nm) is fully overwhelmed by an ESA that extends further to the NIR (till the limit of our probed region, 750 nm). The positive SE signals from 0′ → 0 and 0′ → 1 transitions, at 615 nm (convoluted with PB) and 663 nm respectively, become visible for pump–probe delay times longer than 0.6 ps (Fig. 3b). This overlapping ESA substantially coincides with the spectrum reported earlier and obtained via pump-push-probe experiments in DBOV (Fig. S9, ESI†).47 In such an experiment, a second excitation pulse (push) induces quenching of the SE by shifting the population to a state that absorbs in the same spectral region of SE. Thus, based on past literature on zigzag-edged NGs32,33,43–45 and analogous long acene (i.e. pentacene49,52,53) molecules, also here ESA can be assigned to a CT dark state.54 The TA dynamics presented in Fig. 3b highlight the ultrafast quenching of the SE signal (Fig. 3b inset), and the subsequent partial recovery of the signal that decays following virtually the same trend of the PB (mono-exponential decay with estimated decay time = 2.6 ns). The ultrafast recovery of the SE signal, which occurs within 0.5 ps, can be explained by the very short lifetime of the CT state, as expected in a diluted solution. Transient spectra clearly show that there is also a long-lived ESA that does not directly overlap with SE (700–750 nm). We preliminary assign this transition to absorption from singlet excited states.
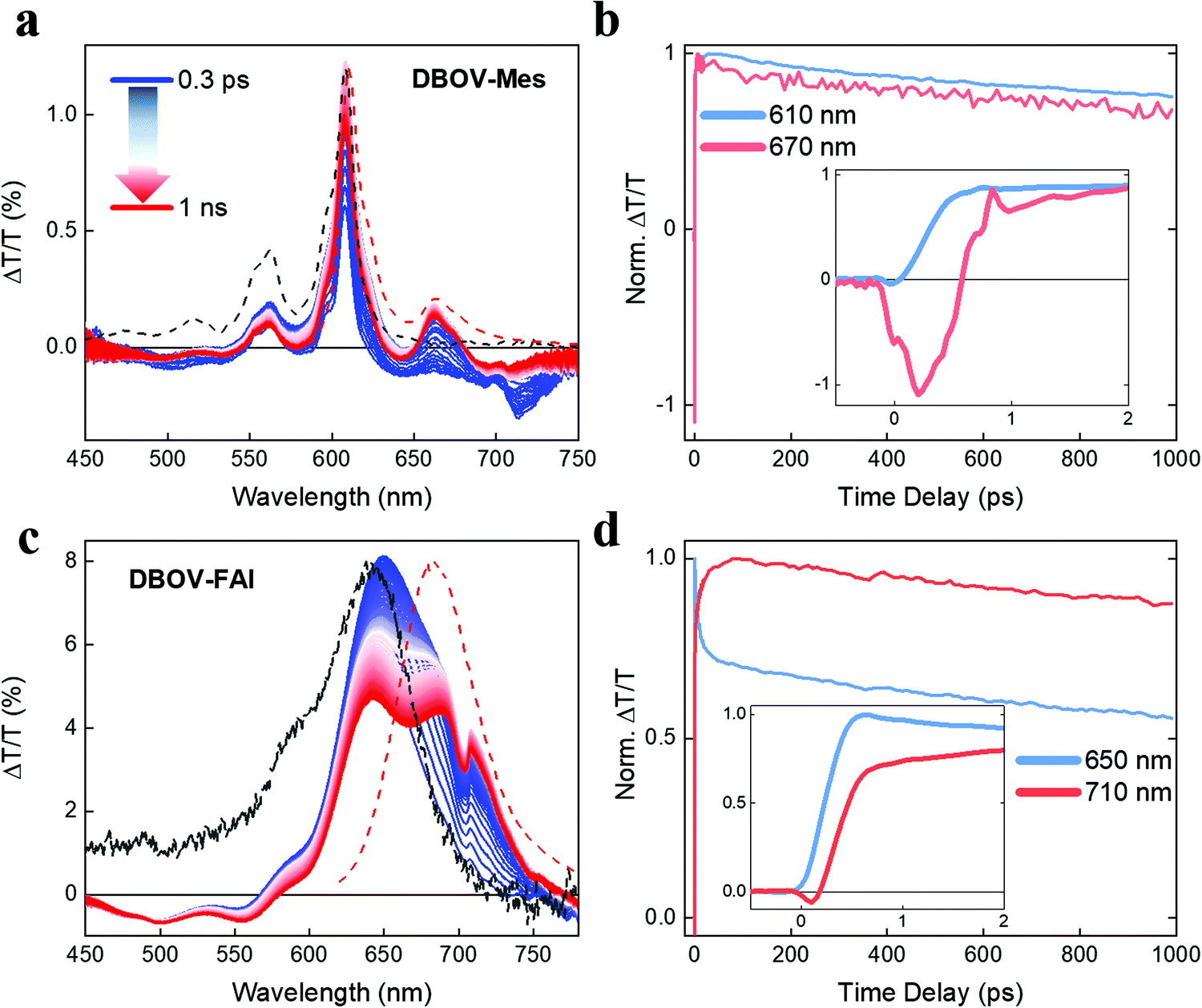 | ||
| Fig. 3 (a) Transient absorption spectra (excitation 390 nm) of DBOV-Mes from 0.3 ps to 1 ns. (b) Transient absorption dynamics at 610 nm (photobleaching signal, PB) and 670 nm (stimulated emission, SE). The inset shows the dynamics at early pump–probe delay time (0–2 ps) highlighting the substantial negative ΔT/T with FWHM of 0.5 ps in the SE signal that can be related to direct generation of CT states, whose absorption quenches the SE signal. (c) Transient absorption spectra of DBOV-FAI from 0.3 ps to 1 ns. (d) Transient absorption dynamics at 650 nm (PB) and 710 nm (SE). In this case, the SE is not appreciably quenched by CT state absorption. The inset shows the dynamics till 2 ps. The concentration of both molecules was 0.02 mg mL−1 in toluene. Dashed black and red lines represent the ground state absorption and photoluminescence spectra, respectively. | ||
With regards to DBOV-FAI transient absorption spectra (Fig. 3c), we note a broad positive feature extending from 570 nm to 770 nm, which can be related to the convolution of PB and SE. In this case, interestingly, we cannot observe any appreciable negative signal competing with gain. However, SE at 680–720 nm grows in time, while the main PB peak at 640 nm is reduced. The dynamical traces reported in Fig. 3c show a clear relation between the PB and SE signal, with the former decaying with time-constant of ∼10 ps and the latter growing with the same dynamics (see Fig. 3c inset). Such a dynamics, which is absent in DBOV-Mes, can be assigned to the conformational rearrangement occurring in the lowest excited state to reach a planar, fully conjugated geometry.55 This process is associated with narrowing of the optical gap due to the enhanced delocalization promoted by the planarization. PB has two contributions, namely ground state depletion and excited state stimulated emission. The latter is reduced by the gap renormalization, explaining the loss in PB. We note that when pumping at the edge (640 nm, Fig. S10, ESI†) we see qualitatively the same decay-rise behaviour, but with a shorter characteristic time (3 ps time constant). This may be explained by the different population initially prepared by the excitation: by pumping at the edge most of the excited molecules are close to a planar configuration, while by pumping at high energy a much broader distribution of conformers could be initially populated, due to the reduced dispersion of the states well above the gap. Starting from a distorted geometry the planarization time becomes longer, introducing a distribution of planarization time constants that accounts for the apparently slower kinetics. To verify this scenario, we calculated the transition energies and HOMO positions as a function of the dihedral angle, noticing that the vertical transition explores essentially distorted states (dihedral angle close to 90°, see Fig. S8 in the DFT section of the ESI†). Furthermore, the TA data taken in a polystyrene (PS) matrix (1 wt%) show indeed a longer rise and decay of all spectral components, while the PB→SE spectral migration due to the conformational relaxation is less evident, due to the freezing of the conformer population in the rigid matrix (Fig. S11, ESI†). Interestingly, we see that in DBOV-FAI the CT absorption state that competes with SE is largely suppressed. This is an important consequence of the new electronic structure and relaxation in the configurational space. Finally, we note that once formed, the planar excited state displays longer lifetime after pumping at high energy. This might be due to the presence of a large vibrational population caused by internal conversion, possibly similar to the phonon bottleneck effect observed in inorganic semiconductors.56
To investigate the possible presence of long-living photogenerated species, i.e. triplets, we also run a TA experiment in the ns–ms regime (Fig. S12, ESI†). Interestingly, in DBOV-Mes we observe the rising (∼20 ns) of two excited state absorption peaks centred at 530 nm and 680 nm, with the latter strongly overlapping with the stimulated emission peak. The long growth of such a signal can be compatible with the formation of triplets from Snvia intersystem crossing, as it has been observed in analogous acenes, such as pentacene.57,58 This is in fact an important quenching mechanism that can hamper the achievement of long pulse excitation lasing.14 In this scenario, the absorption peak in the NIR (750–850 nm) decays with the same dynamics of the photobleaching, and can be indeed attributed to absorption from singlet excited states, as it was inferred from the ps–ns TA data. On the other hand, DBOV FAI simply shows photobleaching peaks alongside the relative excited state absorptions that decay uniformly with the same decay time, while we cannot see any signature of triplet absorption.
To summarize, in the ultrafast time regime (sub-ps), DBOV-Mes undergoes a quenching of the SE that we attribute preliminary to two effects, one being intermolecular and one intramolecular: (i) the CT would be likely stabilised in supramolecular aggregates (intermolecular donor–acceptor). The presence of the FAI groups appended on the bay positions can prevent the intermolecular π–π aggregation and the formation of the CT state; (ii) the electron delocalisation brought about by the substituents will possibly decrease the role of charge transfer excitons connected to edge-states.50 In addition, the long-delay TA data suggest that the introduction of the FAI substituents leads to the suppression of the intersystem crossing pathway that occurs in DBOV-Mes. We speculate that in DBOV-FAI the first singlet energy is lowered by virtue of the electronic delocalisation brought about by the FAI groups (see also the DFT calculations). On the other hand, the triplet energy that does not depend strongly on the degree of delocalisation59 should be less affected by the substitution. This, in turn, would render the spin flip process from S1 to the triplet state less efficient. A pictorial representation of this photophysical scenario is reported in Fig. 4.
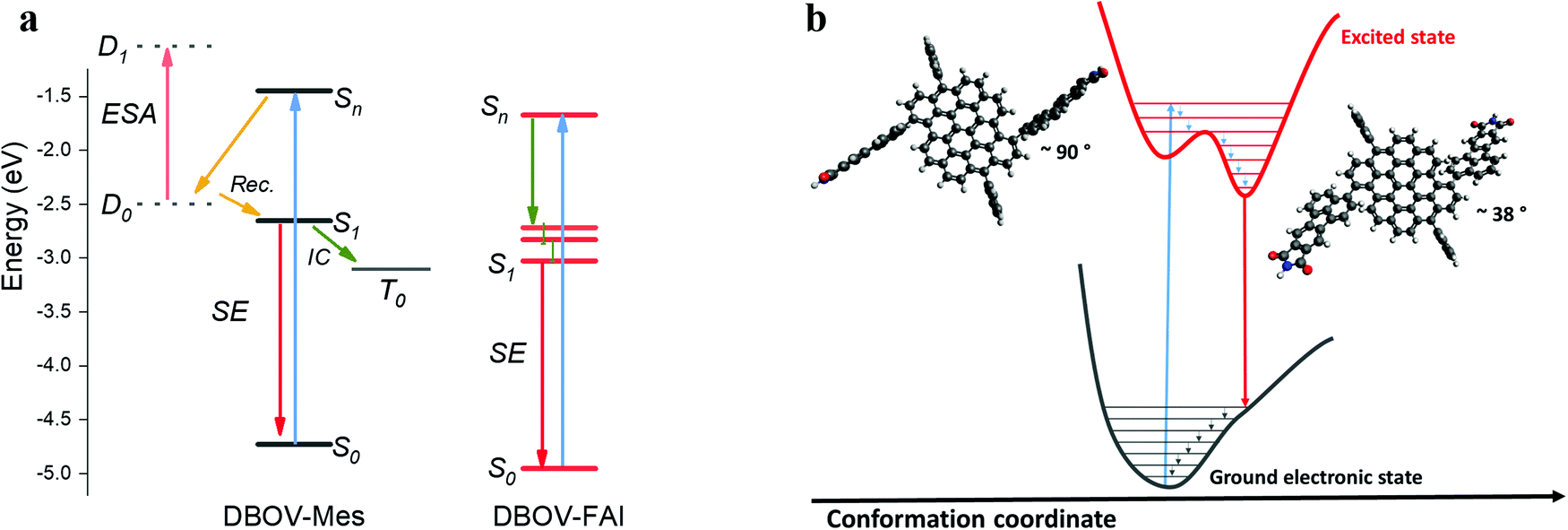 | ||
| Fig. 4 (a) Depiction of the possible excited state processes in DBOV-Mes and DBOV-FAI. The energy levels of the two molecules were computed via the DFT method, while the energetic position of the charge transfer state in DBOV-Mes has been arbitrarily positioned in the diagram based on previous experiments on analogous molecules.43,44,47 (b) Pictorial representation in the configurational space of the planarization process of DBOV-FAI, which arises from TA data. The vertical transition to the higher lying state explores essentially a distorted configuration, with a dihedral angle close to 90°. Radiative relaxation to the ground state occurs after planarization (optimised dihedral angle via DFT method is ∼38°). TA spectroscopy strongly indicates that the excited state relaxation in DBOV-FAI is dominated essentially by geometrical relaxations from Sn to S1. On the other hand, both the ultrafast quenching likely due to the formation of CT excitons and intersystem crossing are suppressed in this molecule. | ||
ASE and lasing action in DBOV-FAI
Next we characterized the ASE performance of DBOV-FAI under nanosecond pulse excitation (and determined the net gain coefficient, gnet). For comparison purposes, experiments with DBOV-Mes were also performed. Here, the NG compound was dispersed in an optically inert PS matrix (1 wt%) to avoid fluorescence self-quenching and deposited as a thin waveguide film, following a procedure used in previous studies.43,44,47 All ASE parameters are collected in Table S6 (ESI†). The DBOV-FAI doped film displayed ASE (under pumping at 415 nm) at λASE = 712 nm (Fig. 5a), above a threshold of 310 μJ cm−2 (Fig. 5b), presenting a linewidth of 15 nm. Remarkably, this threshold value is more than one order of magnitude lower than that reported for the NIR emission of FZ3 NG (λASE = 739 nm and Eth-ASE = 4400 mJ cm−2). ASE measurements with DBOV-FAI containing films were also performed under excitation in resonance with the S0–S1 transition (at 644 nm), observing a 2 nm redshift of the ASE peak, equal linewidth and a similar threshold of 270 μJ cm−2 (Fig. S13, ESI†). Complementarily, we determined the waveguide losses (α) and the net gain coefficient through a recently proposed method,60 which allows us to simultaneously calculate gain and losses through fits of the ASE and PL spectra. For excitation at either 415 nm or 644 nm, we found a similar gain-dependence on the pump fluence behaviour (Fig. 5c) and losses (α ∼5 cm−1, Fig. S14, ESI†). These results are consistent with the similar ASE thresholds observed at both excitation wavelengths. | ||
| Fig. 5 (a) PL spectra vs. excitation fluence for DBOV-FAI:polystyrene film (1 wt%) under 415 nm excitation, showing ASE action above 310 μJ cm−2. (b) Output intensity (Iout, black open squares) and linewidth, defined as full width at half maximum (FWHM, red solid circles), as functions of excitation energy density (Epump). (c) Net gain (gnet) at ASE wavelength versus Epump with the excitation at 415 nm (black squares) and 644 nm (red circles). Lines in (b) and (c) are guides to the eye. Design and properties of the distributed feedback (DFB) laser based on polystyrene:DBOV-FAI (1 wt%) thin films. (d) Device scheme, including a top-layer resonator which consists of a relief grating (Λ, grating period; d, grating depth) over the active film (h, film thickness). (e) Emission spectra for a DBOV-FAI laser under 415 nm excitation. Each mode is associated to a given waveguide mode (TM0 or TE0). The inset shows an image of the laser emission. (f) Output intensity (Iout, left axis, open squares) and linewidth defined as the full width at half maximum (FWHM, right axis, full circles) as functions of pump energy density for TM0 (red) and TE0 (black) laser modes of the device under 415 nm excitation, showing laser thresholds of 250 μJ cm−2 and 60 μJ cm−2, respectively. Lines are guides to the eye. | ||
Distinct features of the ASE performance of DBOV-Mes, with respect to that of DBOV-FAI, are its shorter emission wavelength (λASE = 670 nm) and smaller linewidth (4 nm) (see Table S6, ESI†). Noticeably, the large ASE linewidth of DBOV-FAI, which is presumably related to its large PL linewidth, might be convenient for the purpose of fabricating efficient lasers in a wide spectral range. It should also be noted, that DBOV-FAI is to date, the only NG that has shown ASE under pumping at either higher-lying states or at optical gap. For instance, DBOV-Mes has shown ASE under ns excitation,44 only when pumping in resonance with its S0–S1 transition (613 nm, Fig. 2a, Fig. S15 and S16, ESI†).
With regards to the photostability of the materials under the intense excitation to obtain ASE, it is characterized through the so-called ASE half-life parameter (τ1/2), defined as the number of pulses needed for the output intensity to reach half of its initial value. The experiments were conducted under continuous excitation (3.7 ns pulse duration at λpump = 415 nm; and 4.6 ns at 644 nm, 10 Hz repetition rate) with an intensity around 4 times above the ASE threshold, in air and without any encapsulation. DBOV-FAI showed an excellent performance, at either 415 nm or 644 nm excitation, showing τ1/2 values of 1.3 × 105 pulses and 3.5 × 105 pulses, respectively (Fig. S17, ESI†). Under similar pumping conditions (λpump = 613 nm, 4.5 ns and 10 Hz), DBOV-Mes presents a better photostability performance (τ1/2 = 6.6 × 105 pulses) and, for FZ3, a shorter value was determined (τ1/2 = 1.5 × 105 pulses) though it was measured under a significantly more intense pump (668 nm, 4.6 ns, 10 Hz, 6000 μJ cm−2).20 In order to establish a suitable comparison, we can estimate the total energy density deposited in the sample after the exposition to this number of pump pulses (E1/2) by determining the portion of energy absorbed for each pulse and multiplying by τ1/2. For DVOB-FAI, values of E1/2 = 2 and 5 kJ cm−2 are estimated for the 415 and 644 nm excitations, meanwhile E1/2 = 15 and 30 kJ cm−2 are determined for DBOV-Mes and FZ3, respectively.
Note that these results have been obtained for the NGs dispersed in PS, i.e. an inert matrix. For the prospect of electrical pumping, electroactive matrixes should be used instead. For example, 4,4′-bis(N-carbazikyl)-1,1′-biphenyl has been used in the literature for LEDs and in some cases for electrically-pumped organic lasers.6,61 In this regard, further studies with DBOOV-FAI doped into electroactive matrixes are of great interest and will be the subject of future investigations.
Finally, we demonstrate the relevance of DBOV-FAI for practical applications by manufacturing all-solution processed DFB lasers (Fig. 5d), by following the procedure adopted in previous studies.20,62–64 The DFB resonators are made of a water-soluble dichromated gelatine (DCG) layer, with a relief grating engraved by holographic lithography (HL), deposited on top of the DBOV-FAI:PS film, full details about DFB fabrication are given in the ESI† section. This top-layer architecture presents several advantages with respect to other DFB configurations, e.g. constant thickness of the active film, on-a-chip multi-colour emission62 and preservation of the active film. The grating periods were chosen to obtain devices operating in the second-order of diffraction (m = 2) of the Bragg condition (eqn (1)):15,65
| mλBragg = 2neffΛ | (1) |
DFB devices based on DBOV-FAI:PS films show two very narrow peaks (linewidth < 1.3 nm, limited by instrumental spectral resolution; Fig. 5e and Table S7, ESI†) under 415 nm excitation. These peaks correspond to two laser modes that can be associated with the two propagating modes of the waveguide, i.e. TM0 and TE0. The peak whose light is polarized parallel to the grating lines corresponds to the TE0 waveguide mode, while the peak whose light is polarized perpendicularly to the grating lines, is associated with the TM0 waveguide mode. This assignment can be corroborated through calculations of the neff for each waveguide mode (e.g. with a mode solver software program67) and its corresponding λBragg (eqn (1)).
The inset in Fig. 5e shows an image of the light emitted by the device. The divergence of the laser beam in the direction perpendicular to the grating lines is diffraction limited (θd = 0.9 × 10−3 rad), which is a consequence of its great spatial coherence. Remarkably, laser emission occurs in the NIR at very low thresholds: the two laser modes appear at 716.3 nm (TM0) and 720.3 nm (TE0), displaying lasing thresholds of 250 μJ cm−2 and 50 μJ cm−2, respectively, under 415 nm excitation (Fig. 5f). In line with the ASE results, these threshold values are more than one order of magnitude lower than those previously reported for DFB lasers based on the four-zigzag edged nanographene FZ3, which presented two laser modes at 739.5 nm and 744.5 nm (TM0 and TE0 modes, respectively).45 There have been reports of NIR organic lasers based on molecular aggregates, single crystals and nanowires, whose thresholds are lower, but with more complex device architectures.5 Note that the low-threshold values reported here have been obtained under nanosecond excitation, while usually low-threshold values reported in the literature are obtained under picosecond or even femtosecond excitation. This is an important aspect to take into account for applications. Complementarily, we manufactured a second DBOV-FAI based DFB with a slightly different grating period (device 2, see Table S7, ESI†). This device showed a similar performance, in terms of threshold, to that previously described and showed emission at 711.9 nm and 716.6 nm, for TM0 and TE0 modes, respectively. All the relevant information can be found in Fig. S18 and S20 and Table S7 (ESI†).
Conclusions
We have reported the synthesis of a novel NG, namely DBOV-FAI, which displays ASE (712 nm) and lasing (712–720 nm) in the NIR region. By chemical engineering of the DBOV core with side groups we obtained a molecule, whose Stokes shift between absorption and emission is enhanced (almost 50 nm). This favours optical gain and lasing by reducing intrinsic losses. Furthermore, the absorption from CT states, which usually reduces the optical gain and increases the lasing threshold in the NIR due to spectral overlap, is here strongly minimised. We discussed in detail the DBOV-FAI photophysics, invoking intramolecular torsional motion to explain the kinetics of PB and SE. We conjecture that the molecular conformation, and in particular the dihedral angle at the junction of the side FAI groups with the DBOV core, governs the electron delocalization and thus the inhomogeneous broadening of the electronic transition energies. We estimate that planarization occurs in the lowest excited state, in the time scale ranging from 20 to 80 ps, depending on the excitation energy. Interestingly, we could not find any signature of detrimental CT and triplet absorption in DBOV-FAI, a result that we preliminarily attribute to the delocalisation of the electronic wavefunction in the substituents and suppression of the intermolecular CT excitons, due to the minimisation of the tendency of DBOV-FAI to undergo π-stacking. The large Stokes shift, as well as the reduction of the excited state absorption in the gain region, have allowed achieving ASE and DFB lasing in the NIR at a relatively low pumping threshold. These findings are important for further progress in two relevant aspects: (i) the attainment of NIR lasing with stable NGs, which can be appealing for the development of cheap laser sources useful in telecommunications, night vision and bio-applications; (ii) the spectral disentanglement between optical gain and absorption from dark charged states and triplets, which can open fascinating opportunities in the context of the development of continuous wave- and electrically-pumped organic diode lasers.Author contributions
G. M. P. performed the transient absorption experiments together with M. G., analysed the data and wrote the manuscript. Q. C. synthesised and characterised the molecule and contributed to manuscript writing. R. M. M. performed the ASE and lasing experiments and analysed the data together with V. B., P. G. B., J. M. V, J. A. Q., and contributed to manuscript writing. R. K. carried out the time-resolved photoluminescence experiments and analysed the data. A. J. B. performed the long-delay transient absorption measurements. S. C. and Y. I. synthesised and characterised the FAI molecule. F. S. performed the DFT calculations and contributed to data interpretation and manuscript writing. K. M. supervised the synthesis process. M. A. D. G., A. N. and G. L. conceived and supervised the work, and contributed to data interpretation and manuscript writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. M. P. thanks Fondazione Cariplo for financial support (grant no. 2018-0979). R. M. M., P. G. B., J. M. V., J. A. Q. and M. A.D. G. acknowledge funding from the Ministerio de Economía y Competitividad (MINECO), the European Regional Development Fund (FEDER) and European Social Funds (ESF; grant no. MAT2015-66586-R and FPI fellowship BES-2016-077681), as well as to the Ministerio de Ciencia e Innovación (grant no. PID2020-119124RB-I00). Q. C., K. M. and A. N. are grateful for the financial support by the Max Planck Society and the ANR-DFG NLE Grant GRANAO by DFG 431450789. K. M. acknowledges a fellowship from Gutenberg Research College, Johannes Gutenberg University Mainz. R. K. and A. N. appreciate the support by the Okinawa Institute of Science and Technology Graduate University. Y. I. and A. N. acknowledge financial support from the Japan Society of Promotion Science Program for Advancing Strategic International Networks to Accelerate the Circulation of Talented Researchers. Open Access funding provided by the Max Planck Society.Notes and references
- N. Tessler, G. J. Denton and R. H. Friend, Nature, 1996, 382, 695–696 CrossRef CAS
.
- I. D. W. Samuel and G. A. Turnbull, Chem. Rev., 2007, 107, 1272–1295 CrossRef CAS
- Y. Jiang, Y.-Y. Liu, X. Liu, H. Lin, K. Gao, W.-Y. Lai and W. Huang, Chem. Soc. Rev., 2020, 49, 5885–5944 RSC
- A. L. Kanibolotsky, I. F. Perepichka and P. J. Skabara, Chem. Soc. Rev., 2010, 39, 2695–2728 RSC
- J.-J. Wu, X.-D. Wang and L.-S. Liao, ACS Photonics, 2019, 6, 2590–2599 CrossRef CAS
- D.-H. H. Kim, A. D’Aléo, X.-K. K. Chen, A. D. S. S. Sandanayaka, D. Yao, L. Zhao, T. Komino, E. Zaborova, G. Canard, Y. Tsuchiya, E. Choi, J. W. Wu, F. Fages, J.-L. L. Brédas, J.-C. C. Ribierre, C. Adachi, A. D’Aléo, X.-K. K. Chen, A. D. S. S. Sandanayaka, D. Yao, L. Zhao, T. Komino, E. Zaborova, G. Canard, Y. Tsuchiya, E. Choi, J. W. Wu, F. Fages, J.-L. L. Brédas, J.-C. C. Ribierre and C. Adachi, Nat. Photonics, 2018, 12, 98–104 CrossRef CAS
- G. Hong, A. L. Antaris and H. Dai, Nat. Biomed. Eng., 2017, 1, 0010 CrossRef CAS
- G. Qian and Z. Y. Wang, Chem. – Asian J., 2010, 5, 1006–1029 CrossRef CAS PubMed
- C. Zhang, Y. Yan, Y. S. Zhao and J. Yao, Acc. Chem. Res., 2014, 47, 3448–3458 CrossRef CAS PubMed
- W. Yao, Y. Yan, L. Xue, C. Zhang, G. Li, Q. Zheng, Y. S. Zhao, H. Jiang and J. Yao, Angew. Chem., Int. Ed., 2013, 52, 8713–8717 CrossRef CAS
- C. Zhang, C. L. Zou, Y. Zhao, C. H. Dong, C. Wei, H. Wang, Y. Liu, G. C. Guo, J. Yao and Y. S. Zhao, Sci. Adv., 2015, 1, e1500257 CrossRef
- G. M. Paternò, M. W. A. Skoda, R. Dalgliesh, F. Cacialli and V. García Sakai, Sci. Rep., 2016, 6, 34609 CrossRef PubMed
- R. Englman and J. Jortner, Mol. Phys., 1970, 18, 285–287 CrossRef
- J. Clark and G. Lanzani, Nat. Photonics, 2010, 4, 438–446 CrossRef CAS
- A. J. C. Kuehne and M. C. Gather, Chem. Rev., 2016, 116, 12823–12864 CrossRef CAS
- Y. W. Son, M. L. Cohen and S. G. Louie, Nature, 2006, 444, 347–349 CrossRef CAS
- B. Trauzettel, D. V. Bulaev, D. Loss and G. Burkard, Nat. Phys., 2007, 3, 192–196 Search PubMed
- S. Mishra, D. Beyer, K. Eimre, S. Kezilebieke, R. Berger, O. Gröning, C. A. Pignedoli, K. Müllen, P. Liljeroth, P. Ruffieux, X. Feng and R. Fasel, Nat. Nanotechnol., 2020, 15, 22–28 CrossRef CAS
- S. Mishra, D. Beyer, R. Berger, J. Liu, O. Gröning, J. I. Urgel, K. Müllen, P. Ruffieux, X. Feng and R. Fasel, J. Am. Chem. Soc., 2020, 142, 1147–1152 CrossRef CAS
- V. Bonal, R. Muñoz-Mármol, F. Gordillo Gámez, M. Morales-Vidal, J. M. Villalvilla, P. G. Boj, J. A. Quintana, Y. Gu, J. Wu, J. Casado and M. A. Díaz-García, Nat. Commun., 2019, 10, 1–10 CrossRef CAS
- Z. Jin, P. Owour, S. Lei and L. Ge, Curr. Opin. Colloid Interface Sci., 2015, 20, 439–453 CrossRef CAS
- S. Zhao, J. Lavie, L. Rondin, L. Orcin-Chaix, C. Diederichs, P. Roussignol, Y. Chassagneux, C. Voisin, K. Müllen, A. Narita, S. Campidelli and J.-S. Lauret, Nat. Commun., 2018, 9, 3470 CrossRef
- G. Soavi, S. Dal Conte, C. Manzoni, D. Viola, A. Narita, Y. Hu, X. Feng, U. Hohenester, E. Molinari, D. Prezzi, K. Müllen and G. Cerullo, Nat. Commun., 2016, 7, 11010 CrossRef CAS PubMed
- Y. W. Son, M. L. Cohen and S. G. Louie, Phys. Rev. Lett., 2006, 97, 216803 CrossRef PubMed
- A. Narita, X. Feng, Y. Hernandez, S. A. Jensen, M. Bonn, H. Yang, I. A. Verzhbitskiy, C. Casiraghi, M. R. Hansen, A. H. R. Koch, G. Fytas, O. Ivasenko, B. Li, K. S. Mali, T. Balandina, S. Mahesh, S. De Feyter and K. Müllen, Nat. Chem., 2014, 6, 126–132 CrossRef CAS
- M. Y. Han, B. Özyilmaz, Y. Zhang and P. Kim, Phys. Rev. Lett., 2007, 98, 206805 CrossRef PubMed
- L. Yang, C. H. Park, Y. W. Son, M. L. Cohen and S. G. Louie, Phys. Rev. Lett., 2007, 99, 186801 CrossRef
- L. Cao, M. J. Meziani, S. Sahu and Y. P. Sun, Acc. Chem. Res., 2013, 46, 171–182 CrossRef CAS PubMed
- R. Liu, D. Wu, X. Feng and K. Müllen, J. Am. Chem. Soc., 2011, 133, 15221–15223 CrossRef CAS
- L. A. Ponomarenko, F. Schedin, M. I. Katsnelson, R. Yang, E. W. Hill, K. S. Novoselov and A. K. Geim, Science, 2008, 320, 356–358 CrossRef CAS
- A. Narita, X. Y. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616–6643 RSC
- X. Yao, X.-Y. Wang, C. Simpson, G. M. Paternò, M. Guizzardi, M. Wagner, G. Cerullo, F. Scotognella, M. D. Watson, A. Narita and K. Müllen, J. Am. Chem. Soc., 2019, 141, 4230–4234 CrossRef CAS PubMed
- Y. Hu, G. M. Paternò, X. Y. Wang, X. C. Wang, M. Guizzardi, Q. Chen, D. Schollmeyer, X. Y. Cao, G. Cerullo, F. Scotognella, K. Müllen and A. Narita, J. Am. Chem. Soc., 2019, 141, 12797–12803 CrossRef CAS PubMed
- X.-Y. Wang, A. Narita and K. Müllen, Nat. Rev. Chem., 2018, 2, 0100 CrossRef
- Z. Sun, Q. Ye, C. Chi and J. Wu, Chem. Soc. Rev., 2012, 41, 7857–7889 RSC
- J. Li, K. Zhang, X. Zhang, K.-W. Huang, C. Chi and J. Wu, J. Org. Chem., 2010, 75, 856–863 CrossRef CAS
- J. Liu, P. Ravat, M. Wagner, M. Baumgarten, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2015, 54, 12442–12446 CrossRef CAS PubMed
- A. Konishi, Y. Hirao, K. Matsumoto, H. Kurata, R. Kishi, Y. Shigeta, M. Nakano, K. Tokunaga, K. Kamada and T. Kubo, J. Am. Chem. Soc., 2013, 135, 1430–1437 CrossRef CAS PubMed
- M. R. Ajayakumar, Y. Fu, J. Ma, F. Hennersdorf, H. Komber, J. J. Weigand, A. Alfonsov, A. A. Popov, R. Berger, J. Liu, K. Müllen and X. Feng, J. Am. Chem. Soc., 2018, 140, 6240–6244 CrossRef CAS
- M. R. Ajayakumar, J. Ma, A. Lucotti, K. S. Schellhammer, G. Serra, E. Dmitrieva, M. Rosenkranz, H. Komber, J. Liu, F. Ortmann, M. Tommasini and X. Feng, Angew. Chem., Int. Ed., 2021, 60, 13853–13858 CrossRef CAS PubMed
- D. M. Coles, Q. Chen, L. C. Flatten, J. M. Smith, K. Müllen, A. Narita and D. G. Lidzey, Nano Lett., 2017, 17, 5521–5525 CrossRef CAS PubMed
- Q. Chen, S. Thoms, S. Stöttinger, D. Schollmeyer, K. Müllen, A. Narita and T. Basché, J. Am. Chem. Soc., 2019, 141, 16439–16449 CrossRef CAS
- G. M. Paternò, Q. Chen, X. Y. Wang, J. Liu, S. G. Motti, A. Petrozza, X. Feng, G. Lanzani, K. Müllen, A. Narita and F. Scotognella, Angew. Chem., Int. Ed., 2017, 56, 6753–6757 CrossRef
- G. M. Paternò, L. Nicoli, Q. Chen, K. Müllen, A. Narita, G. Lanzani and F. Scotognella, J. Phys. Chem. C, 2018, 122, 25007–25013 CrossRef
- R. Muñoz-Mármol, V. Bonal, G. M. Paternò, A. M. Ross, P. G. Boj, J. M. Villalvilla, J. A. Quintana, F. Scotognella, C. D’Andrea, S. Sardar, G. Lanzani, Y. Gu, J. Wu and M. A. Díaz-García, Nanomaterials, 2020, 10, 1525 CrossRef
- R. Muñoz-Mármol, F. Gordillo, V. Bonal, J. M. Villalvilla, P. G. Boj, J. A. Quintana, A. M. Ross, G. M. Paternò, F. Scotognella, G. Lanzani, A. Derradji, J. C. Sancho-García, Y. Gu, J. Wu, J. Casado and M. A. Díaz-García, Adv. Funct. Mater., 2021, 2105073, 2105073 CrossRef
- G. M. Paternò, L. Moretti, A. J. Barker, Q. Chen, K. Müllen, A. Narita, G. Cerullo, F. Scotognella and G. Lanzani, Adv. Funct. Mater., 2019, 29, 1805249 CrossRef
- S. Lukman, A. J. Musser, K. Chen, S. Athanasopoulos, C. K. Yong, Z. Zeng, Q. Ye, C. Chi, J. M. Hodgkiss, J. Wu, R. H. Friend and N. C. Greenham, Adv. Funct. Mater., 2015, 25, 5452–5461 CrossRef CAS
- S. Lukman, K. Chen, J. M. Hodgkiss, D. H. P. Turban, N. D. M. Hine, S. Dong, J. Wu, N. C. Greenham and A. J. Musser, Nat. Commun., 2016, 7, 1–13 Search PubMed
- G. M. Paternò, Goudappagouda, Q. Chen, G. Lanzani, F. Scotognella and A. Narita, Adv. Opt. Mater., 2021, 2100508 CrossRef
- Q. Chen, D. Wang, M. Baumgarten, D. Schollmeyer, K. Müllen and A. Narita, Chem. – Asian J., 2019, 14, 1703–1707 CrossRef CAS
- B. J. Walker, A. J. Musser, D. Beljonne and R. H. Friend, Nat. Chem., 2013, 5, 1019–1024 CrossRef CAS PubMed
- V. K. Thorsmølle, R. D. Averitt, J. Demsar, D. L. Smith, S. Tretiak, R. L. Martin, X. Chi, B. K. Crone, A. P. Ramirez and A. J. Taylor, Phys. Rev. Lett., 2009, 102, 017401 CrossRef
- P. M. Zimmerman, Z. Zhang and C. B. Musgrave, Nat. Chem., 2010, 2, 648–652 CrossRef CAS PubMed
- J. Clark, T. Nelson, S. Tretiak, G. Cirmi and G. Lanzani, Nat. Phys., 2012, 8, 225–231 Search PubMed
- J. Urayama, T. B. Norris, J. Singh and P. Bhattacharya, Phys. Rev. Lett., 2001, 86, 4930–4933 CrossRef CAS
- A. Ito, A. Shimizu, N. Kishida, Y. Kawanaka, D. Kosumi, H. Hashimoto and Y. Teki, Angew. Chem., Int. Ed., 2014, 53, 6715–6719 CrossRef CAS
- D. Lubert-Perquel, E. Salvadori, M. Dyson, P. N. Stavrinou, R. Montis, H. Nagashima, Y. Kobori, S. Heutz and C. W. M. Kay, Nat. Commun., 2018, 9, 4222 CrossRef PubMed
- D. Beljonne, J. Cornil, R. H. Friend, R. A. J. Janssen and J. L. Brédas, J. Am. Chem. Soc., 1996, 118, 6453–6461 CrossRef CAS
- L. Cerdán, Opt. Laser Technol., 2020, 121, 105814 CrossRef
- A. S. D. Sandanayaka, T. Matsushima, F. Bencheikh, S. Terakawa, W. J. Potscavage, C. Qin, T. Fujihara, K. Goushi, J. C. Ribierre and C. Adachi, Appl. Phys. Express, 2019, 12, 061010 CrossRef CAS
- J. A. Quintana, J. M. Villalvilla, M. Morales-Vidal, P. G. Boj, X. Zhu, N. Ruangsupapichat, H. Tsuji, E. Nakamura and M. A. Díaz-García, Adv. Opt. Mater., 2017, 5, 1700238 CrossRef
- V. Bonal, J. A. Quintana, J. M. Villalvilla, P. G. Boj and M. A. Díaz-García, Sci. Rep., 2019, 9, 11159 CrossRef PubMed
- V. Bonal, J. M. Villalvilla, J. A. Quintana, P. G. Boj, N. Lin, S. Watanabe, K. Kazlauskas, O. Adomeniene, S. Jursenas, H. Tsuji, E. Nakamura and M. A. Díaz-García, Adv. Opt. Mater., 2020, 8, 2001153 CrossRef CAS
- C. Grivas and M. Pollnau, Laser Photonics Rev., 2012, 6, 419–462 CrossRef
- R. F. Kazarinov and C. H. Henry, IEEE J. Quantum Electron., 1985, 21, 144–150 CrossRef
- M. Hammer, 1-D mode solver for dielectric multilayer slab waveguides, http://www.computational-photonics.eu/oms.html, (accessed 3 December 2020).
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1mh00846c |
| ‡ These authors contributed equally to this work. |
| § Current affiliation Department of Chemistry, University of Oxford, Oxford OX1 3TA, UK. |
| This journal is © The Royal Society of Chemistry 2022 |