
Covalent cannabinoid receptor ligands – structural insight and selectivity challenges†
Ian
Liddle
 a,
Michelle
Glass
b,
Joel D. A.
Tyndall
c and
Andrea J.
Vernall
*a
a,
Michelle
Glass
b,
Joel D. A.
Tyndall
c and
Andrea J.
Vernall
*a
aDepartment of Chemistry, University of Otago, Dunedin, New Zealand. E-mail: andrea.vernall@otago.ac.nz; Tel: +64 3 479 5214
bDepartment of Pharmacology and Toxicology, University of Otago, Dunedin, New Zealand
cSchool of Pharmacy, University of Otago, Dunedin, New Zealand
First published on 4th April 2022
Abstract
X-ray crystallography and cryogenic electronic microscopy have provided significant advancement in the knowledge of GPCR structure and have allowed the rational design of GPCR ligands. The class A GPCRs cannabinoid receptor type 1 and type 2 are implicated in many pathophysiological processes and thus rational design of drug and tool compounds is of great interest. Recent structural insight into cannabinoid receptors has already led to a greater understanding of ligand binding sites and receptor residues that likely contribute to ligand selectivity. Herein, classes of heterocyclic covalent cannabinoid receptor ligands are reviewed in light of the recent advances in structural knowledge of cannabinoid receptors, with particular discussion regarding covalent ligand selectivity and rationale design.
 Ian Liddle | Ian Liddle completed his MSci degree in 2020 in Drug Discovery at the University of Dundee (United Kingdom) having completed an Honour's project working on Proteolysis Targeting Chimeras and his master's project on covalent fragments. He is currently working towards his PhD at the University of Otago (New Zealand) in the Department of Chemistry. His PhD research project focusses on the design and synthesis of selective, covalent fluorescent tools and other ligands for the cannabinoid type 2 receptor. Other research interests include medicinal chemistry and drug discovery. |
 Michelle Glass | Michelle Glass received her Ph.D. in Pharmacology working on describing the localisation of the cannabinoid CB1 receptor in the human brain from the University of Auckland (New Zealand). Her post doctoral studies at the National Institutes of Health (Maryland, USA) focused on the molecular pharmacology of cannabinoid receptor signalling through G proteins. Her current research focus at the University of Otago (New Zealand) is on understanding how molecular pharmacology can be utilised to improve drug therapy through more selective targeting of subsets of receptors or signalling pathways, such as through biased signalling, allosteric modulators or dimer targeted therapies. |
 Joel Tyndall | Joel Tyndall was awarded his PhD in 2000 from the Centre for Drug Design and Development at the University of Queensland (Australia). He spent a year at the University of Edinburgh after which he returned to the University of Queensland. In 2004 he took up a position as lecturer at the School of Pharmacy at the University of Otago. He is now an Associate Professor in Medicinal Chemistry and his research focuses on drug discovery and drug target analysis, in particular, in cancer and infectious diseases as well as GPCRs. |
 Andrea Vernall | Andrea Vernall received her Ph.D. in organic chemistry (metathesis reactions of amino acid derivatives) from the University of Canterbury (New Zealand). She then undertook postdoctoral positions in peptidomimetics at the University of Queensland (Australia) and in GPCR ligand and fluorescent tool development at the University of Nottingham (United Kingdom). Since 2013, she has been a research group leader at the University of Otago (New Zealand). Her current research projects include the development of ligands, fluorescent probes and prodrugs, in particular for cannabinoid and adenosine receptors. |
1. Introduction
G protein-coupled receptors (GPCR) are the largest group of transmembrane signalling proteins and play a crucial role in regular physiology and pathological conditions. GPCRs can be activated by an interaction with a ligand such as a hormone, peptide, ion and/or neurotransmitter.1 Given the important physiological roles of GPCRs, it is not surprising that drugs targeting these receptors account for approximately 35% of all FDA approved drugs and around 27% of the global drug market share.2,3 GPCR structure and ligand-binding site topography has historically been gleaned from extensive receptor mutagenesis, ligand structure–activity-relationship (SAR) data, use of covalent ligands and receptor homology modelling.4–7 Key developments in receptor expression, stabilisation and crystallisation techniques and advancements in X-ray crystallography and single particle cryogenic electron microscopy (cryo-EM) have now accelerated the generation of high resolution GPCR structures.8Cannabinoid receptors (CBRs) are class A GPCRs and form part of the highly controlled endocannabinoid system.9 There are two recognised CBR subtypes – the cannabinoid type 1 receptor (CB1R) and cannabinoid type 2 receptor (CB2R). CB1R is highly expressed in the brain while CB2R is predominantly found peripherally, in particular in immune cells.9 Ligands targeting CB1R may be beneficial for treating conditions such as pain, addiction, obesity and metabolic disorders while ligands targeting CB2R may be beneficial for inflammatory diseases, chronic pain and neurological conditions.9,10 Since 2016, there has been at least eight CB1R and four CB2R X-ray crystallography and cryo-EM structures of CBRs published,11–19 in a range of inactive to active states (e.g.Fig. 1). These recent advancements in CBR structural knowledge have shed light on CBR-ligand receptor interactions, including in terms of different receptor ‘states’ and how CB1R and CB2R differ.20
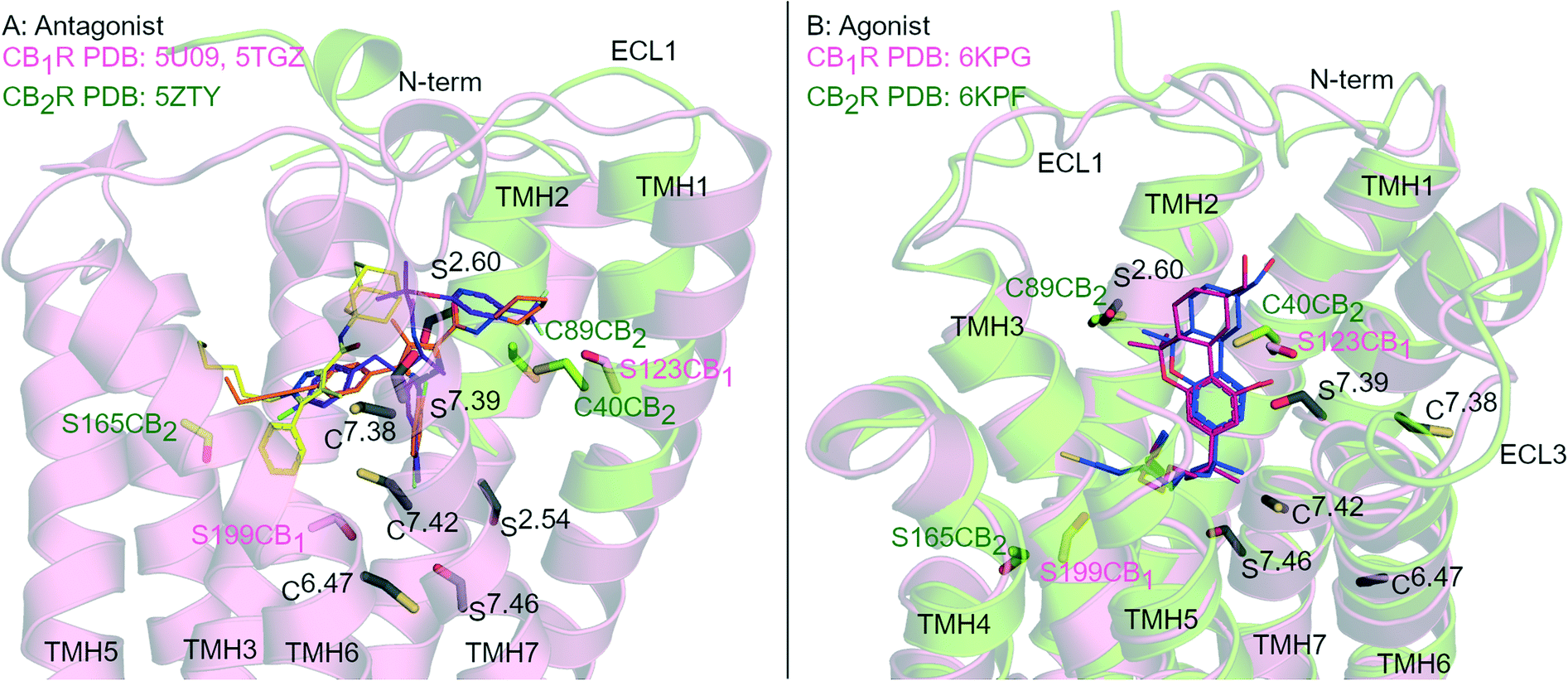 | ||
| Fig. 1 Antagonist- and agonist-bound CBR structures. A: Antagonist-bound overlay of CB1R (pink cartoon) with AM6538 (orange carbons, PDB: 5TGZ) and Taranabant (purple carbons, PDB: 5 U09), and CB2R (green cartoon) with AM10257 (yellow carbons, PDB: 5ZTY). B: Agonist-bound overlay of CB1R (pink cartoon) with AM841 (blue carbons, PDB: 6KPG), and CB2R (green cartoon) with AM12033 (purple carbons, PDB: 6KPF). ECL 2 loop is hidden for clarity, conserved Ser (S) and Cys (C) residues in both CBRs are coloured black and numbered by Ballesteros–Weinstein numbering for GPCRs, Ser and Cys uniquely placed in one receptor subtype are coloured pink (CB1R) or green (CB2R). | ||
Covalent ligands are invaluable tools to investigate GPCRs.21,22 Covalent ligands encompass reversible and irreversible ligands and, depending on the ‘warhead,’ are also termed affinity labels and/or photoaffinity labels.23 Ideally, to limit undesired reactions, a covalent ligand should first bind to the target through high-affinity non-covalent interactions and only then form a covalent bond (with or without photoactivation). Aside from use as tool compounds, covalent ligands are also experiencing a resurgence of interest as a direct drug candidates.24 Covalent ligands are useful tools when ‘biofunctionalised’ – a second functionality can be added such that the covalent ligand-GPCR complex can be ‘tagged’ with a tracer/fluorophore.25–27 Covalent ligands offer enormous insight when paired with GPCR mutagenesis studies,6,28 capture compound mass spectrometry,29,30 and more recently tetra-functionalised covalent ligands have been used to detect low abundance GPCRs.31 Covalent CBR ligands32,33 have been used to identify essential residues in CBR ligand binding sites and are frequently combined with mutagenesis and molecular modelling studies. This methodology paradigm, termed ligand-assisted protein structure (LAPS), takes advantage of bottom-up proteomics to characterise the CBR structural features key to ligand engagement.28
Rational design of subtype selective CBR covalent ligands has proven challenging due to the high sequence similarity and Ser/Cys positioning within the orthosteric pockets of CB1R and CB2R (Fig. 1). Agonists with different core scaffolds bind in near identical poses in CB1R and CB2R13,17 and can interact with similar residues (Fig. 1B), with equivalently positioned serine (Ser2.54, Ser2.60, Ser7.39, Ser7.46) and cysteine (Cys6.47, Cys7.38 and Cys7.42) (superscripts indicate Ballesteros-Weinstein numbering for GPCR34). Different antagonist/inverse agonist scaffolds usually have comparatively divergent binding modes between CBR subtypes20 (Fig. 1A), but again can potentially access conserved nucleophilic serine and cysteine residues. In particular, Cys6.47 is part of the CWxP motif in TMH 6 and is a highly conserved motif in class A GPCRs positioned within the orthosteric binding site (Fig. 1). There is great interest in selective CB2R ligands devoid of CB1R-mediated psychotropic effects, both as potential drugs and as covalent chemical tool compounds. This review examines the binding mode and selectivity of each chemical class of heterocyclic covalent CBR ligands in light of recent X-ray crystallography11,12,14,16,18,19 and cryo-EM studies13,15,17 and provides insight into the future rational design of subtype selective CBR covalent ligands.
2. Pyrazoles
2.1 Biaryl pyrazoles
Biaryl pyrazoles are one of the most well studied CB1R antagonist scaffolds. Widely known non-covalent ligands include the highly CB1R selective inverse agonists AM251 (Ki = 7.49 nM (6.38–8.78 nM rCB1R; Ki = 2290 (1640–3190 nM) mCB2R))35 and SR141716A (Rimonabant) (Ki = 11.5 nM (8.45–13.7 nM) rCB1R; Ki = 1640 nM (1440–1850 nM) mCB2R)35 (Fig. 2A) (refer to ESI† Table S1 for further data on ligands). Several biaryl pyrazoles containing covalent warheads have been developed, e.g.1–4 (ref. 36) and AM6538 (ref. 12) (Fig. 2A). Azide or isothiocyanate-containing 1–4 showed high to moderate apparent binding affinity and selectivity for CB1R, and maintained antagonist/inverse agonist activity similar to the non-covalent inverse agonist SR141716A.36 Azides 1 and 2 (with UV irradiation) and isothiocyanates 3 and 4 were shown to form an irreversible interaction with CB1R, such that subsequent [3H]CP-55940 binding was precluded. AM6538 (Ki = 5.1 ± 0.9 nM hCB1R, CB2R data not reported), containing an acetylenic chain terminating in a nitrate (in place of the iodo of AM251), was designed to facilitate crystallisation of CB1R.12 The nitrate warhead was introduced to act as an electrophile that could be displaced by a nucleophilic CB1R amino acid such as Cys3556.47, or, that could form strong hydrogen bond and π–π interactions with CB1R. Analysis suggested that AM6538 interacts with CB1R as an intact molecule despite wash-resistant binding and evidence of covalent binding was not observed via mass spectrometry (MS).12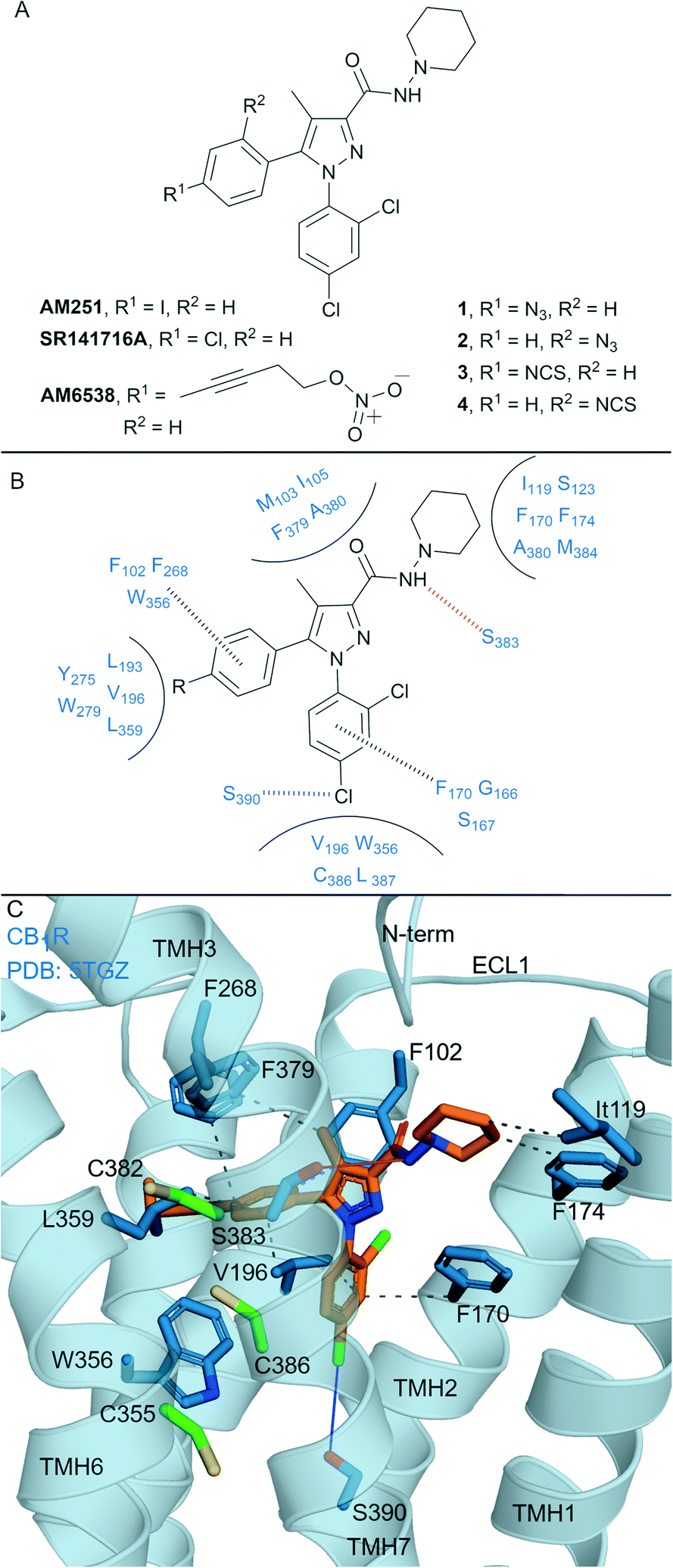 | ||
| Fig. 2 Biaryl pyrazoles. A: Non-covalent and covalent biaryl pyrazoles. B: Key residues for binding in CB1R. C: Co-crystal structure of CB1R (blue cartoon) and AM6538 (orange carbons, PDB ID: 5TGZ), showing key Cys (green carbons) and other key residues (blue). Hydrophobic interactions (black line), π–π stacking (dashed black line), hydrogen bonds (pink), halogen–hydrogen bonds (blue). Nitrate group of AM6538 was not modelled in crystal structure due to low resolution. | ||
The CB1R-AM6538 co-crystal structure12 (Fig. 2C) revealed important interactions for antagonist binding. In line with MS measurements, a covalent bond between CB1R and AM6538 was not observed. The nitrate group could not be modelled due to the lack of electron density as a result of flexibility in the carbon hinge.12 Laprairie et al. suggested AM6538 may essentially be an irreversible but non-covalent binder via strong hydrogen bond interactions between the nitro and Tyr275(5.39) and Trp279(5.43) of CB1R.37 A general binding mode for biaryl pyrazoles at CB1R can be inferred11,12 (Fig. 2B), whereby the azide and isothiocyante groups of 1–4 would be placed in proximity to CB1R nucleophilic residues Cys3827.38 and Cys3867.42 on TMH 7 (Fig. 2C).
This structural knowledge has also reinforced why non-covalent biaryl pyrazoles such as AM251 and SR141716A are CB1R selective, in particular the unique CB1R interactions with residues on TMH 1, TMH 2, ECL2 and the N-terminus,11,12 and hydrophobic interactions with Trp3566.48CB1R that stabilise the receptor inactive state.38,39 Compounds 1–4 were also tested for CB2R affinity36 and were an order of magnitude more selective for CB1R, although probing of whether covalent binding was occurring at CB2R was not undertaken. Although there is a lack of published CB2R biological data for AM6538 and lack of covalent binding investigations for 1–4, the biaryl pyrazole scaffold maintains promise for the development of highly selective CB1R covalent ligands due to the unique initial non-covalent binding mode of the ligand.
2.2 Aryl pyrazoles
In general, non-covalent compounds containing an aryl pyrazole scaffold have been reported as either non-selective or as moderately selective CB2R ligands, depending on the substitution pattern around the pyrazole core.40,41 Non-covalent aryl pyrazoles containing an N1-pentyl or -hexyl are nanomolar affinity ligands for both CB1R and CB2R, e.g. AM263 (Ki = 23 nM rCB1R;40Ki = 26.83 nM mCB2R,41Fig. 3A). AM263 analogues designed as potential covalent ligands have been developed – 5 and 6 (Fig. 3A),36 displaying ∼4 and ∼2 fold selectivity respectively for CB2R over CB1R.36 Compounds 5 and 6 displayed CB2R antagonism and CB1R partial agonism, suggesting a divergent binding mode from biaryl pyrazoles across the CBRs. Covalent labelling experiments for 5 and 6 were not reported.36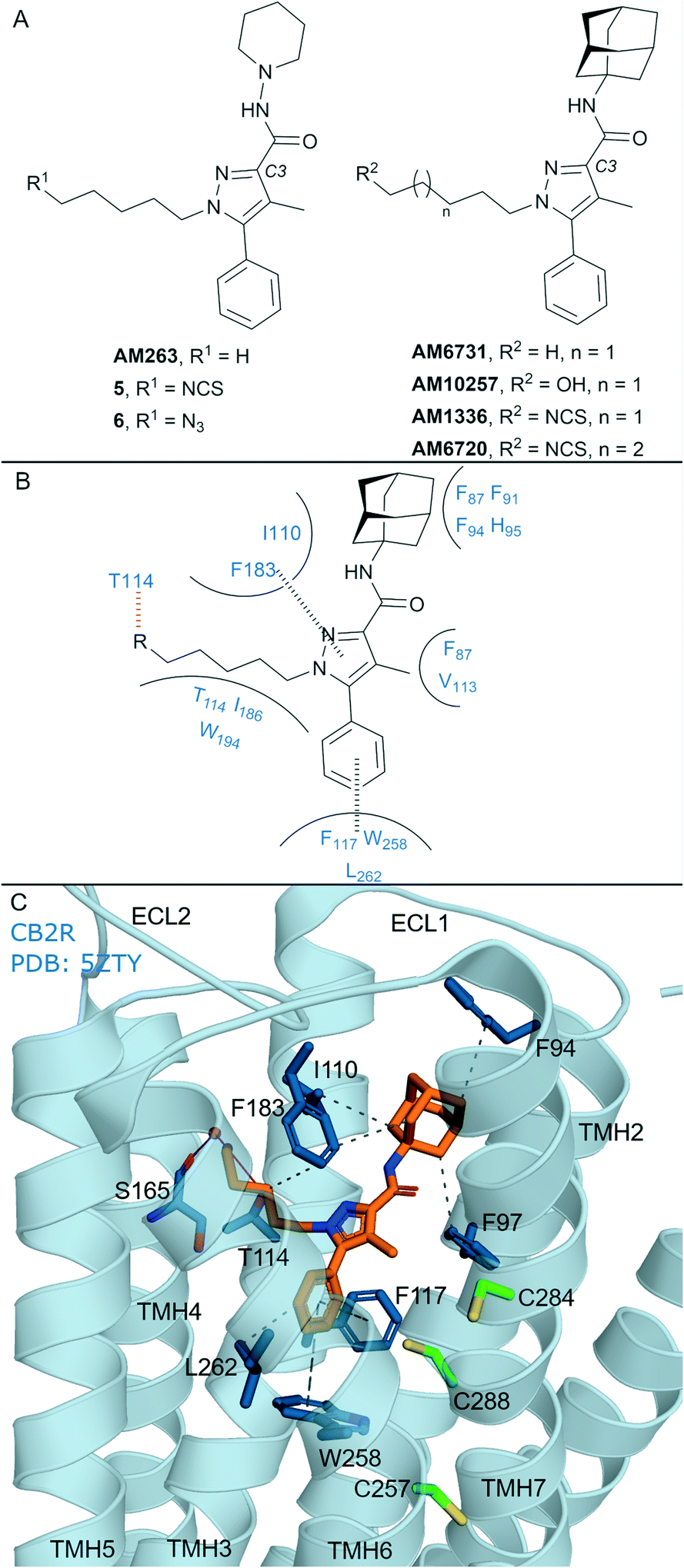 | ||
| Fig. 3 Aryl pyrazoles. A: Non-covalent and covalent aryl pyrazoles. B: Key residues and binding mode in CB2R. C: Co-crystal structure of CB2R (blue cartoon) and AM1025 (orange carbons, PDB ID: 5ZTY), showing key Cys (green carbons) and other key residues (blue). Hydrophobic interactions (black line), π–π stacking (dashed black line) and hydrogen bonds (pink). | ||
Some non-covalent aryl pyrazoles bearing more steric bulk than AM263 at the C3-position such as AM6731 have been reported as a high affinity, inverse agonists for CB2R (Ki = 0.63 nM hCB2R;42 data for CB1R has not been reported).41 The non-covalent very high affinity ligand AM10257 (Ki = 0.075 nM (0.063–0.091 nM) hCB2R; Ki = 13 nM hCB1R),16 with a N1-hydroxypentyl chain, was seen in the first CB2R crystal structure16 (Fig. 3C). Covalent analogues of AM6731 bearing an isothiocyanate group (AM1336, AM6720, Fig. 3A) have been reported as high affinity CB2R antagonists.42 Although washout experiments42 suggested both AM1336 and AM6720 covalently bound to CB2R (approx. 60% bound irreversibly at 10-fold hCB2R Ki concentration) using [3H]-CP-55940, mutagenesis indicated a different residue was involved for each ligand. AM1336 is proposed to bind to Cys2847.38,42,43 while AM6720 is proposed to bind to Cys2887.42,42 that is positioned further down the transmembrane helix than Cys2847.38 (Fig. 3C). Unlike chemoreactive electrophilic warheads such as isothiocyanates that react with nucleophile amino acid residues such as Cys and Ser, photoactivation of alkyl azide leading to a reactive nitrene can be more promiscuous regarding the functional group it can covalently react with. However there are reports of aryl pyrazoles containing alkyl azides (AM1335, structure not shown), that upon photoactivation produce a reactive nitrene that rearranges to an imine and then reacts with a cysteine sulfhydryl.44
The AM10257 – CB2R crystal structure (Fig. 3C, PDB: 5ZTY) revealed the N1 hydroxypentyl chain of AM10257 orientated in the opposite direction to Cys2847.38 and Cys2887.42, formed a hydrogen bond with Thr1143.33, and a water mediated hydrogen bond with Ser1654.57 backbone. The interaction with Ser1654.57 may be a contributing factor to the slightly higher CB2R versus CB1R affinity of AM10257, as Ala2474.57 is present in CB1R in an equivalent position (Fig. 1A). This orientation of AM10257 allows the phenyl arm of the pyrazole core to engage in π–π interaction with Phe1173.36 and Trp2586.48, strong binding interactions with these two aromatic residues have been shown to maintain CB2R antagonist selectivity.16,17 Given the binding mode of AM10257 in CB2R, the isothiocyanate group of covalent aryl pyrazoles (Fig. 3A) may be positioned away from Cys2847.38 and Cys2887.42, precluding covalent binding as viewed from a static model.
The AM10257 CB2R antagonist structure more closely resembles the CB1R agonist AM11542 (Fig. 4A) and AM841 (Fig. 4A and C) structures, with the extracellular portions and orthosteric pocket volume closely matching, with similar interactions.16,20 A ‘yin-yang’ relationship has been proposed for the CBRs whereby overlapping chemical scaffolds can act as CB2R antagonist/CB1R agonist.16,17 This may be related to the relatively restricted movement of the transmembrane helices in CB2R; in contrast, for CB1R, TMH 1 and 2 undergo conformational change to reduce the size of the orthosteric pocket which facilitates agonist/antagonist selectivity.45 An example of this observation is seen with AM10257, 5 and 6, which bind to CB1R, with good affinity and behave as partial agonists. Furthermore, it has been suggested for AM1336 and AM6720 that binding to the CB2R orthosteric binding pocket is relatively restricted. Within patent literature, AM1336 has been reported to bind to CB1R with high affinity (Ki = 5.76 nM),46 however it was noted that AM1336 did not irreversibly bind to CB1R (data not reported)42 suggesting a lack of covalent binding with Cys7.38 and Cys7.42 in CB1R (Fig. 1A). Covalent aryl pyrazoles may be able to form unique, covalent, interactions with Cys2847.38 and Cys2887.42 in CB2R. However, selectivity for CB2R will remain a challenge for this class of ligand given their relative (non-covalent) binding affinity and partial agonism towards CB1R.
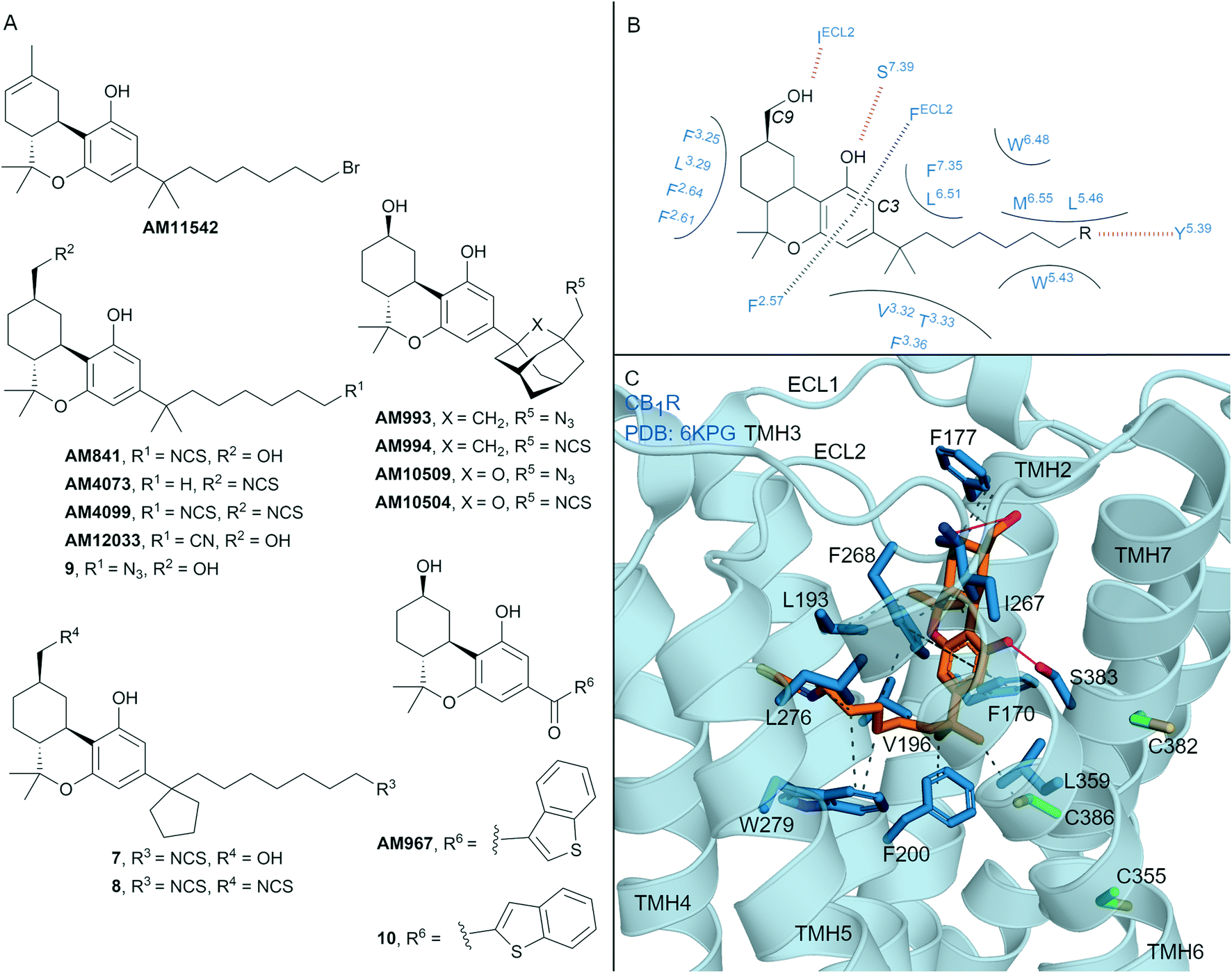 | ||
| Fig. 4 Tricyclic HHC and THC cannabinols. A: Non-covalent and covalent tricyclic cannabinols. B: Key residues and binding mode of ‘classical’ HHC scaffold (e.g. AM841) at CB1R and CB2R, residues are labelled by Ballesteros–Weinstein numbering for ease of showing both CBR interactions; in CB2R Leu3.29 and IleECL2 correspond to Ile3.29 and Leu ECL2 respectively. C: Co-crystal structure of CB1R (blue cartoon) and AM841 (orange carbons, PDB ID: 6KPG), showing key Cys (green carbons) and other key residues (blue). Hydrophobic interactions (black line), π–π stacking (dashed black line) and hydrogen bonds (pink). | ||
3. Tricyclic cannabinols
Many naturally occurring and synthetic non-covalent tricyclic tetrahydrocannabinol (THC) and hexahydrocannabinol (HHC) scaffolds (the ‘classical cannabinoids’) have been reported,47 as either non-selective CBR agonists or selective CB2R agonists, depending on substitution. The large body of structure–activity-relationship (SAR) data for these compound classes has enabled rational design of several series of CBR covalent ligands.47,48 THC derivative AM11542 is a potent CB1R agonist18,49 that contains a pentyl 1′,1′-gem-dimethylheptyl alkyl chain with a terminal bromide (Fig. 4A). AM11542 may have been designed as a potential covalent ligand (electrophilic alkyl halide warhead), however this design rationale and investigations into potential covalent binding have not been reported. Indeed, a non-covalently bound crystal structure of AM11542 and CB1R has been solved.18Several HHC-based ligands have been developed containing covalent warheads – AM841, AM4073, AM4099, 7–9 (Fig. 4A). The non–selective CBR agonist AM841, containing an isothiocyanate at the C3 side chain ω-position, was the first CBR ligand used in combination with site directed mutagenesis.50 These studies indicated that Cys3556.47 of CB1R could be a site of AM841 electrophilic attachment,50 while mutational and proteomic studies with CB2R demonstrated AM841 selectively labelled Cys2576.47 of CB2R.51,52 Compounds AM4073 and AM4099, containing an isothiocyanate (s) at the C3 side chain and/or C9 position, were designed to probe the CB2R orthosteric site.53 Both AM4073 and AM4099 retained nanomolar affinity and displayed CB2R agonism but were 100-fold less potent than AM841 at CB2R. Pre-incubation studies indicated covalent binding to CB2R while mutagenesis suggested Cys892.59 of CB2R was the point of attachment.53 The binding affinity and ability of AM4073 and AM4099 to covalently interact with CB1R has not been reported. More recently, covalent warhead containing 7 and 8 have been reported as non-selective CBR agonists with nanomolar to sub-nanomolar binding affinity.54 Isothiocyanate-containing 7 and 8 showed a modest ability to irreversibly bind in wash out experiments with both CB1R and CB2R, however, the ability to form a covalent bond with the receptor was not probed directly. Within the same paper, one of several azides analogues, 9, showed exceptional pico- to nanomolar binding affinity across both CBRs and upon photoirradiation displayed wash resistance comparable to AM841.54
Several HHC-based ligands containing a C3 adamantane are high affinity, non-covalent ligands with moderate CB1R selectivity.55 Inclusion of covalent warheads and either an adamantyl or oxa-adamantyl, such as ligands AM993, AM994, AM10509 and AM10504, have been reported as high affinity, non-selective CBR ligands (Fig. 4A).56,57 Compounds AM993 and AM994 showed agonist efficacy at CB1R and inverse agonist /antagonist activity at CB2R, supporting the CB1 agonist/CB2R antagonist model as observed with biaryl and aryl pyrazoles. Compounds AM993 (with photoirradiation), AM994 and AM10504 were effective at covalently labelling both CBRs as demonstrated by washout experiments (covalent binding for AM10509 was not reported).56,57 Whilst AM993 and AM994 displayed similar ability to covalently label the CBRs, AM10504 showed a ∼2-fold greater labelling (Bmax) to CB1R despite the similar binding affinity across both receptors.57 HHC-based photoactivable benzophenone derivatives 10 (non-selective CBR binding) and AM967 (10-fold selective binding at CB2R over CB1R) have also been reported.58 Both compounds were effective at covalently labelling CB2R,58 while analogous experiments with CB1R were not reported.
The first two CB1R agonist structures were determined with AM11542 (Fig. 4A, PDB: 5XRA) or AM841 (Fig. 4A and C, PDB: 5XR8)18 bound non-covalently in the CB1R orthosteric site. Given the biochemical evidence for covalent bond formation (CBR Cys6.47)50–52 and that the CBR – HHC/THC crystals were achieved through co-crystallisation (rather than crystal soaking), it was unexpected that a covalent bond was not observed in the co-crystal structure (e.g. to Cys3556.47CB1R, Fig. 4C). More recently, a CB1R cryo-EM structure (PDB: 6KPG) was reported with Gi protein, again with AM841 non-covalently bound.15 In both CB1R-AM11542 (crystal structure not shown) and CB1R-AM841 (Fig. 4C) structures, the C3-alkyl chain points towards TMHs 3, 5 and 6 forming significant hydrophobic interactions (Fig. 4B and C), with various groups at the C3 terminus able to form a hydrogen bond to Tyr5.39CBR (Fig. 4B).18 Notably, in each structure, the C3 terminus is some distance (∼18 Å) from Cys3556.47CB1R, the covalent attachment point proposed using mutagenesis studies.50 Shao et al.11 suggested since Cys3556.47CB1R is facing the bilayer side, for HHC cannabinoids to form a covalent bond to Cys6.47 rotation of TMH 6 at the orthosteric site would be required, leading to possible disruption of the packing around Trp3566.48CB1R. The outcome of the crystal structures versus biochemical experiments50–52 in regards to Cys3556.47CB1R positioning and covalent binding highlights the different factors (e.g. receptor dynamics, ligand entry (discussed in more detail below)) that could influence each method/technique.
In another HHC-scaffold structure (CB2R-AM12033 agonist structure, PDB: 6KPF, Fig. 1B),15 AM12033 is positioned in a near identical position to CB1R-AM841 with similar interacting residues. In considering the binding orientation of THC and HHC analogues in CBR – it is likely the binding poses of AM4073, AM4099, 7–9 match that of the THC/HHC CBR crystal structures (e.g.Fig. 4C).54 This orientation would position the C3 aliphatic chain towards Tyr5.39 (Fig. 4B). For compounds containing two covalent warheads, such as AM4099 and 8, this interaction would position the C9 isothiocyanate group distal from Cys892.59CB2R preventing covalent attachment, as within the AM12033-CB2R crystal structure the C9 hydroxyl forms hydrogen bond with backbone Leu182ECL2 (Fig. 1B).
Although there is experimental evidence of irreversible binding, in the absence of structural and mutagenic data it remains speculative which residues oxa/adamantane HHC derivatives may bind to. Given the benzophenone and oxa/adamantane compounds are CB2R antagonists they may bind in a similar manner to CB2R as aryl pyrazoles (such as AM1336 and AM6720) and be able to access residues Cys2847.38 and/or Cys2887.42CB2R on TMH 7. The bulky adamantane likely forms strong hydrophobic interactions with residues Phe3.36, Trp5.43 and Trp,6.48 with Trp6.48 and Phe3.36 implicated in agonist versus antagonist selectivity across both CBRs.20 Comparatively, for CB1R, the reduction in the orthosteric pocket for agonists of approximately 53% owing to the inwards movement TMH 1 and 2, as Phe1702.57 and Phe1742.61CB1R form additional hydrophobic contacts.18 This greater structural plasticity may contribute to CB1R ability to maintain an active conformation in the presence of bulkier covalent ligands such as AM10504 and AM993. It is expected that the central tricyclic rings for the C3 adamantane/benzophenone covalent compounds (e.g.10 and AM993) would align similarly to the cyclic core of other CB1 agonists (e.g. PDB ID: 6KPG) to maintain the Ser3837.39CB1R hydrogen bond which has been shown to be essential for HHC compounds.13,15 This would place the covalent warhead within the orthosteric pocket of CB1R and it could interact with nucleophilic cysteines such Cys3556.47CB1R. Photoactivation of benzophenones such as 10 or AM967, to generate a radical, favours insertion into C–H bonds such as benzylic positions, amino acid α-positions, hydrogen atoms adjacent to heteroatoms and tertiary carbon centres,59 which are readily accessible within the CBR orthosteric pocket for covalent binding by photoactivated compounds.
The available crystal structures with classical HHC and THC cannabinoids (Fig. 1B, 4C) both have the transmembrane helices, residue rotamers and Cys residues proximal to the ligand in near identical positions, highlighting the challenges faced in designing cannabinoid receptor subtype-selective covalent HHC or THC-based agonists. The major standout difference is residue Cys892.59 that is unique for CB2R (corresponding residue Tyr172 (ref. 89) CB1R, Fig. 1) which is placed on the extrahelical membrane side within the CB2R crystal structure (Fig. 1B). Whereby, AM4073 and AM4099 could indeed be only CB2R covalent ligands; albeit still binding non-covalently to both CBRs. If ligand entry of AM4073 and AM4099 occurs through lateral ligand receptor entry via the phospholipid membrane (which has been proposed for several CBR ligands,13,60 and is discussed in more depth below), the polar headgroup of AM4073 and AM4099 would be positioned towards the phospholipid which would allow the C9 isothiocyanate and Cys892.59CB2R to come in close contact. This orientation would account for the exclusive covalent binding to CB2R and reduces specific interactions with the Trp2586.48CB2R toggle switch accounting for the ligands reduced efficacy.53
The structures of lipid mediated GPCRs reveal an enclosed surface with limited opportunity for ligand entry via the extracellular space,61 with both the N-terminal and ECL 2 covering the orthosteric site (e.g.Fig. 1). For these receptors it has been proposed that ligand entry may occur directly through the plasma membrane, or, and for CB1R, laterally from the extracellular side.61 From the CB1R X-ray and cryo-EM structures this has been suggested to occur via a ligand access tunnel.11,13 Several molecular dynamics simulations have suggested ligand entry occurs between TMH 1 and TMH 7 for lipid mediated GPCRs.60,62,63 For CB2R bitopic ligands, a ligand entry channel has also been proposed between TMH 1 and TMH 7.64 Ligand entry via TMH 1 and 7 has been suggested to induce an N-terminal displacement, which may be part of receptor activation upon agonist binding.13,15,62 Considering that the N-terminus is involved in ligand selectivity between agonist versus antagonists binding,17,20,65 and the significant overlap between the N-terminus of CBR agonist structures,20 there may be a unified activation mechanism involving an N-terminal displacement which is agonist ligand specific.11 Understanding ligand entry remains an important consideration for ligand design, in particular for that of covalent cannabinoid ligands.66
4. Cannabidiols
One of the most well-known non-covalent cannabidiol(CBD)-based CBR ligands is the CB2R selective agonist HU-308 (hCB2R Ki = 22.7 ± 3.9 nM; rCB1R Ki = >10 μM)67 (Fig. 5A). In search of a CB2R selective irreversible ligand, Westphal et al.68 recently designed a series of ligands containing covalent warheads (including 11–13 (ref. 27) and RO7239315,27Fig. 5A) inspired by combining the HU-308 scaffold with features of AM841 (Fig. 4A). Ligands 11–13 and RO7239315 maintained selective (CB2R/CB1R) binding affinity and were CB2R agonists, however when binding of the isothiocyanate-containing 11 was investigated experimental evidence of irreversible binding was not observed.68 The enantiomer of 11 was also synthesised and studied,68 and again, although maintaining affinity for CB2R, irreversible/covalent labelling was not observed in washout experiments. Photoaffinity probe RO7239315, containing a photoreactive diazirine and a C3 terminal azide, successfully labelled CB2R upon irradiation (350 nm)27 as shown by in-gel fluorescence following fluorophore attachment (copper(I)-catalysed click reaction with Cy5-N3).69 This labelling step, carried out after covalent ligand binding, can be described as two-step photoaffinity-based protein profiling (pAfBPP), which can be a powerful approach for many secondary applications.59 Successful labelling of CB2R with RO7239315 with irradiation at 350 nm but unsuccessful labelling with irradiation at 300 nm (ref. 27 and 68) indicated that the photoactivated diazirine was responsible for the reaction with the protein and not the azide.59 Despite success with RO7239315, a probe based on a different scaffold (LEI121, Fig. 6A) was shown to be superior for this two-step photoaffinity labelling approach. | ||
| Fig. 5 Cannabidiols. A: Non-covalent and covalent cannabidiols. B: Proposed binding mode and key interacting residues of 11 in CB2R. C: Docked pose of 11 (refer to footnote in the text) (orange carbons) and CB2R (blue cartoon, PDB ID: 6PT0), showing key Cys (green carbons) and other key residues (blue). Hydrophobic interactions (dashed black line), π–π stacking (dashed black line) and hydrogen bonds (pink). | ||
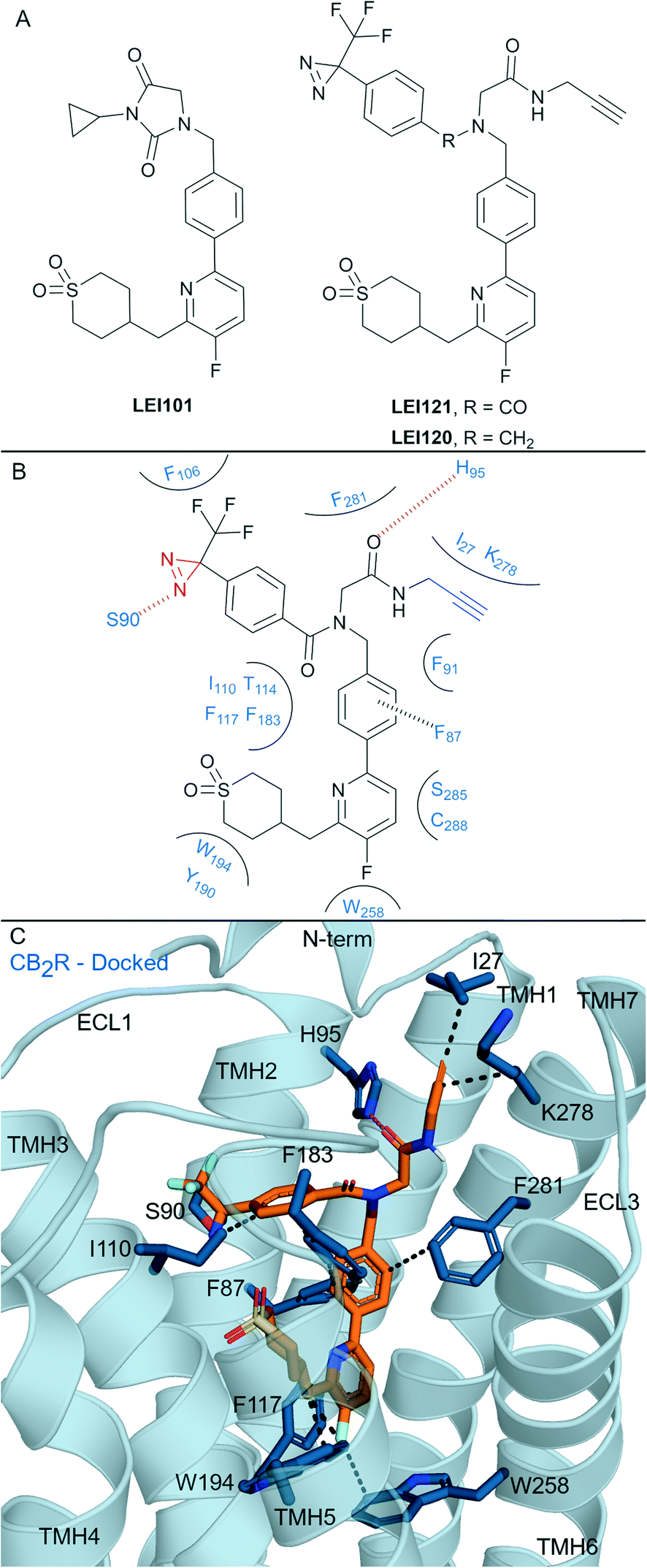 | ||
| Fig. 6 Aryl pyridinyls. A: Non-covalent and covalent aryl pyridinyl ligands. B: Proposed binding mode and key interacting residues with LEI121 in CB2R, photoreactive group and alkyne handle are coloured red and blue respectively. C: Docked pose of LEI121 (refer to footnote in the text) (orange carbons) and CB2R (blue cartoon, PDB: 5ZTY) with key interacting residues (blue carbons). Hydrophobic interactions (black line), π–π stacking (dashed black line) and hydrogen bonds (pink). | ||
Compound 13 had a similarly placed photoreactive diazirine but the C3 terminal azide was replaced with an amide. Comparatively, 13 displayed 10-fold lower binding affinity than RO7239315 and failed to bind covalently to CB2R upon photoactivation,27 highlighting the importance of high affinity non-covalent interactions before photoaffinity labelling. Photoactivation of 12 was not reported.68 Other structurally related cannabidiols (structures not shown) containing a C3 terminal azide have also failed to bind covalently to CB2R upon photoirridation.27
Whilst the cannabidiol-based 11 aimed to mimic covalent ligand AM841 (Fig. 4A), the C3 terminal isothiocyanate failed to bind covalently to CB2R. The central pyran of AM841 may be pivotal in maintaining the octyl chain in the correct orientation for covalent attachment of AM841 to Cys2576.47. We docked‡ cannabidiol 11 into the CB2R crystal structure (PDB: 6PT0, Fig. 5B, C) to rationalise CB2R selectivity (204-fold CB2R over CB1R64) and covalent bond failure. Compared to the non-selective HHC scaffold, the docked position of 11 in CB2R sits closer to ECL 2 and TMH 7, with ligand interactions with these areas implicated for CB2R agonist selectivity.17,65,70 A hydrogen bond is formed between the pinene hydroxyl of 11 and the Leu182ECl2CB2R backbone, which is likely important as ethers and esters in place of this hydroxyl (structures not shown) had a reduction in binding affinity experiments.68 Importantly, for CB2R selectivity, 11 loses the hydrogen bond with Ser2857.39, the corresponding Ser3837.39CB1R residue is critical in maintaining CB1R agonist binding affinity.13,47 The proposed binding pose for 11 forms minimal interactions with the toggle switch residues Trp2586.48CB2R.16 The isothiocyanate of 11 is positioned away from Cys2576.47CB2R, thereby potentially explaining the lack of covalent binding for 11 with CB2R.
Compound RO7239315 with the diazirine may covalently interact with residues on ECL 2 and the N-terminus upon photoirradiation given its relative binding position. The non-covalent cannabidiol HU-308 has been suggested to have costly free energy barriers for binding/entering CB1R,15 poor binding affinity of the cannabidiol ligands means they are unlikely to maintain non-covalent interactions24 to allow for covalent binding.68,71
5. Aryl pyridines
Aryl pyridines have been reported as CBR ligands, often with high CB2R selectivity, e.g. the high affinity non-covalent partial agonist LEI101 that displayed 1000-fold selectivity for CB2R over CB1R (Fig. 6A).72 Several ligands with covalent warheads have been developed based on LEI101. LEI121 (Fig. 6A) was developed as a pAfBPP probe with photoactivable covalent binding utility and the ability to react with a tag/reporter in a second step via the alkyne.27 Compared to LEI101, LEI121 maintained CB2R affinity and was again very selective for CB2R over CB1R.27 However, compared to LEI101, LEI121 resulted in reduced constitutive activity in a G protein activation pathway and reduced β-arrestin recruitment at CB2R and is therefore considered a CB2R inverse agonist. UV-irradiation studies with LEI121 indicated irreversible interaction with hCB2R and covalent bond formation was supported with biotin enrichment through the alkyne handle and observed by MS proteomics.27 LEI121 was successfully reacted with a fluorophore after ligand attachment to CB2R, and could be visualised by SDS-PAGE and in-gel fluorescence imaging. LEI121 was also used to visualise endogenous CB2R expression on human promyelocytic leukaemia and immune cells through fluorescence activated cell sorting. LEI120 (Fig. 6A), a close analogue of LEI121, was also developed as a potential pAfBPP probe, however it showed markedly lower covalent binding compared to LEI121 and was not progressed further as a tool.The structural interaction of aryl pyridines with CB2R (Fig. 6B) can be examined using literature SAR data and a published ligand docking study with a CB2R homology model27 (carried out in 2018,27 before any X-ray or cyroEM CB2R structure). We docked§ LEI121 in a non-photoactivated state into a CB2R crystal structure (Fig. 6C), which closely resembled the previously reported CB2R homology model results.27 The alkyne handle of LEI121 is ideally positioned towards TMH 7 and the N-terminus (Fig. 6C), which could allow an ‘exit vector’ for the alkyne and access for reaction with an ‘extracellular’ tag/reporter. Considering the non-covalent/pre-photoactivation interactions between LEI121-CB2R - the diazirine of LEI121 is sandwiched between TMH 2 and 3, and forms a unique hydrogen bond interaction with Ser902.60CB2R (Fig. 6B and C). Photoactivation of the diazirine will form a reactive carbene, that could be inserted into the C–H protein backbone or a heteroatom-H insertion.73 Trypsinisation and MS identified CB2R specific peptides corresponding to regions in the extracellular side at the N-terminus and TMH 3 associated with LEI121 covalent binding;27 supporting this binding mode of LEI121 (Fig. 6C). Determining the residue of insertion for LEI121 would require crystallographic and/or tandem MS evidence.44,74 The reduced covalent photoactivated labelling of CB2R with LEI120 was thought to be associated with the tertiary amine able to form charged interactions, which are not favoured within the hydrophobic cavity of the receptor (compared to LEI121 with an amide).27
LEI121 is currently the most selective CB2R covalent ligand, achieving this by forming tight binding and selective CB2R non-covalent interactions followed by photoactivation.27 Mainly, LEI121 forms hydrophobic and polar interactions with residues on TMH 2, 3, 4, 6 and 7, with the pyridyl positioned downwards to Trp2586.48 and Phe1173.36CB2R (Fig. 6C). It is anticipated these two residue interactions in particular are contributing to the high CB2R antagonist selectivity.17 These results also highlight the need to ensure correct positioning between the receptor and diazirine-containing ligand, as other photoaffinity compounds have failed to covalently bind to the CBRs.27,75
6. Indole carboxamides
Allosteric ligands interact with GPCRs at a binding site topographically distinct from the orthosteric (endogenous) binding sites. Covalent allosteric modulation of GPCRs is of interest both from a drug discovery and chemical tool perspective.76 Allosteric ligands for CBRs have been reported, more so for CB1R than CB2R.77 The indole-2-carboxamide scaffold (e.g. ORG27569, Fig. 7A) is one of the most widely studied synthetic, CB1R allosteric ligands.78 ORG27569, a non-covalent allosteric ligand, appears to increase orthosteric agonist binding affinity but is considered a negative allosteric modulator (NAM) for efficacy displaying insurmountable antagonism.78 ORG27569 also behaves as an inverse agonist and competitively binds with SR141716A (Rimonabant, Fig. 2A).78,79 Many other non-covalent indole carboxamides (structures not shown) have been evaluated77 as CB1R allosteric ligands, with SAR allowing for the rational design of putative covalent ligands containing isothiocyanate and/or photoactivatable groups.80 Replacement of the chloro in ORG27569 with an isothiocyanate gave GAT100 (Fig. 7A), which showed more efficacious NAM activity compared to ORG27569 and was devoid of inverse agonism.80 CB1R-expressing membranes were preincubated with GAT100 for up to 60 minutes followed by extensive washing and CP-55940 binding was assessed – resulting in increased maximal CP-55940 binding by approximately ∼2.25-fold,80 indicating wash resistance of GAT100. In contrast, the same experiment performed with ORG27569 had no effect on CP-55940 binding.80 Direct evidence for covalent binding between CB1R and GAT100 (such as by MS) has not been reported.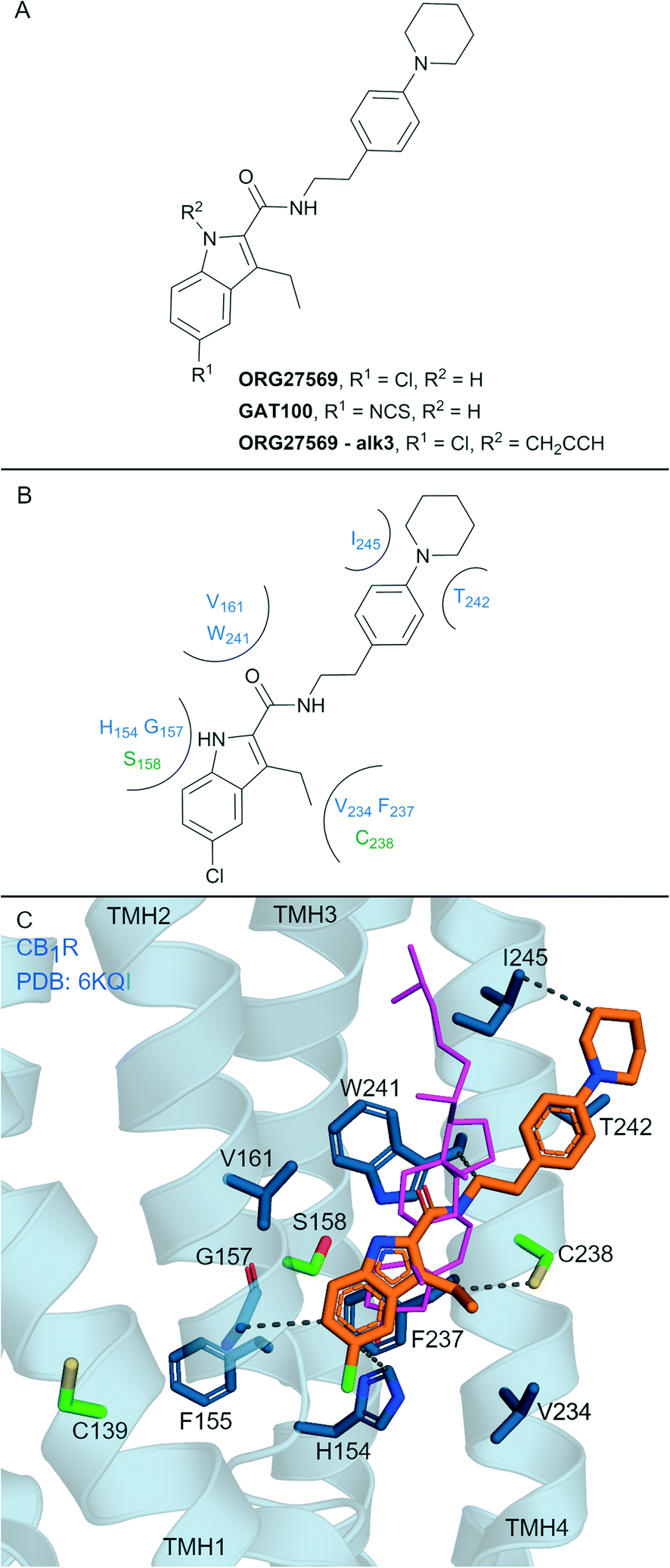 | ||
| Fig. 7 Indole carboxamide allosteric ligands. A: Non-covalent and covalent indole carboxamides. B: Key residues and binding mode in CB2R. C: Co-crystal structure of CB1R (blue cartoon, PDB ID: 6KQI), Org27569 (orange carbons) and CP-55940 (not shown), cholesterol (pink carbon sticks, PDB ID: 5XR8) is overlaid to show similar binding in CB1R, showing key Cys and Ser (green carbons) and other key residues (blue carbons), TMH 5 and 6 are hidden for clarity. Hydrophobic interactions (black line), transmembrane helices 5 and 6 are hidden for clarity. | ||
Several other ORG27569 analogues have been designed with covalent and/or photoreactive warheads (structures not all shown).81,82 Notably, ORG27569-Alk3 (ref. 81) (Fig. 7A) contains a weakly reactive alkyne that was shown via MS to bind to either Ser1582.45 or Ser2063.42CB1R.81 Both Ser1582.45 and Ser2063.42CB1R residues are within the vicinity of a cholesterol consensus motif (CCM), a motif found in 21% of class A GPCRs83 with several GPCRs observed to bind cholesterol by X-ray crystallography at this region.84 Mutagenesis suggested that ORG27569 binds along the extracellular helices at TMH 2, 3 and 4, as mutating to the corresponding CB2R residues (Cys1391.55Tyr, His1542.41Leu and Phe2374.46Leu) abolished ORG27569 binding.81
The crystal structure of CB1R co-crystalised with agonist CP-55940 and non-covalent ligand ORG27569 is an overall antagonist-like structure with some agonist like features (Fig. 7C).14 In this structure, ORG27569 binds on the outside of the TMH domain (within the membrane) along TMH 2, 3 and 4 with significant hydrophobic interactions, with the chloroindole packing against the indole of Trp2414.50 (Fig. 7B). Two of the three residues identified as important by CB1R mutagenesis studies (His1542.41and Phe2374.46CB1R) are in direct contact with ORG27569, and Cys1391.55 is within close proximity (Fig. 7C). Ser1582.45CB1R is a suggested site of attachment for covalent ligand ORG27569-Alk3, and this is situated only 5.2 Å away from the indole nitrogen of ORG27569.14 Prior to the CB1R-ORG27569 crystal structure, modelling studies suggested GAT100 covalently bound to Cys3827.38CB1R,85 close to the orthosteric site (Fig. 1A and B).86 Molecular dynamic trajectories indicated ORG27569 also came into close contact (<5 Å) with Cys1391.55CB1R on the intracellular side of the extrahelical membrane, however, at the time, pharmacological relevance for this site could not be explained.85 Given the new crystallographic evidence (PDB: 6KQI), Cys1391.55 remains a potential site for GAT100 covalent attachment. However, as Cys1391.55 sits distal to the chloroindole, a more plausible site based on structural evidence is Cys2384.47CB1R, which sits closely within the indole carboxamide binding site (Fig. 7C).
Non-covalent CB1R ligand ORG27569 does not have any measurable effect at CB2R.87 To date, CB2R activity has not been reported for irreversible CB1R ligands GAT100 and ORG27569-Alk3. However it is likely these ligands are inactive at CB2R based on the failure of ORG27569 and structurally related indole carboxamides analogues to elicit any activity at CB2R.87,88 The assumed CB1R selectivity of GAT100 and ORG27569-Alk3 therefore stems from the highly selective non-covalent interactions of the CB1R allosteric indole carboxamides. Several residues in the ORG27569 ‘allosteric’ binding region are non-conserved between the CBRs81 – of note are residues Cys1391.55 and Cys2384.47 of CB1R with the corresponding residues being Tyr561.55 and Gly1554.47 in CB2R.
7. Conclusions
Developing subtype selective irreversible CBR ligands has proven challenging – there are very few covalent ligands that can be confidently regarded as having robust-CBR-subtype-selectivity. Many CBR covalent ligands have been reported over the last 20–30 years, however, many lack of comprehensive experimental measurements at both CBR subtypes; based on the recent CBR structural insights (discussed in this review) most of these ligands are not likely to be CBR-subtype-selective. The high CBR sequence similarity, in particular the analogous position of cysteines within the orthosteric ligand binding sites of CB1R and CB2R, is a major factor for the poor selectivity of many reported covalent CBR ligands. Recent CBR structural information revealed by X-ray crystallography and cryo-EM studies provide an opportunity to rationally design covalent, subtype-selective CBR ligands, for example by using alternative electrophilic warheads89 targeted to other nearby nucleophilic residues such as tyrosine and lysine. Development of covalent CBR allosteric ligands is likely to achieve superior subtype selectivity (over orthosteric ligands) because of lower sequence similarity and a greater range of potential allosteric binding ‘sites,’ although the discovery of more allosteric scaffolds, in particular for CB2R, is needed. As the endocannabinoid system continues to be pursued as a drug target, covalent ligands will provide insight into complex pharmacology and aid structural-based drug discovery efforts.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Ian Liddle is supported by a University of Otago Doctoral Scholarship. The authors have received funding for research projects in this area from the Maurice Wilkins Centre for Molecular Biodiscovery.References
- D. Wootten, A. Christopoulos, M. Marti-Solano, M. M. Babu and P. M. Sexton, Nat. Rev. Mol. Cell Biol., 2018, 19, 638–653 CrossRef CAS PubMed
.
- K. Sriram and P. A. Insel, Mol. Pharmacol., 2018, 93, 251–258 CrossRef CAS PubMed
- A. S. Hauser, M. M. Attwood, M. Rask-Andersen, H. B. Schiöth and D. E. Gloriam, Nat. Rev. Drug Discovery, 2017, 16, 829–842 CrossRef CAS PubMed
- J.-Y. Shim, Curr. Top. Med. Chem., 2010, 10, 779–798 CrossRef CAS PubMed
- A. Poso and J. W. Huffman, Br. J. Pharmacol., 2008, 153, 335–346 CrossRef CAS PubMed
- C. Munk, K. Harpsøe, A. S. Hauser, V. Isberg and D. E. Gloriam, Curr. Opin. Pharmacol., 2016, 30, 51–58 CrossRef CAS PubMed
- S. Costanzi, Curr. Opin. Struct. Biol., 2013, 23, 185–190 CrossRef CAS PubMed
-
X. Qu, D. Wang and B. Wu, in GPCRs, ed. B. Jastrzebska and P. S. H. Park, Academic Press, 2020, pp. 3–22 Search PubMed
- V. Di Marzo, Nat. Rev. Drug Discovery, 2018, 17, 623–639 CrossRef CAS PubMed
- P. Morales and N. Jagerovic, Expert Opin. Drug Discovery, 2020, 15, 917–930 CrossRef CAS PubMed
- Z. Shao, J. Yin, K. Chapman, M. Grzemska, L. Clark, J. Wang and D. M. Rosenbaum, Nature, 2016, 540, 602–606 CrossRef CAS PubMed
- T. Hua, K. Vemuri, M. Pu, L. Qu, G. W. Han, Y. Wu, S. Zhao, W. Shui, S. Li, A. Korde, R. B. Laprairie, E. L. Stahl, J.-H. Ho, N. Zvonok, H. Zhou, I. Kufareva, B. Wu, Q. Zhao, M. A. Hanson, L. M. Bohn, A. Makriyannis, R. C. Stevens and Z.-J. Liu, Cell, 2016, 167, 750–762 CrossRef CAS PubMed
- K. K. Kumar, M. Shalev-Benami, M. J. Robertson, H. Hu, S. D. Banister, S. A. Hollingsworth, N. R. Latorraca, H. E. Kato, D. Hilger, S. Maeda, W. I. Weis, D. L. Farrens, R. O. Dror, S. V. Malhotra, B. K. Kobilka and G. Skiniotis, Cell, 2019, 176, 448–458 CrossRef PubMed
- Z. Shao, W. Yan, K. Chapman, K. Ramesh, A. J. Ferrell, J. Yin, X. Wang, Q. Xu and D. M. Rosenbaum, Nat. Chem. Biol., 2019, 15, 1199–1205 CrossRef CAS PubMed
- T. Hua, X. Li, L. Wu, C. Iliopoulos-Tsoutsouvas, Y. Wang, M. Wu, L. Shen, C. A. Johnston, S. P. Nikas, F. Song, X. Song, S. Yuan, Q. Sun, Y. Wu, S. Jiang, T. W. Grim, O. Benchama, E. L. Stahl, N. Zvonok, S. Zhao, L. M. Bohn, A. Makriyannis and Z.-J. Liu, Cell, 2020, 180, 655–665 CrossRef CAS PubMed
- X. Li, T. Hua, K. Vemuri, J. H. Ho, Y. Wu, L. Wu, P. Popov, O. Benchama, N. Zvonok, K. Locke, L. Qu, G. W. Han, M. R. Iyer, R. Cinar, N. J. Coffey, J. Wang, M. Wu, V. Katritch, S. Zhao, G. Kunos, L. M. Bohn, A. Makriyannis, R. C. Stevens and Z. J. Liu, Cell, 2019, 176, 459–467 CrossRef CAS PubMed
- C. Xing, Y. Zhuang, T.-H. Xu, Z. Feng, X. E. Zhou, M. Chen, L. Wang, X. Meng, Y. Xue, J. Wang, H. Liu, T. F. McGuire, G. Zhao, K. Melcher, C. Zhang, H. E. Xu and X.-Q. Xie, Cell, 2020, 180, 645–655 CrossRef CAS PubMed
- T. Hua, K. Vemuri, S. P. Nikas, R. B. Laprairie, Y. Wu, L. Qu, M. Pu, A. Korde, S. Jiang, J.-H. Ho, G. W. Han, K. Ding, X. Li, H. Liu, M. A. Hanson, S. Zhao, L. M. Bohn, A. Makriyannis, R. C. Stevens and Z.-J. Liu, Nature, 2017, 547, 468–471 CrossRef CAS PubMed
- X. Wang, D. Liu, L. Shen, F. Li, Y. Li, L. Yang, T. Xu, H. Tao, D. Yao, L. Wu, K. Hirata, L. M. Bohn, A. Makriyannis, X. Liu, T. Hua, Z.-J. Liu and J. Wang, J. Am. Chem. Soc., 2021, 143, 16320–16325 CrossRef CAS PubMed
- X. Li, L. Shen, T. Hua and Z.-J. Liu, Trends Pharmacol. Sci., 2020, 41, 665–677 CrossRef CAS PubMed
- M. Jörg and P. J. Scammells, ChemMedChem, 2016, 11, 1488–1498 CrossRef PubMed
- D. Weichert and P. Gmeiner, ACS Chem. Biol., 2015, 10, 1376–1386 CrossRef CAS PubMed
- H. Kim, Y. S. Hwang, M. Kim and S. B. Park, RSC Med. Chem., 2021, 12, 1037–1045 RSC
- S. De Cesco, J. Kurian, C. Dufresne, A. K. Mittermaier and N. Moitessier, Eur. J. Med. Chem., 2017, 138, 96–114 CrossRef CAS PubMed
- X. Yang, T. J. M. Michiels, C. de Jong, M. Soethoudt, N. Dekker, E. Gordon, M. van der Stelt, L. H. Heitman, D. van der Es and A. P. Ijzerman, J. Med. Chem., 2018, 61, 7892–7901 CrossRef CAS PubMed
- P. N. H. Trinh, D. J. W. Chong, K. Leach, S. J. Hill, J. D. A. Tyndall, L. T. May, A. J. Vernall and K. J. Gregory, J. Med. Chem., 2021, 64, 8161–8178 CrossRef CAS PubMed
- M. Soethoudt, S. C. Stolze, M. V. Westphal, L. van Stralen, A. Martella, E. J. van Rooden, W. Guba, Z. V. Varga, H. Deng, S. I. van Kasteren, U. Grether, A. P. Ijzerman, P. Pacher, E. M. Carreira, H. S. Overkleeft, A. Ioan-Facsinay, L. H. Heitman and M. van der Stelt, J. Am. Chem. Soc., 2018, 140, 6067–6075 CrossRef CAS PubMed
-
D. R. Janero, A. Korde and A. Makriyannis, in Methods Enzymol., ed. P. H. Reggio, Academic Press, 2017, vol. 593, pp. 217–235 Search PubMed
- C. Blex, S. Michaelis, A. K. Schrey, J. Furkert, J. Eichhorst, K. Bartho, F. Gyapon Quast, A. Marais, M. Hakelberg, U. Gruber, S. Niquet, O. Popp, F. Kroll, M. Sefkow, R. Schülein, M. Dreger and H. Köster, ChemBioChem, 2017, 18, 1639–1649 CrossRef CAS PubMed
- F. M. Müskens, R. J. Ward, D. Herkt, H. van de Langemheen, A. B. Tobin, R. M. J. Liskamp and G. Milligan, Mol. Pharmacol., 2019, 95, 196–209 CrossRef PubMed
- R. Miyajima, K. Sakai, Y. Otani, T. Wadatsu, Y. Sakata, Y. Nishikawa, M. Tanaka, Y. Yamashita, M. Hayashi, K. Kondo and T. Hayashi, ACS Chem. Biol., 2020, 15, 2364–2373 CrossRef CAS PubMed
- A. Cooper, S. Singh, S. Hook, J. D. A. Tyndall and A. J. Vernall, Pharmacol. Rev., 2017, 69, 316–353 CrossRef CAS PubMed
- F. Basagni, M. Rosini and M. Decker, ChemMedChem, 2020, 15, 1374–1389 CrossRef CAS PubMed
-
J. A. Ballesteros and H. Weinstein, in Methods in Neurosciences, ed. S. C. Sealfon, Academic Press, 1995, vol. 25, pp. 366–428 Search PubMed
- R. Lan, Q. Liu, P. Fan, S. Lin, S. R. Fernando, D. McCallion, R. Pertwee and A. Makriyannis, J. Med. Chem., 1999, 42, 769–776 CrossRef CAS PubMed
- A. C. Howlett, G. H. Wilken, J. J. Pigg, D. B. Houston, R. Lan, Q. Liu and A. Makriyannis, J. Neurochem., 2000, 74, 2174–2181 CrossRef CAS PubMed
- R. B. Laprairie, K. Vemuri, E. L. Stahl, A. Korde, J.-H. Ho, T. W. Grim, T. Hua, Y. Wu, R. C. Stevens, Z.-J. Liu, A. Makriyannis and L. M. Bohn, Mol. Pharmacol., 2019, 96, 619–628 CrossRef CAS PubMed
- S. D. McAllister, D. P. Hurst, J. Barnett-Norris, D. Lynch, P. H. Reggio and M. E. Abood, J. Biol. Chem., 2004, 279, 48024–48037 CrossRef CAS PubMed
- S. D. McAllister, G. Rizvi, S. Anavi-Goffer, D. P. Hurst, J. Barnett-Norris, D. L. Lynch, P. H. Reggio and M. E. Abood, J. Med. Chem., 2003, 46, 5139–5152 CrossRef CAS PubMed
- J.-Y. Shim, W. J. Welsh, E. Cartier, J. L. Edwards and A. C. Howlett, J. Med. Chem., 2002, 45, 1447–1459 CrossRef CAS PubMed
- J.-Z. Chen, X.-W. Han, Q. Liu, A. Makriyannis, J. Wang and X.-Q. Xie, J. Med. Chem., 2006, 49, 625–636 CrossRef CAS PubMed
- R. W. Mercier, Y. Pei, L. Pandarinathan, D. R. Janero, J. Zhang and A. Makriyannis, Chem. Biol., 2010, 17, 1132–1142 CrossRef CAS PubMed
- S. Mallipeddi, S. Kreimer, N. Zvonok, K. Vemuri, B. L. Karger, A. R. Ivanov and A. Makriyannis, J. Proteome Res., 2017, 16, 2419–2428 CrossRef CAS PubMed
- D. Szymanski, M. Papanastasiou, L. Pandarinathan, N. Zvonok, D. R. Janero, S. Pavlopoulos, P. Vouros and A. Makriyannis, J. Med. Chem., 2018, 61, 11199–11208 CrossRef CAS PubMed
- F. Shahbazi, V. Grandi, A. Banerjee and J. F. Trant, iScience, 2020, 23, e101301 CrossRef PubMed
-
A. Makriyannis and Q. Liu, University of Connecticut, World Intellectual Property Organization, WO01/29007A1, 2001
- E. W. Bow and J. M. Rimoldi, Perspect. Med. Chem., 2016, 8, 17–39 Search PubMed
- A. D. Khanolkar, S. L. Palmer and A. Makriyannis, Chem. Phys. Lipids, 2000, 108, 37–52 CrossRef CAS PubMed
- B. R. Martin, D. R. Compton, S. F. Semus, S. Lin, G. Marciniak, J. Grzybowska, A. Charalambous and A. Makriyannis, Pharmacol., Biochem. Behav., 1993, 46, 295–301 CrossRef CAS
- R. P. Picone, A. D. Khanolkar, W. Xu, L. A. Ayotte, G. A. Thakur, D. P. Hurst, M. E. Abood, P. H. Reggio, D. J. Fournier and A. Makriyannis, Mol. Pharmacol., 2005, 68, 1623–1635 CrossRef CAS PubMed
- Y. Pei, R. W. Mercier, J. K. Anday, G. A. Thakur, A. M. Zvonok, D. Hurst, P. H. Reggio, D. R. Janero and A. Makriyannis, Chem. Biol., 2008, 15, 1207–1219 CrossRef CAS PubMed
- D. W. Szymanski, M. Papanastasiou, K. Melchior, N. Zvonok, R. W. Mercier, D. R. Janero, G. A. Thakur, S. Cha, B. Wu, B. Karger and A. Makriyannis, J. Proteome Res., 2011, 10, 4789–4798 CrossRef CAS PubMed
- H. Zhou, Y. Peng, A. Halikhedkar, P. Fan, D. R. Janero, G. A. Thakur, R. W. Mercier, X. Sun, X. Ma and A. Makriyannis, ACS Chem. Neurosci., 2017, 8, 1338–1347 CrossRef CAS PubMed
- S. Jiang, C. Iliopoulos-Tsoutsouvas, F. Tong, C. A. Brust, C. M. Keenan, J. G. Raghav, T. Hua, S. Wu, J.-H. Ho, Y. Wu, T. W. Grim, N. Zvonok, G. A. Thakur, Z.-J. Liu, K. A. Sharkey, L. M. Bohn, S. P. Nikas and A. Makriyannis, J. Med. Chem., 2021, 64, 3870–3884 CrossRef CAS PubMed
- D. Lu, Z. Meng, G. A. Thakur, P. Fan, J. Steed, C. L. Tartal, D. P. Hurst, P. H. Reggio, J. R. Deschamps, D. A. Parrish, C. George, T. U. C. Järbe, R. J. Lamb and A. Makriyannis, J. Med. Chem., 2005, 48, 4576–4585 CrossRef CAS PubMed
- G. Ogawa, M. A. Tius, H. Zhou, S. P. Nikas, A. Halikhedkar, S. Mallipeddi and A. Makriyannis, J. Med. Chem., 2015, 58, 3104–3116 CrossRef CAS PubMed
- T. C. Ho, M. A. Tius, S. P. Nikas, N. K. Tran, F. Tong, H. Zhou, N. Zvonok and A. Makriyannis, Bioorg. Med. Chem. Lett., 2021, 38, 127882–127889 CrossRef CAS PubMed
- D. D. Dixon, M. A. Tius, G. A. Thakur, H. Zhou, A. L. Bowman, V. G. Shukla, Y. Peng and A. Makriyannis, Bioorg. Med. Chem. Lett., 2012, 22, 5322–5325 CrossRef CAS PubMed
-
P. P. Geurink, L. M. Prely, G. A. van der Marel, R. Bischoff and H. S. Overkleeft, in Activity-Based Protein Profiling, ed. S. A. Sieber, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012 Search PubMed
- J. Jakowiecki and S. Filipek, J. Chem. Inf. Model., 2016, 56, 2457–2466 CrossRef CAS PubMed
- M. Audet and R. C. Stevens, Protein Sci., 2019, 28, 292–304 CrossRef CAS PubMed
- J. Jakowiecki, U. Orzel, S. Chawanon, P. Miszta and S. Filipek, Molecules, 2020, 25, 1930 CrossRef CAS PubMed
- N. Stanley, L. Pardo and G. D. Fabritiis, Sci. Rep., 2016, 6, 22639 CrossRef CAS PubMed
- P. Morales, G. Navarro, M. Gómez-Autet, L. Redondo, J. Fernandez-Ruiz, L. Perez-Benito, A. Corodomi, L. Pardo, R. Franco and N. Jagerovic, Chem. – Eur. J., 2020, 26, 15839–15842 CrossRef CAS PubMed
- B. Ji, S. Liu, X. He, V. H. Man, X.-Q. Xie and J. Wang, ACS Chem. Neurosci., 2020, 11, 1139–1158 CrossRef CAS PubMed
- J. Payandeh and M. Volgraf, Nat. Rev. Drug Discovery, 2021, 20, 710–722 CrossRef CAS PubMed
- L. Hanuš, A. Breuer, S. Tchilibon, S. Shiloah, D. Goldenberg, M. Horowitz, R. G. Pertwee, R. A. Ross, R. Mechoulam and E. Fride, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 14228–14233 CrossRef PubMed
- M. V. Westphal, R. C. Sarott, E. A. Zirwes, A. Osterwald, W. Guba, C. Ullmer, U. Grether and E. M. Carreira, Chem. – Eur. J., 2020, 26, 1380–1387 CrossRef CAS PubMed
- C. G. Parker and M. R. Pratt, Cell, 2020, 180, 605–632 CrossRef CAS PubMed
- Z. Feng, M. H. Alqarni, P. Yang, Q. Tong, A. Chowdhury, L. Wang and X.-Q. Xie, J. Chem. Inf. Model., 2014, 54, 2483–2499 CrossRef CAS PubMed
- R. C. Sarott, M. V. Westphal, P. Pfaff, C. Korn, D. A. Sykes, T. Gazzi, B. Brennecke, K. Atz, M. Weise, Y. Mostinski, P. Hompluem, E. Koers, T. Miljuš, N. J. Roth, H. Asmelash, M. C. Vong, J. Piovesan, W. Guba, A. C. Rufer, E. A. Kusznir, S. Huber, C. Raposo, E. A. Zirwes, A. Osterwald, A. Pavlovic, S. Moes, J. Beck, I. Benito-Cuesta, T. Grande, S. R. de Martıín Esteban, A. Yeliseev, F. Drawnel, G. Widmer, D. Holzer, T. van der Wel, H. Mandhair, C.-Y. Yuan, W. R. Drobyski, Y. Saroz, N. Grimsey, M. Honer, J. Fingerle, K. Gawrisch, J. Romero, C. J. Hillard, Z. V. Varga, M. van der Stelt, P. Pacher, J. Gertsch, P. J. McCormick, C. Ullmer, S. Oddi, M. Maccarrone, D. B. Veprintsev, M. Nazaré, U. Grether and E. M. Carreira, J. Am. Chem. Soc., 2020, 142, 16953–16964 CrossRef CAS PubMed
- M. van der Stelt, J. Cals, S. Broeders-Josten, J. Cottney, A. A. van der Doelen, M. Hermkens, V. de Kimpe, A. King, J. Klomp, J. Oosterom, I. Pols-de Rooij, J. de Roos, M. van Tilborg, S. Boyce and J. Baker, J. Med. Chem., 2011, 54, 7350–7362 CrossRef CAS PubMed
- J. R. Hill and A. A. B. Robertson, J. Med. Chem., 2018, 61, 6945–6963 CrossRef CAS PubMed
- T. Seifert, M. Malo, J. Lengqvist, C. Sihlbom, E. M. Jarho and K. Luthman, J. Med. Chem., 2016, 59, 10794–10799 CrossRef CAS PubMed
- M. Soethoudt, G. Alachouzos, E. J. van Rooden, M. D. Moya-Garzón, R. J. B. H. N. van den Berg, L. H. Heitman and M. van der Stelt, Cannabis Cannabinoid Res., 2018, 3, 136–151 CrossRef CAS PubMed
- Y. Bian, J. J. Jun, J. Cuyler and X.-Q. Xie, Eur. J. Med. Chem., 2020, 206, 112690 CrossRef CAS PubMed
- F. Gado, S. Meini, S. Bertini, M. Digiacomo, M. Macchia and C. Manera, Future Med. Chem., 2019, 11, 2019–2037 CrossRef CAS PubMed
- M. R. Price, G. L. Baillie, A. Thomas, L. A. Stevenson, M. Easson, R. Goodwin, A. McLean, L. McIntosh, G. Goodwin, G. Walker, P. Westwood, J. Marrs, F. Thomson, P. Cowley, A. Christopoulos, R. G. Pertwee and R. A. Ross, Mol. Pharmacol., 2005, 68, 1484–1495 CrossRef CAS PubMed
- K. H. Ahn, M. M. Mahmoud and D. A. Kendall, J. Biol. Chem., 2012, 287, 12070–12082 CrossRef CAS PubMed
- P. M. Kulkarni, A. R. Kulkarni, A. Korde, R. B. Tichkule, R. B. Laprairie, E. M. Denovan-Wright, H. Zhou, D. R. Janero, N. Zvonok, A. Makriyannis, M. G. Cascio, R. G. Pertwee and G. A. Thakur, J. Med. Chem., 2016, 59, 44–60 CrossRef CAS PubMed
- M. Stornaiuolo, A. Bruno, L. Botta, G. L. Regina, S. Cosconati, R. Silvestri, L. Marinelli and E. Novellino, Sci. Rep., 2015, 5, 15453 CrossRef CAS PubMed
- C.-J. Qiao, H. I. Ali, K. H. Ahn, S. Kolluru, D. A. Kendall and D. Lu, Eur. J. Med. Chem., 2016, 121, 517–529 CrossRef CAS PubMed
- M. A. Hanson, V. Cherezov, M. T. Griffith, C. B. Roth, V.-P. Jaakola, E. Y. T. Chien, J. Velasquez, P. Kuhn and R. C. Stevens, Structure, 2008, 16, 897–905 CrossRef CAS PubMed
-
C. Wang, A. Ralko, Z. Ren, A. Rosenhouse-Dantsker and X. Yang, in Direct Mechanisms in Cholesterol Modulation of Protein Function, ed. A. Rosenhouse-Dantsker and A. Bukiya, Springer International Publishing, Cham, 2019, pp. 67–86 Search PubMed
- R. B. Laprairie, A. R. Kulkarni, P. M. Kulkarni, D. P. Hurst, D. Lynch, P. H. Reggio, D. R. Janero, R. G. Pertwee, L. A. Stevenson, M. E. M. Kelly, E. M. Denovan-Wright and G. A. Thakur, ACS Chem. Neurosci., 2016, 7, 776–798 CrossRef CAS PubMed
- D. M. Shore, G. L. Baillie, D. H. Hurst, F. Navas, 3rd, H. H. Seltzman, J. P. Marcu, M. E. Abood, R. A. Ross and P. H. Reggio, J. Biol. Chem., 2014, 289, 5828–5845 CrossRef CAS PubMed
- F. Piscitelli, A. Ligresti, G. La Regina, A. Coluccia, L. Morera, M. Allarà, E. Novellino, V. Di Marzo and R. Silvestri, J. Med. Chem., 2012, 55, 5627–5631 CrossRef CAS PubMed
- T. Nguyen, N. German, A. M. Decker, J.-X. Li, J. L. Wiley, B. F. Thomas, T. P. Kenakin and Y. Zhang, Bioorg. Med. Chem., 2015, 23, 2195–2203 CrossRef CAS PubMed
- M. Gehringer and S. A. Laufer, J. Med. Chem., 2019, 62, 5673–5724 CrossRef CAS PubMed
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, Aust. J. Chem., 2012, 4, 17 CAS
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, J. Mol. Biol., 1997, 267, 727–748 CrossRef CAS PubMed
- S. Salentin, S. Schreiber, V. J. Haupt, M. F. Adasme and M. Schroeder, Nucleic Acids Res., 2015, 43, 443–447 CrossRef PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2md00006g |
| ‡ 3D model of ligands were generated using Avogadro 1.2 and minimised using the universal force field.90 Compound 11 was docked into the agonist bound crystal structure of CB2R (PDB: 6PT0),17 with G-protein removed. Non – covalent docking was performed using GOLD 2020.2.0 (CCDC software).91 Results were visualised in PyMOL (The PyMOL Molecular Graphics System, Version 2.3.2 Schrödinger, LLC). The highest ranking and most consistent pose containing a cluster of three or more results was chosen for analysis. Further analysis was conducted using the protein-ligand interaction profiler.92 |
| § Compound LEI121 was docked as described for 11 using the antagonist bound crystal structure of CB2R (PDB: 5ZTY),16 with T4 lysozyme removed. |
| This journal is © The Royal Society of Chemistry 2022 |