
The role of adjuvants in overcoming antibacterial resistance due to enzymatic drug modification
Christy
El-Khoury†
a,
Elissar
Mansour†
 *a,
Yori
Yuliandra
a,
Felcia
Lai
a,
Bryson A.
Hawkins
a,
Jonathan J.
Du
b,
Eric J.
Sundberg
b,
Nicolas
Sluis-Cremer
c,
David E.
Hibbs
a and
Paul W.
Groundwater
*a
*a,
Yori
Yuliandra
a,
Felcia
Lai
a,
Bryson A.
Hawkins
a,
Jonathan J.
Du
b,
Eric J.
Sundberg
b,
Nicolas
Sluis-Cremer
c,
David E.
Hibbs
a and
Paul W.
Groundwater
*a
aSydney Pharmacy School, Faculty of Medicine and Health, The University of Sydney, Sydney, NSW 2006, Australia. E-mail: paul.groundwater@sydney.edu.au
bDepartment of Biochemistry, Emory University School of Medicine, Atlanta, GA 30322, USA
cDivision of Infectious Diseases, Department of Medicine, University of Pittsburgh School of Medicine, Pittsburgh, PA 15213, USA
First published on 22nd September 2022
Abstract
Antibacterial resistance is a prominent issue with monotherapy often leading to treatment failure in serious infections. Many mechanisms can lead to antibacterial resistance including deactivation of antibacterial agents by bacterial enzymes. Enzymatic drug modification confers resistance to β-lactams, aminoglycosides, chloramphenicol, macrolides, isoniazid, rifamycins, fosfomycin and lincosamides. Novel enzyme inhibitor adjuvants have been developed in an attempt to overcome resistance to these agents, only a few of which have so far reached the market. This review discusses the different enzymatic processes that lead to deactivation of antibacterial agents and provides an update on the current and potential enzyme inhibitors that may restore bacterial susceptibility.
Introduction
The discovery of antibiotics has resulted in some of the most important innovations in healthcare. Many antibacterial agents were discovered in the decades following Fleming's discovery of penicillin but this era was also accompanied by the rapid emergence of resistance.1The management of infectious diseases caused by bacteria relies on the continuing efficacy of antibacterials, but the rate of bacterial resistance has triggered global healthcare concerns of the advent of a post antibiotic era due to the decreasing availability of effective agents.2 Antimicrobial stewardship programs,3 which aim to ensure that antibacterial agents are used appropriately in order to retard the rate of development of resistance, are mostly restricted to developed nations and hospital rather than community usage.4 From 2017 to 2020, only eleven new antibacterial agents were approved, illustrating that the new antibacterial pipeline alone is insufficient to deal with the decrease in efficacy of current agents due to the steady emergence of drug-resistant infections.2
Antibacterial agents are gradually losing their effectiveness against bacteria (Table 1) and Mycobacterium tuberculosis, Pseudomonas aeruginosa, Acinetobacter baumannii, methicillin-resistant Staphylococcus aureus (MRSA), Enterococcus faecium, Helicobacter pylori, Streptococcus pneumoniae, Haemophilus influenzae, and Enterobacteriaceae have been defined by the World Health Organization (WHO) as priority pathogens.5 These bacteria are responsible for multiple nosocomial and community-acquired infections that can result in serious morbidities or mortality.6,7 Multi-drug resistant (MDR) bacteria are currently estimated to be the cause of 700![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths globally every year and this is forecast to rise to 10 million by 2050.8
000 deaths globally every year and this is forecast to rise to 10 million by 2050.8
| Pathogenic bacterium | Antibacterial resistance |
|---|---|
| S. aureus | β-Lactams9 |
| Aminoglycosides10 | |
| Fosfomycin11 | |
| S. pneumoniae | β-Lactams9 |
| C. difficile | β-Lactams9 |
| Aminoglycosides12 | |
| E. coli | Aminoglycosides13 |
| Chloramphenicol14 | |
| Macrolides15 | |
| Tetracyclines16 | |
| P. aeruginosa | Aminoglycosides17 |
| Macrolides18 | |
| Rifamycin19 | |
| A. baumannii | Aminoglycosides20 |
| Tetracyclines21 | |
| K. pneumoniae | β-Lactams9 |
| Aminoglycosides22 | |
| Rifamycin19 | |
| N. gonorrhoeae | β-Lactams9 |
| M. tuberculosis | Isoniazid23 |
Although resistance is a natural evolutionary process, its spread has been dramatically exacerbated by the overuse and misuse of antibacterial treatments.2 Resistance can be intrinsic (i.e. naturally present) or acquired through horizontal gene transfer or though mutations in chromosomal genes24 and can result in three main outcomes;
• The decreased accumulation of the drug as a result of decreased uptake or increased efflux,
• The modification of the drug target (i.e. enzymes or proteins), and
• Alterations to the drug structure making it inactive,24 for example deactivation by enzymatic hydrolysis (e.g. β-lactams) or enzymatic structural modification (e.g. aminoglycosides) (Table 2).25
| Resistance mechanism | Resistance gene(s) | Enzyme | Resistance phenotype | Ref. |
|---|---|---|---|---|
| Hydrolytic modification | bla | β-Lactamases | β-Lactam antibiotics in Gram positive and Gram negative | 9 |
| ere | Esterases | Macrolides in Gram positive and Gram negative | 26 | |
| Chemical modification | aac | Acetyltransferases | Aminoglycosides in Gram positive and Gram negative | 27 |
| ant | Nucleotidyltransferases | |||
| aph | Phosphotransferases | |||
| fosA | Glutathionetransferase | Fosfomycin in Gram positive and Gram negative | 28 | |
| fosB | Bacillithioltransferase | |||
| fomA | Phosphotransferases | |||
| fomB | ||||
| cat | Acetyltransferases | Chloramphenicol in Gram positive and Gram negative | 29 | |
| orf2 | Phosphotransferase | |||
| nat | Acetyltransferase | Isoniazid in M. tuberculosis | 23 | |
| rgt | Glycosylation | Rifamycin Gram positive and mycobacteria | 30 | |
| rph | Phosphotransferase | |||
| arr | Ribosyltransferase | |||
| tetX | Oxidoreductases | Tetracyclines in Gram negative | 31 |
The discovery of new antibacterial agents is not the only means of overcoming the problems posed by resistance. The development and use of inhibitors of antibacterial deactivating enzymes, termed adjuncts or adjuvants, has had significant clinical effects through the restoration of antibacterial activity and thus improvements to treatment outcomes. Many enzymatic drug modification processes are still unknown or poorly studied, presenting a significant challenge to the development of novel inhibitors.2
The aims of this review are to investigate the range of enzymatic processes that can lead to resistance through antibacterial modification, describe the current and potential adjuvants that can be used to overcome resistance caused by enzymatic drug degradation, and highlight the areas that would benefit from further research.
This review will discuss current and potential inhibitors of enzymes which degrade the antibacterial classes listed in Table 2.
Antibacterial classes
β-Lactams
The β-lactams, consisting of the penicillins, cephalosporins, carbapenems and monobactams (Fig. 1), are among the most prescribed antibacterial agents, particularly those β-lactams which have a broad spectrum of activity and so are often the mainstays of empirical therapy.32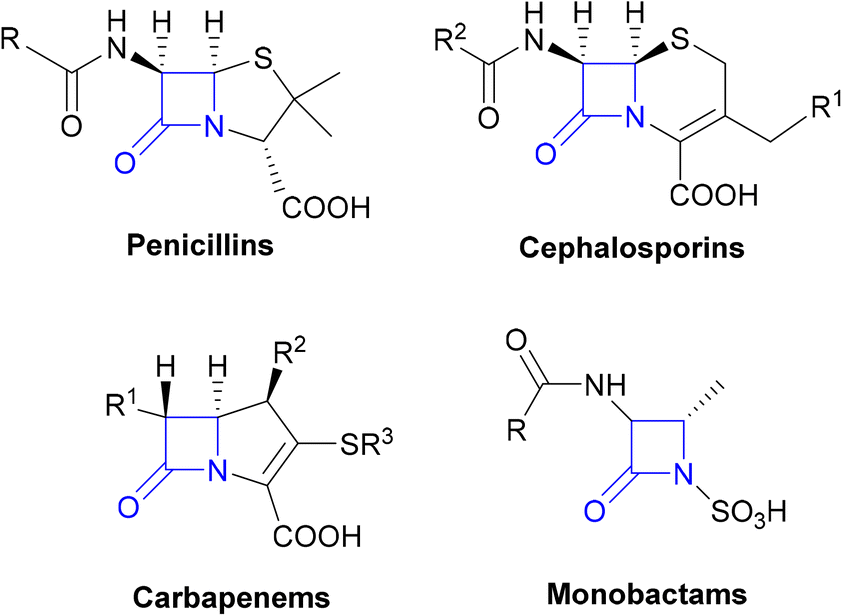 | ||
| Fig. 1 The β-lactam classes of antibiotics. | ||
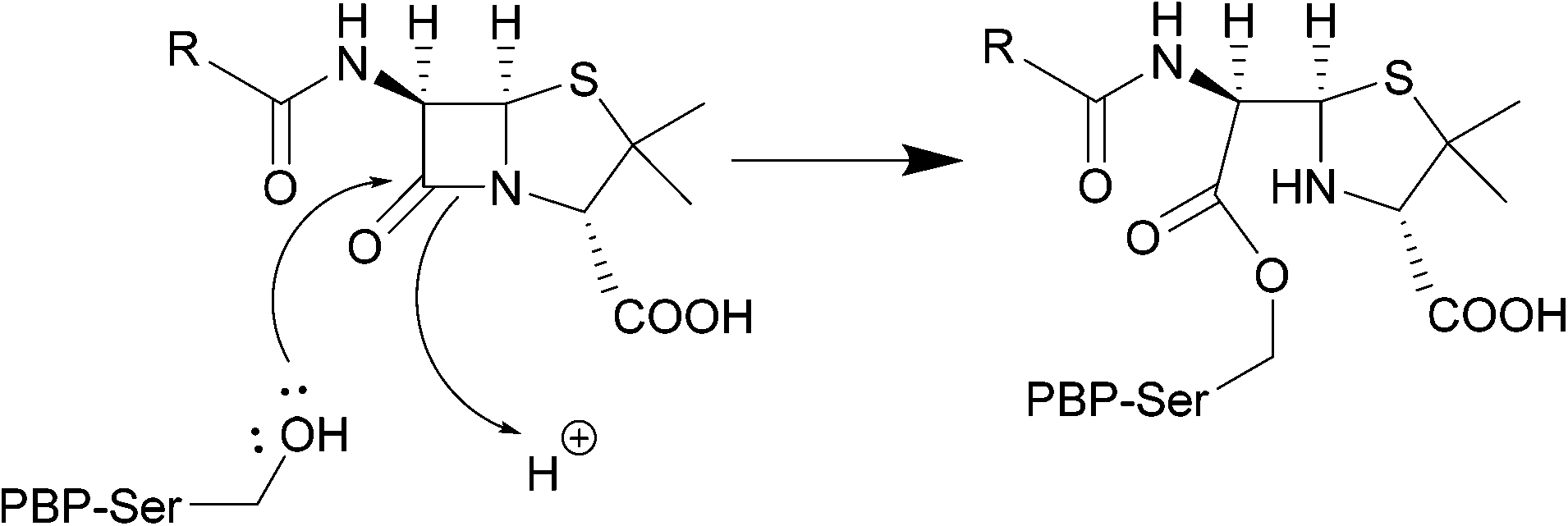 | ||
| Scheme 1 Nucleophilic attack of the hydroxyl group of the PBP active site serine residue on the carbonyl of the β-lactam ring, forming a covalent bond. | ||
| β-Lactamase class | Examples of β-lactamase genes | Host pathogens | Substrates | Ref. |
|---|---|---|---|---|
| Class A | IMI | Enterobacter cloacae | Imipenem | 41 |
| CTX-M | Escherichia coli, K. pneumoniae | Cephalosporins (cefotaxime [CTX] activity greatest) | 42 | |
| Salmonella enterica, P. mirabilis | ||||
| KPC | K. pneumoniae, A. baumannii | Carbapenems, cephalosporins | 43–45 | |
| P. aeruginosa, S. enterica | ||||
| SME | Serratia marcescens | Carbapenems | 46 | |
| BlaZ | S. aureus | Penicillin, cephalosporin, carbapenem | 47 | |
| SHV | K. pneumonia | Penicillins, cephalosporins, carbapenem | 48 | |
| E. coli | ||||
| Class B (metallo-) | IMP | Enteric Gram negative bacteria | Carbapenem, cephalosporins | 49 |
| Pseudomonas spp., Acinetobacter spp. | ||||
| VIM | P. aeruginosa | Penicillins, cephalosporins, carbapenem | 49, 50 | |
| NDM-1 | E. coli, K. pneumoniae, E. cloacae | Carbapenems, cephalosporins, aztreonam, penicillins | 51 | |
| Proteus spp., Citrobacter freundii | ||||
| Class C | AmpC | Enterobacteriaceae | Cephalosporins, penicillin | 52 |
| Class D | OXA | Enterobacter spp., Serratia spp., C. freundii, Aeromonas spp., Proteus spp., Providencia spp., Morganella morganii (ESCAPPMs) | Penicillins, cephalosporins, carbapenems | 53 |
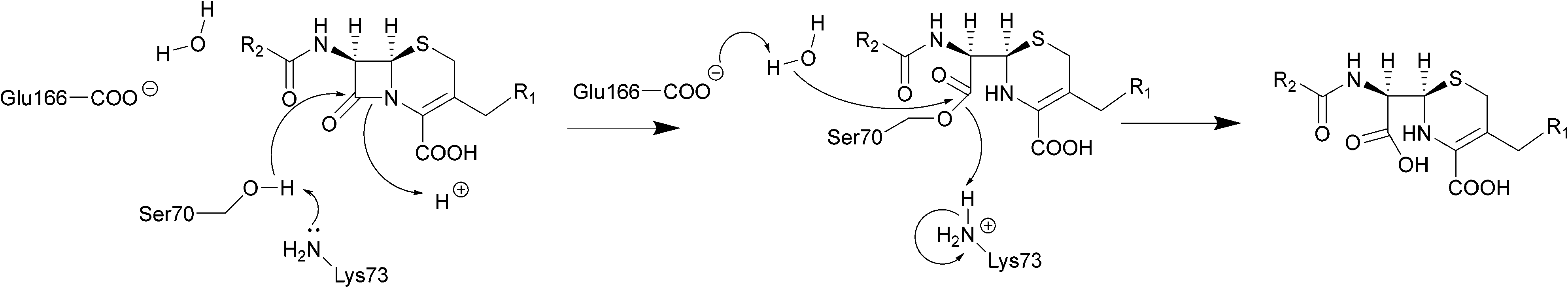 | ||
| Scheme 2 Hydrolysis of cephalosporins by class A β-lactamases. Nucleophilic attack of the hydroxyl group of Ser70 on the amide group of β-lactams results in cleavage of the β-lactam ring. | ||
The serine β-lactamase classes (A, C, D) share a common mechanism of action. The first step of deactivation mimics the first step of PBP inhibition by β-lactams, with the hydroxyl group of the serine residue undergoing nucleophilic attack on the amide bond of the lactam ring, forming a high-energy acyl–enzyme adduct. An activated water molecule then hydrolyses the adduct, breaking open the β-lactam ring and leading to deactivation of the drug. The amino acids which activate the serine residue and the water molecule form the basis of differentiation between the Ambler classes.54–56Scheme 2 illustrates this mechanism using a class A β-lactamase as an example.
