
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImplementation of an AI-assisted fragment-generator in an open-source platform†
Alan E.
Bilsland
a,
Angelo
Pugliese
 *b and
Justin
Bower
a
*b and
Justin
Bower
a
aCancer Research Horizons – Therapeutic Innovation, Cancer Research UK Beatson Institute, Garscube Estate, Switchback Road, Glasgow G61 1BD, UK
bBioAscent Discovery, Bo'Ness Road, Newhouse, Lanarkshire ML1 5UH, UK. E-mail: apugliese@bioascent.com
First published on 15th August 2022
Abstract
We recently reported a deep learning model to facilitate fragment library design, which is critical for efficient hit identification. However, our model was implemented in Python. We have now created an implementation in the KNIME graphical pipelining environment which we hope will allow experimentation by users with limited programming knowledge.
Introduction
Fragment based drug discovery has demonstrated notable successes in the past several years with Sotorasinib and Asciminib being the most recent approvals,1,2 while 21 fragment to lead projects were reported in 2020 alone.3 A central feature of the FBDD paradigm is that well designed fragment libraries allow proportionately greater coverage of chemical space than traditional high throughput screening (HTS) libraries.4 For example, drug-like chemical space is estimated at 1023–1060 molecules, whereas there have been calculated to be 1.66 × 1011 fragments consisting of C, N, O, S, and halogens up to 17 heavy atoms.5–7Fragments are also effective start points for optimisation since binding interactions with protein targets can be more efficient, though of lower affinity, than those of larger molecules.8,9 However, good library design is critical to exploit these potential advantages. Fragment libraries must balance chemical and pharmacophore diversity, molecular complexity, and physicochemical characteristics.
We recently reported a deep learning fragment generator model using a chemical autoencoder.10 Full details of the model are given in that publication. Briefly, however, autoencoders are an encoder/decoder neural network architecture.11 In training, the encoder produces a compressed “latent” representation of its input, while the decoder reconstructs inputs from this. In chemical autoencoders, inputs/outputs are molecular representations. A frequently used approach is based on encoding Daylight SMILES strings.12,13 In generation, the space of latent representations is sampled and, in the aforementioned case, novel molecules can be output in SMILES format.14
Our model is based on Long Short Term Memory Recurrent Neural Networks15 and encodes both SMILES and chemical/pharmacophore fingerprint features. Empirically, merging chemical representations appears to result in improved generative performance, similar to the approach of Bjerrum and colleagues who trained “heteroencoder” models to convert between different representations of the same molecule.14 In our original publication we obtained higher similarity scores at greater sampling distance around known molecules using our model than with others tested.
We also applied transfer learning to the fingerprint decoder branch to develop a model which simultaneously outputs fragment SMILES and predicts the likelihood that generated molecules will be “privileged” fragments, capable of binding to multiple protein targets. We based this training on data from previous in-house fragment screens.10 Unlike the undesirable phenomenon of pan-assay interference compounds in HTS, fragments able to bind to multiple target classes can be useful library members since specificity is built in after screening, during fragment elaboration.16
In generation, our model attempts to optimise this fragment score while also considering synthetic accessibility score,17 and fraction of sp3-hybridised carbons (FSP3), among other features. Previous analyses have indicated poorer DMPK and clinical success rates for highly sp2-hybridised molecules.18,19 Therefore, increasing fragment library 3-dimensionality may be advantageous.20,21 Although greater 3D character in a library may also lead to lower hit rates, hit rate is not the defining quality metric for a library and greater specificity may be obtained with more out of plane interactions in certain target pockets.22
Optimisation of combined scores is performed using particle swarm optimisation (PSO), using the PySwarms library.23,24 In PSO, multiple “particles” move in a search space, each having a “self-belief” (cognitive) component of velocity, and a component influenced by the whole swarm (social). Velocities are iteratively updated as each particle moves, based on scoring the current positions of all particles and comparison with the global best position of the swarm seen so far. In this application, the position of a particle corresponds to a latent embedding that may decode to a valid molecule.
Ultimately, particles are expected to converge around a good solution. An additional inertial weight term controls the magnitude of velocity update and the exploration/convergence behaviour. This sampling approach was previously reported to be an efficient approach for sampling with continuous latent-variable chemical generator models25 and has the advantage that multiple objectives for optimisation are easily combined.
