
Crystal engineering of alkylethynyl fluoroanthradithiophenes†
Karl J.
Thorley
 *ab,
Derek
Dremann
c,
Hamna F.
Iqbal
d,
Sean R.
Parkin
a,
Oana D.
Jurchescu
c and
John E.
Anthony
ab
*ab,
Derek
Dremann
c,
Hamna F.
Iqbal
d,
Sean R.
Parkin
a,
Oana D.
Jurchescu
c and
John E.
Anthony
ab
aDepartment of Chemistry, University of Kentucky, Lexington, KY 40508, USA. E-mail: karl.thorley@uky.edu
bCenter for Applied Energy Research, University of Kentucky, Lexington, KY 40511, USA
cDepartment of Physics and Center for Functional Materials, Wake Forest University, Winston-Salem, NC 27109, USA
dDepartment of Materials Science and Engineering, North Carolina State University, Raleigh, NC 27606, USA
First published on 24th January 2022
Abstract
Fluoroanthradithiophenes are well known organic semiconductors, where alkynyl substituents featuring silicon and germanium exhibit hole mobilities in excess of 5 cm2 V−1 s−1. A key feature to achieve these performance levels is the 2-dimensional brickwork packing of triethylsilyl and triethylgermyl side chains, which direct solid-state packing, increase molecular stability, and increase solution processability for cheap and large scale fabrication. We have recently reported side chains utilising carbon in place of the other group 14 atoms, resulting in less favourable 1-dimensional molecular packing. Here we present the synthesis of new derivatives which adopt 2-D brickwork packing without the use of silicon or germanium to determine substituent effects on charge carrier mobility.
Design, System, ApplicationPolycyclic aromatic hydrocarbons are widely used as organic semiconductors in a range of electronics devices. Charge transport in these materials can be assessed by fabrication of transistor devices, extracting the figure-of-merit charge carrier mobility. Acenes are a class of polycyclic aromatic hydrocarbons that have found great success in organic electronics, where functional groups can direct solid state packing to affect the bulk material charge transport properties. A trend seen in the best performing acene materials is a 2-dimensional brickwork motif, allowing multi-directional charge transport pathways, which has been achieved with side chains containing silicon or germanium. In this work, we expand on our previous success in synthesising similar side chains using only carbon and hydrogen by following a logical design process to achieve the desirable 2-D packing. From device fabrication we aim to understand how this change in chemical structure affects solid-state charge transport properties. |
Introduction
Silyl groups have become synonymous with the design of many molecular organic semiconductors, where their size can play a pivotal role in determining solid state packing as well as providing solubility for increased processability of materials from solution. They further impart molecular stability through steric blocking of radical sites1 and Diels–Alder dienes, as well as promoting reversal of endoperoxide formation.2 While commonly employed on many polycyclic aromatic hydrocarbons,3 they have appeared most often on acenes and heteroacenes,4 notably pentacene5 and fluoroanthradithiophene6 (FADT, Fig. 1) which has resulted in high hole mobility semiconductor materials.7 | ||
| Fig. 1 Alkynyl functionalised fluoroanthradithiophene (FADT) extensively researched as an organic semiconductor. TES-FADT adopts a 2D brickwork packing (blue molecules) while TEC-FADT adopts a 1D slipped stack packing (red molecules). | ||
In particular, high charge mobilities have been recorded in materials where the acene cores adopt a two-dimensional brickwork packing, a lamellar motif which allows for multi-directional charge transport.8 This is achieved with the triisopropylsilyl side group for pentacene and the triethylsilyl side chain for FADT. In the simplest model, the size and shape of the alkylsilyl groups control the packing through a space filling effect. If the side chain is too large or too small for the rest of the packing motif to form, the 2D brickwork polymorph becomes unstable, and other packing configurations are preferable (Fig. 1). In cases where the silyl group is smaller, 1D slipped stacks form with interdigitation between stacks and alkyl–π interactions being formed. In this type of packing, the acene cores remain parallel to one another. When the alkyl silyl groups are too large, the 1D stacks interact with one another through alkyl–π interactions resulting in herringbone-like interactions and non-parallel acene cores. Both 1D slipped stack packings generally result in poor quality thin films,9 which on top of the limited directionality of charge transport and inability for charges to bypass trap sites by orthogonal hopping, result in low charge carrier mobilities.
In addition to the size and shape of the solubilising groups, more recent work has investigated the stabilising forces between molecules within the crystal structures.10 Notably, stabilisation in the interaction of two “π-stacking” molecules is only about 1/3 from direct interaction of the acene cores, while the interactions involving side chains can have a dramatic stabilising effect through dispersion forces.
It is worth considering that along with the desirable 2D brickwork packing, we also need good electronic communication between adjacent molecules such that charges can be transported through the bulk material. It is here that the consideration of small changes in chemical structure, and therefore intermolecular interactions, can play a large role. Furthermore, these intermolecular interactions will likely influence the relative vibrational motions of adjacent molecules, which results in temporal variation in the electronic communication.11 Thus, we are interested in synthesising materials that adopt the 2D brickwork pattern without the use of silyl side chains to determine the effects of atomic identity on charge transport properties.
Literature reported organic semiconductors with alkynyl side groups without silicon consist of aryl-ethynyl groups,12–14 which affect the electronic properties of the acene by extended conjugation, and have very different crystal packings due to the planar aryl rings. We recently developed a synthetic reaction scheme to furnish alkynes made of only carbon and hydrogen.15 Importantly, they bear a branching point adjacent to the triple bond, mimicking the silyl acetylenes that have been extensively used in the synthesis of organic materials. The branching is expected to impart enough steric bulk to prevent acene–acene photodimerization as well as tune the arrangement of molecules in the solid state. We previously synthesised a triethyl carbon alkyne, which we attached to a fluoroanthradithiophene core resulting in 1D slipped-stack packing (TEC-FADT, Fig. 1), whereas silyl7 and germyl16 equivalents adopted the preferred 2D brickwork arrangement. This structural change was rationalised by the smaller side group size due to shorter C–C bond length relative to Si–C, a lower electron count resulting in weaker dispersion forces, and slightly polar Si–C bonds contributing some electrostatic stabilisation. We surmised that larger alkyl chains would be needed to gain the desired packing with carbon side chains. Here, we have utilised variations on our previous synthesis to reach four target alkyl ethynyl anthradithiophenes shown in Fig. 2.
 | ||
| Fig. 2 Chemical structures of FADT derivatives explored in the present study. Synthetic routes are provided in the ESI.† | ||
Functional group selection, synthesis, and structural analysis
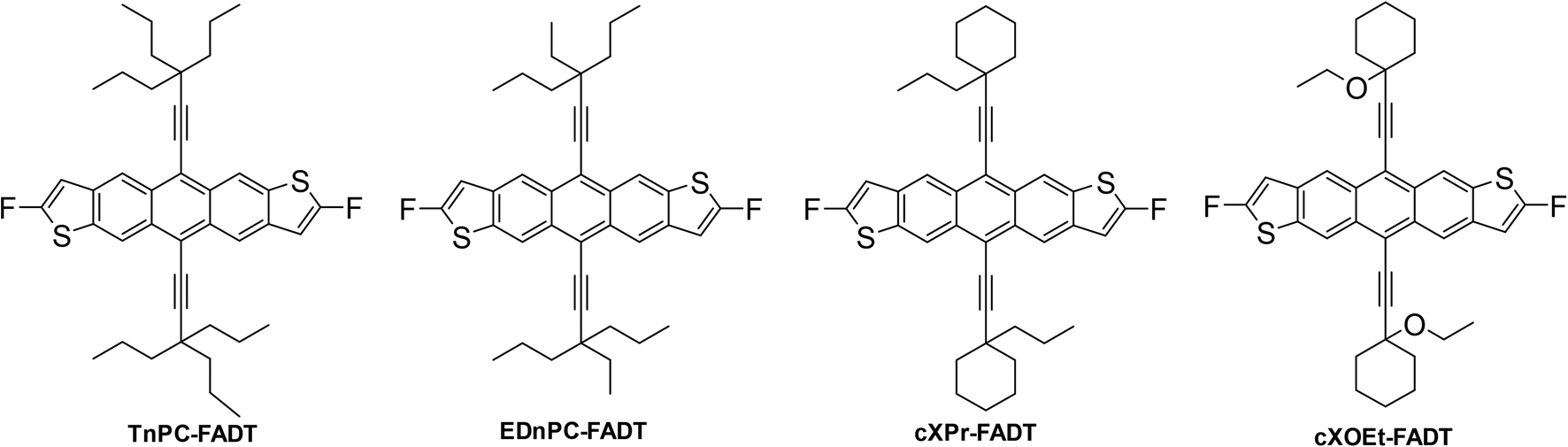
An obvious first synthetic target was the tri-n-propyl derivative TnPC-FADT. The alkyl acetylene was synthesised analogously to our previous triethyl derivative (see ESI†), and was then reacted with FADT quinone according to our established procedure5 to provide TnPC-FADT in good yield. Optoelectronic properties of TnPC-FADT measured in solution were identical to the previously synthesised TEC-FADT, assuring us that the choice of alkyl chains does not affect single molecule properties. Single crystals suitable for X-ray analysis were grown from acetone as thin orange needles. The FADT cores form a 1D slipped stack arrangement (Fig. 3), although the overall packing is quite different from TEC-FADT. In the current case, the alkyl side chains that interact with the FADT π surface do so at an orthogonal angle, resulting in the herringbone arrangement when viewed along the c axis, albeit with no FADT–FADT contact. In contrast, all of the FADT cores in TEC-FADT lay in parallel planes to one another. This change in packing suggests going from too small of a side chain with TEC to too large of one with TnPC.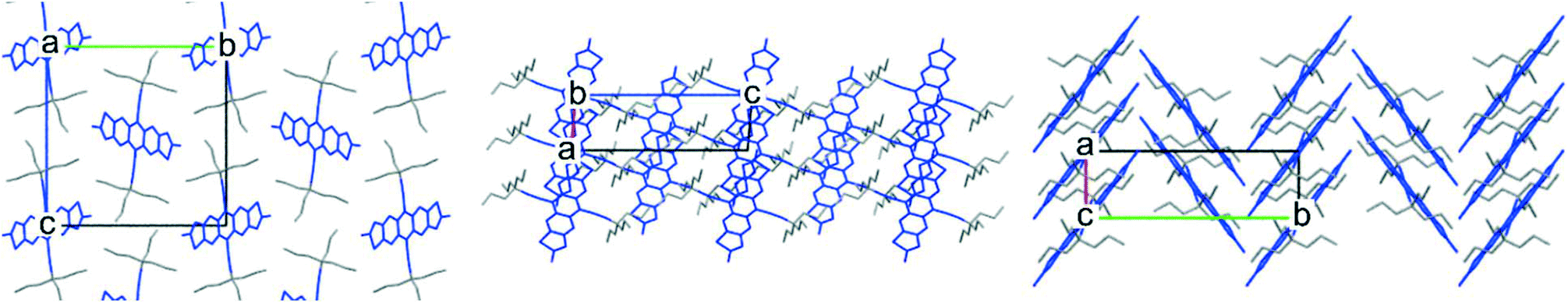 | ||
| Fig. 3 Crystal packing of TnPC-FADT with views along (from left to right) a, b, and c crystallographic axes. | ||