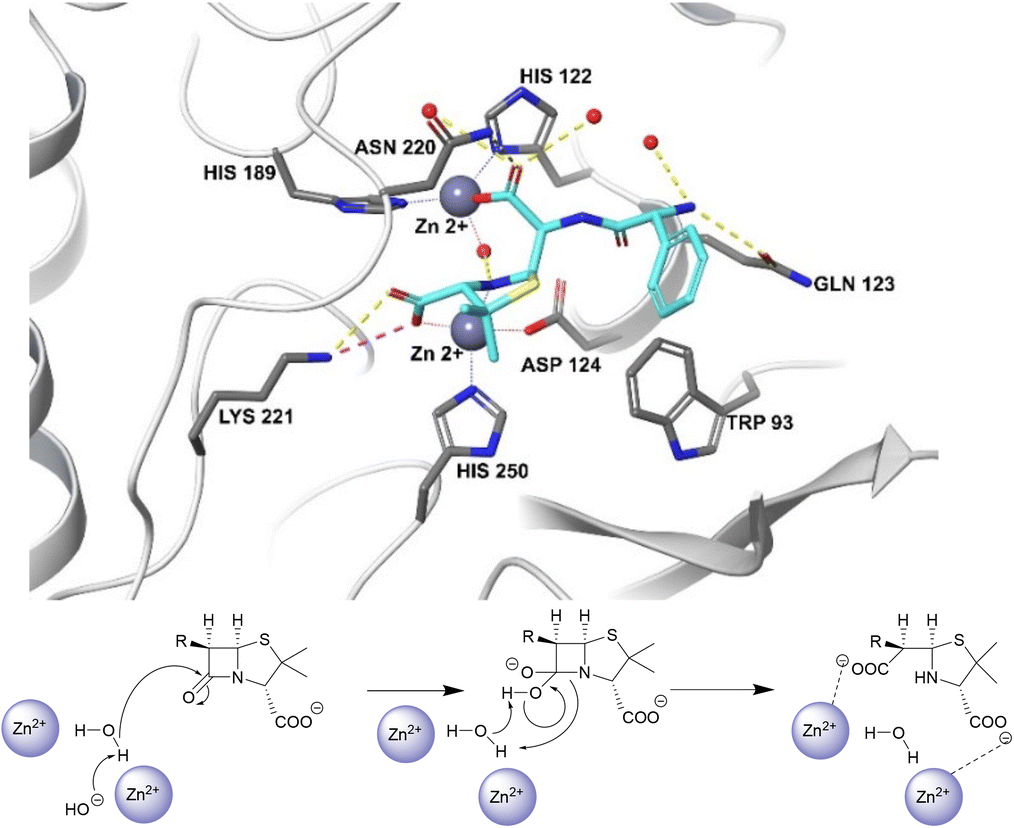
Unlike the serine hydrolases, class B (metallo-)β-lactamases (MBLs) rely on Zn2+ ions in the active site to hydrolyse β-lactams and no amino acid residues are directly involved in the hydrolysis (Fig. 2).57
| Name of inhibitor | β-Lactam | β-Lactamase class inhibited | Ref. |
|---|---|---|---|
| Approved inhibitors | |||
| Clavulanic acid | Amoxicillin or ticarcillin | Class A, some class D | 61, 62 |
| Tazobactam | Piperacillin | Class A, some class D | 63 |
| Sulbactam | Ampicillin or cefoperazone | Class A | 64, 65 |
| Avibactam | Ceftazidime | Class A, C, D | 66 |
| Relebactam | Imipenem/cilastatin | Class A, C | 67, 68 |
| Vaborbactam | Meropenem | Class A, C | 67 |
| Developmental inhibitors in clinical trials | |||
| Nacubactam | Meropenem – phase I trial | Class A, C | 69, 70 |
| Zidebactam | Cefepime – phase III trial | Class A, C | 71 |
| ETX2514 | Sulbactam – phase II trial | Class A, C and some D | 72 |
| ETX0282 | Cefpodoxime – phase I trial | Class A, C | 73 |
| LN-1-255 | Ceftazidime – phase II trial | Class A, D (non-carbapenemases) | 74 |
| Enmetazobactam | Cefepime – phase III trial | Class A | 75, 76 |
| Taniborbactam | Cefepime – phase II trial | Class A, B, C, D | 77 |
| VNRX-7145 | Cefibutin – phase I trial | Class A, C, D | 78 |
| LK-157 | Cefuroxime – phase I trial | Class A, C | 78 |
| BLI-489 | Piperacillin – phase II trial | Class A, C, D | 79 |
| D-Captopril | Meropenem – phase I trial | Class B | 80 |
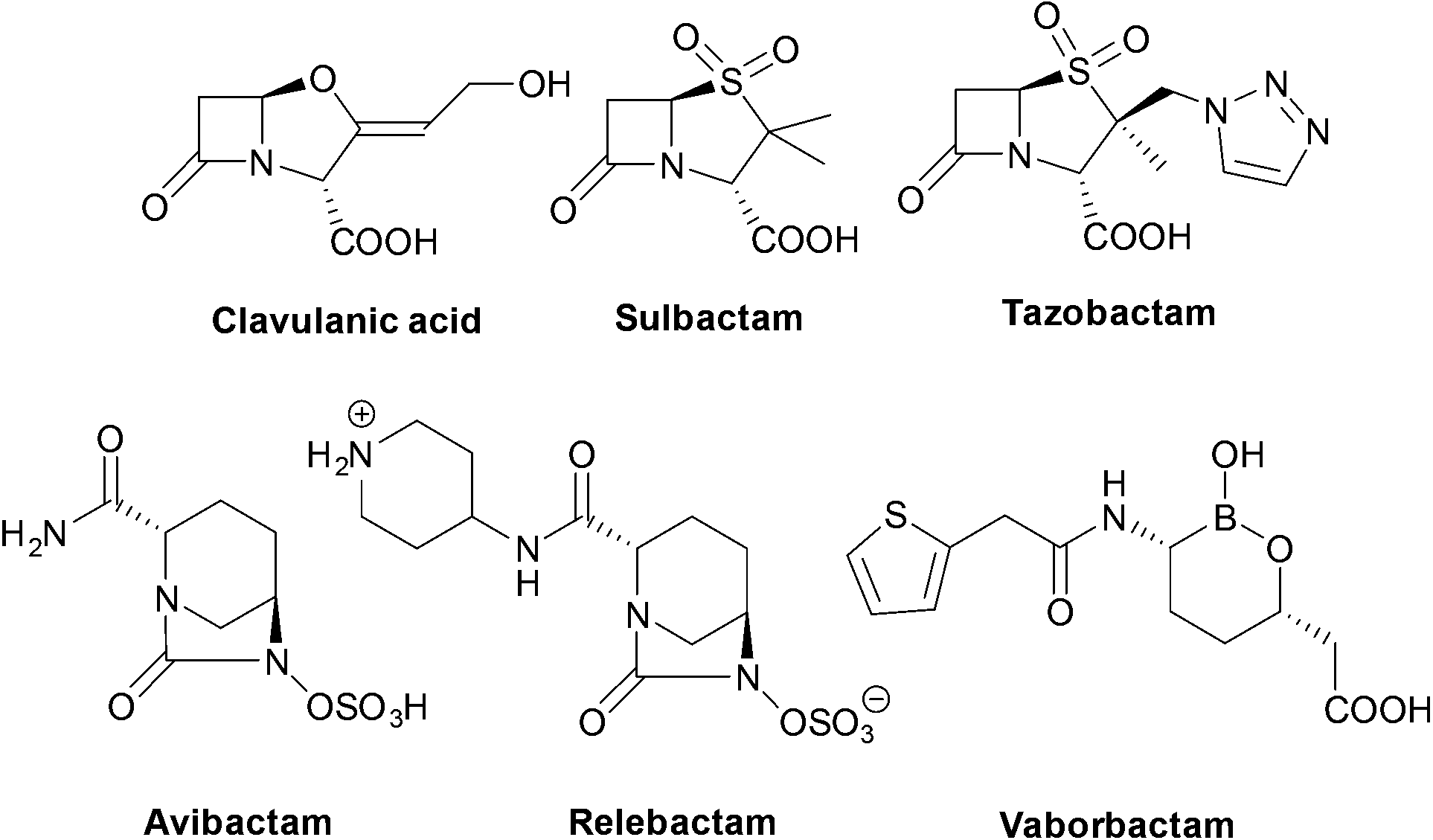 | ||
| Fig. 3 Structures of the six currently approved β-lactamase inhibitors. | ||
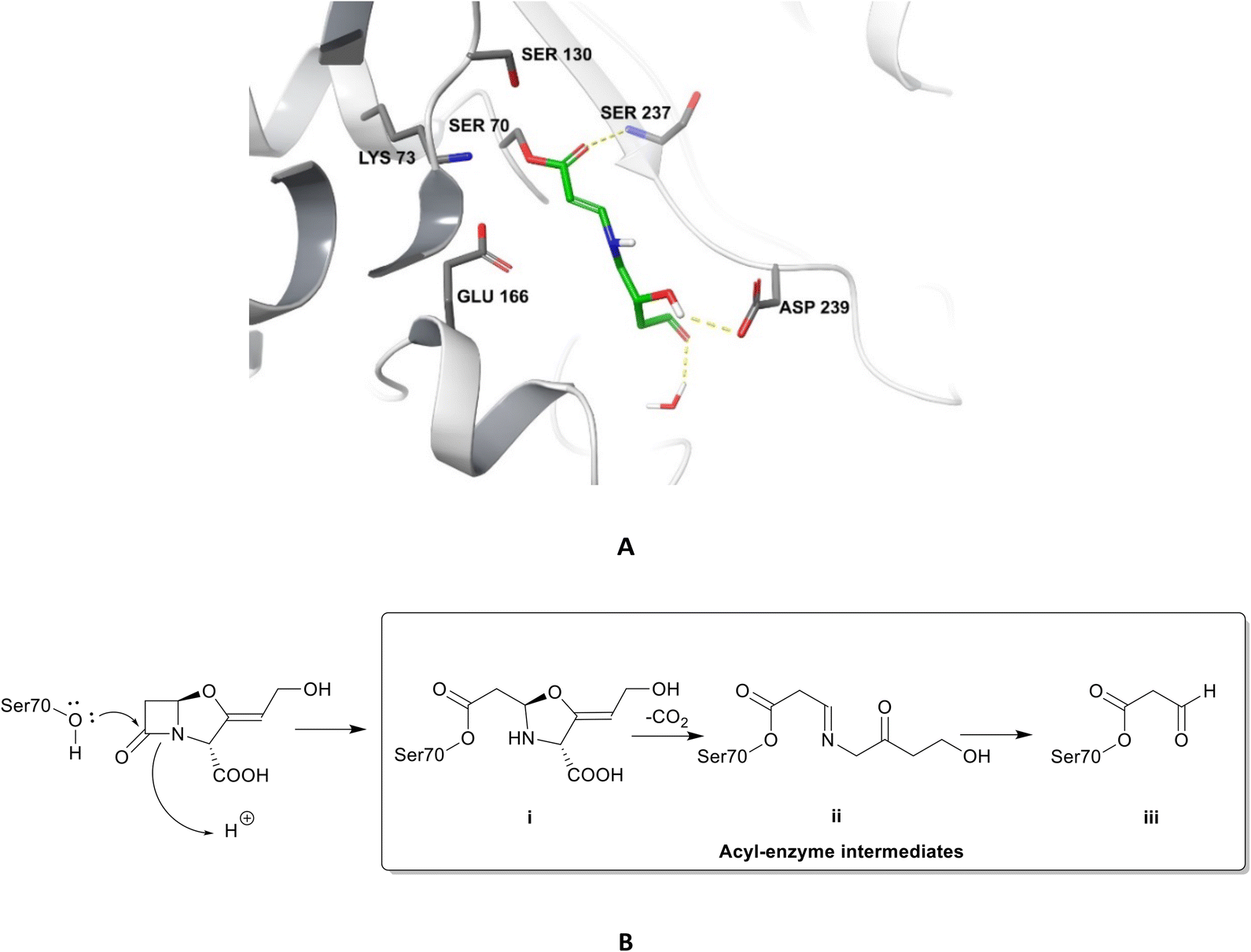 | ||
| Fig. 4 Irreversible binding of clavulanic acid to serine β-lactamase (PDB ID: 6NVU).81 A The clavulanic acid has taken the place of a β-lactam antibiotic in the active site and the hydrogen bond between the carbonyl of the ring opened acyl–enzyme intermediate ii and serine237 is shown. B Schematic representation of the β-lactamase inhibition, involving nucleophilic attack by the hydroxyl group of serine70 on the carbonyl oxygen of the β-lactam ring, decarboxylation of the initially formed acyl–enzyme intermediate i, and hydrolysis of the imine ii, to form a high-energy acyl–enzyme adduct iii. | ||
Clavulanic acid and the other β-lactam containing β-lactamase inhibitors bind to the active site by forming a covalent bond with the hydroxyl group of the serine residue, the same residue that is involved in binding to the β-lactams. The leaving groups of the inhibitors allow the five-membered ring to open readily and form an imine intermediate that further rearranges itself to an enamine intermediate, eventually forming a stable acylated enzyme intermediate. The acyl group is very slowly hydrolysed compared to the acyl group formed by the attack of serine β-lactamases on β-lactams, making this bond quasi-irreversible.7 Avibactam and diazabicyclooctanes (DBOs) also form a covalent acyl–enzyme bond with the key serine residue.2,59 Avibactam is a non-β-lactam β-lactamase inhibitor which is used clinically in combination with ceftazidime and ceftaroline and the ring-opening acylation step is reversible. When bound to KPC-2, avibactam forms hydrogen bonds via its carbonyl oxygen with the nitrogen atoms of Ser70 and Cys237 and the amide of Ala132. The avibactam sulfate moiety forms hydrogen bonds with Asp130, Cys237, Gly235 (Fig. 5).82 Interestingly, the DBO nacubactam (Fig. 5) has been reported to have activity against class A, and B (weak) β-lactamases, as a result of its inhibition of β-lactamases and PBP-2, and a synergistic enhancer effect which is independent of its β-lactamase activity.83
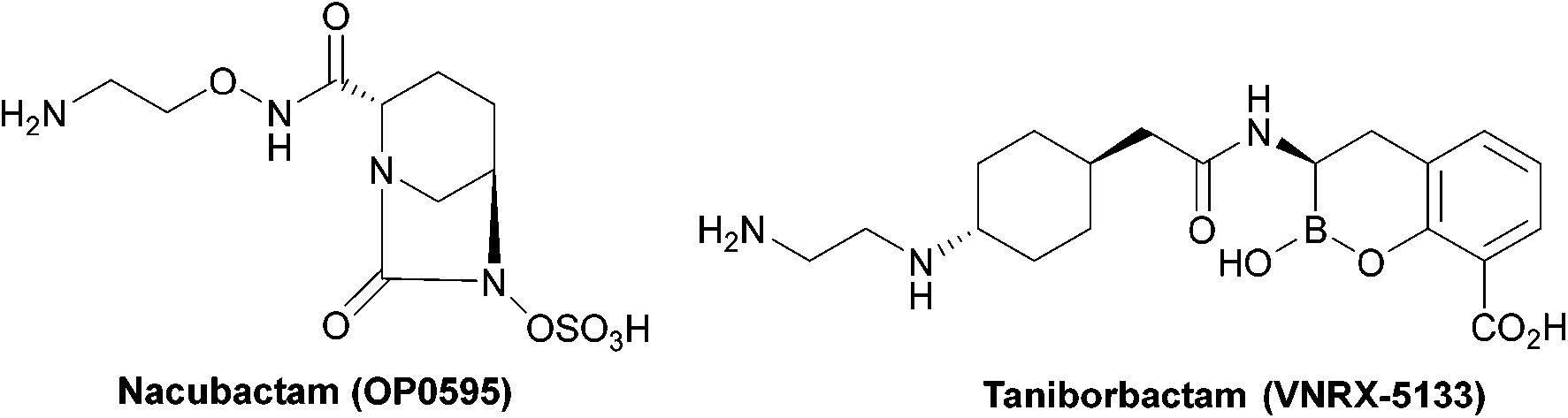 | ||
| Fig. 5 Taniborbactam, the first serine and metallo-β-lactamase inhibitor to enter clinical development, and nacubactam. | ||
The spread of MBLs is increasing in pathogenic species such as Enterobacteriaceae, Pseudomonas and Acinetobacter species; while class B β-lactamases inhibitors are in trials or are the focus of ongoing research (Table 4), there are currently no approved MBL inhibitors, which is a growing concern due to the threat of class B carbapenemases.84
Taniborbactam (Fig. 5) is the first β-lactamase inhibitor active against both serine and metallo-β-lactamases. Taniborbactam is a cyclic boronate able to inhibit serine-β-lactamases as a reversible covalent inhibitor and metallo-β-lactamases as a competitive inhibitor. When used in combination susceptibility to cefepime was restored in all 34 E. coli strains expressing A, B, C, and D β-lactamases, revealing a potent broad-spectrum β-lactamase inhibitor, with early phase II trials demonstrating low toxicity.85
Aminoglycosides
Aminoglycosides are frequently employed in the empirical treatment of infections requiring hospital admission due to their bactericidal activity against Gram positive and negative organisms, as well as mycobacteria; the first, streptomycin, was isolated in 1943.86 Unfavourable side effects prompted the search for less toxic aminoglycosides with the newest agent, plazomicin, gaining FDA approval in 2018 for the treatment of complicated urinary tract infections in adults caused by E. coli, K. pneumoniae, P. mirabilis or E. cloacae.87,88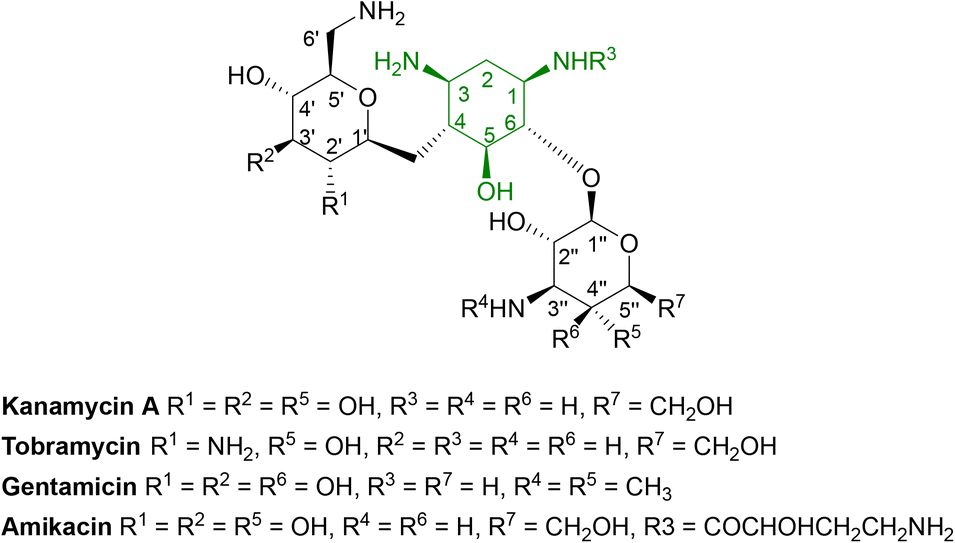 | ||
| Fig. 6 General structure of some clinically used aminoglycosides consisting of free hydroxyl groups, two or more amino groups and an aminocyclitol ring linked to one or more aminosugars.33,88 As the hydroxyl and amino groups are involved in binding to rRNA, they are targeted by aminoglycoside modifying enzymes (AMEs). | ||
| Enzyme | Gene coding enzyme | Pathogenic bacterial expression | Aminoglycoside substrate | Ref. |
|---|---|---|---|---|
| AAC(1)-I | aac(1) | Campylobacter fetus, E. coli | Paromomycin | 97 |
| Neomycin | ||||
| AAC(3)-I | aac(3)-Ib | E. coli, Enterobacter cloacae, K. pneumoniae, P. aeruginosa | Gentamicin | 98 |
| AAC(3)-II | aac(3)-II | A. baumannii, E. coli | Gentamicin | 99 |
| Tobramycin | ||||
| Kanamycin | ||||
| AAC(3)-III | aac(3)-III | P. aeruginosa | Tobramycin | 100 |
| Gentamicin | ||||
| Kanamycin | ||||
| Neomycin | ||||
| AAC(3)-IV | aac(3)-IV | E. coli, Campylobacter jejuni | Netilmicin | 101 |
| Dibekacin | ||||
| Apramycin | ||||
| AAC(3)-VI | aac(3)-VI | E. cloacae, Salmonella enterica | Apramycin | 102 |
| AAC(2′)-I | aac(2′)-I | A. baumannii | Tobramycin | 103 |
| Gentamicin | ||||
| Neomycin | ||||
| AAC(2′)-II | aac(2′)-II | Providencia stuartii | Gentamicin | 104 |
| Tobramycin | ||||
| Netilmicin | ||||
| AAC(6′)-I | aac(6′)-I | P. aeruginosa | Tobramycin | 105 |
| Amikacin | ||||
| Kanamycin | ||||
| AAC(6′)-II | aac(6′)-II | K. pneumoniae | Tobramycin | 106 |
| Gentamicin | ||||
| Kanamycin |
The active site of AACs is aspartate rich, forming a negatively charged area which can accommodate the positively charged aminoglycosides.27 The thioester group of acetyl CoA is subjected to nucleophilic attack by the charged amino group of the drug leading to an acetylated aminoglycoside.92 Each antibiotic binds to the enzyme differently however, the high-resolution X-ray structure of AAC found in M. abscessus shows that some residues are conserved in all subtypes.93