In our implementation, the swarm is initialised around the latent encoding of a seed molecule and searches a bounded neighbourhood in the vicinity of the seed. PSO runs are performed several times for each input. If a higher scoring molecule is found on any run, the swarm reinitialises around the latent encoding of that new molecule for the next run. As the PSO search converges at high scoring molecules, similar molecules can also be found which improve the score. The swarm therefore moves to sample better regions of the search space over successive runs. For each initial input, we perform N runs, which we refer to as sampling depth.
KNIME (Konstanz Information Miner) Analytics Platform (KNIME AG, Zurich, Switzerland) is an open source visual programming platform for data science.26 Within the environment, data processing workflows are built using a pipelining model in which successive processing steps are implemented by connected nodes, each of which executes one step of the overall workflow and processed data flows between nodes.
The interface is intuitive and easy to use, allowing integration with a broad range of data sources, 3rd party tools and software platforms; in particular, a range of cheminformatics tools are available.27 Furthermore, the platform provides integration with several programming languages including Python and libraries such as Tensorflow for deep learning applications.28
Workflows can be exported and shared easily among individual users along with associated data, while the KNIME Server platform which is available via either commercial or academic licensing allows for easy deployment of workflows in a webportal interface. The fragment generator model described above has now been made available as an implementation in KNIME, developed in version 4.1.2 and tested on KNIME Server version 4.10.1 on Ubuntu 16.04. The workflow is available at https://github.com/abilsland/fragmentEncoder_KNIME.
KNIME server implementation
Screenshots of the protocol in KNIME Server webportal are given in Fig. 1. As shown in Fig. 1A, users are prompted to upload a SMILES file containing seed molecules to initiate PSO. A default file is provided in the data folder of the workflow. Our training molecules were stripped of stereochemistry and the workflow performs this step, in addition to removal of SMILES containing certain atoms not in the model vocabulary if present. The complete model vocabulary is available within the workflow annotation and at https://github.com/abilsland/fragmentEncoder. Additional filters can be added easily to a Table Creator node in the workflow if required (Fig. 2A).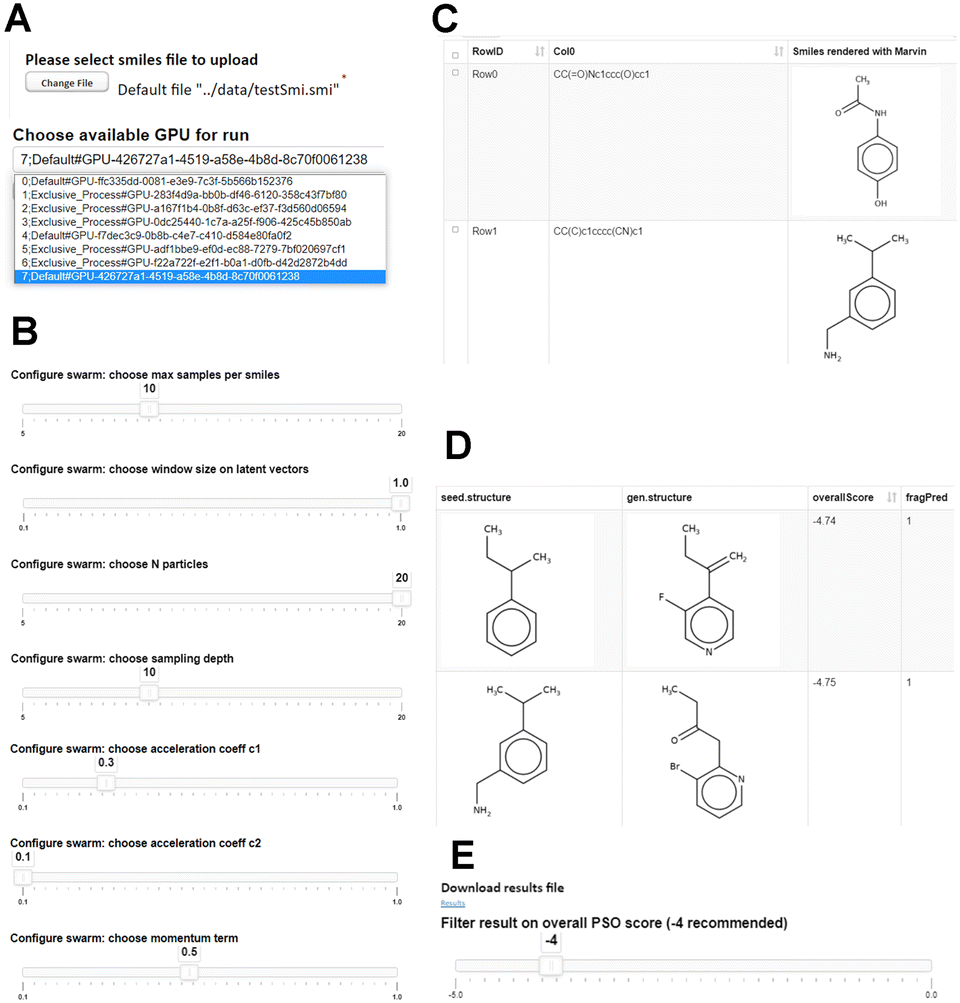 | ||
| Fig. 1 KNIME webportal implementation. A, File upload and GPU choice screens. B, PSO swarm configuration options. C, Display of uploaded PSO seeds. D, PSO results display. E, Download and filtering. | ||
 | ||
| Fig. 2 User editable parts of the workflow. A, Input molecule filters. Top, filter SMILES metanode; bottom, opened metanode. If additional filters are required, these can be added as SMARTS patterns in the Table Creator node. B, Swarm configuration components. Top, the components; bottom, opened component. If different limits on the PSO options are required, the Table Creator configuration can be edited. | ||
Uploaded SMILES are displayed (Fig. 1C), and the user is given the choice of GPUs available on the server to use for the run (Fig. 1A). We note that some potential users may operate in environments with limited resource where different applications compete for GPU resource and have differing compute mode requirements. We therefore display the compute mode. Default (rather than exclusive process) is preferred to allow multiple processes to run on the same GPU.
GPU information is displayed in the format <index>;<compute mode>#<GPU UUID>, where index is the GPU index returned by the Nvidia System Management Interface tool (nvidia-smi; https://developer.nvidia.com/nvidia-system-management-interface). We use the GPU Universally Unique ID (UUID) to set visible devices for Keras Network Executor KNIME nodes and within the Python script that runs PSO. This avoids the possibility for index inconsistencies that might arise in the Network Executor nodes since the default CUDA ordering of GPUs may differ from that returned by nvidia-smi.
After initial processing, Morgan fingerprints (radius 2) and pharmacophore feature fingerprints are generated, and features retained in our original training are extracted.
Subsequently, fingerprints and smiles are encoded by relevant branches of the model, and latent vectors generated for the inputs. In our original implementation, this step would require knowledge of RDKit (RDKit: open-source cheminformatics; https://www.rdkit.org) and use of the utility functions supplied with the code.
A key feature for web users without knowledge of Python is the ability to easily control key parameters of the particle swarm using sliders (Fig. 1B). In our original implementation, control of these would have involved modification of the code and pickled parameters file. The default values for these in the workflow are those values used in the publication.10 Adjustment of the limits of these is easily accomplished by updating the entries in the Table Creator nodes in the “configure swarm” components of the workflow (Fig. 2B).
Users have control of the number of particles, number of iterations over which to perform PSO, boundary distance to search around the current latent vector, sampling depth (number of samples starting from the embedding of each molecule in the input), in addition to swarm velocity and inertia weight parameters. For example, more iterations will increase the time of a run but may lead to more high-scoring outputs while larger bounds may produce greater diversity but also increases the search space which may reduce the chance of finding high-scoring molecules.
After completion of the run, generated molecules are displayed, together with the seed molecule whose latent vector defined the search vicinity on that sampling run (Fig. 1D). Note that seed molecules in the output are not necessarily those present in the input file, since our search strategy recentres the swarm each time a molecule with improved score is found on any sampling round up to sample depth for each input. Therefore, seeds which resulted in high-scoring generated molecules may themselves have been generated on a previous run. Also returned are overall PSO score and the frequent-hitter prediction score.
Finally, users can download the results file or adjust the overall score cutoff for molecules returned by use of the slider in Fig. 1E. To our knowledge there is not yet a dynamically updating Table Viewer in KNIME that can be controlled with a variable. Therefore, it is necessary to use the “next” and “back” buttons in the webportal to update the molecules displayed and available for download. We suggest the default value of −4 as shown. However, some penalties in PSO correspond to features which other users may deem acceptable such as very low or high fsp3 and 7/8 member rings. Thus, lower scores may be acceptable.
One important aspect of scoring is the absence of structural alerts in generated molecules. These are defined by Daylight SMARTS filters (https://www.daylight.com/dayhtml/doc/theory/theory.smarts.html). We supply a default set in a file in the workflow data folder “SMARTS_unwanted_functionalities.txt”. However, these are mainly a publicly available set from SureChembl.org (https://www.surechembl.org/knowledgebase/169485-non-medchem-friendly-smarts). We typically employ a more extensive set internally and it is highly recommended that users augment the supplied set. Without sufficient definition of unsuitable molecules, the swarm may fail to converge in high-scoring regions of the search space.
We direct interested readers to an additional set of filters derived from the pan-assay interference compound filters reported by Baell and Holloway.29 As detailed in ref. 30, these were converted to SMARTS format by Rajarshi Guha. Subsequently, Greg Landrum curated these for optimal performance in RDKit (https://rdkit.blogspot.com/2015/08/curating-pains-filters.html). These curated filters are available at https://github.com/rdkit/rdkit/blob/master/Data/Pains/wehi_pains.csv.
KNIME requirements to execute the workflow are Python, RDKit, and Keras integration. For those unfamiliar with KNIME, RDKit and Keras nodes can be installed from within the software, using File > Install KNIME extensions. Within the available software extension locations, RDKit nodes are found at KNIME Community Extensions – Cheminformatics and Python/Keras nodes at KNIME & Extensions. However, simply opening the workflow will also give the option to automatically search and install required extensions.
On first installation of the Python/KNIME nodes, these node sets also require Anaconda (Anaconda Inc., Austin, TX 78701, USA), available at https://www.anaconda.com. Within KNIME, the path to the Anaconda installation should be set in File > Preferences > Python. Default Anaconda environments can be generated automatically in KNIME preferences, though our model requires a specific environment which we make available as a yml file, along with the workflow. For those not comfortable working in Anaconda at the command line, the required environment can be generated by importing the file in the Anaconda Navigator GUI. KNIME preferences should then be set to use this environment. The same environment should be used within the File > Preferences > Python deep learning setting.
Although our implementation has only been tested on in our internal Linux installation of KNIME Server, the workflow is expected to be portable to Windows. A Windows-specific conda environment file is also provided since the distributions of some required packages obtained under Anaconda are platform-specific.
Use case – generation of a GPCR-focused fragment library
To illustrate use of the workflow, we obtained a set of known GPCR-targeted drugs from ESI† Table S2 of ref. 31. SMILES were obtained from these using the Chemical Identifier Resolver API at the National Cancer Institute Computer Aided Drug Design Group Chemoinformatics Tools and User Services (https://cactus.nci.nih.gov/chemical/structure). Fragments were obtained from these using the retrosynthetic combinatorial analysis procedure (RECAP,32). Existing RDKit KNIME integration includes a molecule fragmenter node, although this is not based on RECAP.We removed dummy atoms, eliminated fragments with heavy atom count less than 8, more than 4 rings, and those with any substructure matches against our SMARTS filters. We then randomised each remaining SMILES 5 times since different SMILES corresponding to the same molecule have different latent encodings by the model.33,34 These were used as input to the model with default settings as above and utilising our in-house SMARTS. These are available in ESI† File S1.
The workflow generated 2847 unique molecules which were further filtered according to our standard in-house fragment-like property filters,35 resulting in a list of 2031 fragments. After filtering further on Tanimoto similarity between generated molecules versus both the input list, and our existing fragment library, using Morgan fingerprints (radius 2), we obtained 340 molecules. 232 were retained after visual inspection. These are available in ESI† File S2.
For all molecules in the input and generated sets, we then calculated 2D pharmacophore fingerprints in RDKit and performed pair-wise similarity comparisons between all generated and input molecules. For each generated molecule we obtained the maximum value of Morgan and pharmacophore fingerprints similarity against all input fragments from the GPCR set. As shown in Fig. 3, despite the relative chemical dissimilarity between generated and input molecules, nevertheless similar 2D pharmacophore features are retained suggesting that these molecules might provide chemically novel starting points for a GPCR-focussed fragment library.
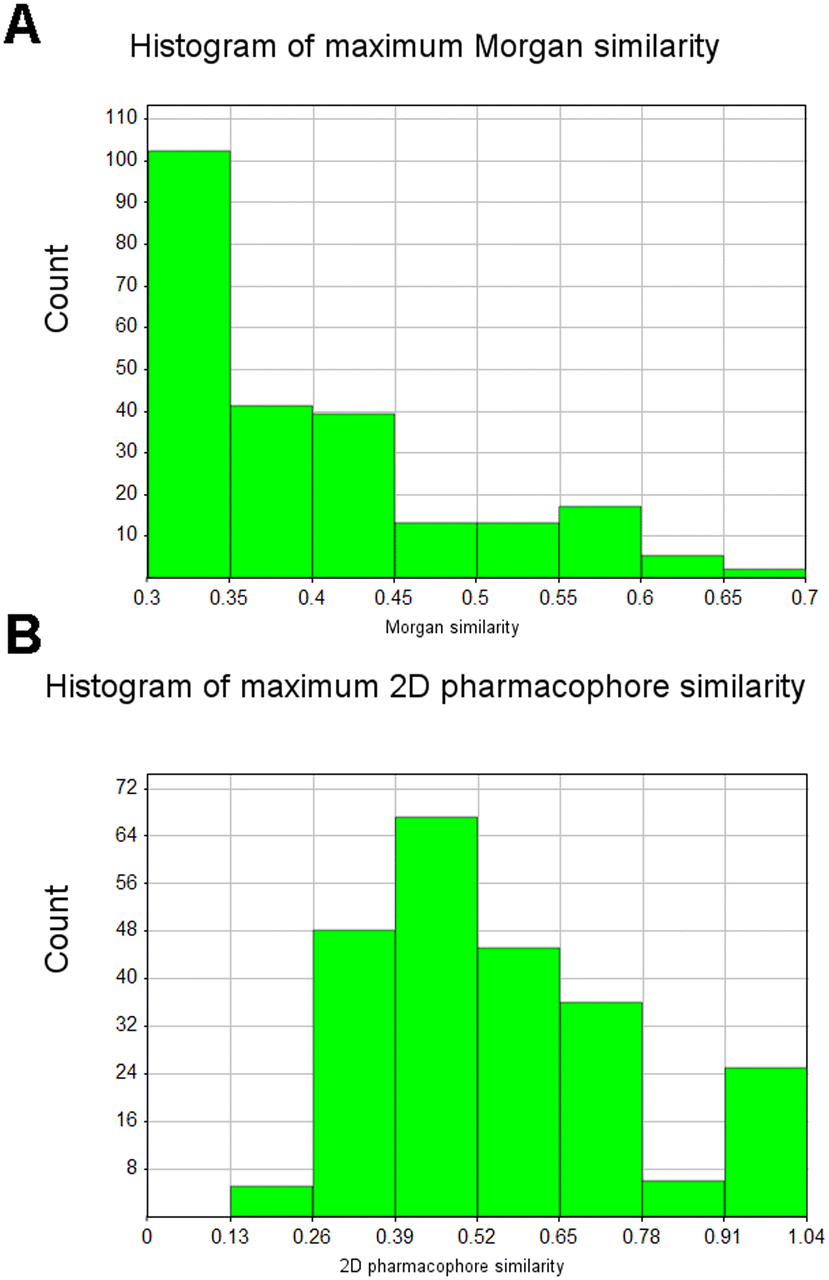 | ||
| Fig. 3 Analysis of generated molecules seeded from the GPCR fragment set. A, Distribution of maximum Morgan fingerprint similarity between generated molecules and input molecules after filtering. B, Distribution of maximum 2D pharmacophore similarity between generated and input molecules. All fingerprints were calculated in RDKit. Histograms were generated in Vortex software (Dotmatics, Bishops Stortford, UK). | ||
Conclusions
In recent years, deep learning applications for de novo design in drug discovery have gained considerable momentum.12,36 However, applications specifically focused on FBDD have received less attention than those focused on drug-like chemical space. Nevertheless, fragment-based approaches are emerging, particularly those with applications in fragment elaboration.Arús-Pous and colleagues developed a “scaffold decorator” model, in which a generator model outputs scaffolds which are modified by the addition of fragments at defined attachment points by a decorator model.37 Lim and colleagues similarly reported a scaffold-based design approach using sequential addition of atoms and bonds to scaffold molecular graphs generated by a variational autoencoder.38 Green and colleagues reported a convolutional neural network “DeepFrag”. Given structural information on a protein/ligand interaction, this model outputs the fingerprint of a fragment from a predefined library that would be predicted to improve binding affinity.39
The latter group subsequently reported an open source browser implementation of their model in order to improve accessibility to a user-base beyond those skilled in programming.40 Alberga and colleagues also recently reported a SMILES generator which was used to generate biased molecule libraries against several targets.41 This model was based on the reinforcement learning approach proposed by Olivecrona in which a previously trained generator (“prior policy”) is guided toward generation of molecules that optimise a new fitness function.42 The authors developed a novel multiobjective fitness function to output molecules with nondominated solutions on the Pareto frontier, considering multiple pairs of features.41 A GUI implementation of the model was made available for end-users.
