
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFragment optimization and elaboration strategies – the discovery of two lead series of PRMT5/MTA inhibitors from five fragment hits†
Christopher R.
Smith
 *a,
Svitlana
Kulyk
*a,
Misbha Ud Din
Ahmad
b,
Valentina
Arkhipova
b,
James G.
Christensen
a,
Robin J.
Gunn
a,
Anthony
Ivetac
a,
John M.
Ketcham
a,
Jon
Kuehler
a,
J. David
Lawson
a,
Nicole C.
Thomas
a,
Xiaolun
Wang
a and
Matthew A.
Marx
a
*a,
Svitlana
Kulyk
*a,
Misbha Ud Din
Ahmad
b,
Valentina
Arkhipova
b,
James G.
Christensen
a,
Robin J.
Gunn
a,
Anthony
Ivetac
a,
John M.
Ketcham
a,
Jon
Kuehler
a,
J. David
Lawson
a,
Nicole C.
Thomas
a,
Xiaolun
Wang
a and
Matthew A.
Marx
a
aMirati Therapeutics, San Diego, California 92121, USA. E-mail: smithc@mirati.com; kulyks@mirati.com
bZoBio BV, J. H. Oortweg 19, 2333 CH Leiden, Netherlands
First published on 27th September 2022
Abstract
Here we describe the early stages of a fragment-based lead discovery (FBLD) project for a recently elucidated synthetic lethal target, the PRMT5/MTA complex, for the treatment of MTAP-deleted cancers. Starting with five fragment/PRMT5/MTA X-ray co-crystal structures, we employed a two-phase fragment elaboration process encompassing optimization of fragment hits and subsequent fragment growth to increase potency, assess synthetic tractability, and enable structure-based drug design. Two lead series were identified, one of which led to the discovery of the clinical candidate MRTX1719.
Introduction
Protein arginine methyl transferase 5 (PRMT5) is an essential class II PRMT that catalyzes the symmetric transfer of two methyl groups to the arginine residues of substrate proteins using S-adenosyl-L-methionine (SAM) as the methyl donor.1 In 2016, several independent research groups reported that loss of the tumor suppressor gene CDKN2A is commonly accompanied by co-deletion of the proximal gene encoding methylthioadenosine phosphorylase (MTAP).2–4 In turn, MTAP deletion causes the accumulation of the cellular metabolite methylthioadenosine (MTA). MTA is an intermediate of the methionine salvage pathway and weakly inhibits PRMT5, competing with SAM to bind in the co-factor binding site to form the PRMT5/MTA complex.4 It was postulated that a compound which binds to and stabilizes the PRMT5/MTA complex would inhibit PRMT5 activity in MTAP-null cancers cells while preserving PRMT5 activity in healthy cells and could potentially increase the therapeutic safety index in CDKN2A/MTAP co-deleted cancer cells relative to first generation PRMT5 inhibitors that are not selective binders of the PRMT5/MTA complex. Recently, we reported the discovery of clinical candidate MRTX1719, a small molecule that potently binds to the PRMT5/MTA complex, and inhibits PRMT5 selectively in MTAP-deleted HCT116 cells.5 Two other groups have published patent applications6,7 that also appear to target the PRMT5/MTA complex, with MRTX1719 and literature examples shown in Fig. 1.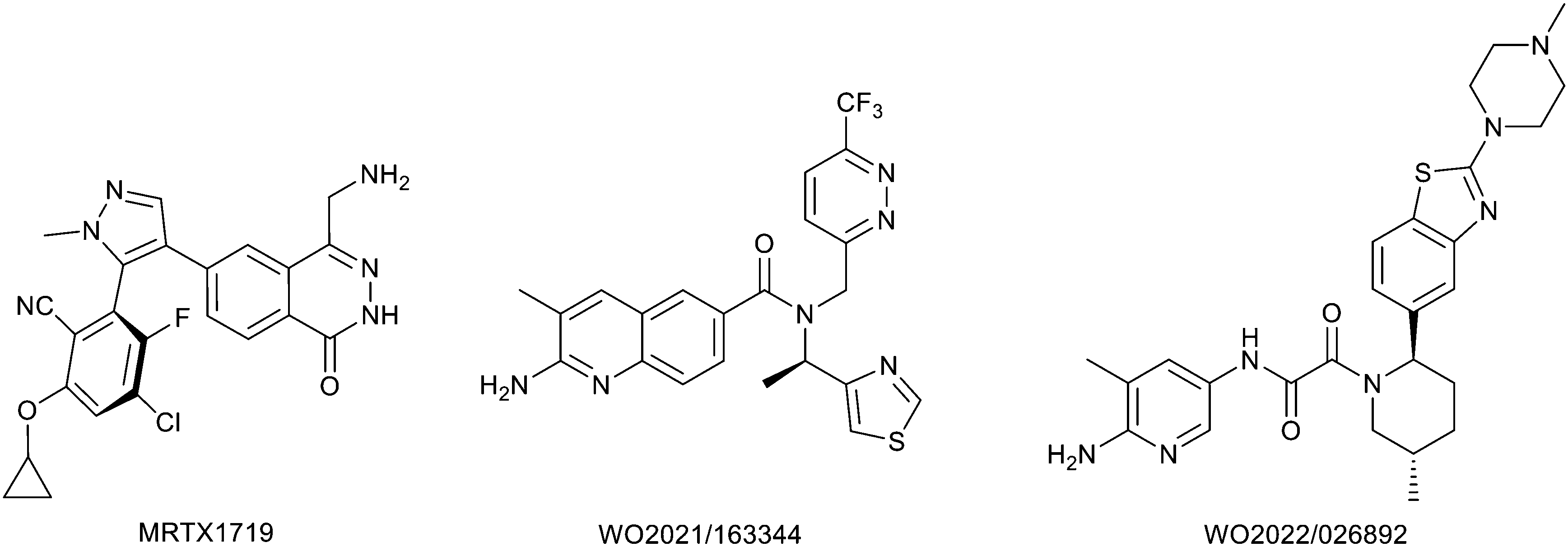 | ||
| Fig. 1 MRTX1719 and representative compound structures of PRMT5/MTA inhibitors from recently published patent applications. | ||
Herein we describe a fragment-based lead discovery (FBLD) program focused specifically on fragment elaboration strategies starting with five fragment hits (1–5, Table 1) supported by structure-based drug design. We outline a two-phase process. Phase 1: optimization of the fragment hit prior to fragment growth strategies, and phase 2: fragment growth to increase potency supported by the development of versatile synthetic chemistry and, exploration of growth vectors aided by structure-based drug design (SBDD). In some instances, phase 1 was skipped and efforts were immediately focused on phase 2. The five fragment hits described in Table 1 led to the discovery of two lead series.

FBLD is a hit finding and lead generation approach where libraries of fragment compounds are screened at high concentration, typically in the range 100 μM to 1 mM.8,9 Libraries typically range in size from 350 to 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compounds and a fragment is typically defined as a compound with between 10 and 17 heavy atoms.10 Due to the small size of the fragments, affinities are weak, thus sensitive screening methods are employed for detection. As a result of testing fragments at high concentration careful attention needs to be paid to factors such as compound aggregation, compound solubility, and pan-assay interference compounds (PAINS).11 Since the first disclosure of an FBLD project in 1996,12 commercially available, well curated fragment libraries are widely available. When we initiated our PRMT5/MTA inhibitor project, there was no evidence in the literature of compounds that were characterized to bind to the PRMT5/MTA complex. We concluded that an FBLD screen of a commercially available fragment library against PRMT5/MTA would be a rapid and cost-effective method to identify hits. Our decision to select a fragment screen rather than a high throughput screen (HTS) or a DNA encoded library (DEL) screen was based on timelines. We did not have immediate access to HTS or DEL, but we had the capability to rapidly run a fragment screen. The details of the fragment screen performed against PRMT5/MTA were previously described.5 To summarize, a commercially available fragment library was screened using surface plasma resonance (SPR) with the PRMT5 protein immobilized on the surface. Formation of the PRMT5/MTA complex was achieved by adding MTA (20 μM) to the running buffer. 24 hits with saturable KDs ≤ 500 μM were identified. The SPR assay was designed to detect affinity for the PRMT5/MTA complex and the affinity of the compounds for PRMT5 alone was not measured. From these 24 hits, five X-ray co-crystal structures were obtained with fragments bound to the PRMT5/MTA complex. The compound structures, SPR data, LE,13 LLE,14 clogP, X-ray co-crystal structures, and PDB codes are shown in Table 1.
000 compounds and a fragment is typically defined as a compound with between 10 and 17 heavy atoms.10 Due to the small size of the fragments, affinities are weak, thus sensitive screening methods are employed for detection. As a result of testing fragments at high concentration careful attention needs to be paid to factors such as compound aggregation, compound solubility, and pan-assay interference compounds (PAINS).11 Since the first disclosure of an FBLD project in 1996,12 commercially available, well curated fragment libraries are widely available. When we initiated our PRMT5/MTA inhibitor project, there was no evidence in the literature of compounds that were characterized to bind to the PRMT5/MTA complex. We concluded that an FBLD screen of a commercially available fragment library against PRMT5/MTA would be a rapid and cost-effective method to identify hits. Our decision to select a fragment screen rather than a high throughput screen (HTS) or a DNA encoded library (DEL) screen was based on timelines. We did not have immediate access to HTS or DEL, but we had the capability to rapidly run a fragment screen. The details of the fragment screen performed against PRMT5/MTA were previously described.5 To summarize, a commercially available fragment library was screened using surface plasma resonance (SPR) with the PRMT5 protein immobilized on the surface. Formation of the PRMT5/MTA complex was achieved by adding MTA (20 μM) to the running buffer. 24 hits with saturable KDs ≤ 500 μM were identified. The SPR assay was designed to detect affinity for the PRMT5/MTA complex and the affinity of the compounds for PRMT5 alone was not measured. From these 24 hits, five X-ray co-crystal structures were obtained with fragments bound to the PRMT5/MTA complex. The compound structures, SPR data, LE,13 LLE,14 clogP, X-ray co-crystal structures, and PDB codes are shown in Table 1.
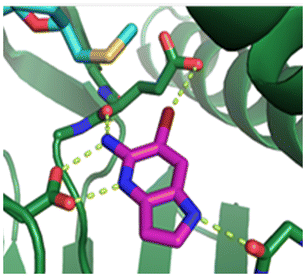
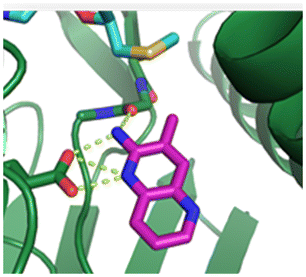
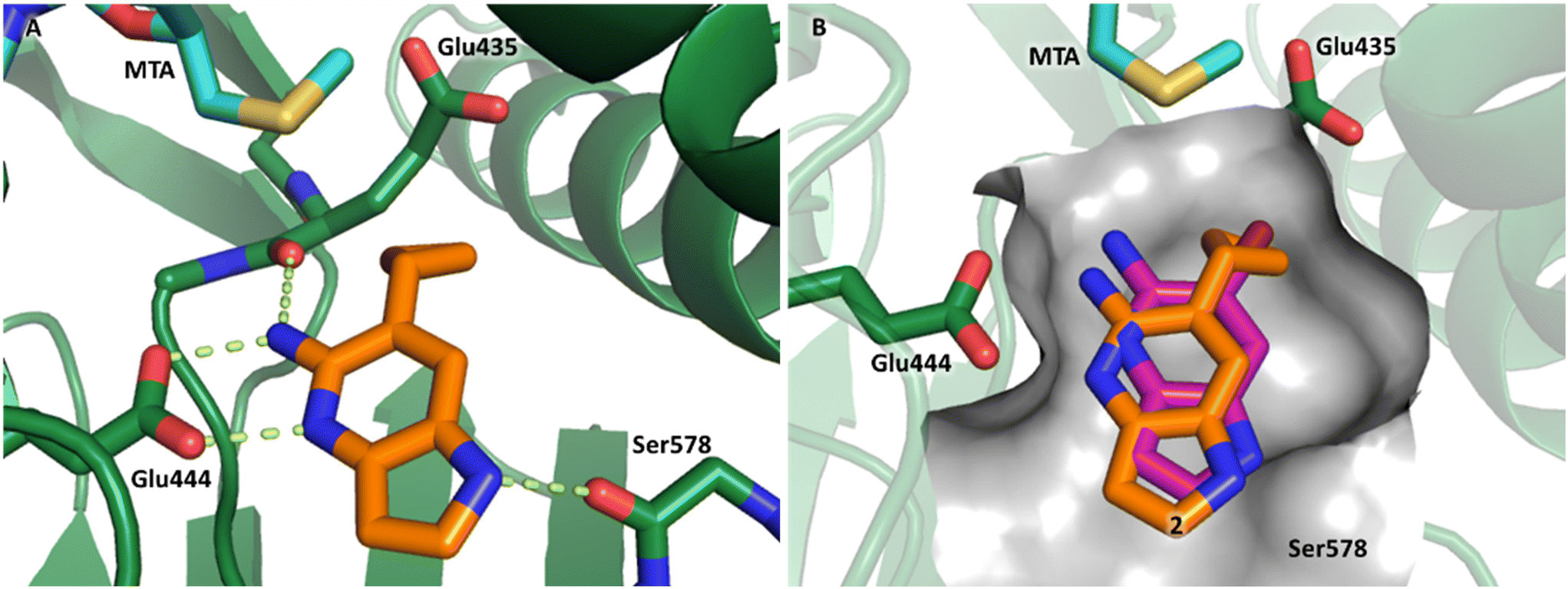
Fragment hit 1, a 1H-pyrrolo[3,2-b]pyridin-5-amine scaffold, was the most potent and efficient binder identified in the screen (PRMT5/MTA KD = 0.74 μM, LE = 0.78, LLE = 4.9). The X-ray co-crystal structure of fragment hit 1 with PRMT5/MTA (PDB: 8CSG) is shown in Fig. 2A. 1 binds in the PRMT5 substrate binding site with a series of specific interactions with the protein and MTA. The productive interactions with the protein include an H-bond between the pyrrolo N–H and the backbone carbonyl of Ser578, a bifurcated H-bond between the –NH2 substituent and the backbone carbonyl of Glu435 and the side chain of Glu444, an ionic interaction between the protonated pyridinyl nitrogen and Glu444 and a halogen bond between the bromo substituent and the Glu435 side chain. Productive van der Waals interactions between MTA and 1 include the sulfur atom interacting with the –NH2 and bromo substituents. The MTA methyl also makes a productive bifurcated van der Waals interaction with the bromo substituent of 1 and the CD atom of Glu435. Fragment hit 1, along with the fragments and analogs described herein, make π-stacking interactions with Phe327 and Trp579 (Fig. 2B).