AAC(6′)-Ib is a plasmid mediated enzyme and is a cause of amikacin and kanamycin resistance, but has no reported effect on gentamicin (Fig. 7).94 Although the aac(6′)-Ib-cr gene is not associated with high levels of resistance, when combined with three or four chromosomal mutations it can cause ciprofloxacin and norfloxacin resistance, both in vitro and in vivo.95 Significant concerns arise from the fact that acc(6′)-Ib is carried by all the ESKAPE pathogens (E. faecium, S. aureus, K. pneumoniae, A. baumannii, P. aeruginosa, and Enterobacter spp.) which are the cause of most hospital infections and have become increasingly resistant to aminoglycosides.96
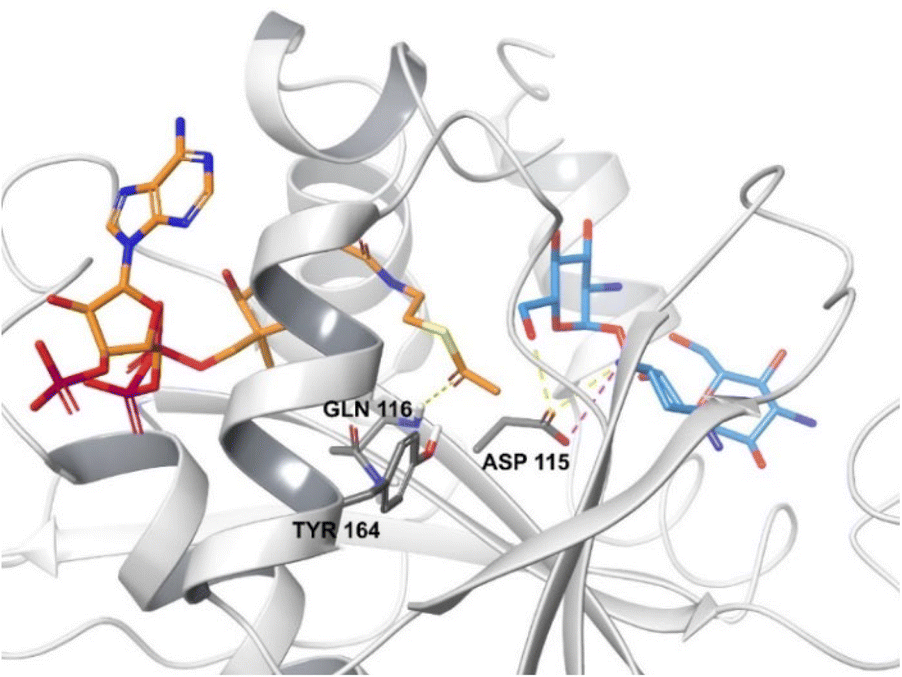
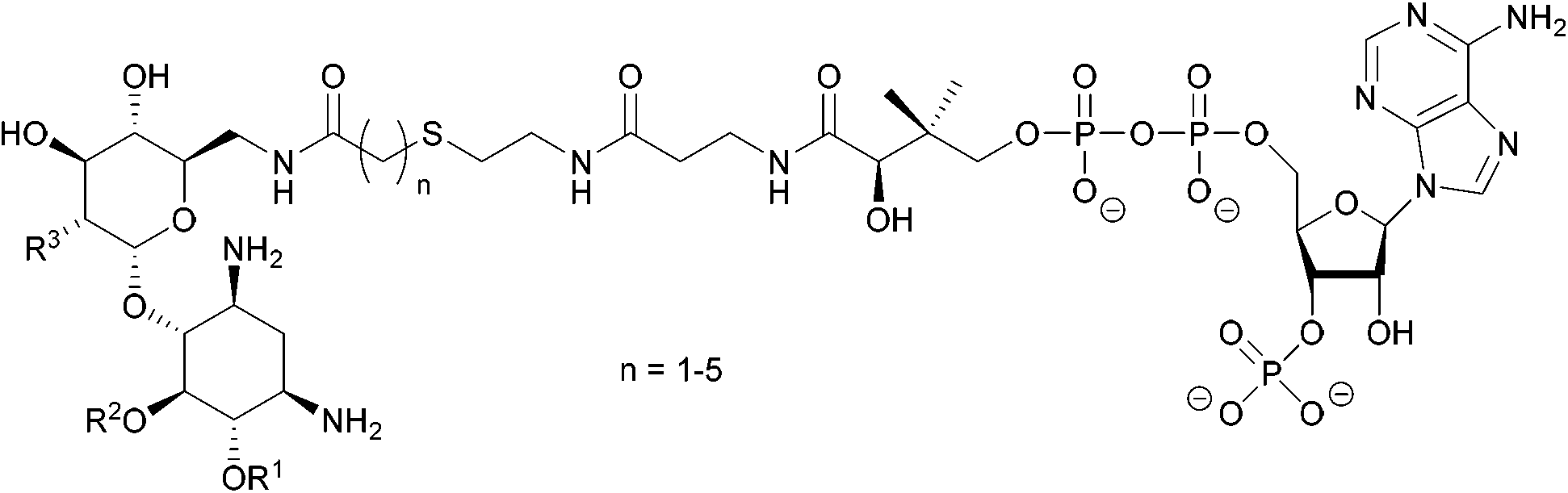 | ||
| Fig. 8 Structure of the a bisubstrate analogue which mimics the tetrahedral transition state of AAC-catalysed acetylation.108 | ||
| Enzyme | Bacterial expression | Aminoglycoside substrates |
|---|---|---|
| ANT(2′′) | P. aeruginosa, E. coli, most ESCAPPM | Tobramycin, gentamicin, kanamycin |
| ANT(3′′)-I | A. baumannii, Pseudomonas spp., Vibrio spp., E. coli, Yersinia enterocolitica | Streptomycin, spectinomycin |
| ANT(4′)-Ia | S. epidermidis, S. aureus, Enterococcus spp., Bacillus spp. | Tobramycin, amikacin, kanamycin |
| ANT(4′)-IIa | P. aeruginosa, Enterobacteriaceae | Tobramycin, amikacin, kanamycin |
| ANT(6′)-I | C. fetus, E. faecium, S. aureus, E. faecalis, Streptococcus spp., Bacillus spp., C. jejuni | Streptomycin |
| ANT(9)-I | S. aureus, Enterococcus spp., Staphylococcus sciuri, E. faecalis | Spectinomycin |
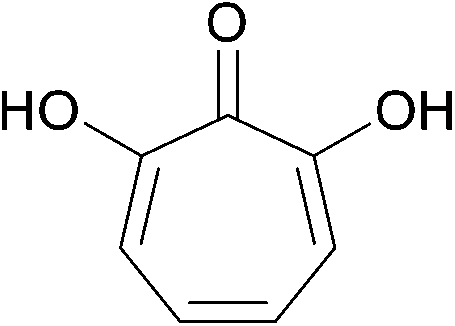 | ||
| Fig. 9 The fermentation product 7-hydroxytropolone. | ||
| Enzyme | Subtype | Pathogenic host | Aminoglycoside substrates |
|---|---|---|---|
| APH(3′) | I | E. coli, S. enterica, K. pneumoniae, A. baumannii, Citrobacter spp. | Kanamycin, neomycin, paromomycin |
| II | E. coli, P. aeruginosa, S. maltophilia | Kanamycin, neomycin, paromomycin | |
| III | S. aureus, Enterococcus spp. | Kanamycin, neomycin, paromomycin, amikacin | |
| IV | B. circulans | Kanamycin, neomycin, paromomycin | |
| V | Streptomyces fradiae, Streptomyces ribosidificus | Neomycin, paromomycin | |
| VI | A. baumannii, K. pneumoniae, S. marcescens | Kanamycin, neomycin, paromomycin, amikacin | |
| VII | C. jejuni | Kanamycin, neomycin | |
| APH(2′′) | I–IV | S. aureus, C. difficile, E. faecium, E. coli, Enterococcus gallinarum | Kanamycin, gentamicin, tobramycin |
| APH(3′′) | Ia, Ib | S. griseus, Enterobacteriaceae, Pseudomonas spp., M. fortuitum | Streptomycin |
| APH(6) | Ia, Ib, Ic, Id | P. aeruginosa, E. coli, Salmonella spp., V. cholerae | Streptomycin |
| APH(9) | Ia, Ib | L. pneumophila | Spectinomycin |
Chloramphenicol
The specific genes coding for class A CATs (catA) have been extensively documented by Schwarz et al.29 The diversity in cat genes prevents the definition of a unified enzymatic model as only five of the seventeen residues are conserved between the different CAT active sites.122 Deactivation of chloramphenicol by the CATI subtype is shown in Scheme 3. CAT genes can be found on chromosomes, plasmids, integrons and transposons.126–128 There are currently no inhibitors available on the market but there have been several studies that have identified potential ligands.129–132
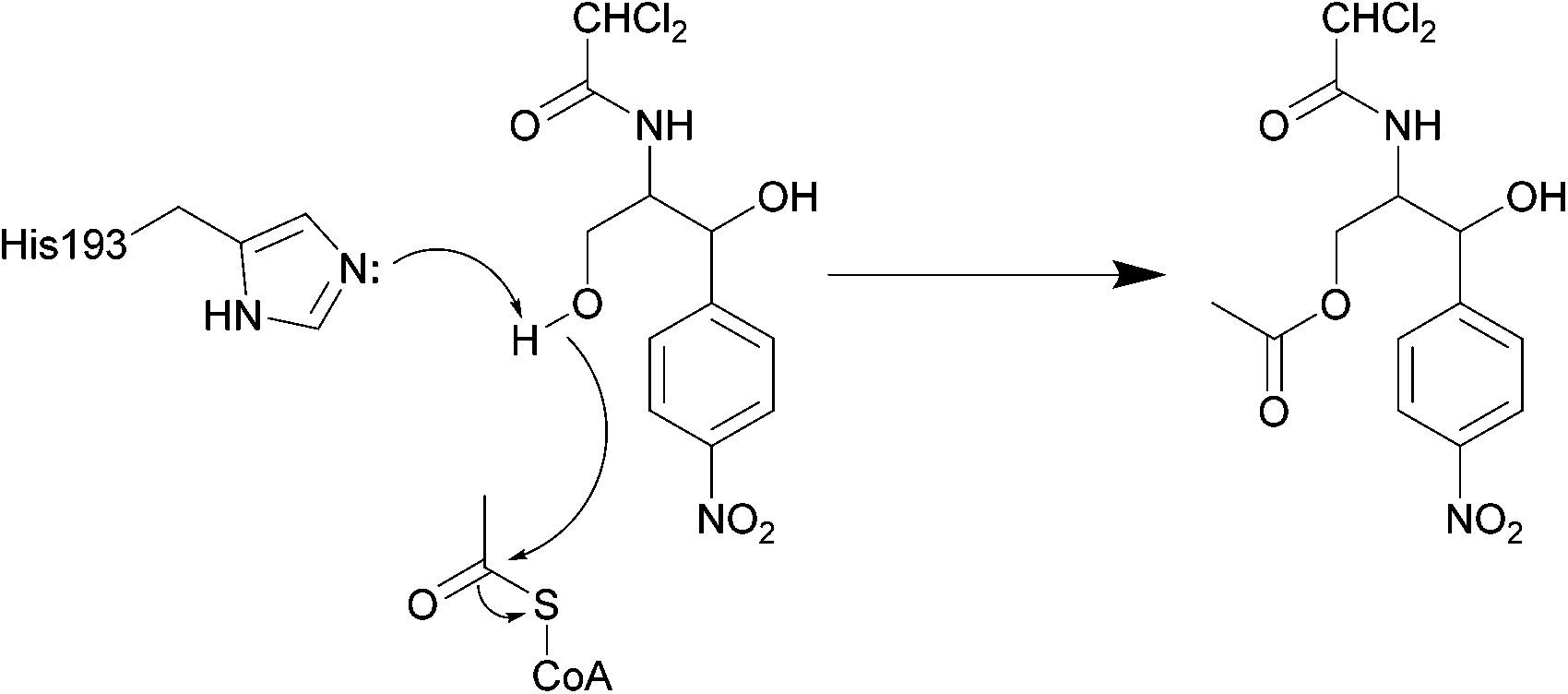 | ||
| Scheme 3 Deactivation of chloramphenicol by CATI.133 Once bound, acetyl CoA reaches the enzyme active site through a tunnel.134 One of the most important residues for catalysis is His193, with the lone pair electrons of the imidazole deprotonating the primary alcohol of chloramphenicol, promoting the attack on the carbonyl of the thioester of acetyl CoA, yielding an acetylated chloramphenicol.134 | ||
 | ||
| Scheme 4 Other chloramphenicol enzymatic modifications. | ||
The active site of CPT is lined with a hydrophobic and electron-dense region. The aromatic ring of chloramphenicol takes part in van der Waals interactions, the carbonyl group undergoes hydrogen bonding and the amide nitrogen undergoes hydrogen bonding with a sulphate anion bound to CPT. Asp37 is thought to deprotonate the primary hydroxyl group which attacks the γ-phosphate yielding a stabilised pentacovalent transition state, facilitating the transfer of a phosphoryl group to 3′-OH of chloramphenicol.123 There are no known inhibitors of CTP, which is mostly found in the chloramphenicol-producing actinomycete S. venezuelae.29
Although there have been no studies investigating the potential of nitroreductase inhibitors as adjuvants, this mechanism of resistance has been found in P. aeruginosa, H. influenzae, and N. meningitidis so adjuvants may have potential in the treatment of infections caused by these MDR organisms.125,138
Macrolides
Due to their broad spectrum of activity, macrolides (Fig. 11) can be used in the treatment of a range of serious infections, and they are also an alternative for patients with penicillin allergies.139,140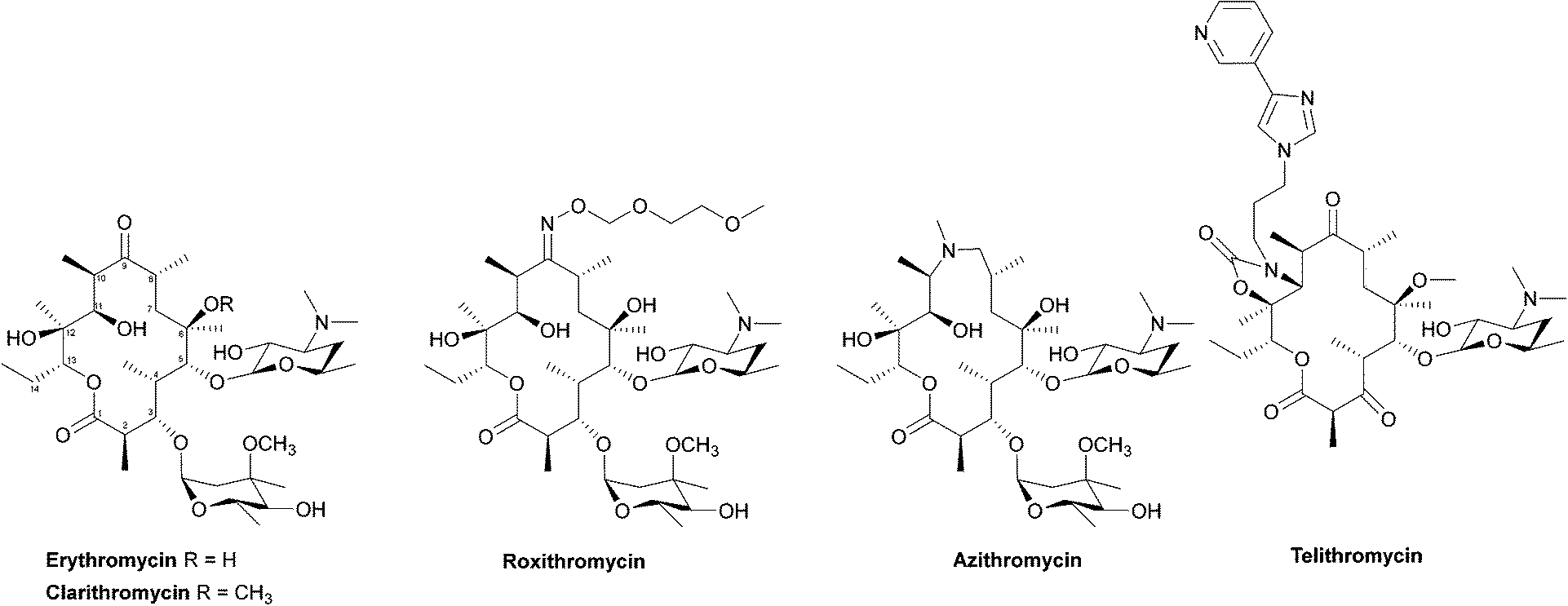 | ||
| Fig. 11 The structures of the macrolide antibiotics. | ||
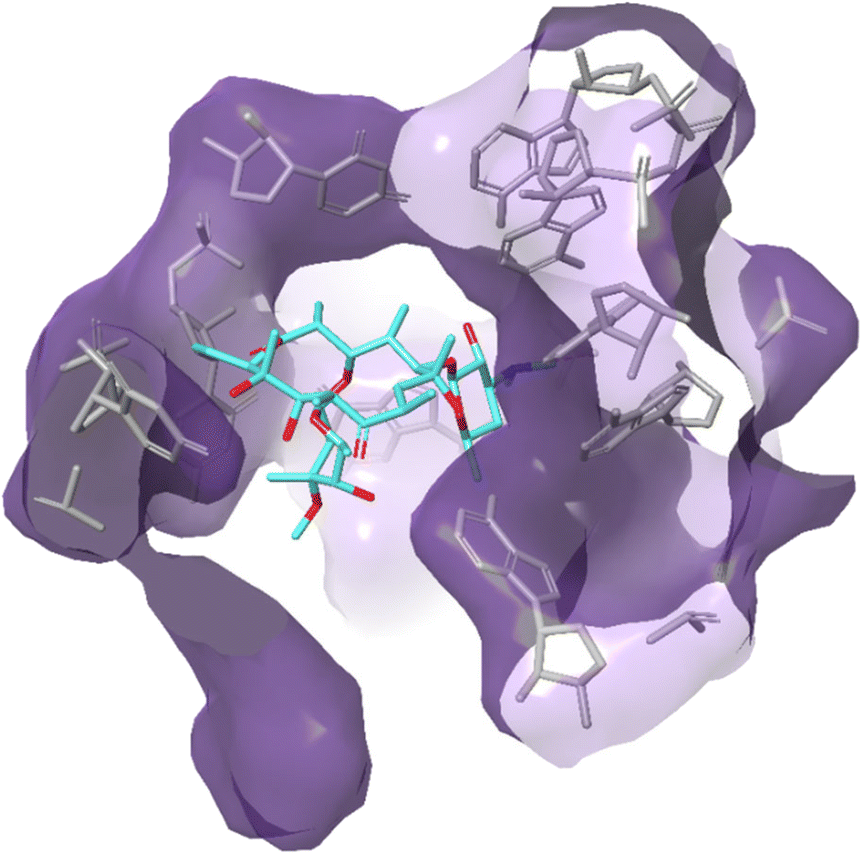 | ||
| Fig. 12 Interaction of erythromycin with the nucleotides in its binding site in the protein exit tunnel (PDB ID 1JZY). In ribosomal RNA nucleotide 2058 is an adenine to which the macrolide antibiotic binds strongly. Hydrogen bonds are formed between the 2′,6,11 and 12-hydroxyl groups of the macrolide and adenine bases 2058 and 2509. An electrostatic interaction exists between the conjugate acid of the tertiary amine of the macrolide and the guanine-2505 phosphate. All these interactions play a key role in macrolides binding to this site, with any modifications in structure disrupting the binding process.144 | ||
| Enzyme | Gene | Host | Substrate | Ref. |
|---|---|---|---|---|
| Esterase (Ere) | ereA | E. coli, K. pneumonia, P. aeruginosa, V. cholera | Erythromycin, roxithromycin, clarithromycin | 155 |
| ereB | E. coli, Salmonella spp. | Erythromycin, roxithromycin, clarithromycin, azithromycin | 148 | |
| ereC | K. pneumoniae | 14-Membered macrolactones (e.g. clarithromycin, erythromycin, oleandomycin, roxithromycin, solithromycin) except telithromycin | 156 | |
| ereD | Riemerella anatipestifer | Erythromycin | 157 | |
| Glycosyltransferase | gimA | Streptomyces ambofaciens | Oleandomycin | 158 |
| ole | Streptomyces antibioticus | Oleandomycin, tylosin | 159 | |
| mgt | S. lividans | Erythromycin, azithromycin | 150, 160 | |
| Phosphotransferase | mphA | E. coli, A. baumannii, ESCAPPM | 14- and 15-membered macrolactones (e.g. erythromycin, roxithromycin, clarithromycin, azithromycin) | 151, 161 |
| mphB | E. coli, K. pneumoniae, S. enterica, E. fergusonii | 14-, 15- and 16-membered macrolactones (e.g. erythromycin, roxithromycin, clarithromycin, azithromycin) | 151, 161 | |
| mphC | S. aureus | Erythromycin, roxithromycin, clarithromycin, azithromycin, oleandomycin | 151, 161, 162 |
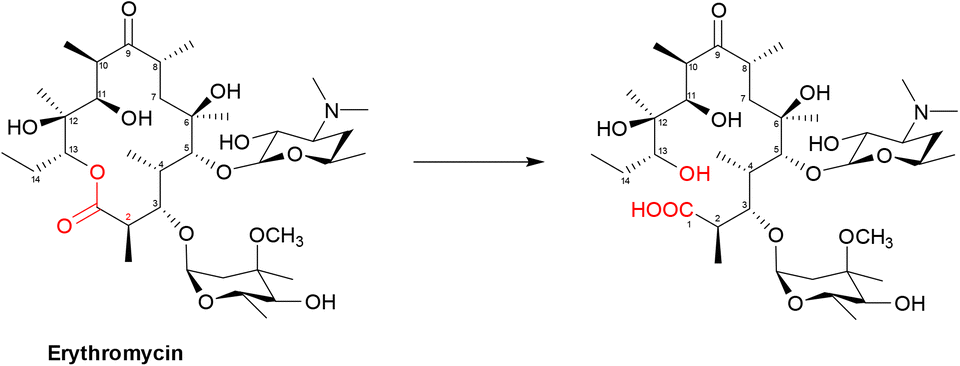 | ||
| Scheme 5 Hydrolysis of macrolides by esterases. Macrolides, such as erythromycin, bind within a buried active site of EreC covered by a glycine and proline rich loop. When the loop is closed, it repositions the ester group of macrolides in close proximity to the catalytic triad, composed of His50, Glu78 and His289. The first step is deprotonation of a water molecule by His50, producing a reactive hydroxide ion which then attacks the carbonyl of the macrolide ester, forming a negatively charged transition-state intermediate which is stabilised by Arg261. The deprotonated hydroxyl group then retrieves the proton previously transferred to His50, releasing the antibiotic in an inactive hydrolysed form.149 | ||
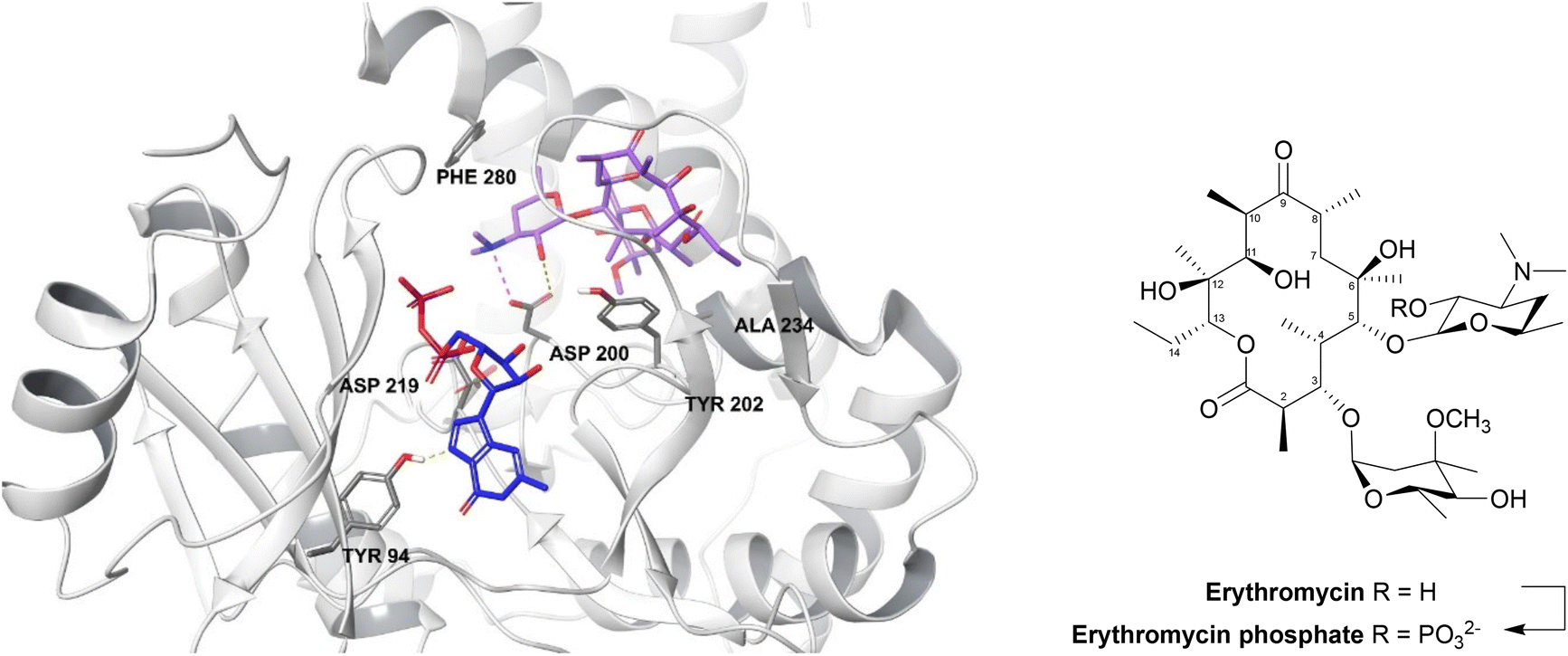 | ||
| Fig. 13 2′-Phosphorylation of erythromycin by an MPH(2′)-I (PDB ID: 5IGP).154 The nucleotide binds within the hydrophobic N-terminus and interacts with Tyr94 and aspartate residues (Asp200, Asp219, Asp234). The macrolide binding site consists of a hydrophobic region, a negatively charged region and an aspartate residue (Asp200), seen in all MPH subtypes, along with Tyr202, Ala234 and Phe280 residues. Asp200 is thought to position the target 2′-OH of macrolides in an orientation which allows proton transfer during catalysis. For MPHs which utilise GTP, a tyrosine residue (Tyr94) acts as a gatekeeper.154 | ||
Macrolide phosphotransferases (MPHs) resemble aminoglycoside phosphotransferases but have a larger interdomain linker which widens the binding pocket. There are over fifteen mph gene subtypes and they can confer resistance to different antibiotics in both Gram positive and negative bacteria (Table 9).152,153 Although these genes are not widely spread, they are found on mobile genetic elements and some are also found to confer resistance to ketolides, an important class of antibiotics derived from macrolides.25,142
Macrolide phosphotransferases have a similar structure to eukaryotic protein kinases which are enzymes that have been studied extensively in drug discovery. One high throughput screening (HTS) study thus investigated the known eukaryotic ATP competitive kinase inhibitors, but none were found to inhibit MPH enzymes.154 There are currently no macrolide adjuvants despite the fact that these enzymes are responsible for conferring resistance in important pathogens (Table 9).147,163,164
Tetracyclines
Tetracyclines are a class of antibiotics that have been shown to bind to the 30S ribosomal subunit near the 16S subunit inhibiting protein synthesis. This occurs through competitive binding to the peptidyl transfer centre A site instead of the charged tRNA.165 Tetracycline binding is reversible, accounting for their bacteriostatic action.166,167Tigecycline has a broad spectrum of activity and is approved for the treatment of serious infections in hospitalised adults, it is used as last resort against serious infections caused by carbapenem-resistant Enterobacteriaceae and Acinetobacter spp.168
Tetracyclines (Fig. 14) were first discovered in 1948, the newest tetracyclines, omadacycline and eravacycline, were approved in 2018 and three other tetracycline derivatives are currently in trials.169
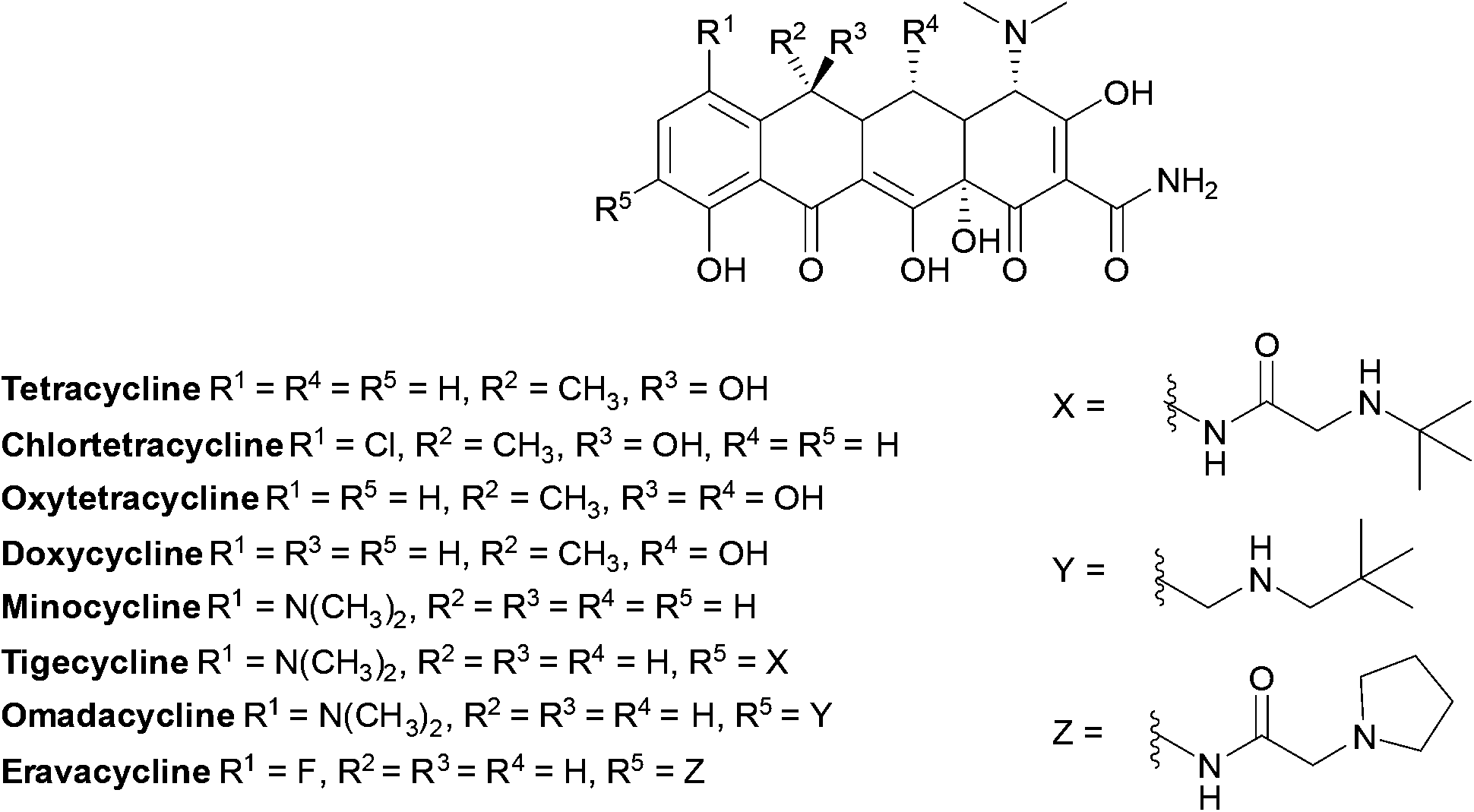 | ||
| Fig. 14 Examples of chemical structures of tetracyclines. Tetracyclines have a common linear fused tetracyclic nucleus; tigecycline is a third-generation tetracycline with an additional side chain on the D ring. | ||
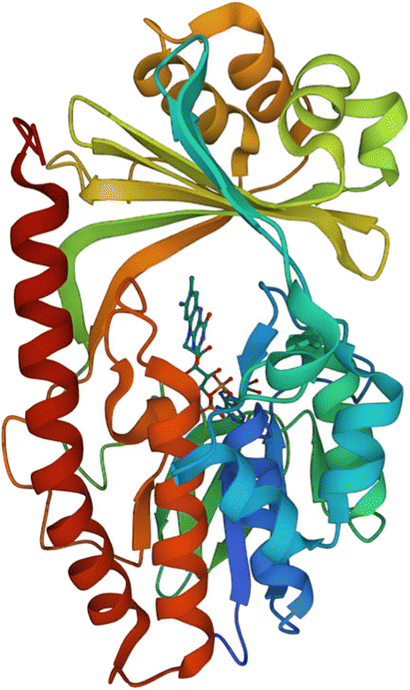 | ||
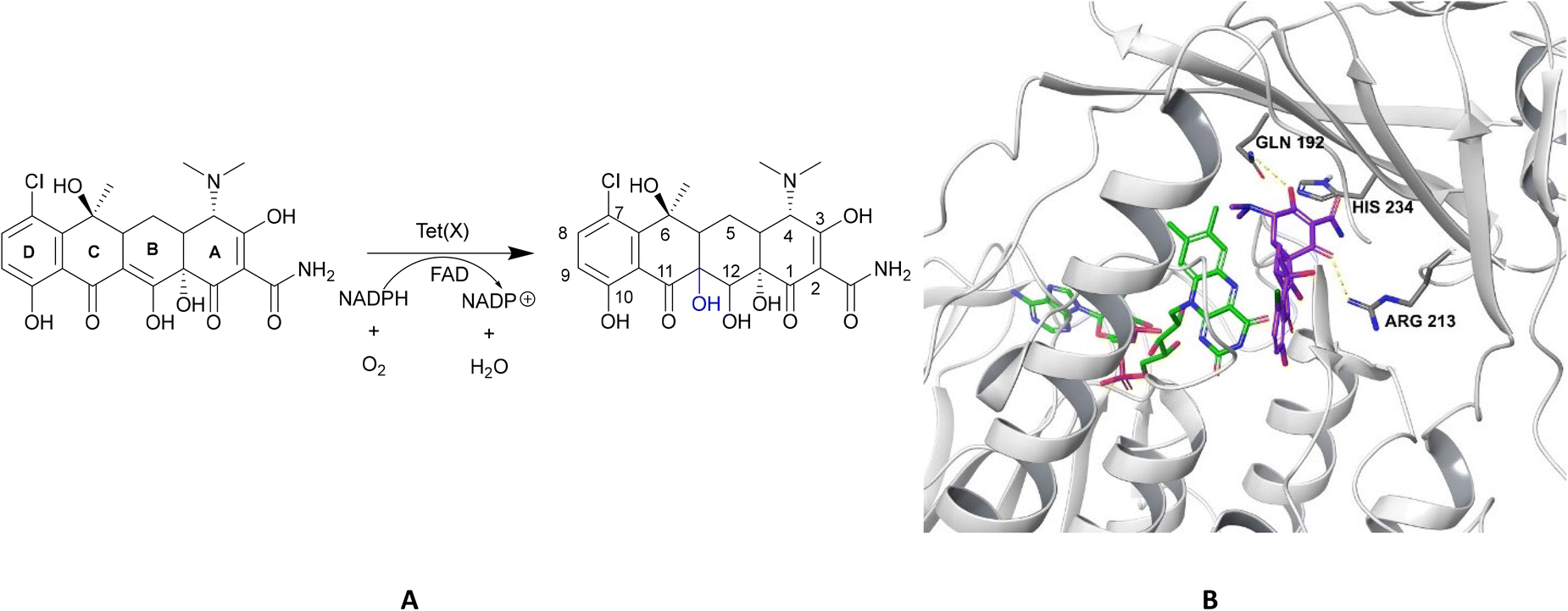
| Fig. 15 X-ray crystal structure of Tet(X) with bound FAD (PDB ID: 2XDO).178 Tet(X) is composed of two domains stabilised by a C-terminal α-helix and another C-terminal α-helix involved in substrate recognition and binding.179 The X-ray crystal structure of Tet(X) show that the first domain contains a non-covalently bound FAD that adopts an IN (pointing towards the binding domain) or OUT (pointing away from the binding domain) conformation. The A ring is positioned near the bound FAD molecule and the D ring lies near the C-terminal α-helix. The OUT confirmation is required for the reduction of FAD to FADH2 by NADPH. The reduced form is then able to bind oxygen in the IN confirmation forming a hydroperoxide able to hydroxylate tetracycline at C11. The second domain covers the FAD-binding site while also containing the substrate recognition pocket178 (image created using Mol*119). | ||
As the enzyme has a requirement for oxygen, this resistance mechanism is only found in aerobic bacteria. Tet(X) recognises the A and B ring of tetracyclines (Fig. 16); the D ring is not involved in binding to the active site which explains why third generation tetracyclines, such as tigecycline, are also susceptible to deactivation by Tet(X).180 The deactivation process can be divided into two steps—hydroxylation at C11 (weakening the binding to Mg2+ and therefore reducing the affinity of the drug for the ribosome) and decomposition of the hydroxylated tetracycline to an inactive polymer.172,173
Tet(X) inhibitors
Anhydrotetracycline, a shunt product involved in the biosynthesis of tetracycline, is structurally similar to tetracycline but dehydration at C6 alters its conformation and makes it more hydrophobic leading to differences in mechanisms of action.21,179 Tet(X) can hydroxylate anhydrotetracycline but at a much slower rate and specific modifications to the structure have been shown to improve stability and potency.21,181 Plumbagin is another potential inhibitor of a wide range of Tet(X) enzymes.182Other tetracycline destructases were discussed in other studies but they will not be discussed in this review due to their clinical insignificance.172,183,184
Rifamycins
Alongside isoniazid, rifampicin is a first-line drug for the treatment of TB. When it was first used, the incidence and mortality due to TB was reduced by 25% and 50% respectively.185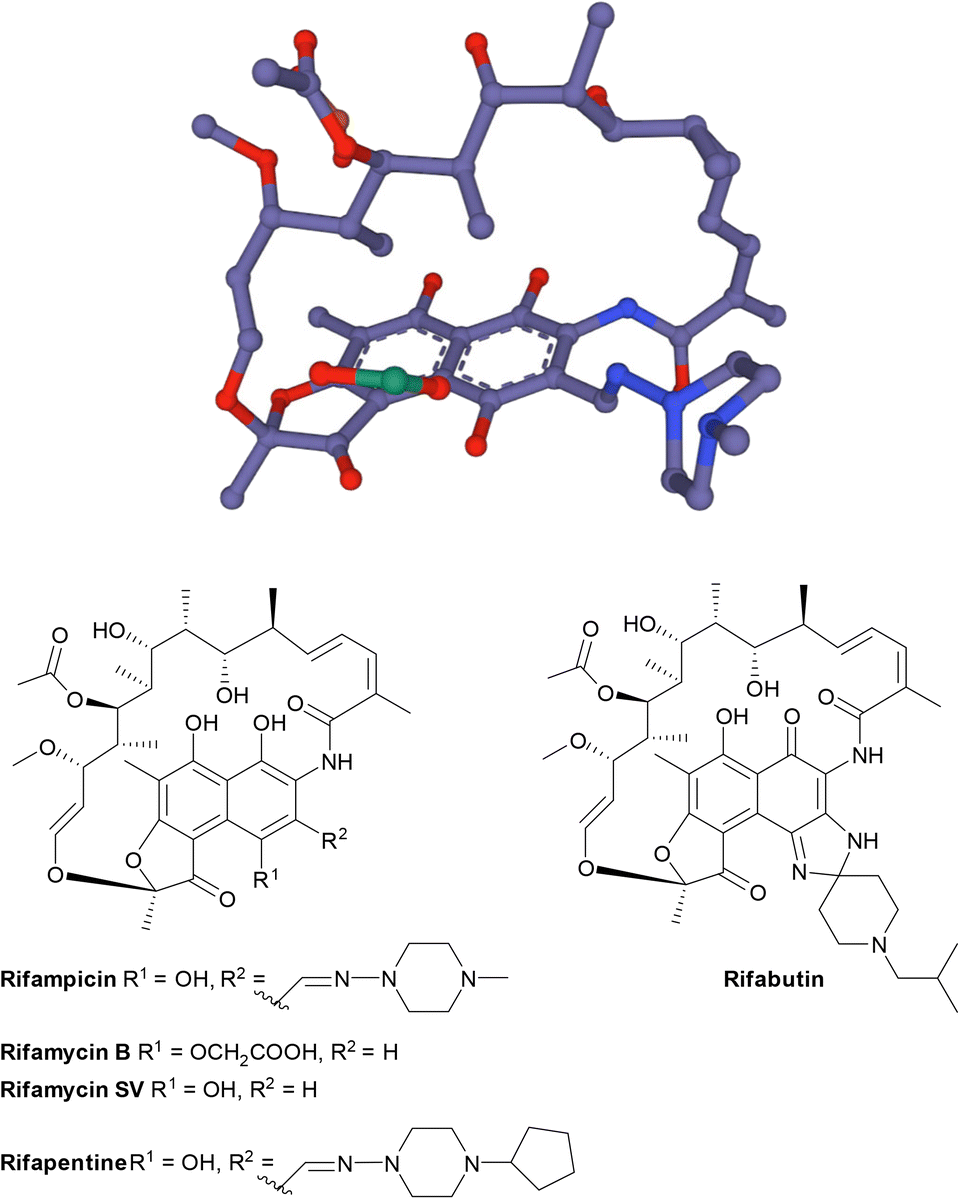 | ||
| Fig. 17 The structure of rifamycins is composed of a naphthalene core and an ansa bridge, which is a basket-like structure.30 | ||
| Enzyme | Host | Site modified | Substrate |
|---|---|---|---|
| Glycosyltransferase (RGT)188,189 | Nocardia brasiliensis, Streptomyces pseudogriseolus | 23-OH | Rifampicin, rifaximin, rifabutin, rifapentine |
| Phosphorylation (RPH)190 | Actinobacteria spp., Clostridia spp., Bacillus anthracis, E. faecalis, L. monocytogenes | 21-OH | Rifampicin, rifaximin, rifabutin, rifapentine |
| ADP-ribosylation (Arr)30,191 | M. smegmatis, P. aeruginosa, A. baumannii, some Enterobacteriaceae | 23-OH | Rifampicin, rifaximin, rifabutin, rifapentine |
| Monooxygenation + decomposition (Rox)192–194 | Nocardia farcinica | Naphthalene moiety | Rifampicin, rifaximin, rifabutin, rifapentine |
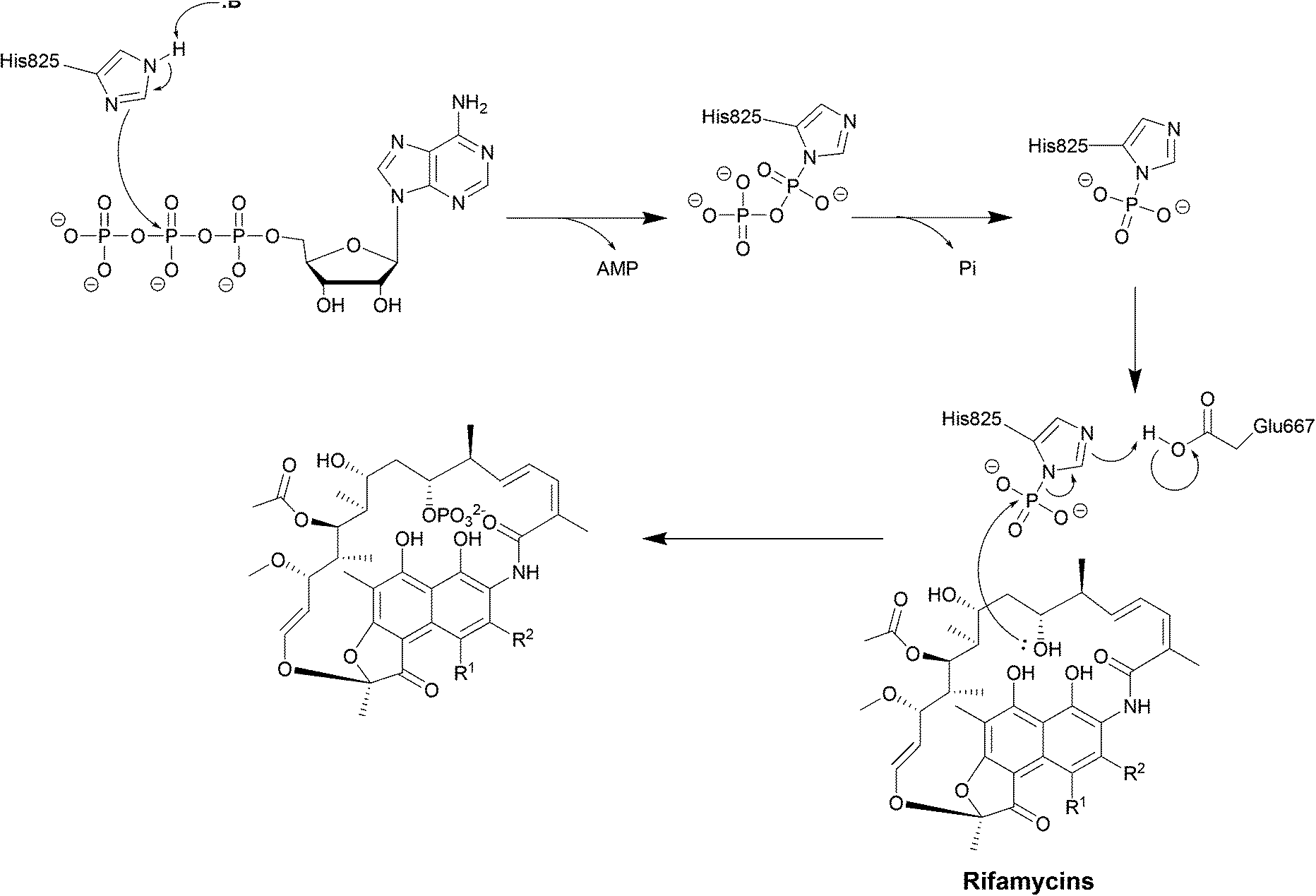 | ||
| Scheme 6 Deactivation of rifamycins by RPH in L. monocytogenes. The enzyme has 3 domains: an N-terminal ATP-grasp domain, a swivel domain containing the catalytic His at the C-terminus and an intermediate domain responsible for binding rifampicin.195 The rifampicin binding domain contains three residues Val333, Met359 and Val368 forming a hydrophobic patch that stabilises rifampicin.196 The naphthol ring of rifampicin binds deep in the pocket through van der Waals interactions while the R groups point towards the opening of the pocket and binds to Pro356 and Phe479.196 The ATP-grasp domain positions the β-phosphate that is then transferred to His825 in the swivel domain. The Glu667–Arg666 residues found in the rifampicin-binding domain then facilitate the transfer of the phosphate group to 21-OH of the polyketide region.30 | ||
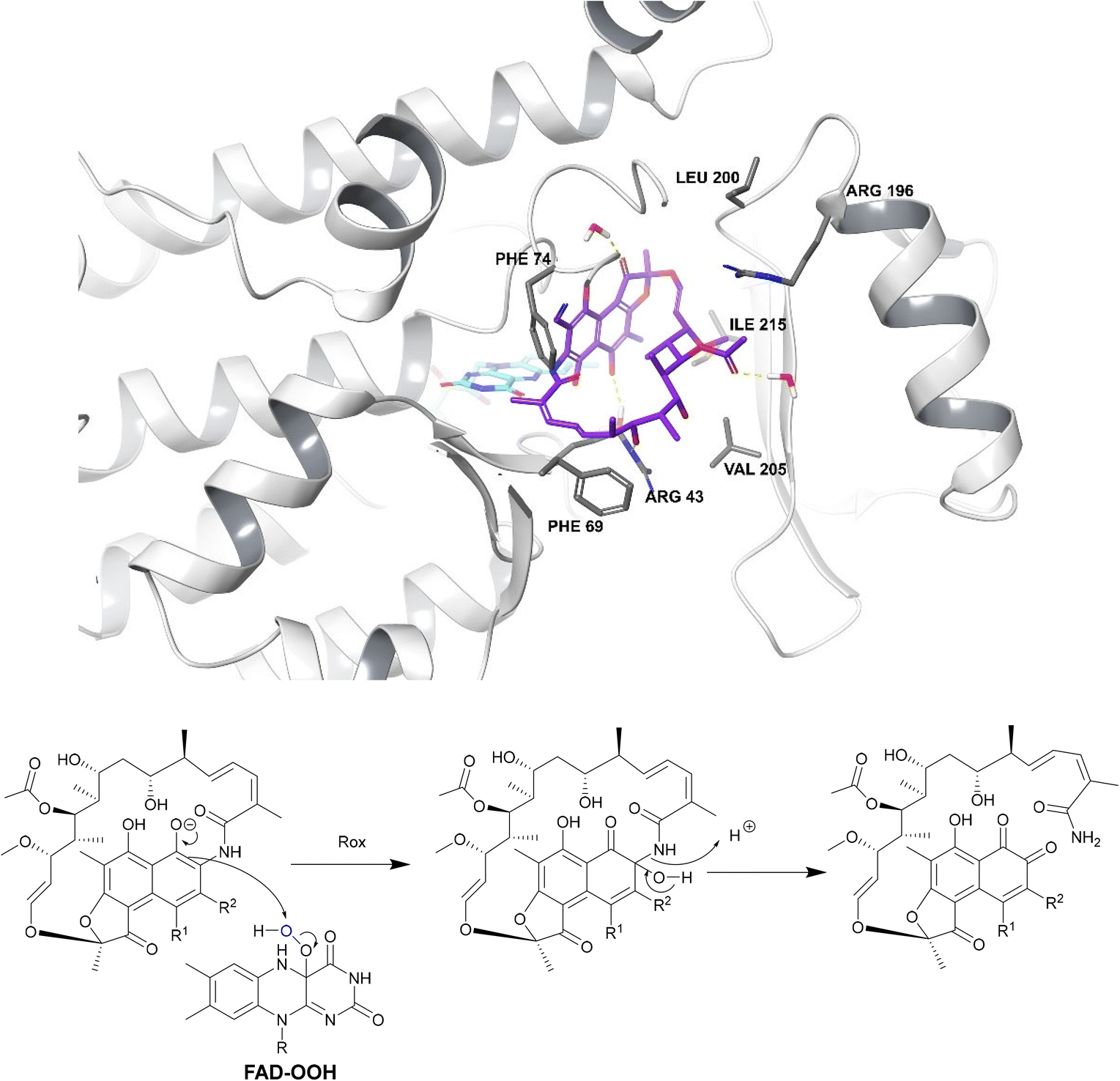 | ||
| Fig. 18 Deactivation of rifamycins by Rox in N. farcinica (5KOX).197 Rifampicin (purple) binds perpendicularly to FAD (light blue) with O4 of FAD undergoing hydrogen bonding with the drug.198 The exact binding interactions differ depending on the species expressing the Rox.198 For example, in S. venezuelae, Arg213 forms a hydrogen bond with 8-OH whereas, in N. farcinica, the 21-OH forms a hydrogen bond with the Arg43 residue.198 In N. farcinica, the drug binds to the enzyme through hydrophobic interactions mainly: the polyketide region interacts with the Val205 residue through hydrophobic interactions.198 In both examples, Rox transfers a hydroxyl group from FAD-OOH to RIF which leads to ring opening, disrupting the ansa bridge structure required for binding. | ||
| Arr enzyme subtype | Gene coding enzyme | Pathogenic bacteria expression | Rifamycin substrate | Ref. |
|---|---|---|---|---|
| Arr-2 | arr-2 | P. aeruginosa, K. pneumoniae, A. baumannii | Rifaximin | 201 |
| Rifabutin | ||||
| Rifapentine | ||||
| Arr-3 | arr-3 | Vibrio fluvialis | Rifampicin | 202 |
| arr-6 | E. coli | Rifaximin | ||
| Arr-4 | arr-4 | P. aeruginosa, A. baumannii | Rifampicin | 203 |
| Rifaximin | ||||
| Rifapentine | ||||
| Arr-5 | arr-5 | P. aeruginosa, K. pneumoniae, S. marcescens | Rifampicin | 203 |
| Rifaximin | ||||
| Rifapentine | ||||
| Arr-7 | arr-7 | P. aeruginosa | Rifampicin | 204 |
| Arr-8 | arr-8 | Klebsiella oxytoca, K. pneumoniae, P. aeruginosa | Rifaximin | 205 |
| Rifabutin | ||||
| Rifapentine |
Isoniazid
M. tuberculosis is a top priority pathogen as TB is one of the leading causes of death in the world due to a single infectious agent. The standard treatment regimen for TB consists of an intensive phase (2 months) involving treatment with rifampicin, isoniazid, pyrazinamide and ethambutol (RIPE), followed by a continuation phase of 4–7 months of rifampicin or isoniazid. Drug-resistant TB often requires even more complex medication regimens and is responsible for 250000 deaths every year.2 Although mutations in KatG and InhA are the major causes for isoniazid resistance, enzymatic modification also occurs in some mycobacterial species.209
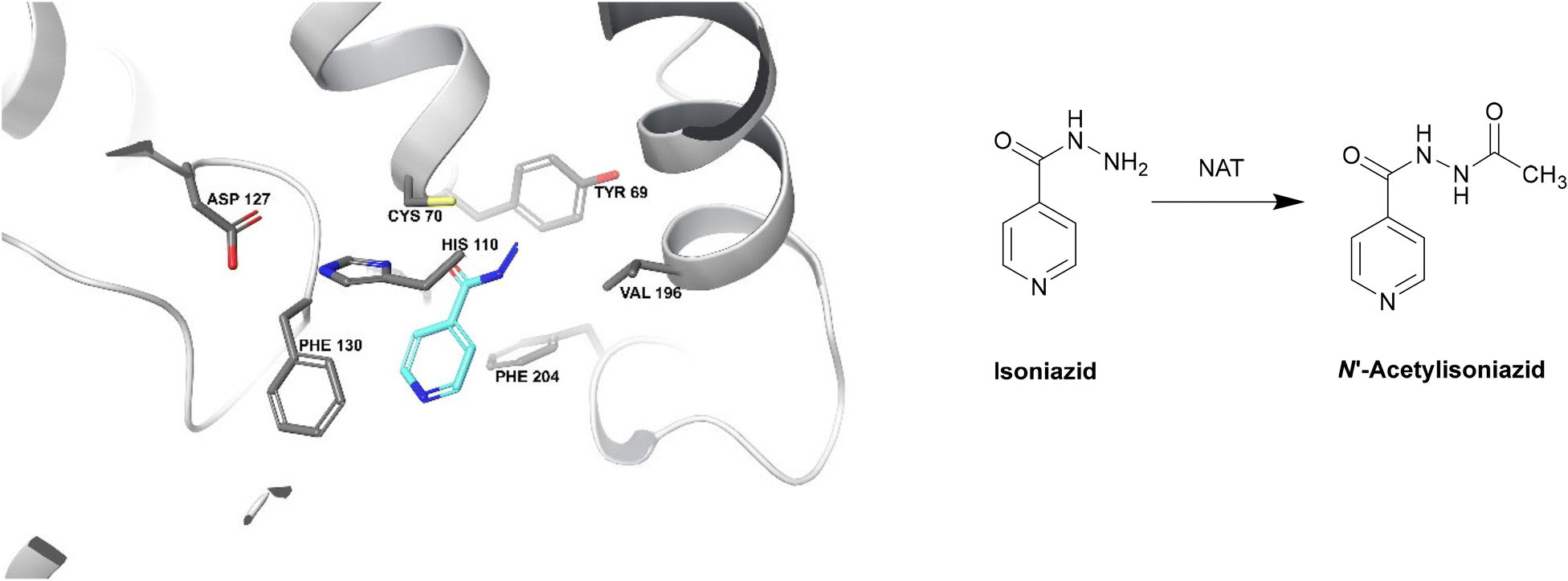 | ||
| Fig. 19 Deactivation of isoniazid by NAT (PDB ID: 1W6F).212 Isoniazid binds to a NAT catalytic triad composed of Cys70, His110 and Asp127. The terminal nitrogen of isoniazid is situated ∼2.4 Å from the γ-S atom of Cys70, making it the most important residue for this catalysis. Isoniazid also undergoes hydrophobic interactions and hydrogen bonding with other residues within the active site. The enzyme is initially acetylated by acetyl CoA and the acetyl group is then transferred onto the hydrazinyl group of isoniazid.212 | ||
Not only does NAT deactivate isoniazid, but it has also been shown to be involved in essential cellular processes and in promoting the survival of M. tuberculosis inside macrophages.213 Inhibition of NAT can thus lead to M. tuberculosis death in different ways, which highlights the potential for inhibitors.209
A novel acetyltransferase, labelled Rv2170, is also capable of acylating the terminal nitrogen of isoniazid, leading to resistance.23
In addition, p-substituted piperidinols with a halide and an ethyl substituent on the nitrogen atom, were shown to increase potency; they can form an irreversible adduct with the cysteine residue of NAT in M. marinum, which is 74% identical in sequence to NAT in M. tuberculosis.215
11α-Hydroxycinnamosmolide from Warburgia salutaris inhibited N-acetyltransferase and weakened the M. bovis cell wall thus was able to inhibit mycobacterial growth (Fig. 20).216
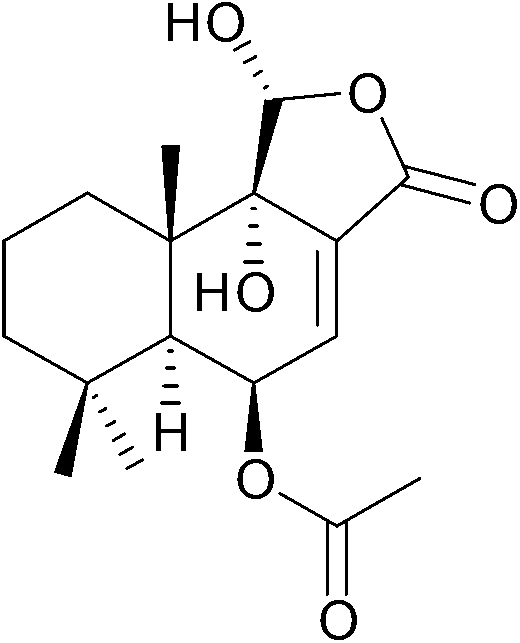 | ||
| Fig. 20 Chemical structure of 11α-hydroxycinnamosmolide an N-acetyltransferase inhibitor.216 | ||
Lincosamides
The first class is observed mostly in Staphylococci and is homologous to aminoglycoside nucleotidyltransferases. The class consists of LinA, LnuC and LnuD.220 LinA (161 amino acids) requires a nucleoside 5′-triphosphate (e.g. ATP, GTP, CTP, UTP) and Mg2+ as a cofactor. The enzyme modifies lincomycin at 3-OH of the sugar and clindamycin at the 4-OH group.221
The second class is found in Enterococci and consists of LinB and LnuF.220 Clindamycin undergoes hydrophobic interactions with Phe104, Tyr27 and Tyr44 of the enzyme, dipole–ion interactions between the 3-OH and Mg2+ and hydrogen bonds between 4-OH and Tyr27. After binding, the α-phosphate group is transferred, forming a pentacoordinate phosphate intermediate product which eventually leads to adenylation at 3-OH of clindamycin (Scheme 7).222
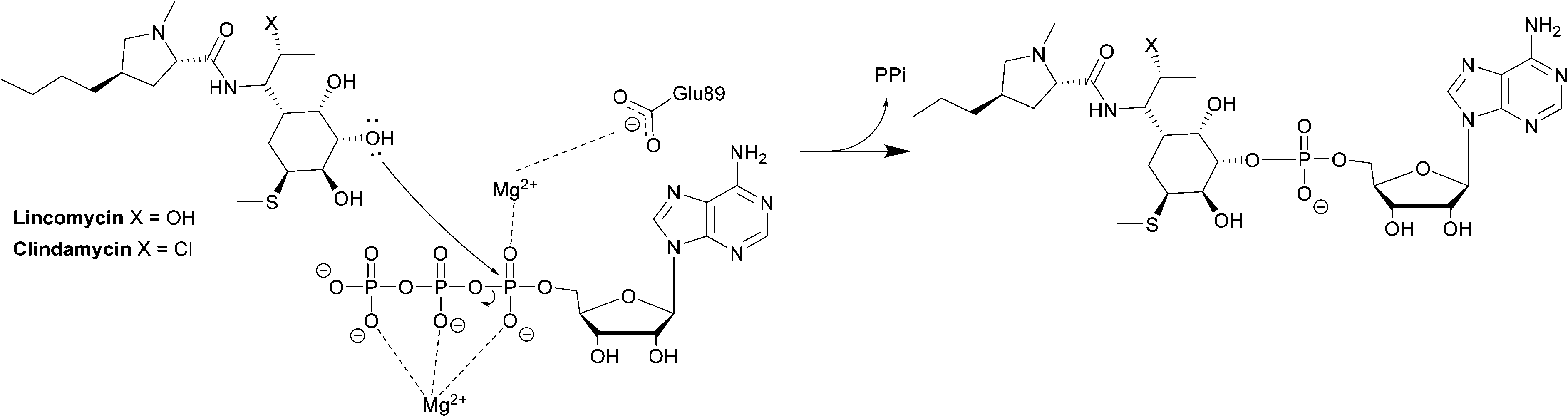 | ||
| Scheme 7 Transfer of an AMP group from ATP to 3-OH of lincosamides by LinB. | ||
These nucleotidyltransferase enzymes are encoded on mobile elements indicating that resistance can spread.223 As LinB and DNA polymerase exhibit many similarities, De Pascale and Wright suggested trialling DNA polymerase inhibitors on nucleotidyltransferases but no follow-up studies have been conducted.224
Fosfomycin
Fosfomycin is often used as an alternative treatment for osteomyelitis or urinary tract infections that are resistant to other antibacterial agents.225,226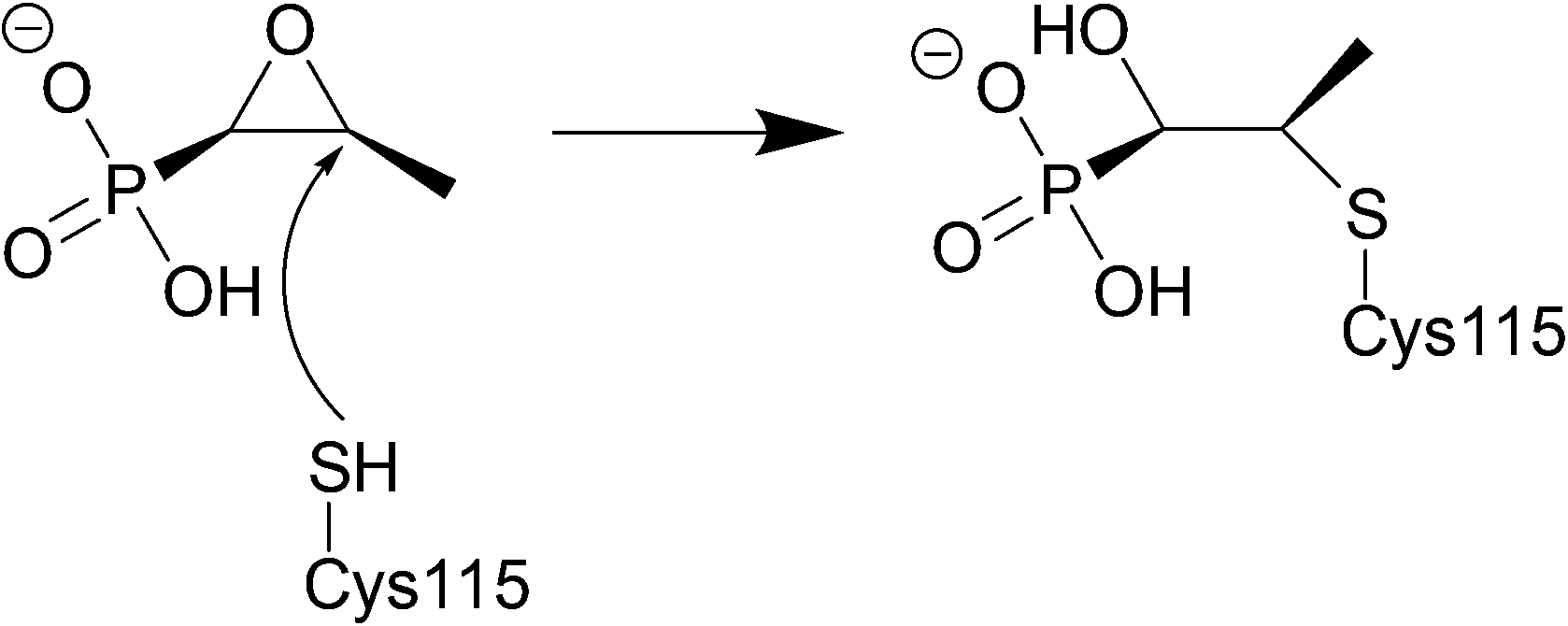 | ||
| Fig. 21 Fosfomycin binding to MurA, which is utilised in the formation of N-acetylmuramic acid, an essential component of the peptidoglycan layer, inhibition by fosfomycin thus leads to cell wall lysis. The catalytic domain of MurA is deep in the cavity of the enzyme's two domains and contains Cys115, which fosfomycin covalently attaches to. Three positively charged residues of MurA, Lys22, Arg120, and Arg397 surround the phosphonate group of fosfomycin creating electrostatic interactions and hydrogen bonds.230 | ||
FosA
FosA is a homodimeric Mn2+- and K+-dependent glutathione S-transferase made up of two active sites and a central cavity.228,229 FosA deactivates fosfomycin by opening the epoxide ring, a critical structure for binding to MurA, via nucleophilic attack (Fig. 22).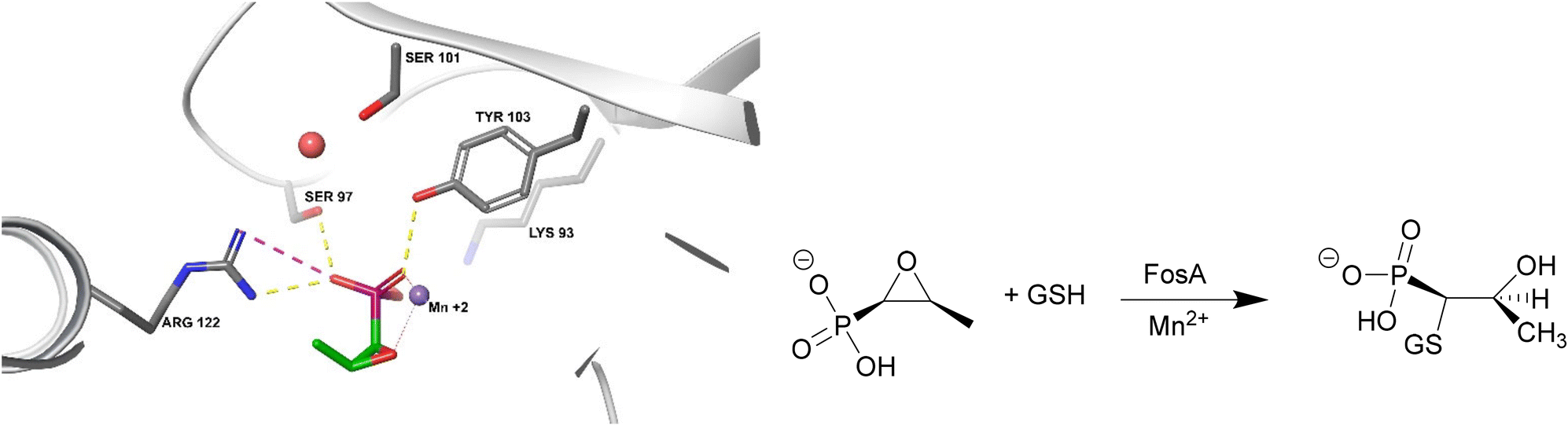 | ||
| Fig. 22 Fosfomycin binding to FosA from K. pneumoniae (PDB ID 5V3D). FosA key residues involved in the binding of fosfomycin include Lys93, Ser97, Ser101, Tyr103, and Arg122. The Mn2+ ion acts as a Lewis acid during the nucleophilic attack by glutathione. A K+ at the active site does not take part in the catalysis reaction but enhances the electrophilicity of the Mn2+ ion, increasing the rate of reaction.229 | ||
FosA genes are found in plasmids (e.g. ESBL E. coli and carbapenemase producing K. pneumonia) or chromosomes (e.g. Klebsiella spp., Enterobacter spp., and P. aeruginosa), highlighting the importance and urgency of developing strategies to overcome fosfomycin resistance.226
Although there are many subtypes of FosA enzymes (Table 12), their active site has been shown to be conserved which makes the development of broad-spectrum FosA inhibitors possible.229 For a long time, fosfomycin resistance was not of concern as it did not significantly affect bacteria which were susceptible to fosfomycin however it has now reached E. coli and other Gram negative pathogens.231
Several phosphonates, such as phosphonoformate, have been shown to be inhibitors of FosA.238 Phosphonoformate can form a five-coordinate complex with the Mn2+ centre with a geometry similar to that of the transition state of the reaction. Phosphonoformate can greatly increase the susceptibility K. pneumoniae, E. cloacae, P. aeruginosa, and to a lesser extent E. coli, to fosfomycin. One disadvantage is the association of phosphonoformate with nephrotoxicity and seizures, thus limiting its use in practice.239
One study showed that compound 19583672 from the Zinc database was a potent inhibitor of FosA variant 7 from K. pneumoniae and could form a stable complex with the enzyme.240 ANY1 (3-bromo-6-[3-(3-bromo-2-oxo-1H-pyrazolo[1,5-a]pyrimidin-6-yl)-4-nitro-1H-pyrazol-5-yl]-1H-pyrazolo[1,5-a]pyrimidin-2-one) is a small-molecule active site non-competitive inhibitor of FosA which exhibits antibacterial activity against K. pneumoniae in combination with fosfomycin (Fig. 23).
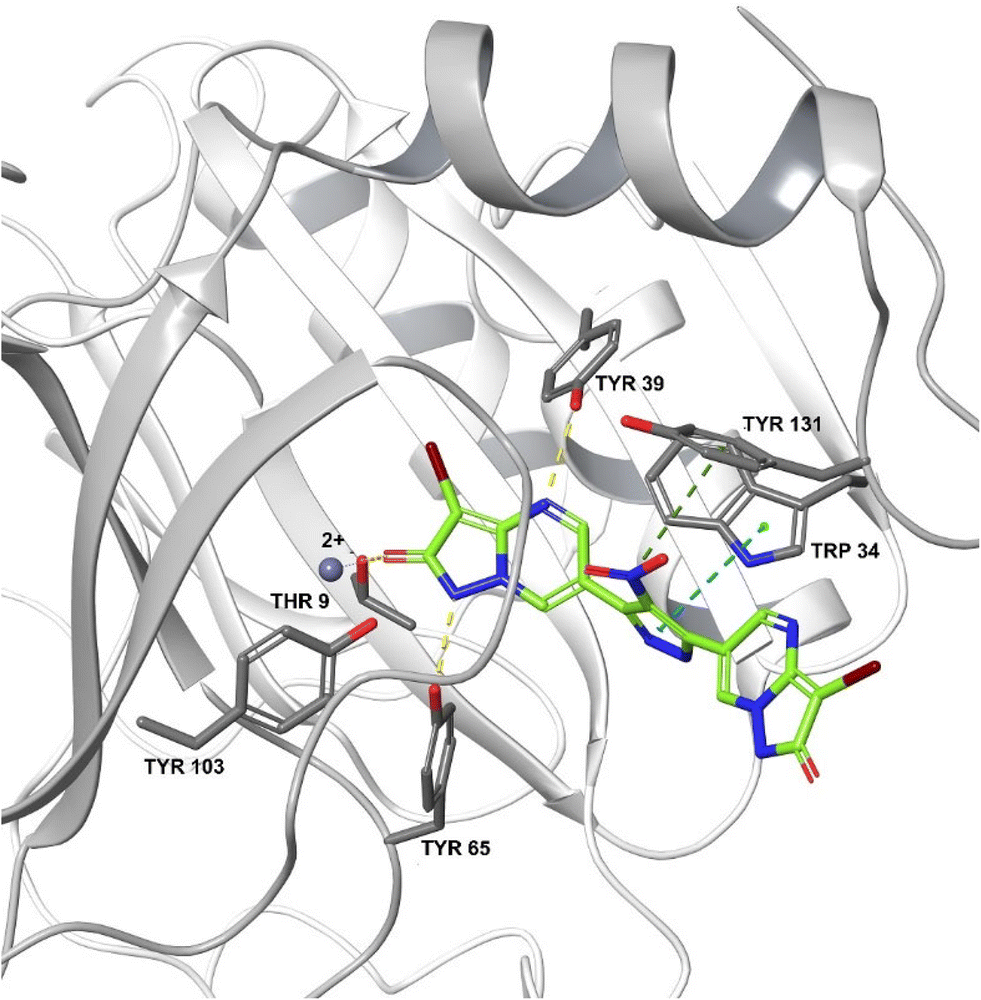 | ||
| Fig. 23 Crystal structure of FosA3 with inhibitor ANY1 bound (PDB ID 5WEP). ANY1 binds to the active site of FosA by interacting with Lys93, Ser97 and Tyr103.239 | ||
These beneficial effects were not seen when ANY1 was given alone or in combination with gentamicin, demonstrating its potential as a fosfomycin adjuvant.239
Susceptibility testing showed that the antibiotic activity of fosfomycin was potentiated by the addition of disulfiram (Fig. 24) or octyl pyrrolidine-1 carbo(dithioperoxo)thioate.
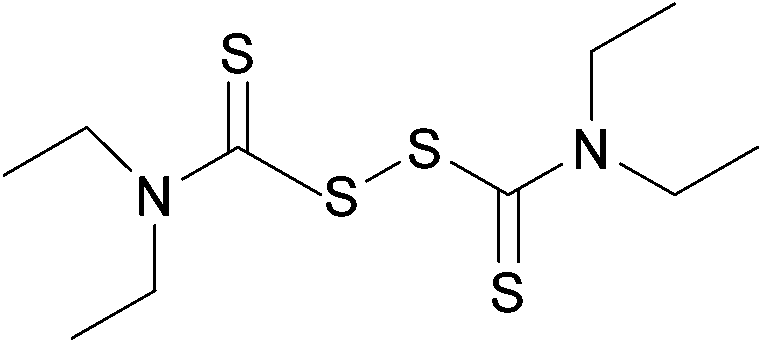 | ||
| Fig. 24 Chemical structure of disulfiram. Disulfiram is able to react with BSH, forming a stable adduct and depleting BSH stores.247 | ||
Conclusions
Enzymatic drug deactivation is an important contributor to the resistance of priority pathogens to many classes of antibacterial agents. The aim of this review has been to describe the enzymatic processes involved in antibacterial deactivation to highlight opportunities for adjuvant development. Serine β-lactamase inhibitors are currently the only adjuvants in clinical use, although other resistant enzymes are encoded on mobile genetic elements, which contribute to a serious spread in resistance. Soon, infections such as sexually transmitted diseases, urinary tract infections and tuberculosis may become impossible to treat without the development of novel drugs or drug-adjuvant combinations, making a post-antibiotic era worryingly possible.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to acknowledge the Gadigal people of the Eora Nation as traditional custodians of the land upon where we work, and where this research was conducted.Notes and references
- R. I. Aminov, Front. Microbiol., 2010, 1, 134 CrossRef PubMed.
- W. H. O. (WHO), 2020 antibacterial agents in clinical and preclinical development: an overview and analysis, https://www.who.int/publications/i/item/9789240021303, (accessed May, 2022) Search PubMed.
- K. D. Leuthner and G. V. Doern, J. Clin. Microbiol., 2013, 51, 3916–3920 CrossRef PubMed.
- D. Nathwani, D. Varghese, J. Stephens, W. Ansari, S. Martin and C. Charbonneau, Antimicrob. Resist. Infect. Control, 2019, 8, 35 CrossRef PubMed.
- W. H. O. (WHO), Antimicrobial resistance, https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance#:~:text=What%20is%20antimicrobial%20resistance%3F,spread%2C%20severe%20illness%20and%20death, (accessed May, 2022) Search PubMed.
- D. Raoult, M. Leone, Y. Roussel and J. Rolain, Lancet Infect. Dis., 2019, 19, 128–129 CrossRef PubMed.
- C. Lim, E. Takahashi, M. Hongsuwan, V. Wuthiekanun, V. Thamlikitkul, S. Hinjoy, N. P. J. Day, S. J. Peacock and D. Limmathurotsakul, eLife, 2016, 5, 180–182 Search PubMed.
- W. H. O. (WHO), New report calls for urgent action to avert antimicrobial resistance crisis, https://www.who.int/news/item/29-04-2019-new-report-calls-for-urgent-action-to-avert-antimicrobial-resistance-crisis, (accessed May, 2022) Search PubMed.
- K. Bush, Antimicrob. Agents Chemother., 2018, 62, e01076-18 CrossRef PubMed.
- G. Zhang, S. O. Leclercq, J. Tian, C. Wang, K. Yahara, G. Ai, S. Liu and J. Feng, PLoS Genet., 2017, 13, e1006602 CrossRef PubMed.
- Z. Fu, Y. Liu, C. Chen, Y. Guo, Y. Ma, Y. Yang, F. Hu, X. Xu and M. Wang, PLoS One, 2016, 11, e0154829 CrossRef PubMed.
- M. S. Ramirez and M. E. Tolmasky, Drug Resistance Updates, 2010, 13, 151–171 CrossRef CAS PubMed.
- L. R. Schwocho, C. P. Schaffner, G. H. Miller, R. S. Hare and K. J. Shaw, Antimicrob. Agents Chemother., 1995, 39, 1790–1796 CrossRef CAS PubMed.
- B. G. Elisha and L. Steyn, Plasmid, 1991, 25, 96–104 CrossRef CAS.
- I. Plante, D. Centrón and P. H. Roy, J. Antimicrob. Chemother., 2003, 51, 787–790 CrossRef CAS PubMed.
- J. Sun, C. Chen, C.-Y. Cui, Y. Zhang, X. Liu, Z.-H. Cui, X.-Y. Ma, Y. Feng, L.-X. Fang, X.-L. Lian, R.-M. Zhang, Y.-Z. Tang, K.-X. Zhang, H.-M. Liu, Z.-H. Zhuang, S.-D. Zhou, J.-N. Lv, H. Du, B. Huang, F.-Y. Yu, B. Mathema, B. N. Kreiswirth, X.-P. Liao, L. Chen and Y.-H. Liu, Nat. Microbiol., 2019, 4, 1457–1464 CrossRef CAS PubMed.
- A. Seupt, M. Schniederjans, J. Tomasch and S. Häussler, Antimicrob. Agents Chemother., 2021, 65, e01166-20 CrossRef PubMed.
- D. Yong, M. A. Toleman, C. G. Giske, H. S. Cho, K. Sundman, K. Lee and T. R. Walsh, Antimicrob. Agents Chemother., 2009, 53, 5046–5054 CrossRef CAS.
- G. Arlet, D. Nadjar, J. L. Herrmann, J. L. Donay, P. H. Lagrange and A. Philippon, Antimicrob. Agents Chemother., 2001, 45, 2971–2972 CrossRef CAS PubMed.
- M. Aghazadeh, M. A. Rezaee, M. R. Nahaei, R. Mahdian, O. Pajand, F. Saffari, M. Hassan and Z. Hojabri, Microb. Drug Resist., 2013, 19, 282–288 CrossRef CAS PubMed.
- J. L. Markley and T. A. Wencewicz, Front. Microbiol., 2018, 9, 1058 CrossRef PubMed.
- D. A. Butler, A. P. Rana, F. Krapp, S. R. Patel, Y. Q. Huang, E. A. Ozer, A. R. Hauser and Z. P. Bulman, J. Antimicrob. Chemother., 2021, 76, 671–679 CrossRef CAS PubMed.
- K. B. Arun, A. Madhavan, B. Abraham, M. Balaji, K. C. Sivakumar, P. Nisha and R. A. Kumar, Antimicrob. Agents Chemother., 2020, 65, e00456-20 CrossRef PubMed.
- W. C. Reygaert, AIMS Microbiol., 2018, 4, 482–501 CAS.
- G. D. Wright, Adv. Drug Delivery Rev., 2005, 57, 1451–1470 CrossRef CAS PubMed.
- M. Miklasińska-Majdanik, Antibiotics, 2021, 10, 1406 CrossRef PubMed.
- G. D. Wright and A. M. Berghuis, in Enzyme-Mediated Resistance to Antibiotics: Mechanisms, Dissemination, and Prospects for Inhibition, 2007, pp. 21–33, DOI:10.1128/9781555815615.ch3.
- M. E. Falagas, F. Athanasaki, G. L. Voulgaris, N. A. Triarides and K. Z. Vardakas, Int. J. Antimicrob. Agents, 2019, 53, 22–28 CrossRef CAS PubMed.
- S. Schwarz, C. Kehrenberg, B. Doublet and A. Cloeckaert, FEMS Microbiol. Rev., 2004, 28, 519–542 CrossRef CAS PubMed.
- M. D. Surette, P. Spanogiannopoulos and G. D. Wright, Acc. Chem. Res., 2021, 54, 2065–2075 CrossRef CAS PubMed.
- M. Linkevicius, L. Sandegren and D. I. Andersson, Antimicrob. Agents Chemother., 2016, 60, 789–796 CrossRef CAS PubMed.
- K. Bush and P. A. Bradford, Cold Spring Harbor Perspect. Med., 2016, 6, a025247 CrossRef PubMed.
- R. Anderson, P. W. Groundwater, A. Todd and A. Worsley, Antibacterial Agents: Chemistry, Mode of Action, Mechanisms of Resistance and Clinical Applications, John Wiley & Sons, Incorporated, New York, 2012 Search PubMed.
- A. Fleming, Penicillin - Nobel Lecture, https://www.nobelprize.org/uploads/2018/06/fleming-lecture.pdf, (accessed May, 2022) Search PubMed.
- D. L. Paterson and R. A. Bonomo, Clin. Microbiol. Rev., 2005, 18, 657–686 CrossRef CAS PubMed.
- S. K. Son, N. R. Lee, J.-H. Ko, J. K. Choi, S.-Y. Moon, E. J. Joo, K. R. Peck and D. A. Park, J. Antimicrob. Chemother., 2018, 73, 2631–2642 CrossRef CAS PubMed.
- T. Naas, S. Oueslati, R. A. Bonnin, M. L. Dabos, A. Zavala, L. Dortet, P. Retailleau and B. I. Iorga, J. Enzyme Inhib. Med. Chem., 2017, 32, 917–919 CrossRef CAS PubMed.
- R. P. Ambler, Philos. Trans. R. Soc., B, 1980, 289, 321–331 CAS.
- C. L. Tooke, P. Hinchliffe, E. C. Bragginton, C. K. Colenso, V. H. A. Hirvonen, Y. Takebayashi and J. Spencer, J. Mol. Biol., 2019, 431, 3472–3500 CrossRef CAS PubMed.
- B. P. Alcock, A. R. Raphenya, T. T. Y. Lau, K. K. Tsang, M. Bouchard, A. Edalatmand, W. Huynh, A.-L. V. Nguyen, A. A. Cheng, S. Liu, S. Y. Min, A. Miroshnichenko, H. Tran, R. E. Werfalli, J. A. Nasir, M. Oloni, D. J. Speicher, A. Florescu, B. Singh, M. Faltyn, A. Hernandez-Koutoucheva, A. N. Sharma, E. Bordeleau, A. C. Pawlowski, H. L. Zubyk, D. Dooley, E. Griffiths, F. Maguire, G. L. Winsor, R. G. Beiko, F. S. L. Brinkman, W. W. L. Hsiao, G. V. Domselaar and A. G. McArthur, Nucleic Acids Res., 2020, 8, 517–525 Search PubMed.
- B. A. Rasmussen, K. Bush, D. Keeney, Y. Yang, R. Hare, C. O'Gara and A. A. Medeiros, Antimicrob. Agents Chemother., 1996, 40, 2080–2086 CrossRef CAS PubMed.
- A. Bauernfeind, I. Stemplinger, R. Jungwirth, S. Ernst and J. M. Casellas, Antimicrob. Agents Chemother., 1996, 40, 509–513 CrossRef CAS PubMed.
- I. E. Robledo, E. E. Aquino and G. J. Vázquez, Antimicrob. Agents Chemother., 2011, 55, 2968–2970 CrossRef CAS PubMed.
- M. A. Jure, M. Duprilot, H. E. Musa, C. López, M. C. D. Castillo, F. X. Weill, G. Arlet and D. Decré, Antimicrob. Agents Chemother., 2014, 58, 6335–6336 CrossRef CAS PubMed.
- D. Shokri, S. Mobasherizadeh, M. N. Baruq and M. Yaran, Maj. Danishkadah-i Pizishki-i Isfahan, 2013, 31, 1247–1256 Search PubMed.
- A. M. Queenan, C. Torres-Viera, H. S. Gold, Y. Carmeli, G. M. Eliopoulos, R. C. Moellering, Jr., J. P. Quinn, J. Hindler, A. A. Medeiros and K. Bush, Antimicrob. Agents Chemother., 2000, 44, 3035–3039 CrossRef CAS.
- J. R. McLaughlin, J. Biol. Chem., 1981, 256, 11283–11291 CrossRef CAS PubMed.
- P. A. Bradford, Antimicrob. Agents Chemother., 1999, 43, 2960–2963 CrossRef CAS PubMed.
- P. Oelschlaeger, N. Ai, K. T. DuPrez, W. J. Welsh and J. H. Toney, J. Med. Chem., 2010, 53, 3013–3027 CrossRef CAS PubMed.
- I. Garcia-Saez, J. D. Docquier, M. Rossolini and O. Dideberg, J. Mol. Biol., 2007, 375, 604–611 CrossRef PubMed.
- K. K. Kumarasamy, M. A. Toleman, T. R. Walsh, J. Bagaria, F. Butt, R. Balakrishnan, U. Chaudhary, M. Doumith, C. G. Giske, S. Irfan, P. Krishnan, A. V. Kumar, S. Maharjan, S. Mushtaq, T. Noorie, D. L. Paterson, A. Pearson, C. Perry, R. Pike, B. Rao, U. Ray, J. B. Sarma, M. Sharma, E. Sheridan, M. A. Thirunarayan, J. Turton, S. Upadhyay, M. Warner, W. Welfare, D. M. Livermore and N. Woodford, Lancet Infect. Dis., 2010, 10, 597–602 CrossRef CAS PubMed.
- J. S. Esterly, C. L. Richardson, N. S. Eltoukhy, C. Qi and M. H. Scheetz, Ann. Pharmacother., 2011, 45, 218–228 CrossRef CAS PubMed.
- B. A. Evans and S. G. B. Amyes, Clin. Microbiol. Rev., 2014, 27, 241–263 CrossRef.
- N. T. Antunes and J. F. Fisher, Antibiotics, 2014, 3, 398–434 CrossRef.
- R. P. Ambler, A. F. W. Coulson, J. M. Frère, J. M. Ghuysen, B. Joris, M. Forsman, R. C. Levesque, G. Tiraby and S. G. Waley, Biochem. J., 1991, 276, 269–270 CrossRef CAS PubMed.
- J. F. Fisher, S. O. Meroueh and S. Mobashery, Chem. Rev., 2005, 105, 395–424 CrossRef CAS PubMed.
- C. M. Rotondo and G. D. Wright, Curr. Opin. Microbiol., 2017, 39, 96–105 CrossRef CAS.
- J. E. Raczynskaa, I. G. Shabalin, W. Minor, A. Wlodawer and M. Jaskolski, Drug Resistance Updates, 2018, 40, 1–12 CrossRef PubMed.
- K. Bush, Int. J. Antimicrob. Agents, 2015, 46, 483–493 CrossRef CAS PubMed.
- S. M. Drawz and R. A. Bonomo, Clin. Microbiol. Rev., 2010, 23, 160–201 CrossRef CAS PubMed.
- H. C. Neu and K. P. Fu, Antimicrob. Agents Chemother., 1978, 14, 650–655 CrossRef CAS PubMed.
- D. M. Shlaes, Ann. N. Y. Acad. Sci., 2013, 1277, 105–114 CrossRef CAS PubMed.
- D. van Duin and R. A. Bonomo, Clin. Infect. Dis., 2016, 63, 234–241 CrossRef CAS PubMed.
- M. Akova, Clin. Microbiol. Infect., 2008, 14, 185–188 CrossRef CAS PubMed.
- A. B. Shapiro, Antimicrob. Agents Chemother., 2017, 61, e01612-17 CrossRef PubMed.
- D. E. Ehmann, H. Jahić, P. L. Ross, R.-F. Gu, J. Hu, T. F. Durand-Réville, S. Lahiri, J. Thresher, S. Livchak, N. Gao, T. Palmer, G. K. Walkup and S. L. Fisher, J. Biol. Chem., 2013, 288, 27960–27971 CrossRef CAS PubMed.
- G. G. Zhanel, C. K. Lawrence, H. Adam, F. Schweizer, S. Zelenitsky, M. Zhanel, P. R. S. Lagacé-Wiens, A. Walkty, A. Denisuik, A. Golden, A. S. Gin, D. J. Hoban, J. P. Lynch and J. A. Karlowsky, Drugs, 2017, 78, 65–98 CrossRef PubMed.
- Y.-A. Heo, Drugs, 2021, 81, 377–388 CrossRef CAS.
- N. L. Mallalieu, E. Winter, S. Fettner, K. Patel, E. Zwanziger, G. Attley, I. Rodriguez, A. Kano, S. M. Salama, D. Bentley and A. M. Geretti, Antimicrob. Agents Chemother., 2020, 64, e02229-19 CrossRef.
- D. M. Livermore, M. Warner, S. Mushtaq and N. Woodford, Antimicrob. Agents Chemother., 2016, 60, 554–560 CrossRef CAS PubMed.
- J. A. Karlowsky, M. A. Hackel, S. K. Bouchillon and D. F. Sahm, Antimicrob. Agents Chemother., 2020, 64, e01432-20 CrossRef PubMed.
- M. D. Barnes, V. Kumar, C. R. Bethel, S. H. Moussa, J. O'Donnell, J. D. Rutter, C. E. Good, K. M. Hujer, A. M. Hujer, S. H. Marshall, B. N. Kreiswirth, S. S. Richter, P. N. Rather, M. R. Jacobs, K. M. Papp-Wallace, F. van den Akker and R. A. Bonomo, MBio, 2019, 10, e00159-19 CrossRef PubMed.
- T. F. Durand-Réville, J. Comita-Prevoir, J. Zhang, X. Wu, T. L. May-Dracka, J. A. C. Romero, F. Wu, A. Chen, A. B. Shapiro, N. M. Carter, S. M. McLeod, R. A. Giacobbe, J. C. Verheijen, S. D. Lahiri, M. D. Sacco, Y. Chen, J. P. O'Donnell, A. A. Miller, J. P. Mueller and R. N. A. Tommasi, J. Med. Chem., 2020, 63, 12511–12525 CrossRef PubMed.
- J. C. Vázquez-Ucha, M. Maneiro, M. Martínez-Guitián, J. Buynak, C. R. Bethel, R. A. Bonomo, G. Bou, M. Poza, C. González-Bello and A. Beceiro, Antimicrob. Agents Chemother., 2017, 61, e01172-17 CrossRef.
- P. A. Lang, R. Raj, A. Tumber, C. T. Lohans, P. Rabe, C. V. Robinson, J. Brem and C. J. Schofield, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2117310119 CrossRef CAS PubMed.
- F. Bernhard, R. Odedra, S. Sordello, R. Cardin, S. Franzoni, C. Charrier, A. Belley, P. Warn, M. Machacek and P. Knechtle, Antimicrob. Agents Chemother., 2020, 64, e00078-20 CrossRef PubMed.
- B. Liu, R. E. L. Trout, G.-H. Chu, D. McGarry, R. W. Jackson, J. C. Hamrick, D. M. Daigle, S. M. Cusick, C. Pozzi, F. De Luca, M. Benvenuti, S. Mangani, J.-D. Docquier, W. J. Weiss, D. C. Pevear, L. Xerri and C. J. Burns, J. Med. Chem., 2020, 63, 2789–2801 CrossRef CAS.
- R. E. Trout, A. Zulli, E. Mesaros, R. W. Jackson, S. Boyd, B. Liu, J. Hamrick, D. Daigle, C. L. Chatwin, K. John, L. McLaughlin, S. M. Cusick, W. J. Weiss, M. E. Pulse, D. C. Pevear, G. Moeck, L. Xerri and C. J. Burns, J. Med. Chem., 2021, 64, 10155–10166 CrossRef CAS PubMed.
- P. J. Petersen, C. Hal Jones, A. M. Venkatesan and P. A. Bradford, Antimicrob. Agents Chemother., 2009, 53, 1698–1700 CrossRef CAS PubMed.
- D. Zhao, H. Li, C. Yue, K. Sun, Y. Dai, H. Zhang, Y. Liu, Y. Gao and J. Li, J. Inorg. Biochem., 2021, 218, 111381 CrossRef CAS PubMed.
- V. Cifuentes-Castro, C. Rodriguez-Almazan, J. Silva-Sanchez and E. Rudino-Pinera, Biochem. Biophys. Res. Commun., 2020, 522, 545–551 CrossRef CAS PubMed.
- N. P. Krishnan, N. Q. Nguyen, K. M. Papp-Wallace, R. A. Bonomo and F. van den Akker, PLoS One, 2015, 10, e0136813 CrossRef PubMed.
- A. Morinaka, Y. Tsutsumi, M. Yamada, K. Suzuki, T. Watanabe, T. Abe, T. Furuuchi, S. Inamura, Y. Sakamaki, N. Mitsuhashi, T. Ida and D. M. Livermore, J. Antimicrob. Chemother., 2015, 70, 2779–2786 CrossRef CAS.
- B. Bedenić, S. Sardelić, J. Luxner, Z. Bošnjak, D. Varda-Brkić, A. Lukić-Grlić, I. Mareković, S. Frančula-Zaninović, M. Krilanović, D. Šijak, A. Grisold and G. Zarfel, Infect., Genet. Evol., 2016, 43, 74–82 CrossRef PubMed.
- J. C. Hamrick, J.-D. Docquier, T. Uehara, C. L. Myers, D. A. Six, C. L. Chatwin, K. J. John, S. F. Vernacchio, S. M. Cusick and R. E. Trout, Antimicrob. Agents Chemother., 2020, 64, e01963-19 CrossRef.
- A. Schatz, E. Bugie and S. A. Waksman, Proc. Soc. Exp. Biol. Med., 1944, 55, 66–69 CrossRef CAS.
- J. A. Clark and D. S. Burgess, Ther. Adv. Infect. Dis., 2020, 7, 1–15 Search PubMed.
- B. Becker and M. A. Cooper, ACS Chem. Biol., 2013, 8, 105–115 CrossRef CAS PubMed.
- A. F. Neuwald and D. Landsman, Trends Biochem. Sci., 1997, 22, 154–155 CrossRef CAS.
- L. P. Samuel, C.-H. Song, J. Wei, E. A. Roberts, J. L. Dahl, C. E. Barry, E.-K. Jo and R. L. Friedman, Microbiology, 2007, 153, 529–540 CrossRef CAS PubMed.
- W. Chen, T. Biswas, V. R. Porter, O. V. Tsodikov and S. Garneau-Tsodikova, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 9804–9808 CrossRef CAS PubMed.
- S. Jana and J. K. Deb, Appl. Microbiol. Biotechnol., 2006, 70, 140–150 CrossRef CAS PubMed.
- K. L. Ung, H. M. A. B. Alsarraf, V. Olieric, L. Kremer and M. Blaise, FEBS J., 2019, 286, 4342–4355 CrossRef CAS PubMed.
- D. Centron and P. H. Roy, Antimicrob. Agents Chemother., 2002, 46, 1402–1409 CrossRef CAS.
- M. W. Vetting, C. H. Park, S. S. Hegde, G. A. Jacoby, D. C. Hooper and J. S. Blanchard, Biochemistry, 2008, 47, 9825–9835 CrossRef CAS PubMed.
- D. M. P. D. Oliveira, B. M. Forde, T. J. Kidd, P. N. A. Harris, M. A. Schembri, S. A. Beatson, D. L. Paterson and M. J. Walker, Clin. Microbiol. Rev., 2020, 33, e00181-19 CrossRef PubMed.
- A. Sunada, M. Nakajima, Y. Ikeda, S. Kondo and K. Hotta, J. Antibiot., 1999, 52, 809–814 CrossRef CAS PubMed.
- L. R. Schwocho, C. P. Schaffner, G. H. Miller, R. S. Hare and K. J. Shaw, Antimicrob. Agents Chemother., 1995, 39, 1790–1796 CrossRef CAS PubMed.
- P.-L. Ho, R. C. Wong, S. W. Lo, K.-H. Chow, S. S. Wong and T.-L. Que, J. Med. Microbiol., 2010, 59, 702–707 CrossRef CAS PubMed.
- J. S. Vliegenthart, P. A. G. Ketelaar-Van Gaalen and J. A. M. Van De Klundert, Antimicrob. Agents Chemother., 1991, 35, 892–897 CrossRef CAS.
- H. Heuer, E. Krögerrecklenfort, E. M. H. Wellington, S. Egan, J. D. van Elsas, L. van Overbeek, J. M. Collard, G. Guillaume, A. D. Karagouni, T. L. Nikolakopoulou and K. Smalla, FEMS Microbiol. Ecol., 2002, 42, 289–302 CrossRef CAS PubMed.
- D. R. Call, R. S. Singer, D. Meng, S. L. Broschat, L. H. Orfe, J. M. Anderson, D. R. Herndon, L. S. Kappmeyer, J. B. Daniels and T. E. Besser, Antimicrob. Agents Chemother., 2010, 54, 590–596 CrossRef CAS PubMed.
- P. N. Rather, E. Orosz, K. J. Shaw, R. Hare and G. Miller, J. Bacteriol., 1993, 175, 6492–6498 CrossRef CAS PubMed.
- A. Yoshii, H. Moriyama and T. Fukuhara, Appl. Environ. Microbiol., 2012, 78, 5555–5564 CrossRef CAS PubMed.
- M. López-Cabrera, J. A. Pérez-González, P. Heinzel, W. Piepersberg and A. Jiménez, J. Bacteriol., 1989, 171, 321–328 CrossRef.
- I. Casin, B. Hanau-Berçot, I. Podglajen, H. Vahaboglu and E. Collatz, Antimicrob. Agents Chemother., 2003, 47, 697–703 CrossRef CAS PubMed.
- J. W. Williams and D. B. Northrop, J. Biol. Chem., 1978, 253, 5908–5914 CrossRef CAS PubMed.
- K. Vong and K. Auclair, MedChemComm, 2012, 3, 397–407 RSC.
- C. A. Smith and E. N. Baker, Curr. Drug Targets: Infect. Disord., 2002, 2, 143–160 CAS.
- S. B. Vakulenko and S. Mobashery, Clin. Microbiol. Rev., 2003, 16, 430–450 CrossRef CAS PubMed.
- D. R. Hirsch, G. Cox, M. P. D'Erasmo, T. Shakya, C. Meck, N. Mohd, G. D. Wright and R. P. Murelli, Bioorg. Med. Chem. Lett., 2014, 24, 4943–4947 CrossRef CAS PubMed.
- H. A. Kirst, G. G. Marconi, F. T. Counter, P. W. Ensminger, N. D. Jones, M. O. Chaney, J. E. Toth and N. E. Allen, J. Antibiot., 1982, 35, 1651–1657 CrossRef CAS PubMed.
- N. E. Allen, W. E. Alborn, Jr., J. N. Hobbs Jr. and H. A. Kirst, Antimicrob. Agents Chemother., 1982, 22, 824–831 CrossRef CAS PubMed.
- K. Shi, S. J. Caldwell, D. H. Fong and A. M. Berghuis, Front. Cell. Infect. Microbiol., 2013, 3, 22 Search PubMed.
- A. Kohl, P. Amstutz, P. Parizek, H. K. Binz, C. Briand, G. Capitani, P. Forrer, A. Plückthun and M. G. Grütter, Structure, 2005, 13, 1131–1141 CrossRef CAS PubMed.
- D. M. Daigle, G. A. McKay and G. D. Wright, J. Biol. Chem., 1997, 272, 24755–24758 CrossRef CAS PubMed.
- T. Shakya, P. J. Stogios, N. Waglechner, E. Evdokimova, L. Ejim, J. E. Blanchard, A. G. McArthur, A. Savchenko and G. D. Wright, Chem. Biol., 2011, 18, 1591–1601 CrossRef CAS PubMed.
- N. Leban, E. Kaplan, L. Chaloin, S. Godreuil and C. Lionne, Biochim. Biophys. Acta, Gen. Subj., 2017, 1861, 3464–3473 CrossRef CAS PubMed.
- D. Sehnal, S. Bittrich, M. Deshpande, R. Svobodová, K. Berka, V. Bazgier, S. Velankar, S. K. Burley, J. Koča and A. S. Rose, Nucleic Acids Res., 2021, 49, W431–W437 CrossRef CAS PubMed.
- J. Ehrilich, Q. R. Bartz, R. M. Smith, D. A. Joslyn and P. R. Burkholder, Science, 1947, 106, 417 CrossRef PubMed.
- G. P. Dinos, C. M. Athanassopoulos, D. A. Missiri, P. C. Giannopoulou, I. A. Vlachogiannis, G. E. Papadopoulos, D. Papaioannou and D. L. Kalpaxis, Antibiotics, 2016, 5, 20 CrossRef.
- W. V. Shaw and A. G. W. Leslie, Annu. Rev. Biophys. Biophys. Chem., 1991, 20, 363–386 CrossRef CAS PubMed.
- T. Izard, Protein Sci., 2008, 10, 1508–1513 CrossRef PubMed.
- V. S. Malik and L. C. Vining, Can. J. Microbiol., 1970, 16, 173–179 CrossRef CAS PubMed.
- A. L. Smith, A. L. Erwin, T. Kline, W. C. T. Unrath, K. Nelson, A. Weber and W. N. Howald, Antimicrob. Agents Chemother., 2007, 51, 2820–2829 CrossRef CAS PubMed.
- N. K. Alton and D. Vapnek, Nature, 1979, 282, 864–869 CrossRef CAS PubMed.
- R. Parent and P. H. Roy, J. Bacteriol., 1992, 174, 2891–2897 CrossRef CAS PubMed.
- D. A. Rowe-Magnus and D. Mazel, Int. J. Med. Microbiol., 2002, 292, 115–125 CrossRef CAS.
- H. Tanaka, K. Izaki and H. Takahashi, J. Biochem., 1974, 76, 1009–1019 CAS.
- S. Miyamura, K. Koizumi and Y. Nakagawa, J. Antibiot., 1979, 32, 1217–1218 CrossRef CAS PubMed.
- J. E. Fitton and W. V. Shaw, Biochemistry, 1979, 177, 575–582 CrossRef CAS PubMed.
- C. Kleanthous, P. M. Cullis and W. V. Shaw, Biochemistry, 1985, 24, 5307–5313 CrossRef CAS PubMed.
- T. Biswas, J. L. Houghton, S. Garneau-Tsodikova and O. V. Tsodikov, Protein Sci., 2012, 21, 520–530 CrossRef CAS PubMed.
- A. G. W. Leslie, P. C. E. Moody and W. V. Shaw, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 4133–4137 CrossRef CAS PubMed.
- R. H. Mosher, D. J. Camp, K. Yang, M. P. Brown, W. V. Shaw and L. C. Vining, J. Biol. Chem., 1995, 270, 27000–270006 CrossRef CAS PubMed.
- E. Cundliffe and A. L. Demain, J. Ind. Microbiol. Biotechnol., 2010, 37, 643–672 CrossRef CAS PubMed.
- M. D. Corbett and B. R. Chipko, Antimicrob. Agents Chemother., 1978, 13, 193–196 CrossRef CAS PubMed.
- A. A. Yunis, Annu. Rev. Pharmacol. Toxicol., 1988, 28, 83–100 CrossRef CAS PubMed.
- D. R. P. Guay, Drugs, 1996, 51, 515–536 CrossRef CAS PubMed.
- E. J. MacLaughlin, J. J. Saseen and D. C. Malone, Arch. Fam. Med., 2000, 9, 722–726 CrossRef CAS.
- S. Douthwaite, Clin. Microbiol. Infect., 2008, 7, 11–17 CrossRef.
- T. Golkar, M. Zielinski and A. M. Berghuis, Front. Microbiol., 2018, 9, 1942 CrossRef PubMed.
- M. S. Svetlov, N. Vázquez-Laslop and A. S. Mankin, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 13673–13678 CrossRef CAS.
- F. Schlünzen, R. Zarivach, J. Harms, A. Bashan, A. Tocilj, R. Albrecht, A. Yonath and F. Franceschi, Nature, 2001, 413, 814–821 CrossRef PubMed.
- M. Bergman, S. Huikko, P. Huovinen, P. Paakkari and H. Seppälä, Antimicrob. Agents Chemother., 2006, 50, 3646–3650 CrossRef CAS PubMed.
- R. A. Hussein, M. T. S. Al-Ouqaili and Y. H. Majeed, Saudi J. Biol. Sci., 2022, 29, 513–520 CrossRef CAS PubMed.
- L. Wondrack, M. Massa, B. V. Yang and J. Sutcliffe, Antimicrob. Agents Chemother., 1996, 40, 992–998 CrossRef CAS PubMed.
- A. Nakamura, K. Nakazawa, I. Miyakozawa, S. Mizukoshi, K. Tsurubuchi, M. Nakagawa, K. O'hara and T. Sawai, J. Antibiot., 2000, 53, 516–524 CrossRef CAS PubMed.
- M. Zieliński, J. Park, B. Sleno and A. M. Berghuis, Nat. Commun., 2021, 12, 1–9 CrossRef PubMed.
- E. Cundliffe, Antimicrob. Agents Chemother., 1992, 36, 348–352 CrossRef CAS.
- A. C. Pawlowski, P. J. Stogios, K. Koteva, T. Skarina, E. Evdokimova, A. Savchenko and G. D. Wright, Nat. Commun., 2018, 9, 112 CrossRef PubMed.
- M. Matsuoka, K. Endou, H. Kobayashi, M. Inoue and Y. Nakajima, FEMS Microbiol. Lett., 1998, 167, 221–227 CrossRef CAS.
- A. Nakamura, I. Miyakozawa, K. Nakazawa and O. H. K. T. Sawai, Antimicrob. Agents Chemother., 2000, 44, 3241–3242 CrossRef CAS PubMed.
- D. H. Fong, D. L. Burk, J. Blanchet, A. Y. Yan and A. M. Berghuis, Structure, 2017, 25, 750–761 CrossRef CAS PubMed.
- M. Morar, K. Pengelly, K. Koteva and G. D. Wright, Biochemistry, 2012, 51, 1740–1751 CrossRef CAS PubMed.
- D. Yong, M. A. Toleman, C. G. Giske, H. S. Cho, K. Sundman, K. Lee and T. R. Walsh, Antimicrob. Agents Chemother., 2009, 53, 5046–5054 CrossRef CAS.
- L. Xing, H. Yu, J. Qi, P. Jiang, B. Sun, J. Cui, C. Ou, W. Chang and Q. Hu, PLoS One, 2015, 10, e0131078 CrossRef PubMed.
- A. Gourmelen, M.-H. L. N. Blondelet-Rouault and J.-L. Pernodet, Antimicrob. Agents Chemother., 1998, 42, 2612–2619 CrossRef CAS.
- L. M. Quirós, I. Aguirrezabalaga, C. Olano, C. Méndez and J. A. Salas, Mol. Microbiol., 2002, 28, 1177–1185 CrossRef.
- J. Sasaki, K. Mizoue, S. Morimoto and S. Omura, J. Antibiot., 1996, 49, 1110–1118 CrossRef CAS PubMed.
- O. Chesneau, K. Tsvetkova and P. Courvalin, FEMS Microbiol. Lett., 2007, 269, 317–322 CrossRef CAS PubMed.
- M. Matsuoka, M. Inoue, Y. Endo and Y. Nakajima, FEMS Microbiol. Lett., 2003, 220, 287–293 CrossRef CAS.
- Y. H. Kim, C. J. Cha and C. E. Cerniglia, FEMS Microbiol. Lett., 2002, 210, 239–244 CrossRef CAS PubMed.
- M. Thungapathra, Amita, K. K. Sinha, S. R. Chaudhuri, P. Garg, T. Ramamurthy, G. B. Nair and A. Ghosh, Antimicrob. Agents Chemother., 2002, 46, 2948–2955 CrossRef CAS PubMed.
- B. Epe, P. Woolley and H. Hornig, FEBS Lett., 1987, 213, 443–447 CrossRef CAS.
- I. Chopra and M. Roberts, Microbiol. Mol. Biol. Rev., 2001, 65, 232–260 CrossRef CAS.
- G. G. Zhanel, K. Homenuik, K. Nichol, A. Noreddin, L. Vercaigne, J. Embil, A. Gin, J. A. Karlowsky and D. J. Hoban, Drugs, 2004, 64, 63–88 CrossRef CAS PubMed.
- H. Tucker, M. Wible, A. Gandhi and A. Quintana, Infect. Drug Resist., 2017, 10, 401–417 CrossRef CAS.
- G. G. Zhanel, J. Esquivel, S. Zelenitsky, C. K. Lawrence, H. J. Adam, A. Golden, R. Hink, L. Berry, F. Schweizer, M. A. Zhanel, D. Bay, P. R. S. Lagacé-Wiens, A. J. Walkty, J. P. Lynch III and J. A. Karlowsky, Int. J. Emerg. Med., 2020, 80, 285–313 CrossRef CAS PubMed.
- M. Pioletti, F. Schlünzen, J. Harms, R. Zarivach, M. Glühmann, H. Avila, A. Bashan, H. Bartels, T. Auerbach, C. Jacobi, T. Hartsch, A. Yonath and F. Franceschi, EMBO J., 2001, 20, 1829–1839 CrossRef CAS PubMed.
- D. E. Brodersen, W. M. Clemons, A. P. Carter, R. J. Morgan-Warren, B. T. Wimberly and V. Ramakrishnan, Cell, 2000, 103, 1143–1154 CrossRef CAS.
- M. Thaker, P. Spanogiannopoulos and G. D. Wright, Cell. Mol. Life Sci., 2010, 67, 419–431 CrossRef CAS PubMed.
- F. Nguyen, A. L. Starosta, S. Arenz, D. Sohmen, A. Dönhöfer and D. N. Wilson, Biol. Chem., 2014, 395, 559–575 CrossRef CAS.
- W. Yang, I. F. Moore, K. P. Koteva, D. C. Bareich, D. W. Hughes and G. D. Wright, J. Biol. Chem., 2004, 279, 52346–52352 CrossRef CAS PubMed.
- B. S. Speer, L. Bedzyk and A. A. Salyers, J. Bacteriol., 1991, 173, 176–183 CrossRef CAS PubMed.
- Y. Xu, L. Liu, J. Sun and Y. Feng, Sci. Bull., 2019, 64, 1478–1481 CrossRef.
- T. A. Leski, U. Bangura, D. H. Jimmy, R. Ansumana, S. E. Lizewski, D. A. Stenger, C. RoweTaitt and G. J. Vora, Int. J. Antimicrob. Agents, 2013, 42, 83–86 CrossRef CAS.
- G. Volkers, G. J. Palma, M. S. Weiss, G. D. Wright and W. Hinrichs, FEBS Lett., 2011, 585, 1061–1066 CrossRef CAS PubMed.
- J. Park, A. J. Gasparrini, M. R. Reck, C. T. Symister, J. L. Elliott, J. P. Vogel, T. A. Wencewicz, G. Dantas and N. H. Tolia, Nat. Chem. Biol., 2017, 13, 730–736 CrossRef CAS PubMed.
- G. Volkers, J. M. Damas, G. J. Palm, S. Panjikar, C. M. Soares and W. Hinrichs, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 1758–1767 CrossRef CAS.
- J. L. Markley, L. Fang, A. J. Gasparrini, C. T. Symister, H. Kumar, N. H. Tolia, G. Dantas and T. A. Wencewicz, ACS Infect. Dis., 2019, 5, 618–633 CrossRef CAS PubMed.
- L. Xu, Y. Zhou, S. Niu, Z. Liu, Y. Zou, Y. Yang, H. Feng, D. Liu, X. Niu, X. Deng, Y. Wang and J. Wang, Lancet, 2022, 78, 103943 CAS.
- L. Nonaka and S. Suzuki, Antimicrob. Agents Chemother., 2002, 46, 1550–1552 CrossRef CAS PubMed.
- M. C. Roberts, FEMS Microbiol. Lett., 2005, 245, 195–203 CrossRef CAS.
- R. A. Adams, G. Leon, N. M. Miller, S. P. Reyes, C. H. Thantrong, A. M. Thokkadam, A. S. Lemma, D. M. Sivaloganathan, X. Wan and M. P. Brynildsen, J. Antibiot., 2021, 74, 786–798 CrossRef CAS PubMed.
- E. A. Campbell, N. Korzheva, A. Mustaev, K. Murakami, S. Nair, A. Goldfarb and S. A. Darst, Cell, 2001, 104, 901–912 CrossRef CAS.
- H. G. Floss and T.-W. Yu, Chem. Rev., 2005, 105, 621–632 CrossRef CAS PubMed.
- K. Yazawa, Y. Mikami, A. Maeda, M. Akao, N. Morisaki and S. Iwasaki, Antimicrob. Agents Chemother., 1993, 37, 1313–1317 CrossRef CAS.
- P. Spanogiannopoulos, M. Thaker, K. Koteva, N. Waglechner and G. D. Wright, Antimicrob. Agents Chemother., 2012, 56, 5061–5069 CrossRef CAS PubMed.
- A. C. Pawlowski, W. Wang, K. Koteva, H. A. Barton, A. G. McArthur and G. D. Wright, Nat. Commun., 2016, 7, 1–10 Search PubMed.
- J. Baysarowich, K. Koteva, D. W. Hughes, L. Ejim, E. Griffiths, K. Zhang, M. Junop and G. D. Wright, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 4886–4891 CrossRef CAS.
- E. R. Dabbs, FEMS Microbiol. Lett., 1987, 44, 395–399 CrossRef CAS.
- E. R. Dabbs, K. Yazawa, Y. Tanaka, Y. Mikami, M. M. Miyaji, S. J. Andersen, N. Morisaki, S. Iwasaki, O. Shida and H. Takagi, J. Antibiot., 1995, 48, 815–819 CrossRef CAS.
- S. J. Andersen, S. Quan, B. Gowan and E. R. Dabbs, Antimicrob. Agents Chemother., 1997, 41, 218–221 CrossRef CAS.
- P. J. Stogios, G. Cox, P. Spanogiannopoulos, M. C. Pillon, N. Waglechner, T. Skarina, K. Koteva, A. Guarné, A. Savchenko and G. D. Wright, Nat. Commun., 2016, 7, 1–12 Search PubMed.
- X. Qia, W. Lin, M. Ma, C. Wang, Y. He, N. He, J. Gao, H. Zhou, Y. Xiao, Y. Wang and P. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2015, 113, 3803–3808 CrossRef.
- L. Liu, H. Abdelwahab, J. S. M. D. Campo, R. Mehra-Chaudhary, P. Sobrado and J. J. Tanner, J. Biol. Chem., 2016, 291, 21553–21562 CrossRef CAS PubMed.
- K. Koteva, G. Cox, J. K. Kelso, M. D. Surette, H. L. Zubyk, L. Ejim, P. Stogios, A. Savchenko, D. Sorensen and G. D. Wright, Cell Chem. Biol., 2018, 25, 403–412 CrossRef CAS PubMed.
- Y. Hoshino, S. Fujii, H. Shinonaga, K. Arai, F. Saito, T. Fukai, H. Satoh, Y. Miyazaki and J. Ishikawa, J. Antibiot., 2010, 63, 23–28 CrossRef CAS PubMed.
- B. Lüscher, M. Bütepage, L. Eckei, S. Krieg, P. Verheugd and B. H. Shilton, Chem. Rev., 2018, 118, 1092–1136 CrossRef.
- C. Tribuddharat and M. Fennewald, Antimicrob. Agents Chemother., 1999, 43, 960–962 CrossRef CAS PubMed.
- G. Chowdhury, G. P. Pazhani, G. B. Nair, A. Ghosh and T. Ramamurthy, Int. J. Antimicrob. Agents, 2011, 38, 169–173 CAS.
- E. Lourenco Da Fonseca, F. Dos Santos Freitas, J. C. De Amorim and A. C. P. Vicente, Antimicrob. Agents Chemother., 2008, 52, 1865–1867 CrossRef CAS PubMed.
- Ø. Samuelsen, M. A. Toleman, T. R. Walsh, C. G. Giske, A. Sundsfjord, J. Rydberg, T. M. Leegaard, M. Walder, A. Lia, T. E. Ranheim, Y. Rajendra and N. O. Hermansen, Antimicrob. Agents Chemother., 2010, 54, 346–352 CrossRef PubMed.
- A. C. S. Almeida, F. L. S. Cavalcanti, W. M. B. Martins, M. A. Vilela, A. C. Gales, M. A. Morais Junior and M. M. C. Morais, Antimicrob. Agents Chemother., 2013, 57, 4077–4078 CrossRef CAS PubMed.
- M. Zheng and T. J. Lupoli, ACS Infect. Dis., 2021, 7, 2604–2611 CrossRef CAS PubMed.
- W. McDermott, J. Infect. Dis., 1969, 119, 678–693 CrossRef CAS PubMed.
- G. S. Timmins, Mol. Microbiol., 2006, 62, 1220–1227 CrossRef CAS.
- A. N. Unissa, S. Subbian, L. E. Hanna and N. Selvakumar, Infect., Genet. Evol., 2016, 45, 474–492 CrossRef CAS.
- M. Payton, R. Auty, R. Delgoda, M. Everett and E. Sim, J. Bacteriol., 1999, 181, 1343–1347 CrossRef CAS PubMed.
- A. M. Upton, A. Mushtaq, T. C. Victor, S. L. Sampson, J. Sandy, D.-M. Smith, P. V. V. Helden and E. Sim, Mol. Microbiol., 2008, 42, 309–317 CrossRef PubMed.
- J. Sandy, S. Holton, E. Fullam, E. Sim and M. Noble, Protein Sci., 2005, 14, 775–782 CrossRef CAS PubMed.
- I. M. Westwood, S. Bhakta, A. J. Russell, E. Fullam, M. C. Anderton, A. Kawamura, A. W. Mulvaney, R. J. Vickers, V. Bhowruth, G. S. Besra, A. Lalvani, S. G. Davies and E. Sim, Protein Cell, 2010, 1, 82–95 CrossRef CAS PubMed.
- N. Agre, N. Tawari, A. Maitra, A. Gupta, T. Munshi, M. Degani and S. Bhakta, Antibiotics, 2020, 9, 368 CrossRef CAS PubMed.
- A. Abuhammad, E. Fullam, S. Bhakta, A. J. Russell, G. M. Morris, P. W. Finn and E. Sim, Molecules, 2014, 19, 16274–16290 CrossRef PubMed.
- V. E. Madikane, S. Bhakta, A. J. Russell, W. E. Campbell, T. D. W. Claridge, B. G. Elisha, S. G. Davies, P. Smith and E. Sim, Bioorg. Med. Chem., 2007, 15, 3579–3586 CrossRef CAS PubMed.
- H. Hoeksema, B. Bannister, R. D. Birkenmeyer, F. Kagan, B. J. Magerlein, F. A. MacKellar, W. Schroeder, G. Slomp and R. R. Herr, J. Am. Chem. Soc., 1964, 86, 4223–4224 CrossRef CAS.
- E. C. Böttger, B. Springer, T. Prammananan, Y. Kidan and P. Sander, EMBO Rep., 2001, 2, 318–323 CrossRef.
- M. J. Mitcheltree, PhD thesis, Harvard University, 2018 Search PubMed.
- E. Petinaki, V. Guérin-Faublée, V. Pichereau, C. Villers, A. Achard, B. Malbruny and R. Leclercq, Antimicrob. Agents Chemother., 2008, 52, 626–630 CrossRef CAS PubMed.
- A. Brisson-Noël, P. Delrieu, D. Samain and P. Courvalin, J. Biol. Chem., 1988, 263, 15880–15887 CrossRef.
- M. Morar, K. Bhullar, D. Hughes, M. Junop and G. Wright, Structure, 2009, 17, 1649–1659 CrossRef CAS PubMed.
- A. M. Egorov, M. M. Ulyashova and M. Y. Rubtsova, Acta Naturae, 2018, 10, 33–48 CrossRef CAS PubMed.
- G. De Pascale and G. D. Wright, ChemBioChem, 2010, 11, 1325–1334 CrossRef CAS.
- M. E. Falagas, K. P. Giannopoulou, G. N. Kokolakis and P. I. Rafailidis, Clin. Infect. Dis., 2008, 46, 1069–1077 CrossRef PubMed.
- M. Diez-Aguilar and R. Canton, Rev. Esp. Quimioter., 2019, 32, 8–18 Search PubMed.
- F. M. Kahan, J. S. Kahan, P. J. Cassidy and H. Kropp, Ann. N. Y. Acad. Sci., 1974, 235, 364–386 CrossRef CAS PubMed.
- S. Pakhomova, C. L. Rife, R. N. Armstrong and M. E. Newcomer, Protein Sci., 2009, 13, 1260–1265 CrossRef.
- E. H. Klontz, A. D. Tomich, S. Günther, J. A. Lemkul, D. Deredge, Z. Silverstein, J. F. Shaw, C. McElheny, Y. Doi and P. L. Wintrode, Antimicrob. Agents Chemother., 2017, 61, e01572-17 CrossRef PubMed.
- Y. Y. Huijong Han, S. H. Olesen, A. Becker, S. Betzi and E. Schönbrunn, Biochemistry, 2010, 49, 4276–4282 CrossRef.
- Y. Li, B. Zheng, Y. Li, S. Zhu, F. Xue and J. Liu, PLoS One, 2015, 10, e0135269 CrossRef PubMed.
- H. Xu, V. Miao, W. Kwong, R. Xia and J. Davies, Lett. Appl. Microbiol., 2011, 52, 427–429 CrossRef CAS PubMed.
- H. Alrowais, C. L. McElheny, C. N. Spychala, S. Sastry, Q. Guo, A. A. Butt and Y. Doi, Emerging Infect. Dis., 2015, 21, 2045 CrossRef CAS PubMed.
- G. Nakamura, J.-i. Wachino, N. Sato, K. Kimura, K. Yamada, W. Jin, K. Shibayama, T. Yagi, K. Kawamura and Y. Arakawa, J. Clin. Microbiol., 2014, 52, 3175–3179 CrossRef CAS PubMed.
- Y. Ma, X. Xu, Q. Guo, P. Wang, W. Wang and M. Wang, Lett. Appl. Microbiol., 2015, 60, 259–264 CrossRef CAS PubMed.
- Q. Guo, A. D. Tomich, C. L. McElheny, V. S. Cooper, N. Stoesser, M. Wang, N. Sluis-Cremer and Y. Doi, J. Antimicrob. Chemother., 2016, 71, 2460–2465 CrossRef CAS PubMed.
- M. A. Rehman, X. Yin, M. G. Persaud-Lachhman and M. S. Diarra, Antimicrob. Agents Chemother., 2017, 61, e00410–e00417 CrossRef CAS PubMed.
- R. E. Rigsby, C. L. Rife, K. L. Fillgrove, M. E. Newcomer and R. N. Armstrong, Biochemistry, 2004, 43, 13666–13673 CrossRef CAS PubMed.
- A. D. Tomich, E. H. Klontz, D. Deredge, J. P. Barnard, C. L. McElheny, M. L. Eshbach, O. A. Weisz, P. Wintrode, Y. Doi, E. J. Sundberg and N. Sluis-Cremer, Antimicrob. Agents Chemother., 2019, 63, e01524-18 CrossRef PubMed.
- D. T. Kumar, P. Lavanya, C. G. P. Doss, I. A. Tayubi, R. N. Kumar, I. F. Yesurajan, R. Siva and V. Balaji, J. Cell. Biochem., 2017, 118, 4088–4094 CrossRef.
- M. K. Thompson, M. Goodman, M. Keithly, N. Hammer, P. Cook, K. Jagessar, J. Harp, E. Skaar and R. N. Armstrong, Biochemistry, 2014, 106, 45a Search PubMed.
- M. Cao, B. A. Bernat, Z. Wang, R. N. Armstrong and J. D. Helmann, J. Bacteriol., 2000, 183, 2380–2383 CrossRef PubMed.
- T. D. Read, S. N. Peterson, N. Tourasse, L. W. Baillie, I. T. Paulsen, K. E. Nelson, H. Tettelin, D. E. Fouts, J. A. Eisen and S. R. Gill, Nature, 2003, 423, 81–86 CrossRef CAS PubMed.
- X. Xu, C. Chen, D. Lin, Q. Guo, F. Hu, D. Zhu, G. Li and M. Wang, PLoS One, 2013, 8, e78106 CrossRef CAS PubMed.
- M. K. Thompson, M. E. Keithly, G. A. Sulikowski and R. N. Armstrong, Perspect. Sci., 2015, 4, 17–23 CrossRef.
- A. P. Lamers, M. E. Keithly, K. Kim, P. D. Cook, D. F. Stec, K. M. Hines, G. A. Sulikowski and R. N. Armstrong, Org. Lett., 2012, 14, 5207–5209 CrossRef CAS PubMed.
- A. D. Lewis, T. M. Riedel, M. B. A. Kesler, M. E. Varney and T. E. Long, J. Antibiot., 2022, 75, 146–154 CrossRef CAS PubMed.
- K. L. Fillgrove, S. Pakhomova, M. R. Schaab, M. E. Newcomer and R. N. Armstrong, Biochemistry, 2007, 46, 8110–8120 CrossRef CAS PubMed.
- F. Zhang, T. Zhai, S. Haider, Y. Liu and Z. J. Huang, J. Am. Chem. Soc., 2020, 5, 7537–7544 CAS.
Footnote |
| † These two authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |