
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of SOT-OH and its application as a building block for the synthesis of new dimeric and trimeric Spiro-OMeTAD materials†
Michele
Cariello‡
 a,
Namrata
Pant‡
a,
Alexander H.
Harkiss‡
a,
Frances M.
Tracey
a,
Joseph
Cameron
a,
Peter J.
Skabara
a,
Peter J.
Holliman
b,
Pablo
Docampo
*a and
Graeme
Cooke
*a
a,
Namrata
Pant‡
a,
Alexander H.
Harkiss‡
a,
Frances M.
Tracey
a,
Joseph
Cameron
a,
Peter J.
Skabara
a,
Peter J.
Holliman
b,
Pablo
Docampo
*a and
Graeme
Cooke
*a
aSchool of Chemistry, Joseph Black Building, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: graeme.cooke@glasgow.ac.uk
bCollege of Engineering, Bay Campus, Swansea University, Swansea, SA1 8EN, UK
First published on 28th April 2022
Abstract
Spiro-OMeTAD has become a ubiquitous hole-transporting material (HTM) in perovskite-based solar cells. However, it has several intrinsic drawbacks including its long-term stability, low conductivity and hole-mobility. Hence, the synthesis of new derivatives with improved charge transporting properties remains an important goal. Here, we have devised a novel synthetic route to prepare unsymmetrically functionalised Spiro-OMeTAD derivatives. We report the synthesis of a mono-demethylated Spiro-OMeTAD derivative, SOT-OH, utilising two different synthetic protocols and show two new derivatives, a dimer (SOT-D) and a trimer (SOT-T) as exemplars. We fully characterise the developed materials and show significantly higher conductivity values than Spiro-OMeTAD when doped with standard ionic salts at the same concentrations. The solar cells fabricated using the mixed composition Cs0.05(FA0.85MA0.15)0.95PbI3 perovskite and the novel SOT-D show power conversion efficiencies of up to 16.4% under standard AM1.5 illumination compared to 16.1% observed when using the benchmark Spiro-OMeTAD.
Design, System, ApplicationWe have designed methodology to reliably and conveniently synthesise a new asymmetric Spiro-OMeTAD building block (SOT-OH) using both top-down and bottom-up procedures. As exemplars, we have synthesised a dimer (SOT-D) and a trimer (SOT-T) from SOT-OH and have shown that these materials have higher conductivities, and in the case of SOT-D a higher power conversion efficiency, than Spiro-OMeTAD, which we attribute to the more porous morphology of their films. This work not only provides new methodology for creating unsymmetrically functionalised Spiro-OMeTAD derivatives, which will allow exciting new HTMs to be prepared for perovskite and solid-state dye-sensitised solar cells, but also indicates that a more porous HTM film architecture may be advantageous in developing better performing devices. |
Introduction
Since its first use as a hole-transporting material (HTM) in solid-state dye-sensitised solar cells (ss-DSSCs)1Spiro-OMeTAD has become the benchmark HTM for ss-DSSCs. Importantly, this molecule has also become the HTM of choice for perovskite-based solar cells (PSCs)2,3 which, since 2012, have surpassed the efficiency of any other emerging solar device by achieving a certified record efficiency of 25.7%.4 Although Spiro-OMeTAD has a relatively poor hole-mobility, it can be improved upon doping,5,6 and has several properties ideal for use as a HTM including energy levels with an appropriate over-potential for typical absorbers (either perovskite or dye), good visible light transparency and electrochemical stability. Furthermore, the high solubility and relatively high glass transition temperature (Tg) contribute to its excellent processability and good morphological properties, which promote pore filling of the mesoporous TiO2 layer.7 This blend of properties arises from the combination of the spiro centre, diphenylamine donor groups and methoxy ether substituents. However, the >20 years of research since its first report have shown it difficult to improve on the Spiro-OMeTAD core design, suggesting this is close to optimal for this HTM family.Nevertheless, Spiro-OMeTAD is not the perfect HTM owing to the high production costs, inconsistent batch-to-batch purity and low conductivities. Even film morphology, a strength in its as-made devices, limits device durability, as device performance of Spiro-OMeTAD-based PSCs has been reported to deteriorate at temperatures above 50 °C.8–10 This has been ascribed to phenomena such as iodine diffusion from the perovskite layer (in PSCs)9 and crystallisation, especially after annealing.10,11 Additionally, additives such as tert-butylpyridine (tBP) which are added to balance the drop in the conduction band edge position of TiO2 after addition of lithium-based salts in ssDSCs or passivate the perovskite layer in PSCs, have been directly shown to evaporate from the Spiro-OMeTAD film causing further device degradation by formation of pinholes.7,12 These changes hint at the importance of molecular packing within HTM films which arise from the key design feature of the spiro unit at the centre of every Spiro-OMeTAD molecule. So, whilst this facilitates ionic movement within films which aids conductivity, dopant loss or indeed rearrangement of Spiro-OMeTAD molecules within HTM films is not desirable for device longevity. In this work, we have explored maintaining the Spiro-OMeTAD motif but doubling or trebling the molecular weight to hinder intra-film molecular movement.