Realising that the TnPC group is too large for a 2D brickwork packing, we took advantage of the flexibility of the synthetic route to carbon alkynes to form a smaller asymmetrically substituted derivative. By starting with commercially available butyronitrile, addition of two propyl chains yielded the ethyl di-n-propyl nitrile which could ultimately be converted to an alkyne. After adding the alkyne to FADT quinone following the procedure described before, the change in solid state packing was dramatic: the crystal packing of ethyldi(n-propyl)ethynyl (EDnPC) FADT shown in Fig. 4 exhibits a number of interactions found in the desired 2D brickwork packing. Each molecule forms π stacking interactions with two other molecules on one side (as per the 2D brickwork) but only one molecule on its other face (as per a 1D slip stack). The result is that electronic communication is limited to only one direction along the molecular stack, confined within narrow channels which are two molecules wide (i.e. pink molecules in Fig. 4). In place of the final π stacking molecule is an alkyl–π interaction as observed in TnPC-FADT, where the adjacent FADT stacks are orthogonal and shielded by alkyl chains. Additionally, each molecule only has one nearest in-plane neighbour stabilised by S–F and H–F interactions, compared to the two found in a 2D brickwork packing. The desirable pairwise interactions involve the longer n-propyl chains, which all align within the 1D slipped stack of FADT molecules. The ethyl chains are found on the outside of this stack (see ESI†), and appear to be too small to sustain further parallel FADT interactions, recalling that the side chains contribute a large amount of stabilising forces to the “π-stacked” interactions.10 Instead, non-parallel alkyl–π interactions are found which disrupt the possibility for a 2D brickwork packing. The observed crystal packing with asymmetric side chains is reminiscent of silylethynyl acenes with asymmetrically substituted aromatic cores.17,18
 | ||
| Fig. 4 Crystal packing of EDnPC-FADT, where molecules of the same color are electronically communicative with one another. | ||
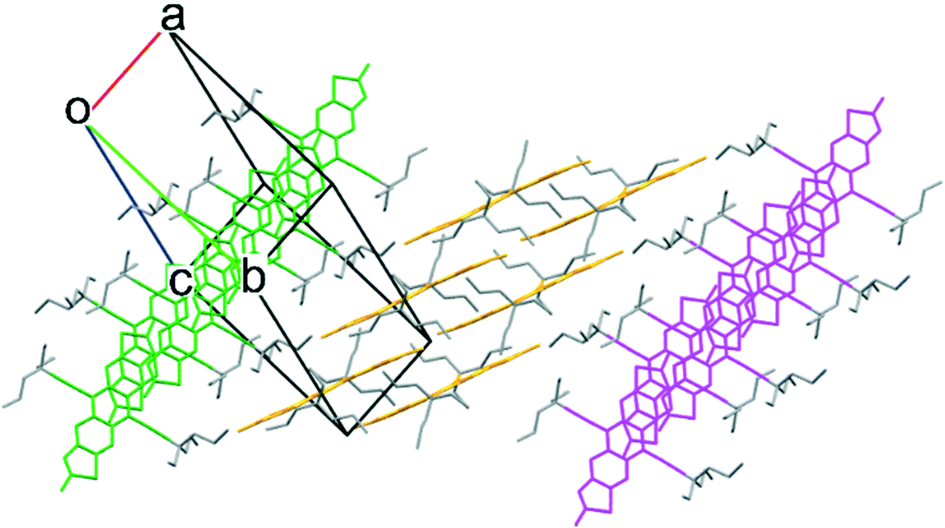
In order to access side chains intermediate in size between TnPC and EDnPC, we explored the use of cycloalkanes, effectively tying up two of the alkyl chains together. Starting from cyclohexane carbonitrile, a further n-propyl chain was added, before following a similar synthetic route as the previous carbon alkynes to yield cyclohexyl-n-propyl alkyne. This alkyne, abbreviated as cXPr alkyne, was used to synthesize the corresponding FADT. Crystals grown from dichloroethane revealed a 2D brickwork packing motif desirable for solid state charge transport (Fig. 5).
 | ||
| Fig. 5 2D brickwork packing in single crystals of cXPr-FADT, viewed along the a, b, and c crystallographic axes, respectively. cXOEt-FADT adopts a very similar packing motif (see ESI†). | ||
An analog of the cXPr side chain was also synthesised where the propyl chain was replaced with an ethoxy group (cXOEt). This was accessible by the addition of trimethylsilyl acetylide to cyclohexanone, followed by trapping of the hydroxyl anion with diethyl sulfate. Removal of the silyl protecting group furnished the terminal acetylene, which could be added to the FADT core as per the general synthetic route. The cXOEt-FADT adopted a very similar crystal packing as the n-propyl derivative, suggesting that the general size and shape of the side chain was an important factor in gaining the 2D brickwork packing.