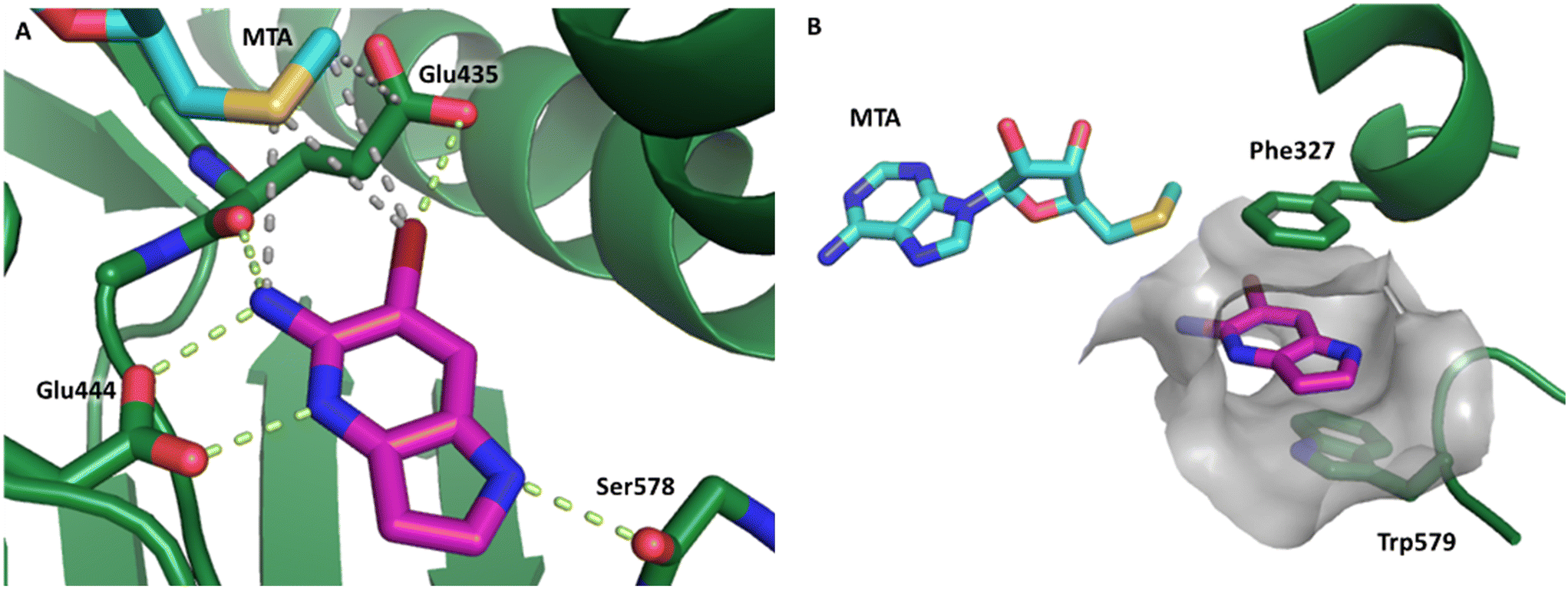 | ||
| Fig. 2 A) The X-ray co-crystal structure of fragment hit 1 bound to PRMT5/MTA (PDB: 8CSG) showing H-bond interactions with Glu444, Glu435 and Ser578, halogen bond interaction with Glu435 and van der Waals interactions between MTA, 1 and Glu435 highlighted in grey. B) A cutaway visual to illuminate the proximity of MTA to 1 and the π-stacking interaction between the bicyclic scaffold and the side chains of Phe327 and Trp579. | ||
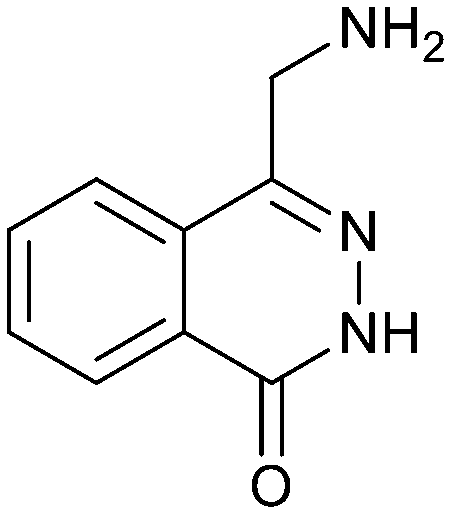
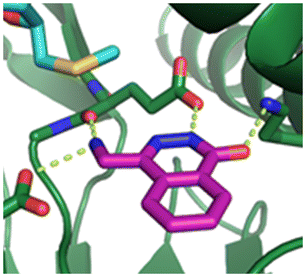
Fragment hit 2, a 4-(aminomethyl)phthalazine-1(2H)-one scaffold, was the second most potent binder and despite the 38-fold lower potency compared to 1, the LLEs for 1 and 2 were identical due to the lower lipophilicity of 2 (PRMT5/MTA KD = 10.2 μM; LE = 0.54, LLE = 4.9 and clogP = 0.1). The X-ray co-crystal structure of fragment hit 2 with PRMT5/MTA (Fig. 3; PDB: 7S0U) was previously reported.5 In addition to the previously described H-bond interactions the van der Waal interactions between the sulfur atom of MTA and the primary amino group of 2 and the MTA methyl with the CD atom of Glu435 are highlighted in Fig. 3.
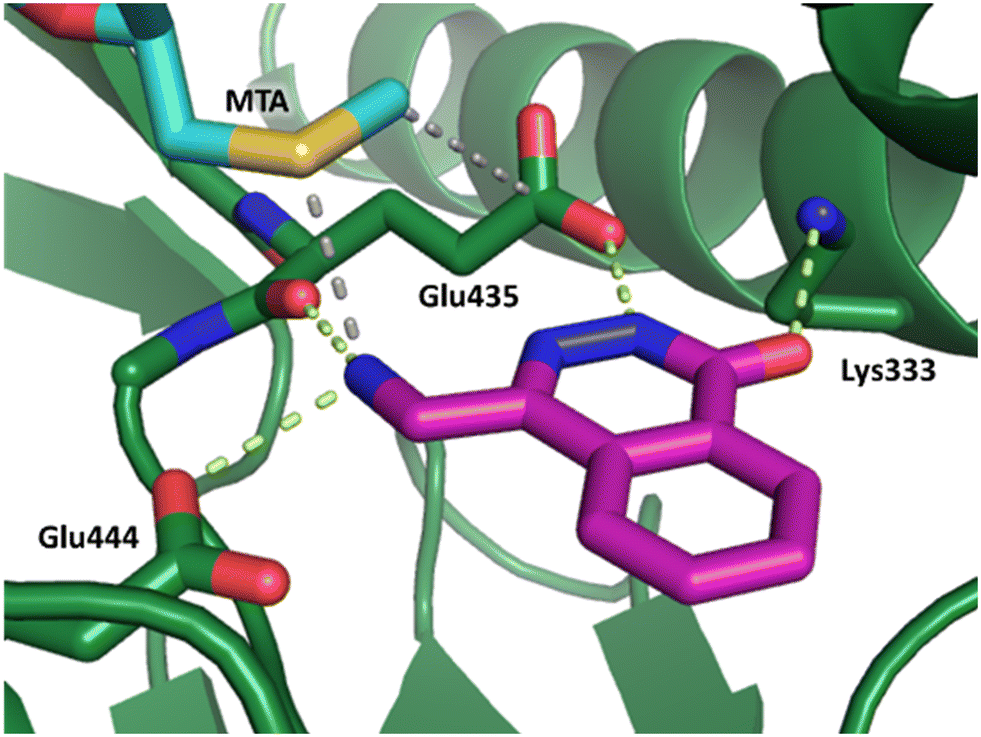 | ||
| Fig. 3 X-ray co-crystal structure of fragment hit 2 bound to PRMT5/MTA (PDB: 7S0U) showing H-bond interactions with Glu444, Glu435 and Lys333 and van der Waals interactions between MTA, 2 and Glu435 highlighted in grey. | ||
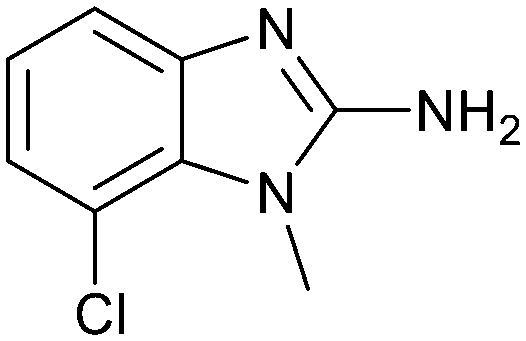
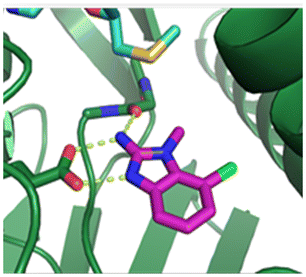
The binding potency of fragment hit 3, a 1-methyl-1H-benzo[d]imidazol-2-amine scaffold, was almost identical to 2 (PRMT5/MTA KD = 12.0 μM). However, due to its higher lipophilicity (clogP = 1.7) the LLE was lower compared to 2 (LLE = 3.2). The X-ray co-crystal structure of 3 with PRMT5/MTA (PDB: 8CTB) is shown in Fig. 4. The 7-chloro-1-methyl-1H-benzo[d]imidazol-2-amine fragment binds in the PRMT5 substrate binding site with a series of specific interactions closely related to fragment hit 1. These include a bifurcated H-bond between the –NH2 substituent and the backbone carbonyl of Glu435 and the side chain of Glu444, and an ionic interaction between the protonated imidazo nitrogen and the Glu444 sidechain. The sulfur atom of MTA makes a productive van der Waals interaction with the methyl and –NH2 substituent of 3. There is a potentially unfavorable interaction between the sigma hole on the chlorine atom and the Lys333 sidechain. This observation highlighted a possible opportunity to optimize the fragment prior to initiating fragment growing strategies.
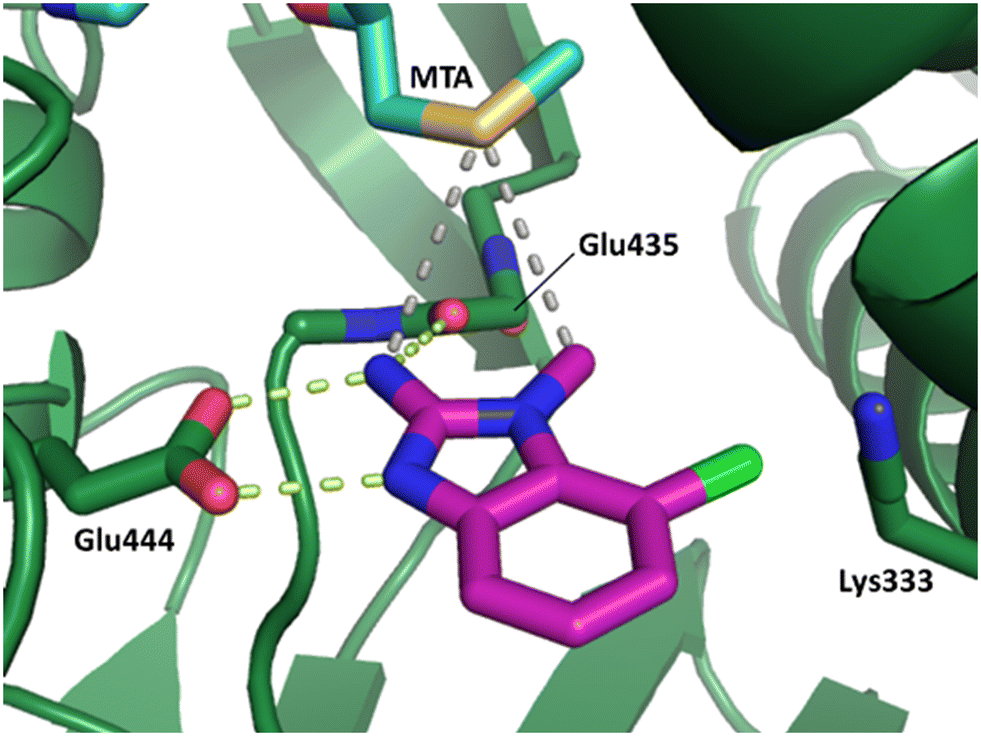 | ||
| Fig. 4 X-ray co-crystal structure of fragment hit 3 bound to PRMT5/MTA (PDB: 8CTB) showing H-bond interactions with Glu444 and Glu435, van der Waals interaction between MTA and 3 in highlighted in grey and a potentially repulsive interaction with Lys333. | ||
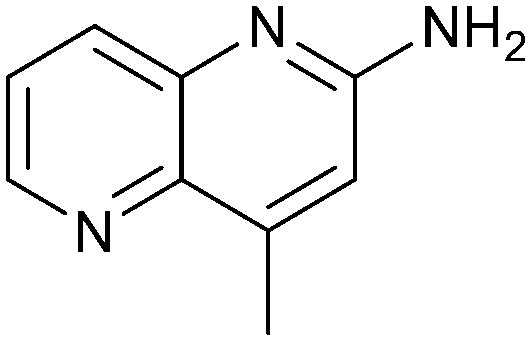
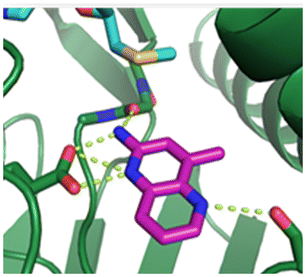
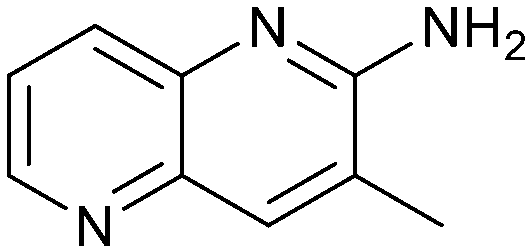
Fragment hits 4 and 5, a 1,5-naphthyridin-2-amine scaffold, are closely related structurally and have similar binding potencies (PRMT5/MTA KD = 62.0 μM and 53.0 μM respectively) and their LLE values (LLE = 3.1 and 3.2 respectively) are almost identical to 3. The X-ray co-crystal structures of 4 and 5 with PRMT5/MTA (PDB: 7UYF and PDB: 7UY1) are shown in Fig. 5. The binding modes and interactions of 4 and 5 are closely related to those observed for fragment hits 1 and 3, with one notable difference—the Ser578 side chain in the fragment hit 4 X-ray co-crystal structure makes a productive H-bond interaction with the 5-naphthyridine nitrogen lone pair (Fig. 5A). Surprisingly, the Ser578 side chain and backbone loop rotate away from the fragment in the PRMT5/MTA X-ray co-crystal structure with 5 and does not make the same Ser578 H-bond interaction as 4 (Fig. 5B). The extent of the side chain and loop movements are visualized in Fig. 5C. Given the high similarity of 4 and 5 it was not clear what was causing the loop and sidechain movement between the two structures.
 | ||
| Fig. 5 A) X-ray co-crystal structure of fragment hit 4 bound to PRMT5/MTA (PDB: 7UYF) showing H-bond interactions with Glu444, Glu435 and Ser578. Van der Waals interactions between MTA and 4 are highlighted in grey. B) X-ray co-crystal structure of fragment hit 5 bound to PRMT5/MTA (PDB: 7UY1). Interactions are closely related to 4 however H-bond interaction with sidechain Ser578 not observed. C) Overlay of fragment hits 4 and 5 showing the movement of Ser578 loop and sidechains. | ||
The output of the fragment screen revealed several important pharmacophoric features of the PRMT5/MTA complex. First, an interconnected H-bond network between the Glu444 side chain, the Glu435 backbone carbonyl and the ligand was common to each fragment hit. In addition, each fragment hit made productive van der Waals interactions with MTA. Second, a “2-aminopyridinyl-like” motif comprised of a 2-amino substituent ortho to an aromatic nitrogen lone pair, is a pharmacophoric feature common to fragments 1, 3, 4 and 5. Third, fragment 2 is a unique pharmacophore that creates the key H-bond network with a 4-(aminomethyl)phthalazine-1(2H)-one motif. A series of other productive protein/ligand interactions were also discovered from the screen. For example, fragment hit 1 picked up an H-bond interaction with the Ser578 backbone carbonyl, fragment hit 2 picked up an H-bond interaction with the Lys333 side chain and fragment hit 4 picked up an H-bond interaction with the side chain of Ser578.
Fragment elaboration
As described above, the hits can be divided into two classes of pharmacophores: the “2-aminopyridinyl-like” pharmacophore class, represented by fragment hits 1, 3, 4 and 5 and the 4-(aminomethyl)phthalazine-1(2H)-one pharmacophore class represented by fragment hit 2. We elected to invest in the exploration of both pharmacophore classes. To explore the SAR of the “2-aminopyridinyl-like” pharmacophore we chose to combine hits 4 and 5 together due to their similarity and elaborate fragments 1, 3 and 4 concurrently. The basis for the decision to explore the SAR of three separate representatives of the “2-aminopyridinyl-like” class was supported by the observation that, despite the similarity of 1, 3 and 4, there were significant differences in the geometries of their growth vectors. We speculated that these differences could lead to divergent SAR. The three divergent growth vector geometries for 1, 3 and 4 are depicted in Fig. 6.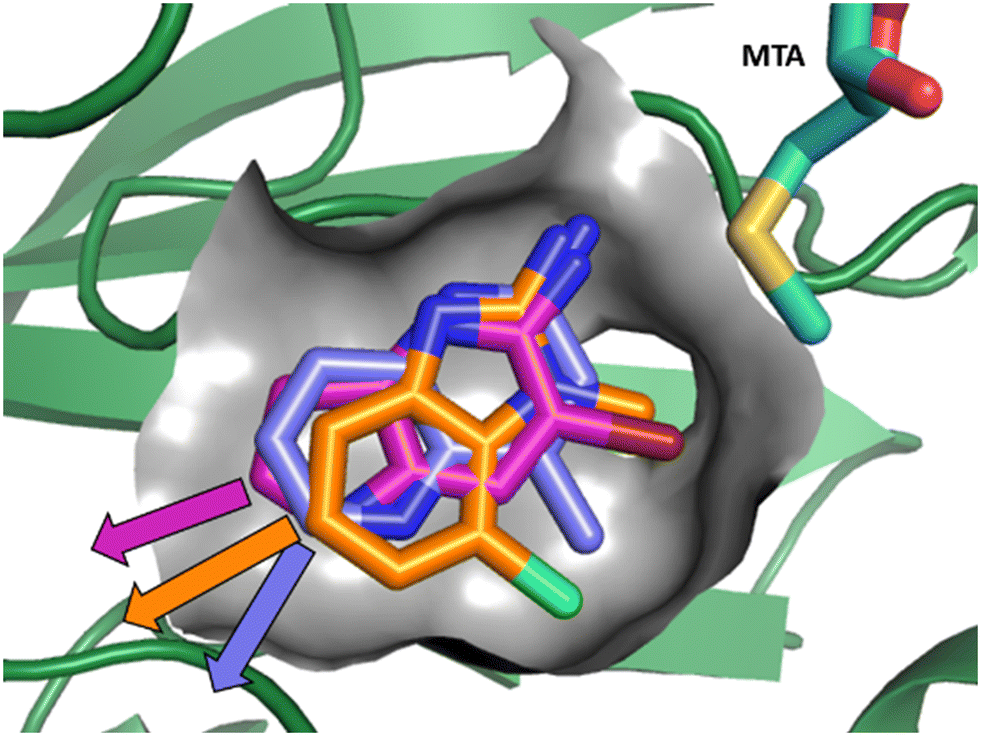 | ||
| Fig. 6 Overlay of fragments 1, 3 and 4 with the divergent growth vectors depicted by magenta, orange and purple arrows, respectively. | ||
We developed several key questions to investigate in the first round of fragment elaboration. First, could binding potency be improved? And, if so, was the potency increase due to adding lipophilicity—the hydrophobic effect,15 or was the increase in potency driven by the ligand making additional productive interactions with the protein? ΔLLE (ΔLLE = LLEanalog − LLEparent) is a useful analysis tool to tease this question apart.14 A positive ΔLLE suggests the analog added favorable interactions with the protein, and a negative ΔLLE suggests the loss of favorable interactions and/or the creation of unfavorable interactions or clashes with the protein. A zero ΔLLE suggests the change in potency is lipophilicity driven. Second, could tractable synthetic chemistry be developed to explore the growth vectors of interest?16 And third, could one or more additional X-ray co-crystal structures be obtained with analogs of the hit to further enable SBDD? And, if so, was the fragment binding mode and the conformation of the protein in the binding pocket reproducible? With data in hand addressing these questions, the goal was to then decide which series to elect as a lead series.
Fragment hit 1 – series 1
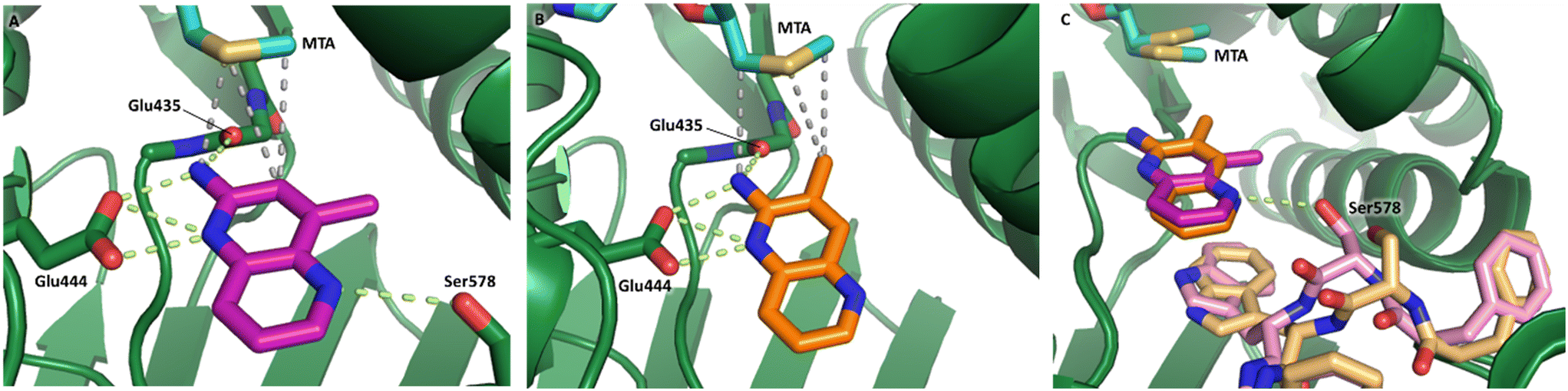
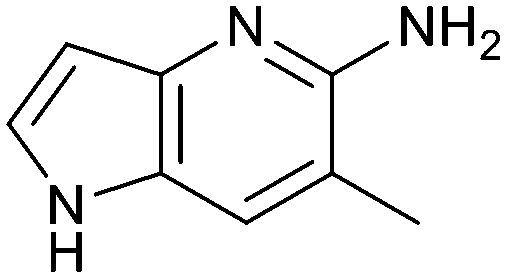
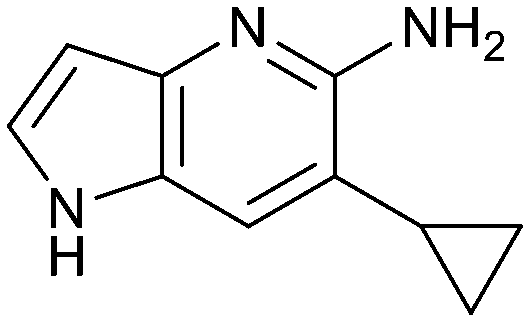
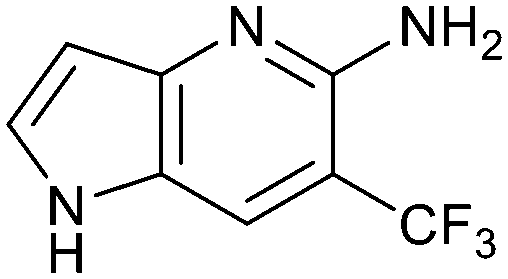
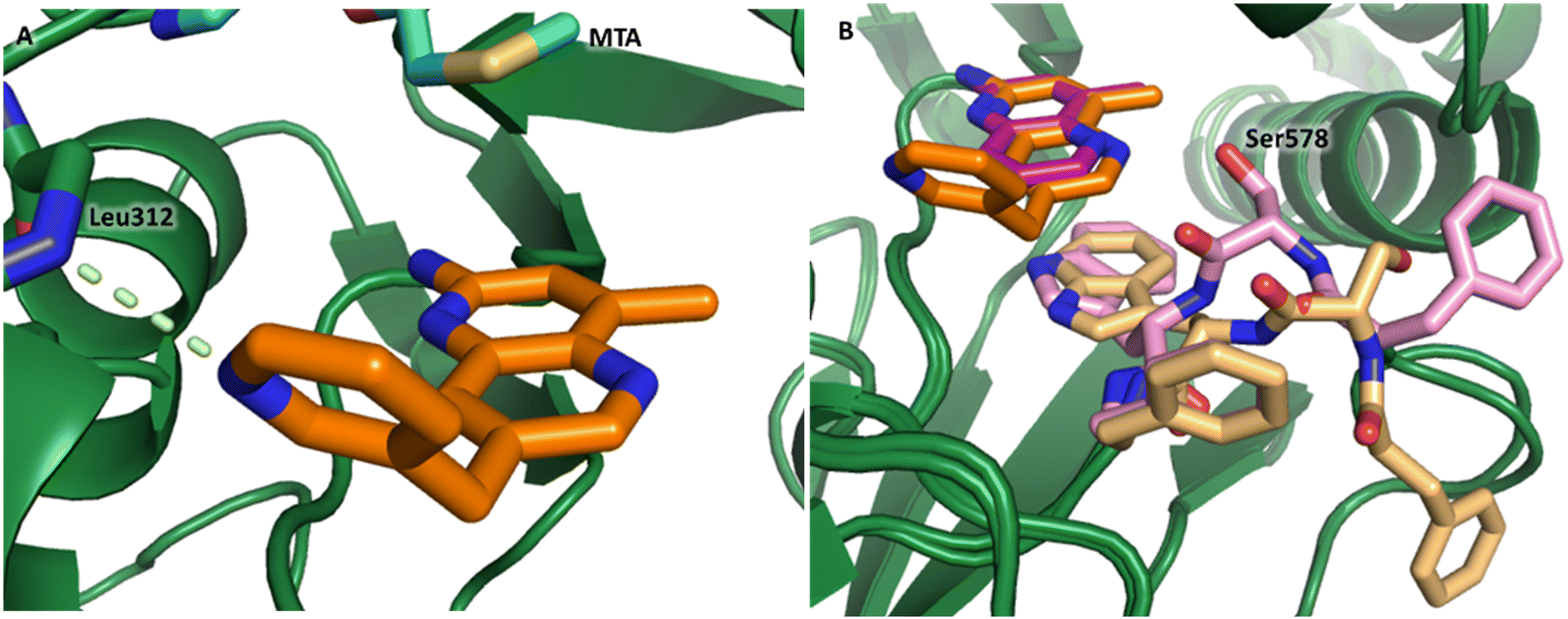
The chemical reactivity of the bromo substituent of 1 created challenges to develop versatile synthetic chemistry; therefore, a series of bromo replacements including-methyl (6), -cyclopropyl (7) and –CF3 (8) were investigated to optimize the fragment, shown in Table 2. The methyl analog 6 increased potency less than two-fold (PRMT5/MTA KD = 0.46 μM), and due to a reduction in clogP, LLE increased (clogP = 0.8; ΔLLE = +0.6). The larger and more lipophilic cyclopropyl analog 7 decreased potency and LLE (PRMT5/MTA KD = 3.3 μM; ΔLLE = −1.0). The –CF3 analog 8 resulted in a 15-fold decrease in potency and a lower LLE (PRMT5/MTA KD = 11.7 μM; ΔLLE = −1.3). Presumably, the electron withdrawing nature of the –CF3 substituent reduced the pKa of the conjugate acid of the pyridinyl nitrogen lone pair, leading to a significant reduction in the potency for 8. An X-ray co-crystal structure of 7 with PRMT5/MTA was generated and is shown in Fig. 7A. The binding mode of 7 with PRMT5/MTA is closely related to the binding mode of fragment hit 1. Indeed, 7 makes the same H-bond interactions with the protein as fragment hit 1. An overlay of fragment hit 1 and 7 is shown in Fig. 7B. Interestingly, the larger cyclopropyl substituent is accommodated in the binding pocket by the ligand shifting out of the pocket by approximately 0.5 Å, as opposed to the protein moving to accommodate the increased steric volume of the cyclopropyl substituent. By the end of phase 1—optimization of the fragment hit prior to fragment growing—we were encouraged to find that the bromo substituent could be effectively replaced by a methyl substituent and that a second X-ray co-crystal structure could be obtained that maintained the original fragment binding mode.
| Ex. | Structure | PRMT5/MTA KD (μM) | LLE | clogP |
|---|---|---|---|---|
| 1 |
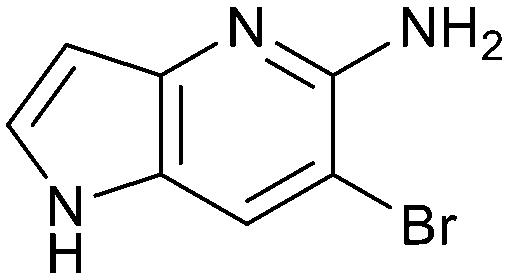
|
0.74 | 4.9 | 1.2 |
| 6 |
|
0.46 | 5.5 | 0.8 |
| 7 |
|
3.3 | 3.9 | 1.6 |
| 8 |
|
11.7 | 3.6 | 1.3 |
 | ||
| Fig. 7 A) X-ray co-crystal structure of 7 bound to PRMT5/MTA (PDB: 7ZVL). B) Overlay of 7 (orange) with fragment hit 1 (magenta). 7 has the same binding mode including H-bond interactions as 1 but is shifted by 0.5 Å to accommodate the cyclopropyl substituent. | ||
Visual inspection of the X-ray co-crystal structure of fragments 1 and 7 revealed that the 2-position of the 1H-pyrrolo[3,2-b]pyridine scaffold was the most suitable position for fragment growth. Identifying a synthetically tractable route to rapidly explore the 2-position vector was a challenge, even with replacement of the 6-bromo substituent with a 6-methyl substituent. Therefore, a less exploratory approach not dependent on rapid parallel synthesis techniques was initiated. To create bespoke molecules for synthesis we employed SBDD molecular modeling with MOE (Molecular Operating Environment).17 An overlay of 1 with the X-ray co-crystal structure of EPZ015666,18,19 a first generation PRMT5 inhibitor bound to the PRMT5/SAM complex (Fig. 8A, PDB: 4X61) inspired a chimeric design, example 9, where the methylene linked pyrimidine-4-carboxamide group of EPZ015666 was fused to the 2-position of 1. A MOE model of 9 bound to PRMT5/MTA is shown in Fig. 8B, where the amide carbonyl group mimics the homologous moiety of EPZ015666, making a productive H-bond interaction with Phe580 backbone N–H. The potency of 9 increased 82-fold (PRMT5/MTA KD = 9 nM) with an increase in LLE (ΔLLE = +2.5), Fig. 8D. This example demonstrates how powerful a chimeric approach with known ligands can be to increase the potency of a fragment. However, what is remarkable in this case is that the chimera was generated between two ligands binding to significantly different conformations of the PRMT5 protein. The X-ray co-crystal structure of 9 in PRMT5/MTA (Fig. 8C, PDB: 7ZUQ) revealed that the binding mode of the initial fragment 1 is conserved but the methylene linker orients the amide in an alternative conformation to the conformation generated by molecular modeling. Indeed, the amide carbonyl group makes a productive H-bond interaction with Leu312 backbone N–H and a water-mediated H-bond with the Ser439 backbone N–H and the sidechain of Asn443. We noted that this result was a fortunate discovery, for it highlighted the interaction with the Leu312 backbone N–H as a favorable interaction. This example illustrates the exploratory—and often serendipitous—nature of fragment growing strategies and highlights the value in obtaining X-ray co-crystal structures during the fragment growing process.
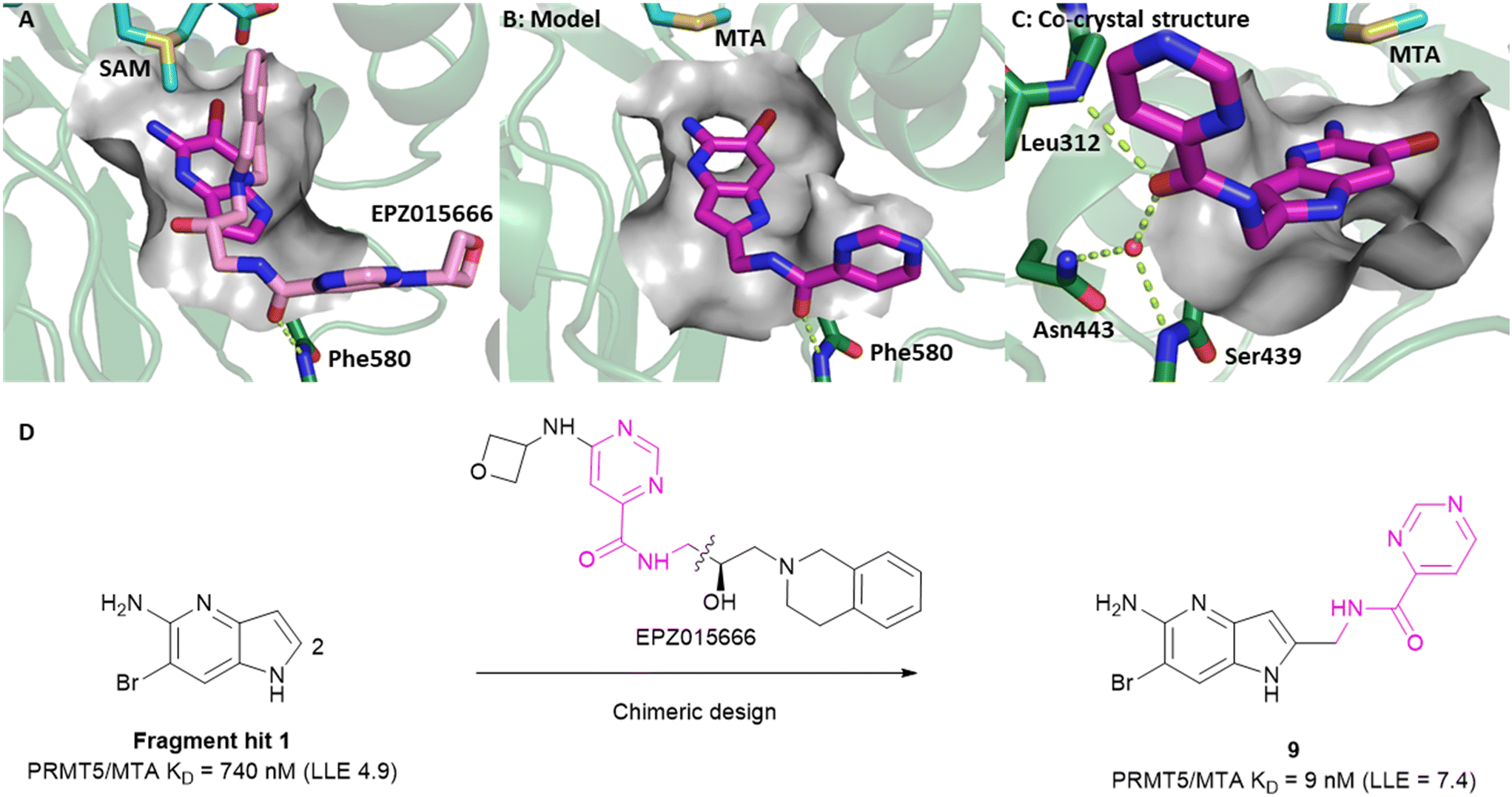 | ||
| Fig. 8 A) Overlay of fragment hit 1 (magenta) onto the EPZ015666 (pink) PRMT5/SAM X-ray co-crystal structure (PDB: 4X61). B) Model of compound 9 bound in PRMT5/MTA conceived by the overlay of fragment hit 1 and EPZ015666. C) X-ray co-crystal structure of 9 bound to PRMT5/MTA (PDB: 7ZUQ). 9 adopts an alternative conformation to the model forming an H-bond between the amide carbonyl of 9 and Leu312 and water mediated H-bonds to Asn443 and Ser439. D) SBDD enabled optimization of fragment hit 1 to give compound 9. | ||
Fragment hit 2 – series 2
A detailed description of the optimization of fragment 2 was previously reported.5 In summary (Fig. 9A), versatile bromo intermediate 10 was prepared, and a series of 20 analogs were made and tested. The rapid exploration of the SAR returned compound 11 with a 2040-fold increase in binding potency and improved LLE (PRMT5/MTA KD = 5 nM; ΔLLE = +3.5). The X-ray co-crystal structure of 11 with PRMT5/MTA (PDB 7S1Q)5 is shown in Fig. 9B. The initial binding mode of 2 was maintained and the N-methylpyrazol-4-yl substituent made a favorable H-bond interaction between the pyrazolo lone pair and the Leu312 backbone N–H. Interestingly, this is the same backbone N–H interaction that was discovered in the chimeric approach with series 1, example 9. This example of fragment growth illustrates how a dramatic increase in binding potency can be achieved with a modest number of analogs when supported by SBDD.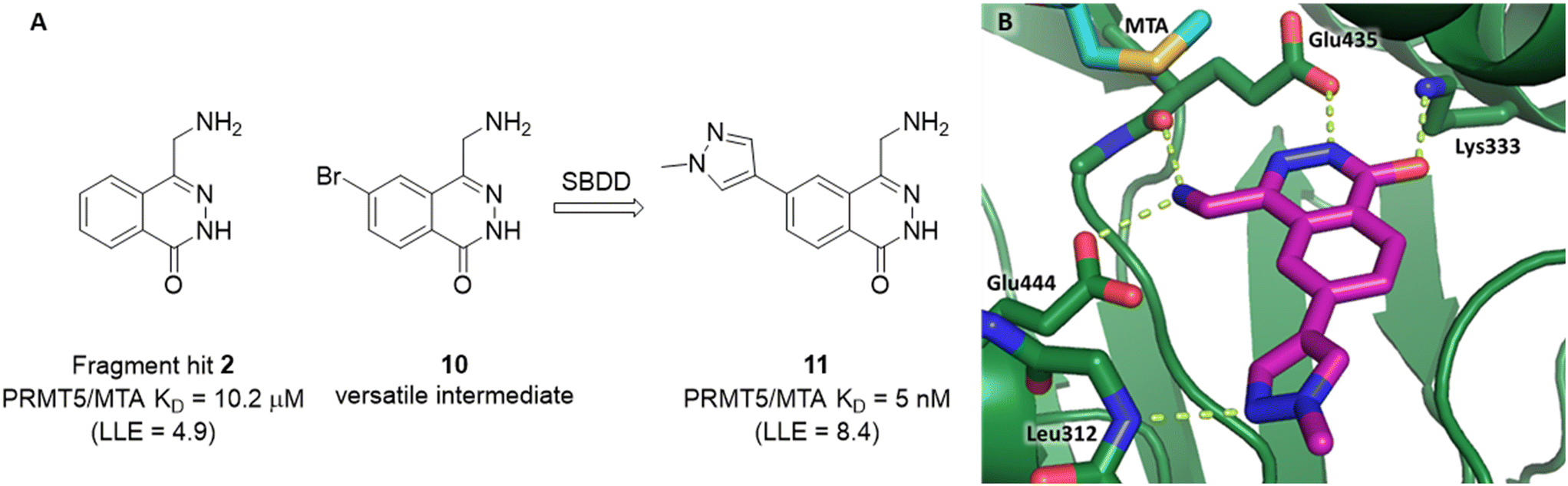 | ||
| Fig. 9 A) Discovery of compound 11 from fragment hit 2via versatile synthetic intermediate 10. B) X-ray co-crystal structure of 11 bound to PRMT5/MTA (PDB: 7S1Q). 11 adopts same binding mode as the original fragment hit and adds an additional H-bond interaction with Leu312. | ||
Fragment hit 3 – series 3
Fragment hit 3 was the most lipophilic (clogP = 1.7) of the five fragment hits. Therefore, before embarking on a fragment growing strategy, optimization of the fragment was investigated. A series of analogs were designed to replace the 7-chloro substituent, reduce clogP and/or make a productive interaction with the Lys333 sidechain, examples 12–18 in Table 3. LLE was tracked to assess progress. Replacement of the 7-chloro substituent with hydrogen (12) reduced the clogP by 0.7 units, while potency dropped approximately 6-fold and the LLE was unchanged (PRMT5/MTA KD = 70.0 μM; ΔLLE = 0, clogP = 1.0). Exchanging the 7-chloro with a 7-nitrile substituent (13) reduced clogP by 0.8, increased potency almost 6-fold and LLE increased significantly (PRMT5/MTA KD = 2.2 μM; ΔLLE = +1.6; clogP = 0.9). The positive LLE suggested the nitrile was making a favorable interaction with the protein. This binding hypothesis was indeed confirmed later by a series of X-ray co-crystal structures with analogs closely related to 13, vide infra. A series of 7-fluoro, 7-methyl and 7-methoxy analogs (14–16) were also investigated. A similar LLE was observed for 14 and 15 while a drop in LLE was observed for 16 (ΔLLE = +0.2; −0.1 and −0.6 respectively). Introduction of an aza into the ring (17) reduced the clogP to 0.4 and LLE increased (PRMT5/MTA KD = 43.0 μM; ΔLLE = +0.8). Homologation of 17 yielded N-ethyl analog 18 (PRMT5/MTA KD IC50 = 28.0 μM). An X-ray co-crystal structure of 18 with PRMT5/MTA (PDB: 7ZUP) revealed that 18 maintains the same binding mode as the initial fragment, and the pyridinyl nitrogen lone pair makes a water-mediated H-bond interaction with the side chains of Lys333 and Ser578, shown in Fig. 10. With the X-ray co-crystal structures of 3 and 18 in hand, a molecular model of 13 bound to PRMT5/MTA was generated using MOE, shown in Fig. 11A. The model indicated that the 7-nitrile substituent made a productive H-bond with the Lys333 sidechain. In summary, optimization of the fragment prior to fragment growth was successful, with an approximate 6-fold increase in potency and a 1.1-unit reduction in clogP. In addition, a second X-ray co-crystal structure was determined with the observation that the new structure maintained the binding mode of the original hit. Encouraged by these findings, an SBDD enabled fragment growing strategy was initiated starting with optimized fragment 13.
| Ex | Structure | PRMT5/MTA KD (μM) | LLE | clogP |
|---|---|---|---|---|
| 3 |
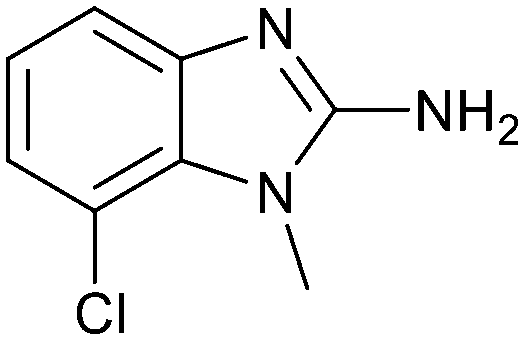
|
12.0 | 3.2 | 1.7 |
| 12 |
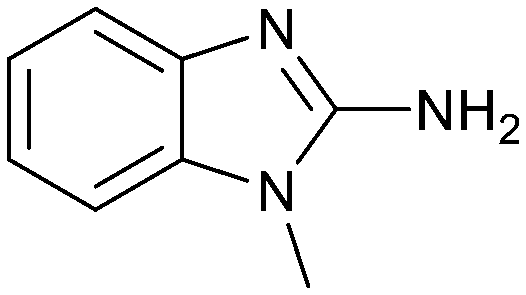
|
70.0 | 3.2 | 1.0 |
| 13 |
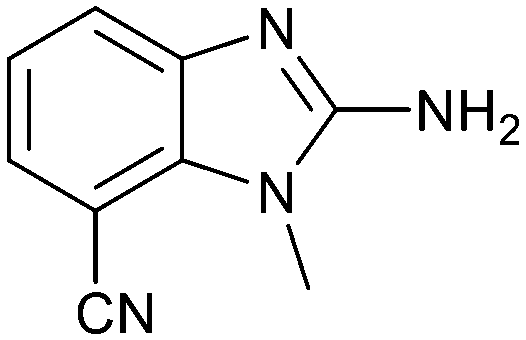
|
2.2 | 4.8 | 0.9 |
| 14 |
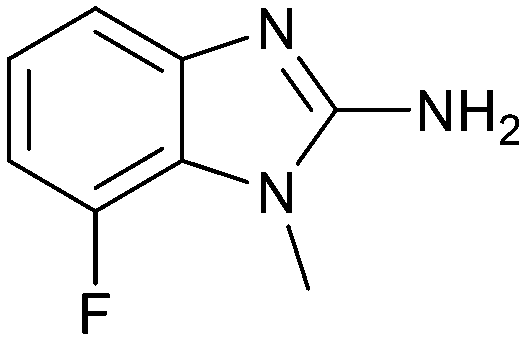
|
23.9 | 3.4 | 1.2 |
| 15 |
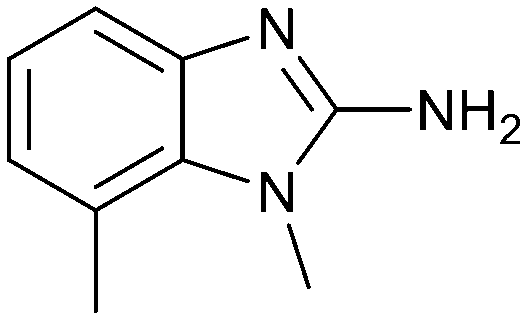
|
35.0 | 3.1 | 1.4 |
| 16 |
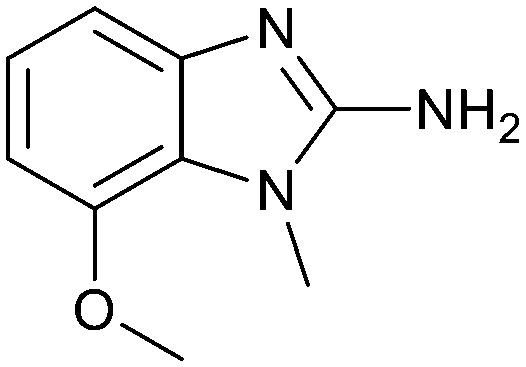
|
240 | 2.6 | 1.0 |
| 17 |
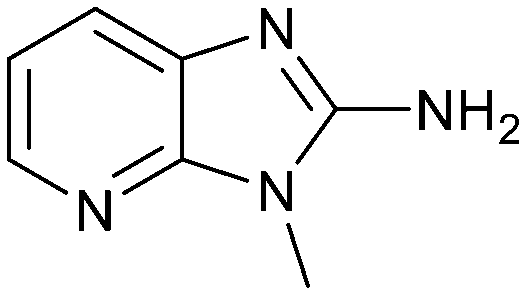
|
43.0 | 4.0 | 0.4 |
| 18 |
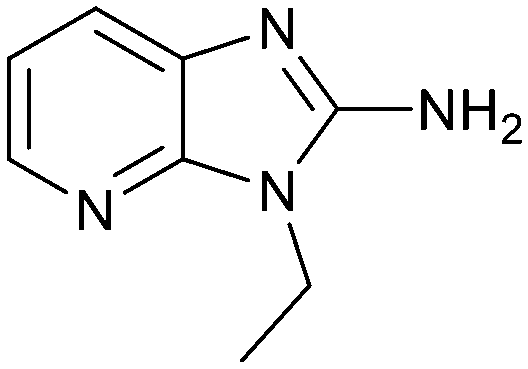
|
28.0 | 4.0 | 0.6 |
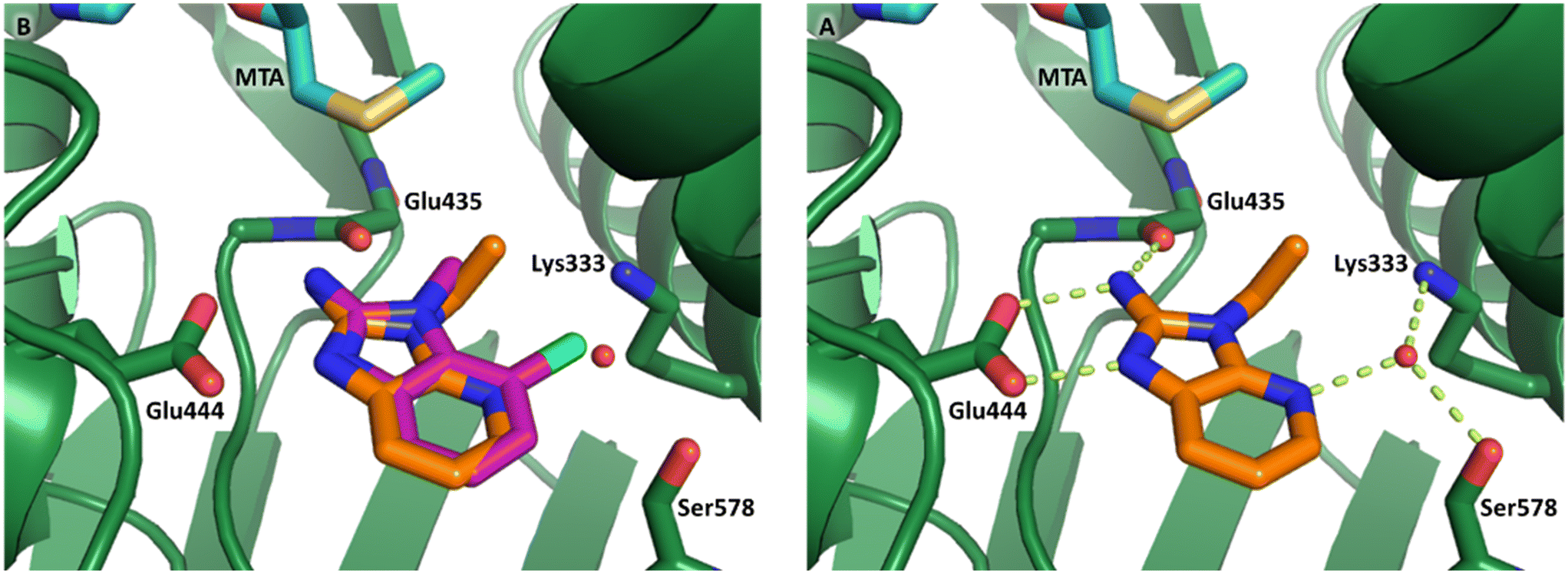 | ||
| Fig. 10 A) X-ray co-crystal structure of 18 bound to PRMT5/MTA (PDB: 7ZUP). 18 adopts the same binding mode as the original fragment hit 3 B) 18 picks up an additional water mediated H-bond to the side chains of Lys333 and Ser578. | ||
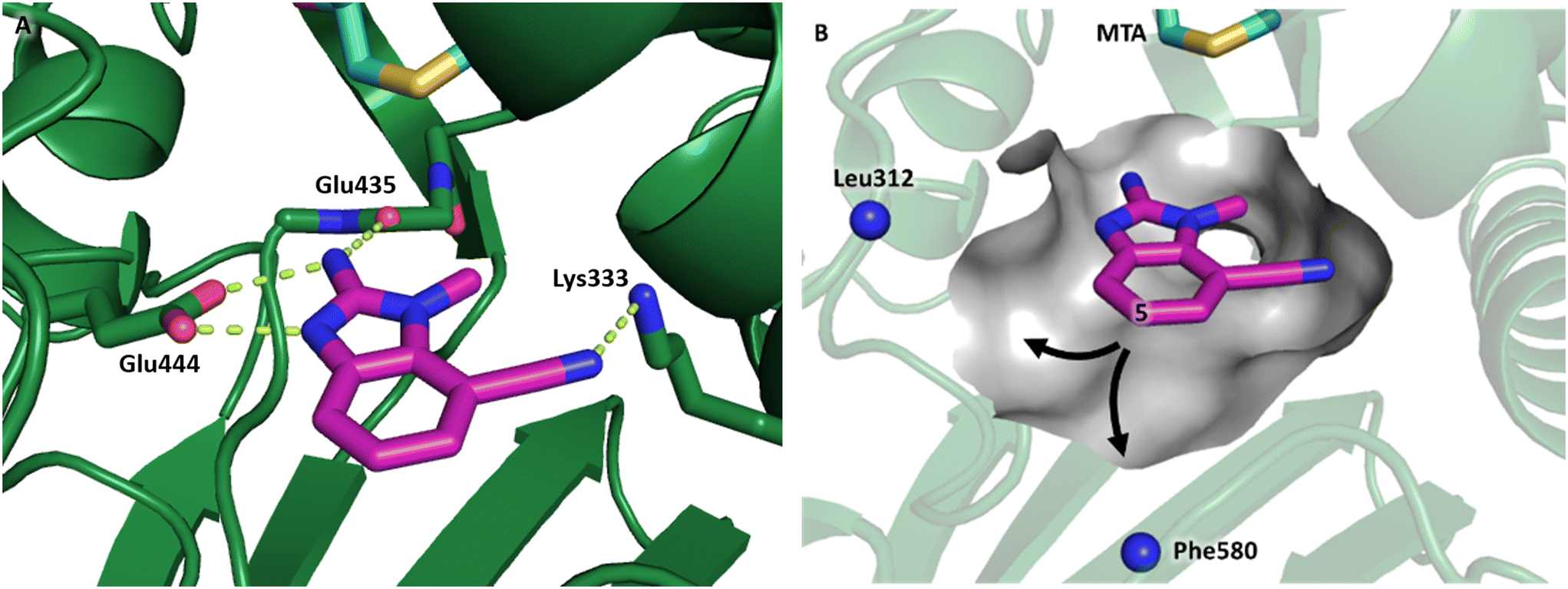 | ||
| Fig. 11 Model of compound 13 bound to PRMT5/MTA (A) H-bond network predicted to mimic parent fragment hit 3 with the addition of an H-bond to Lys-333 (B) backbone N–Hs of Leu312 and Phe580 highlighted as residues to target in SBDD fragment growing designs at the 5-position of the 1-methyl-1H-benzo[d]imidazol-2-amine scaffold. | ||
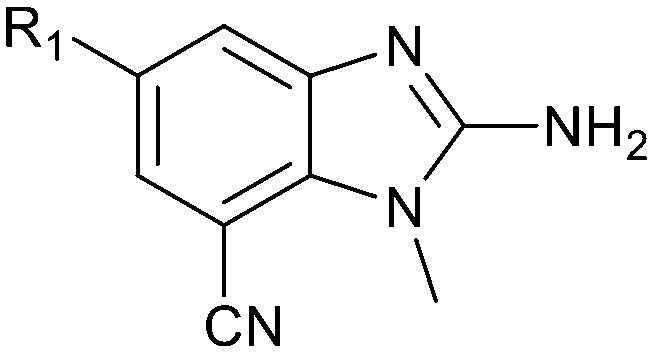
By visual inspection of the PRMT5/MTA X-ray co-crystal structures of 3 and 18, we envisioned a series of analogs at the 5-position of the 2-amino-1-methyl-1H-benzo[d]imidazole-7-carbonitrile scaffold. Further analysis of the model of 13 with PRMT5/MTA identified two unsatisfied backbone N–Hs (Leu312 and Phe580) accessible within 6 Å of the 5-position, shown in Fig. 11B. Thus, a series of analogs incorporating hydrogen bond acceptors designed to target a productive interaction with either of the two available N–Hs were investigated as shown in Table 4.
| Ex |
|
PRMT5/MTA KD (μM) | LLE | clogP |
|---|---|---|---|---|
| 22 |
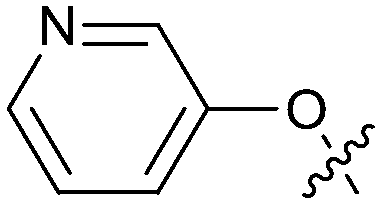
|
1.48 | 4.5 | 1.3 |
| 23 |
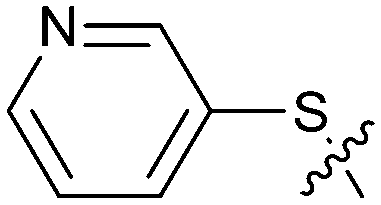
|
1.02 | 4.3 | 1.7 |
| 24 |
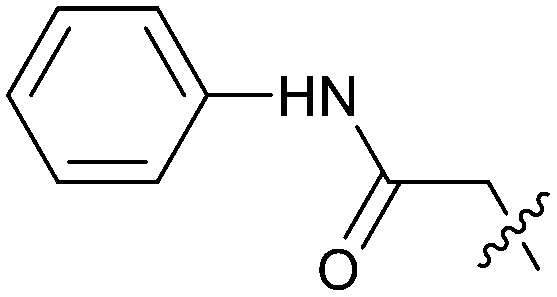
|
0.057 | 5.1 | 2.1 |
| 25 |
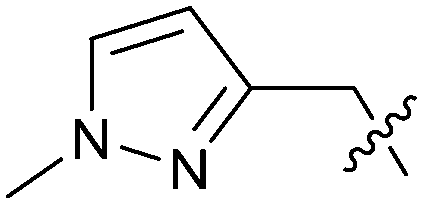
|
0.205 | 5.8 | 0.9 |
| 26 |
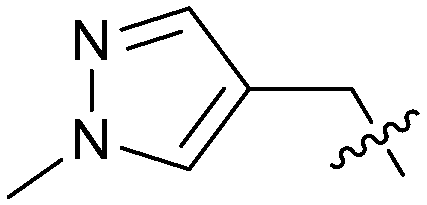
|
0.179 | 5.9 | 0.8 |
| 27 |
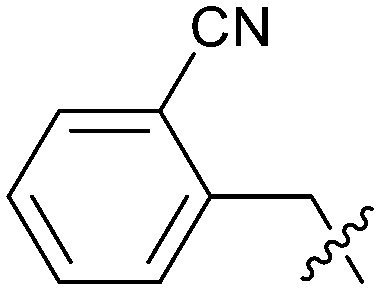
|
0.008 | 5.5 | 2.6 |
To explore the 5-position vector of 13 a series of versatile synthetic routes and intermediates were developed as shown in Scheme 1. Examples 22–24 were prepared from the 6-bromo-2-(2,5-dimethylpyrrol-1-yl)-3-methyl-benzimidazole-4-carbonitrile intermediate 19. Example 22 was synthesized via an Ullman coupling20,21 between pyridin-3-ol and 19 followed by removal of the 2,5-dimethylpyrrole protecting group under acidic conditions. Example 23 was prepared via bromo-lithium exchange of 19 with n-butyl lithium followed by quenching with 3-(3-pyridyldisulfanyl) pyridine. Subsequent removal of the 2,5-dimethylpyrrole protecting group under acidic conditions gave 23. Example 24 was prepared from carboxylic acid 20 which was submitted to standard amide coupling conditions with aniline followed by removal of the 2,5-dimethylpyrrole protecting group under acidic conditions. 20 was prepared by bromo-lithium exchange of 19 with n-butyl lithium followed by quenching with 2-bromoacetic acid. Example 25 was accessed via Suzuki–Miyaura22 coupling between intermediate 21 and 3-(chloromethyl)-1-methyl-pyrazole followed by removal of the 2,5-dimethylpyrrole protecting group under acidic conditions. Examples 26 and 27 were prepared by the same method as example 25.
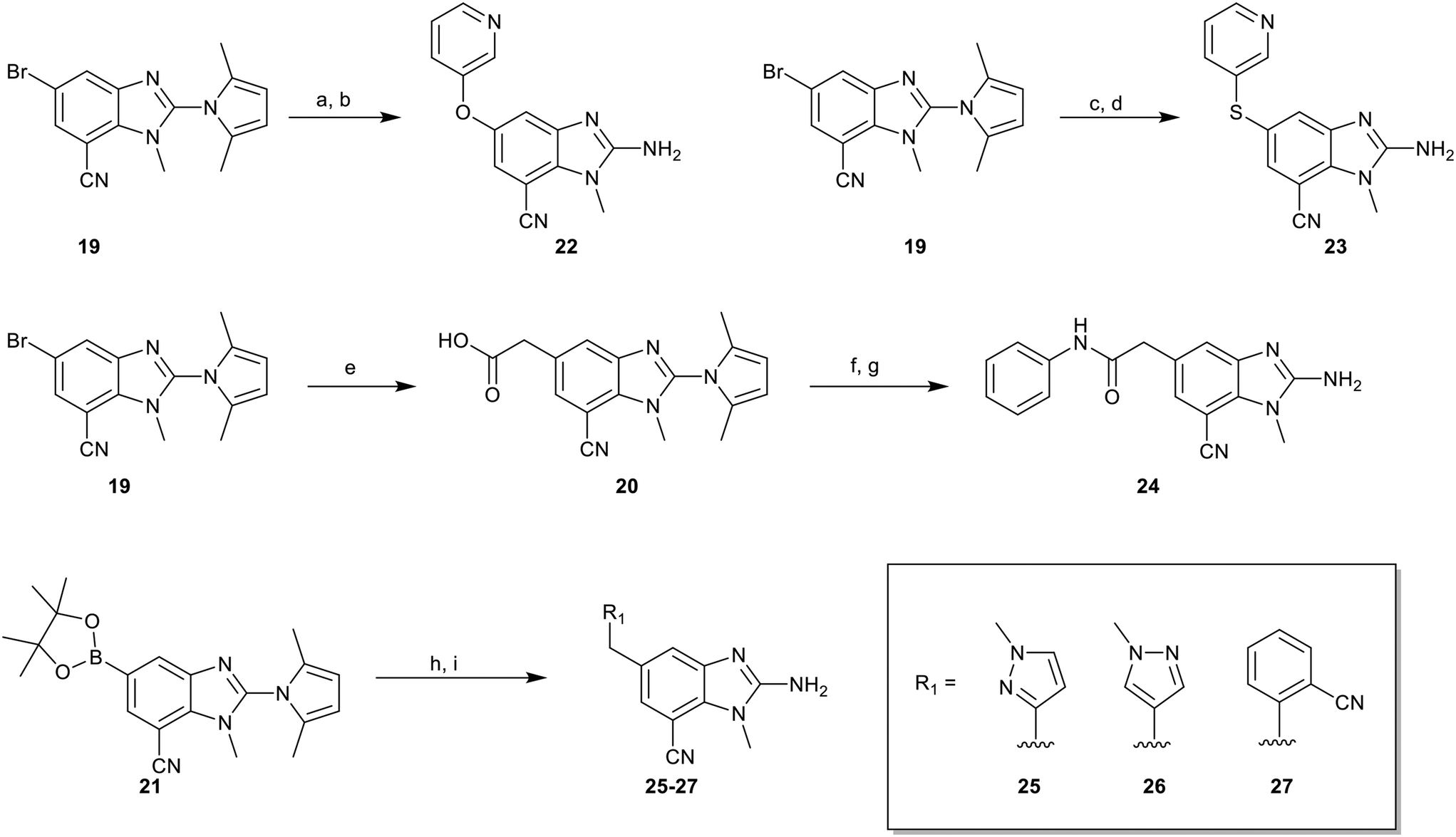 | ||
| Scheme 1 Synthesis of compounds 22–27. (a) Pyridine-3-ol, 2-(dimethylamino)acetic acid CuI, K3PO4, DMSO, 120 °C, 64%; (b) 12 M HCl, EtOH, 120 °C, 21%; (c) 3-(3-pyridyldisulfanyl) pyridine, nBuLi, THF, −65 °C – rt, 28%; (d) 12 M HCl, EtOH, 120 °C, 48%; (e) 20, nBuLi, 2-bromoacetic acid, THF, −65 °C – rt, 12% (f) aniline, EDCI, HOBT, DIEA, DMF, rt, 89%; (g) TFA, 80 °C, 27%; (h) (chloromethyl)–R1, Pd(dppf)Cl2, K2CO3, DMF, 100 °C, 28–71%; (i) 12 M HCl, EtOH, 120 °C, 26–59%. | ||
The synthetic routes described in Scheme 1 enabled the rapid preparation of approximately 150 analogs. Exemplar compounds, 22–27 (Table 4) were selected to highlight key aspects of the SAR for this series. O-linked pyridin-3-yl example 22 did not increase potency significantly and compared to example 13 a slight reduction in LLE was observed (PRMT5/MTA KD = 1.48 μM; ΔLLE = −0.3). S-linked pyridin-3-yl example 23 behaved similarly to 22 (PRMT5/MTA KD = 1.02 μM; ΔLLE = −0.5). Meanwhile, N-phenylacetamide analog 24 increased potency by 39-fold and increased LLE (PRMT5/MTA KD = 0.057 μM; ΔLLE = +0.3). Methylene linked N-methyl pyrazole analogs 25 and 26 gave similar results to each other with a potency increase of 11-fold and together with a reduction in clogP this translated to a significantly higher LLE (PRMT5/MTA KD = 0.205 μM and 0.179 μM; ΔLLE = +1.0 and +1.1 respectively). The methylene linked analog 2-cyanobenzyl 27 was the most potent analog from this cohort (PRMT5/MTA KD = 0.008 μM; ΔLLE = +0.7). Fig. 12A–C show the three PRMT5/MTA X-ray co-crystal structures generated from this cohort of analogs. The X-ray co-crystal structure of 22 (PDB: 7ZV2, Fig. 12A) was the first X-ray co-crystal structure of the 1-methyl-1H-benzo[d]imidazol-2-amine scaffold containing the nitrile substituent. The structure confirmed that the binding mode of 22 maintains the original binding mode of fragment hit 3, and the nitrile substituent indeed makes a favorable H-bond interaction with the Lys333 sidechain residue as predicted by molecular modeling. The pyridinyl nitrogen lone pair of 22 makes a productive H-bond interaction with the Phe580 backbone N–H and the oxygen linker makes a water bridged H-bond to the Leu312 backbone N–H. The X-ray co-crystal structures of 25 and 26 with PRMT5/MTA are shown in Fig. 12B (PDB: 7ZVU) and Fig. 12C (PDB: 7ZVY). The N-methylpyrazole lone pair of 25 makes a productive H-bond with the Leu312 backbone N–H while the N-methylpyrazole lone pair of 26 makes a water bridged H-bond with the Leu312 backbone N–H. It was notable that the 2-amino-1-methyl-1H-benzo[d]imidazole-7-carbonitrile scaffold remains in a constant position while the 5-position substituents scan a range of geometries and make a diverse set of interactions with the protein. This example highlights the value of obtaining X-ray structures during a fragment elaboration campaign. An X-ray co-crystal structure of 27 bound to PRMT5/MTA was not obtained; therefore, 27 was modelled into the binding site using MOE, shown in Fig. 12D. The 2-amino-1-methyl-1H-benzo[d]imidazole-7-carbonitrile scaffold was overlayed with the binding poses of 22, 25 and 26 and molecular modeling indicated the 2-cyano phenyl substituent would make a productive H-bond interaction with the Leu312 backbone N–H.
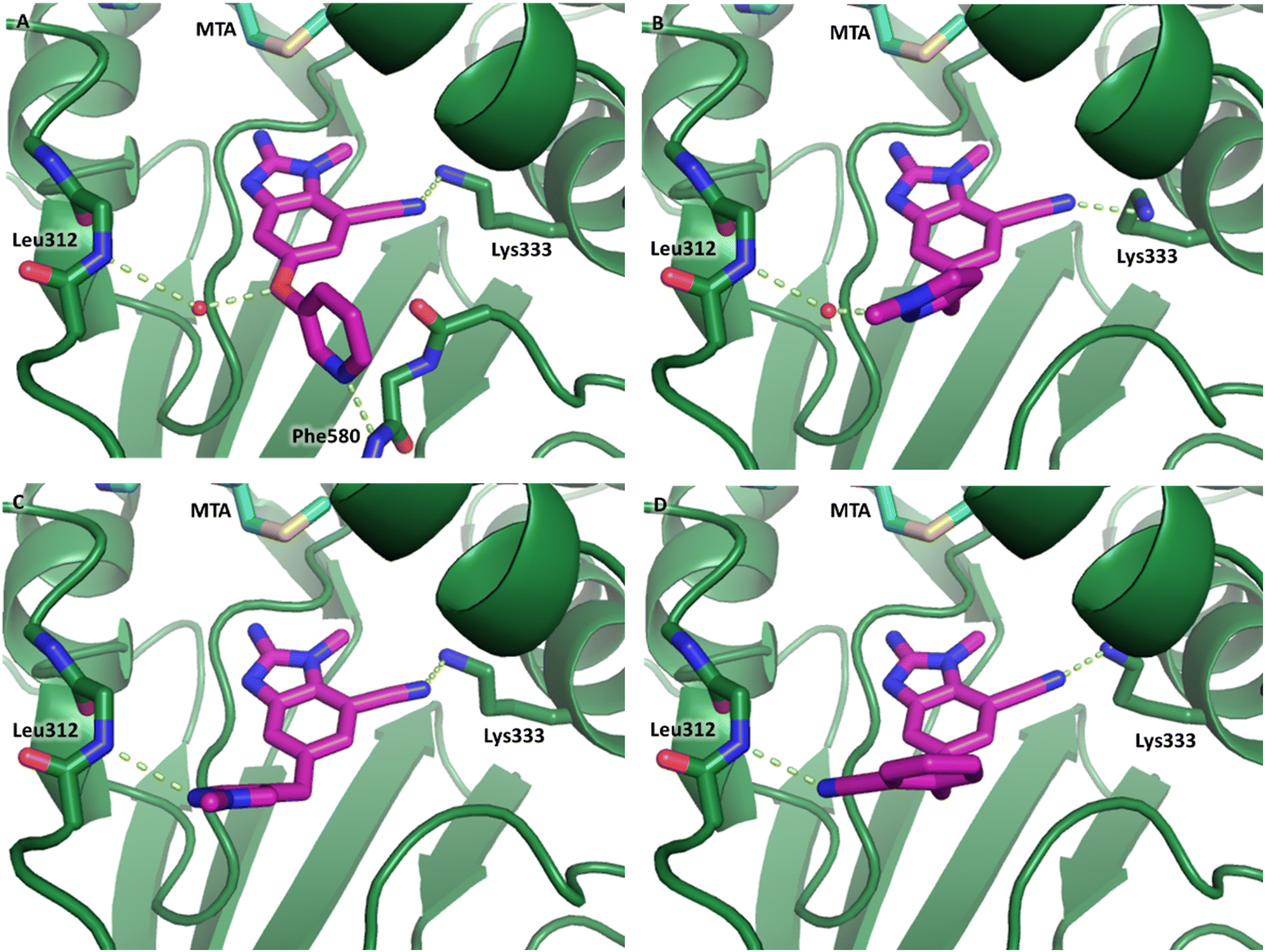 | ||
| Fig. 12 (A) X-ray co-crystal structure of 22 bound to PRMT5/MTA (PDB: 7ZV2). (B) X-ray co-crystal structure of 25 bound to PRMT5/MTA (PDB: 7ZVU). (C) X-ray co-crystal structure of 26 bound to PRMT5/MTA (PDB: 7ZUY). (D) MOE model of compound 27 bound to PRMT5/MTA showing the 2-cyano phenyl substituent making a productive H-bond interaction with the Leu312 backbone N–H. | ||
Fragment hits 4 and 5 – series 4
We elected to focus on hit 4 with the option to transfer SAR to fragment hit 5 at a later timepoint. Therefore, in an analogous approach to the growing strategies of fragment hits 1 and 3 we envisioned a series of analogs branching off the 7-position of the 1,5-naphthyridin-2-amine fragment 4 scaffold. Versatile intermediates 28–31 (Fig. 13) were prepared, and a series of 50 analogs were synthesized using palladium-mediated and copper-mediated coupling chemistry (see ESI† for experimental details).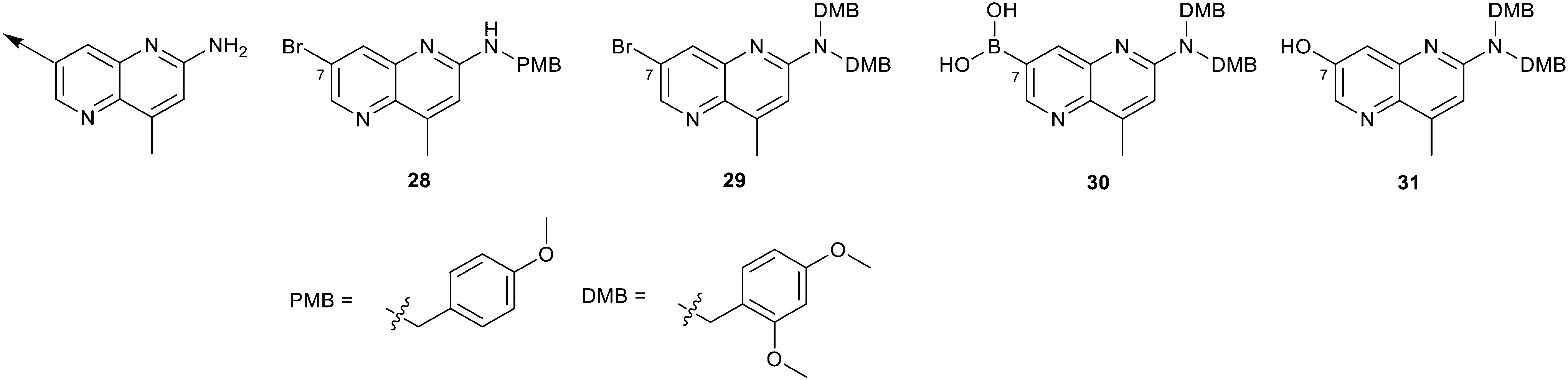 | ||
| Fig. 13 Analogs to explore the SAR of fragment hit 4 were prepared from versatile intermediates 28–31. | ||
Exemplar compounds (32–37, Table 5) were selected to highlight key aspects of the SAR for this series. Thiazol-4-yl analog 32 increased potency 8-fold with no change in LLE indicating the increase in potency was lipophilicity driven (PRMT5/MTA KD = 7.5 μM; ΔLLE = 0.0). Whereas O-linked pyridin-3-yl analog (33) increased potency 25-fold and increased LLE (PRMT5/MTA KD = 1.5 μM; ΔLLE = +1.0). Methylene linked pyridin-3-yl and N-methyl pyrazol-4-yl analogs 34 and 35 increased potency 267-fold and 452-fold respectively and increased LLE (PRMT5/MTA KD = 0.232 μM and 0.137 μM; ΔLLE = +1.6 and +2.8 respectively). An X-ray crystal structure of 34 was solved with PRMT5/MTA (PDB: 7ZUU) and is shown in Fig. 14A. The 1,5-naphthyridine scaffold remains in the same binding mode as the original fragment hit and the methylene linked pyridinyl nitrogen lone pair makes a productive H-bond interaction with the Leu312 backbone N–H. Interestingly the Ser578 loop moves and rotates away from the ligand in a fashion reminiscent of the loop conformation for the fragment hit 5 co-crystal structure, shown in Fig. 14B. Notably, the movement of the Phe577 side chain is more pronounced with 34 than what was previously observed. The movement of the Ser578 loop was not observed with fragment hits 1–3, thus making the properties of the PRMT5 protein structure unique for the 1,5-naphthyridin-2-amine fragment scaffold. The chimeric approach described with fragment hit 1 was also applied to fragment hit 4. This resulted in the identification of 36 and 37 with PRMT5/MTA KD = 0.094 μM and PRMT5/MTA KD = 0.151 μM respectively.
| Ex |
|
clogP |
PRMT5/MTA KD (μM) | LLE |
|---|---|---|---|---|
| 32 |

|
2.1 | 7.50 | 3.0 |
| 33 |
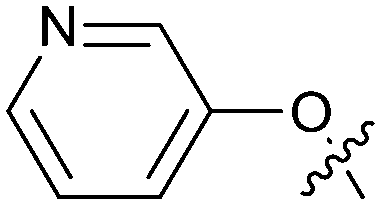
|
1.5 | 2.46 | 4.1 |
| 34 |
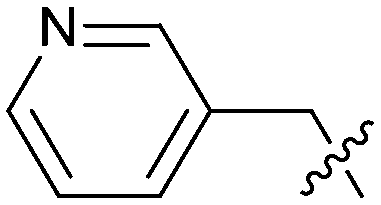
|
1.9 | 0.232 | 4.7 |
| 35 |
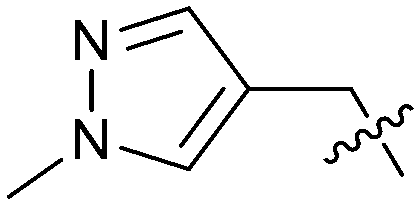
|
1.0 | 0.137 | 5.9 |
| 36 |
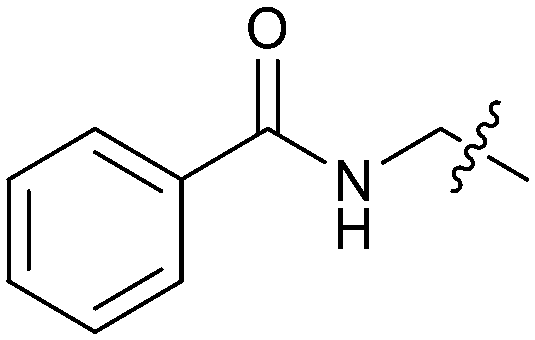
|
2.0 | 0.094 | 5.0 |
| 37 |
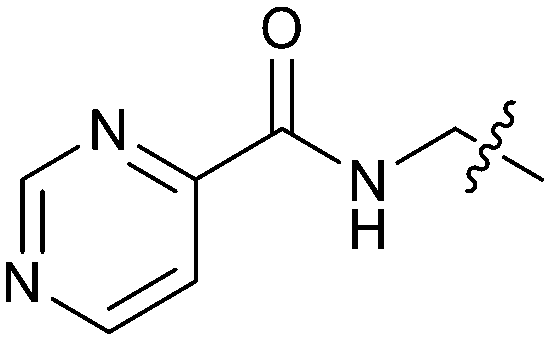
|
0.5 | 0.151 | 6.3 |
 | ||
| Fig. 14 A) X-ray co-crystal structure of 34 bound to PRMT5/MTA (PDB: 7ZUU) showing the methylene linked pyridine-3-yl lone pair making a productive H-bond interaction with the Leu312 backbone N–H. B) Overlay of 4 and 34 highlighting the movement of the Ser578 loop and sidechains. | ||
Discussion
A fragment screen to identify chemical matter for the PRMT5/MTA complex resulted in the discovery of five fragment hits whose binding modes were verified by co-crystal crystallography with PRMT5 in the presence of MTA. The five hits represented four distinct scaffolds. With no validated chemical matter reported in the literature at the time the project was initiated, these hits provided an opportunity to leverage both structure- and synthesis-enabled fragment elaboration strategies to identify potent compounds to further investigate the role of the PRMT5/MTA complex in cancer biology. Fragment elaboration strategies were investigated for each scaffold, ultimately resulting in the identification of two lead series, from fragment hit 2 and fragment hit 3.The chemistry for fragment hit 1 was challenging to develop; therefore, a bespoke set of compounds were designed via a chimeric approach with a known PRMT5 inhibitor EPZ015666, although 666 binds to PRMT5 in an alternative binding mode. Two X-ray crystal structures were obtained via this approach and demonstrated the original fragment binding mode was retained in the elaborated molecules. Compound 9 was the most potent example with PRMT5/MTA KD = 9 nM, LLE = 7.4 and ΔLLE = +2.4, and this molecule highlighted the importance of an H-bond interaction with Leu312 to improve potency.
The chemistry for fragment hit 2 was readily accessible and a viable vector was rapidly identified. Compound 11 was the most potent example identified in the first round of fragment elaborations (PRMT5/MTA KD = 5 nM, LLE = 8.4 and ΔLLE = +3.5). An X-ray co-crystal structure with 11 was obtained demonstrating the binding mode of the original fragment hit was conserved and additional vectors for further growth were clearly identified to support additional rounds of SAR. Ultimately, this series gave rise to the discovery of clinical candidate MRTX1719 (Fig. 1).5
Fragment hit 3 was first optimized before examination of the fragment growth SAR. Fragment analog 13 was identified with improved potency and LLE compared to the starting fragment hit 3. The synthetic chemistry for fragment growth was enabled via several versatile synthetic intermediates developed from fragment 13. A series of 150 analogs were investigated. Compound 27 was the most potent example identified with a PRMT5/MTA KD = 8 nM, LLE = 5.5 and ΔLLE = +2.3. Four additional X-ray structures were discovered in this series, each demonstrating that the original fragment binding mode was conserved and additional vectors for further growth were clearly identified to support further rounds of fragment growing SAR (not reported here).
Finally, the chemistry for fragment hit 4 was also enabled via a series of versatile intermediates 28–31. Example 36 was the most potent example identified, with PRMT5/MTA KD = 94 nM, LLE = 5.0 and ΔLLE = +1.9. An X-ray structure was determined with example 34 which highlighted protein movements in the Ser578 loop. It remains unclear if protein mobility in the Ser578 loop would be an advantage by creating a new pocket to grow into, or a disadvantage by introducing ambiguity into the SBDD molecular modeling. Although the original intention was to examine if the SAR would transfer from fragment 4 to fragment 5 or if the SAR would be divergent, we decided to not perform this exercise but to focus efforts on driving forward series 2 and series 3.
Conclusions
Starting with five fragment hits, each with a clear binding orientation in a PRMT5/MTA co-crystal structure, we embarked on a round of fragment elaboration to generate a dataset with the objective of answering three questions designed to help prioritize the fragment hit series: could the binding potency be improved? Were the series chemically tractable? Could additional co-crystal structures be solved? We discovered that the potency and LLE could be improved for hits 1–4. Indeed, examples with sub-10 nM KDs were discovered for series 1–3, while the most potent example from series 4 was approximately 10-fold weaker. Chemistry enablement and access to versatile intermediates was achieved for hits 2–4. The chemistry was challenging for hit 1, and access to versatile intermediates to probe the vectors of interest was not enabled. Crystallography enablement to support SBDD was excellent for series 1–3, while there were questions raised for series 4 related to protein flexibility and movement of the Ser578 loop. With these data in hand, series 2 and series 3 scored most favorably across the three proposed questions. Thus, these two series were selected as lead series for further optimization. Ultimately, a clinical candidate was discovered from hit series 2 and is currently in a phase 1/2 clinical study in solid tumors with MTAP deletion.23Experimental
Chemistry
Examples 1–5, 12 and 14–18 were commercially available. Intermediate 10 and Example 11 were previously described.5 Preparation of intermediates 19, 21 and 28–31 and examples 6–9, 13 and 32–37 are described in the ESI.† Examples 22–27 were prepared according to Scheme 1 and the procedures are described below.
2-Amino-3-methyl-6-(3-pyridyloxy)benzimidazole-4-carbonitrile (22).
Step a. To a solution of 19 (150 mg, 455 μmol, 1.00 eq.), pyridin-3-ol (130 mg, 1.37 mmol, 3.00 eq.) and 2-(dimethylamino)acetic acid (18.8 mg, 182 μmol, 0.40 eq.) in DMSO (3.0 mL) was added copper iodide (17.4 mg, 91.1 μmol, 0.20 eq.) and potassium phosphate (290 mg, 1.37 mmol, 3.00 eq.). The mixture was stirred at 120 °C for 16 h. After such time the reaction was poured into water (20 mL) and extracted with ethyl acetate (50 mL × 3). The combined organic layers were concentrated and the residue purified by silica gel chromatography (ethyl acetate in petroleum ethergradient 0–100%) to give 2-(2,5-dimethylpyrrol-1-yl)-3-methyl-6-(3-pyridyloxy)benzimidazole-4-carbonitrile (100 mg, 291 μmol, 64% yield) as a yellow solid. LCMS [M + 1]+: 344.2.
Step b. To a solution of 2-(2,5-dimethylpyrrol-1-yl)-3-methyl-6-(3-pyridyloxy)benzimidazole-4-carbonitrile (50 mg, 146 μmol, 1.00 eq.) in ethanol (1.5 mL) was added hydrochloric acid (12 M, 375 μL). The mixture was microwave heated at 120 °C in a sealed microwave vial for 2 h. The cooled mixture was then treated with ammonium hydroxide (0.1 mL) to adjust the pH to 7. The mixture was then concentrated and the residue purified by prep-HPLC (ammonium bicarbonate condition) to give 2-amino-3-methyl-6-(3-pyridyloxy)benzimidazole-4-carbonitrile, 22 (8.3 mg, 30.6 μmol, 21% yield) as a white solid. LCMS [M + 1]+: 266.0; 1H NMR (400 MHz, CD3OD-d4) δ = 8.32–8.24 (m, 2H), 7.41 (d, J = 1.6, 3.2 Hz, 2H), 7.16 (d, J = 2.4 Hz, 1H), 7.05 (d, J = 2.4 Hz, 1H), 3.85 (s, 3H); HRMS (ESI, +ve ion) m/z calcd for C14H11N5O 265.0964; found 265.0949; HPLC Rt 0.582 min, 96.8%.
2-Amino-3-methyl-6-(3-pyridylsulfanyl)benzimidazole-4-carbonitrile hydrochloride (22).
Step c. To a solution of 19 (100 mg, 303 μmol, 1.00 eq.) in THF (2.0 mL) was added n-butyllithium (2.50 M, 145 μL, 1.20 eq.) at −65 °C. The mixture was stirred at −65 °C for 0.5 h. A solution of 3,3′-dithiodipridine (80 mg, 364 μmol, 1.20 eq.) in THF (0.5 mL) was then added at −65 °C. The mixture was stirred at −65 °C for 0.5 h, then warmed up slowly to 25 °C and stirred for an additional 0.5 h. The mixture was diluted with saturated ammonium chloride (50 mL), extracted with ethyl acetate (50 mL), dried over sodium sulfate, filtered, and concentrated. The residue was purified by silica gel chromatography with (ethyl acetate in petroleum ether 0–50%) to afford 2-(2,5-dimethylpyrrol-1-yl)-3-methyl-6-(3-pyridylsulfanyl)benzimidazole-4-carbonitrile (37 mg, 85.4 μmol, 28% yield) as a yellow gum. LCMS [M + 1]+: 360.1.
Step d. To a solution of 2-(2, 5-dimethylpyrrol-1-yl)-3-methyl-6-(3-pyridylsulfanyl)benzimidazole-4-carbonitrile (32 mg, 89.0 μmol, 1.0 eq.) in ethyl alcohol (0.5 mL) was added 12 M hydrochloric acid (0.05 mL, 503 μmol, 5.6 eq.). The mixture was stirred at 120 °C in a microwave reactor for 1 h and then concentrated. The residue was purified by prep-HPLC (HCl condition) to afford 2-amino-3-methyl-6-(3-pyridylsulfanyl)benzimidazole-4-carbonitrile hydrochloride, 23 (13.9 mg, 43.1 μmol, 48% yield) as a white solid. LCMS [M + 1]+: 282.0. 1H NMR (400 MHz, CD3OD-d4) δ = 8.72 (s, 1H), 8.69 (br d, J = 5.2 Hz, 1H), 8.37 (br d, J = 8.0 Hz, 1H), 7.98 (s, 2H), 7.94 (s, 1H), 3.99 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ = 152.40, 147.62, 146.03, 141.23, 134.75, 132.39, 132.18, 131.69, 127.60, 126.12, 120.43, 115.65, 95.29, 30.87; HRMS (ESI, +ve ion) m/z calcd for C14H11N5S 281.0735; found 281.0720; HPLC Rt 0.863 min, 97.5%.
2-(2-Amino-7-cyano-1-methyl-benzimidazol-5-yl)-N-phenyl-acetamide hydrochloride (24).
Step e. To a mixture of 19 (300 mg, 911 μmol, 1.0 eq.) in THF (4.0 mL) at −60 °C under nitrogen atmosphere was added n-butyllithium (2.50 M, 729 μL, 2.0 eq.). The mixture was stirred at −60 °C for 0.5 hour. Then 2-bromoacetic acid (506 mg, 3.65 mmol, 262 μL, 4.0 eq.) in tetrahydrofuran (1.0 mL) was added and the resulting mixture was warmed up to 30 °C and stirred for an additional 0.5 hour. The reaction mixture was quenched with water (1.0 mL) and purified by prep-HPLC (formic acid condition) to afford 2-[7-cyano-2-(2,5-dimethylpyrrol-1-yl)-1-methyl-benzimidazol-5-yl]acetic acid (35 mg, 106 μmol, 12% yield) as a white solid. LCMS [M + 1]+: 309.2. 1H NMR (400 MHz, CD3OD-d4) δ = 7.95 (d, J = 1.2 Hz, 1H), 7.75 (d, J = 1.2 Hz, 1H), 5.98 (s, 2H), 3.83 (s, 2H), 3.77 (s, 3H), 2.04 (s, 6H).
Step f. To a mixture of 2-[7-cyano-2-(2,5-dimethylpyrrol-1-yl)-1-methyl-benzimidazol-5-yl]acetic acid (35 mg, 114 μmol, 1.0 eq.) and aniline (31 μL, 340 μmol, 3.0 eq.) in DMF (1.0 mL) was added N-(3-dimethylaminopropyl)-N-ethylcarbodiimide hydrochloride (33 mg, 170 μmol, 1.50 eq.), 1-hydroxybenzotriazole (23 mg, 170 μmol, 1.5 eq.) and N,N-diisopropylethylamine (340 μmol, 59.3 μL, 3.0 eq.). The mixture was stirred at 30 °C for 16 hours. The reaction mixture was concentrated and purified by prep-TLC (SiO2, petroleum ether/ethyl acetate 3
:1) to afford 2-[7-cyano-2-(2,5-dimethylpyrrol-1-yl)-1-methyl-benzimidazol-5-yl]-N-phenyl-acetamide (40 mg, 100 μmol, 89% yield) as a yellow oil. LCMS [M + 1]+: 384.3. 1H NMR (400 MHz, CDCl3) δ = 8.01 (d, J = 1.2 Hz, 1H), 7.71 (d, J = 1.2 Hz, 1H), 7.48 (d, J = 7.6 Hz, 2H), 7.33–7.29 (m, 3H), 7.17–7.09 (m, 1H), 5.98 (s, 2H), 3.87 (s, 2H), 3.74 (s, 3H), 2.05 (s, 6H).
Step g. A mixture of 2-[7-cyano-2-(2,5-dimethylpyrrol-1-yl)-1-methyl-benzimidazol-5-yl]-N-phenyl-acetamide (30 mg, 78.2 μmol, 1.0 eq.) in trifluoroacetic acid (2.0 mL) was stirred at 80 °C for 16 hours. The mixture was concentrated. The crude was purified by prep-HPLC (HCl condition) to afford 2-(2-amino-7-cyano-1-methyl-benzimidazol-5-yl)-N-phenyl-acetamide hydrochloride, 24 (7.5 mg, 21.4 μmol, 27% yield) as a light green solid. LCMS [M + 1]+: 306.2. 1H NMR (400 MHz, CD3OD-d4) δ = 7.66 (dd, J = 1.2, 14.8 Hz, 2H), 7.55 (dd, J = 1.2, 8.8 Hz, 2H), 7.35–7.25 (m, 2H), 7.13–7.05 (m, 1H), 3.93 (s, 3H), 3.84 (s, 2H); HRMS (ESI, +ve ion) m/z calcd for C17H15N5O 305.1277; found 305.1251; HPLC Rt 2.088 min, 97.8%.
General method for the preparation of examples 25–28 (Scheme 1)
To a solution of 21 (1.00 eq.) and R1–Cl (1.5 eq.) in DMF (2.0 mL) was added potassium carbonate (3.0 eq.), followed by [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II) (0.15 eq.) under a nitrogen atmosphere. The mixture was then stirred at 100 °C for 16 hours. After such time the cooled reaction mixture was concentrated, and the residue purified by flash silica gel chromatography to afford an intermediate product. To a solution of this intermediate product in ethanol (1.0 mL) was added hydrochloric acid (12 M, 7 eq.). The mixture was stirred at 120 °C for 1.5 hours in a microwave reactor. The mixture was basified with ammonium hydroxide until pH 8 and concentrated in vacuum. The residue was purified by prep-HPLC (ammonium bicarbonate condition) to give the desired product.Biology
Methods and procedures for protein expression and purification for crystallography, protein expression and purification for SPR and KD determination by SPR were previously described.5Accession codes
Atomic coordinates for the X-ray structures of 1 (PDB 8CSG) 3 (PDB SCTB), 4 (PDB 7UYF), 5 (PDB 7UY1), 7 (PDB 7ZVL), 9 (PDB 4X61), 18 (PDB 7ZUP) 22 (PDB 7ZV2), 25 (PDB 7ZVU), 26 (PDB 7ZUY) and 34 (PDB 7ZUU) bound to PRMT5·MTA are available from the RCSB Protein Databank (https://www.rcsb.org).Author contributions
CRS and SK coordinated the project and performed compound design and synthesis; MUDA, VA, RJG and NCT designed and performed protein preparation, protein-ligand crystallization, and crystallography; JGC conceived of the project; AI, and JDL performed compound design and computational studies; JMK, JK and XW performed compound design and synthesis; MAM conceived of the project and performed compound design and synthesis.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We would like to thank the following teams/people for their valuable contributions to this work. The IDSU chemistry team (WuXi AppTec in Wuhan, China): Tao Guo, Duan Liu, Song Mo. CSU chemistry team (WuXi AppTec in Wuhan, China): Rongfeng Zhao, Shaojun Song, Wenbing Ruan. In vitro ADME team and the RSD biology team (WuXi Apptec in Shanghai, China). Ronald Jean-Baptiste and Masakazu Kobayashi (ZoBio BV, J.H. Oortweg 19, 2333 CH Leiden, Netherlands) for the SPR PRMT5/MTA binding KD determinations and Wolfgang Komel (ZoBio BV, J.H. Oortweg 19, 2333 CH Leiden, Netherlands) for preparing the PDB scripts and f factor files. Stephan Krapp and Brantley Haigh (Proteros Biostructures GmbH, Bunsenstraße 7a, 82152 Planegg, Germany) for early enablement of X-ray crystallography. The X-ray crystallography work presented herein is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). The Eiger 16M detector on the 24-ID-E beam line is funded by a NIH-ORIP HEI grant (S10OD021527). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. We acknowledge the Paul Scherrer Institute, Villigen, Switzerland for provision of synchrotron radiation beamtime at beamlines PXII/X10SA and PXIII/X06DA of the SLS. The authors would like to thank Diamond Light Source for beamtime and the staff of beamline I03 for assistance with crystal testing and data collection. The research was funded by Mirati Therapeutics.References
- M. T. Bedford and S. G. Clarke, Protein arginine methylation in mammals: who, what, and why, Mol. Cell, 2009, 33(1), 1–13 CrossRef CAS PubMed.
- G. V. Kryukov, F. H. Wilson, J. R. Ruth, J. Paulk, A. Tsherniak, S. E. Marlow, F. Vazquez, B. A. Weir, M. E. Fitzgerald, M. Tanaka, C. M. Bielski, J. M. Scott, C. Dennis, G. S. Cowley, J. S. Boehm, D. E. Root, T. R. Golub, C. B. Clish, J. E. Bradner, W. C. Hahn and L. A. Garraway, MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells, Science, 2016, 351(6278), 1214–1218 CrossRef CAS.
- K. Marjon, M. J. Cameron, P. Quang, M. F. Clasquin, E. Mandley, K. Kunii, M. McVay, S. Choe, A. Kernytsky, S. Gross, Z. Konteatis, J. Murtie, M. L. Blake, J. Travins, M. Dorsch, S. A. Biller and K. M. Marks, MTAP Deletions in Cancer Create Vulnerability to Targeting of the MAT2A/PRMT5/RIOK1 Axis, Cell Rep., 2016, 15(3), 574–587 CrossRef CAS.
- K. J. Mavrakis, E. R. McDonald, 3rd, M. R. Schlabach, E. Billy, G. R. Hoffman, A. deWeck, D. A. Ruddy, K. Venkatesan, J. Yu, G. McAllister, M. Stump, R. deBeaumont, S. Ho, Y. Yue, Y. Liu, Y. Yan-Neale, G. Yang, F. Lin, H. Yin, H. Gao, D. R. Kipp, S. Zhao, J. T. McNamara, E. R. Sprague, B. Zheng, Y. Lin, Y. S. Cho, J. Gu, K. Crawford, D. Ciccone, A. C. Vitari, A. Lai, V. Capka, K. Hurov, J. A. Porter, J. Tallarico, C. Mickanin, E. Lees, R. Pagliarini, N. Keen, T. Schmelzle, F. Hofmann, F. Stegmeier and W. R. Sellers, Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5, Science, 2016, 351(6278), 1208–1213 CrossRef CAS PubMed.
- C. R. Smith, R. Aranda, T. P. Bobinski, D. M. Briere, A. C. Burns, J. G. Christensen, J. Clarine, L. D. Engstrom, R. J. Gunn, A. Ivetac, R. Jean-Baptiste, J. M. Ketcham, M. Kobayashi, J. Kuehler, S. Kulyk, J. D. Lawson, K. Moya, P. Olson, L. Rahbaek, N. C. Thomas, X. Wang, L. M. Waters and M. A. Marx, Fragment-Based Discovery of MRTX1719, a Synthetic Lethal Inhibitor of the PRMT5*MTA Complex for the Treatment of MTAP-Deleted Cancers, J. Med. Chem., 2022, 65(3), 1749–1766 CrossRef CAS PubMed.
- J. R. Allen, A. Amegadzie, D. J. Beylkin, S. Booker, M. P. Bourbeau, J. R. Butler, M. J. Frohn, S. O. S. Glad, B. W. Husemoen, M. R. Kaller, J. R. Butler, M. J. Frohn, S. O. S. Glad, B. W. Husemoen, M. R. Kaller, T. J. Kohn, B. A. Lanman, K. Li, Q. Liu, P. Lopez, V. V. Ma, F. Manoni, J. Medina, A. E. Minatti, J. Peiro Cadahia, L. Pettus, A. J. Pickrell, I. Sarvary, N. A. Tamayo and M. Vestergaard, Preparation of naphthyridinecarboxamide and quinolinecarboxamide compounds as PRMT5 inhibitors, WO2021163344, 2021 Search PubMed.
- K. M. Cottrell and J. P. Maxwell, Piperidin-1-yl-N-pyridin-3-yl-2-oxoacetamide derivatives useful for the treatment of MTAP-deficient and/or MTA accumulating cancers and their preparation, WO2022026892, 2022 Search PubMed.
- D. A. Erlanson, S. W. Fesik, R. E. Hubbard, W. Jahnke and H. Jhoti, Twenty years on: the impact of fragments on drug discovery, Nat. Rev. Drug Discovery, 2016, 15(9), 605–619 CrossRef CAS PubMed.
- A. R. Leach, M. M. Hann, J. N. Burrows and E. J. Griffen, Fragment screening: an introduction, Mol. BioSyst., 2006, 2(9), 430–446 RSC.
- M. Congreve, R. Carr, C. Murray and H. Jhoti, A 'rule of three' for fragment-based lead discovery?, Drug Discovery Today, 2003, 8(19), 876–877 CrossRef.
- J. B. Baell and G. A. Holloway, New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays, J. Med. Chem., 2010, 53(7), 2719–2740 CrossRef CAS PubMed.
- S. B. Shuker, P. J. Hajduk, R. P. Meadows and S. W. Fesik, Discovering high-affinity ligands for proteins: SAR by NMR, Science, 1996, 274(5292), 1531–1534 CrossRef CAS PubMed.
- A. L. Hopkins, C. R. Groom and A. Alex, Ligand efficiency: a useful metric for lead selection, Drug Discovery Today, 2004, 9(10), 430–431 CrossRef PubMed.
- P. D. Leeson and B. Springthorpe, The influence of drug-like concepts on decision-making in medicinal chemistry, Nat. Rev. Drug Discovery, 2007, 6(11), 881–890 CrossRef CAS PubMed.
- D. Chandler, Interfaces and the driving force of hydrophobic assembly, Nature, 2005, 437(7059), 640–647 CrossRef CAS.
- J. D. St Denis, R. J. Hall, C. W. Murray, T. D. Heightman and D. C. Rees, Fragment-based drug discovery: opportunities for organic synthesis, RSC Med. Chem., 2020, 12(3), 321–329 RSC.
- Molecular Operating Environment (MOE), Chemical Computing Group, 2020 Search PubMed.
- K. W. Duncan, N. Rioux, P. A. Boriack-Sjodin, M. J. Munchhof, L. A. Reiter, C. R. Majer, L. Jin, L. D. Johnston, E. Chan-Penebre, K. G. Kuplast, M. Porter Scott, R. M. Pollock, N. J. Waters, J. J. Smith, M. P. Moyer, R. A. Copeland and R. Chesworth, Structure and Property Guided Design in the Identification of PRMT5 Tool Compound EPZ015666, ACS Med. Chem. Lett., 2016, 7(2), 162–166 CrossRef CAS PubMed.
- E. Chan-Penebre, K. G. Kuplast, C. R. Majer, P. A. Boriack-Sjodin, T. J. Wigle, L. D. Johnston, N. Rioux, M. J. Munchhof, L. Jin, S. L. Jacques, K. A. West, T. Lingaraj, K. Stickland, S. A. Ribich, A. Raimondi, M. P. Scott, N. J. Waters, R. M. Pollock, J. J. Smith, O. Barbash, M. Pappalardi, T. F. Ho, K. Nurse, K. P. Oza, K. T. Gallagher, R. Kruger, M. P. Moyer, R. A. Copeland, R. Chesworth and K. W. Duncan, A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models, Nat. Chem. Biol., 2015, 11(6), 432–437 CrossRef CAS PubMed.
- F. Ullmann and J. Bielecki, Ueber Synthesen in der Biphenylreihe, Chem. Ber., 1901, 34(2), 2174–2185 CrossRef CAS.
- S. V. Ley and A. W. Thomas, Modern synthetic methods for copper-mediated C(aryl)[bond]O, C(aryl)[bond]N, and C(aryl)[bond]S bond formation, Angew. Chem., Int. Ed., 2003, 42(44), 5400–5449 CrossRef CAS PubMed.
- N. Miyaura and A. Suzuki, Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- NCT05245500, https://clinicaltrials.gov.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2md00163b |
| This journal is © The Royal Society of Chemistry 2022 |