
Covalent sortase A inhibitor ML346 prevents Staphylococcus aureus infection of Galleria mellonella†
Xiang-Na
Guan‡
 ab,
Tao
Zhang‡
a,
Teng
Yang
ac,
Ze
Dong
a,
Song
Yang
c,
Lefu
Lan
abd,
Jianhua
Gan
e and
Cai-Guang
Yang
*abd
ab,
Tao
Zhang‡
a,
Teng
Yang
ac,
Ze
Dong
a,
Song
Yang
c,
Lefu
Lan
abd,
Jianhua
Gan
e and
Cai-Guang
Yang
*abd
aCenter for Chemical Biology, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China. E-mail: yangcg@simm.ac.cn
bUniversity of the Chinese Academy of Sciences, Beijing 100049, China
cState Key Laboratory Breeding Base of Green Pesticide and Agricultural Bioengineering, Key Laboratory of Green Pesticide and Agricultural Bioengineering, Ministry of Education, Center for R&D of Fine Chemicals, Guizhou University, Guiyang 550025, China
dSchool of Pharmaceutical Science and Technology, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, Hangzhou 310024, China
eSchool of Life Sciences, Fudan University, Shanghai 200433, China
First published on 1st November 2021
Abstract
The housekeeping sortase A (SrtA), a membrane-associated cysteine transpeptidase, is responsible for anchoring surface proteins to the cell wall peptidoglycan in Gram-positive bacteria. This process is essential for the regulation of bacterial virulence and pathogenicity. Therefore, SrtA is considered to be an ideal target for antivirulence therapy. In this study, we report that ML346, a compound with a barbituric acid and cinnamaldehyde scaffold, functions as an irreversible inhibitor of Staphylococcus aureus SrtA (SaSrtA) and Streptococcus pyogenes SrtA (SpSrtA) in vitro at low micromolar concentrations. According to our X-ray crystal structure of the SpSrtAΔN81/ML346 complex (Protein Data Bank ID: 7V6K), ML346 covalently modifies the thiol group of Cys208 in the active site of SpSrtA. Importantly, ML346 significantly attenuated the virulence phenotypes of S. aureus and exhibited inhibitory effects on Galleria mellonella larva infection caused by S. aureus. Collectively, our results indicate that ML346 has potential for development as a covalent antivirulence agent for treating S. aureus infections, including methicillin-resistant S. aureus.
Introduction
Staphylococcus aureus is a major human pathogen that causes a variety of infectious diseases, including skin and soft tissue infections, and invasive infections such as bacteraemia, infective endocarditis, pneumonia, and toxic shock syndrome.1,2 The emergence of multi-drug resistant strains, including methicillin-resistant S. aureus (MRSA), represents a great challenge in the treatment of S. aureus infections.3 Given that the antibiotics currently available for clinical use are insufficient to treat such infections,4 it is imperative to develop alternative antibacterial strategies to address the crisis of antibiotic resistance. Antivirulence therapies interfere with bacterial virulence factors instead of central growth pathways to treat infection; thus, evolutionary pressure is reduced and the emergence of drug resistance is less likely.5–7 In addition, antivirulence drugs cause minimal perturbation of the healthy microbiota. Therefore, they represent an attractive alternative strategy to antibiotics. Indeed, antivirulence drugs against Clostridium botulinum, Bacillus anthracis, and Clostridium difficile have been approved by the US Food and Drug Administration for treatment of bacterial toxin-mediated diseases,5 and several antivirulence drugs for antibiotic-resistant S. aureus have entered phase-II clinical trials. However, none of these drugs are small molecules.5Sortase A (SrtA), a membrane-associated cysteine transpeptidase, is essential for bacterial pathogenesis but not for bacterial growth and survival.8–14 The housekeeping SrtA specifically recognizes the conserved LPXTG motif of surface proteins and anchors them to the cell wall peptidoglycan.9 Some surface proteins in the cell envelope are essential for adherence of pathogenic bacteria to host tissues and thus for establishing infections.15 The srtA mutant of S. aureus fails to anchor surface proteins into the cell envelope, which hinders its ability to cause lethal infections,11,16 whereas the mutation minimally affects the growth of S. aureus.11 In addition, SrtA is conveniently accessible in the cell membrane. Owing to these advantages, SrtA has been considered to be an ideal target for antivirulence therapy.
Over the past two decades, several classes of SrtA inhibitors have been developed, including natural products,17–22 synthetic small molecules,23–27 and peptidomimetic inhibitors.28–30 Although studies have revealed that chemical inhibition of SrtA transpeptidation may be a useful antiinfective strategy to prevent S. aureus infection without the side-effects of antibiotics, very few SrtA inhibitors have been assessed using in vivo models, and no such inhibitors have entered clinical trials.28,31–34 Therefore, there is still an urgent need to discover more promising small-molecule inhibitors to target SrtA.
Herein, we report that ML346, which was previously identified as an activator of heat shock protein Hsp70,35,36 functions as a selective irreversible SrtA inhibitor. Compound ML346 interferes in the transpeptidation activity of SrtA, by which it anchors surface proteins into the staphylococcus envelope. The therapeutic effect of ML346 on S. aureus-induced infection was further evaluated in a Galleria mellonella model. In addition, we determined the X-ray crystal structure of SrtAΔN81 from Streptococcus pyogenes (SpSrtAΔN81) bound to ML346. To the best of our knowledge, this is the first report of the structure of SpSrtAΔN81 bound to an inhibitor.
Results and discussion
Identification of SrtA inhibitors
Previously, we developed [3-(4-pyridinyl)-6-(3-sodiumsulfonatephenyl)[1,2,4]triazolo[3,4-b][1,3,4]thiadiazole] (6e) and Tideglusib (TD) as reversible SrtA inhibitors that are effective to against S. aureus infection in vivo (Fig. 1A).31,32 To identify SrtA inhibitors with novel scaffolds and good efficacy in vivo, we performed two rounds of screening on a collected compound library containing more than 4000 drug candidates and clinical drugs (Fig. S1A†). The initial high-throughput screening was performed using a fluorescence resonance energy transfer (FRET) assay.8,37 N-Terminal truncation of 24 residues which function as membrane anchor segment do not affect the transpeptidation activity of S. aureus SrtA but significantly increased its solubility.9 Therefore, SaSrtAΔN24 was expressed and purified and the inhibitory activity of compounds was monitored by measuring the transpeptidation activity of the recombinant SaSrtAΔN24 on the peptide substrate (Abz-LPATG-Dnp). Compounds with inhibition ratio higher than 90% was identified as preliminary hits; this led to 230 hits (Fig. S1B†). And the statistical confidence of the FRET-based assay was evaluated by calculating the Z-factor from the fluorescence of sample and control plotted in Fig. S1C.† Herein, the Z-factor of this assay was 0.59, which indicated a stable and good system. To exclude false positives resulting from fluorescence interference, hit compounds were subsequently validated by a polyacrylamide gel electrophoresis (PAGE)-based assay in which the recombinant surface protein IsdA64-323 was used as a substrate for SaSrtAΔN24. ML346, a compound with a barbituric acid and cinnamaldehyde scaffold (Fig. 1A),35,36 showed an outstanding half-maximal inhibitory concentration (IC50) of 0.37 μM against SaSrtAΔN24 in the FRET-based assay, and blocked the cleavage of surface protein IsdA64-323 by SaSrtAΔN24 in a dose-dependent manner in the PAGE-based assay (Fig. 1B and C). In addition, ML346 exhibited an inhibitory activity on SpSrtAΔN81 comparable with that observed for SaSrtAΔN24, with an IC50 value of 1.37 μM (Fig. 1B). ML346 also inhibited the cleavage of IsdA64-323 by the recombinant SpSrtAΔN81 in the PAGE-based assay (Fig. 1C). The IC50 values of ML346 against SaSrtAΔN24 and SpSrtAΔN81 in PAGE-based assay were calculated as 2.65 μM and 5.88 μM, respectively, by quantifying the band intensity (Fig. S1D†). These data demonstrated the inhibitory activity of ML346 on SrtA transpeptidation in vitro.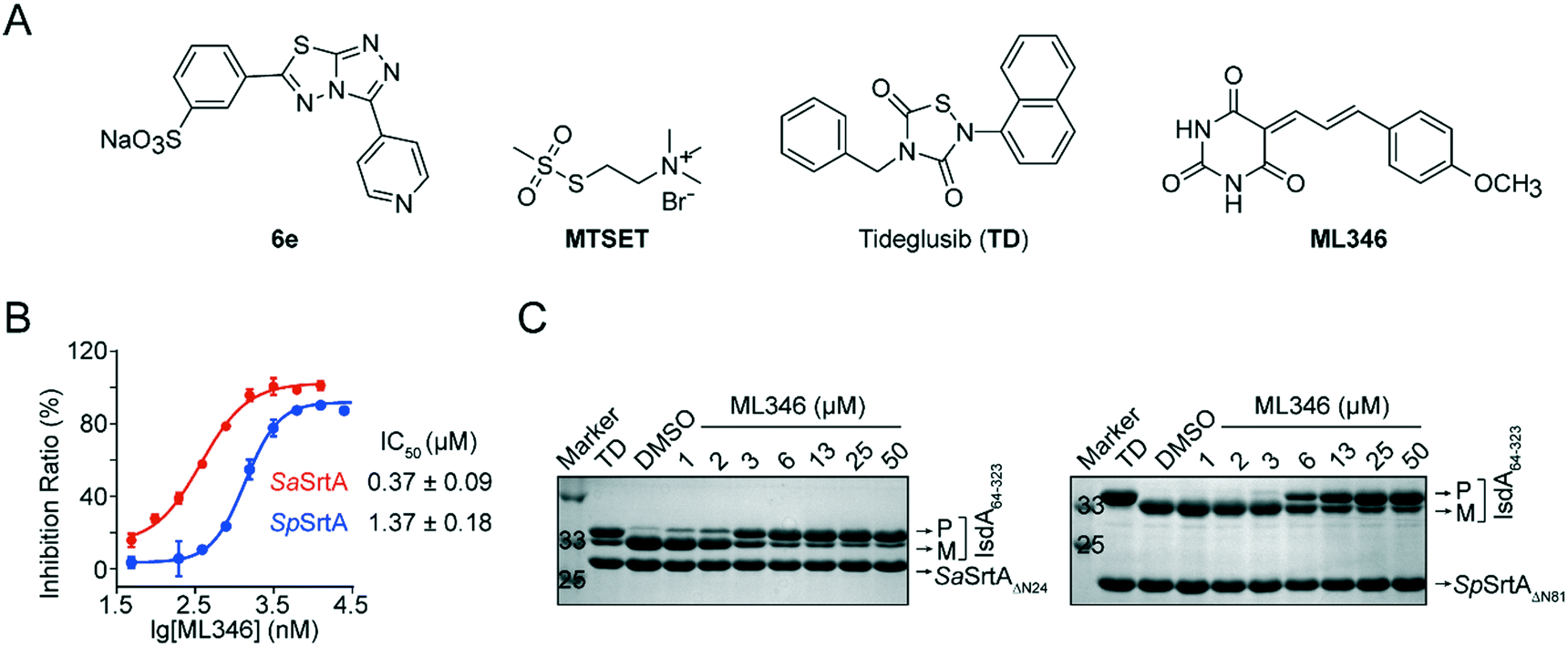 | ||
| Fig. 1 Identification of SrtA inhibitors. (A) Chemical structures of compounds used in this study. (B) Determination of IC50 values of ML346 against SaSrtAΔN24 and SpSrtAΔN81 by FRET based assay in which fluorogenic peptide was assayed as substrate. Data were presented as mean ± SEM. (C) Inhibition of SaSrtAΔN24 and SpSrtAΔN81 transpeptidations on IsdA64-323 protein with gradient concentrations of ML346 in PAGE based assay. Migratory positions of IsdA64-323 substrate precursor (P), mature transpeptidation product (M), SaSrtAΔN24, and SpSrtAΔN81 are indicated. Tideglusib (TD) at 200 μM was used as a positive control. | ||
Pan-assay interference compounds (PAINS) that attenuate multiple targets via non-specific mechanisms are the main sources of false positives in drug discovery.37,38 Hence, we investigated whether ML346 was a PAINS inhibitor of SrtA. First, ML346 did not show intrinsic fluorescence in an assay buffer (Fig. S1E†). Second, the presence of 0.01% freshly prepared nonionic detergent Triton X-100 in the assay buffer minimally altered the IC50 values of ML346 against both SaSrtAΔN24 and SpSrtAΔN81 (Fig. S1F†), demonstrating that ML346 does not act as an universal aggregator for proteins.38 Moreover, the circular dichroism (CD) spectra of SaSrtAΔN24 showed no obvious differences in the presence of a 10-fold excess of inhibitor compared with a methanol (MeOH) control (Fig. S1G†), indicating that ML346 did not disrupt the secondary folding properties of SaSrtAΔN24.39 Collectively, these results indicate that ML346 is not a PAINS inhibitor of SrtA.
ML346 is an irreversible inhibitor of SrtA
We then characterized the mode of action of inhibitor ML346. After pre-incubation of ML346 with SaSrtAΔN24 for different times (10 min, 20 min, 40 min, and 60 min), we quantified the IC50 values of ML346 using a FRET-based assay. The inhibitory activity of ML346 appeared to increase in a time-dependent manner, suggesting irreversible binding between ML346 and SaSrtAΔN24 (Fig. 2A, left panel). We further investigated the effects of ML346 on the Km values of SaSrtAΔN24. Similar to the covalent alkylator N,N,N-trimethyl-2-[(methylsulfonyl)sulfanyl]ethanaminium bromide (MTSET) (Fig. 1A),40 ML346 increased the Km value of SaSrtAΔN24 compared with that in the dimethyl sulfoxide (DMSO) treated group, whereas the Km value minimally changed after ML346 was removed from the reaction system (Fig. 2A, right panel). These results provide additional evidence to support the irreversible interaction between ML346 and SrtA. As SaSrtA is a cysteine protease bearing only one cysteine C184, we constructed a C184A SaSrtAΔN24 mutant to test whether C184 was the covalent modification residue. Given that C184A SaSrtAΔN24 mutant is catalytically inactive,9 we evaluated the effects of ML346 on the thermal stability of SaSrtAΔN24 and the C184A SaSrtAΔN24 mutant by nano differential scanning fluorimetry (nano DSF) assay. The results showed that ML346 significantly thermally stabilized the SaSrtAΔN24 protein by increasing its melting temperature by 4.1 °C, whereas it exhibited a minimal effect on the thermal stability of the C184A SaSrtAΔN24 mutant protein (Fig. 2B). These results indicated that ML346 may function as an irreversible inhibitor of SaSrtA, and that C184 is likely to be the covalent modification site. In addition, ML346 caused similar time-dependent inhibition of SpSrtAΔN81, and the Km values showed no significant difference before and after buffer exchange (Fig. 2C), suggesting that ML346 inhibited SaSrtA and SpSrtA via a similar mechanism.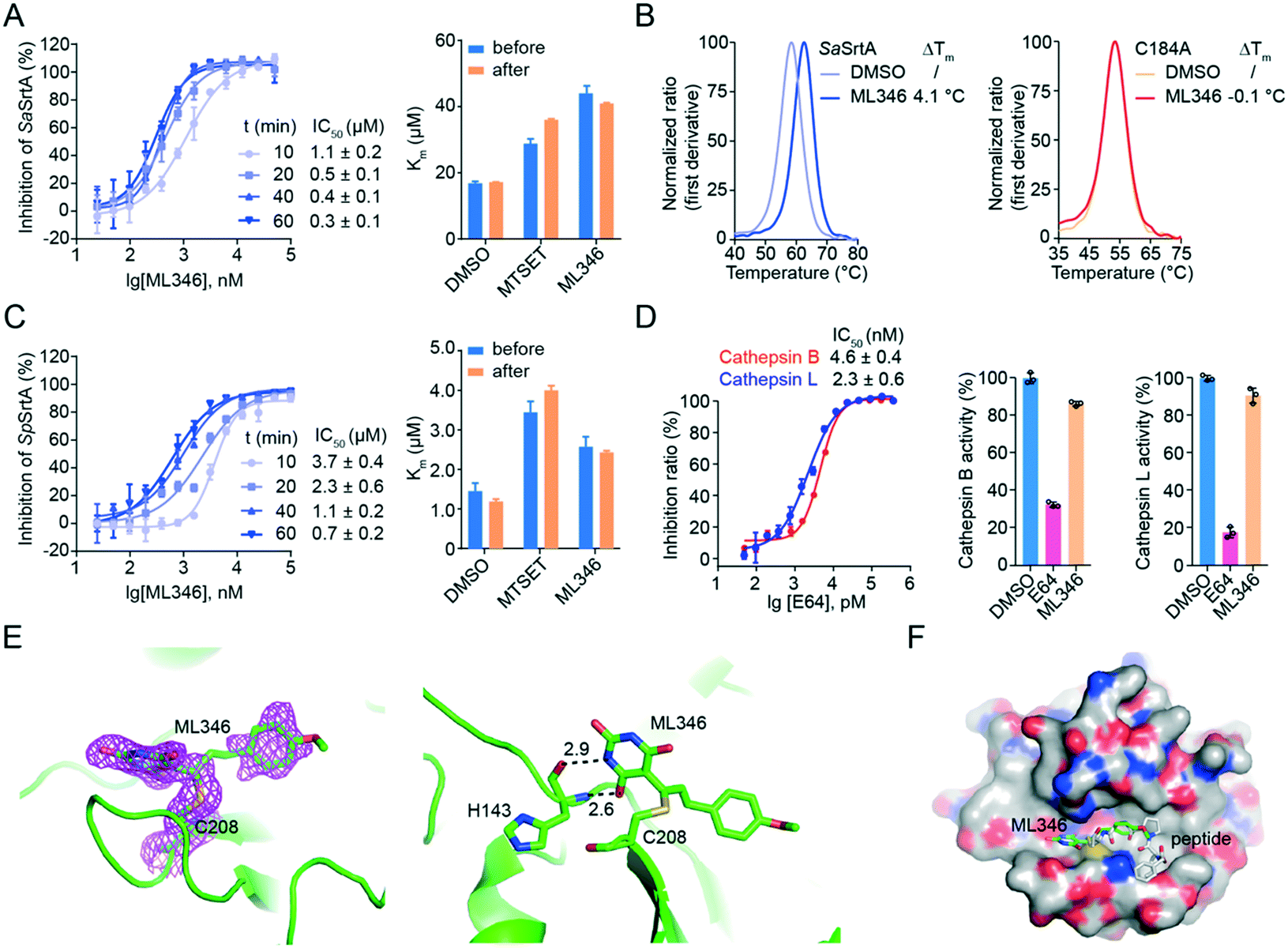 | ||
| Fig. 2 Characterization of ML346 as an irreversible inhibitor of SrtA. (A) Irreversible inhibition on SaSrtAΔN24 by ML346. IC50 values of ML346 against SaSrtAΔN24 under varying incubation time was shown in left panel. Right panel displayed Km values of SaSrtAΔN24 enzyme in the presence of DMSO, MTSET, and ML346. (B) Effect of ML346 on the thermal stability of SaSrtAΔN24 (left panel) and C184A SaSrtAΔN24 (right panel) in nano DSF assay. (C) Irreversible inhibition on SpSrtAΔN81 by ML346. Left panel showed effect of incubation time on IC50 values of ML346 against SpSrtAΔN81. Right panel showed Km values of SpSrtAΔN81 incubated with DMSO, MTSET, and ML346. (D) Selectivity of the SrtA inhibitor ML346 on cathepsin B and cathepsin L proteases. Inhibitor E64 was assayed as a positive control, and inhibitory effects of SrtA inhibitor on cysteine proteases activity of cathepsins B and L were tested in the presence of 20 μM ML346. (E) X-ray crystal structure of SpSrtAΔN81/ML346 complex (PDB ID: 7V6K). The protein is colored in green, and 2Fo-Fc omit map is colored in magenta. Hydrogen bonding is indicated by black dashed lines, and the distance is shown in Å. (F) Structural alignment of SpSrtAΔN81/ML346 complex with SaSrtAΔN59/LPAT* complex. Surface representation of SrtA is shown with inhibitor ML346 and substrate peptide LPAT* covalently bound in the active site. The substrate peptide LPAT* is colored in grey. | ||
Having observed the time-dependent inhibition of SrtA by the irreversible inhibitor ML346, we further performed kinetic analysis of the enzyme inactivation by determining the inactivation constant (KI) and maximum inactivation rate constant (kinact). The KI and kinact values of SaSrtAΔN24 were measured to be 2.453 μM and 0.185 min−1, respectively, giving a kinact/KI value of 0.075 μM−1 min−1 (Fig. S2A†). In addition, ML346 had a KI value of 0.686 μM for the first step of reversible binding to SpSrtAΔN81, indicating an approximately four-fold lower potency compared with SaSrtAΔN24. Similarly, the maximum potential rate of the second step, covalent bond formation (kinact = 0.044 min−1) between ML346 and SpSrtAΔN81, was also an approximately four-fold lower than that of SaSrtAΔN24. The kinact/KI value of SpSrtAΔN81 was 0.064 μM−1 min−1 (Fig. S2A†), indicating a comparable overall covalent modification rate of ML346 to the two SrtA enzymes.
Mammalian cathepsin B, cathepsin L, and SrtA are similarly related to the papain-like cysteine proteases with respect to both structure and mechanism.41 To investigate the selectivity of our SrtA inhibitor to other cysteine proteases, we tested the inhibitory effects of ML346 on cathepsin B and cathepsin L. A broad-spectrum inhibitor of cysteine proteases, L-trans-epoxysuccinyl-leucylamido(4-guanidino)butane (E64), was assayed as a positive control (Fig. S2B†); it showed similar inhibition of the two cathepsin enzymes, with IC50 values of 2.3–4.6 nM (Fig. 2D).27,42 Interestingly, ML346 minimally inhibited cathepsin B and L at a concentration of 20 μM (Fig. 2D, Table S1†), whereas E64 at 6.25 nM exhibited a significant inhibitory effect; these results suggest that ML346 is likely to function as a selective irreversible inhibitor of bacterial SrtA rather than mammalian cysteine proteases.
To gain insight into the mechanism of SrtA inhibition by ML346, we determined the X-ray crystal structure of the SpSrtAΔN81/ML346 complex (Fig. S2C, Table S2†). The electron densities of the 2Fo-Fc map and Fo-Fc omit simulation map confirmed the existence of ML346 in the SpSrtAΔN81 active site (Fig. 2E and S2C†). Compound ML346 was covalently added to the thiol group of Cys208, the key catalytic residue of SpSrtA.43 The barbiturate motif in ML346 was involved in hydrogen binding to the main chain of His143 in SpSrtAΔN81, which enhanced the interaction between ML346 and SrtA. ML346 may undergo a Michael addition reaction with Cys208 of SpSrtA, in which α,β-unsaturated carbonyl groups of ML346 as Michael acceptor was attacked by thiol group of Cys208, resulting in covalent adduct (Scheme 1). Both SaSrtA and SpSrtA have the typical eight-stranded β-barrel fold and their overall structures are very similar.44 Structural alignment of the SaSrtAΔN59/peptide complex (Protein Data Bank [PDB] ID: 2KID)45 and SpSrtAΔN81/ML346 complex (PDB ID: 7V6K) revealed that ML346 was positioned in the peptide-binding cavity of SrtA (Fig. 2F and S2D†). Notably, ML346 occupied only part of the pocket in SaSrtAΔN59 and SpSrtAΔN81, whereas the peptide LPAT* substrate occupied the entire pocket. These results lay the groundwork for structure-guided design of more potent SrtA inhibitors derived from the chemical scaffold of ML346.
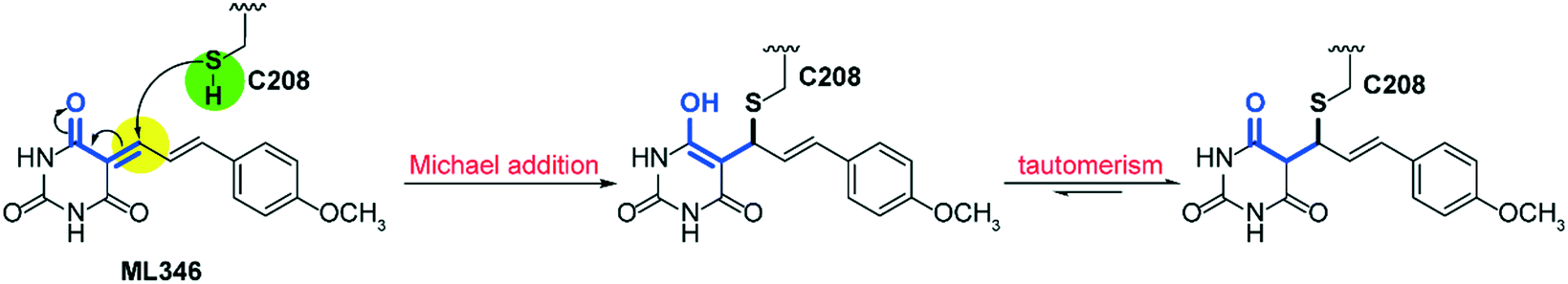 | ||
| Scheme 1 Mechanism of SpSrtA inhibition by ML346. | ||
ML346 attenuates the virulence of S. aureus
All S. aureus isolates encode 17–21 surface proteins that determine bacterial infectivity and pathogenicity.46,47 These surface proteins, including SpA, are covalently anchored to the cell envelope by SrtA transpeptidation.15,40 As shown by the western blot results, the S. aureus Newman strain cultured with ML346 displayed decreased SpA on the staphylococcus envelope; this decrease also occurred in a dose-dependent manner in the USA300 strain, a community-associated MRSA strain (Fig. 3A). Because surface protein SpA in cell envelope is covalently linked to peptidoglycan,8 the molecular weight of SpA observed in western blot assay was higher than the theoretical value and was a wide band. Given that SpA specifically binds to the Fcγ fragment of human IgG, we used fluorescein isothiocyanate (FITC)-labelled IgG and further estimated the abundance of the cell-wall anchored SpA by measuring fluorescence (Fig. 3B).46,48,49 Consistent with the results of the western blot assay, the S. aureus Newman ΔsrtA mutant strain almost completely lost its ability to anchor SpA to the bacterial cell envelope. Both the S. aureus Newman and USA300 strains showed reduced abundance of cell-wall anchored SpA in the presence of ML346, and the inhibitory activity of ML346 on SpA incorporation was better than that of MESET. Collectively, the SrtA inhibitor ML346 significantly suppressed the display of virulence factors on the cell surface in S. aureus.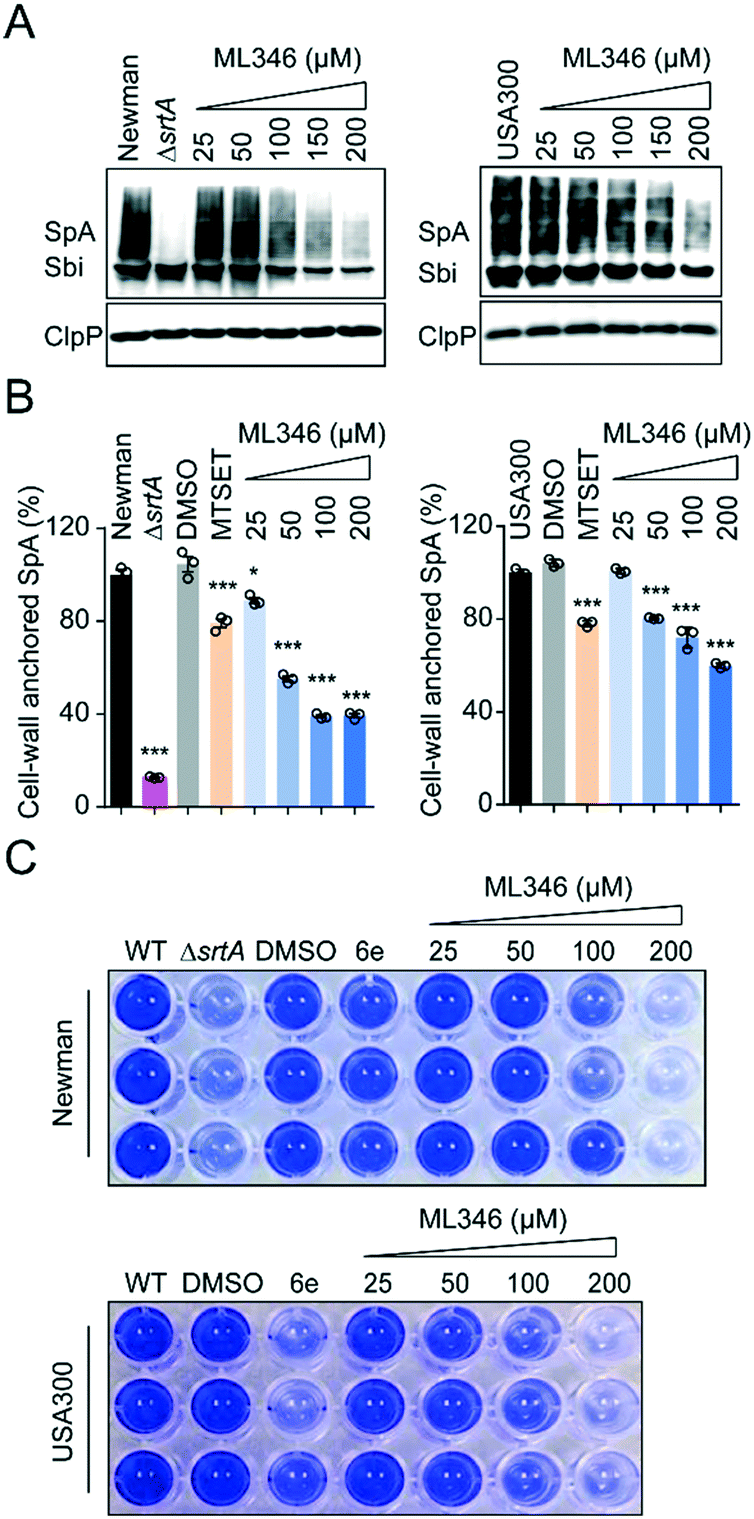 | ||
| Fig. 3 SrtA inhibitor ML346 attenuated virulence of S. aureus Newman and USA300 strains. (A) Western blot analysis of cell-wall anchored SpA in S. aureus Newman and USA300 strains in the presence of ML346. Sbi, the IgG binding protein of S. aureus, was also detected. TD at 100 μM was assayed as positive control. (B) Effect of ML346 on the abundance of cell-wall anchored SpA estimated by FITC-labelled human IgG in S. aureus Newman and USA300 strains. Statistical significance (*P < 0.05, **P < 0.01, ***P < 0.001) was determined using the unpaired, two-tailed Student's t test (n = 3). Data were presented as mean ± SEM. MTSET at 200 μM was assayed as positive control. (C) Inhibitory effect of ML346 on biofilm formation of S. aureus Newman and USA300 strains. 6e at 100 μM was assayed as positive control. | ||
Biofilm formation is critical in the establishment of bacterial infections associated with medical devices,50 which are usually dormant and resistant to antibiotic treatment.51 Biofilm formation in S. aureus is dependent on surface proteins such as ClfB, FnBPs, SasC, and SpA.52 Both srtA gene deletion and SrtA inhibitors have been found to reduce the ability of S. aureus to form biofilms.53,54 To investigate whether ML346 suppressed biofilm formation, we determined the effect of ML346 on staphylococci biofilm formation in a crystal violet staining assay. The known SrtA inhibitor 6e was assayed as a positive control (Fig. 1A).31 As shown in Fig. 3C, the S. aureus ΔsrtA mutant strain produced much less biofilm biomass than the wild-type (WT) strain in the crystal violet staining assay. The biofilm biomass of the WT strain decreased gradually in the presence of ML346 (Fig. 3C and S3A†). A similar inhibition of biofilm formation by ML346 treatment was observed in the MRSA strain USA300 (Fig. 3C and S3A†). Overall, our SrtA inhibitor significantly inhibited the anchoring of surface proteins to cell envelopes, thereby impairing the biofilm formation capability of S. aureus.
Antiinfective therapy with compound ML346 in vivo
Deletion of the srtA gene does not inhibit the in vitro growth of S. aureus.55 Similarly, a genuine SrtA inhibitor should ideally have no inhibitory effect on the survival and growth of bacteria. Indeed, ML346 treatment slightly reduced the in vitro growth of S. aureus Newman and USA300 strains at concentrations up to 200 μM (Fig. S4A†), and the determined minimum inhibitory concentration (MIC) values of ML346 against S. aureus Newman, USA300, and Escherichia coli AB1157 were all above 512 μg mL−1 (Fig. S4B, Table S3†), suggesting that ML346 is unlikely to function as a conventional antibiotic. We next tested the toxicity of ML346 in G. mellonella. All G. mellonella larvae were alive 120 h after ML346 administration and were normal in appearance, with the same creamy color as the vehicle-treated larvae (Fig. S4C†). Together, these results indicate that the non-bactericidal inhibitor ML346 is safe to G. mellonella larvae.Melanization, an innate immune response in G. mellonella, involves the synthesis and deposition of melanin to encapsulate pathogens at a wound site, followed by hemolymph coagulation and opsonization, and is a major indicator of the health status of larvae.56,57 To test the pathogenic effect of S. aureus, we injected S. aureus Newman at a dose of 1 × 105 colony-forming units (CFUs) into larvae at the last proleg and observed the melanization response. S. aureus Newman infection caused larvae to acquire a brown color, whereas no melanization was observed in larvae groups injected with the ΔsrtA S. aureus or the ML346 pre-treated WT S. aureus strains (Fig. 4A). Given the promising capability of ML346 to reduce the virulence and pathogenicity of S. aureus, we assessed whether ML346 could function as an antiinfective agent in vivo. A G. mellonella infection model was established by inoculation with S. aureus Newman at a lethal dose of 1 × 106 CFUs.58 The G. mellonella that received ML346 administration exhibited significantly prolonged survival times, with a survival rate of 46.7% at the end of monitoring (Fig. 4B). Similarly, ML346 was effective in protecting G. mellonella from infection with MRSA strain USA300 at a lethal dose of 1 × 107 CFUs. Together, these data support our SrtA inhibitor ML346 as a promising antivirulence agent to prevent S. aureus infection without the side-effects of antibiotics.
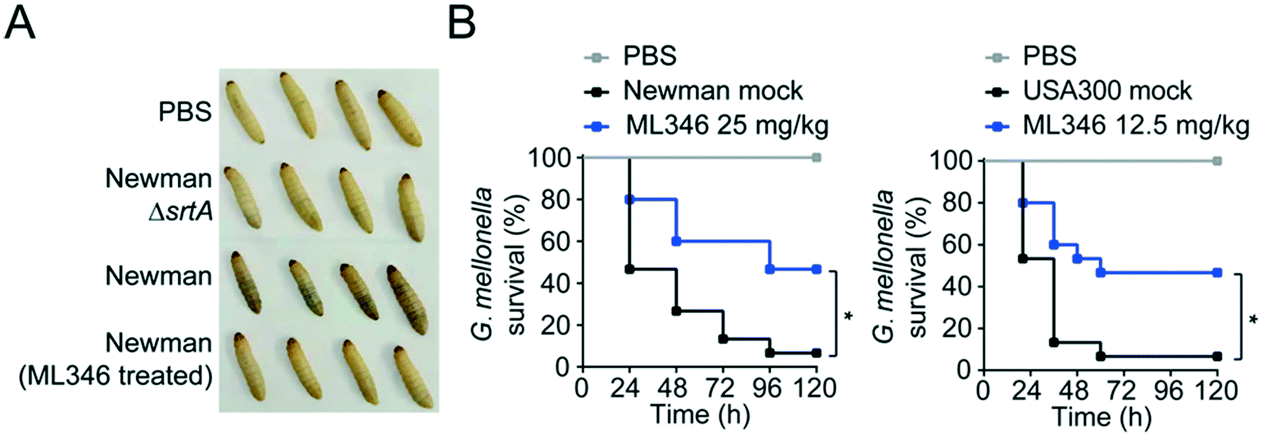 | ||
| Fig. 4 Antiinfective therapy with SrtA inhibitor ML346 in G. mellonella infection model. (A) Appearances of G. mellonella larvae infected with S. aureus strains. The ΔsrtA S. aureus Newman was assayed as a positive control. (B) The survival rate of G. mellonella larvae (n = 15) infected with S. aureus Newman (left panel) or USA300 (right panel). Larvae were concomitantly treated with either mock or ML346 at indicated dosage. Statistical significance was determined from a Mantel–Cox test using Prism (*p < 0.05). | ||
Conclusions
In this study, we identified ML346 as a new covalent inhibitor of SrtA with antivirulence effects on Gram-positive bacteria. ML346 inhibited the transpeptidation of SaSrtAΔN24 and SpSrtAΔN81in vitro at low micromolar concentrations but exhibited weak inhibitory effects on human cysteine proteases cathepsin B and cathepsin L, suggesting that ML346 could be a selective inhibitor of SrtA. In addition to the biochemical characterization of ML346 as a covalent inhibitor, an X-ray crystal structure of the SpSrtAΔN81/ML346 complex at a high resolution was determined; this showed that ML346 was indeed covalently bound to the thiol group of Cys208 in the active site. As expected, ML346 blocked the anchoring and display of the surface protein SpA to the cell envelope of S. aureus, that is, it suppressed a key virulence factor that contributes to staphylococci pathogenesis. The biofilm formation ability of S. aureus was also significantly suppressed in the presence of ML346. Furthermore, ML346 was shown to be effective in protecting G. mellonella from S. aureus infection.Though ML346 exhibited SrtA inhibitory activity in vitro and in vivo, its toxicity against HeLa-luc cells have been reported previously.35 This reminds us that the toxicity of ML346 against mammalian cells should not be overlooked. Inspired by the determined SpSrtAΔN81/ML346 complex crystal structure in which ML346 only occupied part of the binding pocket, structure-based optimization with ML346 as a covalent warhead may be an effective strategy to develop more promising SrtA inhibitor with excellent in vivo activity and safety. In summary, our identification of ML346 as a covalent inhibitor of SrtA provides a new chemical scaffold that has potential for further structure-based development of antivirulence agents.
Experimental section
General method
ML346, MTSET, and E64 used in this study were purchased from MedChemExpress, Toronto Research Chemicals, and TargetMol, respectively. TD and 6e was synthesized in house.Protein expression and purification
S. aureus SrtAΔN24 (SaSrtAΔN24) and S. pyogenes SrtAΔN81 (SpSrtAΔN81) were expressed and purified as described previously.31 Briefly, the S. aureus srtAΔN24 and S. pyogenes srtAΔN81 genes were cloned into the pET28a vector. The recombinant plasmids were transformed into E. coli BL21(DE3) cells and cultured in lysogeny broth (LB) medium in the presence of 30 μg mL−1 kanamycin at 37 °C. Protein expression was induced with 1 mM isopropyl-beta-D-thiogalactopyranoside when the A600 reached 0.6–0.8, followed by culture at 30 °C for another 4 h. The culture was centrifuged, and the pellet was collected and resuspended in 40 mL buffer A (50 mM Tris–HCl, pH 7.5, 40 mM imidazole, 200 mM NaCl, 1 mM dithiothreitol [DTT]). The cells were lysed by high pressure, and the lysate was cleared by centrifugation at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 min at 4 °C. Proteins were purified from the supernatant by Ni-NTA (HisTrap HP 5 mL column, GE Healthcare) with an elution gradient using buffer B (50 mM Tris–HCl, pH 7.5, 300 mM imidazole, 200 mM NaCl, 1 mM DTT). Eluted proteins were subsequently subjected to gel-filtration (HiLoad 16/60 Superdex 75 column, GE Healthcare) and eluted with desalting buffer (20 mM Tris–HCl, pH 7.50, 50 mM NaCl, 1 mM DTT). Protein purity was assessed by 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Purified proteins were concentrated, shock frozen in liquid nitrogen, and stored at −80 °C.
000 rpm for 30 min at 4 °C. Proteins were purified from the supernatant by Ni-NTA (HisTrap HP 5 mL column, GE Healthcare) with an elution gradient using buffer B (50 mM Tris–HCl, pH 7.5, 300 mM imidazole, 200 mM NaCl, 1 mM DTT). Eluted proteins were subsequently subjected to gel-filtration (HiLoad 16/60 Superdex 75 column, GE Healthcare) and eluted with desalting buffer (20 mM Tris–HCl, pH 7.50, 50 mM NaCl, 1 mM DTT). Protein purity was assessed by 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Purified proteins were concentrated, shock frozen in liquid nitrogen, and stored at −80 °C.
The C184A SaSrtAΔN24 mutant was expressed and purified in a similar way. The S. aureus isdA64-323 gene was cloned into the pET28b vector and transformed into E. coli BL21(DE3) cells. SaIsdA64-323 protein was then purified in the same way as SaSrtAΔN24.
FRET-based assay
Stock concentrations of all compounds were prepared in DMSO at a concentration of 2 mM, and 1 μL was added to a 384-well black plate for high-throughput screening. Before the screening, 25 μL reaction buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 5 mM CaCl2) containing 1 μM recombinant SaSrtAΔN24 was added to each well, followed by incubation at room temperature for 20 min. The transpeptidation reaction was initiated by adding 24 μL peptide substrate (Abz-LPATG-Dnp) to a concentration of 10 μM. The fluorescence intensity was measured every 1 min for 15 min using a Flex Station 3 (Molecular Devices), with the excitation and emission wavelengths set to 309 and 420 nm, respectively. The inhibition ratio was calculated according to the following equation: inhibition ratio (%) = (1 − Vi/V0) × 100%, where V0 and Vi are the initial velocities of the enzymatic reaction in the absence or presence of inhibitors, respectively. Two technical replicates were performed for each compound.To determine IC50 values, 1 μM SaSrtAΔN24 or SpSrtAΔN81 in reaction buffer was incubated with a series of concentrations of the appropriate compound (0.05–50 μM) at room temperature for 20 min. The IC50 values were analysed using GraphPad Prism software applying the sigmoidal dose–response equation. Three technical replicates were taken for each data point, and two independent experiments were performed for each compound. Data are presented as mean ± SEM.
To evaluate the statistical confidence the FRET-based assay, Z-factor was calculated from a single black 384-well microplate including the samples (TD at 50 μM, n = 192) and the controls (DMSO, n = 192). The Z-factor was calculated by the following equation:59
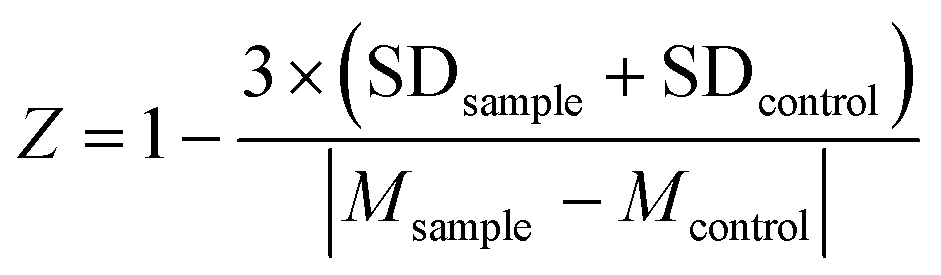
PAGE-based transpeptidation reaction
The PAGE assay was performed in 50 μL reaction buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 5 mM CaCl2). SaSrtAΔN24 or SpSrtAΔN81 (200 μg mL−1) was incubated with various concentrations of inhibitor (1–50 μM) at room temperature for 20 min. Substrate protein IsdA64-323 and (Gly)3 were subsequently added to a final concentration of 300 μg mL−1 and 3 mM, respectively. The reaction was quenched with 12.5 μL 5× SDS loading buffer after incubation at 37 °C for 1.5 h. The samples were then subjected to 12% SDS-PAGE and photographed using a Tanon 2500 Gel Image System. Inhibition activity of inhibitors was analysed by quantifying the IsdA64-323 substrate precursor (P) and mature transpeptidation product (M). Tideglusib (TD) was used as a positive control. Two independent experiments were performed.CD spectra
CD spectra of samples were acquired at wavelengths ranging from 190 to 250 nm using a JASCO J-815-150 s spectropolarimeter. Measurements were taken in 0.1 cm path length quartz cuvettes, and the baseline was calibrated with phosphate-buffered saline (PBS) before each measurement series. Compounds dissolved in methanol were incubated with 10 μM SaSrtAΔN24 at room temperature for 30 min before CD analysis. The spectra displayed were averaged from three consecutive scans. Two independent experiments were performed.Irreversible inhibition assay
The purified SaSrtAΔN24 and SpSrtAΔN81 (5 μM) were incubated with ML346 at final concentration equivalent to IC50 or DMSO at room temperature for 1 h. Then, centrifugal filtration was performed to remove the inhibitors from the reaction system. Protein aliquots (25 μL) before and after centrifugal filtration were respectively added to 25 μL of various concentrations (0.78–200 μM for SaSrtAΔN24 and 0.2–50 μM for SpSrtAΔN81) of Abz-LPATG-Dnp substrate. The fluorescence intensity was monitored every 1 min for 15 min using a Flex Station 3 (Molecular Devices) with the excitation and emission wavelengths set to 309 and 420 nm, respectively. Km values were calculated by GraphPad Prism software. MTSET was assayed as a positive control. Three replicates were taken for each data point. The experiments were performed in duplicate. Data are presented as mean ± SEM.To determine kinact (maximum inactivation rate constant) and KI (inactivation constant) values, variable concentrations of inhibitor were incubated with SaSrtAΔN24 or SpSrtAΔN81 for different times, and enzyme kinetics curves were constructed. The Napierian logarithms of the remaining relative activity of SrtA were plotted against the incubation time. Then, linear regression analysis was conducted to determine the first-order rate constants (kobs). The values of kobs were subsequently plotted against inhibitor concentration and fitted to the following equation to obtain kinact and KI values:60
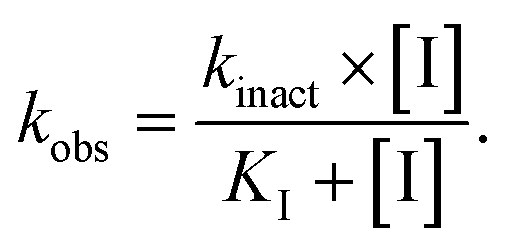
Nano DSF assay
Forty micrograms of SaSrtAΔN24 protein or C184A SaSrtAΔN24 mutant protein in 100 μL buffer (50 mM Tris–HCl, pH 9.0, 150 mM NaCl, 5 mM CaCl2) was incubated with 0.5 μL compound stock in DMSO at the appropriate concentration at room temperature for 15 min. Then, the mixture was centrifuged for 2 min at 3000 rpm to remove any precipitates. Capillaries filled with 10 μL of sample were simultaneously scanned at 330/350 nm wavelengths by label-free native nano DSF (NanoTemper, Prometheus NT.48). The temperature was increased from 30 °C to 90 °C at a rate of 2 °C per min. The fluorescence intensity ratio and its first derivative were calculated using the manufacturer's software (PR. ThermControl, version 2.1.2). Data were normalized in GraphPad Prism software.Inhibition of cysteine proteases
Fluorometric assays for cathepsin B and cathepsin L (Sigma-Aldrich) were performed as described previously with minor modifications.61 Briefly, Cbz-Phe-Arg-AMC in assay buffer (20 mM MES, pH 6.0, 1 mM EDTA, 200 mM NaCl, 0.005% Brij35) was used as a substrate. Twenty-five microliters of 0.2 ng μL−1 cathepsin B or 0.02 ng μL−1 cathepsin L was loaded into a 96-well black plate and pre-incubated with the appropriate compounds at 40 °C for 5 min. The reaction was initiated by adding 25 μL of Cbz-Phe-Arg-AMC substrate (40 μM for cathepsin B and 80 μM for cathepsin L, respectively), which had been pre-incubated at 40 °C for 5 min. The product released by the substrate hydrolysis was determined continuously by reading the fluorescence intensity at excitation and emission wavelengths of 380 nm and 460 nm, respectively, over a period of 10 min. L-trans-Epoxysuccinyl-leucylamido(4-guanidino)butane (E64) at 6.25 nM was assayed as a positive control. Three technical replicates were measured for each data point, and two independent experiments were performed. Data are presented as mean ± SEM.Crystallization, data collection and structure determination
Ten milligrams per millilitre of SpSrtAΔN81 protein was incubated with ML346 at two-fold concentration and mixed with a reservoir solution containing 0.2 M magnesium chloride hexahydrate, 0.1 M Bis–Tris (pH 5.5), and 25% (w/v) polyethylene glycol 3350. Crystals were grown at 16 °C using the hanging-drop vapor diffusion method for 4 weeks. The crystals were cryo-protected using 20% (v/v) glycerol. Diffraction data were collected on the BL17U1 beamline at the Shanghai Synchrotron Research Facility.62 All X-ray data were processed using the HKL2000 program and converted to structure factors within the CCP4 program.63,64 The structure was solved by molecular replacement in Phaser using the structure of SpSrtAΔN81 (PDB: 3FN5) as the search model. The structural model of the SpSrtAΔN81/ML346 complex was computationally refined using REFMAC5. The statistics for the data collection and refinement are summarized in Table S2.†Western blot
Overnight cultures of S. aureus were diluted 1:1000 with fresh Tryptic soy broth (TSB) medium and further cultured with various concentrations of inhibitors until A600 reached 3.0. One millilitre of the culture was pipetted, and cells were collected by centrifuging at 12000 rpm for 5 min. The pellet was washed three times with PBS and resuspended in 500 μL PBS containing 10 μg mL−1 lysostaphin. After incubation at 37 °C for 15 min, samples were centrifuged at 4 °C for 30 min (12000 rpm) and protoplasts were precipitated. The supernatant containing cell-wall anchored proteins was pipetted and mixed with 5× SDS loading buffer. Samples were separated by SDS-PAGE and subsequently transferred onto nitrocellulose membranes (Millipore, USA). After blocking with 5% skim milk at room temperature for 1 h, the membranes were incubated with SpA antibody and ClpP antibody at 4 °C overnight. Protein ClpP was used as a loading control. Horseradish peroxidase-labelled goat anti-rabbit IgG was used as the secondary antibody. Immune-reactive bands were visualized with a chemiluminescence reagent (Tanon), and the images were obtained with an enhanced chemiluminescence detection system (GE Healthcare Bioscience, USA). Three independent experiments were performed for each compound.
FITC-IgG based assay
Overnight cultures of S. aureus were diluted 1:1000 with fresh TSB medium and further cultured with inhibitors at varying concentrations until A600 reached 1.0. Six hundred microliters of culture were pipetted, and cells were collected by centrifuging at 12000 rpm for 5 min. The pellet was washed three times with PBS and resuspended in 400 μL PBS supplemented with 4 μL 0.5 mg mL−1 FITC-labelled human IgG. Samples were incubated at room temperature for 30 min with shaking in dark. Cells were subsequently collected by centrifuging and rinsed three times with PBS. The fluorescence intensity of bacteria was monitored using a Flex Station 3 (Molecular Devices) at emission and excitation wavelengths of 495 and 520 nm, respectively. Three technical replicates were measured for each data point, and three independent experiments were performed for each compound. Data are expressed as mean ± SEM.
Biofilm formation
Overnight cultures of S. aureus were diluted 1:1000 with fresh TSB medium. Two hundred microliter aliquots were added to the wells of 96-well microtiter plates with various concentrations of inhibitors and cultured without shaking at 37 °C for 18 h. The medium was then discarded and the wells were gently rinsed two times with PBS. The biofilm was immobilized with 200 μL methanol for 10 min and subsequently stained with 0.1% (w/v) crystal violet solution for 15 min. Excess stain was discarded, and the plates were washed three times with sterile distilled water before being photographed. For quantification, the absorbance of crystal violet stain dissolved in 33% (v/v) acetic acid solution was measured at 600 nm using a microplate reader. Compound 6e was assayed as a positive control. Three independent experiments were performed for each compound. Data are reported as mean ± SEM.
Bacterial growth curve
Overnight cultures of S. aureus were diluted 1:1000 with fresh TSB medium and further cultured with various concentrations of inhibitors until A600 reached 0.6. The culture was diluted again 1:400 with fresh TSB medium. Two hundred microliter aliquots were added to 96-well microtiter plates with variable concentrations of the test compounds. The A600 values of each well were measured every 1 h for 18 h using a Flex Station 3 (Molecular Devices). The growth curve was drawn using GraphPad Prism software. Three technical replicates were measured for each data point, and three independent experiments were performed.
MIC assay
S. aureus Newman and USA300 strains were cultured in TSB medium, and E. coli was cultured in LB medium. Overnight cultures of these strains were diluted 1:1000 and cultured for another 2–3 h at 37 °C until A600 reached 0.6. After diluting the cultures 1:400, the inoculum was added to the wells of 96-well microtiter plates containing two-fold dilutions of compounds and cultured at 37 °C for 16–18 h without shaking. The MIC of each compound was defined as the lowest concentration of compound that resulted in no visible growth. Vancomycin, tetracycline, and kanamycin were used as the positive controls. Two independent experiments were performed.
Infection, toxicity, and efficacy in the G. mellonella model
The 2–2.5 cm G. mellonella larvae with creamy color were selected for use in the in vivo studies. The larvae were kept at room temperature and starved for 24 h before being assayed. All injections were administered using a sterile 50 μL syringe with a 9 mm-long 30 gauge needle, and the bacterial suspension was delivered into the last proleg of the larvae. After injection, groups of 15 larvae were placed in a Petri dish at 37 °C. To evaluate the pathogenicity of the ML346-treated S. aureus to G. mellonella, overnight cultures of S. aureus were diluted 1:1000 with or without ML346 and cultured continuously until A600 reached 1.0. Ten microliter bacterial suspensions prepared in PBS to a density of 1 × 107 CFU mL−1 were subsequently injected into larvae. The ΔsrtA S. aureus Newman strain was used as a positive control. The larvae were anesthetized on ice and photographed 24 h after injection for observation of melanization. To evaluate the efficacy of compounds, overnight cultures of S. aureus were diluted 1:1000 in fresh TSB and grown at 37 °C until A600 reached 1.0. The bacteria were centrifuged at 3000 g, washed, and resuspended in PBS to an appropriate density. Suspensions of S. aureus were pre-mixed with compounds dissolved in the vehicles (5% DMSO, 20% PEG300, 75% PBS [containing 6.6% solutol HS-15]) prior to injection. The survival rate of larvae was recorded for 120 h. Death of larvae was assessed by visual inspection of the lack of movement. Aliquots of the inoculum were plated, and the CFUs were counted. The inoculation dose was approximately 1 × 106 CFU per larva for S. aureus Newman and 1 × 107 CFU per larva for S. aureus USA300. Statistical significance was determined by Mantel–Cox test using GraphPad Prism, and p < 0.05 was deemed statistically significant. For toxicity assessment, 10 μL of compounds dissolved in the vehicle were injected into larvae. Survival rates of larvae were recorded for 120 h, and they were photographed as described before. Three independent experiments were performed.
Author contributions
C.-G. Y. conceived the project and designed the research. X.-N. G. performed the experiments with help from T. Z., T. Y., Z. D., and J. G., S. Y., L. L., and C.-G. Y. contributed data analyses and/or grant support. X.-N. G. and C.-G. Y. wrote the paper. All authors reviewed the results and approved the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We thank the staff of the BL17U1 beamlines at Shanghai Synchrotron Radiation Facility for data collection support. This study was supported by the National Natural Science Foundation of China (22107109 to T. Z. and 81861138046 and 21725801 to C.-G. Y.).Notes and references
- F. D. Lowy, Staphylococcus aureus infections, N. Engl. J. Med., 1998, 339, 520–532 CrossRef CAS PubMed.
- S. Y. Tong, J. S. Davis, E. Eichenberger, T. L. Holland and V. G. Fowler, Jr., Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management, Clin. Microbiol. Rev., 2015, 28, 603–661 CrossRef CAS PubMed.
- Centers for Disease Control and Prevention (CDC), Outbreaks of community-associated methicillin-resistant Staphylococcus aureus skin infections—Los Angeles County, California, 2002–2003, MMWR Morb Mortal Wkly Rep., 2003 Search PubMed.
- World Health Organization, 2020 Antibacterial agents in clinical and preclinical development: an overview and analysis, Report 978–92–4-002130-3, 2021 Search PubMed.
- S. W. Dickey, G. Y. C. Cheung and M. Otto, Different drugs for bad bugs: antivirulence strategies in the age of antibiotic resistance, Nat. Rev. Drug Discovery, 2017, 16, 457–471 CrossRef CAS PubMed.
- M. Totsika, Disarming pathogens: benefits and challenges of antimicrobials that target bacterial virulence instead of growth and viability, Future Med. Chem., 2017, 9, 267–269 CrossRef CAS PubMed.
- F. Sun, L. Zhou, B. C. Zhao, X. Deng, H. Cho, C. Yi, X. Jian, C. X. Song, C. H. Luan, T. Bae, Z. Li and C. He, Targeting MgrA-mediated virulence regulation in Staphylococcus aureus, Chem. Biol., 2011, 18, 1032–1041 CrossRef CAS PubMed.
- S. K. Mazmanian, G. Liu, H. Ton-That and O. Schneewind, Staphylococcus aureus sortase, an enzyme that anchors surface proteins to the cell wall, Science, 1999, 285, 760–763 CrossRef CAS PubMed.
- H. Ton-That, G. Liu, S. K. Mazmanian, K. F. Faull and O. Schneewind, Purification and characterization of sortase, the transpeptidase that cleaves surface proteins of Staphylococcus aureus at the LPXTG motif, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 12424–12429 CrossRef CAS.
- P. Cossart and R. Jonquieres, Sortase, a universal target for therapeutic agents against Gram-positive bacteria?, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 5013–5015 CrossRef CAS.
- S. K. Mazmanian, G. Liu, E. R. Jensen, E. Lenoy and O. Schneewind, Staphylococcus aureus sortase mutants defective in the display of surface proteins and in the pathogenesis of animal infections, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 5510–5515 CrossRef CAS.
- L.-L. Zhou and C.-G. Yang, Chemical Intervention on Staphylococcus aureusVirulence, Chin. J. Chem., 2019, 37, 183–193 CrossRef CAS.
- S. Cascioferro, M. Totsika and D. Schillaci, Sortase A: An ideal target for anti-virulence drug development, Microb. Pathog., 2014, 77, 105–112 CrossRef CAS.
- S. Cascioferro, D. Raffa, B. Maggio, M. V. Raimondi, D. Schillaci and G. Daidone, Sortase A Inhibitors: Recent Advances and Future Perspectives, J. Med. Chem., 2015, 58, 9108–9123 CrossRef CAS.
- S. K. Mazmanian, H. Ton-That and O. Schneewind, Sortase-catalysed anchoring of surface proteins to the cell wall of Staphylococcus aureus, Mol. Microbiol., 2001, 40, 1049–1057 CrossRef CAS PubMed.
- T. C. Bolken, C. A. Franke, K. F. Jones, G. O. Zeller, C. H. Jones, E. K. Dutton and D. E. Hruby, Inactivation of the srtA gene in Streptococcus gordonii inhibits cell wall anchoring of surface proteins and decreases in vitro and in vivo adhesion, Infect. Immun., 2001, 69, 75–80 CrossRef CAS PubMed.
- X. C. Hou, M. N. Wang, Y. Wen, T. F. Ni, X. N. Guan, L. F. Lan, N. X. Zhang, A. Zhang and C. G. Yang, Quinone skeleton as a new class of irreversible inhibitors against Staphylococcus aureus sortase A, Bioorg. Med. Chem. Lett., 2018, 28, 1864–1869 CrossRef CAS.
- J. Wang, M. Song, J. Pan, X. Shen, W. Liu, X. Zhang, H. Li and X. Deng, Quercetin impairs Streptococcus pneumoniae biofilm formation by inhibiting sortase A activity, J. Cell. Mol. Med., 2018, 22, 6228–6237 CrossRef CAS PubMed.
- M. Song, Z. Teng, M. Li, X. Niu, J. Wang and X. Deng, Epigallocatechin gallate inhibits Streptococcus pneumoniae virulence by simultaneously targeting pneumolysin and sortase A, J. Cell. Mol. Med., 2017, 21, 2586–2598 CrossRef CAS PubMed.
- H. Li, Y. Chen, B. Zhang, X. Niu, M. Song, Z. Luo, G. Lu, B. Liu, X. Zhao, J. Wang and X. Deng, Inhibition of sortase A by chalcone prevents Listeria monocytogenes infection, Biochem. Pharmacol., 2016, 106, 19–29 CrossRef CAS PubMed.
- J. Wang, B. Liu, Z. Teng, X. Zhou, X. Wang, B. Zhang, G. Lu, X. Niu, Y. Yang and X. Deng, Phloretin Attenuates Listeria monocytogenes Virulence Both In vitro and In vivo by Simultaneously Targeting Listeriolysin O and Sortase A, Front. Cell. Infect. Microbiol., 2017, 7, 9 Search PubMed.
- L. Wang, G. Wang, H. Qu, K. Wang, S. Jing, S. Guan, L. Su, Q. Li and D. Wang, Taxifolin, an Inhibitor of Sortase A, Interferes With the Adhesion of Methicillin-Resistant Staphylococcal aureus, Front. Microbiol., 2021, 12, 686864 CrossRef.
- M. W. Ha, S. W. Yi and S. M. Paek, Design and Synthesis of Small Molecules as Potent Staphylococcus aureus Sortase A Inhibitors, Antibiotics, 2020, 9, 706 CrossRef CAS.
- J. E. Gosschalk, C. Y. Chang, C. K. Sue, S. D. Siegel, C. G. Wu, M. D. Kattke, S. W. Yi, R. Damoiseaux, M. E. Jung, H. Ton-That and R. T. Clubb, A Cell-based Screen in Actinomyces oris to Identify Sortase Inhibitors, Sci. Rep., 2020, 10, 8520 CrossRef CAS PubMed.
- F. Barthels, G. Marincola, T. Marciniak, M. Konhauser, S. Hammerschmidt, J. Bierlmeier, U. Distler, P. R. Wich, S. Tenzer, D. Schwarzer, W. Ziebuhr and T. Schirmeister, Asymmetric Disulfanylbenzamides as Irreversible and Selective Inhibitors of Staphylococcus aureus Sortase A, ChemMedChem, 2020, 15, 839–850 CrossRef CAS PubMed.
- A. H. Chan, S. W. Yi, E. M. Weiner, B. R. Amer, C. K. Sue, J. Wereszczynski, C. A. Dillen, S. Senese, J. Z. Torres, J. A. McCammon, L. S. Miller, M. E. Jung and R. T. Clubb, NMR structure-based optimization of Staphylococcus aureus sortase A pyridazinone inhibitors, Chem. Biol. Drug Des., 2017, 90, 327–344 CrossRef CAS.
- B. A. Frankel, M. Bentley, R. G. Kruger and D. G. McCafferty, Vinyl sulfones: Inhibitors of SrtA, a transpeptidase required for cell wall protein anchoring and virulence in Staphylococcus aureus, J. Am. Chem. Soc., 2004, 126, 3404–3405 CrossRef CAS PubMed.
- J. F. Wang, H. E. Li, J. Pan, J. Dong, X. Zhou, X. D. Niu and X. M. Deng, Oligopeptide Targeting Sortase A as Potential Anti-infective Therapy for Staphylococcus aureus, Front. Microbiol., 2018, 9, 00245 CrossRef.
- K. M. Connolly, B. T. Smith, R. Pilpa, U. Ilangovan, M. E. Jung and R. T. Clubb, Sortase from Staphylococcus aureus does not contain a thiolate-imidazolium ion pair in its active site, J. Biol. Chem., 2003, 278, 34061–34065 CrossRef CAS PubMed.
- C. J. Scott, A. McDowell, S. L. Martin, J. F. Lynas, K. Vandenbroeck and B. Walker, Irreversible inhibition of the bacterial cysteine protease-transpeptidase sortase (SrtA) by substrate-derived affinity labels, Biochem. J., 2002, 366, 953–958 CrossRef CAS.
- J. Zhang, H. C. Liu, K. K. Zhu, S. Z. Gong, S. Dramsi, Y. T. Wang, J. F. Li, F. F. Chen, R. H. Zhang, L. Zhou, L. F. Lan, H. L. Jiang, O. Schneewind, C. Luo and C. G. Yang, Antiinfective therapy with a small molecule inhibitor of Staphylococcus aureus sortase, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 13517–13522 CrossRef CAS.
- T. Yang, T. Zhang, X. N. Guan, Z. Dong, L. F. Lan, S. Yang and C. G. Yang, Tideglusib and Its Analogues As Inhibitors of Staphylococcus aureus SrtA, J. Med. Chem., 2020, 63, 8442–8457 CrossRef CAS PubMed.
- L. Wang, Q. Li, J. Li, S. Jing, Y. Jin, L. Yang, H. Yu, D. Wang, T. Wang and L. Wang, Eriodictyol as a Potential Candidate Inhibitor of Sortase A Protects Mice From Methicillin-Resistant Staphylococcus aureus-Induced Pneumonia, Front. Microbiol., 2021, 12, 635710 CrossRef.
- L. Wang, S. Jing, H. Qu, K. Wang, Y. Jin, Y. Ding, L. Yang, H. Yu, Y. Shi, Q. Li and D. Wang, Orientin mediates protection against MRSA-induced pneumonia by inhibiting Sortase A, Virulence, 2021, 12, 2149–2161 CrossRef CAS.
- B. Calamini, M. C. Silva, F. Madoux, D. M. Hutt, S. Khanna, M. A. Chalfant, S. A. Saldanha, P. Hodder, B. D. Tait, D. Garza, W. E. Balch and R. I. Morimoto, Small-molecule proteostasis regulators for protein conformational diseases, Nat. Chem. Biol., 2011, 8, 185–196 CrossRef.
- B. Calamini, M. C. Silva, F. Madoux, D. M. Hutt, S. Khanna, M. A. Chalfant, C. Allais, S. Ouizem, S. A. Saldanha, J. Ferguson, B. A. Mercer, C. Michael, B. D. Tait, D. Garza, W. E. Balch, W. R. Roush, R. I. Morimoto and P. Hodder, ML346: A Novel Modulator of Proteostasis for Protein Conformational Diseases, in Probe Reports from the NIH Molecular Libraries Program, National Center for Biotechnology Information (US), Bethesda (MD), 2010, ch. ML346: A Novel Modulator of Proteostasis for Protein Conformational Diseases Search PubMed.
- J. Baell and M. A. Walters, Chemistry: Chemical con artists foil drug discovery, Nature, 2014, 513, 481–483 CrossRef CAS PubMed.
- C. Aldrich, C. Bertozzi, G. I. Georg, L. Kiessling, C. Lindsley, D. Liotta, K. M. Merz, Jr., A. Schepartz and S. Wang, The Ecstasy and Agony of Assay Interference Compounds, ACS Med. Chem. Lett., 2017, 8, 379–382 CrossRef CAS PubMed.
- N. J. Greenfield, Using circular dichroism spectra to estimate protein secondary structure, Nat. Protoc., 2007, 1, 2876–2890 CrossRef.
- H. Ton-That and O. Schneewind, Anchor structure of staphylococcal surface proteins IV. Inhibitors of the cell wall sorting reaction, J. Biol. Chem., 1999, 274, 24316–24320 CrossRef CAS.
- W. J. Bradshaw, A. H. Davies, C. J. Chambers, A. K. Roberts, C. C. Shone and K. R. Acharya, Molecular features of the sortase enzyme family, FEBS J., 2015, 282, 2097–2114 CrossRef CAS.
- A. J. Barrett, A. A. Kembhavi, M. A. Brown, H. Kirschke, C. G. Knight, M. Tamai and K. Hanada, L-trans-Epoxysuccinyl-leucylamido(4-guanidino)butane (E-64) and its analogues as inhibitors of cysteine proteinases including cathepsins B, H and L, Biochem. J., 1982, 201, 189–198 CrossRef CAS.
- P. R. Race, M. L. Bentley, J. A. Melvin, A. Crow, R. K. Hughes, W. D. Smith, R. B. Sessions, M. A. Kehoe, D. G. McCafferty and M. J. Banfield, Crystal Structure of Streptococcus pyogenes Sortase A IMPLICATIONS FOR SORTASE MECHANISM, J. Biol. Chem., 2009, 284, 6924–6933 CrossRef CAS.
- A. W. Jacobitz, M. D. Kattke, J. Wereszczynski and R. T. Clubb, Sortase Transpeptidases: Structural Biology and Catalytic Mechanism, Adv. Protein Chem. Struct. Biol., 2017, 109, 223–264 CAS.
- N. Suree, C. K. Liew, V. A. Villareal, W. Thieu, E. A. Fadeev, J. J. Clemens, M. E. Jung and R. T. Clubb, The structure of the Staphylococcus aureus sortase-substrate complex reveals how the universally conserved LPXTG sorting signal is recognized, J. Biol. Chem., 2009, 284, 24465–24477 CrossRef CAS PubMed.
- T. J. Foster, J. A. Geoghegan, V. K. Ganesh and M. Hook, Adhesion, invasion and evasion: the many functions of the surface proteins of Staphylococcus aureus, Nat. Rev. Microbiol., 2014, 12, 49–62 CrossRef CAS PubMed.
- H. Ton-That, S. K. Mazmanian, K. F. Faull and O. Schneewind, Anchoring of surface proteins to the cell wall of Staphylococcus aureus - Sortase catalyzed in vitro transpeptidation reaction using LPXTG peptide and NH2-Gly(3) substrates, J. Biol. Chem., 2000, 275, 9876–9881 CrossRef CAS PubMed.
- L. Cedergren, R. Andersson, B. Jansson, M. Uhlen and B. Nilsson, Mutational analysis of the interaction between staphylococcal protein A and human IgG1, Protein Eng., 1993, 6, 441–448 CrossRef CAS.
- F. Falugi, H. K. Kim, D. M. Missiakas and O. Schneewind, Role of protein A in the evasion of host adaptive immune responses by Staphylococcus aureus, MBio, 2013, 4, e00575-00513 CrossRef.
- H. McCarthy, J. K. Rudkin, N. S. Black, L. Gallagher, E. O'Neill and J. P. O'Gara, Methicillin resistance and the biofilm phenotype in Staphylococcus aureus, Front. Cell. Infect. Microbiol., 2015, 5, 1 Search PubMed.
- R. M. Donlan and J. W. Costerton, Biofilms: survival mechanisms of clinically relevant microorganisms, Clin. Microbiol. Rev., 2002, 15, 167–193 CrossRef CAS PubMed.
- D. E. Moormeier and K. W. Bayles, Staphylococcus aureus biofilm: a complex developmental organism, Mol. Microbiol., 2017, 104, 365–376 CrossRef CAS.
- E. O'Neill, C. Pozzi, P. Houston, H. Humphreys, D. A. Robinson, A. Loughman, T. J. Foster and J. P. O'Gara, A novel Staphylococcus aureus biofilm phenotype mediated by the fibronectin-binding proteins, FnBPA and FnBPB, J. Bacteriol., 2008, 190, 3835–3850 CrossRef.
- B. Zhang, Z. Teng, X. Li, G. Lu, X. Deng, X. Niu and J. Wang, Chalcone Attenuates Staphylococcus aureus Virulence by Targeting Sortase A and Alpha-Hemolysin, Front. Microbiol., 2017, 8, 01715 CrossRef.
- S. K. Mazmanian, G. Liu, E. R. Jensen, E. Lenoy and O. Schneewind, Staphylococcus aureus sortase mutants defective in the display of surface proteins and in the pathogenesis of animal infections, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 5510–5515 CrossRef CAS.
- H. Tang, Regulation and function of the melanization reaction in Drosophila, Fly, 2014, 3, 105–111 CrossRef PubMed.
- C. J. Tsai, J. M. Loh and T. Proft, Galleria mellonella infection models for the study of bacterial diseases and for antimicrobial drug testing, Virulence, 2016, 7, 214–229 CrossRef CAS.
- A. P. Desbois and P. J. Coote, Wax moth larva (Galleria mellonella): an in vivo model for assessing the efficacy of antistaphylococcal agents, J. Antimicrob. Chemother., 2011, 66, 1785–1790 CrossRef CAS.
- J. H. Zhang, T. D. Chung and K. R. Oldenburg, A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays, J. Biomol. Screening, 1999, 4, 67–73 CrossRef PubMed.
- J. M. Strelow, A Perspective on the Kinetics of Covalent and Irreversible Inhibition, SLAS Discovery, 2017, 22, 3–20 CrossRef CAS.
- R. Ettari, E. Nizi, M. E. Di Francesco, M. A. Dude, G. Pradel, R. Vicik, T. Schirmeister, N. Micale, S. Grasso and M. Zappala, Development of peptidomimetics with a vinyl sulfone warhead as irreversible falcipain-2 inhibitors, J. Med. Chem., 2008, 51, 988–996 CrossRef CAS.
- F. Yu, Q. Wang, M. Li, H. Zhou, K. Liu, K. Zhang, Z. Wang, Q. Xu, C. Xu, Q. Pan and J. He, Aquarium: an automatic data-processing and experiment information management system for biological macromolecular crystallography beamlines, J. Appl. Crystallogr., 2019, 52, 472–477 CrossRef CAS.
- G. Winter, C. M. C. Lobley and S. M. Prince, Decision making inxia2, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 1260–1273 CrossRef CAS PubMed.
- P. Emsley and K. Cowtan, Coot: model-building tools for molecular graphics, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2126–2132 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1md00316j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |