
Advances in research on 3C-like protease (3CLpro) inhibitors against SARS-CoV-2 since 2020
Roufen
Chen
a,
Yali
Gao
b,
Han
Liu
a,
He
Li
a,
Wenfa
Chen
*b and
Junjie
Ma
 *a
*a
aSchool of Medicine, Huaqiao University, Quanzhou, 362000, China. E-mail: majunjie3612@hqu.edu.cn
bPharmacy Department, The Second Affiliated Hospital of Fujian Medical University, Quanzhou, 362000, China. E-mail: 67942074@qq.com
First published on 27th October 2022
Abstract
COVID-19 caused by SARS-CoV-2 in late 2019 is still threatening global human health. Although some vaccines and drugs are available in the market, controlling the spread of the SARS-CoV-2 virus remains a huge challenge. 3C-like protease (3CLpro) is a highly conserved key protease for SARS-CoV-2 replication, and no relevant homologous protein with a similar cleavage site to 3CLpro has been identified in humans, highlighting that development of 3CLpro inhibitors exhibits great promise for treatment of COVID-19. In this review, the authors describe the structure and function of 3CLpro. To better understand the characteristics of SARS-CoV-2 3CLpro inhibitors, the SARS-CoV-2 3CLpro inhibitors reported since 2020 are classified into peptidomimetic covalent inhibitors, non-peptidomimetic covalent inhibitors and non-covalent small molecule inhibitors, and the representative inhibitors, their biological activities and binding models are highlighted. Collectively, we hope that all the information presented here will provide new insights into the design and development of more effective 3CLpro inhibitors against SARS-CoV-2 as novel anti-coronavirus drugs.
1. Introduction
Coronavirus disease (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is having a major impact on public health worldwide. Statistics from the World Health Organization (WHO) show that as of 14 October 2022, 620![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 878405 coronavirus cases have been diagnosed, including 6543138 deaths.1 Common symptoms of COVID-19 include cough, loss of smell or taste, nasal congestion, fever, etc.,2–4 and may leave multiple sequelae such as fatigue, breathing difficulties, chest pain and mental health problems.5,6
878405 coronavirus cases have been diagnosed, including 6543138 deaths.1 Common symptoms of COVID-19 include cough, loss of smell or taste, nasal congestion, fever, etc.,2–4 and may leave multiple sequelae such as fatigue, breathing difficulties, chest pain and mental health problems.5,6
The coronavirus is approximately 80–120 nM in diameter and has a linear single-stranded positive-sense RNA (+ssRNA) genome with a full length of 27–32 kb, which is the RNA virus with the largest genome ever discovered.7,8 In 2003, the virus causing severe acute respiratory syndrome (SARS) was known as SARS-CoV-1 as a member of the genus coronavirus in the coronaviridae family.9–11 The virus causing SARS in 2019 belongs to the same beta-coronavirus as SARS-CoV-1 and shares 89.1% gene sequence similarity with SARS-CoV-1 and is known as SARS-COV-2.12–15
As shown in Fig. 1A, the genome of SARS-CoV-2 consists of 14 open reading frames (ORFs). The first ORFs (ORF1a and ORF1b) are located at the 5′ end of the genome and occupy approximately two-thirds of the genome, while the other ORFs are located at the 3′ end of the genome and encode four common structural proteins, namely spike (S) protein, envelope (E) protein, membrane (M) protein and nucleocapsid (N) protein, as shown in Fig. 1B.16,17 SARS-CoV-2 invades the host cells by manipulating the host serum protease (TMPRSS2 and TMPRSS11D) to cleave the spike proteins.18,19 After entering the host cell, the viral RNA is released into the host cytoplasm and translated by the host ribosome, i.e. ORF1a and ORF1b are translated into polyprotein 1a (pp1a) and polyprotein 1ab (pp1ab), respectively. pp1a and pp1ab are precursors to a variety of factors involved in the viral replication cycle and need to be hydrolyzed and released by the virus' own encoded protease to function.20 There are two types of key proteases responsible for the hydrolysis of these polyproteins (pp1a and pp1ab) in the coronavirus, namely papain-like protease (PLpro) and 3C-like protease (3CLpro).21–23 PLpro cleaves pp1a and pp1ab to produce non-structural proteins (nsp)1–3. 3CLpro, also known as the main protease (Mpro), cleaves pp1a and pp1ab to produce nsp4–16 and is highly conserved in different types of coronaviruses; the 3CLpro of SARS-CoV-2 shares 96.1% gene sequence similarity with SARS-CoV-1 and the RMSD of their three-dimensional structures is only 0.41 Å (Fig. 1C).24–26
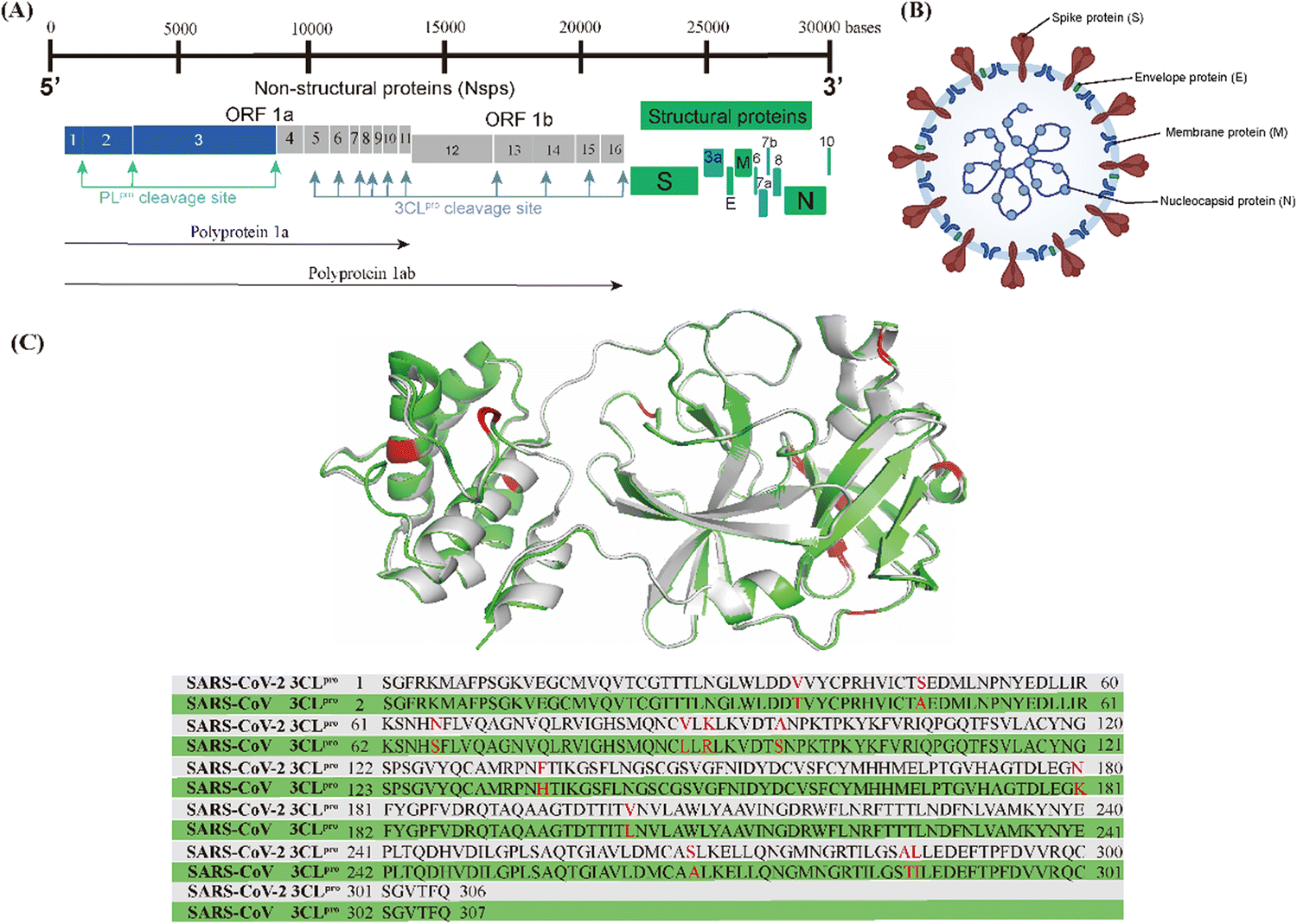 | ||
| Fig. 1 (A) Genome of SARS-CoV-2. (B) Morphological structure of SARS-CoV-2. (C) 3CLpro superposition and gene sequence comparison of SARS-CoV-2 (grey, PDB code: 6XHM) and SARS-CoV-1 (green, PDB code: 6XHO). | ||
2. 3CL protease
3CLpro is a cysteine protease consisting of three structural domains (Fig. 2).27,28 A catalytic dyad of Cys145–His41 residues in the cleft between structural domain I and structural domain II is responsible for cleaving pp1a and pp1ab, producing 12 functional proteins (nsp4–16) that execute functions such as the replication and transcription of the viral genome.29–32 Therefore, the catalytic pocket of 3CLpro is a potential site for the development of inhibitors against SARS-CoV-2 and other coronaviruses. Moreover, no relevant homologous protein with a similar cleavage site to 3CLpro has been identified in humans, which reduces the risk of off-target and makes the development of 3CLpro inhibitors a very attractive strategy for the treatment of COVID-19.33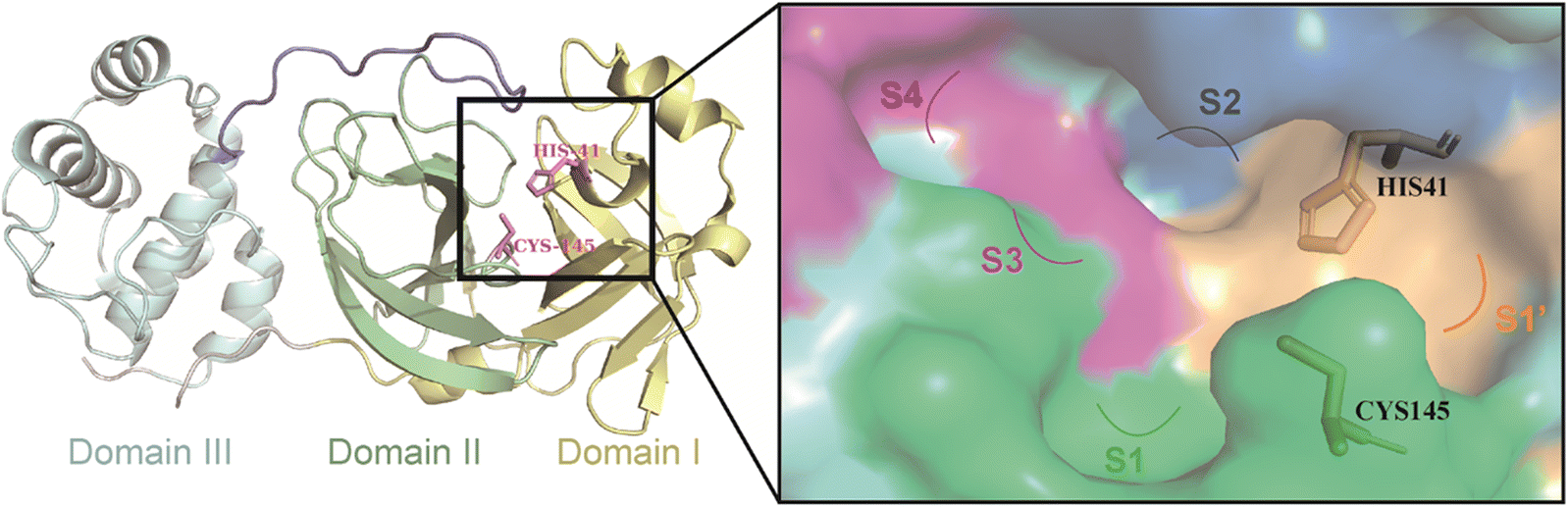 | ||
| Fig. 2 The structure of 3CLpro (PDB code: 6UL7) and the surface of its catalytic pocket. | ||
The peptide substrate of SARS-CoV-2 3CLpro is numbered as P4-P3-P2-P1-P1′ (Fig. 3) according to the Schechter–Berger nomenclature from the N to C-terminal amino acids.34 The binding sites of the substrate in the catalytic pocket are numbered correspondingly as S4-S3-S2-S1-S1′ (Fig. 3). The cleavage site is located between P1 and P1′. The substrate hydrolysis mechanism of 3CLpro is shown in Fig. 3. His41 captures the hydrogen proton of the thiol group on Cys145, generating a nucleophilic sulfur negative ion, which attacks the carbonyl group of the amide bond on the peptide substrate, forming a tetrahedral intermediate. Then the tetrahedral intermediate is decomposed to give a thiol ester and a corresponding amine. Next, the electrophilic thiol ester is hydrolyzed to produce a corresponding carboxylic acid, thereby completing the cleavage of the peptide substrate, and regenerating the active protease.35–37
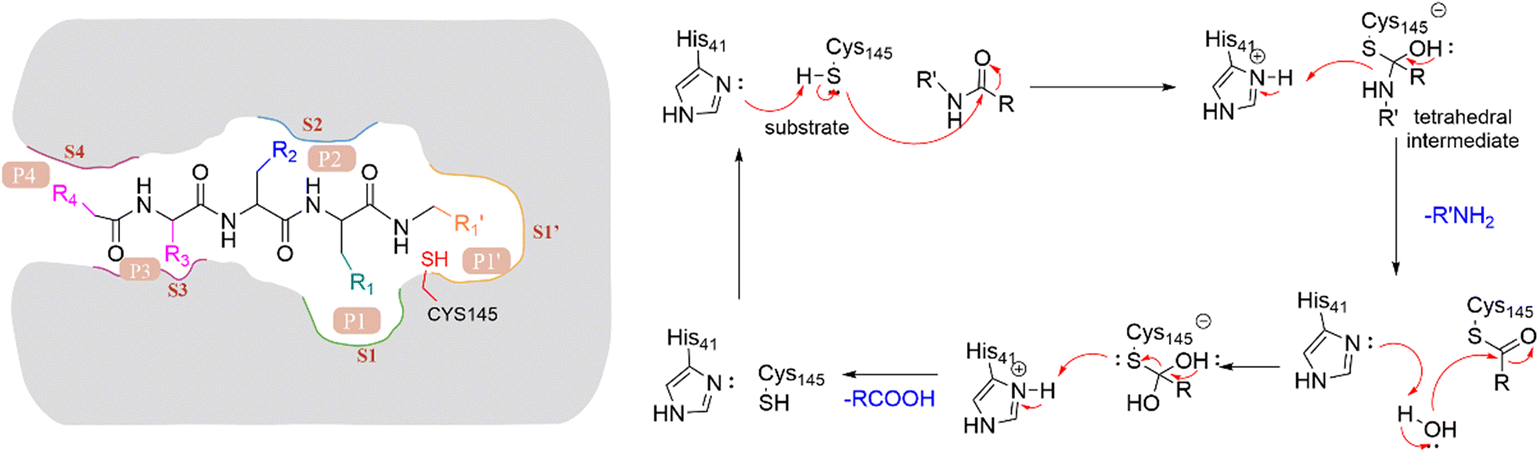 | ||
| Fig. 3 Nomenclature of 3CLpro hydrolysis substrates and mechanism of substrate proteolysis. | ||
Currently, the reported 3CLpro inhibitors include covalent and non-covalent inhibitors. Covalent inhibitors reversibly or irreversibly inhibit the activity of 3CLpro by forming a covalent adduct with Cys145 in the catalytic pocket through an electrophilic group (warhead), and according to their structural characteristics, covalent inhibitors mainly include peptidomimetic and non-peptidomimetic covalent inhibitors. Most non-covalent inhibitors are substrate competitive inhibitors that exhibit reversible inhibitory activity by competing with the substrate of 3CLpro for the catalytic pocket, while a few non-covalent inhibitors bind to the dimerization surface of 3CLpro.38–40
3. Peptidomimetic covalent inhibitors
Peptidomimetic covalent inhibitors are designed by mimicking the SARS-CoV-2 3CLpro substrate. Based on the structure of the warhead in the P1′ site, peptidomimetic covalent inhibitors include aldehydes, sulfonates, nitriles, ketones and Michael acceptors.3.1 Aldehydes
After the outbreak of COVID-19, Dai et al. reported two peptidomimetic covalent 3CLpro inhibitors using an aldehyde group as the warhead (1, 2, Fig. 4), which showed outstanding inhibitory activity against SARS-CoV-2 3CLpro with IC50 values of 53 nM and 40 nM and potent antiviral activity against SARS-CoV-2 infected Vero E6 cells with EC50 values of 0.53 μM and 0.72 μM, respectively. The co-crystallization of 1 and 2 with 3CLpro revealed that the aldehyde group acts as an electrophilic group to form a covalent bond with the thiol group of Cys145, and the (S)-γ-butyrolactam group and the indole-2-formyl group occupy the S1 pocket and S4 pocket, respectively, establishing multiple hydrogen bonds with His163 and Glu166. The cyclohexyl group of 1 and the 3-fluorophenyl group of 2 occupy the S2 pocket and form important hydrophobic interactions, in which the fluorine atom of the phenyl group further creates a hydrogen bond interaction with Gln189 (Fig. 4).41 In the continuing study, Dai et al. further reported compound 3 with a phenyl group at the P2 site, which inhibited SARS-CoV-2 3CLpro comparably to 1 and 2 with an IC50 value of 34 nM. The further studies revealed that 3 displayed potent antiviral activity against SARS-CoV-2 in Vero E6 cells with an EC50 value of 0.29 μM and a longer half-life (T1/2) for intraperitoneal, subcutaneous and intravenous administration compared to 1 and 2. But the clearance rate of 3 was faster by intravenous injection than that of compound 2, which might be due to the absence of the fluorine atom increasing the electron cloud density of the benzene ring at the P2 position, thereby reducing the metabolic stability.42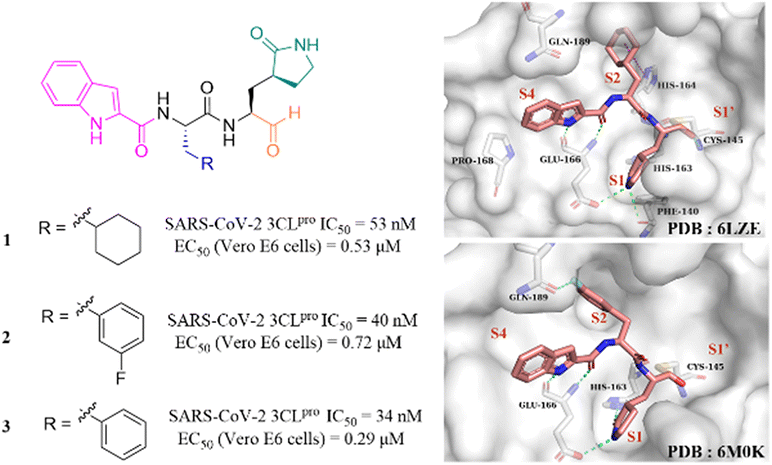 | ||
| Fig. 4 Peptidomimetic covalent inhibitors containing an aldehyde moiety (1–3) (PDB code: 6LZE and 6M0K). | ||
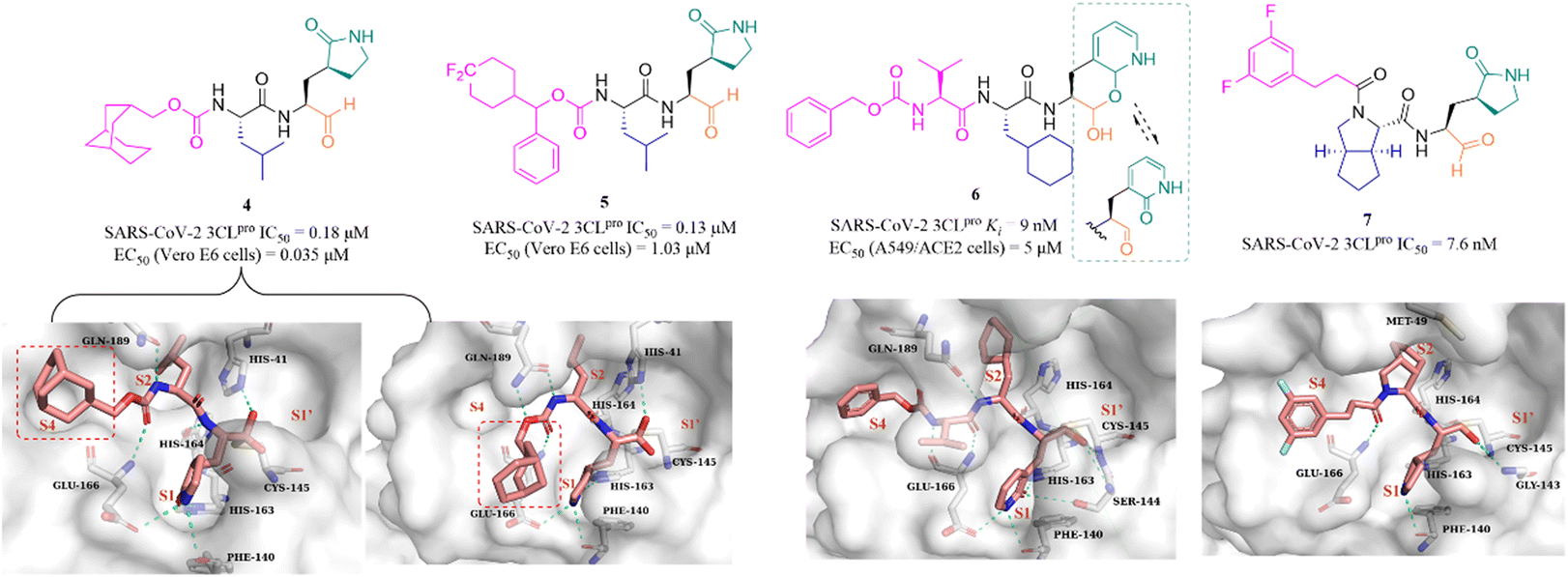
In recent years, many groups have designed a variety of peptidomimetic inhibitors using the aldehyde group as a warhead by introducing different types of groups at P1, P2, P3 and P4 sites. Dampalla et al. designed compound 4 by introducing a conformationally restricted cyclohexyl group at the P3 site, which showed significant inhibition against SARS-CoV-2 3CLpro and SARS-CoV-2 infected Vero E6 cells with IC50 and EC50 values of 0.18 μM and 0.035 μM, respectively (Fig. 5).43 X-ray crystallography studies confirmed that 4 adopts two conformations in which the bicyclic ring is projected away from the S4 pocket in subunit A and is positioned in the S4 pocket in subunit B. Subsequently, a range of structural optimizations around the structure of GC376 were carried out by Dampalla et al. including introduction of a gem-dimethyl group or stereocenter, deuteration and fluorine, at the P4 site, leading to the identification of compound 5, with an IC50 value of 0.13 μM against SARS-CoV-2 3CLpro and an EC50 value of 1.03 μM against SARS-CoV-2 in Vero E6 cells. X-ray crystallography demonstrated the feasibility of using a chiral center to attain directional control at the P4 site and augment binding interactions; for instance, in compound 5, the 4,4-difluorocyclohexane methyl linking the benzyl carbon at the P4 site, together with the directional control provided by the chiral center, produced a near-optimal combination.44
 | ||
| Fig. 5 Peptidomimetic covalent inhibitors containing an aldehyde moiety (4–7) (PDB code: 7LKR, 7M2P, and 7D3I). | ||
To improve the safety and metabolic instability caused by the high electrophilicity of the aldehyde group, Li et al. designed a peptidomimetic self-masked aldehyde covalent inhibitor (6) by replacing γ-lactam at the P1 with 2-pyridone (Fig. 5). Compound 6 had a ki value of 9 nM against SARS-CoV-2 3CLpro. The NMR spectra of 6 showed that there was no apparent aldehydic peak in CDCl3 as the solvent, but the exact type (lactol/aldehyde and pyridone/hydroxypyridine) of 6 inside cells is unclear due to various factors such as solvent, temperature, pH, etc. The crystal structure of SARS-CoV-2 3CLpro in complex with 6 revealed that the γ-lactam can be bioisosterically replaced by the 2-pyridone moiety, and 3CLpro is capable of catalyzing the ring-opening of the putative masked aldehyde of 6, forming a hemithioacetal with Cys145. The safety and pharmacokinetic properties of 6 were not further validated in this work.45 Inspired by the co-crystal structures of SARS-CoV-2 3CLpro in complex with the approved antivirals, boceprevir (PDB: 7COM) and telaprevir (PDB: 7C7P), a series of 3CLpro inhibitors containing a bicycloproline moiety at the P2 site and a medium size hydrophobic group at the P3 site, were designed by Qiao et al. The most active compound, 7, had an IC50 value of 7.6 nM against SARS-CoV-2 3CLpro. The cocrystal structure of 3CLpro with 7 confirmed that 7 binds to the catalytic pocket of 3CLpro. The warhead aldehyde forms a covalent bond with Cys145, and the γ-lactam ring inserts deeply into the S1 pocket, which is like other peptidomimetic covalent inhibitors. The 1-ethyl-3,5-difluorobenzene moiety in an extended conformation and the rigid bicycloproline occupy the S4 and S2 pockets, respectively, and form multiple hydrophobic interactions with the residues of the catalytic pocket.46
3.2 Sulfonates