Since the crystallisation is facilitated by the symmetry of the molecule, Getautis and co-workers demonstrated that making Spiro-OMeTAD more unsymmetric improves its mechanical stability without significantly affecting its optoelectronic properties.11 The introduction of a methyl group to each diphenylamine peripheral unit significantly increased device stability, with 90% of the efficiency retained after 1000 hours of light exposure at 60 °C. This result was followed by more systematic studies of unsymmetric Spiro-OMeTAD analogues, changing both the position13,14 and the type of peripheral heteroatoms.15 These changes also improved thermal properties and hydrophobicity, with minimal impact on the spectro-electrochemical properties of the HTM.13,14 More complex modifications involving the central core of Spiro-OMeTAD have been investigated by several research groups.16–19
Small molecules having completely different properties,20,21 which sometimes outperform Spiro-OMeTAD, have been reported, while significant improvements have been made using polymeric HTMs,22–25 metal–organic26–28 or inorganic HTMs.29,30 Here, we report the synthesis of SOT-OH, which serves as a novel building block, due to its reactive hydroxyl group, for the synthesis of new derivatives. Two synthetic approaches using top-down and bottom-up synthetic protocols to furnish SOT-OH are described. Furthermore, we report the synthesis of two new Spiro-OMeTAD derivatives, SOT-D and SOT-T, having identical optoelectronic properties to the parent material but different thermal properties and circa double or treble the molecular mass, respectively.
Results and discussion
Two different synthetic routes were developed. In our top-down approach (route A, Scheme 1 and ESI†), SOT-OH was synthesised from commercially available Spiro-OMeTAD. Various demethylation procedures were attempted (Table S1, ESI†), with the most successful conditions being a sodium bis(trimethylsilyl)amide (NaN(SiMe3)2) mediated demethylation in 1,3-dimethyl-2-imidazolidinone (DMI) at 185 °C.31 This convenient one-step approach proved effective for the small scale (∼50 mg) synthesis of SOT-OH, whereas for large scales, our bottom-up protocol (route B, Scheme 1) proved to be very robust. This route commenced with inexpensive 4-bromophenol 1, which underwent quantitative hydroxy-protection using a tert-butyldimethylsilyl (TBDMS) group under standard conditions to produce compound 2. A Buchwald–Hartwig cross-coupling reaction with p-anisidine was then employed to obtain the TBDMS-protected diphenylamine 3. Next, a one-pot Buchwald–Hartwig cross-coupling between 3, 4,4′-dimethyoxydiphenylamine 4, and 2,2′,7,7′-tetrabromo-9,9′-spirobifluorene 6 produced consistent 32% yields of mono-TBDMS-protected SOT-OTBDMS. Despite the moderate yield, this process is scalable (>1 g) and highly reproducible. The other major product of this reaction is Spiro-OMeTAD which suggests that symmetrical diphenylamine 4 is more active under the reaction conditions in comparison to the TBDMS-protected diphenylamine 3. The final step to form SOT-OH involved the removal of the protecting group via tetrabutylammonium fluoride (TBAF). Full experimental details are reported in the ESI.† | ||
| Scheme 1 Synthesis of SOT-OHvia route A (1 step) and route B (4 steps). | ||
Using the desired building block SOT-OH, we investigated its reactivity by synthesising exemplar dimer and trimer derivatives, SOT-D and SOT-T, respectively. Assemblies of this type were selected to both indicate the potential synthetic applications of SOT-OH and provide new materials that will retain the advantages of Spiro-OMeTAD whilst providing materials with potentially enhanced thermal and morphological properties. Hydroquinone 7 and phloroglucinol 9 were chosen as linker groups due to their commercial availability and structural simplicity (Scheme 2). Both compounds were subjected to standard Williamson ether synthesis conditions using sodium hydride and 1,6-dibromohexane to install the brominated hexyl chain on each derivative. Hexyl chains were selected in this study as it was hypothesised that, if shorter alkyl chains were used, then steric hindrance between the bulky spiro units could lead to poor conversion towards the target materials. The obtained alkyl bromide intermediates 8 and 10 underwent Williamson ether synthesis with SOT-OH, to yield the desired dimer SOT-D and trimer SOT-T materials in 51% and 57% yields, respectively (detailed synthesis provided in the ESI†).
 | ||
| Scheme 2 Synthesis of dimer SOT-D and trimer SOT-T materials. | ||
To maximise the light harvesting of perovskite/dye absorbers, HTM light absorption between 400–800 nm should be minimised. The absorption and emission spectra for SOT-D and SOT-T were recorded in dichloromethane and compared with Spiro-OMeTAD (Fig. 1). The λmax for SOT-D and SOT-T are both observed at 386 nm, which is similar to the λmax for Spiro-OMeTAD (384 nm) and correspond to their π–π* electron transitions.32,33 The extinction coefficients increase on going from Spiro-OMeTAD to SOT-T due to the increased number of Spiro-OMeTAD units per molecule. However, importantly for light harvesting, no red shift into the visible range is observed and all three molecules have an identical cut-off wavelength of 420 nm, corresponding to an optical energy gap (Eg,opt) of ∼2.95 eV. This phenomenon arises from the molecular design of these new HTMs whereby separate Spiro-OMeTAD units are connected through non-conjugated linkers, which effectively means each Spiro-OMeTAD unit behaves as a separate HTM unit regardless of whether present within the monomeric Spiro-OMeTAD, dimeric SOT-D or trimeric SOT-T. The emission spectra of Spiro-OMeTAD, SOT-D and SOT-T originate from the π*–π transition. The maximum emission wavelength of the three materials is again very similar at 424, 427 and 427 nm, respectively. Due to the analogous absorption and emission spectra, the Stokes shift values were almost identical (40 nm), for the same reasons described above.
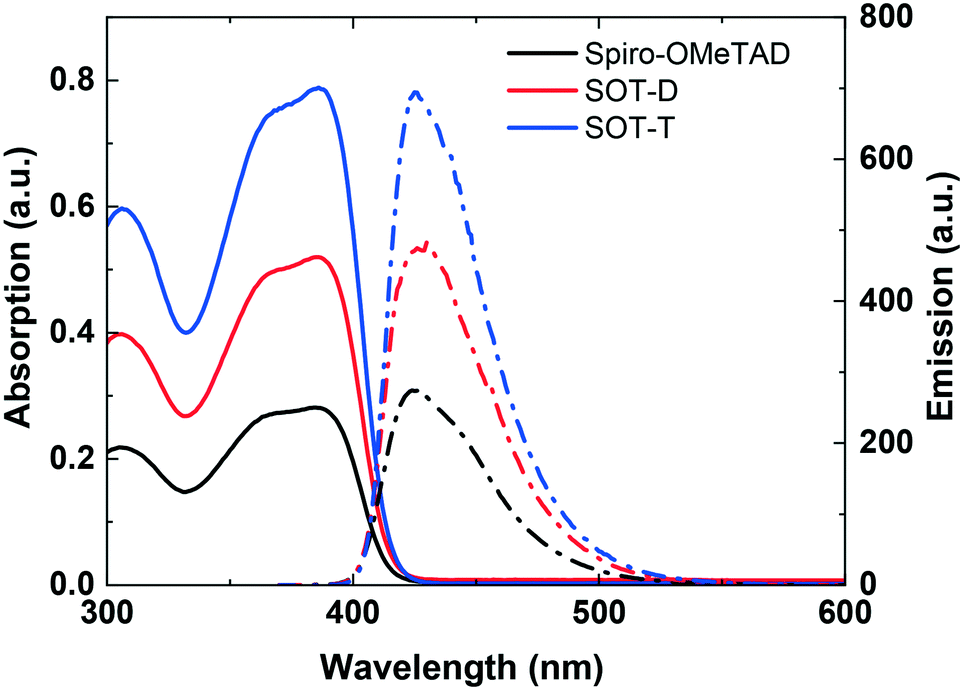 | ||
| Fig. 1 UV-vis absorption (solid lines) and fluorescence (dashed lines) spectra of Spiro-OMeTAD, SOT-D and SOT-T (CH2Cl2, 1.0 × 10−5 M). Excitation at λ = 386 nm for fluorescence spectra. | ||
Electrochemical analysis of SOT-D and SOT-T in dichloromethane was carried out via cyclic voltammetry (CV) and square wave voltammetry (SWV) and the results were compared with Spiro-OMeTAD,34 under the same conditions. As the CVs indicate (Fig. 2), SOT-D and SOT-T both undergo three main oxidations, identical to Spiro-OMeTAD,35 confirming that the alkyl chains that connect the terminal units are long enough to prevent intramolecular charge transfer between spiro units. Although the peak currents increase in line with increasing spiro units on going from Spiro-OMeTAD, SOT-D to SOT-T, the shape of the curves are almost identical suggesting that, similarly to Spiro-OMeTAD, the first oxidation events (grey shaded area, Fig. 2) are likely the result of two reversible one-electron oxidations of the diarylamine subunits. Finally, the third more prominent reversible oxidation event corresponds to a two-electron oxidation from the di-cation to form the fully quinoidal tetra-cation.33 SWV (Fig. S1, ESI†) allows more accurate determination of the oxidation peaks and ionisation potentials (IP), which for the three compounds are −4.9 eV, which is in good agreement with reported data for Spiro-OMeTAD.36 Similar results were observed by computational studies. The structures of SOT-D and SOT-T were calculated using density functional theory (DFT) calculations, using the B3LYP hybrid functional and the 3-21G basis set. The results were compared to those of Spiro-OMeTAD, obtained with the same level of theory. The highest occupied molecular orbital (HOMO) levels of −4.27 eV, −4.29 eV and −4.26 eV for SOT-D, SOT-T and Spiro-OMeTAD, respectively, were obtained. Although the use of more robust basis sets on Spiro-OMeTAD has provided more accurate results,37 these performance-demanding methods could not easily be applied on the structurally more complex SOT-D and SOT-T. Nonetheless, this DFT study shows how the HOMO levels of the three molecules are very close to each other, highlighting again the electronic similarities of the three systems, thereby auguring well for future device applications of SOT-D and SOT-T. The HOMO–LUMO maps (Fig. S2, ESI†) give an idea of the spatial distribution of the frontier molecular orbitals of the three molecules. As expected, the symmetry gives rise to nearly degenerate HOMOs (Table S2, ESI†), two for Spiro-OMeTAD, four for SOT-D and six for SOT-T, all separated by less than 10 meV and spatially delocalised over the side arms and the spiro-centres. The same phenomenon applies to the lowest unoccupied molecular orbitals (LUMOs) which are however spatially more localised over the spiro-centres. A summary of electro-optical properties of SOT-D and SOT-T compared with Spiro-OMeTAD is provided in Table S3 (ESI†).
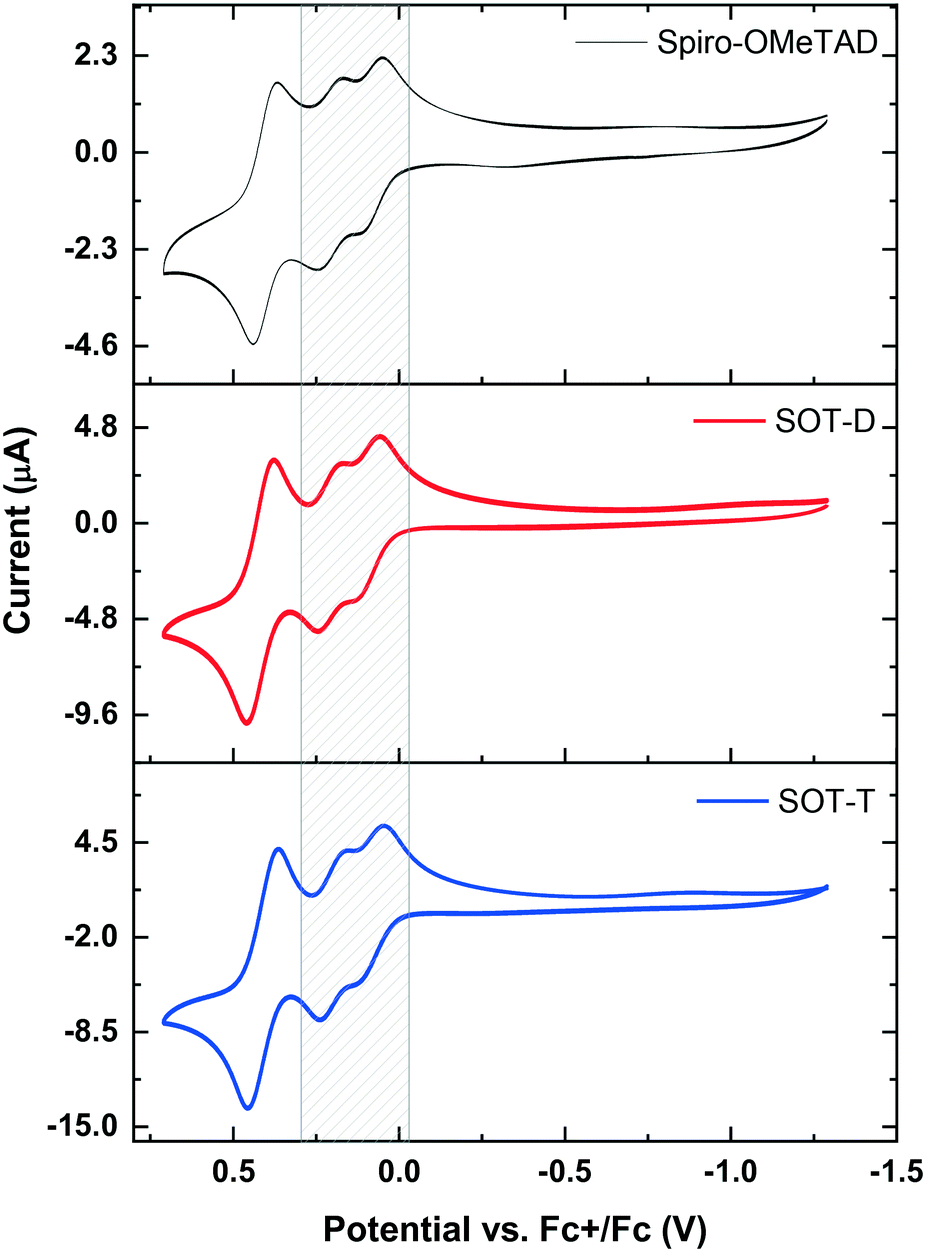 | ||
| Fig. 2 CVs of Spiro-OMeTAD, SOT-D and SOT-T (CH2Cl2, 5 × 10−4 M). Conditions: Pt disc working electrode, Pt wire counter electrode, Ag wire pseudo-reference electrode, 0.1 M TBAPF6 electrolyte. Fc+/Fc was used as external reference. | ||
The thermal properties of SOT-D and SOT-T were studied by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC), and compared with Spiro-OMeTAD. The results (Fig. 3 and 4) indicate that SOT-D behaves very similarly to Spiro-OMeTAD, with a mass loss <5%, below 400 °C. SOT-T is even more robust with <1% mass lost below 400 °C. Above this temperature they all start to decompose.
 | ||
| Fig. 3 TGA of Spiro-OMeTAD, SOT-D and SOT-T. Heating rate: 10 °C min−1. | ||
 | ||
| Fig. 4 DSC of Spiro-OMeTAD, SOT-D and SOT-T (first heating wave). Heating rate: 10 °C min−1. Heat flow values were offset for better visualisation. | ||
The DSC analysis of Spiro-OMeTAD indicates that it can exist in both amorphous and crystalline forms,38 as suggested by the presence of a Tg of 117 °C and a crystallisation temperature (Tc) of 162 °C. The melting point (Tm) of the material is identified by the peak at 238 °C. The second heating wave confirmed the presence of the Tg, shifted at 125 °C, while the Tc and Tm were not observed, suggesting that the amorphous phase is more stable than the crystalline (Fig. S3, ESI†). A similar behaviour is shown by SOT-D, which exhibits a post crystallisation peak at 170 °C during the first heating cycle, that is not observed during the second heating cycle (Fig. S4, ESI†). A more pronounced Tg is observed at 79 °C, during the first heating wave, and at 121 °C during the second. A different thermal behaviour is shown by SOT-T and in particular, the lack of Tc in all cycles suggests that the material is highly amorphous. A Tg at 137 °C is appreciable only during the second heating cycle (Fig. S5, ESI†). This value is higher than that of Spiro-OMeTAD, indicating that, in principle, SOT-T should have better morphological stability under continuous sunlight exposure.39
To verify their suitability as hole-transporting materials for solar cells, thin films of these materials were spin coated on patterned ITO substrates including typical additive concentrations and deposited following state-of-the-art perovskite solar cell protocols (detailed device fabrication provided in the ESI†). Their conductivity was extracted from the resistance values of standard current–voltage curves employing an interdigitated electrode pattern, as shown in Fig. 5. To ensure that the resulting HTM films had completed the oxidation process and therefore reach maximum conductivity, the films were left in a desiccator for one week in the dark prior to measuring. The conductivity values obtained for Spiro-OMeTAD ((0.4 ± 0.1) × 10−4 S cm−1) match well with the values reported in the literature.40,41 Interestingly, SOT-D and SOT-T demonstrated better conductivity than Spiro-OMeTAD throughout. The steeper curves for the SOT-D and SOT-T clearly demonstrate lower resistance, and conductivity values about 4 times higher ((1.6 ± 0.1) × 10−4 S cm−1). The higher values could be attributed to higher oxidation rate in these materials as compared to Spiro-OMeTAD, as shown by the increase in the oxidised band present at 520 nm in the light absorption spectra (Fig. S6, ESI†).42,43 We attribute this difference to the more porous nature of our synthesised spiro derivatives, as evidenced in AFM images, (Fig. 6, line plots in fig. S7, ESI†), which increases the available surface area for the oxidation process and thus results in a higher oxidation rate, leading to the observed increase in conductivity. The larger peak-to-trough lateral distances in SOT-D films, may be explained by the presence of the spacer groups which may prevent tighter intermolecular packing. Additionally, the increased molecular weight of the dimer, relative to Spiro-OMeTAD, can cause reduced solubility, meaning a rougher film is formed due to reduced drying time of solutions undergoing spin-coating.
 | ||
| Fig. 5 Current (I)–voltage (V) characteristics of Spiro-OMeTAD, SOT-D and SOT-T. | ||
 | ||
| Fig. 6 Tapping mode AFM micrographs of (a) Spiro-OMeTAD (Rq = 2.7 nm) and (b) SOT-D (Rq = 7.5 nm) films on a glass substrate. Scan area = 10 × 10 μm. | ||
Further, perovskite solar cells were fabricated to investigate the suitability of these materials as HTMs. Conventional n-i-p device architecture was adopted with FTO/SnO2 nanoparticles/FAMACs perovskite/HTM/Au stack (experimental details can be found in the ESI†). A mixed cation perovskite with composition Cs0.05(FA0.85MA0.15)0.95PbI3 was used as the light absorbing layer. The HTMs were doped with tBP and lithium bis(trifluoromethanesulfonyl)imide (LiTFSi) to increase the conductivity and mobility of these materials. The devices were characterised through current–voltage measurements under standard AM1.5 sun illumination. The current density/voltage (J/V) curves of the best devices are shown in Fig. 7. The devices displayed good performance with 16.47% and 16.01% efficiency for the best SOT-D and Spiro-OMeOTAD based devices, respectively. The slightly enhanced efficiency value for devices employing SOT-D can be attributed to faster oxidation and increased p-doping of the material which can be corroborated with the increased surface area as seen in the AFM images. The data also show slightly enhanced fill factor for the SOT-D devices. This suggests that slightly less recombination may occur for SOT-D due to its improved conductivity, which is a further benefit of the dimeric HTM. It is often assumed that smoother surfaces are desirable for HTMs, but this study has shown that a more porous film can be used to improve the device performance.
 | ||
| Fig. 7 Current density (J)–voltage (V) characteristics of devices under one sun illumination (AM 1.5) employing Spiro-OMeTAD and SOT-D as HTMs. | ||
Conclusions
In summary, we report a convenient top-down and a scalable bottom-up procedure for synthesising SOT-OH. Furthermore, we report the use of this building block to synthesise SOT-D and SOT-T by exploiting the reactivity of the hydroxyl group of SOT-OH. We have investigated the optical, redox, thermal and optoelectronic properties of SOT-D and SOT-T, which have shown that these derivatives retain the redox and optical properties of Spiro-OMeTAD, whilst in the case of SOT-T improved thermal and optoelectronic properties are displayed. We have also shown that SOT-D and SOT-T have higher conductivity values compared to the parent Spiro-OMeTAD which we have attributed to the higher porosity of the dimer and trimer. Likewise, the power conversion efficiencies of perovskite-based devices fabricated using SOT-D are higher than those obtained using Spiro-OMeTAD, thereby indicating that HTM porosity may also be an important factor affecting device performance. Therefore, this study paves the way for further development of SOT-OH for creating a range of new Spiro-OMeTAD-based materials for high-performance ss-DSSCs and PSCs, and our progress in this area will be reported in due course.Author contributions
MC, AHH and FMT synthesised and characterised the compounds and drafted parts of the manuscript and ESI.† PJH and GC devised the synthesis of the materials and contributed to writing the manuscript and ESI.† NP carried out the electronic characterisation of the materials and fabricated the perovskite solar cells, and drafted parts of the manuscript and ESI.† PD oversaw all aspects of the electronic characterisation and device fabrication, interpreted the data and contributed to writing the manuscript and ESI.† JC and PS acquired the AFM images and analysed the resulting data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC for funding (GC and AHH EP/P030106/1, PJH EP/P030068/1 and PD EP/T010568/1). We also thank Dr Logan Mackay of the Scottish Instrumentation and Resource Centre for Advanced Mass Spectrometry (University of Edinburgh; Edinburgh, Scotland, UK), for running some of the HRMS. We acknowledge Dr Affar Karimullah for measuring the thickness of the films.Notes and references
- U. Bach, D. Lupo, P. Comte, J. E. Moser, F. Weissörtel, J. Salbeck, H. Spreitzer and M. Grätzel, Nature, 1998, 395, 583–585 CrossRef CAS.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- H.-S. Kim, C.-R. Lee, J.-H. Im, K.-B. Lee, T. Moehl, A. Marchioro, S.-J. Moon, R. Humphry-Baker, J.-H. Yum, J. E. Moser, M. Grätzel and N.-G. Park, Sci. Rep., 2012, 2, 591 CrossRef PubMed.
- NREL Best Research Cell Efficiency Chart, https://www.nrel.gov/pv/cell-efficiency.html, (accessed March 2022).
- T. H. Schloemer, J. A. Christians, J. M. Luther and A. Sellinger, Chem. Sci., 2019, 10, 1904–1935 RSC.
- Z. Hawash, L. K. Ono and Y. Qi, Adv. Mater. Interfaces, 2018, 5, 1700623 CrossRef.
- C. D. Bailie, E. L. Unger, S. M. Zakeeruddin, M. Grätzel and M. D. McGehee, Phys. Chem. Chem. Phys., 2014, 16, 4864–4870 RSC.
- G. Divitini, S. Cacovich, F. Matteocci, L. Cinà, A. Di Carlo and C. Ducati, Nat. Energy, 2016, 1, 15012 CrossRef CAS.
- Y. Kato, L. K. Ono, M. V. Lee, S. Wang, S. R. Raga and Y. Qi, Adv. Mater. Interfaces, 2015, 2, 1500195 CrossRef.
- N. Rolston, B. L. Watson, C. D. Bailie, M. D. McGehee, J. P. Bastos, R. Gehlhaar, J.-E. Kim, D. Vak, A. T. Mallajosyula, G. Gupta, A. D. Mohite and R. H. Dauskardt, Extreme Mech. Lett., 2016, 9, 353–358 CrossRef.
- T. Malinauskas, D. Tomkute-Luksiene, R. Sens, M. Daskeviciene, R. Send, H. Wonneberger, V. Jankauskas, I. Bruder and V. Getautis, ACS Appl. Mater. Interfaces, 2015, 7, 11107–11116 CrossRef CAS PubMed.
- L. K. Ono, S. R. Raga, M. Remeika, A. J. Winchester, A. Gabe and Y. Qi, J. Mater. Chem. A, 2015, 3, 15451–15456 RSC.
- D. Tomkute-Luksiene, M. Daskeviciene, T. Malinauskas, V. Jankauskas, R. Degutyte, R. Send, N. G. Pschirer, H. Wonneberger, I. Bruder and V. Getautis, RSC Adv., 2016, 6, 60587–60594 RSC.
- M.-D. Zhang, D.-X. Zhao, L. Chen, N. Pan, C.-Y. Huang, H. Cao and M.-D. Chen, Sol. Energy Mater. Sol. Cells, 2018, 176, 318–323 CrossRef CAS.
- Z. Hu, W. Fu, L. Yan, J. Miao, H. Yu, Y. He, O. Goto, H. Meng, H. Chen and W. Huang, Chem. Sci., 2016, 7, 5007–5012 RSC.
- M. Saliba, S. Orlandi, T. Matsui, S. Aghazada, M. Cavazzini, J.-P. Correa-Baena, P. Gao, R. Scopelliti, E. Mosconi, K.-H. Dahmen, F. De Angelis, A. Abate, A. Hagfeldt, G. Pozzi, M. Graetzel and M. K. Nazeeruddin, Nat. Energy, 2016, 1, 15017 CrossRef CAS.
- Y. Shi, Y. Xue, K. Hou, G. Meng, K. Wang, R. Chi, F. Chen, H. Ren, M. Pang and C. Hao, RSC Adv., 2016, 6, 96990–96996 RSC.
- M.-H. Li, C.-W. Hsu, P.-S. Shen, H.-M. Cheng, Y. Chi, P. Chen and T.-F. Guo, Chem. Commun., 2015, 51, 15518–15521 RSC.
- Y.-K. Wang, Z.-C. Yuan, G.-Z. Shi, Y.-X. Li, Q. Li, F. Hui, B.-Q. Sun, Z.-Q. Jiang and L.-S. Liao, Adv. Funct. Mater., 2016, 26, 1375–1381 CrossRef CAS.
- L. Yang, F. Cai, Y. Yan, J. Li, D. Liu, A. J. Pearson and T. Wang, Adv. Funct. Mater., 2017, 27, 1702613 CrossRef.
- A. Magomedov, S. Paek, P. Gratia, E. Kasparavicius, M. Daskeviciene, E. Kamarauskas, A. Gruodis, V. Jankauskas, K. Kantminiene, K. T. Cho, K. Rakstys, T. Malinauskas, V. Getautis and M. K. Nazeeruddin, Adv. Funct. Mater., 2018, 28, 1704351 CrossRef.
- X. Jiang, Z. Yu, Y. Zhang, J. Lai, J. Li, G. G. Gurzadyan, X. Yang and L. Sun, Sci. Rep., 2017, 7, 42564 CrossRef CAS PubMed.
- N. Y. Nia, F. Matteocci, L. Cina and A. Di Carlo, ChemSusChem, 2017, 10, 3854–3860 CrossRef CAS PubMed.
- Z. Zhou, X. Li, M. Cai, F. Xie, Y. Wu, Z. Lan, X. Yang, Y. Qiang, A. Islam and L. Han, Adv. Energy Mater., 2017, 7, 1700763 CrossRef.
- M. Li, Z.-K. Wang, T. Kang, Y. Yang, X. Gao, C.-S. Hsu, Y. Li and L.-S. Liao, Nano Energy, 2018, 43, 47–54 CrossRef CAS.
- J.-M. Wang, Z.-K. Wang, M. Li, K.-H. Hu, Y.-G. Yang, Y. Hu, X.-Y. Gao and L.-S. Liao, ACS Appl. Mater. Interfaces, 2017, 9, 13240–13246 CrossRef CAS PubMed.
- H.-H. Chou, Y.-H. Chiang, M.-H. Li, P.-S. Shen, H.-J. Wei, C.-L. Mai, P. Chen and C.-Y. Yeh, ACS Energy Lett., 2016, 1, 956–962 CrossRef CAS.
- M. Freitag, Q. Daniel, M. Pazoki, K. Sveinbjörnsson, J. Zhang, L. Sun, A. Hagfeldt and G. Boschloo, Energy Environ. Sci., 2015, 8, 2634–2637 RSC.
- S. Pitchaiya, M. Natarajan, A. Santhanam, V. M. Ramakrishnan, V. Asokan, P. Palanichamy, B. Rangasamy, S. Sundaram and D. Velauthapillai, Mater. Lett., 2018, 221, 283–288 CrossRef CAS.
- H. Zhang, H. Wang, W. Chen and A. K.-Y. Jen, Adv. Mater., 2017, 29, 1604984 CrossRef PubMed.
- J. R. Hwu, F. F. Wong, J.-J. Huang and S.-C. Tsay, J. Org. Chem., 1997, 62, 4097–4104 CrossRef CAS.
- H. Ashassi-Sorkhabi and P. Salehi-Abar, RSC Adv., 2018, 8, 18234–18242 RSC.
- W. Zhang, L. Wang, Y. Guo, B. Zhang, V. Leandri, B. Xu, Z. Li, J. M. Gardner, L. Sun and L. Kloo, Chem. Commun., 2020, 56, 1589–1592 RSC.
- S. Fantacci, F. De Angelis, M. K. Nazeeruddin and M. Grätzel, J. Phys. Chem. C, 2011, 115, 23126–23133 CrossRef CAS.
- J. García-Cañadas, F. Fabregat-Santiago, H. J. Bolink, E. Palomares, G. Garcia-Belmonte and J. Bisquert, Synth. Met., 2006, 156, 944–948 CrossRef.
- W. H. Nguyen, C. D. Bailie, E. L. Unger and M. D. McGehee, J. Am. Chem. Soc., 2014, 136, 10996–11001 CrossRef CAS PubMed.
- Y. Li, H. Li, C. Zhong, G. Sini and J.-L. Brédas, npj Flexible Electron., 2017, 1, 2 CrossRef.
- G. Tumen-Ulzii, C. Qin, T. Matsushima, M. R. Leyden, U. Balijipalli, D. Klotz and C. Adachi, Sol. RRL, 2020, 4, 2000305 CrossRef CAS.
- L. Calió, S. Kazim, M. Grätzel and S. Ahmad, Angew. Chem., Int. Ed., 2016, 55, 14522–14545 CrossRef PubMed.
- L. Caliò, M. Salado, S. Kazim and S. Ahmad, Joule, 2018, 2, 1800–1815 CrossRef.
- S. Kim, S. Bae, S.-W. Lee, K. Cho, K. D. Lee, H. Kim, S. Park, G. Kwon, S.-W. Ahn, H.-M. Lee, Y. Kang, H.-S. Lee and D. Kim, Sci. Rep., 2017, 7, 1200 CrossRef PubMed.
- J. H. Noh, N. J. Jeon, Y. C. Choi, M. K. Nazeeruddin, M. Grätzel and S. I. Seok, J. Mater. Chem. A, 2013, 1, 11842–11847 RSC.
- U. Bach, Y. Tachibana, J.-E. Moser, S. A. Haque, J. R. Durrant, M. Grätzel and D. R. Klug, J. Am. Chem. Soc., 1999, 121, 7445–7446 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2me00038e |
| ‡ These authors contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2022 |