
The identification of highly efficacious functionalised tetrahydrocyclopenta[c]pyrroles as inhibitors of HBV viral replication through modulation of HBV capsid assembly†
Andrew G.
Cole
 *a,
Steven G.
Kultgen
a,
Nagraj
Mani
a,
Andrzej
Ardzinski
a,
Kristi Yi
Fan
a,
Emily P.
Thi
a,
Bruce D.
Dorsey
a,
Kim
Stever
a,
Tim
Chiu
a,
Sunny
Tang
a,
Owen
Daly
a,
Janet R.
Phelps
a,
Troy
Harasym
a,
Andrea
Olland
b,
Robert K.
Suto
b and
Michael J.
Sofia
a
*a,
Steven G.
Kultgen
a,
Nagraj
Mani
a,
Andrzej
Ardzinski
a,
Kristi Yi
Fan
a,
Emily P.
Thi
a,
Bruce D.
Dorsey
a,
Kim
Stever
a,
Tim
Chiu
a,
Sunny
Tang
a,
Owen
Daly
a,
Janet R.
Phelps
a,
Troy
Harasym
a,
Andrea
Olland
b,
Robert K.
Suto
b and
Michael J.
Sofia
a
aArbutus Biopharma, Inc., 701 Veterans Circle, Warminster, PA 18974, USA. E-mail: acole@arbutusbio.com
bXtal BioStructures Inc., 12 Michigan Drive, Natick, MA 01760, USA
First published on 19th January 2022
Abstract
Disruption of the HBV viral life cycle with small molecules that prevent the encapsidation of pregenomic RNA and viral polymerase through binding to HBV core protein is a clinically validated approach to inhibiting HBV viral replication. Herein we report the further optimisation of clinical candidate AB-506 through core modification with a focus on increasing oral exposure and oral half-life. Maintenance of high levels of anti-HBV cellular potency in conjunction with improvements in pharmacokinetic properties led to multi-log10 reductions in serum HBV DNA following low, once-daily oral dosing for key analogues in a preclinical animal model of HBV replication.
Introduction
Hepatitis B virus (HBV) represents a significant unmet medical need despite the availability of an effective vaccine. The World Health Organization estimates that there are in excess of 290 million people living with chronic hepatitis B (CHB), resulting in almost one million deaths per year primarily from cirrhosis and hepatocellular carcinoma.1 Currently approved small-molecule drugs for the treatment of CHB are nucleos(t)ide-based viral polymerase inhibitors that suppress the virus through inhibition of replication,2 however, they do not suppress viral replication completely and do not provide a cure for CHB patients.3 They can also be ineffective against nucleos(t)ide resistant variants.4 As a result, there is a great deal of interest in identifying inhibitors of viral replication that operate via a different mechanism to the polymerase inhibitors. Furthermore, the potential of combining such agents with other antivirals that target different processes involved in the viral life cycle provides an opportunity for a curative regimen.5 HBV core protein (Cp) is essential to the HBV life cycle. Cp encapsidates the viral polymerase and pregenomic RNA (pgRNA) in a spheroid of protein comprising 240 units of Cp in dimeric pairs, enabling the formation of relaxed circular DNA (rcDNA) and ultimately, either producing new virions or translocating into the nucleus of a hepatocyte to replenish the pool of covalently closed circular DNA (cccDNA). Small molecules that interfere with this essential encapsidation process can inhibit rcDNA synthesis, and represent a viable opportunity for the development of therapeutics.6HBV Cp is a 183–185 (depending on genotype) amino acid protein where the first 149 amino acids (N-terminal domain) encompass the region associated with nucleocapsid assembly, and amino acids 150 to 183–5 (C-terminal domain) is an arginine rich portion of the protein associated with nucleic acid binding. A number of small-molecule capsid assembly modulators that elicit their effect through binding to the N-terminal domain of Cp dimers have been reported6,7 and can be categorised into two distinct classes. Class I inhibitors, which are commonly represented by a dihydropyrimidine core structure incorporating a stereochemically defined phenyl substituent at the 4-position, have the consequence of essentially polymerising Cp into aberrant tube-like structures which cannot encapsidate the viral polymerase and pgRNA.8 The second class of compounds (class II) has been exemplified with inhibitors displaying a far broader range of chemical diversity and have been shown to accelerate capsid assembly into morphologically regular capsids which are devoid of the encapsidated pgRNA, thus inhibiting viral replication and the production of rcDNA.9 Interestingly, X-ray crystallographic studies with capsid assembly inhibitors in conjunction with a single point mutation (Y132A) to the assembly domain of Cp have indicated that although these two classes of compounds have a different consequence on the way in which the dimeric units of Cp assemble, they bind within the same pocket at the Cp dimer:dimer interface.10,11
Since Cp plays a critical role during the viral life cycle, there has been considerable interest in identifying agents that inhibit HBV viral replication through interaction with Cp,12–14 and in recent years, a number of HBV capsid assembly modulators have entered clinical development (Fig. 1), resulting in the demonstration of significant reductions in serum HBV DNA in HBV infected patients. Small molecules GLS4 (ref. 15 and 16) and HAP_R10,17,18 which are based on the aforementioned dihydropyrimidine core, represent class I capsid assembly modulators and inhibit viral replication through the prevention of formation of nucleocapsids. NVR 3-778,19,20 ABI-H0731,21,22 JNJ-56136379 (ref. 23 and 24) and AB-506 (ref. 25) are representative class II capsid assembly modulators and disrupt the viral life cycle through accelerating the formation of nucleocapsids which cannot produce rcDNA and subsequently cannot produce infectious virions capable of infecting other hepatocytes. NVR 3-778, representing the well-studied sulfamoyl benzamide chemotype,9,19,26 was the first capsid assembly modulator to enter clinical studies and provided proof of concept for this class of antiviral agent.
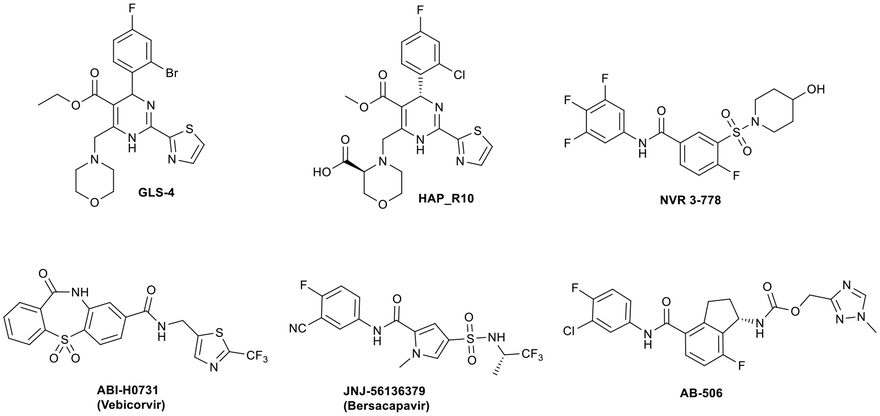 | ||
| Fig. 1 Examples of class I (GLS4 and HAP_R10) and class II (NVR 3-778, ABI-H0731, JNJ-56136379 and AB-506) capsid assembly modulators with clinical data. | ||
We have previously reported the pre-clinical antiviral profile of the potent class II capsid inhibitor AB-506 (ref. 25 and 27) (Fig. 1). In vitro antiviral activity of AB-506 was determined by measuring rcDNA inhibition in a HepDE19 cell line28,29 and demonstrated an EC50 of 77 nM.27 This compared favourably with the EC50 values determined in the same HepDE19-based cellular assay for the other clinical candidates detailed in Fig. 1 (GLS-4 EC50 = 110 nM, HAP_R10 EC50 = 50 nM, NVR 3-778 EC50 = 650 nM, ABI-H0731 EC50 = 250 nM and JNJ 56136379 EC50 = 45 nM). In addition, AB-506 was effective in dose dependently reducing HBV DNA in an in vivo hydrodynamic injection (HDI)‡ mouse model of HBV replication.28 In this model, HBV DNA was reduced by 0.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log10, 2.1log10 and 2.7log10vs. control when AB-506 was dosed orally at 10, 30 and 100 mg kg−1 BID, respectively, for 7 days. A 1.0log10 reduction in HBV DNA vs. control could also be achieved with QD oral dosing for the same period of time but required a high dose of 100 mg kg−1.27 To further optimise the in vivo efficacy of this chemical series, we embarked on additional structural modifications of the chemotype with the aim of further improving anti-HBV potency and the pharmacokinetic profile to elicit a robust reduction in HBV DNA following a low, once daily dosing schedule.
log10, 2.1log10 and 2.7log10vs. control when AB-506 was dosed orally at 10, 30 and 100 mg kg−1 BID, respectively, for 7 days. A 1.0log10 reduction in HBV DNA vs. control could also be achieved with QD oral dosing for the same period of time but required a high dose of 100 mg kg−1.27 To further optimise the in vivo efficacy of this chemical series, we embarked on additional structural modifications of the chemotype with the aim of further improving anti-HBV potency and the pharmacokinetic profile to elicit a robust reduction in HBV DNA following a low, once daily dosing schedule.
Evident from published X-ray crystallographic data of capsid inhibitors binding at the core protein dimer:dimer interface, key amino acid residues within the targeted binding site are W102 and T128, providing a hydrogen-bond donor and a hydrogen-bond acceptor, respectively. These residues have been shown to interact with a secondary amido-carbonyl hydrogen-bond acceptor and the associated NH donor within bound molecules. Additionally, a hydrophobic pocket exists that accommodates the 4-position phenyl group of HAP-based compounds similar to GLS-4,10 and also the anilide phenyl group of NVR 3-778.11 The carboxamide functionality at the 4-position of the aminoindane core of AB-506 satisfies this simplistic pharmacophore model and represents a functional group that would be maintained in further analogue synthesis. Opportunity does, however, exist to modify the central portion of the molecule to provide subtle differences in the three-dimensional presentation of the aminoindane carbamate functionality relative to the carboxamide. Structural comparison of NVR 3-778 and JNJ-56136379, which both contain a carboxamide and a sulfonamide separated by an aromatic core, would suggest that 1-methyl pyrrole is a tolerated replacement for phenyl. To evaluate this hypothesis, a synthesis was devised to generate the 1-methyl pyrrole-based analogue of AB-506 (Scheme 1).
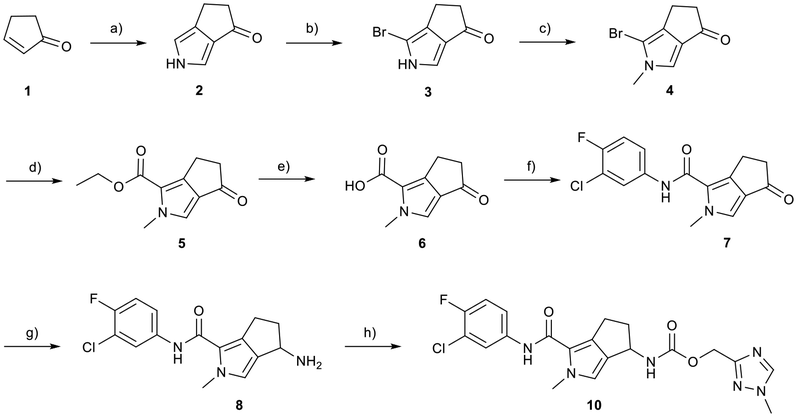 | ||
| Scheme 1 Synthesis of 1-methyl pyrrole analogue of AB-506. Reagents and conditions: a) NaH, TosMIC, Et2O, DMSO, 0 °C to RT, 16 h, 34%; b) NBS, THF, −78 °C, 2 h, 45%; c) Cs2CO3, MeI, THF, 0 °C to RT, 16 h, 75%; d) Pd(OAc)2, dppp, CO (200 psi), Et3N, EtOH, 100 °C, 16 h, 72%; e) LiOH monohydrate, THF, EtOH, H2O, RT, 16 h, 95%; f) 3-chloro-4-fluoro aniline, HATU, iPr2NEt, DMF, 60 °C, 16 h, 72%; g) NH4OAc, NaCNBH3, EtOH, 90 °C (μwave), 2 h, 79%; h) (1-methyl-1H-1,2,4-triazol-3-yl)methyl 1H-imidazole-1-carboxylate (9), iPr2NEt, THF, 65 °C, 16 h, 68%. | ||
Synthesis
Conversion of cyclopentenone (1) to the bicyclic pyrrole 2 was achieved by reaction with tosylmethyl isocyanide (TosMIC).30 Reaction of 2 with N-bromosuccinimide afforded regioselective bromination of the pyrrole31 to generate 3 which was then N-methylated with methyl iodide. Palladium acetate/1,3-bis(diphenylphosphino) propane (dppp) catalysed carbonylation of 4 under 200 psi of carbon monoxide in ethanol provided ethyl ester 5, and ester saponification with lithium hydroxide allowed for the desired carboxamide functionality to be incorporated through HATU mediated coupling to provide 7. Amino incorporation providing 8 was conducted in a racemic fashion through reductive amination with ammonium acetate/sodium cyanoborohydride under microwave conditions. Generation of 10 was achieved through functionalisation of the primary amine with pre-formed (1-methyl-1H-1,2,4-triazol-3-yl)methyl 1H-imidazole-1-carboxylate (9), which was synthesised through the reaction of (1-methyl-1H-1,2,4-triazol-3-yl)methanol with 1,1′-carbonyldiimidazole in acetonitrile. Racemic 10 was subsequently separated into the individual enantiomers (10a and 10b) by chiral SFC chromatography.Results and discussion
Following chiral SFC resolution of the individual enantiomers of 10 (10a and 10b), the compounds were evaluated in the HepDE19 cellular assay to determine levels of inhibition of HBV DNA. The first eluting enantiomer based on the chromatographic conditions employed (arbitrarily assigned as 10a) demonstrated an EC50 value of 3.5 μM whereas the second eluting enantiomer (arbitrarily assigned as 10b) demonstrated an EC50 value of 52 nM. These data are consistent with the aminoindane analogues where the (S)-enantiomer of AB-506 was markedly more potent than its (R)-enantiomer.25 As with AB-506, neither 10a nor 10b showed any significant cytotoxicity in the HepDE19 cell line at concentrations up to 25 μM. The observed anti-HBV activity of this initial example supports the hypothesis that the N-methyl pyrrole is a suitable replacement for the phenyl group within the aminoindane and provides a comparative level of potency. Further evaluation was conducted with the more potent 10b to determine the consequence of the core replacement on its in vitro microsomal stability and its pharmacokinetic profile. Comparative in vitro metabolic stability for 10bvs. AB-506 was assessed through incubation employing liver microsomes from human (HLM) and mouse (MLM, CD-1). Stability was evaluated at 0.3 μM concentration by monitoring the compound levels at various time points up to 1 h to determine the T1/2 and CLint (Table 1).| Compound | Species | CLInt (μL min−1 mg−1) | T 1/2 (min) | % remaining@1 h |
|---|---|---|---|---|
| AB-506 | HLM | 6.2 | 193 | 83 |
| MLM | 20.6 | 74 | 56 | |
| 10b | HLM | 5.4 | 256 | 87 |
| MLM | 12.2 | 114 | 81 |
Both compounds demonstrate high levels of stability in the HLM incubations and a slightly improved profile for 10b in the MLM incubations. The initial pharmacokinetic assessment was conducted in CD-1 mice incorporating a 10 mg kg−1 oral arm and a 2 mg kg−1 i.v. arm using a dosing vehicle of 40%PEG400/5%EtOH/55%H2O and compared with historical data for AB-506 (Table 2).
| Compound | p.o. AUC0−∞ (ng mL−1 h) | p.o. Cmax (ng mL−1) | T 1/2 (h) | CLobs (mL min−1 kg−1) |
|---|---|---|---|---|
| AB-506 | 9870 | 1610 | 2.5 | 18.6 |
| 10b | 46730 |
4270 | 2.6 | 4.1 |
In comparison to AB-506, 10b demonstrates an improved AUC and Cmax with lower clearance. While the slightly improved mouse microsomal stability is potentially a contributing factor to the improved PK profile in mice, improved absorption is also likely. Despite the reduced clearance, the half-life remains unchanged when compared to AB-506.
With the primary goal of improving the pharmacokinetic profile of the AB-506 series of compounds realised through incorporation of the fused pyrrole core, compound 10b was evaluated in the in vivo HDI mouse model of HBV replication.§32,33 Prior to the initiation of dosing of 10b, 10 μg of a plasmid encoding a 1.3-fold-overlength copy of an HBV genotype D sequence was administered to six week old female NOD.CB17-Prkdcscid/J mice via hydrodynamic injection.‡ Seven days after HDI administration, either test article or vehicle control was administered via oral gavage at the selected doses either once daily or twice daily for 7 consecutive days starting on day 0 (6 animals per study group). Blood was collected on day 0 (predose) and on day 7 for serum HBV DNA quantitation. Total extracted nucleic acid was measured using quantitative polymerase chain reaction (qPCR) as described previously.34 Based on the improved mouse PK relative to AB-506, and taking into account the fact that the half-life remains unchanged, 10b was dosed in the HDI HBV mouse model at 3, 10 and 30 mg kg−1 BID with an additional arm included involving QD dosing at 30 mg kg−1.
When dosed BID for 7 days at doses of 3, 10 and 30 mg kg−1, 10b demonstrated a dose dependent reduction in HBV DNA of 0.8log10, 1.9log10 and 2.5log10, respectively, vs. vehicle control (Fig. 2). This represented a significant improvement compared to AB-506, which provided 0.6log10 and 2.1log10 reductions at the overlapping doses of 10 and 30 mg kg−1, respectively. Furthermore, the level of serum HBV DNA reduction observed for 10b when dosed once daily at 30 mg kg−1 (1.8log10) far exceeded the 1.0log10 reduction in HBV DNA for AB-506 when administered QD at more than triple the dose (100 mg kg−1). Since the cellular antiviral potencies of the two compounds are very similar, the increased efficacy of 10b can be directly attributed to the enhanced pharmacokinetic profile.
 | ||
| Fig. 2 Quantitated log10 reduction in serum HBV DNA vs. vehicle control of 10b in a mouse hydrodynamic injection model of HBV. ****p < 0.0001 compared to vehicle group by one-way ANOVA with Dunnett's multiple comparisons test. | ||
To confirm direct interaction with the target and to determine stereochemical configuration, X-ray crystallographic analysis of the assembly domain of a Cp Y132A mutant soaked with 10b was conducted. This Y132A variant is an assembly deficient protein that results in the formation of hexameric units (A–F) and has been used to assess key interactions of capsid inhibitors with Cp at the dimer:dimer interface.10,11 Protein isolation and crystallisation were conducted according to published methods.10 Soaking was conducted in a solution containing 10% PEG 3350, 14% (+/−)-2-methyl-2,4-pentanediol, 9% isopropanol, and 100 mM ammonium citrate/citric acid at pH 6.5 at a 1 mM concentration of 10b for 60 hours at 16 °C. Following diffraction, the structure in complex with 10b was determined at 2.50 Å resolution (PDB ID 7S76) showing binding at the B–C interface of the hexameric unit (Fig. 3). In the structure of 10b bound to the Y132A protein, and consistent with other published capsid inhibitors, the halogenated phenyl group fits into a deep hydrophobic pocket formed by P25, D29, L30, T33 and I105 from chain B, as well as V124 and R127 from chain C. The key hydrogen bonding interactions of the secondary amide carbonyl of 10b with the hydrogen bond donor provided by W102, and the secondary amide NH of 10b with the hydrogen bond acceptor provided by the oxygen of T128 (vide supra) are maintained. In addition, the presentation of (S)-absolute stereochemistry of the amino substituent in 10b, as present in AB-506, orients the carbamate group such that it provides an additional electrostatic interaction with the backbone carbonyl of L140 through a hydrogen bond with the NH of the (methyltriazolyl)methyl carbamate. The methyltriazole ring is located at the extreme of the binding site and is relatively solvent exposed.
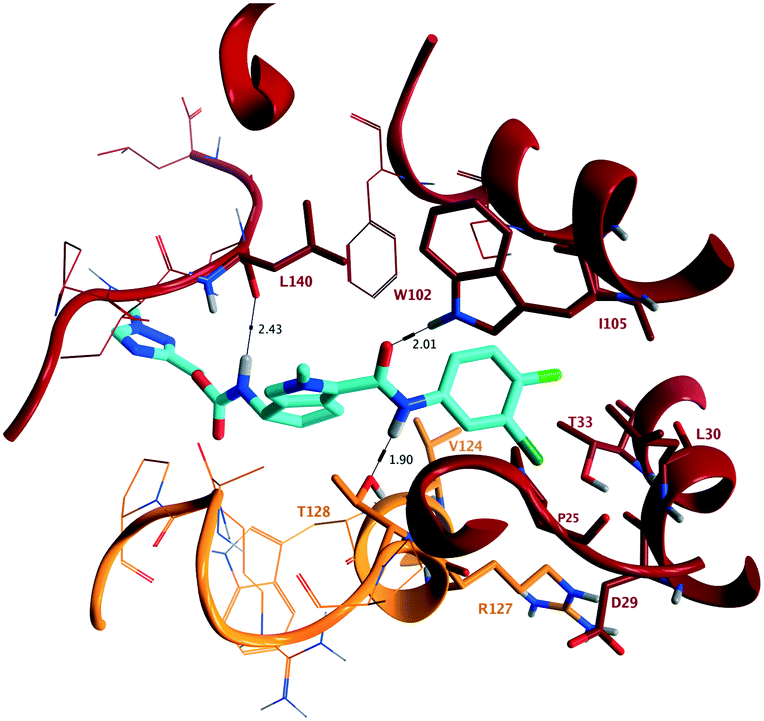 | ||
| Fig. 3 (PDB ID 7S76) Representation of the X-ray crystallographic analysis of 10b bound at the B–C interface of the hexameric Y132A mutant of the assembly domain of Cp. Key hydrogen bonding interactions are identified with W102 (2.01 Å) and T128 (1.90 Å), interacting with the acceptor and donor provided by the secondary amide, and the (S)-configuration of 10b provides an additional hydrogen bond with the backbone carbonyl of L140 (2.43 Å). | ||
To assess the steric and electronic effect of further substitution of the pyrrole core, analogues involving chlorination, bromination and methylation at the 2-position were synthesised. Chlorine substitution was achieved through NCS halogenation of 5. Target molecule synthesis was subsequently completed in a similar manner as outlined in Scheme 1 to generate 11 (Table 3). Similar 2-position bromination of the pyrrole ring system could be achieved with NBS to ultimately generate 12, and 2-position methylation (13) was achieved through palladium acetate/RuPhos coupling of 12 with methyl boronic acid under microwave conditions. The increased antiviral potency of the 2-chloro- (11, EC50 = 13 nM) and 2-bromo- (12, EC50 = 12 nM) analogues indicate that substitution can be tolerated in this position from a steric perspective, even with the highly sterically demanding bromine and that electron withdrawing substituents are potentially beneficial. Methylation, however, resulted in a reduction in cellular potency, supporting the electronic rationale.
|
|
|||||
|---|---|---|---|---|---|
| Compound | R | HepDE19 EC50 (nM) | HLM CLInt (μL min−1 mg−1) | MLM CLInt (μL min−1 mg−1) | Aq. solubility (μM) |
| 10b | H | 52 ± 23 | 5.4 | 12.2 | 24 |
| 11 | Cl | 13 ± 4 | 8.8 | 33.8 | 5 |
| 12 | Br | 12 ± 2 | 8.5 | 31.4 | <4 |
| 13 | Me | 114 ± 40 | 12.7 | 52.8 | 5 |
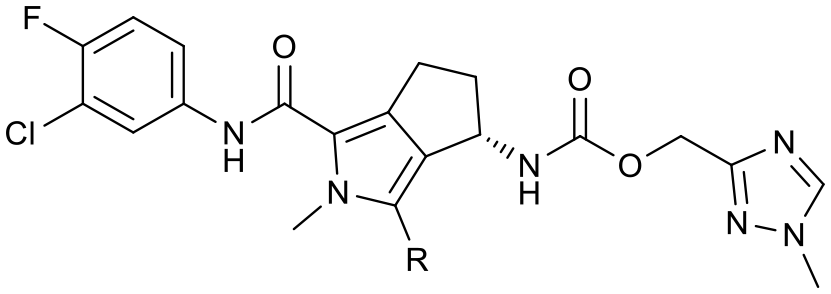
The 2-position analogues were also assessed in the in vitro microsomal stability and kinetic aqueous solubility assays. While the measured human intrinsic clearance showed modest decreases in stability, intrinsic clearance in an MLM environment was significantly increased. Substitution at the 2-position also had the effect of significantly reducing aqueous solubility (Table 3). As is evident from the Cp-Y132A bound crystal structure of 10b, the methyltriazole is positioned towards the edge of the binding site and is somewhat solvent exposed. While modification of this region of the molecule may not provide an avenue to improved potency, it does potentially provide an opportunity for additional optimisation of the chemical series through modulation of physicochemical properties to further improve the pharmacokinetic profile.35 To investigate this hypothesis, a number of compounds based on minor modification to the isomeric orientation of the triazole or on the reduction of lipophilicity through removal of the sp3 carbon were designed. Synthesis was achieved in an analogous manner as described previously. Instances incorporating a 1,2,4-NH-triazole were generated using the corresponding N-trityl protected (triazolyl)methanol and 1,1′-carbonyldiimidazole with an acid mediated trityl deprotection (HCl in p-dioxane) being employed following carbamate formation. Instances incorporating a 1,2,3-NH-triazole were generated from the propargylic carbamate through reaction with trimethylsilyl azide.
As anticipated, the minor modifications made to the nature of the triazole have little effect on the anti-HBV potency of the analogues, with activity being consistent with the nature of the pyrrole substitution (Table 4). Comparison of the N-methyl 1,2,4 triazole (10b) with the corresponding N-methyl 1,2,3 triazole (14), however, does show a reduction in both human and mouse in vitro microsomal stability and a loss of aqueous solubility. Demethylation of 10b to provide 15 maintains comparable potency and microsomal stability to the parent but significantly, reduces the overall lipophilicity of the compound through removal of a saturated carbon and the introduction of the NH hydrogen bond donor. The NH 1,2,3 triazole 17 also provides an increase in the hydrophilicity of the analogue although the in vitro properties are slightly less desirable. Consistent with previous results, 2-chloro-substituted compounds 16 and 18 provide a 4 to 5-fold potency increase in comparison to the corresponding des-chloro analogues 15 and 17, respectively. The increase in clogP for compounds 16 and 18 due to chlorine incorporation is somewhat offset by the absence of the methyl group in comparison to compound 11, however, the aqueous solubility remains low for the chlorinated analogues.
|
|
||||||
|---|---|---|---|---|---|---|
| Cmpd | R | R′ | Hep DE19 EC50 (nM) | HLM CLInt (μL min−1 mg−1) | MLM CLInt (μL min−1 mg−1) | Aq. solubility (μM) |
| 10b | H |
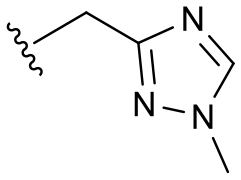
|
52 ± 23 | 5.4 | 12.2 | 24 |
| 14 | H |

|
52 ± 11 | 24.4 | 38.4 | <4 |
| 15 | H |

|
50 ± 19 | 4.1 | 12.3 | 23 |
| 16 | Cl |

|
11 ± 1 | 8.6 | 29.7 | <4 |
| 17 | H |

|
57 ± 15 | 7.9 | 21.6 | 10 |
| 18 | Cl |
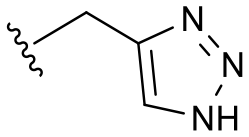
|
14 ± 5 | 11.2 | 20.2 | <4 |
Based on this in vitro data, compounds 15, 17 and 18 were selected to advance into in vivo mouse pharmacokinetic studies. Similar to the evaluation of 10b, pharmacokinetic assessment was conducted in CD-1 mice incorporating a 10 mg kg−1 oral arm and a 2 mg kg−1 i.v. arm using the same dosing vehicle (Table 5).
| Compound | p.o. AUC0−∞ (ng mL−1 h) | p.o. Cmax (ng mL−1) | T 1/2 (h) | CLobs (mL min−1 kg−1) |
|---|---|---|---|---|
| 10b | 46730 |
4270 | 2.6 | 4.1 |
| 15 | 40740 |
3860 | 4.8 | 6.1 |
| 17 | 60620 |
4530 | 5.8 | 2.6 |
| 18 | 30560 |
2510 | 3.5 | 5.5 |
All of the compounds included within this study demonstrated a favourable pharmacokinetic profile with high AUC and Cmax and low clearance. Limitations in solubility is likely a contributor to the slightly reduced AUC and Cmax for compound 18. A consistent theme to the data, however, is that the half-life of the more hydrophilic NH-triazoles is significantly increased in comparison to the N-methyl triazole 10b.
Based on these data compound 17, demonstrating the most pronounced improvement in in vivo half-life, was selected for dose dependent analysis in the HBV HDI in vivo mouse model. As a consequence of the extended half-life, the initial mouse PK had indicated that the 24 h plasma concentration following a single oral 10 mg kg−1 dose was at a high multiple over the cellular EC50 so administration of 17 was conducted using the preferred dosing schedule of once daily at 3, 10 and 30 mg kg−1. For comparison, an additional dosing arm of compound 15 at 10 mg kg−1 QD was also included. The log10 reduction in HBV DNA vs. vehicle control are represented in Fig. 4.
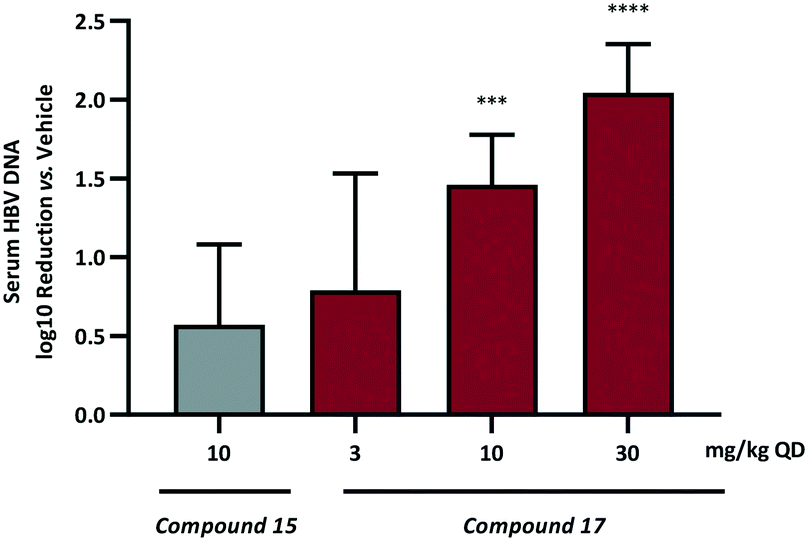 | ||
| Fig. 4 Quantitated log10 reduction in serum HBV DNA vs. vehicle control based on QD evaluation of 15 and 17 in a HDI mouse of HBV. ***p < 0.001 or ****p < 0.0001 compared to vehicle group by one-way ANOVA with Dunnett's multiple comparisons test. | ||
Once daily dosing of compound 15 resulted in a 0.6log10 DNA reduction at 10 mg kg−1. More prominent, however, are the results from the QD dose escalation of 17. This analogue demonstrated the highest AUC and Cmax based on single oral dosing in addition to the lowest observed clearance and the longest half-life. Repeat dosing for 7 days in the HDI model provided a 0.8log10 drop at the low dose (3 mg kg−1) and further inhibition of HBV replication at 10 mg kg−1 (1.5log10 reduction) and 30 mg kg−1 (2.0log10 reduction). This represents a significant progression in the chemical series when it is considered that AB-506 required a significantly higher 100 mg kg−1 QD dose to achieve a 1.0log10 reduction.
Conclusions
The results presented herein provide a detailed evaluation of the continued optimisation of an already promising clinical candidate (AB-506). The combination of key structural features associated with distinct chemical series that elicit the same pharmacological effect through modulation of HBV capsid assembly and the demonstrated value of structural biology in determining regions of a molecule that can be modified to optimise not only potency but to also modulate physicochemical properties have been described. The core modifications incorporated into the analogues have allowed for the significant improvement in pharmacokinetic profiles which were further enhanced through the modification of a solvent exposed region of the lead molecules. The in vivo characterisation of 10b, 15 and 17 in a relevant preclinical animal model has demonstrated that a combination of potent cellular activity with high and sustained oral exposure can result in inhibition of HBV viral replication following a low once daily dosing schedule.Conflicts of interest
AGC, SGK, NM, AA, KYF, EPT, KS, TH and MJS are current employees of Arbutus Biopharma, Inc. and may own company stock.Acknowledgements
The authors would like to thank Angela M. Lam and Eugen F. Mesaros for valuable discussion and Boya Liu for the acquisition of high-resolution mass spectroscopy data.Notes and references
- WHO Hepatitis B Fact Sheet, July 2021, www.who.int Search PubMed.
- F. Zoulim, F. Lebosse and M. Levrero, Curr. Opin. Virol., 2016, 18, 109–116 CrossRef CAS PubMed.
- S. Naggie and A. S. Lok, Annu. Rev. Med., 2021, 72, 93–105 CrossRef CAS PubMed.
- L. Menendez-Arias, M. Alvarez and B. Pacheco, Curr. Opin. Virol., 2014, 8, 1–9 CrossRef CAS PubMed.
- T. M. Block, H. Alter, N. Brown, A. Brownstein, C. Brosgart, K. M. Chang, P. J. Chen, C. Cohen, H. El-Serag, J. Feld, R. Gish, J. Glenn, T. F. Greten, J. T. Guo, Y. Hoshida, K. V. Kowdley, W. Li, A. S. Lok, B. McMahon, A. Mehta, R. Perrillo, C. M. Rice, J. Rinaudo, R. F. Schinazi and K. Shetty, Antiviral Res., 2018, 150, 93–100 CrossRef CAS PubMed.
- A. G. Cole, Curr. Opin. Pharmacol., 2016, 30, 131–137 CrossRef CAS PubMed.
- S. D. Kudak and A. M. Lam, Med. Chem. Rev., 2017, 52, 305–317 Search PubMed.
- S. J. Stray, C. R. Bourne, S. Punna, W. G. Lewis, M. G. Finn and A. Zlotnick, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 8138–8143 CrossRef CAS PubMed.
- M. R. Campagna, F. Liu, R. Mao, C. Mills, D. Cai, F. Guo, X. Zhao, H. Ye, A. Cuconati, H. Guo, J. Chang, X. Xu, T. M. Block and J. T. Guo, J. Virol., 2013, 87, 6931–6942 CrossRef CAS PubMed.
- K. Klumpp, A. M. Lam, C. Lukacs, R. Vogel, S. Ren, C. Espiritu, R. Baydo, K. Atkins, J. Abendroth, G. Liao, A. Efimov, G. Hartman and O. A. Flores, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 15196–15201 CrossRef CAS PubMed.
- Z. Zhou, T. Hu, X. Zhou, S. Wildum, F. Garcia-Alcalde, Z. Xu, D. Wu, Y. Mao, X. Tian, Y. Zhou, F. Shen, Z. Zhang, G. Tang, I. Najera, G. Yang, H. C. Shen, J. A. Young and N. Qin, Sci. Rep., 2017, 7, 42374 CrossRef CAS PubMed.
- B. Nijampatnam and D. C. Liotta, Curr. Opin. Chem. Biol., 2019, 50, 73–79 CrossRef CAS PubMed.
- S. Senaweera, H. Du, H. Zhang, K. A. Kirby, P. R. Tedbury, J. Xie, S. G. Sarafianos and Z. Wang, Viruses, 2021, 13, 770 CrossRef CAS PubMed.
- Y. Wang, Z. Wang, J. Liu, Y. Wang, R. Wu, R. Sheng and T. Hou, Bioorg. Med. Chem., 2021, 36, 116096 CrossRef CAS PubMed.
- G. Wu, B. Liu, Y. Zhang, J. Li, A. Arzumanyan, M. M. Clayton, R. F. Schinazi, Z. Wang, S. Goldmann, Q. Ren, F. Zhang and M. A. Feitelson, Antimicrob. Agents Chemother., 2013, 57, 5344–5354 CrossRef CAS PubMed.
- H. Zhang, F. Wang, X. Zhu, Y. Chen, H. Chen, X. Li, M. Wu, C. Li, J. Liu, Y. Zhang, Y. Ding and J. Niu, Clin. Infect. Dis., 2021, 73, 175–182 CrossRef CAS PubMed.
- X. Lin, H. Shi, W. Zhang, Z. Qiu, Z. Zhou, F. Dey, S. Zhong, H. Qiu, J. Xie, X. Zhou, G. Yang, G. Tang, H. C. Shen and W. Zhu, J. Med. Chem., 2019, 62, 10352–10361 CrossRef CAS PubMed.
- N. A. Kratochwil, M. Triyatni, M. B. Mueller, F. Klammers, B. Leonard, D. Turley, J. Schmaler, A. Ekiciler, B. Molitor, I. Walter, P. A. Gonsard, C. A. Tournillac, A. Durrwell, M. Marschmann, R. Jones, M. Ullah, F. Boess, G. Ottaviani, Y. Jin, N. J. Parrott and S. Fowler, J. Pharmacol. Exp. Ther., 2018, 365, 237–248 CrossRef CAS PubMed.
- A. M. Lam, C. Espiritu, R. Vogel, S. Ren, V. Lau, M. Kelly, S. D. Kuduk, G. D. Hartman, O. A. Flores and K. Klumpp, Antimicrob. Agents Chemother., 2019, 63, e01734 CAS.
- M. F. Yuen, E. J. Gane, D. J. Kim, F. Weilert, H. L. Yuen Chan, J. Lalezari, S. G. Hwang, T. Nguyen, O. Flores, G. Hartman, S. Liaw, O. Lenz, T. N. Kakuda, W. Talloen, C. Schwabe, K. Klumpp and N. Brown, Gastroenterology, 2019, 156, 1392–1403 CrossRef CAS PubMed , e1397.
- Q. Huang, D. Cai, R. Yan, L. Li, Y. Zong, L. Guo, A. Mercier, Y. Zhou, A. Tang, K. Henne and R. Colonno, Antimicrob. Agents Chemother., 2020, 64, e01463 CAS.
- M. F. Yuen, K. Agarwal, E. J. Gane, C. Schwabe, S. H. Ahn, D. J. Kim, Y. S. Lim, W. Cheng, W. Sievert, K. Visvanathan, E. Ruby, S. Liaw, R. Yan, Q. Huang, R. Colonno and U. Lopatin, Lancet Gastroenterol. Hepatol., 2020, 5, 152–166 CrossRef PubMed.
- J. Vandenbossche, W. Jessner, M. van den Boer, J. Biewenga, J. M. Berke, W. Talloen, L. De Zwart, J. Snoeys and J. Yogaratnam, Adv. Ther., 2019, 36, 2450–2462 CrossRef CAS PubMed.
- T. Verbinnen, M. Hodari, W. Talloen, J. M. Berke, D. Blue, J. Yogaratnam, J. Vandenbossche, U. Shukla, S. De Meyer and O. Lenz, J. Viral Hepatitis, 2020, 27, 1127–1137 CrossRef CAS PubMed.
- N. Mani, A. G. Cole, J. R. Phelps, A. Ardzinski, R. Burns, T. Chiu, A. Cuconati, B. D. Dorsey, E. Evangelista, K. Fan, F. Guo, T. O. Harasym, S. Kadhim, R. Kowalski, S. G. Kultgen, A. C. H. Lee, A. H. Li, S. A. Majeski, A. Miller, C. Pasetka, S. P. Reid, R. Rijnbrand, H. M. Micolochick Steuer, K. Stever, S. Tang, X. Teng, X. Wang and M. J. Sofia, Antiviral Res., 2021, 197, 105211 CrossRef PubMed.
- Y. H. Lee, H. M. Cha, J. Y. Hwang, S. Y. Park, A. G. Vishakantegowda, A. Imran, J. Y. Lee, Y. S. Yi, S. Jun, G. H. Kim, H. J. Kang, S. J. Chung, M. Kim, H. Kim and S. B. Han, ACS Med. Chem. Lett., 2021, 12, 242–248 CrossRef CAS PubMed.
- N. Mani, A. G. Cole, J. R. Phelps, C. Abbott, A. Ardzinski, J. Bechard, R. Burns, T. Chiu, A. Cuconati, B. D. Dorsey, E. Evangelista, L. Fu, H. Guo, T. O. Harasym, A. Jarosz, S. Kadhim, S. Kultgen, K. Kwak, A. C. Lee, A. H. Li, S. Majeski, K. McClintock, A. Miller, C. J. Pasetka, S. P. Reid, R. Rijnbrand, A. Shapiro, K. Stever, S. Tang, X. Teng, X. Wang, H. Zhang and M. J. Sofia, Hematology, 2017, 66(S1), 510A Search PubMed.
- N. Mani, A. G. Cole, J. R. Phelps, A. Ardzinski, K. D. Cobarrubias, A. Cuconati, B. D. Dorsey, E. Evangelista, K. Fan, F. Guo, H. Guo, J. T. Guo, T. O. Harasym, S. Kadhim, S. G. Kultgen, A. C. H. Lee, A. H. L. Li, Q. Long, S. A. Majeski, R. Mao, K. D. McClintock, S. P. Reid, R. Rijnbrand, N. M. Snead, H. M. Micolochick Steuer, K. Stever, S. Tang, X. Wang, Q. Zhao and M. J. Sofia, Antimicrob. Agents Chemother., 2018, 62, e00082 CAS.
- H. Guo, D. Jiang, T. Zhou, A. Cuconati, T. M. Block and J. T. Guo, J. Virol., 2007, 81, 12472–12484 CrossRef CAS PubMed.
- J. M. Kelly and F. J. Leeper, Tetrahedron Lett., 2012, 53, 819–821 CrossRef CAS.
- J. S. Y. Kyung, A. Yu, D. K. Eom, Y. I. Lee, M. R. Han, J. H. Lee, H. N. Seo, J. D. Kim and S. H. Lee, PCT Patent Application, WO2015030514, 2015.
- L. G. Guidotti, B. Matzke, H. Schaller and F. V. Chisari, J. Virol., 1995, 69, 6158–6169 CrossRef CAS PubMed.
- P. L. Yang, A. Althage, J. Chung and F. V. Chisari, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 13825–13830 CrossRef CAS PubMed.
- T. Tanaka, K. Inoue, Y. Hayashi, A. Abe, K. Tsukiyama-Kohara, H. Nuriya, Y. Aoki, R. Kawaguchi, K. Kubota, M. Yoshiba, M. Koike, S. Tanaka and M. Kohara, J. Med. Virol., 2004, 72, 223–229 CrossRef PubMed.
- W. J. Egan, K. M. Merz, Jr. and J. J. Baldwin, J. Med. Chem., 2000, 43, 3867–3877 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures and characterisation data, assay details and materials for EC50 and CC50, microsomal stability and solubility determinations. Protocols for in vivo pharmacokinetic and efficacy determinations. See DOI: 10.1039/d1md00318f |
| ‡ HDI (hydrodynamic injection) describes a rapid injection of a high volume of plasmid into the tail vein of subject animals within a time period of 5 seconds. |
| § All animal-related procedures were conducted according to written operating procedures, in accordance with Canadian Council on Animal Care (CCAC) Guidelines on Good Animal Practices or the In-Life Group Standard Operating Procedures of Inotiv (St. Louis, Missouri, USA). Protocols were approved by Arbutus' Institutional Animal Care and Use Committee (IACUC) or the St. Louis University Animal Care and Use Committee. |
| This journal is © The Royal Society of Chemistry 2022 |