Rationalisation for different packing
Thermal analysis of the new derivatives, as well as the previously reported TEC-FADT, was carried out using differential scanning calorimetry (DSC, Table 1). The melting enthalpies of the straight chain FADTs are essentially the same as each other, although melting points vary range over 20 °C. TEC-FADT has the highest melting point while EDnPC-FADT has the lowest, which would make sense from an entropic point of view. The longer flexible propyl chains in TnPC-FADT versus TEC would lead to a greater change in disorder upon melting, reducing the melting point. EDnPC-FADT also has more possible side chain alignments due to its asymmetry and therefore the lowest melting point. Both cyclic side chains exhibit higher melting enthalpies than any of the straight chain side chains contributing to higher melting points. The tying together of two of the alkyl chains into a cyclic structure might result in a more ordered system, although the ring itself is still quite flexible and the side chain as a whole is asymmetric, so the entropic contribution is perhaps not so clear. Solid-state thermal data was also obtained through computational analysis. Plane-wave density functional theory (DFT) was used to optimise the geometries of the unit cells of each of the obtained crystal structures. Comparison to approximations of gas phase molecules at the same level of theory allows the derivation of the lattice binding energies of the unit cells (Table 1). These computed energies are all quite similar in magnitude to one another, and do not show the same variation as the experimentally measured enthalpies.Computational analysis of the structures
Using plane-wave DFT, it is also possible to explore hypothetical structures which cannot be obtained experimentally. We first took the experimental 2D brickwork packing structures of cXPr- and cXOEt-FADT and optimised the geometries by DFT. Symmetry adapted perturbation theory (SAPT0)19 analysis of pairwise interactions in the optimised crystal structures are summarised in Fig. 6, where filled circles represent the cXPr and cXOEt optimised structures, cXPr- and cXOEt-FADT exhibit strong pairwise interactions between the main overlapping stacked molecules (black data points), with stabilisations of around 30 kcal mol−1. Interestingly, cXPr-FADT has some asymmetry within this stack, where a slight rotation of the n-propyl chain results in unequal molecular spacing along this main π-stacking direction (black arrow). This is evident when comparing the unit cells of the two materials, as cXOEt-FADT contains one molecule per unit cell whereas cXPr-FADT contains two. The difference in molecular spacing results in a decreased SAPT0 interaction energy (−28.0 vs. −29.1 kcal mol−1) compared to the closer packed neighbours, which appears to be mostly due to a reduced dispersion stabilisation. Comparing the decomposed SAPT0 energies for these strongest interactions in the two different derivatives, there is an increased electrostatic stabilisation for cXOEt-FADT (−10.7 kcal mol−1) compared with cXPr-FADT (−8.5 or −8.9 kcal mol−1). This is likely originating from the partial charge on the oxygen of the ether chain interacting with adjacent alkyl chains through a non-traditional hydrogen bond. Other interactions within the crystals include the second π-stacking direction which is symmetrical for both cXOEt- and cXPr-FADTs, with SAPT0 energies around −10 kcal mol−1 (red data points in Fig. 6). Analysis of another in-stack pair made up of only sidechain-to-sidechain interactions was also performed (blue data points).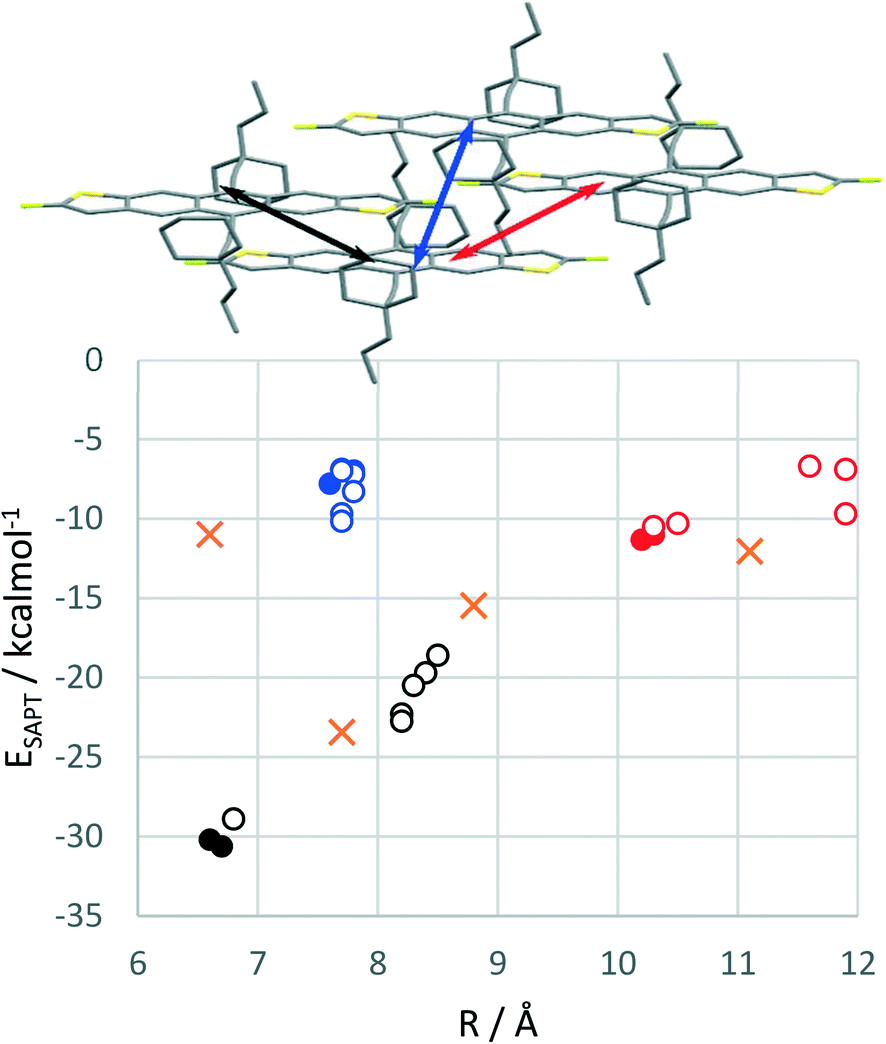 | ||
| Fig. 6 SAPT0 interaction energies versus molecular displacement for all FADT derivatives in 2D brickwork packing. Filled circles represent structures optimised from experimental crystal structures of cXOEt- and cXPr-FADTs. Open circles represent six different hypothetical structures based on inserting TnPC- or EDnPC-FADTs into similar 2D motifs. Colours of circles correspond to molecular pairs in the figure. Orange crosses denote similar molecular pair interactions from optimised EDnPC-FADT crystal structure. | ||
TnPC- and EDnPC-FADT were next placed into a 2D brickwork packing environment by editing the side chains of the cXOEt or cXPr structures and re-optimising. It was quite apparent while building these structures that the starting point would be influential, where the direction chosen to extend the side chains leads to different final local minima, and that these could potentially be very different from any experimentally derived 2D brickwork polymorphs for these materials. Furthermore, in the case of EDnPC there is the additional problem of the position of the ethyl chain versus the other two propyl chains, with each starting conformer resulting in different packing and different lattice energies. In total, three different 2D brickwork structures were generated for both EDnPC- and TNPC-FADT to act as representative structures of what a 2D brickwork packing with these side chains might look like. In each case, these show weaker lattice energies than those optimised from their experimental 1D crystal structures.
The starting alignment of the alkyl groups in these hypothetical structures strongly influences pairwise interaction energies, particularly the interaction with major molecular overlap (black open circles, Fig. 6). Changing the side chain from cyclic to linear pushes the molecules further apart and weakens this interaction by up to 10 kcal mol−1. The second π-stack exhibits similar changes on a smaller scale, while the alkyl-only interactions seem less affected by cyclic versus straight alkyl chains. These changes can mostly be attributed to the increased separation of the molecules by breaking the cyclic chain and increasing side group volume. This forces the FADT molecules further apart, with resulting loss of dispersion stabilisation which is only partially compensated by reduced steric exchange repulsion. These effects can be observed in the component energies of the SAPT decomposition analysis (see ESI†). There is one form of EDnPC-FADT packing where all three pairwise interactions are close to that of cXPr- and cXOEt-FADT, with SAPT0 energies of −28.9, −10.3 and −8.3 kcal mol−1, suggesting that under the correct conditions growth of a 2D brickwork polymorph might be possible. However, the computed lattice energy of this hypothetical structure is still smaller than the relaxed experimental packing structure.
The experimental structure of EDnPC also contains a number of the interactions found in the 2D brickwork packing (Fig. 4). SAPT0 energies and intermolecular displacements for these molecular pairs are shown in Fig. 6 as orange crosses. The π-stacking interactions occur at 7.7 and 8.8 Å with interaction energies of −23.4 and −15.5 kcal mol−1. Despite these interactions looking quite similar to the 2D brickwork, the energetics are quite different, with a long axis displacement making the stronger pair weaker, but the secondary π-stacked pair more tightly bound. This lateral shift also impacts the alkyl-only interaction, which is now at a closer centre-to-centre molecular distance and slightly strengthened relative to the blue data points. Finally, an alkyl-to-π interaction was also investigated, with 11.1 Å between the centres of the molecules due to the length of the alkylethynyl group. Stabilisation energies are on the same order as the weaker π-stacking interaction. As we have seen in past studies,10 the dispersion stabilisation gained from interactions of side chains should not be underestimated, and is likely a large factor on EDnPC-FADT not forming a true 2D brickwork packing.
Computed transfer integrals for the π-stacking interactions show similar magnitudes for electronic coupling in the two materials. Due to the presence of two isomers, and the relatively symmetrical shape of the FADT molecules, different disordermers20–23 are possible due to different positions of the thiophene atoms in the solid state. Both cXPr- and cXOEt-FADT exhibit around 65% occupation of the major atomic positions and 35% of the minor positions. The highest couplings are found in the most tightly bound molecular pairs (black arrow, Fig. 6), where the most abundant disordermer interaction exhibits hole transfer integrals of 35 and 37 meV for cXPr and cXOEt-FADTs respectively. This coupling increases for minor disordermers due to more overlap of the diffuse sulfur orbitals with the adjacent π system. The increased molecular distance due to side chain rotation in cXPr results in lower electronic coupling for all disordermer pairs. The transfer integrals in the alternative charge transport direction (red arrow, Fig. 6) are much smaller, and exhibit the opposite disordermer dependence. These couplings are also higher in cXOEt-FADT (26 meV) than in cXPr-FADT (16 meV). Based on the static molecular alignments, cXOEt-FADT is expected to exhibit higher charge mobilities than cXPr-FADT.
Assessment of charge transport properties
Heteroacenes exhibiting 2D brickwork π-stacking are typically well suited to organic field-effect transistor (OFET) fabrication, and the calculated intermolecular electronic couplings suggest that, while outstanding charge carrier mobilities are not to be expected, these molecules should yield reasonably functioning devices to assess the impact of the removal of the Si atom on film-forming capabilities and device properties. To that end, both cXOEt-FADT and cXPr-FADT were successfully used to form solution-cast bottom-contact, bottom gate OFETs on Si/SiO2 substrates and the charge carrier mobility was extracted.24 We were quite surprised by the low solubility of these materials compared to TES-FADT: while appropriate solution concentrations could be obtained, dissolution required chlorinated aromatic solvents and extensive heating. Fig. 7a shows the images of the cXPr-FADT films obtained by casting from a solution of chlorobenzene (CB), 1,2 dichlorobenzene (DCB) and n-butylbenzene (NBB), respectively. The DCB solution was heated at 60 °C for 5 minutes before deposition to ensure that the starting material was dissolved well, while the NBB solution was heated at 160 °C for 15 minutes. All samples were annealed 100 °C for 10 minutes prior to testing to remove any residual solvent. At least 30 devices of each type have been analysed from both spin-casted and drop casted samples. Electrical characterisation was performed in ambient atmosphere and typical transfer (drain current IDversus gate–source voltage VGS at constant drain–source voltage VDS) and transport curves (IDversus VDS at constant VGS) are illustrated in Fig. 7b, along with the schematic of the device structure included in the inset. All contacts were treated with pentafluorobenzene thiol (PFBT) to improve injection.7 Additional current–voltage characteristics obtained under other processing conditions, as well as on cXOEt-FADT are included in Fig. S46–S49 in the ESI.† The mobility was extracted from the transfer curve in the saturation regime using standard procedures,24 and we found that it varies greatly depending on the processing conditions, with values between μ = 10−6 cm2 V−1 s−1 and μ = 10−2 cm2 V−1 s−1 being recorded for cXPr-FADT and between μ = 10−8 cm2 V−1 s−1 and μ = 10−3 cm2 V−1 s−1 for cXOEt-FADT samples. These mobilities are comparable with the values obtained using space charge limited current (SCLC) measurements with the same types of contacts (Fig. S50, ESI†), and that, unlike OFET measurements which probe the transport at the interface between the dielectric and the semiconductor, can access the bulk properties. The lack of uniformity in the morphology of the films was reflected in high variations in charge carrier mobility even on the same substrate. Polymorphism25 has also not been ruled out as X-ray diffraction was not performed on the thin films. Additionally, the current–voltage curves point to the fact that the contact effects are not negligible in these OFETs, thus we expect that higher mobilities are possible with further improvements of the interface between the semiconductor and source/drain electrodes. We attempted to extract the value of the contact resistance using the gated transmission line method,26 but the non-uniformity of the films made the analysis challenging and the resulting values were unreliable.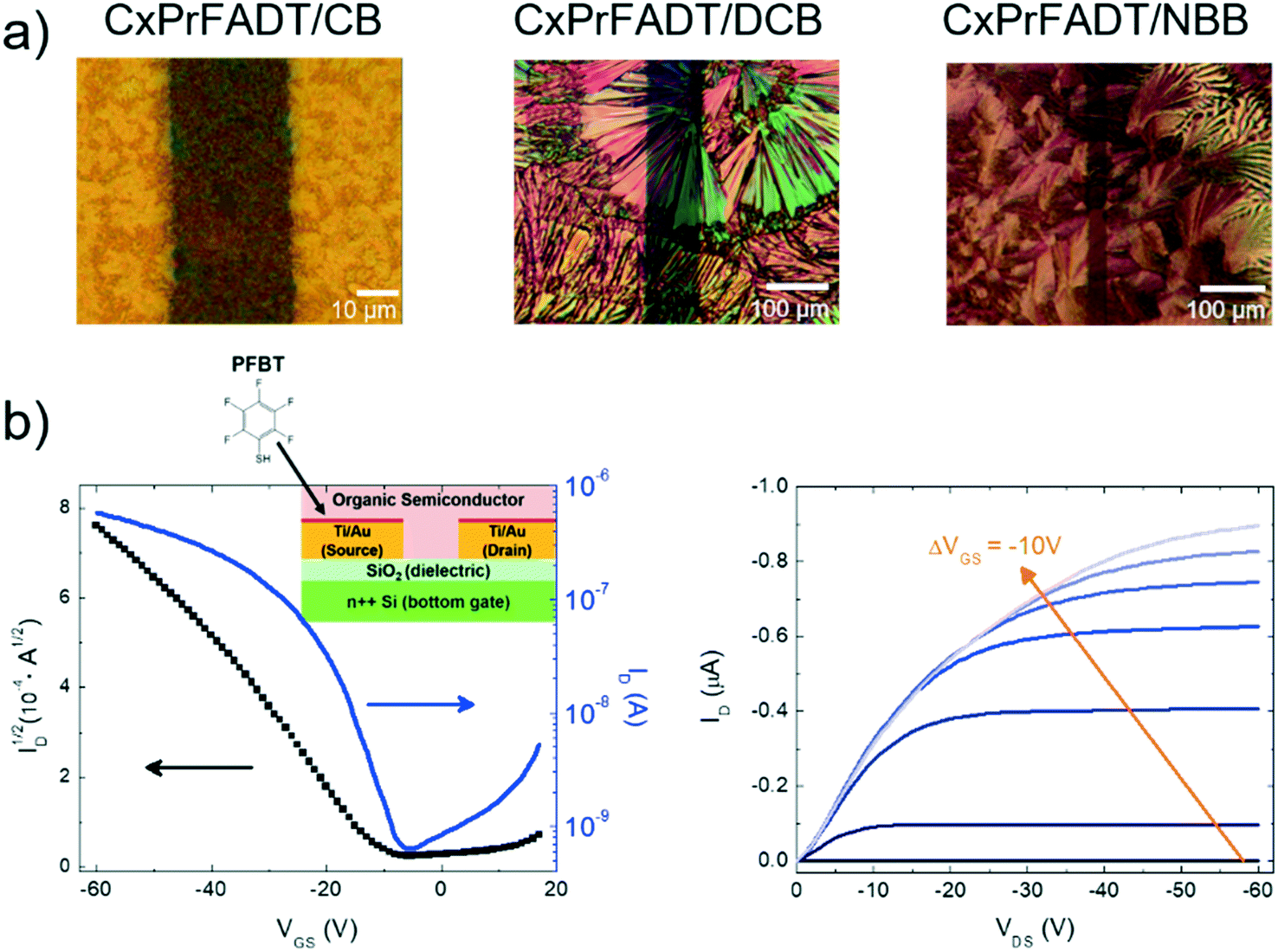 | ||
| Fig. 7 a) From left to right, top view image of the OFETs based on cXPr-FADT films obtained by casting from a solution of chlorobenzene (CB), 1,2 dichlorobenzene (DCB) and n-butylbenzene (NBB), respectively. b) Transfer (left) and transport (right) plots measured to characterise the OFETs, with the device structure in the inset. | ||
Conclusions
While the use of silylethynyl functional groups to impart solubility and stability to organic semiconductor chromophores has been widespread for nearly twenty years, very few reports of similar carbon alkynes can be found in the literature. We previously reported synthesis of the hydrocarbon analog of diF TES ADT, and showed that it adopted a completely different crystal packing compared to the silyl and germyl derivatives.15 Here, we have followed a logical design process, synthesizing a series of carbon-based substituents to arrive at two derivatives with 2-dimensional crystal packing suitable for transistor studies. Crystal packing seems to be impacted by the overall size and shape of the side chains, which affect intermolecular interactions by a balance of dispersion and exchange repulsion energies, as well as entropic effects introduced by elongated alkyl chains and asymmetry. In both derivatives, hole mobility was low and device fabrication was hampered by poor solubility. Optimisation of fabrication conditions may yet yield consistently high performing devices from these materials which may need drastically different approaches, for example blending with binder polymers,27 to overcome their low solubilities. While the use of an all-carbon functional group allows for a much wider array of structural modifications to be explored, the beneficial properties of the silicon atom (higher electron density, weak electron-withdrawing characteristics, and length and flexibility of the C–Si bond) may outweigh the synthetic advantages. Future small molecule organic semiconductor design should consider the solubility and processability of the material in addition to obtaining desirable crystal packing structures with good charge transport networks. The new alkylethynyl side chains can clearly be adapted in many ways to control the size, shape, and functional group incorporation that can influence solid state packing, and may be more suitable for other organic semiconductors whose electronically active cores have higher intrinsic solubilities than fluoroanthradithiophene.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Synthetic efforts were supported by the National Science Foundation under Cooperative Agreement No. 1849213. Computational analysis was supported by NSF DMREF (DMR-1627428). Transistor fabrication and testing was also supported by NSF DMREF (DMR-1627925).References
- C. K. Frederickson, B. D. Rose and M. M. Haley, Acc. Chem. Res., 2017, 50, 977–987 CrossRef CAS PubMed.
- W. Fudickar and T. Linker, J. Am. Chem. Soc., 2012, 134, 15071–15082 CrossRef CAS PubMed.
- L. Zhang, A. Fonari, Y. Liu, A.-L. M. Hoyt, H. Lee, D. Granger, S. Parkin, T. P. Russell, J. E. Anthony, J.-L. Brédas, V. Coropceanu and A. L. Briseno, J. Am. Chem. Soc., 2014, 136, 9248–9251 CrossRef CAS PubMed.
- U. H. F. Bunz, Chem. – Eur. J., 2009, 15, 6780–6789 CrossRef CAS PubMed.
- J. E. Anthony, J. S. Brooks, D. L. Eaton and S. R. Parkin, J. Am. Chem. Soc., 2001, 123, 9482–9483 CrossRef CAS PubMed.
- O. D. Jurchescu, S. Subramanian, R. J. Kline, S. D. Hudson, J. E. Anthony, T. N. Jackson and D. J. Gundlach, Chem. Mater., 2008, 20, 6733–6737 CrossRef CAS.
- Z. A. Lamport, K. J. Barth, H. Lee, E. Gann, S. Engmann, H. Chen, M. Guthold, I. McCulloch, J. E. Anthony, L. J. Richter, D. M. DeLongchamp and O. D. Jurchescu, Nat. Commun., 2018, 9, 5130 CrossRef PubMed.
- J. E. Anthony, D. L. Eaton and S. R. Parkin, Org. Lett., 2002, 4, 15–18 CrossRef CAS PubMed.
- J. E. Anthony, S. Subramanian, S. R. Parkin, S. K. Park and T. N. Jackson, J. Mater. Chem., 2009, 19, 7984–7989 RSC.
- K. J. Thorley, T. W. Finn, K. Jarolimek, J. E. Anthony and C. Risko, Chem. Mater., 2017, 29, 2502–2512 CrossRef CAS.
- A. Troisi, G. Orlandi and J. E. Anthony, Chem. Mater., 2005, 17, 5024–5031 CrossRef CAS.
- R. Schmidt, S. Göttling, D. Leusser, D. Stalke, A. M. Krause and F. Würthner, J. Mater. Chem., 2006, 16, 3708–3714 RSC.
- Y. Li, Y. Wu, P. Liu, Z. Prostran, S. Gardner and B. S. Ong, Chem. Mater., 2007, 19, 418–423 CrossRef CAS.
- H. G. Kim, H. H. Choi, E. Song, K. Cho and E. J. Choi, RSC Adv., 2015, 5, 8070–8076 RSC.
- K. J. Thorley, M. Benford, Y. Song, S. R. Parkin, C. Risko and J. E. Anthony, Mater. Adv., 2021, 2, 5415–5421 RSC.
- Y. Mei, M. A. Loth, M. Payne, W. Zhang, J. Smith, C. S. Day, S. R. Parkin, M. Heeney, I. McCulloch, T. D. Anthopoulos, J. E. Anthony and O. D. Jurchescu, Adv. Mater., 2013, 25, 4352–4357 CrossRef CAS PubMed.
- Y. Shu, Y. F. Lim, Z. Li, B. Purushothaman, R. Hallani, J. E. Kim, S. R. Parkin, G. G. Malliaras and J. E. Anthony, Chem. Sci., 2011, 2, 363–368 RSC.
- Y. F. Lim, Y. Shu, S. R. Parkin, J. E. Anthony and G. G. Malliaras, J. Mater. Chem., 2009, 19, 3049–3056 RSC.
- E. G. Hohenstein, R. M. Parrish, C. D. Sherrill, J. M. Turney and H. F. Schaefer, J. Chem. Phys., 2011, 135, 174107 CrossRef PubMed.
- H. F. Iqbal, E. K. Holland, J. E. Anthony and O. D. Jurchescu, Mater. Horiz., 2020, 7, 2390–2398 RSC.
- C.-W. Huang, X. You, P. J. Diemer, A. J. Petty, J. E. Anthony, O. D. Jurchescu and J. M. Atkin, Commun. Chem., 2019, 2, 22 CrossRef.
- P. J. Diemer, J. Hayes, E. Welchman, R. Hallani, S. J. Pookpanratana, C. A. Hacker, C. A. Richter, J. E. Anthony, T. Thonhauser and O. D. Jurchescu, Adv. Electron. Mater., 2017, 3, 1600294 CrossRef PubMed.
- K. J. Thorley and C. Risko, J. Mater. Chem. C, 2016, 4040–4048 RSC.
- Z. A. Lamport, H. F. Haneef, S. Anand, M. Waldrip and O. D. Jurchescu, J. Appl. Phys., 2018, 124, 071101 CrossRef.
- J. Chen, M. Shao, K. Xiao, A. J. Rondinone, Y. L. Loo, P. R. C. Kent, B. G. Sumpter, D. Li, J. K. Keum, P. J. Diemer, J. E. Anthony, O. D. Jurchescu and J. Huang, Nanoscale, 2014, 6, 449–456 RSC.
- M. Waldrip, O. D. Jurchescu, D. J. Gundlach and E. G. Bittle, Adv. Funct. Mater., 2020, 30, 1904576 CrossRef CAS.
- T. Salzillo, F. D'Amico, N. Montes, R. Pfattner and M. Mas-Torrent, CrystEngComm, 2021, 23, 1043–1051 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2119595–2119598. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1me00158b |
| This journal is © The Royal Society of Chemistry 2022 |