Peptidomimetic sulfonate inhibitors are prodrugs of aldehyde inhibitors that improve water solubility and pharmacokinetic properties. The sulfonate in the inhibitors can be rapidly hydrolyzed and converted to the corresponding aldehyde in a physiological environment.47 In 2020, Vuong et al. demonstrated two peptidomimetic covalent inhibitors, the parent GC373 and its prodrug GC376, which were previously used to treat feline coronavirus infection, that could effectively inhibit SARS-CoV-2 3CLpro with IC50 values of 0.40 μM and 0.19 μM, respectively. The co-crystallization of GC373 (PDB: 6WTK) and GC376 (PDB: 6WTJ) with 3CLpro confirmed that the prodrug GC376 converted to the parent drug GC373 resulting in identical ligands in the structures, and the aldehyde group forms a covalent bond with Cys145 of the catalytic pocket (Fig. 6).48 In the further study, to enhance the efficacy of GC376 against SARS-CoV-2, Vuong et al. carried out the structural optimization of GC376 in the P2 and P3 sites, and identified compounds 8 and 9 (Fig. 6) with IC50 values of 0.07 μM and 0.08 μM against SARS-CoV-2 3CLpro, respectively. The antiviral activity study in SARS-CoV-2 infected Vero E6 cells showed that 8 and 9 exhibited more potent inhibition with EC50 values of 0.57 μM and 0.70 μM than GC376, respectively.49 Crystallographic structures of inhibitor-3CLpro complexes reveal that the cyclopropyl group fills the S2 pocket in a more compact fashion compared to GC373, and the substitution of Cbz by a 3-fluoro/chlorobenzyl group forces a relocation of the P3 substituent into the S4 pocket, allowing for additional interactions with the protease.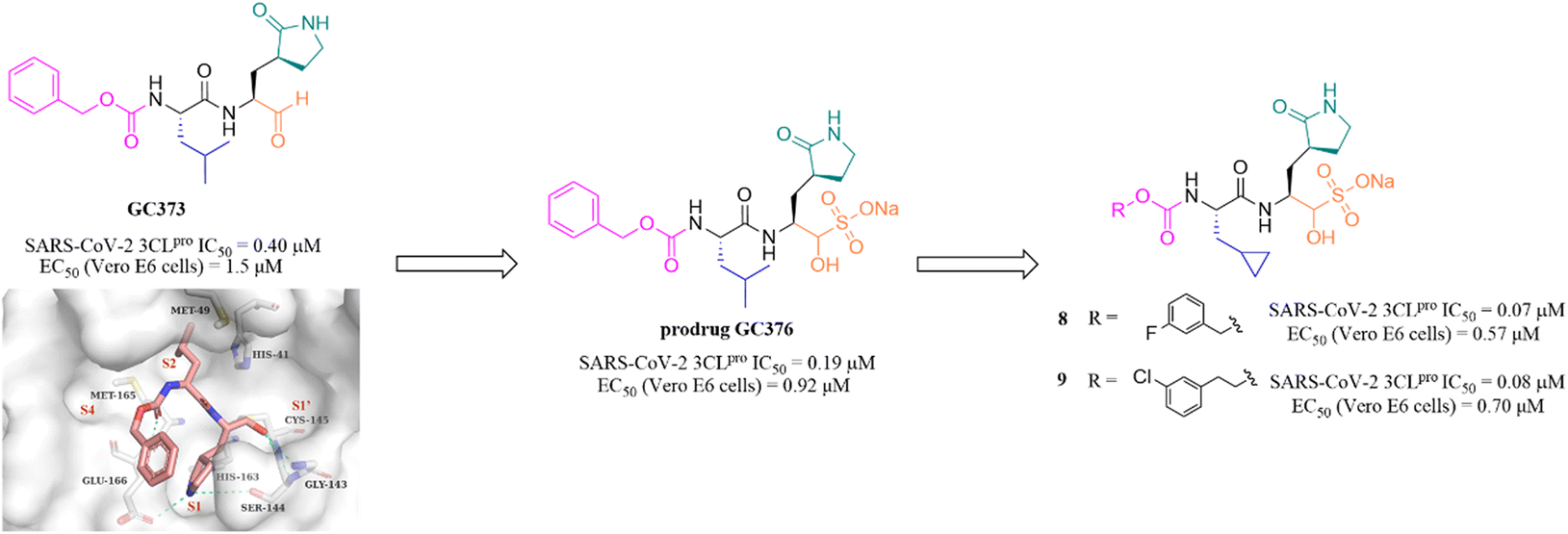 | ||
| Fig. 6 Peptidomimetic covalent inhibitors bearing a sulfonate moiety (GC373, GC376, 8 and 9) (PDB code: 6WTK). | ||
3.3 Nitriles
Nitrile has been widely used in drug design as a covalent reversible warhead of protease inhibitors. Although the nitrile group exhibits relative inertness, the electrophilicity of which can be enhanced by linkage to an electron-withdrawing group.50–52PF-07321332 (Nirmatrelvir, Fig. 7) is a reversible peptidomimetic covalent inhibitor bearing a nitrile warhead developed by Pfizer Inc., which effectively inhibited SARS-CoV-2 3CLpro with a Ki value of 3.11 nM, and exhibited potent antiviral activity in SARS-CoV-2 infected Vero E6 cells with an EC50 value of 74.5 nM. The co-crystallization of PF-07321332 with 3CLpro confirmed that the nitrile group forms a covalent bond with the thiol group of Cys145 and two hydrogen bonds with Cys145 and Gly143. The bicyclic tetrahydropyrrole moiety and the trifluoroacetyl occupy the S2 and S4 pockets, respectively. The γ-lactam at the P1 site and the amide skeleton at P3–P4 sites establish multiple hydrogen bonds with His163, Glu166, and Gln189, respectively. The in vivo antiviral activity in a mouse-adapted SARS-CoV-2 (SARS-CoV-2 MA10) model showed that PF-07321332 was effective at reducing the amount of SARS-CoV-2 MA10 virus in the lungs of mice. The pharmacokinetic property study showed that the CYP3A4 enzyme oxidatively metabolizes multiple sites of PF-07321332 (e.g., tert-butyl, azabicyclic and pyrrolidone), resulting in rapid clearance. Thus, PF-07321332 was co-administered with the potent CYP3A4 inhibitor (Ritonavir) to improve the therapeutic concentration of PF-07321332 in clinical trials.53 Currently, PAXLOVID (PF-07321332 + Ritonavir) is approved in the USA, UK and China as the first oral antiviral treatment for COVID-19.54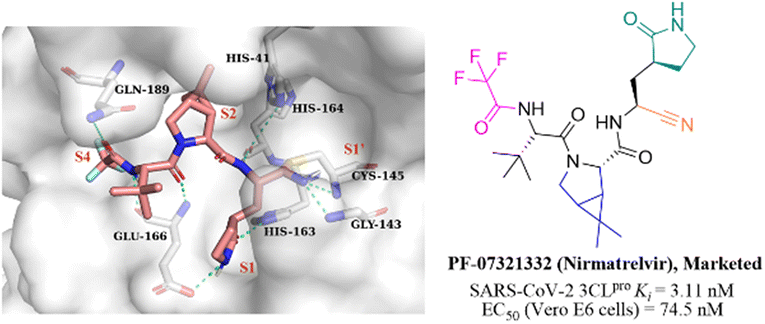 | ||
| Fig. 7 The co-crystallization of the peptidomimetic covalent inhibitor bearing a nitrile moiety (PF-07321332) with 3CLpro (PDB code: 7RFW). | ||
3.4 Ketones
PF-00835231 is a keto-based 3CLpro inhibitor designed by Pfizer Inc. for the treatment of SARS CoV-1 that occurred in 2003.55 Following the COVID-19 outbreak, PF-00835231 (Fig. 8) was demonstrated to be a potent SARS CoV-2 3CLpro inhibitor with a Ki value of 0.27 nM. The co-crystallization of PF-00835231 with SARS CoV-2 3CLpro revealed that the carbonyl group of the hydroxymethyl ketone forms a covalent bond with the thiol of Cys145, generating a hydroxyl group, which further establishes hydrogen bonds with the backbone NH of Cys145 and with the amide NH of Gly143 via a bridging water molecule. The terminal primary alcohol group forms a hydrogen bond with His41 (Fig. 8).55 In continuing research, to enhance the aqueous solubility of PF-00835231, Pfizer Inc. further designed a phosphate prodrug (PF-07304814) of PF-00835231. Preclinical studies showed that PF-07304814 exhibited excellent ADME properties, safety, and was rapidly converted to the active moiety (PF-00835231) in vivo.56 Currently, PF-07304814 is in phase I clinical trial (NCT04627532 and NCT04535167). Furthermore, inspired by the leaving ability of the α-acyloxy group, Bai et al. reported a series of peptidomimetic covalent inhibitors bearing an α-acetoxymethyl ketone warhead based on the structure of peptidomimetic hydroxymethyl ketone inhibitors. The activity study led to the identification of compound 10 (Fig. 8) with a 2,4,6-trimethyl-substituted pyridyl moiety, which not only showed more potent inhibitory activity against SARS-CoV-2 3CLpro with an IC50 value of 19 nM than GC376, but also displayed significant antiviral activity in SARS-CoV-2 infected Vero E6 cells with an EC50 value of 0.30 μM. The co-crystallization of 10 with SARS-CoV-2 3CLpro showed that the 2,4,6-trimethylnicotinate group has been left off, allowing 10 to form an irreversible covalent bond with the sulphur of Cys145 via the methylene carbon, rather than the carbonyl carbon, which is different from other ketone-based inhibitors. Notably, the six-membered lactam in the P1 site mimics well the five-membered glutamine in previously reported peptidomimetic inhibitors, demonstrating that these two groups are comparable in activity (Fig. 8).57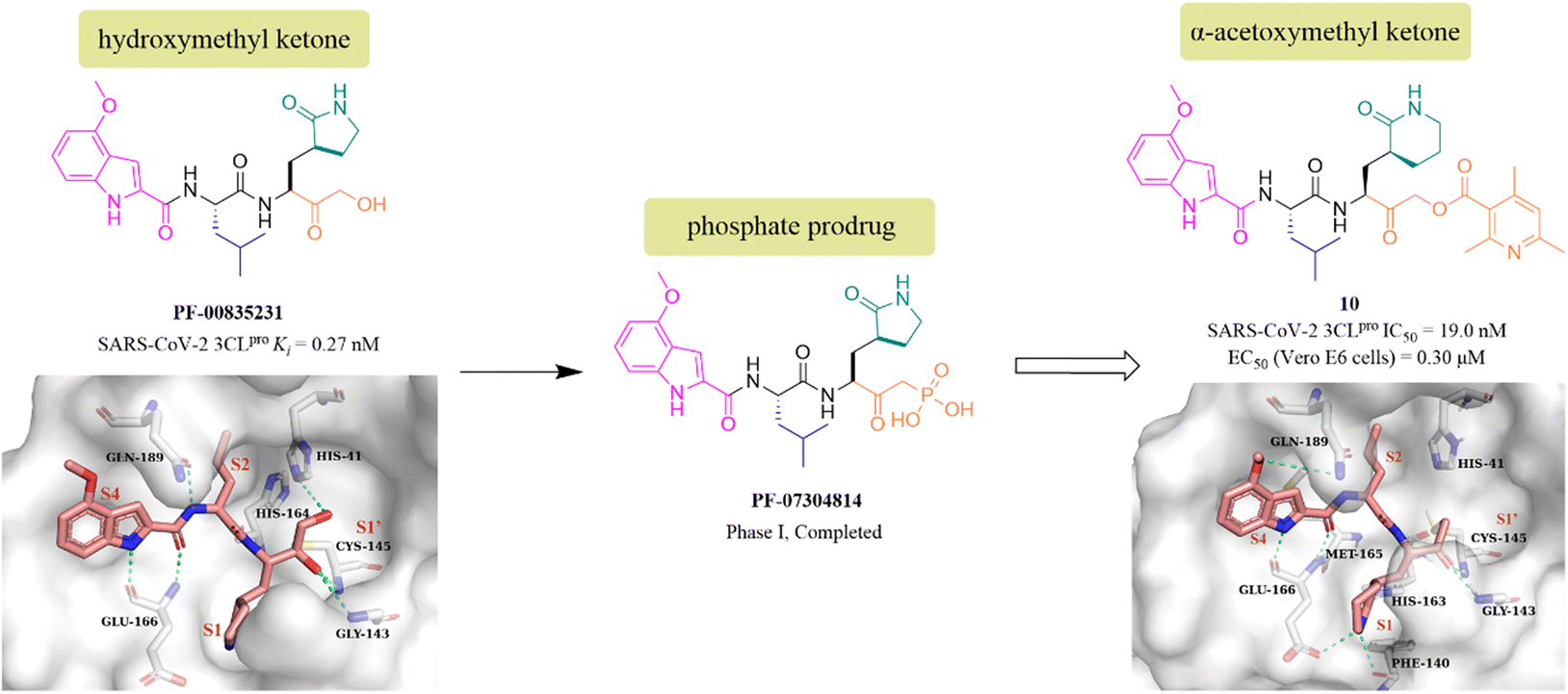 | ||
| Fig. 8 Peptidomimetic covalent inhibitors bearing ketones (PF-07304814, PF-00835231, and 10) and their co-crystallizations with 3CLpro (PDB code: 6XHM, 7MBI). | ||
YH-53 is a peptidomimetic covalent inhibitor containing a unique benzothiazolyl ketone warhead designed by Hayashi's team after the outbreak of SARS in 2003. In 2021, Konno et al. of Hayashi's team investigated the potential of YH-53 as an anti-SARS-CoV-2 drug, and found that YH-53 showed potent inhibitory activity against SARS-CoV-2 3CLpro with a Ki value of 34.7 nM and completely blocked the replication of the virus in Vero cells at a concentration of 10 μM (Fig. 9). The co-crystallization of YH-53 with SARS-CoV-2 3CLpro revealed that the carbonyl carbon at the P1′ site forms a covalent bond with the thiol group of Cys145 (Fig. 9). The preclinical pharmacokinetic property study revealed that the first-pass effect in the liver caused by the hydrolysis of the amide bond between P1 and P2 was the main reason for the low bioavailability (3.5%) of YH-53 in rats, which provided an important guide for further design of potent 3CLpro inhibitors with good pharmacokinetic properties.58
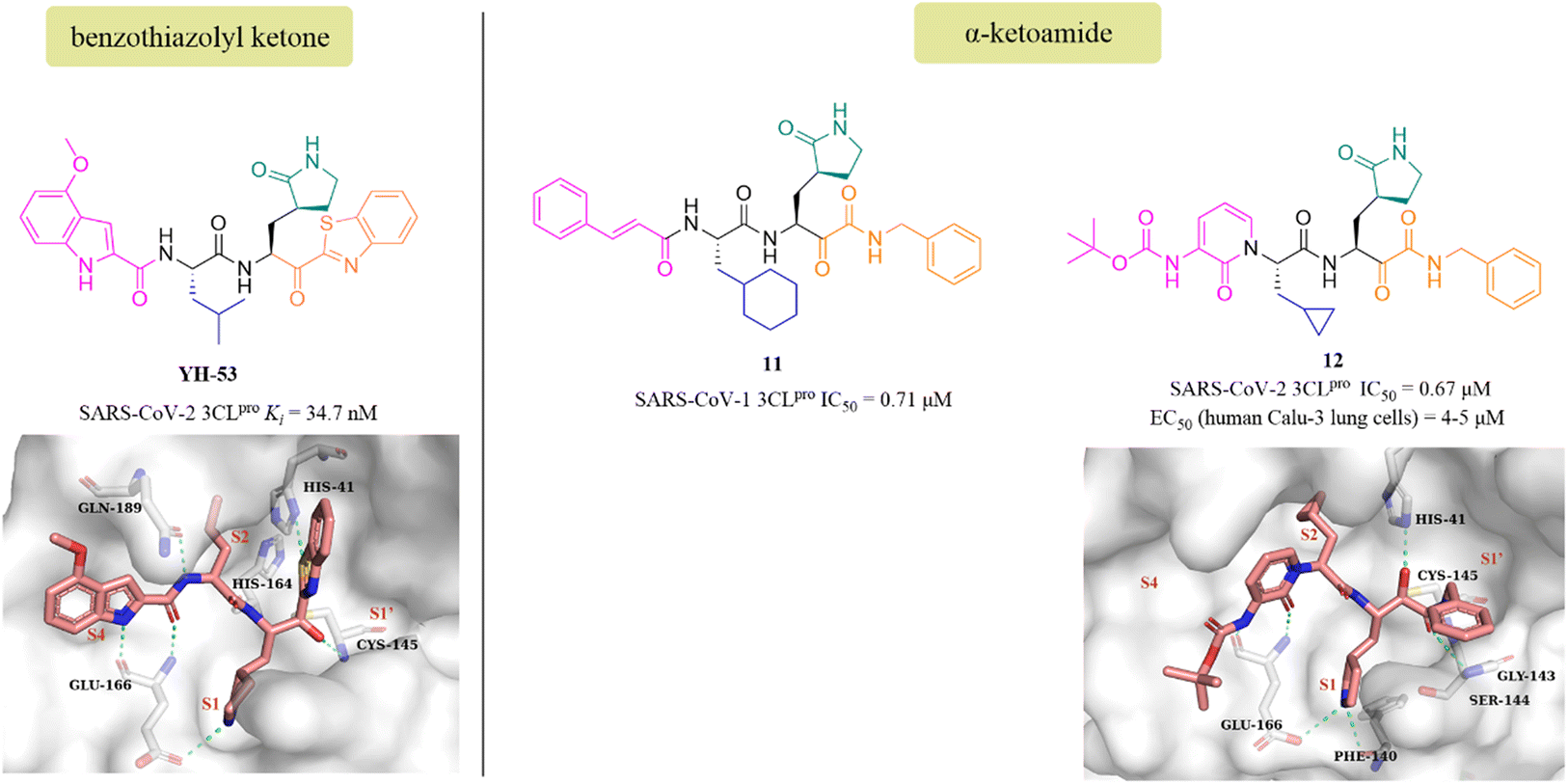 | ||
| Fig. 9 Peptidomimetic covalent inhibitors bearing ketones (YH-53, 11, and 12) and their co-crystallizations with 3CLpro (PDB code: 7E18, 6Y2G). | ||
The advantage of α-ketoamide as a warhead is that it provides two receptors for hydrogen bonding interactions rather than just one, as with other warheads such as aldehyde, mono-ketone or nitrile. Compound 11 is a peptidomimetic α-ketoamide broad-spectrum inhibitor of the 3CLpro of β-coronaviruses and α-coronaviruses as well as the 3Cpro of enteroviruses. Zhang et al. carried out a range of structural optimizations in the P2 and P3–P4 sites of 11, and identified compound 12, with an IC50 value of 0.67 μM against SARS-CoV-2 3CLpro (Fig. 9). Notably, the pharmacokinetic study showed that 12 had a pronounced lung tropism, highlighting the potential of administration by the inhalation route. The co-crystallization of 12 with SARS-CoV-2 3CLpro confirmed that the α-keto group forms a covalent bond with the thiol group of Cys145, and the α-ketoamide creates multiple hydrogen bonds with His41, Gly143, Cys145 and Ser144, respectively. The γ-lactam at the P1 site and the pyridone at the P3 site establish multiple hydrogen bonds with Phe140 and Glu166, respectively (Fig. 9).59,60
3.5 Michael acceptors
N3 (Fig. 10) is a broad-spectrum 3CLpro inhibitor containing the Michael receptor as a warhead that is effective against multiple coronaviruses, including SARS-CoV and MERS-CoV.60 Following the outbreak of COVID-19, Jin et al. further demonstrated that N3 could effectively inhibit SARS-CoV-2 3CLpro. The crystal structure of SARS-CoV-2 3CLpro in complex with N3 revealed that N3 binds in the catalytic pocket in an extended conformation, and the β-C atom of α,β-unsaturated ester forms a covalent bond with the thiol group of Cys145, indicating that Michael addition has occurred. In addition, the lactam occupies the S1 pocket and forms a hydrogen bond with His163, and the isobutyl of Leu and the methyl of Ala insert into the S2 and S4 pockets. The isopropyl of Val and the 3-methylisoxazole-5-formyl extend to the solvent-exposed area, suggesting that these parts may not be essential for inhibitory activity, and structural optimization of these parts can be carried out to improve potency and pharmacokinetic properties (Fig. 10).61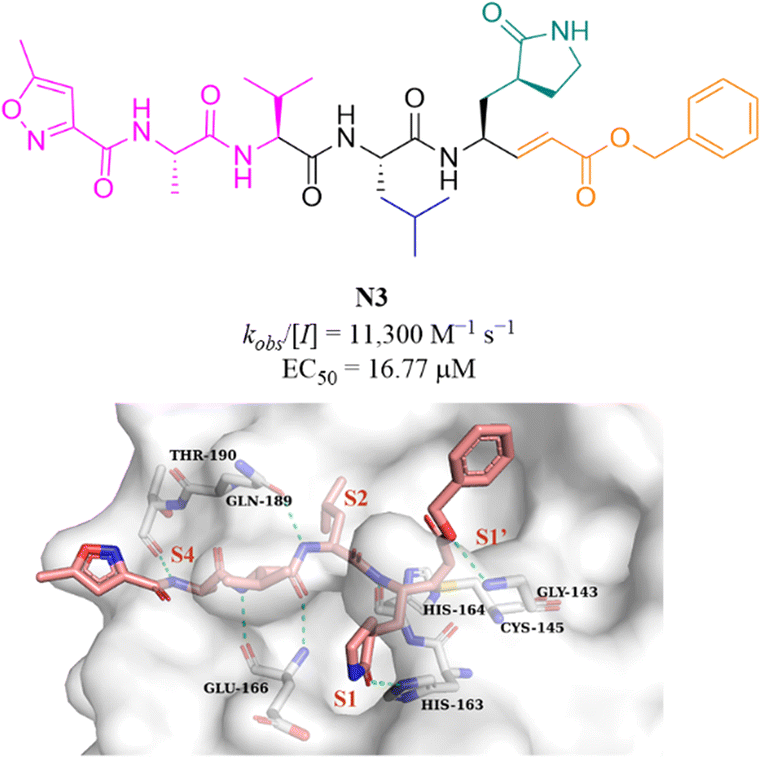 | ||
| Fig. 10 Peptidomimetic covalent inhibitor bearing a Michael receptor (N3) and its co-crystallization with 3CLpro (PDB code: 6LU7). | ||
4. Non-covalent small molecule inhibitors
Covalent inhibitors that form a covalent bond with the amino acid residue in the active pocket of the target protein often have potential toxic side effects and off-target problems, while non-covalent inhibitors that competitively bind to the target protein through weak reversible binding can greatly reduce these risks.62,63Drayman et al. screened a library of 1900 clinically safe drugs against OC43, a human beta coronavirus that causes the common cold, and identified masitinib, an orally bioavailable tyrosine kinase inhibitor, with an IC50 of 2.5 μM against SARS-CoV-2 3CLpro and an EC50 of 3.2 μM in SARS-CoV-2 infected A549/ACE2 cells (Fig. 11). The co-crystallization of masitinib with SARS-CoV-2 3CLpro revealed that masitinib noncovalently binds to the catalytic pocket, and the pyridine ring inserts into the S1 pocket and forms a hydrogen bond with His163; the aminothiazole ring forms two hydrogen bonds with His164 and the key residue Cys145. The toluene ring occupies the hydrophobic S2 pocket and creates an important π–π stacking interaction with another key residue His41. The N-methylpiperaze group is located outside of the catalytic pocket and the absence of masitinib is observed in S1′ and S4 pockets, suggesting that structural optimization can be carried out at these areas to further improve the inhibitory activity of 3CLpro, while decreasing the tyrosine kinase inhibitory activity to reduce the associated side effects (Fig. 11).64 Besides, Liu et al. and Cui et al. reported an N-substituted isatin derivative (13) and a juglone derivative (14) as non-covalent SARS-CoV-2 3CLpro inhibitors, with IC50 values of 45 nM and 72.07 nM, respectively (Fig. 11). 14 inhibited SARS-CoV-2 replication in Vero E6 cells with an EC50 value of 4.55 μM, whereas the high cytotoxicity of 13 limited the testing of anti-SARS-CoV-2 activity at the cellular level. The binding models of 13 and 14 to 3CLpro are unclear due to the lack of co-crystal structure (Fig. 11).65,66
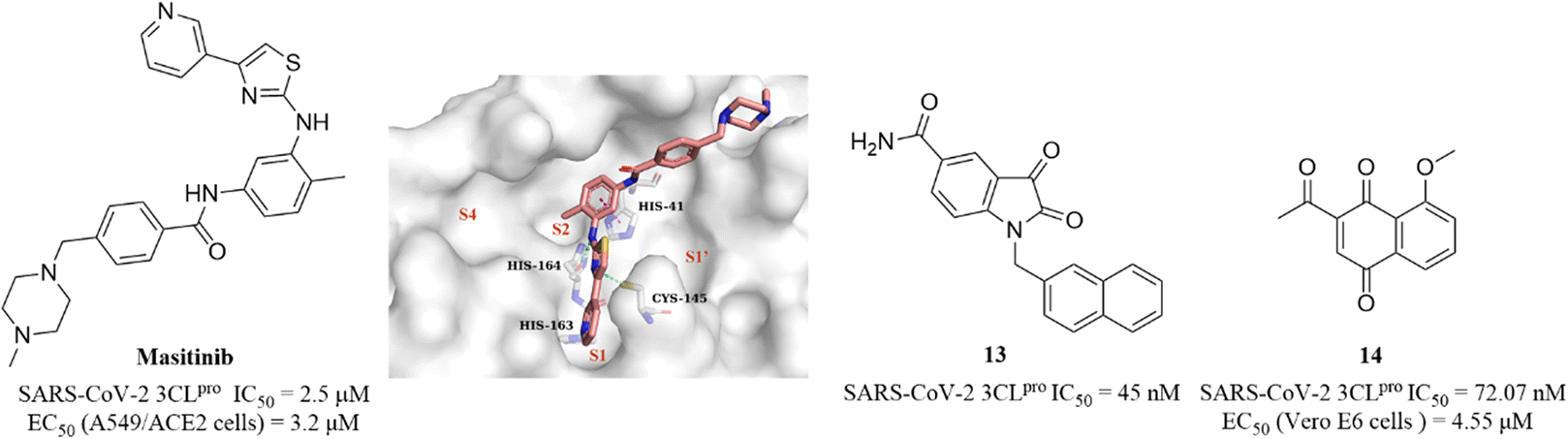 | ||
| Fig. 11 Non-covalent SARS-CoV-2 3CLpro inhibitor (masitinib and 13–14) (PDB code: 7JU7). | ||
Next, based on the co-crystallization of GC376, calpain inhibitor XII and ML188(R) with 3CLpro, Kitamura et al. discovered a non-covalent small molecule inhibitor (15, Fig. 12) using the one-pot Ugi-4CR synthetic methodology, which had an IC50 value of 0.20 μM against SARS-CoV-2 3CLpro and an EC50 value of 1.27 μM against SARS-CoV-2 in Vero E6 cells. The co-crystallization of 15 with 3CLpro revealed a ligand-induced binding pocket between the S2 and S4 pockets, which was not previously identified, and the benzyl group inserts into this binding pocket, enhancing the binding to 3CLpro (Fig. 12). The novel binding model of 15 can be applied in the further design of 3CLpro inhibitors.67 In addition, a structural optimization of SARS-CoV-1 3CLpro inhibitor ML300 was carried out by Han et al., leading to the identification of CCF0058981, with an IC50 value of 68 nM against SARS-CoV-2 3CLpro. CCF0058981 exhibited potent antiviral activity with EC50 values of 0.497 μM and 0.558 μM against SARS-CoV-2 infected Vero E6 cells in the cytopathic effect (CPE) inhibition assay and plaque reduction assay, respectively. The co-crystallization of an analogue (16) of CCF0058981 showed that the thiophene and benzotriazole moieties occupy the S1 and S2sp (S2) pockets, respectively. Significantly, the imidazole group occupies a newly formed channel (S2c) located between the canonical S1′ and S2sp pockets and forms two H-bonding interactions with Cys44 and The25. However, the high clearance and strong inhibition on CYP enzymes of CCF0058981 need to be improved in further studies (Fig. 12).68
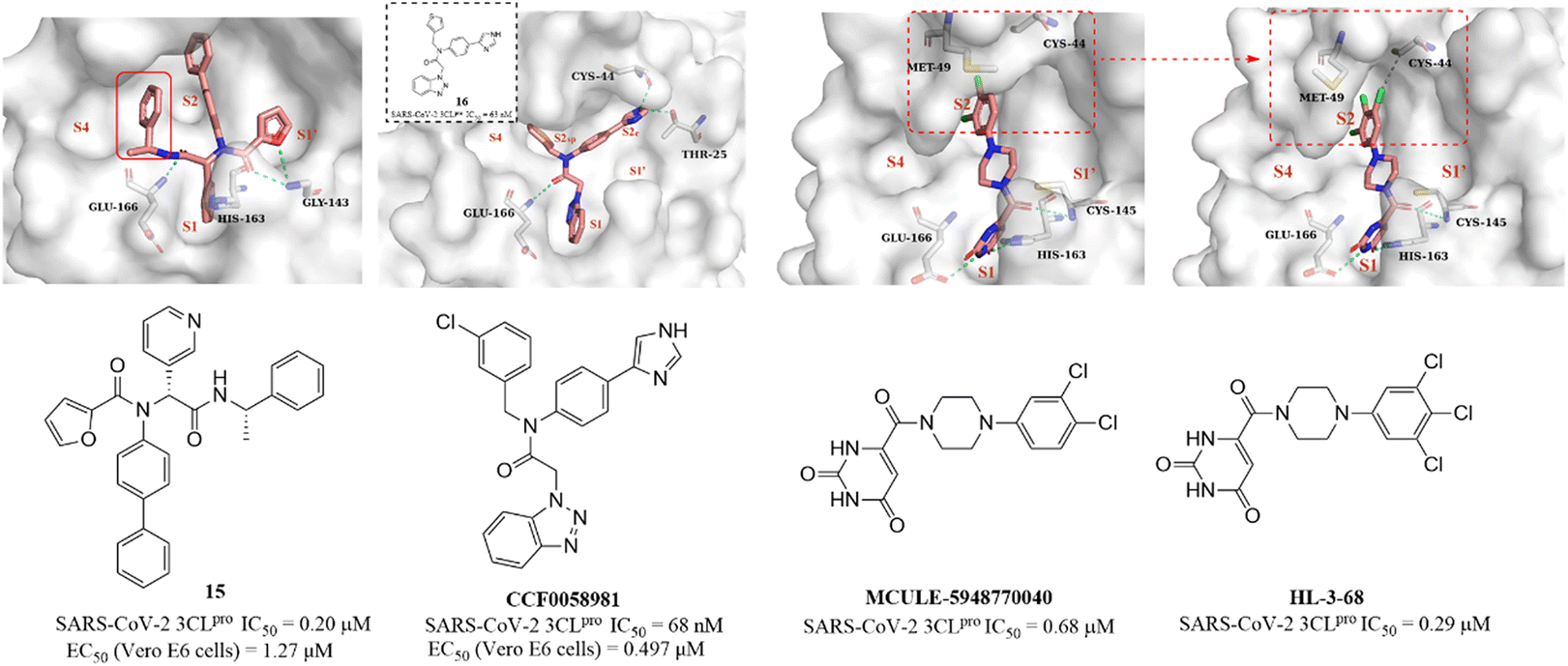 | ||
| Fig. 12 Non-covalent SARS-CoV-2 3CLpro inhibitors (15, 16, CCF0058981, MCULE-5948770040, HL-3-68) and their co-crystallizations with 3CLpro (PDB code: 7KX5, 7LMF, 7LTJ, 7RLS). | ||
Clyde et al. conducted a high-throughput virtual screening (HTVS) of over 6.5 million compounds in the Mcule database and identified MCULE-5948770040 (Fig. 12), with an IC50 value of 0.68 μM against SARS-CoV-2 3CLpro. The co-crystallization showed that the uracil moiety of MCULE-5948770040 occupies the S1 pocket and forms a hydrogen bond with His163, and the dichlorophenyl moiety occupies the S2 pocket (Fig. 12).69 In the continuing study, Kneller et al. carried out the structural optimization of MCULE-5948770040 to obtain HL-3-68 (Fig. 12), which exhibited more potent inhibition on SARS-CoV-2 3CLpro with an IC50 value of 0.29 μM than MCULE-5948770040. The co-crystallization of HL-3-68 with 3CLpro further revealed that the S2 pocket is sensitive to small changes in the ligand properties and the introduction of a chlorine atom at the 5-position of the phenyl group rotates Met49 and creates a van der Waals interaction with Cys44, enhancing the binding to 3CLpro (Fig. 12).70
Unoh et al. combined virtual screening, activity validation and structure-based drug design strategies to discover a non-covalent inhibitor with a triazine skeleton (S-217622, Fig. 13), with an IC50 value of 13 nM against SARS-CoV-2 3CLpro and an EC50 value of 0.37 μM in SARS-CoV-2 infected Vero E6 cells (Fig. 13). S-217622 showed promising DMPK properties and higher bioavailability in monkeys and dogs, suggesting that S-217622 might be used for the once-daily oral treatment of COVID-19 without requiring co-administration with a PK booster. The co-crystallization of S-217622 with 3CLpro revealed that the two carbonyls of the center triazine moiety form multiple hydrogen bonds with Glu166, Gly143 and Cys145, the 1-methyl-1H-1,2,4-triazolyl moiety occupies the S1 pocket and forms a hydrogen bonding interaction with His163, and the 2,4,5-trifluorobenzyl moiety occupies the hydrophobic S2 pocket. The 6-chloro-2-methyl-2H-indazole at the P1′ site establishes hydrogen bonding with Thr26 and hydrophobic interaction with Met49, respectively (Fig. 13).71 These above multiple interactions and the high fit with the catalytic pocket contribute to the excellent inhibitory activity of S-217622 against 3CLpro. Currently, S-217622 is in phase II/III clinical trial (JPRN-jRCT2031210350). Recently, Hou et al. screened more than 49 billion compounds by DNA encoded library (DEL) technology to identify a non-covalent small molecule SARS-CoV-2 3CLpro inhibitor, WPV01 (Fig. 13), with an IC50 value of 72 nM and an EC50 value of 12 nM against SARS-CoV-2 in A549-hACE2 cells. In vivo antiviral activity showed that WPV01 exhibited comparable anti-SARS-CoV-2 activity to PF-07321332 in K18-hACE2 mice. The co-crystallization of WPV01 with 3CLpro revealed that the isoquinoline ring inserts well into the S1 pocket and forms a hydrogen bond with His163. The carbonyl group at the 3-position of the isoquinoline ring and the methylcarbamoyl group form two hydrogen bonds with Asn142 and Glu166. The phenyl ring forms an amino–π interaction with Gln189, and the 4-bromo of the phenyl ring extends to the S4 pocket, establishing a halogen bond with Thr190. Notably, the strongly electron-withdrawing 6-nitro group extending to the S2 pocket provides an important contribution to the potency by further enhancing the amino–π and halogen bond interactions, which explains the decrease or loss of inhibitory activity when the nitro is replaced by other electron-withdrawing groups or hydrogen (Fig. 13). WPV01 has been approved for clinical trials.72
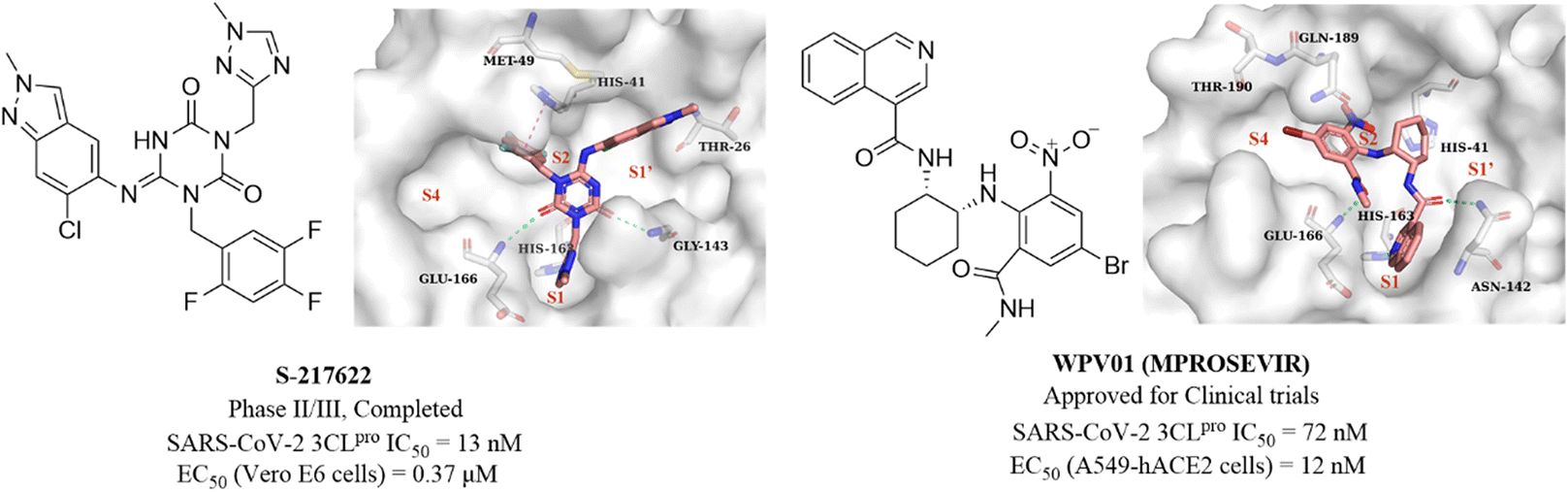 | ||
| Fig. 13 Non-covalent SARS-CoV-2 3CLpro inhibitors (S-217622 and WPV01) and their co-crystallizations with 3CLpro (PDB code: 7VU6 and 7EN8). | ||
5. Non-peptidomimetic covalent inhibitors
Although the presence of an electrophilic warhead in 3CLpro inhibitors contributes a longer duration of action in vivo, improving the oral bioavailability of peptidomimetic inhibitors remains a major challenge. Therefore, the development of non-peptidomimetic covalent inhibitors containing an electrophilic warhead provides the opportunity to discover 3CLpro inhibitors with better druggability.In 2021, a small-molecule compound (GRL-1720) containing an indoline moiety was reported by Hattori et al. as a SARS-CoV-2 3CLpro inhibitor with an IC50 value of 0.32 μM.73 In the continuing study, a comprehensive structure–activity relationship study of GRL-1720 was carried out by Ghosh et al., leading to the identification of compound 17, which showed more potent inhibitory activity against SARS-CoV-2 3CLpro with an IC50 value of 73 nM than GRL-1720. The co-crystallization of GRL-1720 with 3CLpro revealed that the formate ester at the P1′ site could react with the thiol group of Cys145 in 3CLpro to form an important covalent adduct (Fig. 14).74
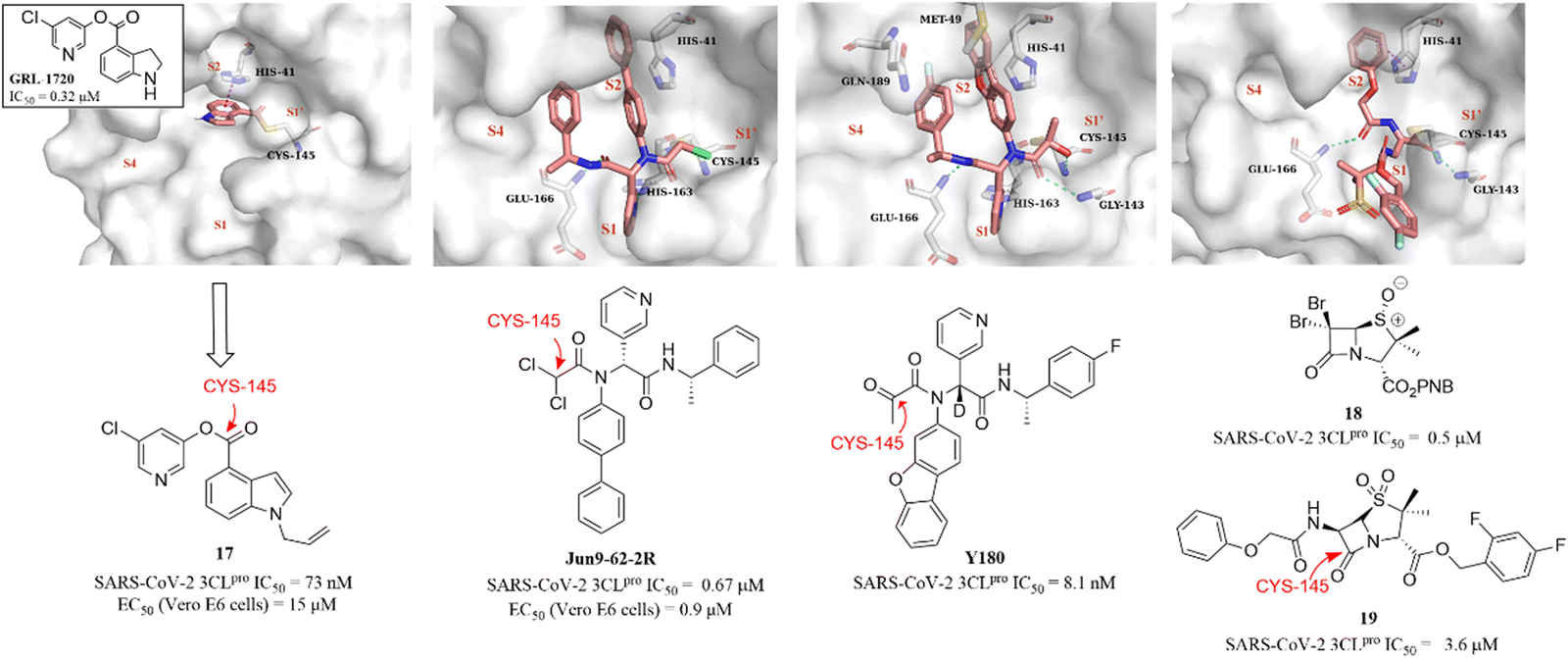 | ||
| Fig. 14 Non-peptidomimetic covalent inhibitors (GRL-1720, Jun9-62-2R, Y180, and 17–19) and their co-crystallizations with 3CLpro (PDB code: 7RBZ, 7RN1, 7FAZ, and 7Z59). | ||
Ma et al. explored a series of novel electrophiles for replacing the P1′ furyl substitution in compound 15, a non-covalent small molecule 3CLpro inhibitor discovered in their group's previous work (Fig. 14). Of these, Jun9-62-2R, containing dichloroacetamide as a warhead, obtained using the Ugi four-component reaction (Ugi-4CR), showed not only significantly 3CLpro inhibition (IC50 = 0.67 μM) and antiviral activity (EC50 = 0.90 μM in Vero E6 cells; EC50 = 2.05 μM in Caco2-hACE2 cells) but also remarkably improved target specificity over caplain, cathepsins, caspase-3, and trypsin (IC50 > 20 μM) (Fig. 14). The crystal structure of SARS-CoV-2 3CLpro in complex with Jun9-62-2R revealed that the binding pose is very similar to that of 15, and the key distinction between Jun9-62-2R and 15 is that the dichloroacetamide moiety forms a covalent bond with the thiol group of Cys145, which is consistent with the expectation (Fig. 14).75 In addition, Quan et al. further used the Ugi-4CR to synthesize a series of α-ketoamide-containing non-peptidomimetic covalent 3CLpro inhibitors. The promising compound, Y180, inhibited SARS-CoV-2 3CLpro with an IC50 value of 8.1 nM and showed broad antiviral activity in VeroE6-TMPRSS2 cells against WT SARS-CoV-2, B.1.1.7, B.1.617.1 and P.3, respectively. Moreover, Y180 displayed satisfying PK properties and safety with oral bioavailability of 92.9%, 31.9% and 85.7% in mice, rats and dogs, respectively. The co-crystallization revealed that the terminal carbonyl moiety of Y180 acts as an electrophilic warhead to form a covalent bond with Cys145 in the S1′ pocket, and the binding model of pyridyl, dibenzo(b,d)furanyl and 4-fluorophenyl of Y180 to the catalytic pocket is similar to that of 15 and Jun9-62-2R (Fig. 14).76
Recently, inspired by the β-lactam scaffold, a series of penicillin derivatives as non-peptidomimetic covalent inhibitors were reported by Malla et al. Of these, compound 18 had the strongest inhibitory activity against SARS-CoV-2 3CLpro with an IC50 value of 0.5 μM. The co-crystallization of an analogue (19) of 18 with SARS-CoV-2 3CLpro revealed that the β-lactam ring can be cleaved by reacting with Cys145 to form an important covalent adduct with 3CLpro (Fig. 14), highlighting the potential of β-lactams for use as a warhead of 3CLpro inhibitors.77
6. Conclusions
3CLpro has emerged as one of the most promising targets for the treatment of COVID-19. In recent years, many SARS-CoV-2 3CLpro inhibitors have been reported, mainly including peptidomimetic covalent inhibitors, non-peptidomimetic covalent inhibitors and non-covalent small molecule inhibitors. Peptidomimetic covalent inhibitors designed by mimicking the 3CLpro hydrolysis substrate are the most widely reported and exhibit significant inhibitory activity against 3CLpro, some of which have a nanomolar level of inhibitory activity. However, due to the presence of a highly reactive electrophilic warhead, there is a potential safety issue caused by off-target for covalent inhibitors, as well as the disadvantage of poor oral bioavailability associated with the peptide structure, e.g.Nirmatrelvir requires co-administration with a PK booster (Ritonavir) to improve oral bioavailability. Non-covalent small molecule inhibitors exhibit inhibitory activity mainly through weak reversible interactions with amino acid residues in the S1, S2 and S4 pockets of 3CLpro. Compared to the covalent inhibitors, non-covalent small molecule inhibitors avoid the safety issue associated with the off-target of the electrophilic warhead, and their oral bioavailabilities have also been significantly improved without the combination with a PK booster; S-217622 and WPV01 are in clinical trial. Nevertheless, the lack of strong interaction with 3CLpro results in the risk that non-covalent small molecule inhibitors may require prolonged or heavy dosing, potentially leading to drug resistance. Non-peptidomimetic covalent inhibitors that combine small molecule features and the advantages of covalent inhibitor warheads show the potential for better druggability than peptidomimetic covalent inhibitors, some of which were designed by introducing the warhead at the P1′ site based on the structures of reported non-covalent small molecule inhibitors, such as Jun9-62-2R. However, there are few reports of these inhibitors and no compounds are currently in clinical trials. Moreover, there is no known cysteine protease in humans with a similar cleavage site to 3CLpro, suggesting that the development of proteolysis-targeting chimera (PROTAC) degraders targeting the catalytic pocket of 3CLpro as anti-coronavirus drugs would be an attractive and promising strategy. SARS-CoV-2 is still spreading and mutating around the world, the highly conserved 3CLpro will continuously attract more attention from scientists and more novel and potent anti-coronavirus drugs targeting 3CLpro against SARS-CoV-2 will be reported in the future.Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
This work was supported by Fujian Provincial Health Technology Project (2020QNA060, China), the Startup Fund for scientific research, Fujian Medical University (2019QH1123, China) and the Fujian Province Natural Science Foundation (2021J01309, China).References
- WHO COVID-19 Explorer, World Health Organization, Geneva, https://worldhealthorg.shinyapps.io/covid/, (accessed August 2020) Search PubMed.
- R. C. Gerkin, K. Ohla, M. G. Veldhuizen, P. V. Joseph, C. E. Kelly, A. J. Bakke, K. E. Steele, M. C. Farruggia, R. Pellegrino, M. Y. Pepino, C. Bouysset, G. M. Soler, V. Pereda-Loth, M. Dibattista, K. W. Cooper, I. Croijmans, A. Di Pizio, M. H. Ozdener, A. W. Fjaeldstad, C. Lin, M. A. Sandell, P. B. Singh, V. E. Brindha, S. B. Olsson, L. R. Saraiva, G. Ahuja, M. K. Alwashahi, S. Bhutani, A. D'Errico, M. A. Fornazieri, J. Golebiowski, L. Dar Hwang, L. Öztürk, E. Roura, S. Spinelli, K. L. Whitcroft, F. Faraji, F. P. S. Fischmeister, T. Heinbockel, J. W. Hsieh, C. Huart, I. Konstantinidis, A. Menini, G. Morini, J. K. Olofsson, C. M. Philpott, D. Pierron, V. D. C. Shields, V. V. Voznessenskaya, J. Albayay, A. Altundag, M. Bensafi, M. A. Bock, O. Calcinoni, W. Fredborg, C. Laudamiel, J. Lim, J. N. Lundström, A. Macchi, P. Meyer, S. T. Moein, E. Santamaría, D. Sengupta, P. Rohlfs Dominguez, H. Yanik, T. Hummel, J. E. Hayes, D. R. Reed, M. Y. Niv, S. D. Munger, V. Parma and G. G. Author, Chem. Senses, 2021, 46, bjaa081 CrossRef PubMed.
- A. Giacomelli, L. Pezzati, F. Conti, D. Bernacchia, M. Siano, L. Oreni, S. Rusconi, C. Gervasoni, A. L. Ridolfo, G. Rizzardini, S. Antinori and M. Galli, Clin. Infect. Dis., 2020, 71, 889–890 CrossRef CAS PubMed.
- G. U. Kim, M. J. Kim, S. H. Ra, J. Lee, S. Bae, J. Jung and S. H. Kim, Clin. Microbiol. Infect., 2020, 26, 948 Search PubMed.
- N. Ferreira, A. Mikocka-Walus, M. A. L. van Tilburg, L. A. Graff, P. Apputhurai, M. Barreiro-de Acosta, F. B. Evertsz, J. Burisch, B. Lo, M. Petrik, I. A. Trindade, S. Jedel, G. Moser, A. Mokrowiecka, C. N. Bernstein, D. Dumitrascu, A. C. Ford, A. Stengel, R. Gearry and S. R. Knowles, J. Psychosom. Res., 2021, 148, 110561 CrossRef.
- A. Nalbandian, K. Sehgal, A. Gupta, M. V. Madhavan, C. McGroder, J. S. Stevens, J. R. Cook, A. S. Nordvig, D. Shalev, T. S. Sehrawat, N. Ahluwalia, B. Bikdeli, D. Dietz, C. Der-Nigoghossian, N. Liyanage-Don, G. F. Rosner, E. J. Bernstein, S. Mohan, A. A. Beckley, D. S. Seres, T. K. Choueiri, N. Uriel, J. C. Ausiello, D. Accili, D. E. Freedberg, M. Baldwin, A. Schwartz, D. Brodie, C. K. Garcia, M. S. V. Elkind, J. M. Connors, J. P. Bilezikian, D. W. Landry and E. Y. Wan, Nat. Med., 2021, 27, 601–615 CrossRef CAS PubMed.
- S. Ludwig and A. Zarbock, Anesth. Analg., 2020, 131, 93–96 CrossRef CAS.
- J. Cui, F. Li and Z. L. Shi, Nat. Rev. Microbiol., 2019, 17, 181–192 CrossRef CAS PubMed.
- M. A. Marra, S. J. Jones, C. R. Astell, R. A. Holt, A. Brooks-Wilson, Y. S. Butterfield, J. Khattra, J. K. Asano, S. A. Barber, S. Y. Chan, A. Cloutier, S. M. Coughlin, D. Freeman, N. Girn, O. L. Griffith, S. R. Leach, M. Mayo, H. McDonald, S. B. Montgomery, P. K. Pandoh, A. S. Petrescu, A. G. Robertson, J. E. Schein, A. Siddiqui, D. E. Smailus, J. M. Stott, G. S. Yang, F. Plummer, A. Andonov, H. Artsob, N. Bastien, K. Bernard, T. F. Booth, D. Bowness, M. Czub, M. Drebot, L. Fernando, R. Flick, M. Garbutt, M. Gray, A. Grolla, S. Jones, H. Feldmann, A. Meyers, A. Kabani, Y. Li, S. Normand, U. Stroher, G. A. Tipples, S. Tyler, R. Vogrig, D. Ward, B. Watson, R. C. Brunham, M. Krajden, M. Petric, D. M. Skowronski, C. Upton and R. L. Roper, Science, 2003, 300, 1399–1404 CrossRef CAS.
- P. A. Rota, M. S. Oberste, S. S. Monroe, W. A. Nix, R. Campagnoli, J. P. Icenogle, S. Peñaranda, B. Bankamp, K. Maher, M. H. Chen, S. Tong, A. Tamin, L. Lowe, M. Frace, J. L. DeRisi, Q. Chen, D. Wang, D. D. Erdman, T. C. Peret, C. Burns, T. G. Ksiazek, P. E. Rollin, A. Sanchez, S. Liffick, B. Holloway, J. Limor, K. McCaustland, M. Olsen-Rasmussen, R. Fouchier, S. Günther, A. D. Osterhaus, C. Drosten, M. A. Pallansch, L. J. Anderson and W. J. Bellini, Science, 2003, 300, 1394–1399 CrossRef CAS PubMed.
- K. Stadler, V. Masignani, M. Eickmann, S. Becker, S. Abrignani, H. D. Klenk and R. Rappuoli, Nat. Rev. Microbiol., 2003, 1, 209–218 CrossRef CAS PubMed.
- M. E. Killerby, H. M. Biggs, C. M. Midgley, S. I. Gerber and J. T. Watson, Emerging Infect. Dis., 2020, 26, 191–198 CrossRef.
- J. F. Chan, S. K. Lau, K. K. To, V. C. Cheng, P. C. Woo and K. Y. Yuen, Clin. Microbiol. Rev., 2015, 28, 465–522 CrossRef CAS PubMed.
- E. de Wit, N. van Doremalen, D. Falzarano and V. J. Munster, Nat. Rev. Microbiol., 2016, 14, 523–534 CrossRef CAS PubMed.
- Y.-Z. Zhang and E. C. Holmes, Cell, 2020, 181, 223–227 CrossRef CAS.
- R. Arya, S. Kumari, B. Pandey, H. Mistry, S. C. Bihani, A. Das, V. Prashar, G. D. Gupta, L. Panicker and M. Kumar, J. Mol. Biol., 2021, 433, 166725 CrossRef CAS PubMed.
- Z. Ke, J. Oton, K. Qu, M. Cortese, V. Zila, L. McKeane, T. Nakane, J. Zivanov, C. J. Neufeldt, B. Cerikan, J. M. Lu, J. Peukes, X. Xiong, H. G. Kräusslich, S. H. W. Scheres, R. Bartenschlager and J. A. G. Briggs, Nature, 2020, 588, 498–502 CrossRef CAS.
- C. B. Jackson, M. Farzan, B. Chen and H. Choe, Nat. Rev. Mol. Cell Biol., 2022, 23, 3–20 CrossRef CAS.
- M. Hoffmann, H. Kleine-Weber, S. Schroeder, N. Krüger, T. Herrler, S. Erichsen, T. S. Schiergens, G. Herrler, N.-H. Wu, A. Nitsche, M. A. Müller, C. Drosten and S. Pöhlmann, Cell, 2020, 181, 271–280.e278 CrossRef CAS.
- A. C. Brant, W. Tian, V. Majerciak, W. Yang and Z.-M. Zheng, Cell Biosci., 2021, 11, 136 CrossRef CAS.
- W. Rut, Z. Lv, M. Zmudzinski, S. Patchett, D. Nayak, S. J. Snipas, F. El Oualid, T. T. Huang, M. Bekes, M. Drag and S. K. Olsen, Sci. Adv., 2020, 6, eabd4596 CrossRef CAS.
- S. A. Amin, S. Banerjee, K. Ghosh, S. Gayen and T. Jha, Bioorg. Med. Chem., 2021, 29, 115860 CrossRef CAS.
- J. Osipiuk, S. A. Azizi, S. Dvorkin, M. Endres, R. Jedrzejczak, K. A. Jones, S. Kang, R. S. Kathayat, Y. Kim, V. G. Lisnyak, S. L. Maki, V. Nicolaescu, C. A. Taylor, C. Tesar, Y. A. Zhang, Z. Zhou, G. Randall, K. Michalska, S. A. Snyder, B. C. Dickinson and A. Joachimiak, Nat. Commun., 2021, 12, 743 CrossRef CAS.
- J. F. Chan, K. H. Kok, Z. Zhu, H. Chu, K. K. To, S. Yuan and K. Y. Yuen, Emerging Microbes Infect., 2020, 9, 221–236 CrossRef CAS.
- R. Channappanavar and S. Perlman, Semin. Immunopathol., 2017, 39, 529–539 CrossRef CAS PubMed.
- W.-H. Chen, L. Du, S. M. Chag, C. Ma, N. Tricoche, X. Tao, C. A. Seid, E. M. Hudspeth, S. Lustigman, C.-T. K. Tseng, M. E. Bottazzi, P. J. Hotez, B. Zhan and S. Jiang, Hum. Vaccines Immunother., 2014, 10, 648–658 CrossRef CAS.
- K. Anand, J. Ziebuhr, P. Wadhwani, J. R. Mesters and R. Hilgenfeld, Science, 2003, 300, 1763–1767 CrossRef CAS PubMed.
- V. Mody, J. Ho, S. Wills, A. Mawri, L. Lawson, M. Ebert, G. M. Fortin, S. Rayalam and S. Taval, Commun. Biol., 2021, 4, 93 CrossRef CAS PubMed.
- J. C. Ferreira and W. M. Rabeh, Sci. Rep., 2020, 10, 22200 CrossRef CAS.
- J. Lee, L. J. Worrall, M. Vuckovic, F. I. Rosell, F. Gentile, A.-T. Ton, N. A. Caveney, F. Ban, A. Cherkasov, M. Paetzel and N. C. J. Strynadka, Nat. Commun., 2020, 11, 5877 CrossRef CAS PubMed.
- A. Paasche, A. Zipper, S. Schäfer, J. Ziebuhr, T. Schirmeister and B. Engels, Biochemistry, 2014, 53, 5930–5946 CrossRef CAS.
- D. W. Kneller, G. Phillips, H. M. O'Neill, R. Jedrzejczak, L. Stols, P. Langan, A. Joachimiak, L. Coates and A. Kovalevsky, Nat. Commun., 2020, 11, 3202 CrossRef CAS PubMed.
- H. M. Mengist, T. Dilnessa and T. Jin, Front. Chem., 2021, 9, 622898 CrossRef CAS PubMed.
- I. Schechter and A. Berger, Biochem. Biophys. Res. Commun., 1967, 27, 157–162 CrossRef CAS.
- J. Yin, C. Niu, M. M. Cherney, J. Zhang, C. Huitema, L. D. Eltis, J. C. Vederas and M. N. James, J. Mol. Biol., 2007, 371, 1060–1074 CrossRef CAS.
- T. Pillaiyar, M. Manickam, V. Namasivayam, Y. Hayashi and S.-H. Jung, J. Med. Chem., 2016, 59, 6595–6628 CrossRef CAS.
- S. Zhang, M. Krumberger, M. A. Morris, C. M. T. Parrocha, A. G. Kreutzer and J. S. Nowick, Eur. J. Med. Chem., 2021, 218, 113390 CrossRef CAS PubMed.
- Y. Liu, C. Liang, L. Xin, X. Ren, L. Tian, X. Ju, H. Li, Y. Wang, Q. Zhao, H. Liu, W. Cao, X. Xie, D. Zhang, Y. Wang and Y. Jian, Eur. J. Med. Chem., 2020, 206, 112711 CrossRef CAS PubMed.
- J. He, L. Hu, X. Huang, C. Wang, Z. Zhang, Y. Wang, D. Zhang and W. Ye, Int. J. Antimicrob. Agents, 2020, 56, 106055 CrossRef CAS PubMed.
- B. Goyal and D. Goyal, ACS Comb. Sci., 2020, 22, 297–305 CrossRef CAS.
- W. Dai, B. Zhang, X. M. Jiang, H. Su, J. Li, Y. Zhao, X. Xie, Z. Jin, J. Peng, F. Liu, C. Li, Y. Li, F. Bai, H. Wang, X. Cheng, X. Cen, S. Hu, X. Yang, J. Wang, X. Liu, G. Xiao, H. Jiang, Z. Rao, L. K. Zhang, Y. Xu, H. Yang and H. Liu, Science, 2020, 368, 1331–1335 CrossRef CAS.
- W. Dai, D. Jochmans, H. Xie, H. Yang, J. Li, H. Su, D. Chang, J. Wang, J. Peng, L. Zhu, Y. Nian, R. Hilgenfeld, H. Jiang, K. Chen, L. Zhang, Y. Xu, J. Neyts and H. Liu, J. Med. Chem., 2021, 65, 2794–2808 CrossRef PubMed.
- C. S. Dampalla, Y. Kim, N. Bickmeier, A. D. Rathnayake, H. N. Nguyen, J. Zheng, M. M. Kashipathy, M. A. Baird, K. P. Battaile, S. Lovell, S. Perlman, K.-O. Chang and W. C. Groutas, J. Med. Chem., 2021, 64, 10047–10058 CrossRef CAS PubMed.
- C. S. Dampalla, A. D. Rathnayake, K. D. Perera, A.-R. M. Jesri, H. N. Nguyen, M. J. Miller, H. A. Thurman, J. Zheng, M. M. Kashipathy, K. P. Battaile, S. Lovell, S. Perlman, Y. Kim, W. C. Groutas and K.-O. Chang, J. Med. Chem., 2021, 64, 17846–17865 CrossRef CAS PubMed.
- L. Li, B. C. Chenna, K. S. Yang, T. R. Cole, Z. T. Goodall, M. Giardini, Z. Moghadamchargari, E. A. Hernandez, J. Gomez, C. M. Calvet, J. A. Bernatchez, D. M. Mellott, J. Zhu, A. Rademacher, D. Thomas, L. R. Blankenship, A. Drelich, A. Laganowsky, C.-T. K. Tseng, W. R. Liu, A. J. Wand, J. Cruz-Reyes, J. L. Siqueira-Neto and T. D. Meek, J. Med. Chem., 2021, 64, 11267–11287 CrossRef CAS PubMed.
- J. Qiao, Y.-S. Li, R. Zeng, F.-L. Liu, R.-H. Luo, C. Huang, Y.-F. Wang, J. Zhang, B. Quan, C. Shen, X. Mao, X. Liu, W. Sun, W. Yang, X. Ni, K. Wang, L. Xu, Z.-L. Duan, Q.-C. Zou, H.-L. Zhang, W. Qu, Y.-H.-P. Long, M.-H. Li, R.-C. Yang, X. Liu, J. You, Y. Zhou, R. Yao, W.-P. Li, J.-M. Liu, P. Chen, Y. Liu, G.-F. Lin, X. Yang, J. Zou, L. Li, Y. Hu, G.-W. Lu, W.-M. Li, Y.-Q. Wei, Y.-T. Zheng, J. Lei and S. Yang, Science, 2021, 371, 1374–1378 CrossRef CAS PubMed.
- S. R. Mandadapu, M. R. Gunnam, K.-C. Tiew, R. A. Z. Uy, A. M. Prior, K. R. Alliston, D. H. Hua, Y. Kim, K.-O. Chang and W. C. Groutas, Bioorg. Med. Chem. Lett., 2013, 23, 62–65 CrossRef CAS PubMed.
- W. Vuong, M. B. Khan, C. Fischer, E. Arutyunova, T. Lamer, J. Shields, H. A. Saffran, R. T. McKay, M. J. van Belkum, M. A. Joyce, H. S. Young, D. L. Tyrrell, J. C. Vederas and M. J. Lemieux, Nat. Commun., 2020, 11, 4282 CrossRef CAS PubMed.
- W. Vuong, C. Fischer, M. B. Khan, M. J. van Belkum, T. Lamer, K. D. Willoughby, J. Lu, E. Arutyunova, M. A. Joyce, H. A. Saffran, J. A. Shields, H. S. Young, J. A. Nieman, D. L. Tyrrell, M. J. Lemieux and J. C. Vederas, Eur. J. Med. Chem., 2021, 222, 113584 CrossRef CAS PubMed.
- J. Wang and H. Liu, Chin. J. Org. Chem., 2012, 32, 1643–1652 CrossRef CAS.
- S. Brogi, R. Ibba, S. Rossi, S. Butini, V. Calderone, S. Gemma and G. Campiani, Molecules, 2022, 27, 2561 CrossRef CAS PubMed.
- R. M. Oballa, J.-F. Truchon, C. I. Bayly, N. Chauret, S. Day, S. Crane and C. Berthelette, Bioorg. Med. Chem. Lett., 2007, 17, 998–1002 CrossRef CAS PubMed.
- D. R. Owen, C. M. N. Allerton, A. S. Anderson, L. Aschenbrenner, M. Avery, S. Berritt, B. Boras, R. D. Cardin, A. Carlo, K. J. Coffman, A. Dantonio, L. Di, H. Eng, R. Ferre, K. S. Gajiwala, S. A. Gisbson, S. E. Greasley, B. L. Hurst, E. P. Kadar, A. S. Kalgutkar, J. C. Lee, J. Lee, W. Liu, S. W. Mason, S. Noell, J. J. Novak, R. S. Obach, K. Ogilvie, N. C. Patel, M. Pettersson, D. K. Rai, M. R. Reese, M. F. Sammons, J. G. Sathish, R. S. P. Singh, C. M. Steppan, A. E. Stewart, J. B. Tuttle, L. Updyke, P. R. Verhoest, L. Wei, Q. Yang and Y. Zhu, Science, 2021, 374, 1586–1593 CrossRef CAS.
- Y. N. Lamb, Drugs, 2022, 82, 585–591 CrossRef CAS PubMed.
- R. L. Hoffman, R. S. Kania, M. A. Brothers, J. F. Davies, R. A. Ferre, K. S. Gajiwala, M. He, R. J. Hogan, K. Kozminski, L. Y. Li, J. W. Lockner, J. Lou, M. T. Marra, L. J. Mitchell, B. W. Murray, J. A. Nieman, S. Noell, S. P. Planken, T. Rowe, K. Ryan, G. J. Smith, J. E. Solowiej, C. M. Steppan and B. Taggart, J. Med. Chem., 2020, 63, 12725–12747 CrossRef CAS PubMed.
- B. Boras, R. M. Jones, B. J. Anson, D. Arenson, L. Aschenbrenner, M. A. Bakowski, N. Beutler, J. Binder, E. Chen, H. Eng, H. Hammond, J. Hammond, R. E. Haupt, R. Hoffman, E. P. Kadar, R. Kania, E. Kimoto, M. G. Kirkpatrick, L. Lanyon, E. K. Lendy, J. R. Lillis, J. Logue, S. A. Luthra, C. Ma, S. W. Mason, M. E. McGrath, S. Noell, R. S. Obach, M. N. O'Brien, R. O'Connor, K. Ogilvie, D. Owen, M. Pettersson, M. R. Reese, T. F. Rogers, R. Rosales, M. I. Rossulek, J. G. Sathish, N. Shirai, C. Steppan, M. Ticehurst, L. W. Updyke, S. Weston, Y. Zhu, K. M. White, A. Garcia-Sastre, J. Wang, A. K. Chatterjee, A. D. Mesecar, M. B. Frieman, A. S. Anderson and C. Allerton, Nat. Commun., 2021, 12, 6055 CrossRef CAS PubMed.
- B. Bai, A. Belovodskiy, M. Hena, A. S. Kandadai, M. A. Joyce, H. A. Saffran, J. A. Shields, M. B. Khan, E. Arutyunova, J. Lu, S. K. Bajwa, D. Hockman, C. Fischer, T. Lamer, W. Vuong, M. J. van Belkum, Z. Gu, F. Lin, Y. Du, J. Xu, M. Rahim, H. S. Young, J. C. Vederas, D. L. Tyrrell, M. J. Lemieux and J. A. Nieman, J. Med. Chem., 2022, 65, 2905–2925 CrossRef PubMed.
- S. Konno, K. Kobayashi, M. Senda, Y. Funai, Y. Seki, I. Tamai, L. Schäkel, K. Sakata, T. Pillaiyar, A. Taguchi, A. Taniguchi, M. Gütschow, C. E. Müller, K. Takeuchi, M. Hirohama, A. Kawaguchi, M. Kojima, T. Senda, Y. Shirasaka, W. Kamitani and Y. Hayashi, J. Med. Chem., 2022, 65, 2926–2939 CrossRef CAS PubMed.
- L. Zhang, D. Lin, Y. Kusov, Y. Nian, Q. Ma, J. Wang, A. von Brunn, P. Leyssen, K. Lanko, J. Neyts, A. de Wilde, E. J. Snijder, H. Liu and R. Hilgenfeld, J. Med. Chem., 2020, 63, 4562–4578 CrossRef CAS PubMed.
- L. Zhang, D. Lin, X. Sun, U. Curth, C. Drosten, L. Sauerhering, S. Becker, K. Rox and R. Hilgenfeld, Science, 2020, 368, 409–412 CrossRef CAS PubMed.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang, C. Peng, Y. Duan, J. Yu, L. Wang, K. Yang, F. Liu, R. Jiang, X. Yang, T. You, X. Liu, X. Yang, F. Bai, H. Liu, X. Liu, L. W. Guddat, W. Xu, G. Xiao, C. Qin, Z. Shi, H. Jiang, Z. Rao and H. Yang, Nature, 2020, 582, 289–293 CrossRef CAS PubMed.
- M. Gehringer, Future Med. Chem., 2020, 12, 1363–1368 CrossRef CAS PubMed.
- A. Aljoundi, I. Bjij, A. El Rashedy and M. E. S. Soliman, Protein J., 2020, 39, 97–105 CrossRef CAS PubMed.
- N. Drayman, J. K. DeMarco, K. A. Jones, S.-A. Azizi, H. M. Froggatt, K. Tan, N. I. Maltseva, S. Chen, V. Nicolaescu, S. Dvorkin, K. Furlong, R. S. Kathayat, M. R. Firpo, V. Mastrodomenico, E. A. Bruce, M. M. Schmidt, R. Jedrzejczak, M. Á. Muñoz-Alía, B. Schuster, V. Nair, K. Han, A. O'Brien, A. Tomatsidou, B. Meyer, M. Vignuzzi, D. Missiakas, J. W. Botten, C. B. Brooke, H. Lee, S. C. Baker, B. C. Mounce, N. S. Heaton, W. E. Severson, K. E. Palmer, B. C. Dickinson, A. Joachimiak, G. Randall and S. Tay, Science, 2021, 373, 931–936 CrossRef CAS PubMed.
- P. Liu, H. Liu, Q. Sun, H. Liang, C. Li, X. Deng, Y. Liu and L. Lai, Eur. J. Med. Chem., 2020, 206, 112702 CrossRef CAS PubMed.
- J. Cui and J. Jia, Eur. J. Med. Chem., 2021, 225, 113789 CrossRef CAS PubMed.
- N. Kitamura, M. D. Sacco, C. Ma, Y. Hu, J. A. Townsend, X. Meng, F. Zhang, X. Zhang, M. Ba, T. Szeto, A. Kukuljac, M. T. Marty, D. Schultz, S. Cherry, Y. Xiang, Y. Chen and J. Wang, J. Med. Chem., 2022, 65, 2848–2865 CrossRef CAS PubMed.
- S. H. Han, C. M. Goins, T. Arya, W.-J. Shin, J. Maw, A. Hooper, D. P. Sonawane, M. R. Porter, B. E. Bannister, R. D. Crouch, A. A. Lindsey, G. Lakatos, S. R. Martinez, J. Alvarado, W. S. Akers, N. S. Wang, J. U. Jung, J. D. Macdonald and S. R. Stauffer, J. Med. Chem., 2022, 65, 2880–2904 CrossRef CAS PubMed.
- A. Clyde, S. Galanie, D. W. Kneller, H. Ma, Y. Babuji, B. Blaiszik, A. Brace, T. Brettin, K. Chard, R. Chard, L. Coates, I. Foster, D. Hauner, V. Kertesz, N. Kumar, H. Lee, Z. Li, A. Merzky, J. G. Schmidt, L. Tan, M. Titov, A. Trifan, M. Turilli, H. Van Dam, S. C. Chennubhotla, S. Jha, A. Kovalevsky, A. Ramanathan, M. S. Head and R. Stevens, J. Chem. Inf. Model., 2022, 62, 116–128 CrossRef CAS PubMed.
- D. W. Kneller, H. Li, S. Galanie, G. Phillips, A. Labbé, K. L. Weiss, Q. Zhang, M. A. Arnould, A. Clyde, H. Ma, A. Ramanathan, C. B. Jonsson, M. S. Head, L. Coates, J. M. Louis, P. V. Bonnesen and A. Kovalevsky, J. Med. Chem., 2021, 64, 17366–17383 CrossRef CAS PubMed.
- Y. Unoh, S. Uehara, K. Nakahara, H. Nobori, Y. Yamatsu, S. Yamamoto, Y. Maruyama, Y. Taoda, K. Kasamatsu, T. Suto, K. Kouki, A. Nakahashi, S. Kawashima, T. Sanaki, S. Toba, K. Uemura, T. Mizutare, S. Ando, M. Sasaki, Y. Orba, H. Sawa, A. Sato, T. Sato, T. Kato and Y. Tachibana, J. Med. Chem., 2022, 65, 6499–6512 CrossRef CAS PubMed.
- N. Hou, L. Shuai, L. Zhang, X. Xie, K. Tang, Y. Zhu, Y. Yu, W. Zhang, Q. Tan, G. Zhong, Z. Wen, C. Wang, X. He, H. Huo, H. Gao, Y. Xu, J. Xue, C. Peng, J. Zou, C. Schindewolf, V. Menachery, W. Su, Y. Yuan, Z. Shen, R. Zhang, S. Yuan, H. Yu, P.-Y. Shi, Z. Bu, J. Huang and Q. Hu, bioRxiv, 2022, preprint, DOI:10.1101/2022.08.10.503531.
- S.-I. Hattori, N. Higashi-Kuwata, H. Hayashi, S. R. Allu, J. Raghavaiah, H. Bulut, D. Das, B. J. Anson, E. K. Lendy, Y. Takamatsu, N. Takamune, N. Kishimoto, K. Murayama, K. Hasegawa, M. Li, D. A. Davis, E. N. Kodama, R. Yarchoan, A. Wlodawer, S. Misumi, A. D. Mesecar, A. K. Ghosh and H. Mitsuya, Nat. Commun., 2021, 12, 668 CrossRef CAS PubMed.
- A. K. Ghosh, J. Raghavaiah, D. Shahabi, M. Yadav, B. J. Anson, E. K. Lendy, S.-I. Hattori, N. Higashi-Kuwata, H. Mitsuya and A. D. Mesecar, J. Med. Chem., 2021, 64, 14702–14714 CrossRef CAS PubMed.
- C. Ma, Z. Xia, M. D. Sacco, Y. Hu, J. A. Townsend, X. Meng, J. Choza, H. Tan, J. Jang, M. V. Gongora, X. Zhang, F. Zhang, Y. Xiang, M. T. Marty, Y. Chen and J. Wang, J. Am. Chem. Soc., 2021, 143, 20697–20709 CrossRef CAS PubMed.
- B.-X. Quan, H. Shuai, A.-J. Xia, Y. Hou, R. Zeng, X.-L. Liu, G.-F. Lin, J.-X. Qiao, W.-P. Li, F.-L. Wang, K. Wang, R.-J. Zhou, T. T.-T. Yuen, M.-X. Chen, C. Yoon, M. Wu, S.-Y. Zhang, C. Huang, Y.-F. Wang, W. Yang, C. Tian, W.-M. Li, Y.-Q. Wei, K.-Y. Yuen, J. F.-W. Chan, J. Lei, H. Chu and S. Yang, Nat. Microbiol., 2022, 7, 716–725 CrossRef CAS PubMed.
- T. R. Malla, L. Brewitz, D.-G. Muntean, H. Aslam, C. D. Owen, E. Salah, A. Tumber, P. Lukacik, C. Strain-Damerell, H. Mikolajek, M. A. Walsh and C. J. Schofield, J. Med. Chem., 2022, 65, 7682–7696 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |