
Reactivity-based chemical-genetic study of protein kinases
Renata
Rezende Miranda
 ac and
Chao
Zhang
*ab
ac and
Chao
Zhang
*ab
aDepartment of Chemistry, Loker Hydrocarbon Research Institute, University of Southern California, Los Angeles, California 90089, USA. E-mail: zhang.chao@usc.edu
bUSC Norris Comprehensive Cancer Center, University of Southern California, Los Angeles, California 90089, USA
cThomas H. Gosnell School of Life Sciences, Rochester Institute of Technology, Rochester, New York 14623, USA
First published on 30th March 2022
Abstract
The human protein kinase superfamily comprises over 500 members that operate in nearly every signal transduction pathway and regulate essential cellular processes. Deciphering the functional roles of protein kinases with small-molecule inhibitors is essential to enhance our understanding of cell signaling and to facilitate the development of new therapies. However, it is rather challenging to identify selective kinase inhibitors because of the conserved nature of the ATP binding site. A number of chemical-genetic approaches have been developed during the past two decades to enable selective chemical perturbation of the activity of individual kinases. Herein, we review the development and application of chemical-genetic strategies that feature the use of covalent inhibitors targeting cysteine residues to dissect the cellular functions of protein kinases.
 Renata Rezende Miranda | Renata Rezende Miranda received her PhD in Chemistry from the University of Southern California in 2016. She worked with Prof. Chao Zhang in the structure-based design, synthesis, and biological evaluation of covalent small-molecule inhibitors and clickable probes targeting protein kinases from the FGFR subfamily. She is currently pursuing postdoctoral research at the Rochester Institute of Technology with Prof. André Hudson. Her research now focuses on the development of chemical biology and genomic approaches to study antimicrobial resistance mechanisms, as well as on the discovery, identification, and characterization of novel antibiotic lead compounds. |
 Chao Zhang | Chao Zhang received his BS in Chemistry from Peking University in 1998. He then conducted his PhD study in Chemistry from Princeton University in 2004. His graduate study focused on the development and application of chemical-genetic approaches to dissect the functions of protein kinases in cell signaling. This was followed by a postdoctoral study at UC San Francisco, where his research focused on developing innovative approaches to generate covalent kinase inhibitors and to investigate the mechanism of drug actions. Chao started his own lab at the University of Southern California (USC) in 2011. His research at USC has focused on the discovery of covalent small-molecule inhibitors using structure-based design and chemical proteomics. |
Introduction
Protein kinases participate in central signaling pathways by controlling reversible phosphorylation of substrate proteins in eukaryotic cells. Due to the large number of kinases and their ubiquitous presence in physiological processes, these enzymes represent a major class of targets for drug discovery and development.1 While significant progress has been achieved in understanding the regulation and function of numerous kinases, a large portion of the “kinome” remains understudied, with less than 10% of all protein kinases being targeted by FDA-approved small-molecule drugs.1,2 Identification of potent and selective kinase inhibitors is a challenge because of the highly conserved nature of the kinase active site where the majority of kinase inhibitors bind. Having selective kinase inhibitors available, however, is critical to expand our understanding of kinase functions and mechanisms in biological processes, which in turn will accelerate the development of therapeutics for this protein family. About twenty years ago, chemical genetics emerged as an innovative means for deciphering the roles of kinases in biology and disease and, therefore, aid in the discovery and development of selective kinase inhibitors. Chemical genetics is a valuable and powerful tool that marries genetic selectivity to rapid pharmacological perturbation. As an emerging discipline, it incorporates diverse areas of research, including genetics, protein engineering, medicinal chemistry, and cell biology, that together allow the generation of potent and specific small-molecule inhibitors capable of selectively and rapidly perturbing the functions of one or few protein targets at a time.3,4A number of chemical-genetic approaches have been successfully developed and applied to the study of protein kinases over the past 25 years. They involve engineering a kinase for specific recognition and inhibition by a rationally designed inhibitor. Ideally, these inhibitors are not recognized by wild-type (WT) kinases to diminish off-target effects and confidently attribute the observed phenotypes to inhibition of the mutant proteins. The selective inhibition of the engineered kinase is based on either shape or reactivity complementarities between the mutant kinase and the inhibitor afforded by these chemical-genetic approaches. The first approach, termed “bump-hole”, was developed earlier, and found wide applications in the functional characterization of protein kinases. The latter approach has found complementary and unique applications compared to bump-hole using electrophilic compounds to afford covalent inhibition of the engineered kinases. In this review, we highlight structural and functional features of protein kinases that are important for inhibitor development and then describe various chemical-genetic strategies that have been used to study protein kinases in cells, with a focus on those featuring covalent inhibitors targeting cysteines.
Protein kinases
The human kinome contains 538 distinct protein kinases, accounting for approximately 3% of all human genes.5,6 Protein kinases are enzymes that catalyze the transfer of the γ-phosphoryl group of ATP to serine, threonine, and tyrosine residues on a substrate protein. Reversible phosphorylation is an essential post-translational modification that plays key roles in cellular signal transduction and metabolism. Phosphorylation catalyzed by protein kinases is involved in nearly all eukaryotic cellular pathways and is crucial to normal cell growth and development.7,8 Dysregulation of kinase function, therefore, contributes to cancers and a variety of other human disorders, including neurological, metabolic, and immunological diseases. As a result, protein kinases represent an important class of drug targets.9Structurally, serine/threonine and tyrosine kinases share a highly conserved active site that is located in a deep cleft between two lobes, the N-terminal and the C-terminal domains (Fig. 1A). These are connected by a short and flexible conserved loop known as the hinge region. The N-lobe is composed of β-sheets and one α-helix, termed helix αC, as well as a glycine-rich phosphate-binding loop (P-loop). The C-lobe is predominantly α-helical and contains the conserved DFG motif, the activation loop (A-loop), and the catalytic loop (C-loop).10,11
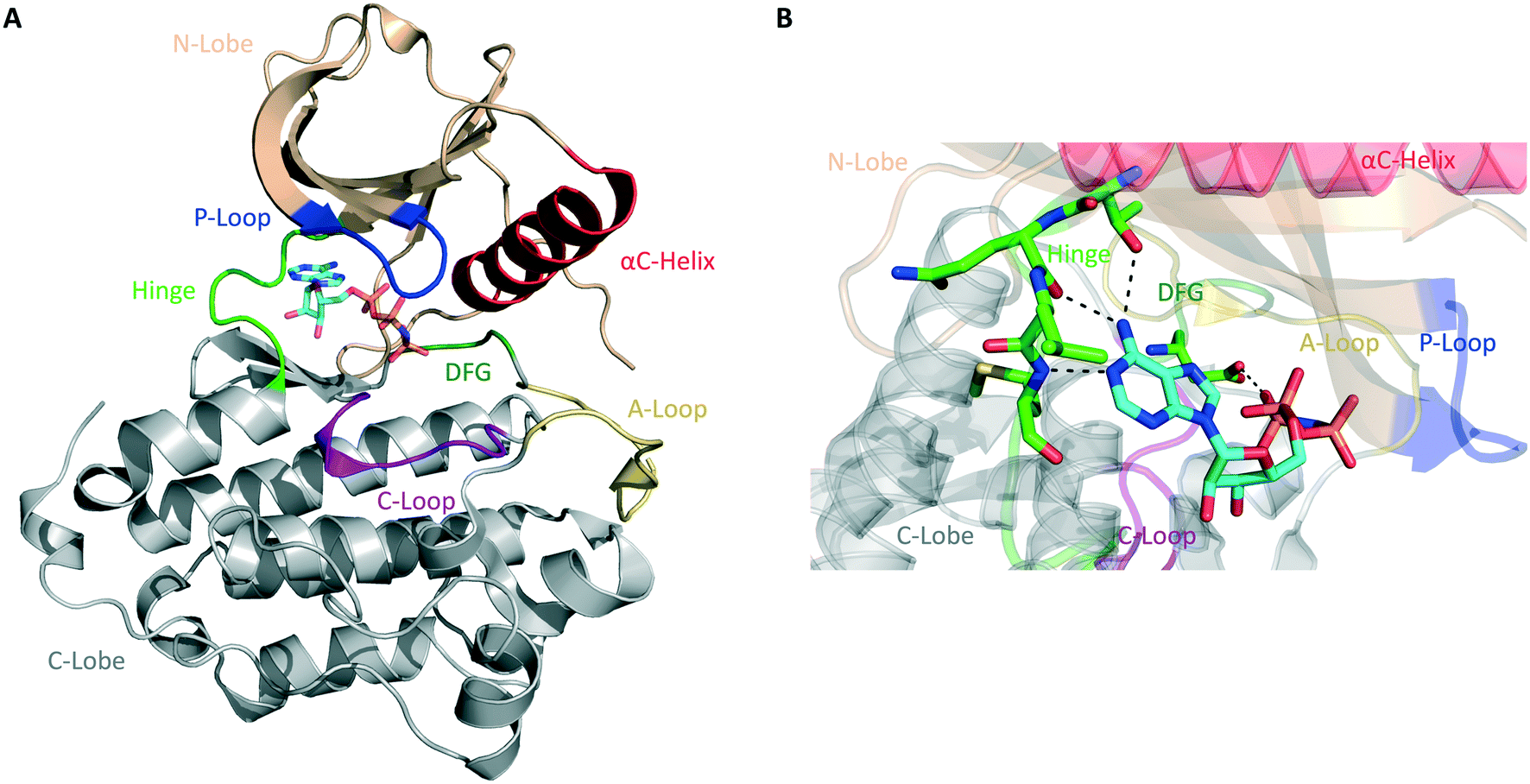 | ||
| Fig. 1 General structure of the catalytic domain of protein kinases. (A) Ribbon representation of the kinase domain of the epidermal growth factor receptor (EGFR) in its active state with key structural elements highlighted in different colors as follows: activation loop (A-loop), yellow; αC helix, red; phosphate-binding loop (P-loop), blue; catalytic loop (C-loop), pink; hinge region, green; DGF motif, dark green. Adenylyl-imidodiphosphate (AMP-PNP) is bound to the ATP-binding pocket (PDB ID 2GS6). (B) Hydrogen bonds between the ATP-binding site of EGFR and AMP-PNP are indicated by black dashed lines. | ||
Most protein kinases adopt similar active conformations upon activation, while inactive kinases adopt distinct conformations, reflecting the diversity in their regulation. Active kinases require a specific alignment of key elements in the kinase domain for a successful phosphoryl transfer. ATP binding is stabilized by interactions with conserved amino acid residues located on the hinge region, αC helix, DFG motif, and P-loop (Fig. 1B). The adenine ring of ATP forms hydrogen bonds with the peptide backbone of the hinge region, with the N6 position binding to a large and conserved amino acid side chain, termed gatekeeper, located N-terminal to this region. The phosphate groups are stabilized by interactions with the glycine-rich P-loop and a conserved lysine residue in the N-lobe (not shown in Fig. 1B). A peptide substrate binds close to the γ-phosphate of ATP near the A-loop. This loop is phosphorylated on either serine/threonine or tyrosine residues when the kinase is in its active state. The activation segment varies its conformations substantially depending on the state of the kinase. In the unphosphorylated state (inactive), it collapses into the active site and blocks the binding of both ATP and peptide substrate. When the kinase is activated by phosphorylation, it moves outwards from the active site, exposing the phosphorylated residues and allowing substrate binding and catalysis.10–12
The majority of kinase inhibitors are ATP-competitive small molecules that lack specificity due to the highly homologous nature of the ATP-binding pocket within the protein superfamily. They interact with the kinase active site, often mimicking the hydrogen bonds that the adenine group of ATP forms with the protein main-chain atoms located in the hinge region (Fig. 1B). These compounds can bind to either the active/DFG-in (type I) or the inactive/DFG-out (type II) conformations of the kinase. Other compounds bind to a nonconserved allosteric site that can be adjacent to (type III) or remote from (type IV) the kinase active site.13,14 Small molecules that target the same conserved ATP-binding pocket usually exhibit cross-reactivity, resulting in promiscuous inhibitors. Allosteric inhibitors can circumvent this hurdle by binding to distinct sites involved in regulatory mechanisms that are unique to an individual kinase, thus presenting higher selectivity.9 Only a fraction of protein kinases have been shown to be amenable to allosteric inhibition, however.
Another way to achieve selectivity is with covalent inhibitors – compounds that form an irreversible bond with a nucleophile on the kinase, usually a cysteine residue.15,16 The efficiency of a covalent inhibitor is determined by the initial reversible interactions with the protein and the rate of subsequent covalent bond formation with a nearby nucleophilic amino acid residue in the target kinase.16,17 The former depends on its reversible binding kinetics, while the latter depends on the reactivity of the electrophile and its accurate positioning.18,19 Using structure-guided design, one can predict the optimal position to attach an electrophile to a known non-covalent inhibitor. The initial step of reversible binding serves to orient the reactive warhead in the proximity of a cysteine residue for covalent bond formation. This combination of noncovalent and covalent binding factors often results in a highly specific permanent interaction between the irreversible inhibitor and the target kinase.20–22 Many kinases have cysteines in and near the ATP-binding site, which are collectively known as the cysteinome of protein kinases.23 Different studies have mapped these residues, revealing the positions of the cysteines, which are largely distributed among the hinge region, the glycine-rich P-loop, and near the DFG motif.9,24–26 These cysteines are all non-catalytic residues that do not directly participate in phosphoryl transfer, hence being well-suited for covalent targeting with electrophilic inhibitors.23
Genetic techniques including knockout/knock-in and transgenic animals, gene mutations or deletions, and RNA interference (RNAi) technology have been instrumental in functional characterization of kinases.27 The advantages of genetic approaches are the high specificity and portability, given that a single gene can be modified in different organisms. Despite its usefulness in dissecting biological functions in lower organisms, traditional genetics presents difficulties when applied in mammals due to their large physical size, large diploid genome, prolonged gestation periods, and slow reproduction rate. Another limitation is the functional compensation effect produced when related genes mask a phenotype caused by the knockout of a non-essential gene. Gene deletions can also impair the study of protein functions that are modulated by post-translational modifications and, in the case of an essential gene, can be lethal, compromising the further study of the organism. Furthermore, most genetic mutations are not conditional, i.e., they cannot be turned on and off as needed. Although conditional mutations, like temperature-sensitive (TS) alleles, can be employed to achieve reversibility, they can induce undesirable side effects, such as the heat shock response, making the resulting phenotypes difficult to interpret.3,28,29
Pharmacological approaches to study protein kinases present advantages when compared to genetic techniques, such as rapid and conditional target inhibition. The poor selectivity of small-molecule kinase inhibitors, however, is a limitation of these methods because it is always possible that the observed pharmacological phenotypes reflect an off-target effect of the drug. Combining pharmacological and genetic approaches, chemical genetics surfaced as an alternative to overcome the difficulties found in classical genetics and pharmacological methods. This approach allows the study of genes and/or proteins by using small molecules to induce a particular phenotype.30 The use of chemical genetics to dissect complex signaling pathways offers several advantages over traditional genetics. First, small molecules act rapidly and reversibly, usually functioning in different cell types and organisms. Second, this approach is conditional; ligands can be added or removed at will as well as used at different concentrations, allowing for real-time temporal control and direct kinetic analysis of the protein modulation by the chemical compound. Third, small molecules can act as either gain-of-function or loss-of-function mutations, depending on whether they act as agonists or inhibitors of the target protein. Finally, rather than shutting down entire pathways (as with classical genetics), specific binders make it possible to observe the effects of perturbing individual activities of multifunctional enzymes and receptors.27,29,31
Dissecting protein kinase functions with chemical-genetic approaches
The bump-hole approach
The use of small molecules to perturb biological functions is limited to compounds that are available and characterized. Although this was an issue when the field of chemical genetics first appeared, the number of small molecules that became accessible through commercial libraries and public repositories increased dramatically in the past two decades.29,32 A more significant issue, however, is to find compounds that specifically interact with a protein of interest. This is especially challenging when the target protein shares a high degree of homology with others, as it happens with protein kinases. As a means to overcome this problem, a combination of medicinal chemistry with molecular biology to generate non-natural ligand/protein pairs can be used.33 Using genetic techniques, it is possible to introduce point mutations or even entire domains in proteins to confer specificity to a high-affinity promiscuous inhibitor.3 Shokat and colleagues have previously established a combined chemical-genetic approach to afford highly specific inhibition of kinases by modifying the protein to exclusively recognize rationally designed inhibitors.34,35 The approach is known as “bump-hole”, with the engineered kinase referred to as analog-sensitive (AS) allele (Fig. 2). In this context, a functionally silent but structurally significant mutation is introduced to the kinase active site, specifically, to the gatekeeper residue. The gatekeeper is a conserved, bulky, and mostly hydrophobic amino acid (methionine, leucine, threonine, phenylalanine, among others) located near the hinge region (Fig. 2A). Replacement of this residue with glycine or alanine (smaller residues) creates an extended pocket (or “hole”) that can be uniquely accessed by a bulky (or “bumped”) inhibitor (Fig. 2B). The inhibitor is designed by modifying non-specific kinase inhibitors with substituents that are complementary to the new pocket only, thus not inhibiting WT kinases due to steric clash.36–39 To expand the kinome coverage of the AS-kinase technology, different heterocyclic scaffolds have been derivatized, such as quinazoline40 and indazole.41 In the attempt of producing more potent and selective AS-kinase inhibitors, however, pyrazolopyrimidine-based (PP) analogs (Fig. 2C) were found to be the ones with not only the best potency and selectivity but also generality toward protein kinases.42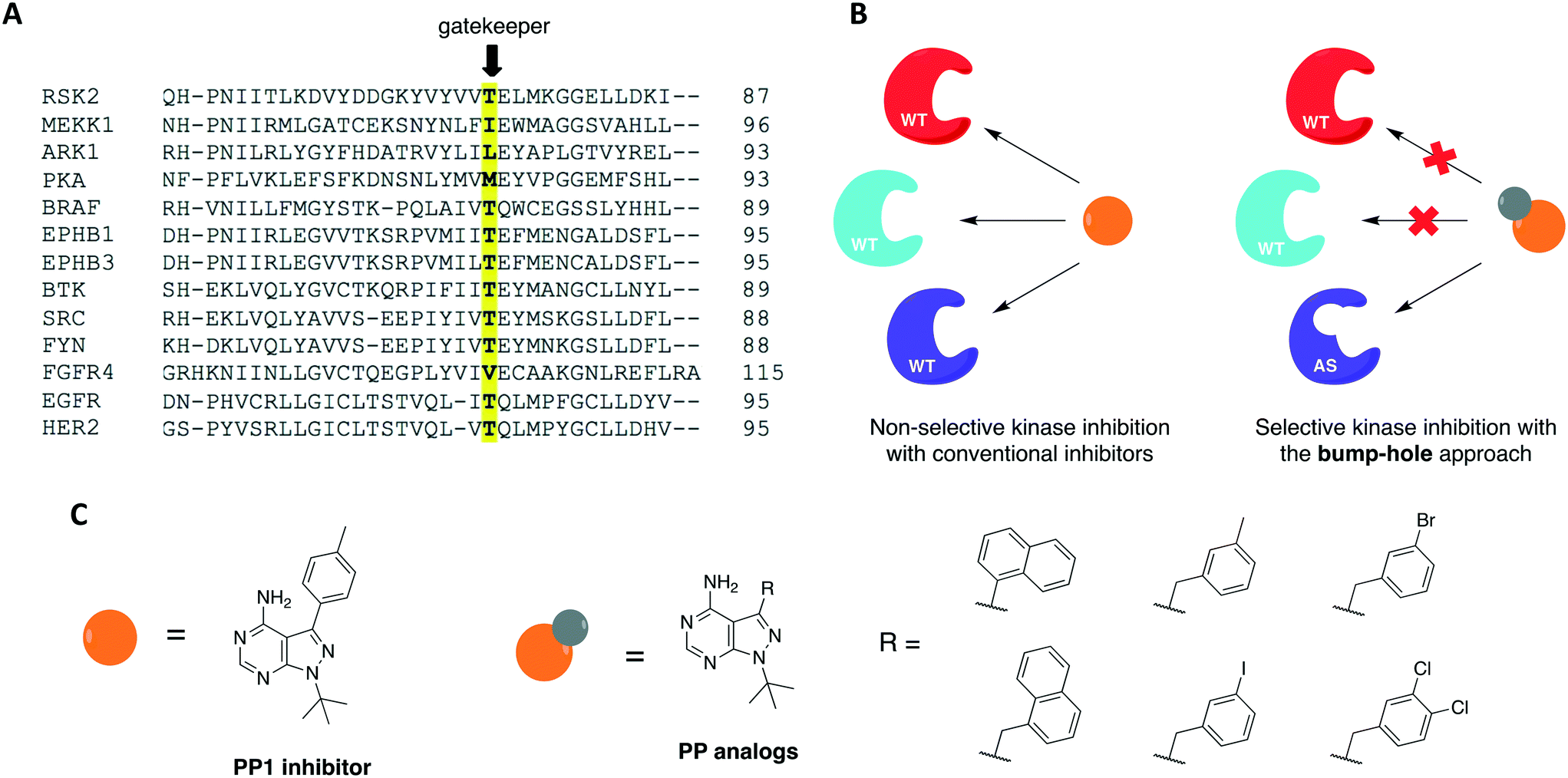 | ||
| Fig. 2 Bump-hole approach allows for selective inhibition of a single engineered protein kinase. (A) Partial sequence alignment of different kinase domains highlighting the conserved gatekeeper residue. (B) Mutation of this residue to glycine or alanine creates an extra pocket (“hole”) in the active site of the analog-sensitive kinase (AS-allele) that can be specifically targeted by a bulky (“bumped”) inhibitor. (C) Chemical structures of non-selective kinase inhibitor PP1 and rationally designed PP analogs. | ||
The bump-hole method has been applied to the study of numerous kinases in diverse organisms including mammals and yeast.43–51 Its use allowed the dissection of kinase functions and the elucidation of novel signaling mechanisms as well as the discovery of highly selective and potent kinase inhibitors. For a kinase of interest to be amenable to the approach, however, some requirements must be met. First, the stability and activity of the mutant protein should be comparable to that of the WT. Second, it must be possible to introduce the mutant allele into the organism of interest. Third, a potent, bioavailable, and orthogonal inhibitor analog to the protein of interest should be identified.3,42 Despite the successful application of this approach to dozens of kinases from diverse organisms, there are two limitations in its applications. First, some enzymes do not tolerate the required gatekeeper mutation.52 The gatekeeper residue is in direct contact with the N6 group of ATP. It governs inhibitor and substrate specificity in the kinase active site by controlling access to a deep hydrophobic pocket that is not in contact with ATP (thus the name “gatekeeper”).53 Upon mutation of this residue to glycine or alanine, certain kinases suffer a significant or severe loss of catalytic activity and cellular function. Second, although tolerant to the gatekeeper mutation, some kinases are not effectively inhibited by available bumped inhibitors.
Over the past two decades, researchers have developed complementary techniques to overcome these limitations. For instance, introduction of second-site suppressor mutations (or suppressors of glycine gatekeeper (sogg)) was able to restore the catalytic activity or cellular function of weakened AS-kinase alleles.52 This strategy has been successfully applied to kinases that play important roles in diverse signaling pathways, such as stress-activated MAPK signaling (MEKK1), mitotic regulation and cytokinesis (Cdc5), G protein-coupled receptor signaling (GRK2), and plant disease resistance (Pto). Although these strategies improved the tolerance of some kinases to bump-hole, this technique still does not work for all kinases. As a result, a significant number of the kinases are not amenable to the bump-hole method, leaving researchers with the challenging quest to discover alternative chemical-genetic approaches that could fill in this gap.
Cysteine-targeting chemical-genetic approaches
For protein kinases that were not amenable to the aforementioned methods, the use of covalent inhibition has proven effective in achieving selectivity without compromising kinase activity or biological function. These covalent chemical-genetic approaches involve the use of electrophilic compounds to target a nucleophilic cysteine residue, natural or engineered, that is in or near the active site of the target kinase. They differ in the positions of the covalently targeted cysteine residue, however. These covalent chemical-genetic studies are grouped based on the position of the covalently targeted cysteine and reviewed in detail below.The successful application of these compounds in oncology triggered more research efforts not only into the generation of novel covalent kinase inhibitors to treat an increased number of tumor types but also into other areas of research involving protein kinases, such as chemical genetics. In covalent chemical genetics, a cysteine residue can be irreversibly targeted by an electrophilic inhibitor. Covalent inhibition can be employed alone or in combination with bump-hole as a second filter to further improve selectivity (Fig. 3A). While bump-hole works with shape complementarity, cysteine-targeting approaches are based on reactivity complementarity. Some advantages of this approach over bump-hole are the possibility of targeting cysteine residues located at different positions other than the gatekeeper (Fig. 3B), achievement of a more complete inhibition due to the formation of an irreversible bond between cysteine and electrophile, higher selectivity due to targeting of rare or even unique cysteine residues on protein kinases, and the potential of developing orthogonal chemical-genetic systems to inhibit distinct kinases in the same cell (Fig. 3C).
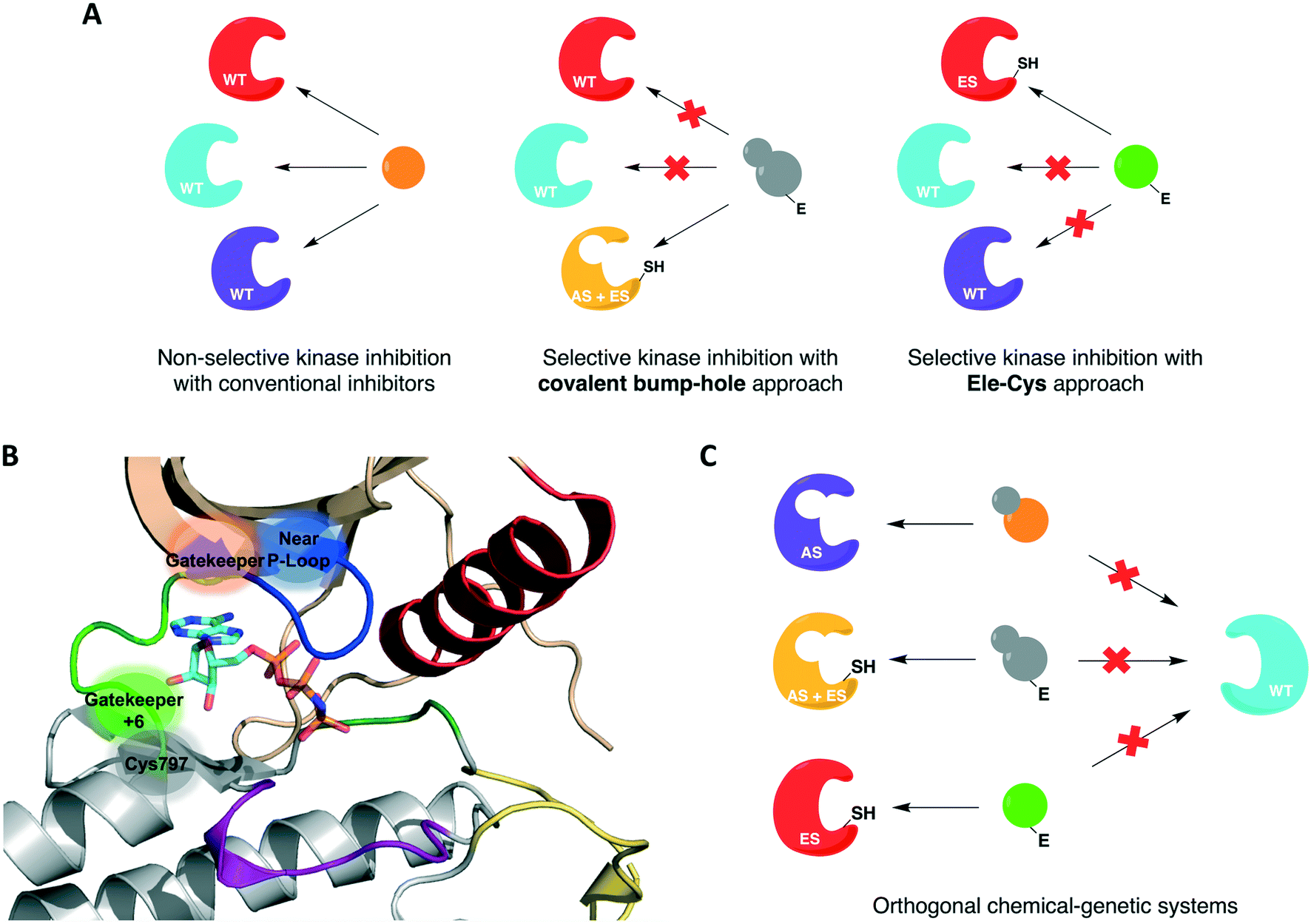 | ||
| Fig. 3 Cysteine-targeting covalent approaches to selectively inhibit protein kinases. (A) Covalent chemical-genetic approaches achieve selectivity by relying on the covalent complementarity between a native or engineered cysteine and an electrophilic inhibitor. (B) Cysteine residues at different positions can be used instead of relying on just the gatekeeper. A few previously reported positions are highlighted in and near the ATP-binding pocket in the kinase domain of EGFR (PDB ID 2GS6). (C) Covalent complementarity makes it possible to develop orthogonal pairs of engineered kinases and electrophilic inhibitors that allow the study of signaling pathways involving multiple kinases in the same cell. | ||
One covalent chemical-genetic strategy to identify specific inhibitors of protein kinases combines shape complementarity with covalent complementarity. Here, kinases are rationally designed to bear a space-creating mutation at the gatekeeper position and a cysteine at a position analogous to Cys797 of EGFR in the active site (native residue or engineered), which functions as an irreversible anchor point for a space-filling kinase inhibitor (Fig. 4A and B). The first admits binding of bulky small molecules that WT kinases do not tolerate due to steric clash, and the latter increases potency and target specificity. Some protein kinases may already have one or both selectivity filters, requiring either none or only one mutation to be introduced in its active site for this method to work. Using this strategy, c-Src was sensitized to 6-acrylamido-4-quinazoline analogs containing bulky aryl groups (Fig. 4C) by engineering the tyrosine kinase with both a glycine gatekeeper and a cysteine residue at a position analogous to Cys797 of EGFR, located at the C-terminus of the hinge region (Fig. 4A).62 These analogs were designed based on the structure of EGFR's reversible inhibitor erlotinib. The rationally designed compounds showed higher potency against analog-sensitive c-Src alleles (both AS-kinase allele and double mutant) while being less effective against a c-Src-cys mutant that did not have the expanded binding pocket. After confirming the inhibitors' selectivity for kinases containing both specificity elements, an irreversible fluorescent affinity probe was also designed to specifically label analog-sensitive EGFR in cells (Fig. 4D). With a covalent probe in hand, it was possible to measure target engagement after probe treatment and subsequent downstream signaling activation specifically caused by the kinase allele. In another study, fission yeast Aurora kinase (Ark1) was also genetically engineered for selective inhibition by covalent anilinoquinazolines.63 Both alanine gatekeeper and a cysteine residue analogous to EGFR Cys797 were introduced in the protein's active site, but additional suppressor mutations were necessary to rescue kinase function.
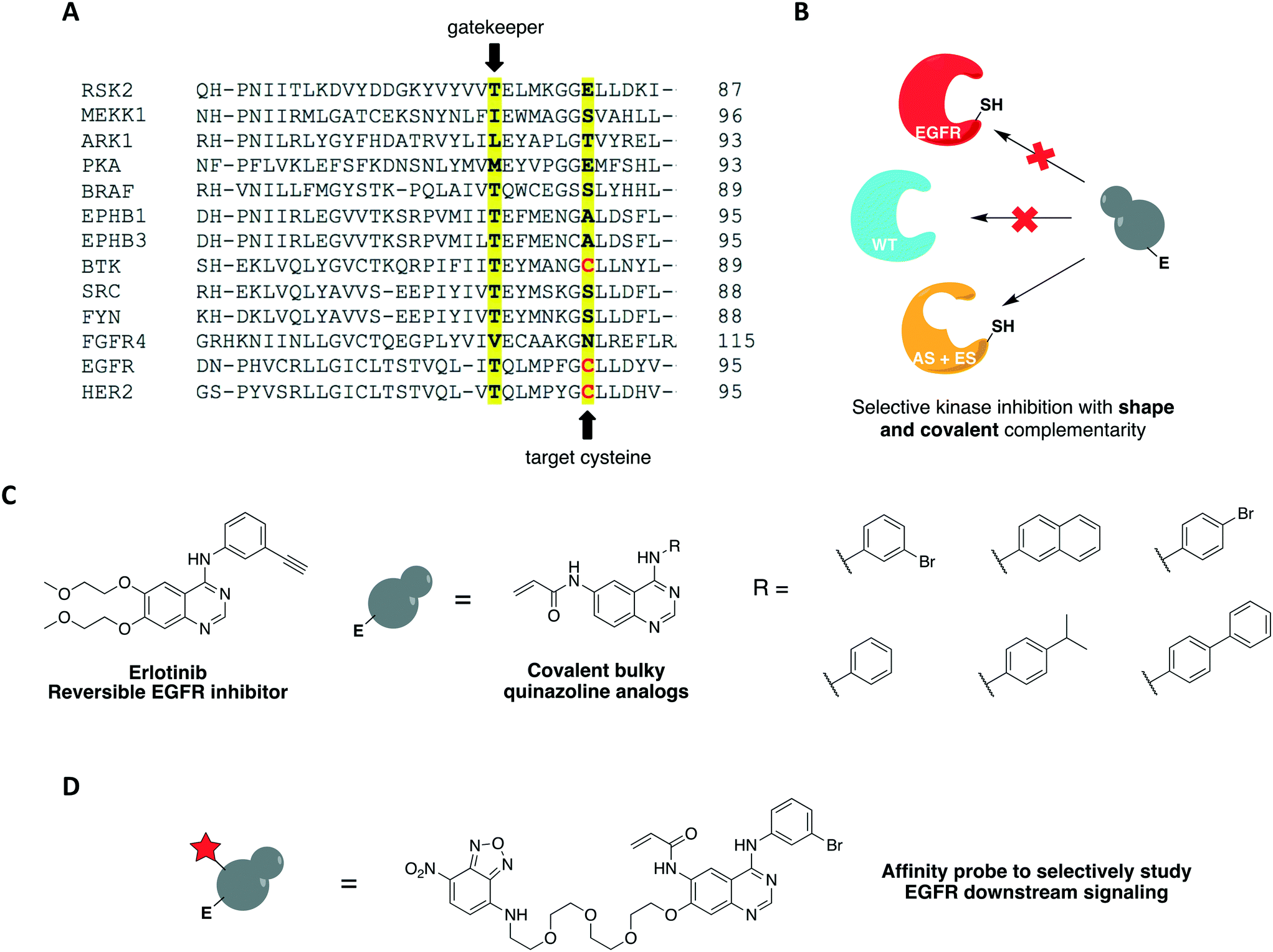 | ||
| Fig. 4 Covalent chemical-genetic approach to specifically target tyrosine kinases. (A) Partial sequence alignment of several protein kinases showing the chosen sites for the two-point mutations: the gatekeeper and a rare cysteine residue (Cys797 in EGFR). (B) By introducing two mutations into its active site, only the sensitized kinase (AS + ES) will bind to the rationally designed inhibitor since these features are not found together in any WT kinase. (C) Chemical structures of 6-acrylamido-4-anilinoquinazoline irreversible inhibitors used in the studies. The 4-anilinoquinazoline core is common in many EFGR inhibitors, such as erlotinib. (D) Chemical structure of covalent affinity probe to measure target engagement and study the downstream signaling of EGFR allele in cells. A nitrobenzoxadiazole (NBD) fluorophore is linked to the quinazoline scaffold at C7 via a PEG chain. | ||
A previous work by Poulikakos and coworkers utilized covalent complementarity with other chemical-genetic systems in a mechanistical study of RAF kinases. Here, a panel of engineered RAF kinases were used to investigate the mechanism behind an observed MEK/ERK signaling increase in cells expressing WT BRAF but not mutant BRAF(V600E) when treated with ATP-competitive RAF inhibitors (Fig. 5A and B).64 This finding was observed with six ATP-competitive RAF inhibitors, including PLX4032 and PLX4720 (vemurafenib and its precursor) (Fig. 5E). The authors proposed that the paradoxical activation of the enzyme by inhibitors was due to drug-mediated transactivation of RAF dimers. To test this hypothesis, RAF mutants with different kinase properties, such as catalytic inactivity, disrupted drug binding, electrophile sensitivity, and inability to dimerize, were created. The first series of experiments were performed with the drug PLX4720 to analyze the requirements for the paradoxical activation. Using CRAF mutants that could not bind to either RAS or the drug, the study showed that MEK/ERK activation by PLX4720 is RAS dependent and requires direct drug binding to the catalytic domain of RAF. Besides, knockout of Braf or Craf proved that only CRAF expression is necessary for significant signaling induction while BRAF is not required. The paradoxical activation was further confirmed by the observed increase on phosphorylation of both CRAF WT and a kinase-dead mutant upon binding of PLX4720, but not of the drug-resistant CRAF mutant. This induced phosphorylation led to activation of the enzyme and, subsequently, ERK signaling. After confirming the activation hypothesis, a covalent chemical-genetic approach was introduced to study the mechanism by which RAF inhibitors activate RAF dimers. For this, a cysteine residue was introduced into RAF at the position that is homologous to EGFR Cys797 (Fig. 5C) to achieve isoform selectivity through covalent inhibition by electrophilic quinazolines (JAB analogs) (Fig. 5E), which would not be possible with the available reversible inhibitors of the kinase. Co-expression of JAB34-sensitive, kinase-dead CRAF and JAB34-insensitive, catalytically active CRAF proteins in 293H cells led to pronounced induction of ERK phosphorylation, showing that drug binding to the kinase-dead mutant transactivated the catalytically competent RAF. In contrast, when the drug bound to a catalytically active mutant that dimerized with another that was kinase-dead but JAB34-insensitive, high concentration of the drug (10 μM) inhibited ERK signaling. Transactivation from CRAF to BRAF was also confirmed when co-expressing BRAF and the JAB34-sensitive, kinase-dead CRAF mutant in 293H cells. This model suggested a dependency on RAF dimerization for transactivation to occur. This was further validated with JAB34 failing to induce ERK signaling in cells co-expressing catalytically competent kinase and a JAB34-sensitive CRAF mutant with compromised ability to dimerize.
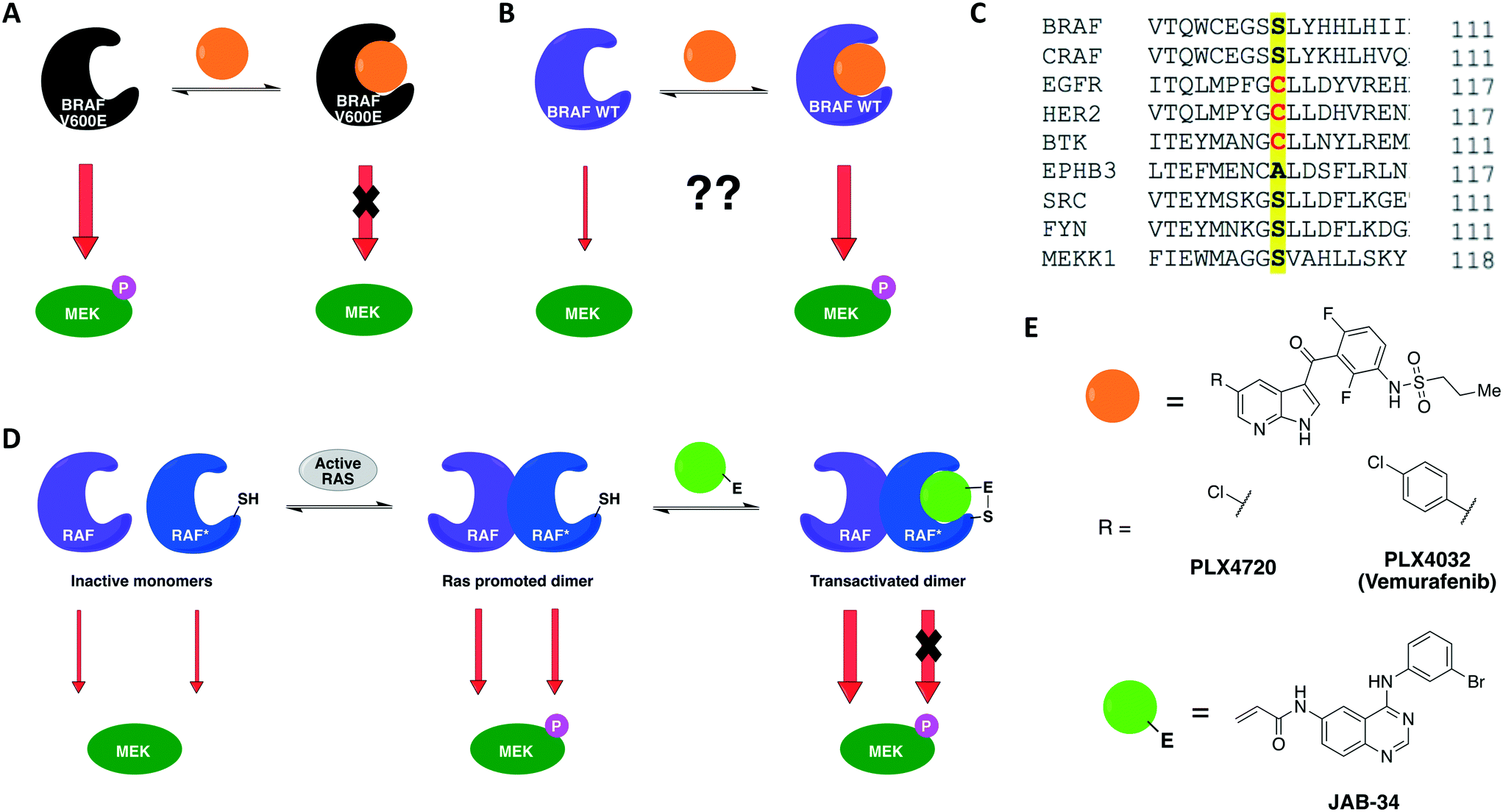 | ||
| Fig. 5 Constitutively active BRAF(V600E) is inhibited by ATP-competitive drugs (A) but not BRAF(WT) (B). (C) CRAF kinase was engineered with a cysteine residue analogous to EGFR Cys797 to achieve isoform selectivity through covalent inhibition by electrophilic quinazolines. (D) Covalent chemical genetics explained the paradoxical activation of BRAF(WT). Binding of the covalent inhibitor to the electrophile-sensitive protomer* within a RAF dimer produced both abolition of the catalytic activity of the inhibitor-bound RAF and transactivation of the drug-free RAF. (E) Reversible and irreversible ATP-competitive inhibitors that were employed in the chemical-genetic study. | ||
Together, these chemical-genetic tools successfully showed that induction of RAF signaling in cells with WT BRAF is caused by the paradoxical transactivation of RAF dimers by ATP-competitive RAF inhibitors. Binding of the drug to the ATP-binding site of one promoter within a RAF dimer (either CRAF–CRAF or CRAF–BRAF) caused both abolition of the catalytic activity of the inhibitor-bound RAF and transactivation of the drug-free RAF (Fig. 5D). The use of EGFR inhibitors in this specific study was not ideal because EGFR is situated upstream to MAPK kinases in this cellular signaling pathway, which might interfere with the RAF–MEK–ERK pathway outputs. Nonetheless, the specificity of the cysteine-targeting approach granted orthogonality to the combined systems and the rigorous use of WT RAF as a control in most experiments still allowed the authors to successfully test their dimer transactivation hypothesis. These findings contributed profoundly to the development of RAF inhibitors for use in the clinics. The transactivation model reported in this study predicted in which cases RAF inhibitors would be effective or not, and when they could cause toxicity or drug resistance. Some of these predictions were later confirmed in the clinical trial of vemurafenib, the first RAF-selective inhibitor, and pointed directions for the development of second-generation BRAF inhibitors, such as pan-RAF inhibitors and dimer breakers (or paradox breakers).
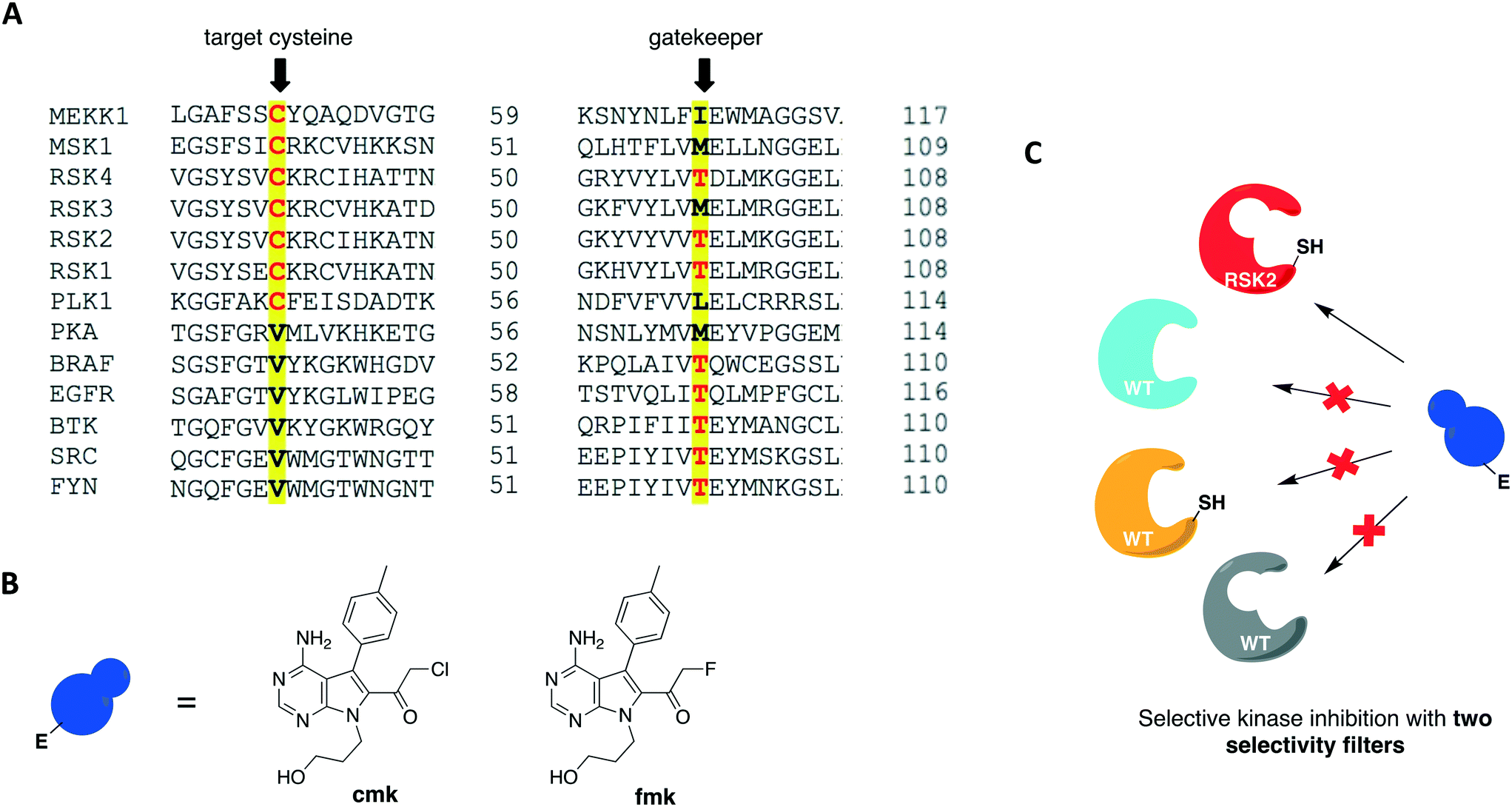 | ||
| Fig. 6 Covalent chemical-genetic approach exploiting two selectivity filters to target RSK kinases. (A) RSK2 is amongst the few protein kinases bearing a reactive cysteine at the P-loop C-terminal end and a small threonine gatekeeper residue. (B) Structures of the rationally designed halomethylketone pyrrolopyrimidines, cmk and fmk. (C) The two filters combined afforded selective targeting of RSK2 with the bulky electrophilic inhibitor. | ||
A more recent work reported a chemical-genetic strategy named cysteine installation for modulating allostery and targeted inhibition of kinases (CystIMATIK).67 This method combines shape complementarity and covalent complementarity with comprehensive mutagenesis of Src's catalytic domain to probe the conformation of Src kinases. Using deep mutation scanning, they identified amino acid residues that are important for regulating the protein's activity and studied the phenotypes corresponding to gain-of-function mutations. For example, the E381T mutant led to increased phosphotyrosine levels in multiple mammalian cell lines and promoted non-apoptotic membrane blebbing. The chemical-genetic method was then used to study the mechanism that caused a mutant's specific observed phenotype. CystIMATIK consisted of the installation of a cysteine mutation at the homologous position to RSK2 Cys436 into the kinase active site for selective targeting with a set of electrophilic probes. These compounds can stabilize different ATP-binding conformations, such as helix αC-out or DFG-out, that are responsible for modulating intramolecular regulatory interactions and global conformations of the kinase. They are also ATP-competitive inhibitors that will block phosphotransferase activity to allow the investigation of phosphotransferase-independent phenotypic effects. Using inhibitor 1 as a starting point, a panel of covalent pyrrolopyrimidine derivatives were designed to contain different bulky substituents at the C-5 position to specifically bind to a certain kinase conformation, and an α-cyano-substituted acrylamide as the warhead (Fig. 7A). Probe 1 is known to bind to kinases containing both a cysteine on the P-loop and a threonine at the gatekeeper position, which are residues V284 and T341 in Src, respectively (Fig. 7B). The rationally designed inhibitors exhibited potent and selective activity against the V284C mutant Src kinase. They were also shown to control the confirmation of Src's catalytic domain precisely and specifically, allowing for the study of the protein's global conformation separately from its phosphotransferase activity. As expected, 1 caused minimal perturbation to Src's global conformation. Helix αC-out-stabilizing inhibitor 2 promoted a closed global conformation of Src. Finally, DFG-out-stabilizing inhibitor 3 produced an open Src global conformation and consequently an increased SH3 domain accessibility (Fig. 7C). The study showed that promoting Src's open global conformation is responsible for the previously observed membrane blebbing, which requires Src's membrane interaction but not its phosphotransferase activity. Moreover, it unveiled a direct regulatory interaction in Src caused by a previously unknown intramolecular interaction between Src's N-terminal SH4 domain and the αF pocket in the C-terminal lobe of the catalytic domain. This engagement seems to be responsible for modulating the conformation, activity, localization, and effect of the kinase in cells.
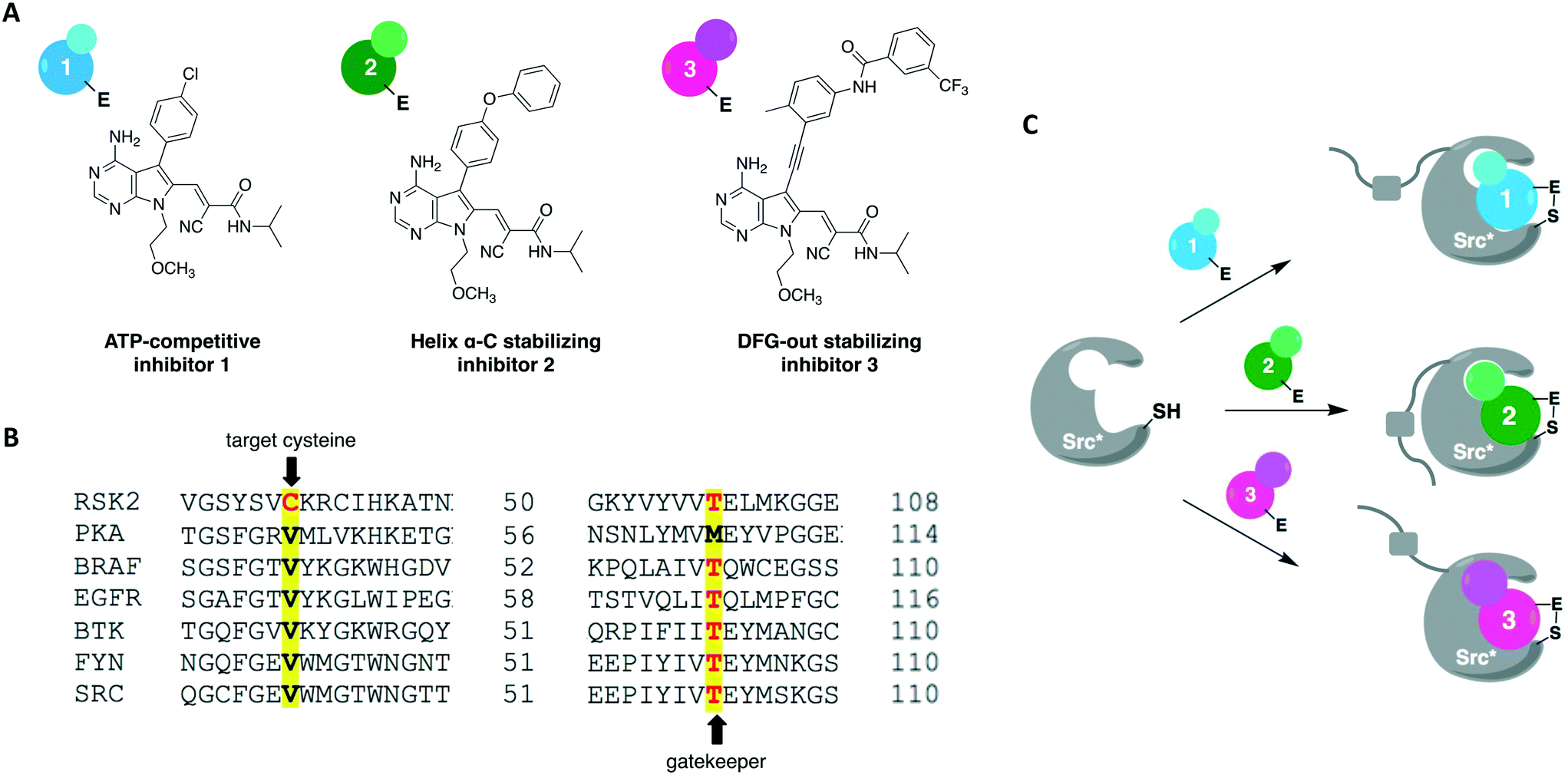 | ||
| Fig. 7 Cysteine installation for modulating allostery and targeted inhibition of kinases (CystIMATIK) method to probe kinase conformation. (A) Compound 1 was used as a starting point to generate conformation-selective probes 2 and 3. By changing the C5 substituent on the pyrrolopyrimidine scaffold, CystIMATIK probes stabilize different ATP-binding site conformations. (B) Cysteine residue at the P-loop C-terminal end (V284 in Src) and a threonine gatekeeper residue (T341 in Src) were chosen as the two selectivity filters to generate CystIMATIK-sensitive kinases. (C) CystIMATIK was used to modulate Src's global conformation to investigate phosphotransferase-independent activity. Mutant Src* (V284C), that has a small gatekeeper residue, was selectively targeted by the inhibitors while sparing most wild-type protein kinases, allowing for the specific dissection of the engineered kinase's cellular activities. | ||
In 2016, Kung and collaborators identified a cysteine residue at a position six residues after the gatekeeper (gatekeeper +6), Cys717, that is unique to receptor tyrosine kinase EphB3 among all human kinases.70 The authors tested a panel of electrophilic quinazolines against EphB3 and found that one quinazoline-chloroacetamide compound inhibited Cys717 with high potency and selectivity. As an alternative to overcome the aforementioned limitations, a recent study reported a novel covalent chemical-genetic approach, termed Ele-Cys, which yielded potent and specific interactions between an electrophilic small molecule and an engineered cysteine in the target protein.71 A position homologous to Cys717 of EphB3 was chosen to introduce the cysteine mutation on EphB1 (Fig. 3B and 8A). Because only EphB3 contains a native cysteine at the same location, covalent targeting of a thiol group at this position should allow for potent and specific inhibition of an EphB1 Cys mutant (G703C) by compound 1 (Fig. 8B and C), which caused the most potent inhibition of the kinase amongst a panel of electrophilic quinazolines. Furthermore, EphB3 expression is limited to a few tissues, maximizing inhibitor selectivity in most cell types. The authors also created an AS allele (T697G) of EphB1, to be targeted by the bumped inhibitor 3 MB-PP1, as a comparison to the Ele-Cys approach in their experiments (Fig. 8B and C). The cysteine mutation caused less perturbation of the enzyme's catalytic activity than the gatekeeper mutation, suggesting that the first is more well tolerated than the latter. Both compounds inhibited their respective allele kinase target with great potency in vitro and in cells. While the two chemical-genetic systems showed cross-inhibition in vitro, this effect was significantly lower in situ likely due to an increased efficacy of covalent inhibition in a cellular environment. Other protein kinases, such as FGFR4 and RAF (unpublished data), were amenable to the Ele-Cys approach when introduced with a homologous cysteine mutation. Finally, the study revealed that it is possible to control two separate kinases within the same cell by combining the two approaches (Fig. 8C). Covalent inhibitor 1 and bumped inhibitor 3 MB-PP1 targeted EphB3(WT) and EphB1(T697G), respectively, and specifically inhibited their autophosphorylation. With these results, the authors discovered not only that there is little trans-phosphorylation occurring between the two kinases but also that they have a preference for homodimerization over heterodimerization (Fig. 8D). Overall, this approach demonstrated generality when different kinases with homologous cysteine mutations were sensitive to the inhibitors, and orthogonality to the established bump-hole method when WT EphB3 and a Gly-gatekeeper mutant of EphB1 were expressed in the same cell and separately inhibited by the covalent small-molecule and a bulky PP1 analog, respectively.
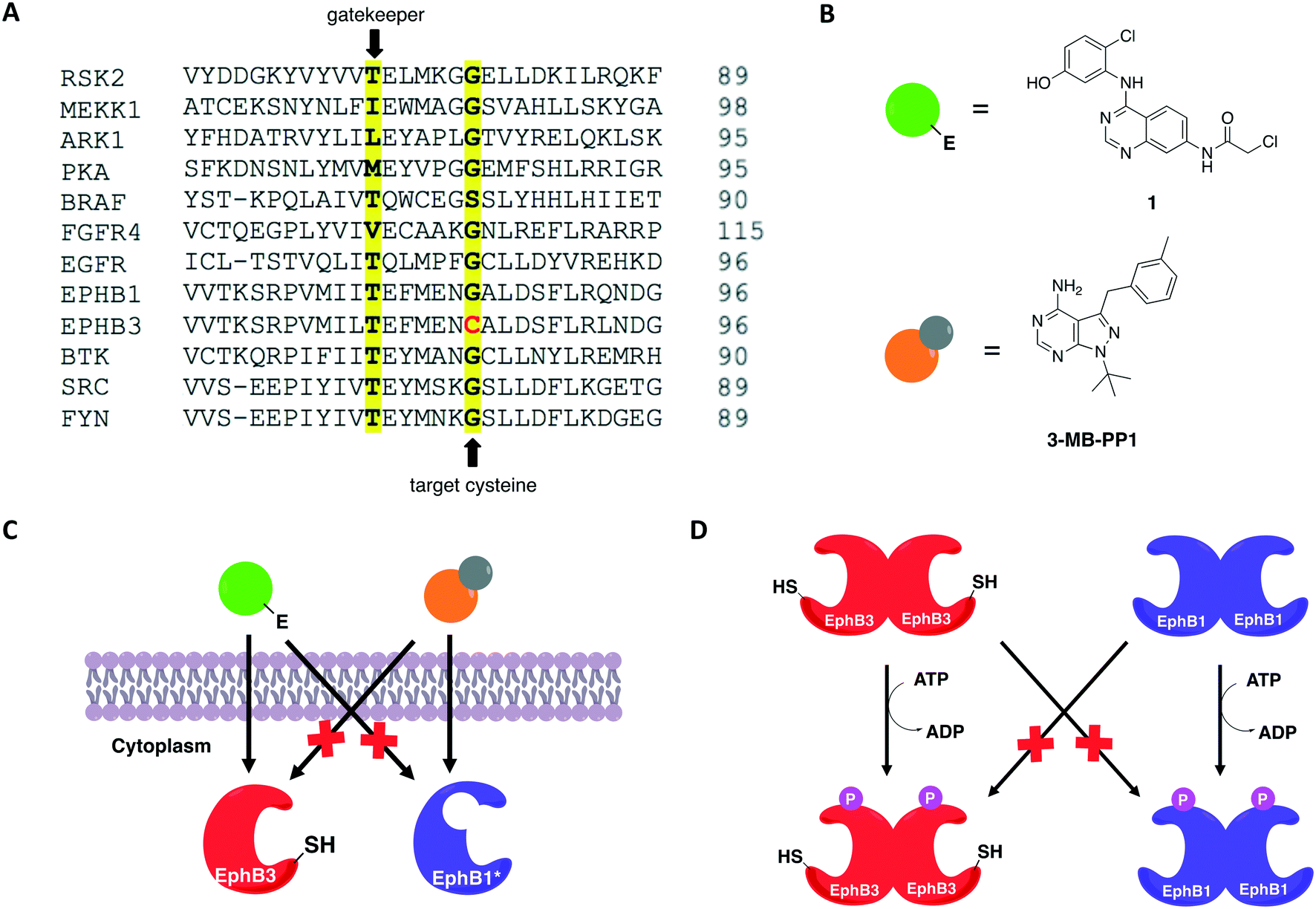 | ||
| Fig. 8 Covalent chemical-genetic approach named Ele-Cys to identify selective covalent inhibitors of protein kinases. (A) A position six residues C-terminal to the gatekeeper was chosen to install the cysteine, which is only found in a single human kinase, EphB3. (B) Chemical structures of the inhibitors utilized in the study. Electrophilic quinazoline 1 and bumped inhibitor 3-MB-PP1 serve as a comparison. (C) Both AS and Ele-Cys chemical-genetic approaches afforded orthogonal inhibition of two kinases in the same cell. (D) The two strategies were employed together to examine the communications between two distinct Eph kinases: EphB3 and EphB1. Selective inhibition of EphB3-WT and EphB1-AS upon treatment with the two inhibitors shown in (B) caused selective abolishment of autophosphorylation of only the respective Eph kinase, indicating that there is little trans-phosphorylation between them. | ||
Conclusions and perspectives
Protein kinases are an important class of drug targets for the treatment of several diseases. Although protein kinases comprise one of the largest enzyme families being encoded by the human genome,72,73 only a small portion (<10%) of the kinome is targeted by current FDA-approved kinase inhibitors. As of January 2021, about two dozen distinct protein kinases were targets of 62 therapeutic agents, with the majority being prescribed as anticancer therapies, and only 7 being used in the treatment of non-malignant disorders such as rheumatoid arthritis and pulmonary fibrosis.74,75 Protein kinases have been strongly linked to cancer since the 1970s when vSrc was identified as the first oncogene.76 The complexity of kinase signaling and the successful history of kinase inhibitors in cancer therapy since then have perhaps biased kinase drug discovery towards anticancer drug development.77 Nonetheless, the physiological functions of the remaining untargeted kinases and their direct substrates are still poorly understood. Consequently, the untargeted kinome represents a major opportunity to expand the range of kinase targets for the development of novel therapies for other disease areas.A key step in the process of drug development is preclinical target validation.78 This involves target engagement studies to monitor the interaction of an inhibitor with its target protein and correlate it to pharmacological and phenotypical effects in the cell. Quantifying protein–ligand binding is also important to determine the inhibitor's target occupancy, i.e., the extent of target engagement. This is crucial to differentiate the doses that produce efficacy while limiting side effects from those that generate toxicity as well as to attribute the physiological effect to the perturbation of the protein of interest as opposed to other targets.79,80 The controlled manipulation of proteins with chemical probes through chemical genetics is a powerful approach to study target engagement and uncover protein functions in biological systems. Great progress has been made in the field of chemical genetics and its applications in the past two decades. Chemical genetics has proven to be a more advantageous tool to interrogate proteins and pathways than genetic and pharmacological models because it offers a combination of higher target specificity with rapid and reversible spatiotemporal control of kinase activity.81 Besides, engineered protein kinases and their ligands have been successfully applied to a variety of scenarios to dissect the functions of these important biological regulators. For instance, unique findings from chemical-genetic studies include the identification of new functions and endogenous substrates of several protein kinases45,62,63,66 and the elucidation of dimerization and phosphorylation mechanisms between distinct isoforms of the Eph kinase subfamily,70 as well as of intramolecular interactions between different domains on the Src protein that regulate its global conformation and cellular activity.67 A chemical-genetic study of RAF kinases had significant clinical implications on the development of RAF inhibitors predicting in which cases they would be effective or cause toxicity and drug resistance.64 Additionally, in a recent work, Remenyi and colleagues described a chemical-genetic model in which a C905S mutant of JAK3 in mice was created to study the function of this kinase and its inhibitors in vivo.82 A summary of the different cysteine residues that have been explored thus far in covalent chemical genetics with the respective protein kinases can be found in Fig. 9.
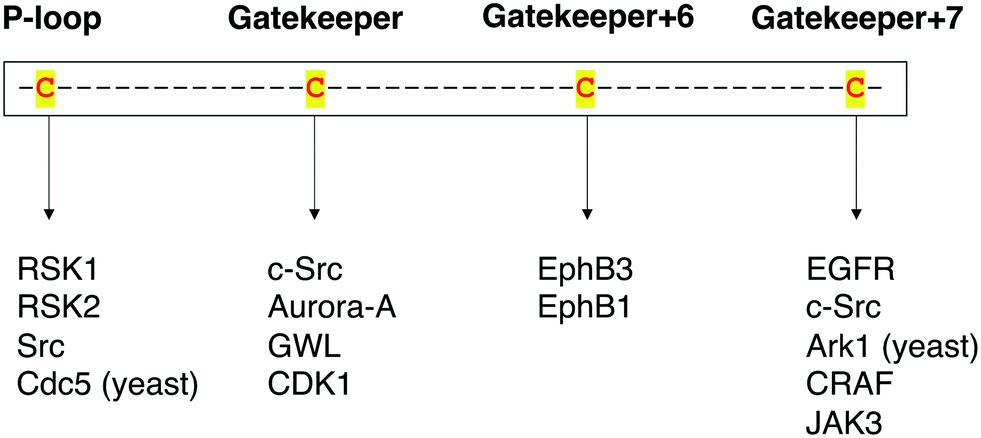 | ||
| Fig. 9 Summary of the protein kinases that have been studied by covalent chemical-genetic methods according to the targeted cysteine positions (either native or mutated). | ||
These unique chemical-genetic findings were made possible with the use of chemical probes with a covalent, irreversible mode of action. The initial chemical-genetic studies employed the bump-hole approach to introduce shape or steric complementarity between the engineered kinase–ligand pair and achieve selectivity. Although this method proved effective in some cases, it later showed drawbacks such as impaired kinase activity and function, and non-specific interactions of the bumped inhibitor with WT proteins. These difficulties inspired scientists to search for different approaches and introduce the concept of covalent chemical genetics. Based on reactivity or covalent complementarity, inhibitors containing an electrophilic warhead were rationally designed to specifically and irreversibly bind to a non-catalytic cysteine residue, native or mutated, on the ATP-binding pocket of a target protein kinase. Different from bump-hole, which involves engineering the kinase in only one site (the gatekeeper) for the method to work, cysteine residues can be introduced at multiple positions in covalent chemical-genetics. Consequently, covalent complementarity, either alone or combined with steric components, has demonstrated superior generality and orthogonality across the kinome than bump-hole, thereby becoming the approach of choice in the majority of chemical-genetic studies since its discovery.
In most examples of the covalent approach, the unique nucleophilicity of the thiol group of cysteine has been explored. While this strategy has been successful so far, it restricts the number of proteins that can be targeted by electrophilic inhibitors. Cysteine residues, although highly reactive, are among the lowest naturally abundant amino acids found in proteins and, in several cases, are present within disulfide bonds, thus reducing their accessibility for covalent modification.83 This shortcoming has encouraged researchers to develop novel methodologies that target other amino acid residues for covalent inhibition. Lysine, tyrosine and serine are amidst the residues with increasing interest in this context and hold the potential to be employed as chemical warheads in the development of novel covalent probes for chemical-genetic studies. For example, a sulfonyl fluoride probe (XO44) was employed in the first broad-spectrum chemoproteomic study of the human kinome in intact cells.84 Probe XO44 was shown to covalently label lysine residues in the active sites of about 133 diverse endogenous kinases, and was used to monitor the intracellular target engagement by dasatinib, an FDA-approved BCR-ABL inhibitor for chronic myeloid leukemia. More recently, a sulfonyl-triazole analog of XO44 (KY-26) was designed to modify both lysine and tyrosine residues on protein kinases in another chemoproteomic analysis.85 This shows that irreversible inhibitors are well-suited not only for chemical genetics but they also serve as ideal scaffolds for the development of chemical probes for use in other applications such as activity-based protein profiling (ABPP) or high-throughput screens (HTS).
Another current advancement in this field is the use of gene editing technologies such as CRISPR/Cas9. So far, reported chemical-genetic studies relied on the transient or stable overexpression of the engineered kinase instead of its endogenous expression. However, overexpression systems can disturb kinase signaling pathways86 and potentially produce misleading results. In a recent work, CRISPR/Cas9 was used to mutate a serine residue to cysteine at the DFG-1 position in the ATP-binding pocket of the non-receptor tyrosine kinase feline sarcoma (FES) oncogene, a potential target for cancer and immune disorder therapies.87 A complementary fluorescent covalent probe was also developed to specifically target and label the mutant kinase. With a small change at the genome and protein level, this strategy showed that regulation at transcriptional and (post-)translational level activity were minimally affected when CRISPR/Cas9-generated mutant cells behaved comparably to WT cells in downstream signaling assays. This combined chemical-genetic strategy was developed as a target validation method for FES kinase, but the rapid advancements in gene editing technologies will considerably accelerate the application of chemical-genetic approaches to a larger number of protein kinases.
Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
This work was supported by National Science Foundation (CHE-1455306), American Cancer Society (IRG-58-007-51), and the University of Southern California.References
- F. M. Ferguson and N. S. Gray, Nat. Rev. Drug Discovery, 2018, 17, 353–376 CrossRef CAS PubMed.
- R. Santos, O. Ursu, A. Gaulton, A. P. Bento, R. S. Donadi, C. G. Bologa, A. Karlsson, B. Al-Lazikani, A. Hersey, T. I. Oprea and J. P. Overington, Nat. Rev. Drug Discovery, 2017, 16, 19–34 CrossRef CAS PubMed.
- P. J. Alaimo, M. A. Shogren-knaak and K. M. Shokat, Curr. Opin. Chem. Biol., 2001, 5, 360–367 CrossRef CAS PubMed.
- L. M. Elphick, S. E. Lee, A. A. Anderson, E. S. Child, L. Bonnac, V. Gouverneur and D. J. Mann, Future Med. Chem., 2009, 1, 1233–1241 CrossRef CAS PubMed.
- D. Fabbro, S. W. Cowan-Jacob and H. Moebitz, Br. J. Pharmacol., 2015, 172, 2675–2700 CrossRef CAS PubMed.
- N. Berndt, R. M. Karim and E. Schönbrunn, Curr. Opin. Chem. Biol., 2017, 39, 126–132 CrossRef CAS PubMed.
- M. Huse and J. Kuriyan, Cell, 2002, 109, 275–282 CrossRef CAS PubMed.
- P. Cohen, Nat. Rev. Drug Discovery, 2002, 1, 309–315 CrossRef CAS PubMed.
- J. Zhang, P. L. Yang and N. S. Gray, Nat. Rev. Cancer, 2009, 9, 28–39 CrossRef CAS PubMed.
- M. Huse and J. Kuriyan, Cell, 2002, 109, 275–282 CrossRef CAS PubMed.
- J. A. Endicott, M. E. M. Noble and L. N. Johnson, Annu. Rev. Biochem., 2012, 81, 587–613 CrossRef CAS PubMed.
- M. A. Lemmon and J. Schlessinger, Cell, 2010, 141, 1117–1134 CrossRef CAS PubMed.
- A. C. Dar and K. M. Shokat, Annu. Rev. Biochem., 2011, 80, 769–795 CrossRef CAS PubMed.
- Z. Zhao, H. Wu, L. Wang, Y. Liu, S. Knapp, Q. Liu and N. S. Gray, ACS Chem. Biol., 2014, 9, 1230–1241 CrossRef CAS PubMed.
- Q. Liu, Y. Sabnis, Z. Zhao, T. Zhang, S. J. Buhrlage, L. H. Jones and N. S. Gray, Chem. Biol., 2013, 20, 146–159 CrossRef CAS PubMed.
- A. Aljoundi, I. Bjij, A. El Rashedy and M. E. S. Soliman, Protein J., 2020, 39, 97–105 CrossRef CAS PubMed.
- I. Miyahisa, T. Sameshima and M. S. Hixon, Angew. Chem., Int. Ed., 2015, 54, 14099–14102 CrossRef CAS PubMed.
- J. S. Martin, C. J. MacKenzie, D. Fletcher and I. H. Gilbert, Bioorg. Med. Chem., 2019, 27, 2066–2074 CrossRef CAS PubMed.
- M. E. Flanagan, J. A. Abramite, D. P. Anderson, A. Aulabaugh, U. P. Dahal, A. M. Gilbert, C. Li, J. Montgomery, S. R. Oppenheimer, T. Ryder, B. P. Schuff, D. P. Uccello, G. S. Walker, Y. Wu, M. F. Brown, J. M. Chen, M. M. Hayward, M. C. Noe, R. S. Obach, L. Philippe, V. Shanmugasundaram, M. J. Shapiro, J. Starr, J. Stroh and Y. Che, J. Med. Chem., 2014, 57, 10072–10079 CrossRef CAS PubMed.
- R. A. Bauer, Drug Discovery Today, 2015, 20, 1061–1073 CrossRef CAS PubMed.
- Z. Zhao and P. E. Bourne, Drug Discovery Today, 2018, 23, 727–735 CrossRef CAS PubMed.
- A. Abdeldayem, Y. S. Raouf, S. N. Constantinescu, R. Moriggl and P. T. Gunning, Chem. Soc. Rev., 2020, 49, 2617–2687 RSC.
- M. Gehringer, in Topics in Medicinal Chemistry, Springer Nature, Berlin, Heidelberg, 2020, pp. 1–52 Search PubMed.
- E. Leproult, S. Barluenga, D. Moras, J. M. Wurtz and N. Winssinger, J. Med. Chem., 2011, 54, 1347–1355 CrossRef CAS PubMed.
- Z. Zhao, Q. Liu, S. Bliven, L. Xie and P. E. Bourne, J. Med. Chem., 2017, 60, 2879–2889 CrossRef CAS PubMed.
- A. Chaikuad, P. Koch, S. A. Laufer and S. Knapp, Angew. Chem., Int. Ed., 2018, 57, 4372–4385 CrossRef CAS PubMed.
- M. Kawasumi and P. Nghiem, J. Invest. Dermatol., 2007, 127, 1577–1584 CrossRef CAS PubMed.
- B. R. Stockwell, Nat. Rev. Genet., 2000, 1, 116–125 CrossRef CAS PubMed.
- C. J. O'connor, L. Laraia and D. R. Spring, Chem. Soc. Rev., 2011, 40, 4332–4345 RSC.
- K. M. Specht and K. M. Shokat, Curr. Opin. Cell Biol., 2002, 14, 155–159 CrossRef CAS PubMed.
- B. R. Stockwell, Trends Biotechnol., 2000, 18, 449–455 CrossRef CAS PubMed.
- I. Smukste and B. R. Stockwell, Annu. Rev. Genomics Hum. Genet., 2005, 6, 261–286 CrossRef CAS PubMed.
- L. M. Elphick, S. E. Lee, V. Gouverneur and D. J. Mann, ACS Chem. Biol., 2007, 2, 299–314 CrossRef CAS PubMed.
- A. C. Bishop, K. Shah, Y. Liu, L. Witucki, C. Y. Kung and K. M. Shokat, Curr. Biol., 1998, 8, 257–266 CrossRef CAS PubMed.
- A. C. Bishop and K. M. Shokat, Pharmacol. Ther., 1999, 82, 337–346 CrossRef CAS PubMed.
- Y. Liu, K. Shah, F. Yang, L. Witucki and K. M. Shokat, Bioorg. Med. Chem., 1998, 6, 1219–1226 CrossRef CAS PubMed.
- A. Bishop, O. Buzko, I. Jung, B. Kraybill, Y. Liu, K. Shah, S. Ulrich, L. Witucki, F. Yang, C. Zhang and K. M. Shokat, Annu. Rev. Biophys. Biomol. Struct., 2000, 29, 577–606 CrossRef CAS PubMed.
- J. Blethrow, C. Zhang, K. M. Shokat and E. L. Weiss, Curr. Protoc. Mol. Biol., 2004, 66, 18.11.1–18.11.19 Search PubMed.
- M. S. Lopez, J. I. Kliegman and K. M. Shokat, in Methods in Enzymology, Elsevier Inc., 1st edn, 2014, vol. 548, pp. 189–213 Search PubMed.
- C. Zhang and K. M. Shokat, Tetrahedron, 2007, 63, 5832–5838 CrossRef CAS.
- T. Okuzumi, G. S. Ducker, C. Zhang, B. Aizenstein, R. Hoffman and K. M. Shokat, Mol. BioSyst., 2010, 6, 1389–1402 RSC.
- C. Zhang, M. S. Lopez, A. C. Dar, E. Ladow, S. Finkbeiner, C. H. Yun, M. J. Eck and K. M. Shokat, ACS Chem. Biol., 2013, 8, 1931–1938 CrossRef CAS PubMed.
- A. C. Bishop, C. Y. Kung, K. Shah, L. Witucki, K. M. Shokat and Y. Liu, J. Am. Chem. Soc., 1999, 121, 627–631 CrossRef CAS.
- A. C. Bishop, J. A. Ubersax, D. T. Petsch, D. P. Matheos, N. S. Gray, J. Blethrow, E. Shimizu, J. Z. Tsien, P. G. Schultz, M. D. Rose, J. L. Wood, D. O. Morgan and K. M. Shokat, Nature, 2000, 407, 395–401 CrossRef CAS PubMed.
- E. L. Weiss, A. C. Bishop, K. M. Shokat and D. G. Drubin, Nat. Cell Biol., 2000, 2, 677–685 CrossRef CAS PubMed.
- T. Okuzumi, D. Fiedler, C. Zhang, D. C. Gray, B. Aizenstein, R. Hoffman and K. M. Shokat, Nat. Chem. Biol., 2009, 5, 484–493 CrossRef CAS PubMed.
- Y. Liu, L. Warfield, C. Zhang, J. Luo, J. Allen, W. H. Lang, J. Ranish, K. M. Shokat and S. Hahn, Mol. Cell. Biol., 2009, 29, 4852–4863 CrossRef CAS PubMed.
- B. B. Au-Yeung, S. E. Levin, C. Zhang, L. Y. Hsu, D. A. Cheng, N. Killeen, K. M. Shokat and A. Weiss, Nat. Immunol., 2010, 11, 1085–1092 CrossRef CAS PubMed.
- L. Cipak, C. Zhang, I. Kovacikova, C. Rumpf, E. Miadokova, K. M. Shokat and J. Gregan, Cell Cycle, 2011, 10, 3527–3532 CrossRef CAS PubMed.
- X. Chen, R. Wang, X. Liu, Y. Wu, T. Zhou, Y. Yang, A. Perez, Y. C. Chen, L. Hu, J. P. Chadarevian, A. Assadieskandar, C. Zhang and Q. L. Ying, Dev. Cell, 2017, 43, 563–576.e4 CrossRef CAS PubMed.
- M. Hannaford, N. Loyer, F. Tonelli, M. Zoltner and J. Januschke, Dev., 2019, 146, 1–11 Search PubMed.
- C. Zhang, D. M. Kenski, J. L. Paulson, A. Bonshtien, G. Sessa, J. V. Cross, D. J. Templeton and K. M. Shokat, Nat. Methods, 2005, 2, 435–441 CrossRef CAS PubMed.
- M. Azam, M. A. Seeliger, N. S. Gray, J. Kuriyan and G. Q. Daley, Nat. Struct. Mol. Biol., 2008, 15, 1109–1118 CrossRef CAS PubMed.
- A. Wissner and T. S. Mansour, Arch. Pharm., 2008, 341, 465–477 CrossRef CAS PubMed.
- P. A. Schwartz, P. Kuzmic, J. Solowiej, S. Bergqvist, B. Bolanos, C. Almaden, A. Nagata, K. Ryan, J. Feng, D. Dalvie, J. C. Kath, M. Xu, R. Wani and B. W. Murray, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 173–178 CrossRef CAS PubMed.
- E. D. Deeks, Drugs, 2017, 77, 1695–1704 CrossRef CAS PubMed.
- Z. Zhao and P. E. Bourne, Drug Discovery Today, 2018, 23, 727–735 CrossRef CAS PubMed.
- V. Nelson, J. Ziehr, M. Agulnik and M. Johnson, OncoTargets Ther., 2013, 6, 135–143 CAS.
- Z. Pan, H. Scheerens, S.-J. Li, B. E. Schultz, P. A. Sprengeler, L. C. Burrill, R. V. Mendonca, M. D. Sweeney, K. C. K. Scott, P. G. Grothaus, D. A. Jeffery, J. M. Spoerke, L. A. Honigberg, P. R. Young, S. A. Dalrymple and J. T. Palmer, ChemMedChem, 2007, 2, 58–61 CrossRef CAS PubMed.
- R. A. Ward, M. J. Anderton, S. Ashton, P. A. Bethel, M. Box, S. Butterworth, N. Colclough, C. G. Chorley, C. Chuaqui, D. A. E. Cross, L. A. Dakin, J. É. Debreczeni, C. Eberlein, M. R. V. Finlay, G. B. Hill, M. Grist, T. C. M. Klinowska, C. Lane, S. Martin, J. P. Orme, P. Smith, F. Wang and M. J. Waring, J. Med. Chem., 2013, 56, 7025–7048 CrossRef CAS PubMed.
- C. Saura, M. Oliveira, Y.-H. Feng, M.-S. Dai, S.-W. Chen, S. A. Hurvitz, S.-B. Kim, B. Moy, S. Delaloge, W. Gradishar, N. Masuda, M. Palacova, M. E. Trudeau, J. Mattson, Y. S. Yap, M.-F. Hou, M. De Laurentiis, Y.-M. Yeh, H.-T. Chang, T. Yau, H. Wildiers, B. Haley, D. Fagnani, Y.-S. Lu, J. Crown, J. Lin, M. Takahashi, T. Takano, M. Yamaguchi, T. Fujii, B. Yao, J. Bebchuk, K. Keyvanjah, R. Bryce and A. Brufsky, J. Clin. Oncol., 2020, 38, 3138–3149 CrossRef CAS PubMed.
- J. A. Blair, D. Rauh, C. Kung, C. H. Yun, Q. W. Fan, H. Rode, C. Zhang, M. J. Eck, W. A. Weiss and K. M. Shokat, Nat. Chem. Biol., 2007, 3, 229–238 CrossRef CAS PubMed.
- A. Koch, H. B. Rode, A. Richters, D. Rauh and S. Hauf, ACS Chem. Biol., 2012, 7, 723–731 CrossRef CAS PubMed.
- P. I. Poulikakos, C. Zhang, G. Bollag, K. M. Shokat and N. Rosen, Nature, 2010, 464, 427–430 CrossRef CAS PubMed.
- M. S. Cohen, C. Zhang, K. M. Shokat and J. Taunton, Science, 2005, 308, 1318–1321 CrossRef CAS PubMed.
- J. L. Snead, M. Sullivan, D. M. Lowery, M. S. Cohen, C. Zhang, D. H. Randle, J. Taunton, M. B. Yaffe, D. O. Morgan and K. M. Shokat, Chem. Biol., 2007, 14, 1261–1272 CrossRef CAS PubMed.
- E. Ahler, A. C. Register, S. Chakraborty, L. Fang, E. M. Dieter, K. A. Sitko, R. S. R. Vidadala, B. M. Trevillian, M. Golkowski, H. Gelman, J. J. Stephany, A. F. Rubin, E. A. Merritt, D. M. Fowler and D. J. Maly, Mol. Cell, 2019, 74, 393–408.e20 CrossRef CAS PubMed.
- A. L. Garske, U. Peters, A. T. Cortesi, J. L. Perez and K. M. Shokat, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 15046–15052 CrossRef CAS PubMed.
- C. A. Ocasio, A. A. Warkentin, P. J. McIntyre, K. J. Barkovich, C. Vesely, J. Spencer, K. M. Shokat and R. Bayliss, ACS Chem. Biol., 2018, 13, 2956–2965 CrossRef CAS PubMed.
- A. Kung, Y. C. Chen, M. Schimpl, F. Ni, J. Zhu, M. Turner, H. Molina, R. Overman and C. Zhang, J. Am. Chem. Soc., 2016, 138, 10554–10560 CrossRef CAS PubMed.
- A. Kung, M. Schimpl, A. Ekanayake, Y. C. Chen, R. Overman and C. Zhang, ACS Chem. Biol., 2017, 12, 1499–1503 CrossRef CAS PubMed.
- M. Kostich, J. English, V. Madison, F. Gheyas, L. Wang, P. Qiu, J. Greene and T. M. Laz, Genome Biol., 2002, 3, 1–12 CrossRef PubMed.
- H. Zhang, X. Cao, M. Tang, G. Zhong, Y. Si, H. Li, F. Zhu, Q. Liao, L. Li, J. Zhao, J. Feng, S. Li, C. Wang, M. Kaulich, F. Wang, L. Chen, L. Li, Z. Xia, T. Liang, H. Lu, X.-H. Feng and B. Zhao, Elife, 2021, 10, 1–31 Search PubMed.
- F. Grimminger, R. T. Schermuly and H. A. Ghofrani, Nat. Rev. Drug Discovery, 2010, 9, 956–970 CrossRef CAS PubMed.
- R. Roskoski, Pharmacol. Res., 2021, 165, 105463 CrossRef CAS PubMed.
- P. K. Vogt, Nat. Rev. Cancer, 2012, 12, 639–648 CrossRef CAS PubMed.
- O. Fedorov, S. Müller and S. Knapp, Nat. Chem. Biol., 2010, 6, 166–169 CrossRef CAS PubMed.
- J. Stefaniak and K. V. M. Huber, ACS Med. Chem. Lett., 2020, 11, 403–406 CrossRef CAS PubMed.
- G. M. Simon, M. J. Niphakis and B. F. Cravatt, Nat. Chem. Biol., 2013, 9, 200–205 CrossRef CAS PubMed.
- M. Schürmann, P. Janning, S. Ziegler and H. Waldmann, Cell Chem. Biol., 2016, 23, 435–441 CrossRef PubMed.
- K. Islam, ACS Chem. Biol., 2015, 10, 343–363 CrossRef CAS PubMed.
- J. Remenyi, R. J. Naik, J. Wang, M. Razsolkov, A. Verano, Q. Cai, L. Tan, R. Toth, S. Raggett, C. Baillie, R. Traynor, C. J. Hastie, N. S. Gray and J. S. C. Arthur, Sci. Rep., 2021, 11, 10093 CrossRef CAS PubMed.
- H. Mukherjee and N. P. Grimster, Curr. Opin. Chem. Biol., 2018, 44, 30–38 CrossRef CAS PubMed.
- Q. Zhao, X. Ouyang, X. Wan, K. S. Gajiwala, J. C. Kath, L. H. Jones, A. L. Burlingame and J. Taunton, J. Am. Chem. Soc., 2017, 139, 680–685 CrossRef CAS PubMed.
- R. L. McCloud, K. Yuan, K. E. Mahoney, D. L. Bai, J. Shabanowitz, M. M. Ross, D. F. Hunt and K.-L. Hsu, Anal. Chem., 2021, 93, 11946–11955 CrossRef CAS PubMed.
- G. Prelich, Genetics, 2012, 190, 841–854 CrossRef CAS PubMed.
- T. van der Wel, R. Hilhorst, H. den Dulk, T. van den Hooven, N. M. Prins, J. A. P. M. Wijnakker, B. I. Florea, E. B. Lenselink, G. J. P. van Westen, R. Ruijtenbeek, H. S. Overkleeft, A. Kaptein, T. Barf and M. van der Stelt, Nat. Commun., 2020, 11, 1–20 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2022 |