With similar intent, we report here an implementation of our previous model that can be used directly in KNIME Analytics platform or deployed for use in a user-friendly webportal via KNIME Server. We hope that this may encourage experimentation by users without significant knowledge of Python programming. A significant advantage of deployment in KNIME is the considerable code-free integration with applications by various 3rd party vendors and the wide range of pre-built workflows available via KNIME Hub (https://hub.knime.com/). This would allow users to easily augment the existing workflow, adding functionality downstream for various tasks.
For example, a straightforward modification would be to include selection or filtering based on fragments with known activity against targets of interest, or on the members of an existing library for applications such as that shown above. Falcón-Cano and colleagues also reported a KNIME workflow for prediction of aqueous solubility using gradient boosting models which requires only SMILES as input.43 We did not consider solubility directly in our model, though it is a parameter of critical importance in fragment library design due to high fragment concentrations required to detect binding in fragment screens.35,44 Notably, Simulations Plus (Lancaster, CA, USA) provide KNIME integration for their ADMET Predictor software for prediction of a wide range of properties including solubility and log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D.
D.
The 3D-e-Chem node set also provides a range of functionality for structural cheminformatics, including ligand and pharmacophore alignment, and docking.45 Another key commercial vendor with substantial KNIME integration is Schrödinger, LLC (New York, NY, USA). These nodes provide access to a wide range of ligand- and structure-based functionality from their Glide, Prime, Phase, MacroModel, and Jaguar applications. Thus, one might also envisage workflows coupling molecular generation to virtual screening against specific targets.
In this respect, it is interesting to note that, in our original publication, we demonstrated the potential for our model to generate structures not in the FDB-17 screening subset of the GDB-17 database, which comprises a virtual screening set of 10 M fragments selected for even coverage of lower complexity fragment space suitable for hit progression.46 Thus, generative approaches can suggest novel chemical matter that may not be found even in relatively large virtual screens with predefined libraries.
While we cannot provide a comprehensive review of KNIME cheminformatics capabilities here, we hope the above examples may provide readers unfamiliar with the platform some insight into the potential value of combining molecular de novo design approaches based on deep learning, such as our model, with the wide range of additional tools available and deployable in the KNIME webportal.
Author contributions
Author roles: A. B., data curation, formal analysis, investigation, methodology, software, validation, visualization, writing–initial draft, writing–review and editing; A. P., conceptualization, formal analysis, methodology, project administration, supervision, visualization, writing–initial draft, writing–review and editing; J. B., formal analysis, funding acquisition, project administration, resources, writing–review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All authors were supported by Cancer Research UK Core Grant Numbers A17096 (core funding the CRUK Beatson Institute Drug Discovery Unit) and A17196 (core funding to the CRUK Beatson Institute).References
- B. A. Lanman, et al., Discovery of a Covalent Inhibitor of KRASG12C (AMG 510) for the Treatment of Solid Tumors, J. Med. Chem., 2020, 63(1), 52–65 CrossRef CAS PubMed.
- J. Schoepfer, et al., Discovery of Asciminib (ABL001), an Allosteric Inhibitor of the Tyrosine Kinase Activity of BCR-ABL1, J. Med. Chem., 2018, 61(18), 8120–8135 CrossRef CAS PubMed.
- W. Jahnke, et al., Fragment-to-Lead Medicinal Chemistry Publications in 2019, J. Med. Chem., 2020, 63(24), 15494–15507 CrossRef CAS PubMed.
- C. W. Murray and D. C. Rees, The rise of fragment-based drug discovery, Nat. Chem., 2009, 1(3), 187–192 CrossRef CAS PubMed.
- R. S. Bohacek, C. McMartin and W. C. Guida, The art and practice of structure-based drug design: a molecular modeling perspective, Med. Res. Rev., 1996, 16(1), 3–50 CrossRef CAS PubMed.
- P. Ertl, Cheminformatics analysis of organic substituents: identification of the most common substituents, calculation of substituent properties, and automatic identification of drug-like bioisosteric groups, J. Chem. Inf. Comput. Sci., 2003, 43(2), 374–380 CrossRef CAS PubMed.
- L. Ruddigkeit, et al., Enumeration of 166 billion organic small molecules in the chemical universe database GDB-17, J. Chem. Inf. Model., 2012, 52(11), 2864–2875 CrossRef CAS PubMed.
- A. L. Hopkins, C. R. Groom and A. Alex, Ligand efficiency: a useful metric for lead selection, Drug Discovery Today, 2004, 9(10), 430–431 CrossRef PubMed.
- I. D. Kuntz, et al., The maximal affinity of ligands, Proc. Natl. Acad. Sci. U. S. A., 1999, 96(18), 9997–10002 CrossRef CAS PubMed.
- A. E. Bilsland, et al., Automated Generation of Novel Fragments Using Screening Data, a Dual SMILES Autoencoder, Transfer Learning and Syntax Correction, J. Chem. Inf. Model., 2021, 61(6), 2547–2559 CrossRef CAS PubMed.
- D. Bank, N. Koenigstein and R. Giryes, Autoencoders, ArXiv, 2021, preprint, arXiv:2003.05991, DOI:10.48550/arXiv.2003.05991.
- D. C. Elton, et al., Deep learning for molecular design-a review of the state of the art, Mol. Syst. Des. Eng., 2019, 4(4), 828–849 RSC.
- D. Weininger, SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules, J. Chem. Inf. Comput. Sci., 1988, 28(1), 31–36 CrossRef CAS.
- E. J. Bjerrum and B. Sattarov, Improving Chemical Autoencoder Latent Space and Molecular De Novo Generation Diversity with Heteroencoders, Biomolecules, 2018, 8(131), 1–17 Search PubMed.
- S. Hochreiter and J. Schmidhuber, Long Short-Term Memory, Neural Comput., 1997, 9(8), 1735–1780 CrossRef CAS PubMed.
- P. S. Kutchukian, et al., Large Scale Meta-Analysis of Fragment-Based Screening Campaigns: Privileged Fragments and Complementary Technologies, J. Biomol. Screening, 2015, 20(5), 588–596 CrossRef CAS PubMed.
- P. Ertl and A. Schuffenhauer, Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions, J. Cheminf., 2009, 1(8), 1–11 Search PubMed.
- F. Lovering, J. Bikker and C. Humblet, Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success, J. Med. Chem., 2009, 52(21), 6752–6756 CrossRef CAS PubMed.
- T. J. Ritchie and S. J. F. Macdonald, The impact of aromatic ring count on compound developability – are too many aromatic rings a liability in drug design?, Drug Discovery Today, 2009, 14(21–22), 1011–1020 CrossRef CAS PubMed.
- A. D. Morley, et al., Fragment-based hit identification: thinking in 3D, Drug Discovery Today, 2013, 18(23–24), 1221–1227 CrossRef PubMed.
- N. Fuller, et al., An improved model for fragment-based lead generation at AstraZeneca, Drug Discovery Today, 2016, 21(8), 1272–1283 CrossRef PubMed.
- J. A. Johnson, et al., Evaluating the Advantages of Using 3D-Enriched Fragments for Targeting BET Bromodomains, ACS Med. Chem. Lett., 2019, 10(12), 1648–1654 CrossRef CAS PubMed.
- J. Kennedy and R. Eberhart, Particle swarm optimization, in Proceedings of ICNN'95 – International Conference on Neural Networks, 1995 Search PubMed.
- L. J. V. Miranda, PySwarms: a research toolkit for particle swarm optimization in Python, J. Open Source Softw., 2018, 3(433), 1–2 Search PubMed.
- R. Winter, et al., Efficient multi-objective molecular optimization in a continuous latent space, Chem. Sci., 2019, 10(34), 8016–8024 RSC.
- M. R. Berthold, et al., KNIME: The Konstanz Information Miner, Data Analysis, Machine Learning and Applications, 2008, pp. 319–326 Search PubMed.
- A. Afantitis and G. Melagraki, Cheminformatics Toolboxes and Workflows within KNIME Analytics, Curr. Med. Chem., 2020, 27(38), 6442–6443 CrossRef CAS PubMed.
- M. Abadi, et al., TensorFlow: A system for large-scale machine learning. Proceedings of Osdi'16: 12th Usenix Symposium on Operating Systems Design and Implementation, 2016, pp. 265–283 Search PubMed.
- J. B. Baell and G. A. Holloway, New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays, J. Med. Chem., 2010, 53(7), 2719–2740 CrossRef CAS PubMed.
- S. Saubern, R. Guha and J. B. Baell, KNIME Workflow to Assess PAINS Filters in SMARTS Format. Comparison of RDKit and Indigo Cheminformatics Libraries, Mol. Inf., 2011, 30(10), 847–850 CrossRef CAS PubMed.
- K. Sriram and P. A. Insel, G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs?, Mol. Pharmacol., 2018, 93(4), 251 CrossRef CAS PubMed.
- X. Q. Lewell, et al., RECAP--retrosynthetic combinatorial analysis procedure: a powerful new technique for identifying privileged molecular fragments with useful applications in combinatorial chemistry, J. Chem. Inf. Comput. Sci., 1998, 38(3), 511–522 CrossRef CAS PubMed.
- J. Arús-Pous, et al., Randomized SMILES strings improve the quality of molecular generative models, J. Cheminf., 2019, 11(71), 1–13 Search PubMed.
- E. J. Bjerrum, SMILES enumeration as data augmentation for neural network modeling of molecules, arXiv, 2017, reprint, arXiv:1703.07076v2, pp. 1–7, DOI:10.48550/arXiv.1703.07076.
- M. Bon, et al., Fragment-based drug discovery—the importance of high-quality molecule libraries, Mol. Oncol., 2022 DOI:10.1002/1878-0261.13277.
- F. Palazzesi and A. Pozzan, Deep Learning Applied to Ligand-Based De Novo Drug DesignDe novo drug design, in Artificial Intelligence in Drug Design, ed. A. Heifetz, Springer US, New York, NY, 2022, pp. 273–299 Search PubMed.
- J. Arús-Pous, et al., SMILES-based deep generative scaffold decorator for de novo drug design, J. Cheminf., 2020, 12(1), 38 Search PubMed.
- J. Lim, et al., Scaffold-based molecular design with a graph generative model, Chem. Sci., 2020, 11(4), 1153–1164 RSC.
- H. Green, D. R. Koes and J. D. Durrant, DeepFrag: a deep convolutional neural network for fragment-based lead optimization, Chem. Sci., 2021, 12(23), 8036–8047 RSC.
- H. Green and J. D. Durrant, DeepFrag: An Open-Source Browser App for Deep-Learning Lead Optimization, J. Chem. Inf. Model., 2021, 61(6), 2523–2529 CrossRef CAS PubMed.
- D. Alberga, et al., De Novo Drug Design of Targeted Chemical Libraries Based on Artificial Intelligence and Pair-Based Multiobjective Optimization, J. Chem. Inf. Model., 2020, 60(10), 4582–4593 CrossRef PubMed.
- M. Olivecrona, et al., Molecular de novo design through deep reinforcement learning, J. Cheminf., 2017, 9(48), 1–14 Search PubMed.
- G. Falcón-Cano, C. Molina and M. A. Cabrera-Perez, ADME prediction with KNIME: A retrospective contribution to the second “Solubility Challenge”, ADMET DMPK, 2021, 9(3), 209–218 Search PubMed.
- M. J. Harner, A. O. Frank and S. W. Fesik, Fragment-based drug discovery using NMR spectroscopy, J. Biomol. NMR, 2013, 56(2), 65–75 CrossRef CAS PubMed.
- A. J. Kooistra, et al., 3D-e-Chem: Structural Cheminformatics Workflows for Computer-Aided Drug Discovery, ChemMedChem, 2018, 13(6), 614–626 CrossRef CAS PubMed.
- R. Visini, M. Awale and J. L. Reymond, Fragment Database FDB-17, J. Chem. Inf. Model., 2017, 57(4), 700–709 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2md00152g |
| This journal is © The Royal Society of Chemistry 2022 |