
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInterlocking dendritic fibrous nanosilica into microgranules by polyethylenimine assisted assembly: in situ neutron diffraction and CO2 capture studies†
Jitendra
Bahadur
 *ab,
Swati
Mehta
ab,
Saideep
Singh
c,
Avik
Das
ab,
Ayan
Maity
c,
Tristan
Youngs
d,
Debasis
Sen
ab and
Vivek
Polshettiwar
*c
*ab,
Swati
Mehta
ab,
Saideep
Singh
c,
Avik
Das
ab,
Ayan
Maity
c,
Tristan
Youngs
d,
Debasis
Sen
ab and
Vivek
Polshettiwar
*c
aSolid State Physics Division, Bhabha Atomic Research Centre, Mumbai, 400085, India. E-mail: jbahadur@barc.gov.in
bHomi Bhabha National Institute, Anushaktinagar, Mumbai, 400094, India
cDepartment of Chemical Sciences, Tata Institute of Fundamental Research, Mumbai-400005, India
dDisordered Materials Group (ISIS), STFC Rutherford Appleton Laboratory, Oxfordshire, UK. E-mail: vivekpol@tifr.res.in
First published on 5th July 2022
Abstract
Solid amine-based nano-adsorbents have shown tremendous potential for mitigating CO2 emissions. The conventional approaches for achieving these nano-adsorbents utilize the loading of the amines in well-defined mesopores, which face several challenges, including pore blocking and slow adsorption kinetics. In this work, we report an evaporation induced assembly approach to achieve dendritic fibrous nano-silica (DFNS)–polyethylenimine (PEI) microgranules using a mixed colloidal dispersion of nanometer-sized DFNS particles and PEI. The PEI incorporated DFNS microgranules were studied using small-angle X-ray scattering, electron microscopy, and N2 gas adsorption techniques in a detailed fashion. Two-dimensional fast Fourier transform (2D FFT) of the high-resolution micrographs shows an intriguing order to disorder transition in the jamming of DFNS in the presence of PEI. This disordered jamming of DFNS led to the formation of voids, causing increased accessibility of DFNS internal pores. Furthermore, the interstices between the jammed DFNS in microgranules provided additional space to immobilize more PEI molecules, which was not possible in bare DFNS particles. The CO2 adsorption characteristics have been found to be excellent with good regeneration capability up to 50 cycles due to the unique morphology of the DFNS microgranules and strong PEI confinement. The CO2 specific interaction of amine sites in PEI allows high selectivity of CO2 adsorption against N2 and H2O except at low temperatures. The fast kinetics of DFNS–PEI was attributed to the connectivity between mesopores and macropores as evident from in situ neutron diffraction studies, which provided crucial experimental evidence of the connectivity of mesopores and macropores for the first time, refining the enigmatic DFNS structure.
1. Introduction
The burning of fossil fuels remains a major energy source across the world despite the tremendous efforts in the area of renewable energy sources. The anthropogenic carbon dioxide emission from fossil fuel-based sources is causing global climate change.1,2 CO2 capture and sequestration (CCS) are one of the major strategies for mitigating climate change caused by the increasing concentration of CO2 in the atmosphere.3–5 The post-combustion carbon capture at the large-scale sources is considered the best approach to reduce the CO2 footprint. The current state-of-art technologies based on liquid amine solutions are prevalent on industrial scales due to their selectivity and ability to operate at a low partial pressure of CO2 even in the presence of water which exists in the flue gases of the power plants.6 However, liquid amine-based technologies possess several drawbacks such as corrosion, thermal degradation, volatility, and high heat capacity, causing a large amount of energy for regenerations.7 To overcome the issues related to liquid amines, solid adsorbents loaded with amines have become a viable, attractive alternative for post-combustion carbon capture.8–13 The reduced heat capacities of solid adsorbents lead to a significant decrease in the energy penalty, keeping the excellent selectivity for CO2 even in the presence of moisture and thus, suitable for carbon capture at low-pressure prevalent in the flue gases. Due to these unique properties of solid-adsorbents, they are still at the forefront of the CCS technologies.14Several approaches15,16 exist to achieve amine-based solid adsorbents, among which physical impregnation17,18 and grafting methods19,20 are extensively used. The impregnation and grafting of amines are carried out in porous materials such as SBA-1521 and MCM-41.18 Although these solid adsorbents show good CO2 adsorption characteristics such as fast kinetics and reduced operating temperature, several issues remain unresolved with regard to their practical applications.22,23 The major issue in the physical impregnation is the blockage of cylindrical pores leading to a decrease in the accessibility of active amine sites for CO2 capture.24,25 Another issue is caused by thermal degradation of the amine molecules in the case of physical impregnation, although covalent grafting methods offer to tackle this issue but with reduced loading capacity.26
To address the accessibility issues in the amine incorporated porous materials, one needs to tune the pore characteristics in such a way that both mesopores and macropores exist simultaneously. The recently discovered dendritic fibrous nano-silica (DFNS)27 possesses a unique bimodal pore size distribution consisting of both mesopores and macropores.28 Using DFNS, we recently developed an amine based sorbent with good CO2 capture capacity, kinetics, and stability.29 However, due to the nano sizes of sorbents, their practical application was limited. In most practical CO2 capture applications, the fluid-bed or fixed-bed reactor configurations are used where a significant pressure drop in the flow is observed because of the significant low permeability of the powered column due to the smaller size of the grains.30,31 Furthermore, the mechanical strength and bulk density of the highly porous small-sized grains pose another hindrance to optimal use. Also, the finite pore volume of the DFNS defines the theoretical loading capacity of amines. The loading beyond actual pore capacity leads to amine loading at the external surface of the pores, which is responsible for poor thermal stability. To solve this, one needs to devise novel approaches where the amine molecules present on the external pore surface are sufficiently immobilized to avoid degradation at elevated temperatures. Therefore, a one-step one-pot process is needed to immobilize the PEI in porous nanoparticles like DFNS and their granulation to micron-size powder particles, using techniques like evaporation-induced assembly (EIA).
EIA has been an established technique to immobilize organic components such as polymers,32 surfactants,33 drug molecules, and cells34,35 in the inorganic matrix formed by jammed nanoparticles. This process also results in the granulation of the smaller nanoparticles into micron-sized grains. The micrometric droplets of an aqueous dispersion of nanoparticles along with the organic components undergo drying and subsequent shrinkage of the droplets in the course of drying leads to microgranules where the organic part is immobilized. The jamming behaviour of the nanoparticles and the morphology of the microgranules depend on the interaction between the different components, drying time, and the diffusion of nanoparticles during drying36,37 EIA offers great tunability in nanoparticle jamming and also in the immobilization of organic molecules.
In the present work, we used EIA to convert DFNS nanoparticles into PEI-incorporated microgranules in a one-step process. The effect of PEI loading on the morphology of the DFNS microgranules and their internal structures has been probed using electron microscopy and small-angle X-ray scattering. The pore characteristics of microgranules with and without PEI loading are studied using N2 adsorption isotherms. The CO2 capture performance of PEI-loaded microgranules was determined using volumetric adsorption and thermogravimetric measurements under CO2 flow. Furthermore, in situ neutron diffraction was carried out to probe the connectivity of the mesopores and macropores and the confinement behavior of physically adsorbed CO2 in the mesopores.
2. Experimental
2.1 DFNS preparation
To see the effect of size and pore size distribution of the DFNS particles on the PEI incorporation during assembly, two distinct-sized DFNS particles were chosen for the experiments. The DFNS synthesis procedure and the important synthesis parameters are shown in the ESI† (Table S1). The size distributions of the DFNS particles are shown in Fig. S1 (ESI†). The typical sizes of the synthesized DFNS particles were estimated and found to be ∼50 nm and 170 nm and accordingly, these were named DFNS-S and DFNS-L depending on their smaller or larger size, respectively.2.2 Spray drying
Spray drying of 1 wt% dispersions of DFNS-S and DFNS-L was carried out using a laboratory spray dryer, LU228 (Lab Ultima, Mumbai, India). The inlet temperature was fixed at 170 °C and the feed rate was maintained at 2 mL min−1. The aspiration rate was fixed at 50 m3 h−1. A two-fluid jet nozzle was used for generating micron size droplets. The atomization pressure was set to 2 kg cm−2. The obtained microgranules consisting of DFNS-S and DFNS-L particles are labelled as DFNS-SM and DFNS-LM, respectively. A schematic diagram of the spray dryer is shown in schematic S1 of the ESI.†Again, the 1 wt% dispersion of DFNS-S and DFNS-L were prepared. After that 1 wt% branched PEI of molecular weight 800 kg mol−1 (Sigma Aldrich) was added and sonication was performed. The resulting dispersions were spray-dried at the identical parameters used for previous experiments. The PEI incorporated spray-dried microgranules of DFNS-S and DFNS-L were named DFNS-SM-PEI and DFNS-LM-PEI, respectively.
2.3 Fourier transform infrared spectroscopy and thermogravimetric analysis
FTIR spectra of the DFNS–PEI microgranules were recorded in transmission mode using a JASCO FT/IR-4700 instrument. Thermogravimetric measurements of the DFNS–PEI microgranules were performed on Mettler Toledo TGA/DSC2 apparatus. Prior to the measurements the sample was heated for 15 minutes at 90 °C in order to remove moisture from the sample. Then 15 mg of sample was heated in an alumina pan from ambient temperature to 500 °C at a heating rate of 10 °C min−1 and an air flow rate of 100 mL min−1.2.4 Field emission scanning electron microscopy
Field emission scanning electron microscopy (FESEM) was carried out using a GEMINISEM 300 (Zeiss make) microscope to probe the morphology of the microgranules. To observe the surface morphology of the microgranules, imaging was carried out at a relatively low accelerating voltage of 2 kV.2.5 Small-angle X-ray scattering (SAXS)
Small-angle X-ray scattering (SAXS) measurements on the DFNS microgranules have been carried out using an SWAXS beamline (BL-18)38 at Indus-2 synchrotron, India. The experiments were carried out with an X-ray energy of 12 keV and sample to detector distance was tuned to nearly 6 m for the desired accessible angular range. The scattering intensity is represented as a function of wave vector transfer q that is defined as 4π sin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ/λ, 2θ is the scattering angle and λ is the wavelength of the X-rays. SAXS measurements on the DFNS samples before and after spray drying was measured using a laboratory-based SAXS facility with an accessible q-range of 0.1 to 2.5 nm−1.
θ/λ, 2θ is the scattering angle and λ is the wavelength of the X-rays. SAXS measurements on the DFNS samples before and after spray drying was measured using a laboratory-based SAXS facility with an accessible q-range of 0.1 to 2.5 nm−1.
2.6 N2 sorption studies
The nitrogen adsorption–desorption isotherms were measured at −196 °C with a Micromeritics Flex 3 surface area and porosity analyzer. Before the measurements, the samples were degassed under vacuum at 80 °C for 12 h in an external degasser and then for 2 h in situ.2.7 In situ small and wide-angle neutron diffraction
In situ measurements were carried out on spray-dried DFNS-LM and DFNS-LM-PEI microspheres during CO2 adsorption at various gas pressures using a Near and Inter Mediate Range Order Diffractometer (NIMROD) at the ISIS Pulsed Neutron and Muon Facility, STFC Rutherford Appleton Laboratory, UK.39,40 This instrument covers both the small and wide angular ranges of 0.5 to 40 degrees. The accessible q-range in the instrument is 0.02 to 50 Å−1 which provides both traditional SANS and wide-angle neutron scattering data. The samples were held in a null scattering Ti0.676Zr0.324 pressure cell with a rating of ∼100 bar and sealed using indium wire. CO2 gas is introduced into the evacuated sample in a controlled manner using a gas handling panel and the neutron scattering profiles were recorded as a function of pressure. All the measurements were performed at 22 °C. The samples were outgassed overnight at ∼100 °C before the measurements. The outgassed sample under vacuum was measured first. Subsequently, the scattering data are recorded under CO2 pressurization as a function of pressure. The scattering patterns were corrected for instrument and container backgrounds, absorption, and multiple scattering, and finally normalized to standard vanadium using the Gudrun software.412.8 Volumetric CO2 adsorption
The volumetric CO2 adsorption measurements on the outgassed PEI-loaded DFNS microgranules were carried out using a Micromeritics Flex 3 at various temperatures. 200 mg of DFNS–PEI was taken in a glass tube and degassed at 120 °C for 12 h.2.9 Thermogravimetric CO2 adsorption–desorption
Thermogravimetric CO2 adsorption–desorption measurements were performed using a Mettler Toledo TGA/DSC2 system. In a typical experiment, ∼50 mg of the DFNS–PEI were placed in a 450 μL Alumina pan (modified for shorter pan-wall height). The DFNS–PEI microgranules were heated to 110 °C in N2 (100 mL min−1) for 2 h to remove the adsorbed ambient moisture and/or CO2. Then, the temperature was reduced to 50 °C, which was maintained for 30 min to stabilize the weight. Subsequently, the gas flow rate was switched to 150 mL min−1 of 15% CO2 (in nitrogen) and maintained for 1 h. For desorption and regeneration, the DFNS–PEI sample was heated to 110 °C under a N2 flow rate of 150 mL min−1. The experimental methodology for the adsorption–desorption measurement is depicted in schematic S2 in the ESI.† For CO2 capture in the presence of moisture, CO2 is passed through a bubbler during TGA measurements under flow. An experimental setup is shown in the ESI.† For the recyclability study, the adsorption at 90 °C was done for 60 min under 15% CO2 in N2 (150 mL min−1). Desorption at 110 °C was done for 10 min under N2 (100 mL min−1). Adsorption–desorption (at 110 °C) cycles were repeated 50 times.3. Results and discussion
3.1 Structure of the DFNS–PEI microgranules
Fig. 1 shows the electron micrographs of the DFNS-SM, DFNS-SM-PEI, and DFNS-LM, DFNS-LM-PEI microgranules at different magnifications. Spherical-shaped microgranules with jammed DFNS particles are observed. The jamming behaviour of the DFNS in the presence of PEI is significantly different compared to the jamming of the DFNS in the absence of PEI. Jamming of the DFNS particles is highly ordered in the bare DFNS microgranules, whereas disordered jamming is observed for PEI incorporated microgranules. This order to disorder transition during jamming is seen for both smaller and larger DFNS particles. Fig. 2 depicts the 2D fast Fourier transform (FFT) analysis of the high-resolution micrographs using the ImageJ software.42 The 2D FFT of the ordered structure shows a ring-like feature indicating strong ordering of the DFNS particles whereas this feature disappears in PEI loaded microspheres, and instead, a diffuse pattern is seen in the low momentum regime due to the clustering of the DFNS particles. The 2D FFT profile of the high-resolution micrographs of DFNS-S microgranules shows a similar trend (Fig. 2). | ||
| Fig. 1 FESEM micrographs of (a–c) DFNS-LM microspheres, (d–f) DFNS-LM-PEI microspheres, (g–i) DFNS-SM microspheres and (j–l) DFNS-SM-PEI microspheres. | ||
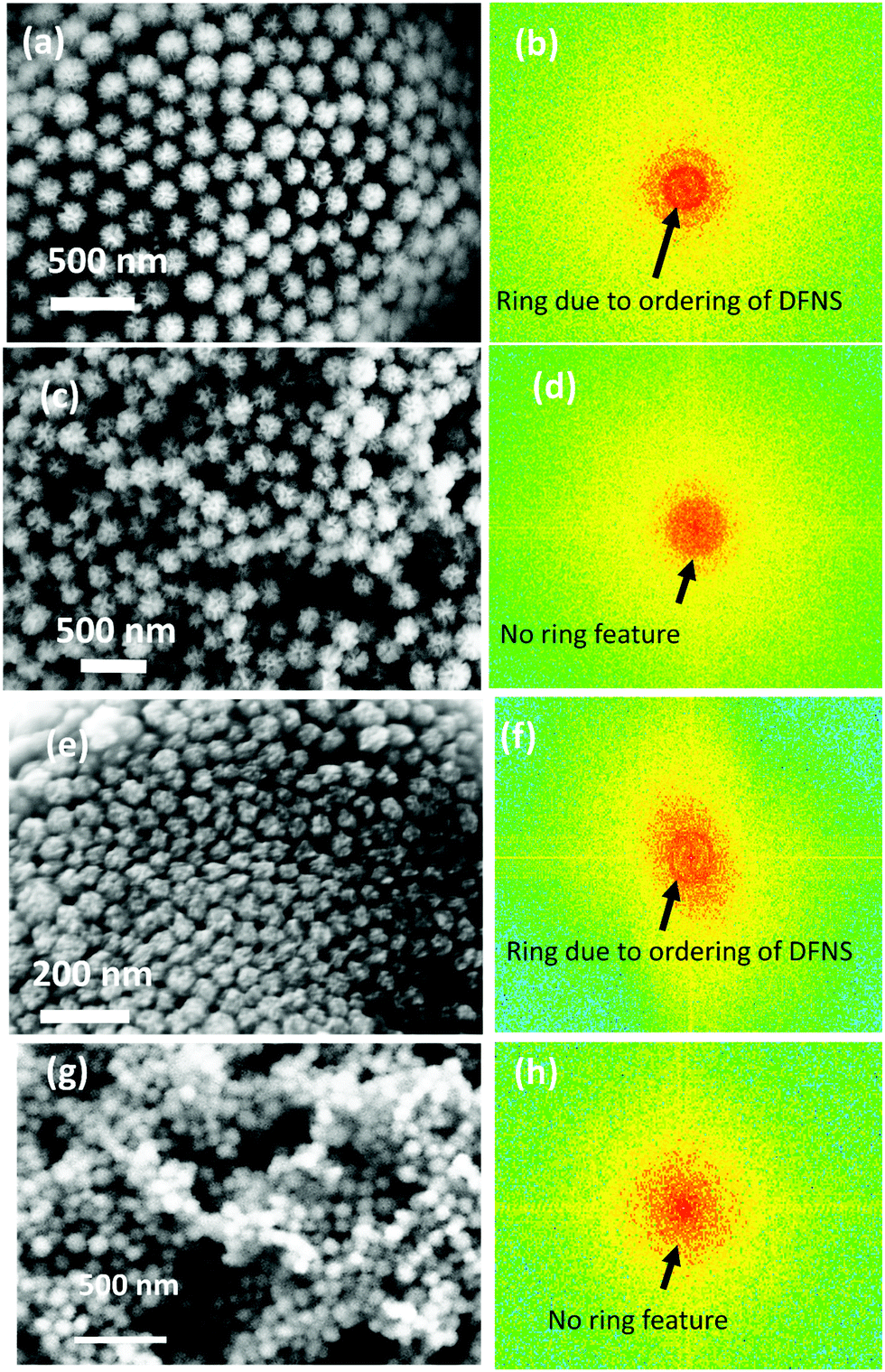 | ||
| Fig. 2 Magnified FESEM micrographs of the (a) DFNS-LM microspheres, (c) DFNS-LM-PEI microspheres, (e) DFNS-SM microspheres, and (g) DFNS-SM-PEI microspheres. (b, d, f and h) Fast Fourier transform (FFT) analysis of the respective DFNS particles in the microspheres. | ||
3.2 Physical and chemical properties of DFNS–PEI microgranules
The N2 adsorption isotherm and pore size distribution of the DFNS particles prior to spray drying are shown in Fig. S2 (ESI†). The Brunauer–Emmett–Teller (BET) surface area and pore volume of the DFNS particles are shown in Table S2 (ESI†). The BJH (Barrett, Joyner, and Halenda) pore size distributions of the DFNS-S and DFNS-L particles (Fig. S2, ESI†) show distinct functionality, although their specific surface area and pore volume are not much different. The number density of macropores in DFNS-L particles was more compared to that in DFNS-S particles. The N2 adsorption isotherms of the DFNS microgranules obtained after spray drying is shown in Fig. 3. As there is no plateau or inflection point indicating the adsorption saturation in the high-pressure region of the curve, that means the isotherms tend to be more type-II rather than type-IV. Type-II with a hysteresis loop of the type H3 adsorption–desorption isotherm suggests the presence of macropores in the microspheres. Furthermore, the presence of hysteresis loops, as evident in Fig. S3 of the ESI,† indicates the existence of mesopores.43 The shape of the adsorption branch indicates unrestricted monolayer-multilayer adsorption. The existence of the hysteresis loop in the multilayer physisorption range can be attributed to capillary condensation. The estimated BET-specific surface areas for the DFNS-S and DFNS-L particles before spray drying are ∼841 and 807 m2 g−1, which are reduced slightly to 746 and 738 m2 g−1 for the respectively assembled microgranules. Similarly, a slight reduction in the pore volume is also observed in the DFNS microgranules. The small reduction in the specific surface area and pore volume in the microgranules is caused by the contacts formed between the DFNS particles during assembly.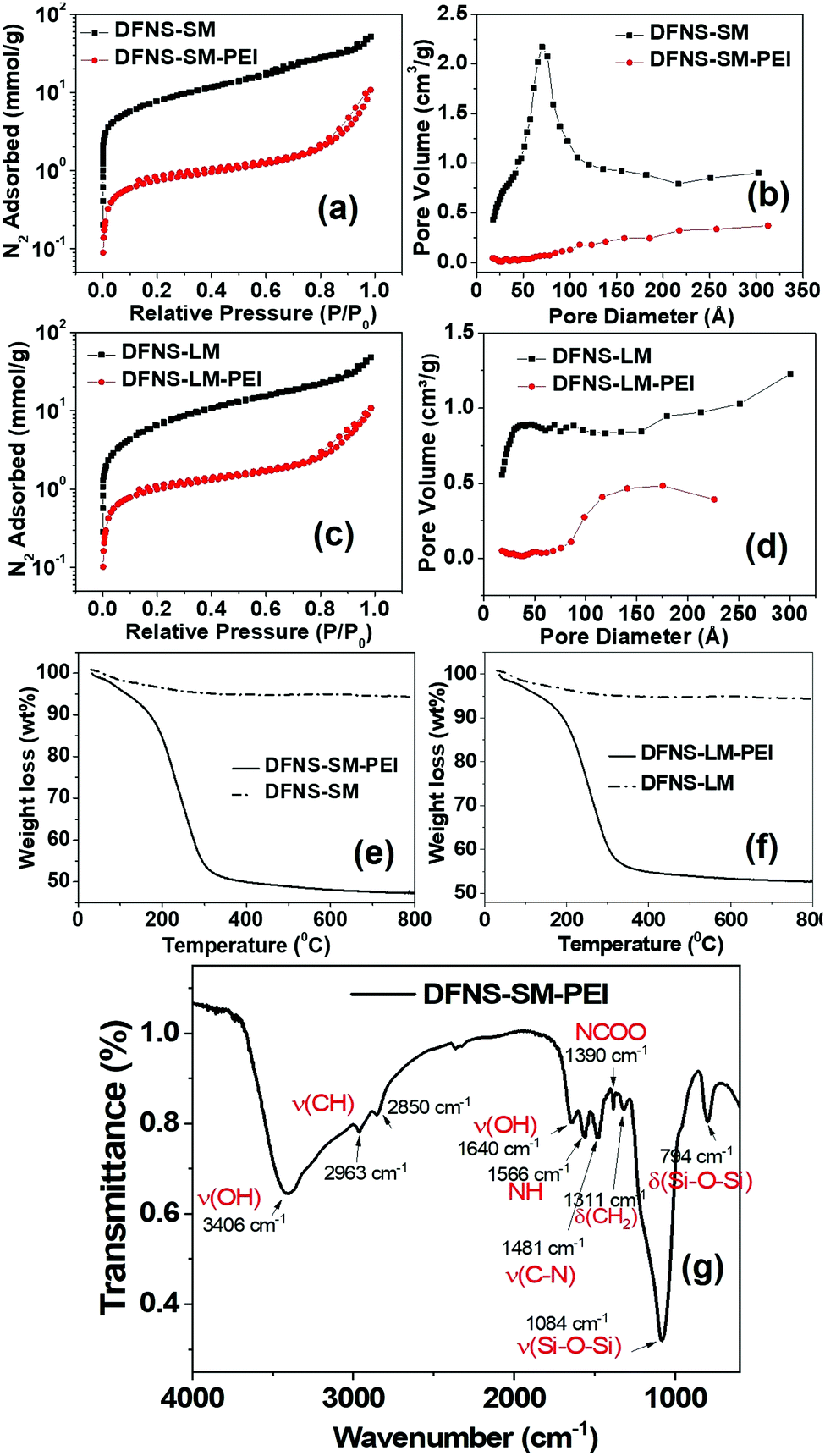 | ||
| Fig. 3 (a and c) N2 adsorption isotherm and (b and d) pore size distribution of DFNS-SM and DFNS-LM, with and without PEI incorporation. TGA profile of (e) DFNS-SM-PEI and (f) DFNS-LM-PEI microgranules, (g) FTIR spectra of the DFNS-SM-PEI microgranules depicting the functional groups of the silica and PEI. | ||
The N2 adsorption isotherms alter significantly for PEI incorporated microgranules due to the incorporation of PEI molecules in the pores (Fig. 3). The BET-specific surface area for the DFNS-S-PEI and DFNS-L-PEI microgranules decreases significantly to 60 and 80 m2 g−1 after PEI incorporation. The BJH pore size distribution shows a significant reduction in porosity as depicted in Fig. 3. The pore volumes of the DFNS-S and DFNS-L microgranules without PEI are found to be 1.30 and 1.15 cm3 g−1, respectively. After PEI incorporation, the pore volumes are reduced to 0.18 and 0.23 cm3 g−1, respectively. The thermogravimetric analysis of the PEI incorporated microgranules is shown in Fig. 3(e and f). DFNS-SM and DFNS-LM show an initial ∼3% loss (below 100 °C) which was attributed to the evaporation of the water confined in the interstices of jammed nanoparticles. The weight loss of ∼50% at higher temperatures in both the microgranules loaded with PEI was due to the volatilization of PEI, which corroborates the weight of the DFNS and PEI used during spray drying.
The Fourier transform infrared (FTIR) spectrum of DFNS-SM-PEI microgranules showing the functional groups of the silica and PEI is depicted in Fig. 3(g). The bands at 794 cm−1 and 1084 cm−1 arise from the bending and stretching of the silanol groups. Due to the incorporation of PEI, the presence of asymmetric and symmetric stretching vibrations of CH2 is observed at 2963 cm−1 and 2850 cm−1, whereas bending is observed at 1311 cm−1. The bands arising at 1566 cm−1 and 1390 cm−1 are due to the NH and NCOO vibrations.44 The bands from –OH groups44 are observed at 3400 cm−1 and 1640 cm−1. The presence of characteristic amine groups and carbon groups in FTIR spectra confirms the incorporation of PEI in the microgranules.
The SAXS profiles of the DFNS before and after spray drying are shown in Fig. S4 of the ESI.† The profiles remain identical, indicating that the internal pore structure of the DFNS does not change during spray drying. The SAXS profiles of DFNS microspheres before and after PEI incorporation are shown in Fig. 4. A complex DFNS pore structure has been recently unravelled using time-resolved SAXS and N2 adsorption isotherms and it has been established that the DFNS structure originated by modulation of the bicontinuous microemulsion during synthesis.28 The scattering profile from a bicontinuous structure is described by the Teubner–Strey phenomenological model45 as shown in the ESI.† The estimated particle size of DFNS-S particles matches quite well with that obtained by SEM image analysis, as shown in Fig. S2 (ESI†). The important parameters obtained from the analysis of the SAXS profiles are shown in Table S3 (ESI†). The internal pore structure of DFNS does not change much on PEI loading except for the contrast factors η2 which confirm the incorporation of PEI molecules inside pores.
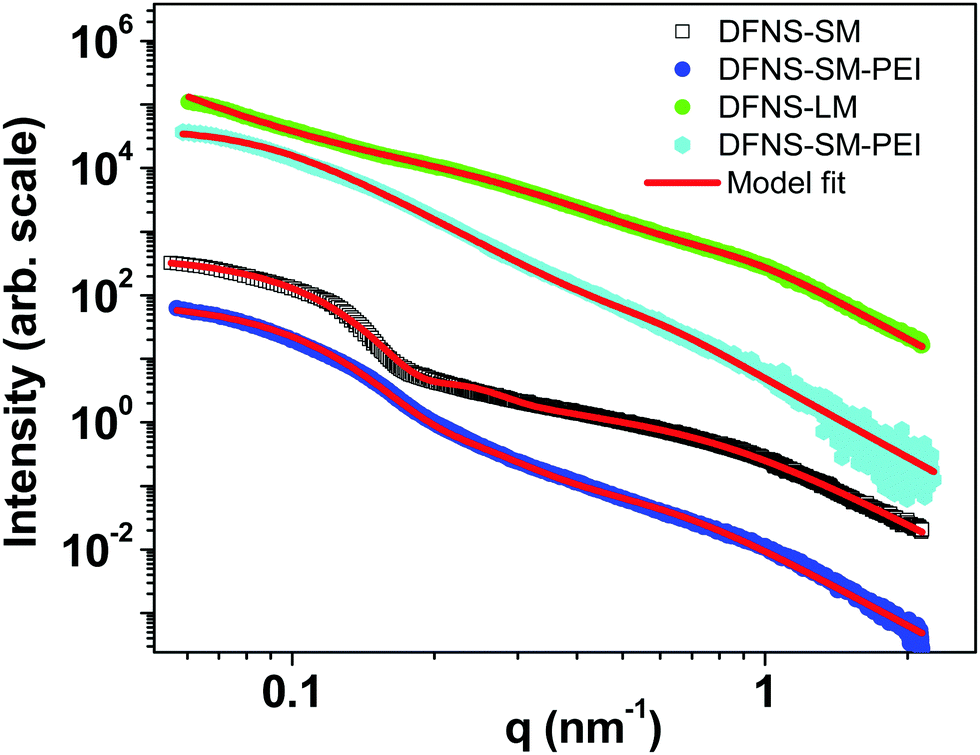 | ||
| Fig. 4 SAXS profiles of the DFNS microspheres with and without PEI loading. | ||
3.3 Formation mechanism of DFNS–PEI microgranules via evaporation-induced assembly
Fig. 5 shows the schematic of the formation mechanism of DFNS microgranules and PEI incorporation in the evaporation-induced assembly process. DFNS particles contain significant porosity. Thus, PEI adsorbs into the pores in the aqueous dispersion through diffusion. Some fraction of molecules also adsorbed on the outer surface of the DFNS, while some PEI molecules remained non-adsorbed. The DFNS particles assembled in micron-sized microspheres during shrinkage of the droplet in the drying step of evaporation-induced assembly.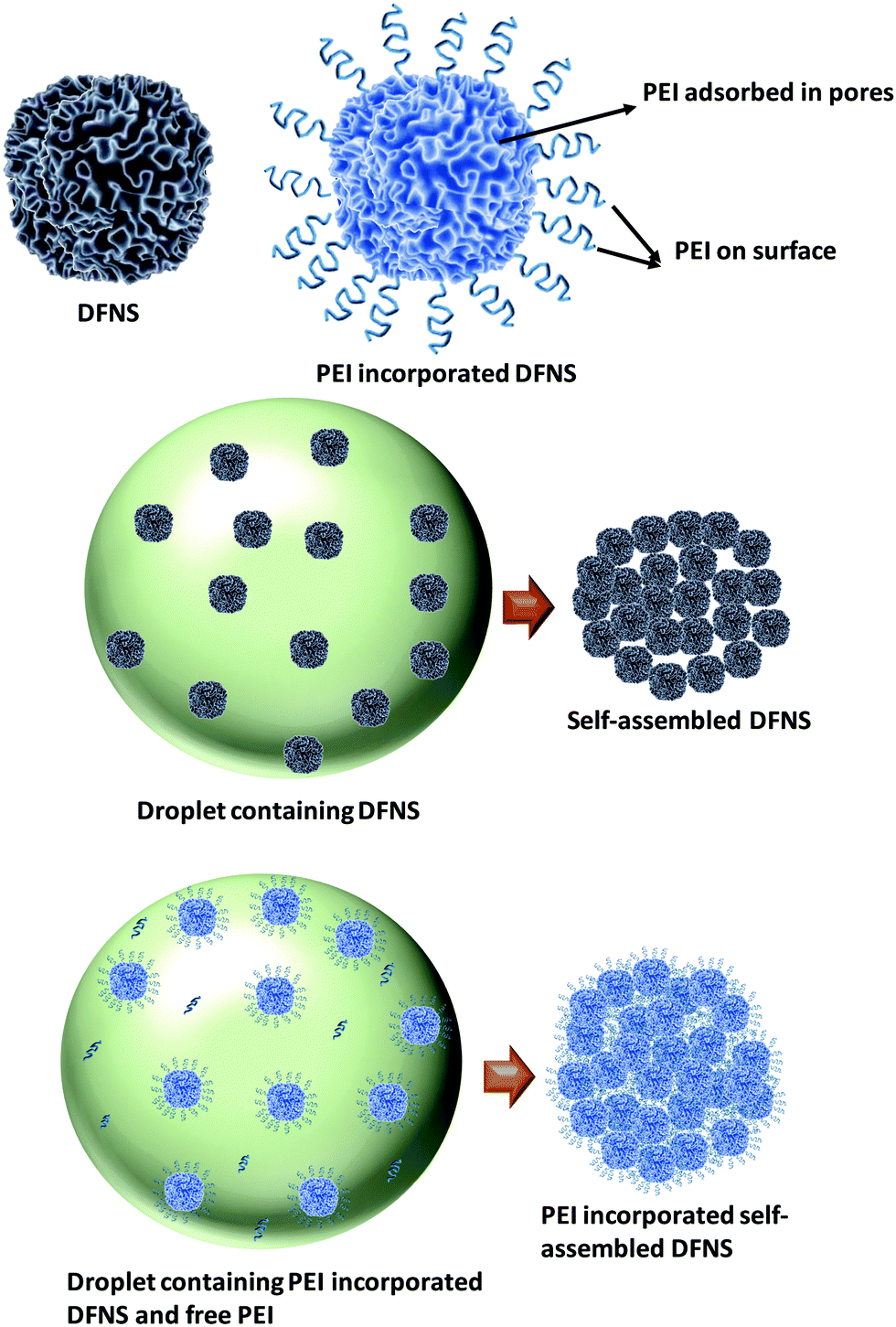 | ||
| Fig. 5 Schematic showing PEI incorporation through evaporation-induced assembly. | ||
The DFNS particles are assembled in an ordered manner due to their well-defined size and shape. The PEI incorporated DFNS also undergoes an assembly process during drying; however, their jamming behaviour is random, as seen in the FFT analysis of the electron micrograph (Fig. 2). The jamming behaviour is governed by the interaction between the DFNS nanoparticles. The DFNS particles in an aqueous solution acquires a negative charge46 at pH > pHPZC [pHPZC = 3.0] for silica where PZC is defined as the point of zero charge, leading to a stable dispersion. The pH and zeta potential of the PEI solution are estimated to be ∼11 and 5 mV, respectively. In the presence of PEI molecules, the net repulsion forces between the DFNS nanoparticles decrease due to the screening of the charge by PEI molecules. The morphology of the drying particles is governed by the interplay of interfacial and diffusive forces during the drying. As the drying is fast, therefore, shrinkage of the droplet is large compared to the diffusion of the nanoparticles causing an increase in the concentration at the air–water interface.47 The capillary force between the nanoparticles at the air–water interface brings particles together during drying due to meniscus formation, whereas the coulombic repulsion force repels nanoparticles. The strong repulsion force between nanoparticles is responsible for ordered assembly, whereas the reduced charge on the DFNS nanoparticles in the presence of PEI leads to random jamming. Thus, the residual negative charge on the DFNS nanoparticles offers weak coulombic repulsion and the van der Waals attraction locks the particles in the primary minima of the van der Waals potential.37 PEI adsorbed in the internal pores of the DFNS are immobilized even before their assembly; however, the remaining unadsorbed PEI molecules are immobilized due to jamming of the DFNS after assembly, which is otherwise not possible in the conventional approach of impregnation. The loosely bound PEI molecules at the external surface of the DFNS also get strongly immobilized during the jamming process. Furthermore, the disordered jamming of DFNS nanoparticles in PEI incorporated microgranules offers the advantage of increased permeability due to the presence of voids of a few hundred nanometres.
3.4 CO2 capture performance of DFNS–PEI microgranules
CO2 capture performance (capacity, kinetics and stability/regenerability) of the DFNS microgranules and PEI incorporated DFNS microgranules has been evaluated using temperature-dependent volumetric adsorption studies. Fig. 6(a–d) shows the CO2 adsorption isotherms of the bare DFNS and PEI incorporated microgranules at different temperatures. The capture capacity of PEI incorporated microgranules is large even at very low pressure compared to the bare DFNS microgranules. Furthermore, the capture capacity increases with temperature in PEI incorporated microgranules, whereas an opposite trend is observed for microgranules without PEI incorporation. This indicates the suitability of these DFNS–PEI microgranules for CO2 capture from the air, where the CO2 concentration is quite low. The CO2 capture in PEI incorporated microgranules occurs through the chemisorption process at active amine sites, whereas physisorption is the primary mechanism in bare DFNS microgranules.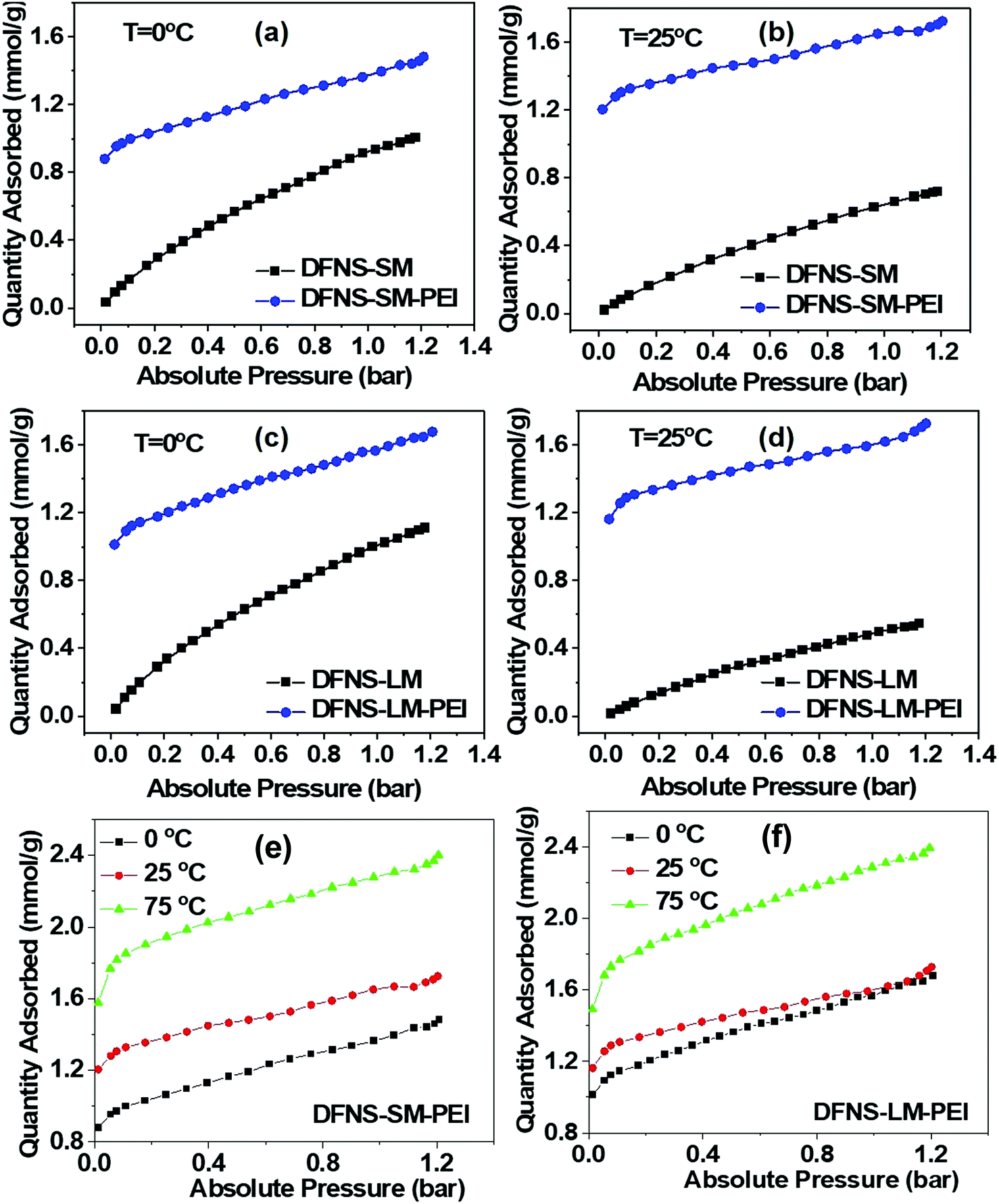 | ||
| Fig. 6 Comparison of CO2 adsorption of the DFNS microgranules with and without PEI loading at 0 °C (a and c) and 25 °C (b and d). Temperature-dependent volumetric CO2 adsorption of (e) DFNS-SM-PEI and (f) DFNS-LM-PEI microgranules. | ||
Fig. 6(e and f) shows the volumetric adsorption of the PEI-loaded DFNS microspheres. Both PEI-loaded DFNS-SM-PEI and DFNS-LM-PEI microspheres show increased CO2 capture at a higher temperature. DFNS-LM-PEI shows higher adsorption at a low temperature and pressure compared to DFNS-SM-PEI microgranules due to the larger residual surface area and porosity after PEI incorporation providing increased accessibility to the active amine sites for CO2 molecules. The maximum sorption capacity was found to be ∼2.4 mmol g−1 which is quite good8,29,30,48–52 for practical application as a solid adsorbent. A table for the comparison of various sorbents with the conditions matching those in the present study is shown in the ESI† (Table S4). The temperature dependence on adsorption capacity shows Arrhenius type behavior as illustrated in Fig. S5 (ESI†).
To evaluate the performance of PEI-loaded microspheres for capturing CO2 from flue gases, thermogravimetric measurements under the flow of CO2 have been carried out. Fig. 7 shows the adsorption kinetics of the CO2 at various temperatures. It is evident from Fig. 7b and d that adsorption kinetics is quite fast and the saturation adsorption capacity is achieved in less than one minute. Furthermore, the uptake capacity is also quite significant under flow conditions. The entire thermal cycle during TGA analysis for the CO2 capture of PEI incorporated microspheres is shown in Fig. S6 (ESI†). The heat flow during the adsorption and desorption processes of CO2 has been illustrated in the differential thermogravimetric analysis (DTA) profiles. The peak height corresponding to the desorption peak is significantly less than the peak height of the adsorption peak. This shows that the desorption process is less energy-intensive, which is an important requirement for practical application of solid adsorbents.
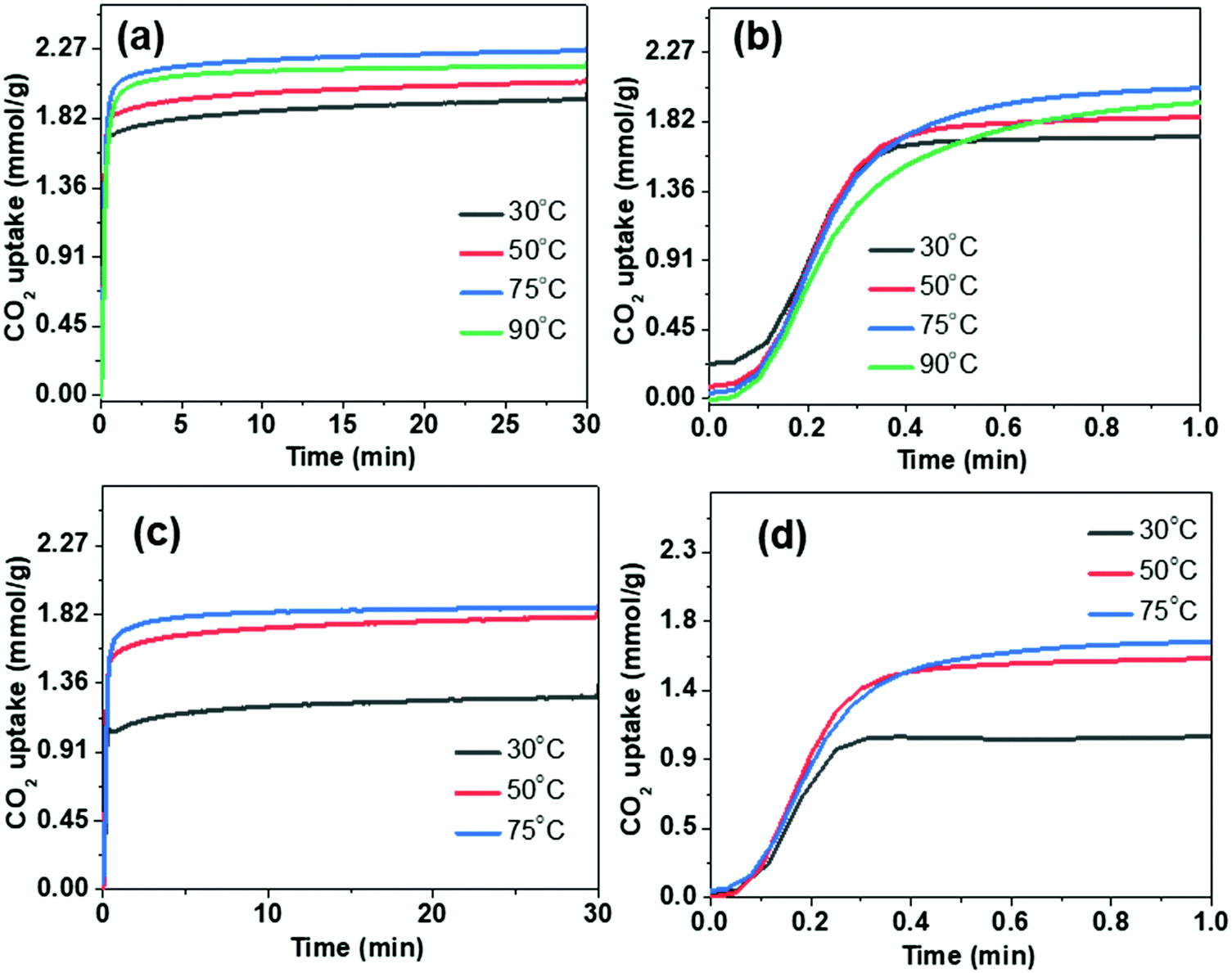 | ||
| Fig. 7 Thermogravimetric CO2 capture study under the flow conditions using DFNS–PEI microspheres: (a and b) DFNS-SM-PEI and (c and d) DFNS-LM-PEI. | ||
Furthermore, the effect of moisture and N2 on the CO2 capture study was carried out using TGA under flow conditions. The adsorption of N2 for both DFNS–PEI-SM and DFNS–PEI-LM samples at all the temperatures is very negligible, as shown in Fig. S7a and b in the ESI.†Fig. 8a shows the effect of moisture on CO2 capture. The moisture uptake is carried out by studying the adsorption of N2 gas bubbled through water and subtracting the uptake of dry N2. It is found that moisture uptake is significantly high at a low temperature of 30 °C. The moisture uptake decreases rapidly at higher temperatures of 50, 75 and 90 °C (Fig. 8b). CO2 uptake is affected only at 30 °C, while at higher temperatures (typical flue gas conditions)10,53 CO2 uptake did not change significantly.
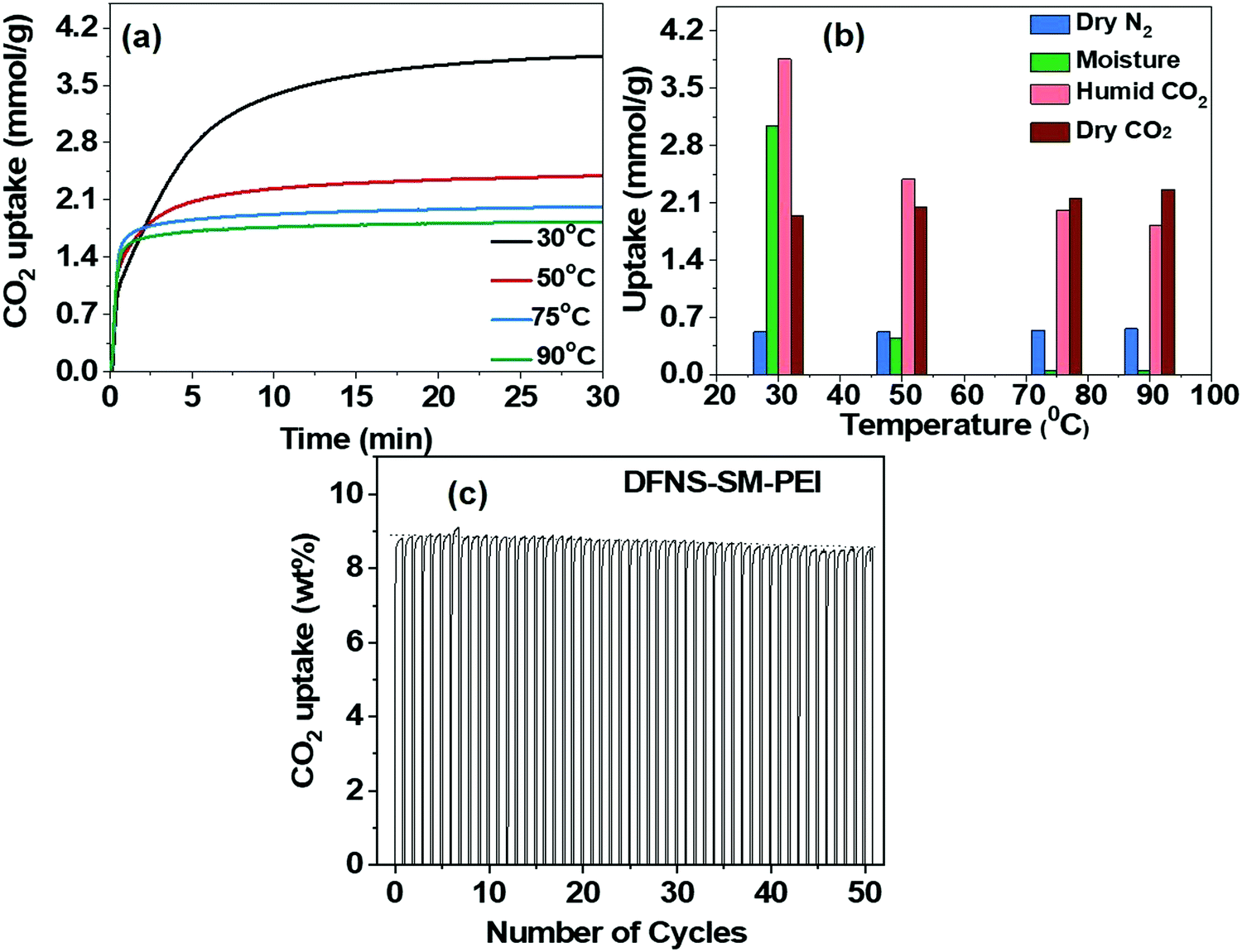 | ||
| Fig. 8 (a) Effect of moisture on CO2 capture capacity. (b) selectivity between N2, CO2 and H2O, using DFNS-SM-PEI microspheres. (c) Cycling stability and regeneration characteristics of DFNS-LM-PEI. | ||
The cyclic stability study of the PEI incorporated microgranules was evaluated over 50 adsorption–desorption cycles. The capture capacity has not shown any appreciable degradation until 50 cycles and a mere 2% reduction is seen, indicating good thermal stability of the microspheres against PEI degradation. Furthermore, IR and thermogravimetric analysis of the sorbent after 50 cycles were carried out to study the thermal degradation of PEI incorporated DFNS (Fig. S9, ESI†). We observed minimum loss of PEI and hence, the sorbent was showing nearly the same CO2 capture performance (capacity and kinetics). We believe that the formation of microgranules is restricting the loss of PEI by trapping them in their hierarchal pores of DFNS as well as between jammed DFNS particles in the spray-dried microgranules. The origin of high CO2 capacity with fast kinetics in the PEI-loaded microspheres is attributed to the distinct pore size distribution of the DFNS granules, which consists of well-defined mesopores and macropores originating due to their fibrous texture. The well-defined mesopores give rise to a high pore volume, whereas ample macropores provide accessibility to these mesopores. The fast adsorption kinetics of CO2 in the PEI incorporated microgranules is attributed to the connectivity between the mesopores and macropores; however, the experimental evidence of such connectivity has not been explored so far.
3.5 In situ neutron diffraction studies of DFNS–PEI microgranules
Understanding the intricate pore structure of the DFNS is a subject of immense importance. Although recent time-resolved small-angle X-ray measurements revealed the complex structure,28 there remains an issue of probing connectivity of the macropores and mesopores in DFNS granules. The phase behaviour of the adsorbed CO2 in the DFNS pores could provide important clues related to the connectivity of the macropores and mesopores. Moreover, the physisorption of the CO2 contributes significantly to the overall capture at low temperatures. To study the connectivity of the mesopores and macropores and phase behaviour of the physisorbed CO2 in DFNS microspheres, in situ neutron scattering has been performed over a wide angular range covering both small and wide regimes as a function of CO2 pressure. Furthermore, neutron scattering experiments were also performed on PEI incorporated DFNS.Fig. 9a shows the neutron scattering data of the DFNS-LM microspheres in the q-range of ∼0.02 to 50 Å−1. Scattering profiles show a significant change in both small and wide-angle regimes at different pressures of CO2. The overall high-q intensity increases with increasing CO2 pressure in the high-q regime as depicted in the inset of Fig. 9a. Furthermore, the intensity of the peak at q ∼ 1.8 Å−1, which coincides with the intermolecular structure peak between the CO2 molecules, increases with pressure. Fig. 9b shows the scattering profiles in the small-angle regime consisting of two distinct regimes, namely level-1 where the scattering from macropores dominates, and level-2 is dominated by scattering from mesopores. Interestingly, the functionality of the scattering profiles in the Kratky representation shows a shape with two broad peaks similar to the BJH pore size distribution. The scattering intensity in the level-1 regime shows a monotonic increase with pressure, whereas the intensity decreases in level-2. To understand the contrasting variation of intensity in level-1 and level-2, the Porod invariant is determined.
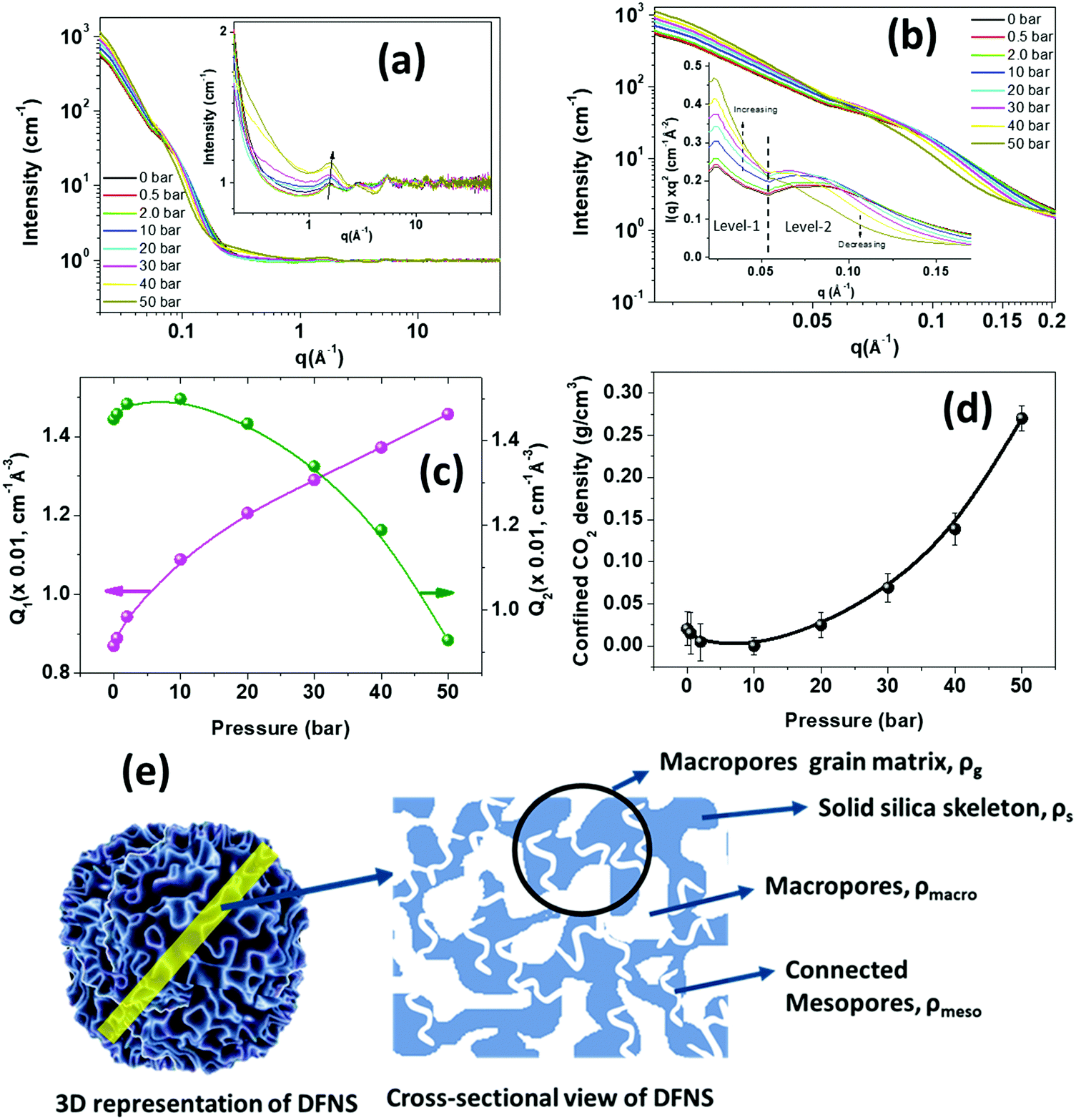 | ||
| Fig. 9 In situ neutron diffraction on the DFNS-LM microspheres as a function of CO2 pressure (a) in the q-range of 0.020 to 50 Å−1. The inset shows the zoom view of high-q data in the q range of 0.20 to 50 Å−1. (b) The scattering data in the small-angle regime in the q range of 0.020 to 0.20 Å−1 is depicted. The inset shows the small-angle scattering data in the Kratky representation [I(q)q2vs. q], which shows two levels of pore structures consisting of mesopores and macropores. (c) The variation of the Porod invariant of the scattering data in the small-angle regime is shown as a function of pressure. (d) The variation of the Porod invariant of level-1 (macropores) and level-2 (mesopores) is depicted as a function of pressure. (e) Schematic showing connected meso/macropore structures in DFNS. The scattering length density of each component is also illustrated. | ||
The Porod invariant Q is defined as:
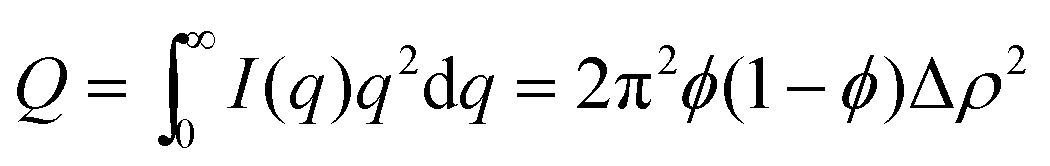
The Porod invariant for level-1 and level-2 regimes displays the opposite trend as shown in Fig. 9c. The Porod invariant for level-1, Q1, increases with an increase in CO2 pressure, whereas the Porod invariant at level-2, Q2, decreases with an increase in CO2 pressure.
Level-2 corresponds to the structure consisting of mesopores that are embedded in a solid silica matrix. A schematic diagram showing the mesoporous and macroporous structures in DFNS is depicted in Fig. 9e. The scattering contrast for level-2 varies as Δρ = ρs − ρmeso, where ρs is the SLD of the solid silica skeleton and ρmeso is the SLD of the mesopores and in the case of CO2 filled mesopores, ρmeso = (ρCO2)m, where (ρCO2)m is the SLD of CO2 in the mesopores. The density of the confined CO2 depends on the size of the pores and their interaction with the pore wall. With pressure, the density of the CO2 increases leading to an increase in (ρCO2)m while ρs remains constant and therefore, the scattering contrast Δρ2 decreases, leading to a reduction in the scattering intensity and Porod invariant Q2.
The SLD of the confined fluid in the mesopores can be estimated using the equation below:
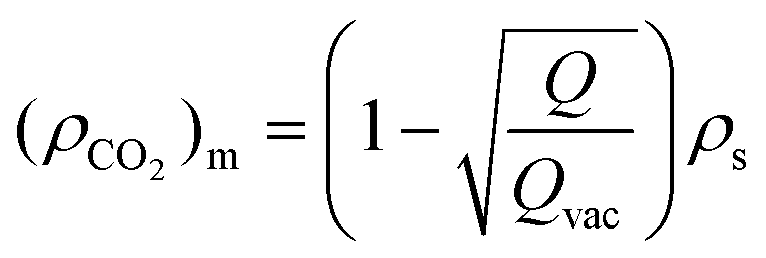
Level-1 corresponds to the structure consisting of macropores surrounded by the silica matrix (Fig. 9e) and the contrast factor for level-1 is defined as Δρ1 = ρg − ρmacro, where ρg is the SLD of the grain surrounding macropores and ρmcro is the SLD of the CO2 present in the macropore. If the mesopores are not embedded in the grain matrix of the macropores and there is no connectivity between the macropores and mesopores, then the SLD of the macropore matrix, ρg, will not be affected by the CO2 injection and therefore in this case contrast factor Δρ1 should decrease with pressure. However, the opposite trend is observed, i.e., Δρ1 increases with CO2 pressure. This behaviour can only be possible if the mesopores are embedded in the grain matrix surrounding the macropores and both macropores and mesopores are connected. In this case, the SLD of the macropore matrix, ρg, may be written as ρg = (1 − ϕmeso)ρs + ϕmesoρmeso, where ϕmeso is the volume fraction of the mesopores. ρmeso becomes (ρCO2)m after injection of CO2. (ρCO2)m increases with CO2 pressure leading to an increase in the SLD of the macropore matrix. Due to the larger size of the macropores, ρmacro is considered to be equal to the SLD of bulk CO2, which is less than the SLD of the confined CO2 in mesopores. Therefore, the increase in ρg is more compared to ρmacro as CO2 is injected, leading to an effective increase in the contrast factor of the macropores.
Fig. S10 (ESI†) shows the neutron scattering profiles of the PEI incorporated DFNS microspheres in the q-range of 0.02 to 50 Å−1. The scattering profiles do not show much change with CO2 pressure due to the reduced surface area and low porosity caused by immobilized PEI molecules. The scattering intensity and, therefore, the contrast factor in the small-angle regime (Fig. S10b, ESI†) also do not show much change with CO2 pressure indicating the absence of physisorption of CO2 in the PEI incorporated microspheres. This fact is further evident in Fig. S10c (ESI†), which shows that the scattering profile (Fig. S10c, ESI†) in the small-angle regime of 0.02 to 0.2 Å−1 displays a dramatic reduction in the intensity due to the filling of the mesopores and macropores with PEI molecules. The chemisorption-led CO2 absorption at active PEI sites does not affect the scattering profiles much. The Porod invariant of the high-q data, as shown in Fig. S10d (ESI†) does not show appreciable change with pressure confirming the lack of physisorption of CO2 in PEI-loaded microgranules. The neutron diffraction studies provided an indirect method for probing the interconnectivity of the mesopores and macropores in the DFNS microgranules and this analysis approach is applicable for exploring the pore connectivity in other porous materials.
4. Conclusions
PEI-loaded microgranules were achieved by a one-step evaporation-induced assembly process of the DFNS–PEI dispersion. The cooperative assembly of the DFNS and PEI leads to the incorporation of the PEI in the microgranules. The pore morphology of DFNS–PEI microgranules was extensively characterized by SAXS, electron microscopy and N2 gas adsorption techniques. The incorporation of PEI is confirmed by infrared spectroscopy and thermogravimetric analysis. The SAXS scattering intensity decreases without changing its functionality upon PEI incorporation whereas N2 adsorption isotherms showed a reduction in the pore volume and surface area after PEI incorporation. The CO2 adsorption capacity and kinetics are found to be significantly high due to high porosity and distinct pore size distribution consisting of mesopores and macropores with good interconnectivity, as evidenced by in situ neutron scattering results. The regeneration capability of the DFNS–PEI microgranules is found to be excellent, with negligible reduction in the adsorption capacity up to 50 cycles, which is attributed to the strong immobilization of the PEI during the assembly process as confirmed by SAXS and FESEM studies. A plausible mechanism for the assembly process is illustrated, which describes the order of the disordered assembly of the DFNS in the presence of PEI. The in situ neutron diffraction studies provided crucial experimental evidence for the first time about the connectivity of mesopores and macropores in DFNS responsible for fast CO2 adsorption kinetics. Furthermore, the specific interaction of CO2 with amine groups of PEI shows the highly selective capture characteristics of DFNS–PEI microspheres for CO2.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the funding support of the Department of Atomic Energy (under project no. 12-R&D-TFR-RTI4003). We acknowledge the XRD and EM facility of TIFR, Mumbai. JB, AD, and DS thank Dr S. M. Yusuf for his support for the SWAXS beamline activity. Neutron measurements were performed as part of ISIS beamtime allocation RB 1910045 (DOI: http://10.5286/ISIS.E.RB1910045).References
- N. J. Lenssen, G. A. Schmidt, J. E. Hansen, M. J. Menne, A. Persin, R. Ruedy and D. Zyss, J. Geophys. Res.: Atmos., 2019, 124, 6307–6326 CrossRef
.
-
J. Hansen, M. Sato, R. Ruedy, G. A. Schmidt, K. Lo and A. Persin, Global Temperature in 2018 and beyond, Earth Institute, Columbia University, New York, 2019 Search PubMed
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed
- R. S. Haszeldine, Science, 2009, 325, 1647–1652 CrossRef CAS PubMed
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed
- M. E. Boot-Handford, J. C. Abanades, E. J. Anthony, M. J. Blunt, S. Brandani, N. Mac Dowell, J. R. Fernández, M.-C. Ferrari, R. Gross and J. P. Hallett, Energy Environ. Sci., 2014, 7, 130–189 RSC
- A. Goeppert, M. Czaun, G. S. Prakash and G. A. Olah, Energy Environ. Sci., 2012, 5, 7833–7853 RSC
- A. Goeppert, H. Zhang, M. Czaun, R. B. May, G. S. Prakash, G. A. Olah and S. Narayanan, ChemSusChem, 2014, 7, 1386–1397 CrossRef CAS PubMed
- X. Ma, X. Wang and C. Song, J. Am. Chem. Soc., 2009, 131, 5777–5783 CrossRef CAS PubMed
- J. C. Hicks, J. H. Drese, D. J. Fauth, M. L. Gray, G. Qi and C. W. Jones, J. Am. Chem. Soc., 2008, 130, 2902–2903 CrossRef CAS PubMed
- M. B. Yue, Y. Chun, Y. Cao, X. Dong and J. H. Zhu, Adv. Funct. Mater., 2006, 16, 1717–1722 CrossRef CAS
- C. Chen, S.-T. Yang, W.-S. Ahn and R. Ryoo, Chem. Commun., 2009, 3627–3629 RSC
- S. Araki, H. Doi, Y. Sano, S. Tanaka and Y. Miyake, J. Colloid Interface Sci., 2009, 339, 382–389 CrossRef CAS PubMed
- Y. Jiang, P. Tan, S. C. Qi, X. Q. Liu, J. H. Yan, F. Fan and L. B. Sun, Angew. Chem., Int. Ed., 2019, 58, 6600–6604 CrossRef CAS PubMed
- A. Heydari-Gorji, Y. Belmabkhout and A. Sayari, Langmuir, 2011, 27, 12411–12416 CrossRef CAS PubMed
- A. Semenova, M. L. P. Vidallon, B. Follink, P. L. Brown and R. F. Tabor, Langmuir, 2021, 38, 191–202 CrossRef PubMed
- X. Xu, C. Song, J. M. Andresen, B. G. Miller and A. W. Scaroni, Energy Fuels, 2002, 16, 1463–1469 CrossRef CAS
- X. Xu, C. Song, J. M. Andresen, B. G. Miller and A. W. Scaroni, Microporous Mesoporous Mater., 2003, 62, 29–45 CrossRef CAS
- S.-N. Kim, W.-J. Son, J.-S. Choi and W.-S. Ahn, Microporous Mesoporous Mater., 2008, 115, 497–503 CrossRef CAS
- W. Long, N. A. Brunelli, S. A. Didas, E. W. Ping and C. W. Jones, ACS Catal., 2013, 3, 1700–1708 CrossRef CAS
- A. Holewinski, M. A. Sakwa-Novak, J.-M. Y. Carrillo, M. E. Potter, N. Ellebracht, G. Rother, B. G. Sumpter and C. W. Jones, J. Phys. Chem. B, 2017, 121, 6721–6731 CrossRef CAS PubMed
- J.-M. Y. Carrillo, M. A. Sakwa-Novak, A. Holewinski, M. E. Potter, G. Rother, C. W. Jones and B. G. Sumpter, Langmuir, 2016, 32, 2617–2625 CrossRef CAS PubMed
- G. Rother, U. Tumuluri, K. Huang, W. T. Heller, S. Dai, J.-M. Carrillo and B. G. Sumpter, Langmuir, 2021, 37, 4622–4631 CrossRef CAS PubMed
- X. Wang and C. Song, Catal. Today, 2012, 194, 44–52 CrossRef CAS
- P. Sharma, S. Chakrabarty, S. Roy and R. Kumar, Langmuir, 2018, 34, 5138–5148 CrossRef CAS PubMed
- Y. Zhao, Y. Shen and L. Bai, J. Colloid Interface Sci., 2012, 379, 94–100 CrossRef CAS PubMed
- V. Polshettiwar, D. Cha, X. Zhang and J. M. Basset, Angew. Chem., Int. Ed., 2010, 49, 9652–9656 CrossRef CAS PubMed
- D. Sen, A. Maity, J. Bahadur, A. Das and V. Polshettiwar, Microporous Mesoporous Mater., 2021, 111234 CrossRef CAS
- B. Singh and V. Polshettiwar, J. Mater. Chem. A, 2016, 4, 7005–7019 RSC
- F. Akhtar, L. Andersson, S. Ogunwumi, N. Hedin and L. Bergström, J. Eur. Ceram. Soc., 2014, 34, 1643–1666 CrossRef CAS
- A. Mosca, J. Hedlund, F. N. Ridha and P. Webley, Adsorption, 2008, 14, 687–693 CrossRef CAS
- J. Bahadur, D. Sen, S. Mazumder, B. Paul, H. Bhatt and S. Singh, Langmuir, 2012, 28, 1914–1923 CrossRef CAS PubMed
- D. Sen, J. Bahadur, S. Mazumder, G. Verma, P. Hassan, S. Bhattacharya, K. Vijai and P. Doshi, Soft Matter, 2012, 8, 1955–1963 RSC
- S. Mukundan, J. S. Melo, D. Sen and J. Bahadur, Colloids Surf., B, 2020, 195, 111245 CrossRef CAS PubMed
- D. Sen, J. S. Melo, J. Bahadur, S. Mazumder, S. Bhattacharya, S. F. D'Souza, H. Frielinghaus, G. Goerigk and R. Loidl, Soft Matter, 2011, 7, 5423–5429 RSC
- J. Bahadur, A. Das, J. Prakash, P. Singh, A. Khan and D. Sen, J. Appl. Phys., 2019, 126, 204301 CrossRef
- J. Bahadur, D. Sen, S. Mazumder, S. Bhattacharya, H. Frielinghaus and G. Goerigk, Langmuir, 2011, 27, 8404–8414 CrossRef CAS PubMed
- J. Bahadur, A. Das and D. Sen, J. Appl. Crystallogr., 2019, 52, 1169–1175 CrossRef CAS
- D. Bowron and A. Soper, Neutron News, 2011, 22, 12–14 CrossRef
- D. Bowron, A. Soper, K. Jones, S. Ansell, S. Birch, J. Norris, L. Perrott, D. Riedel, N. Rhodes and S. Wakefield, Rev. Sci. Instrum., 2010, 81, 033905 CrossRef CAS PubMed
- P. Day, J. Enderby, W. Williams, L. Chapon, A. Hannon, P. Radaelli and A. Soper, Neutron News, 2004, 15, 19–23 CrossRef
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS
- Z. Bacsik, N. Ahlsten, A. Ziadi, G. Zhao, A. E. Garcia-Bennett, B. Martín-Matute and N. Hedin, Langmuir, 2011, 27, 11118–11128 CrossRef CAS PubMed
- M. Teubner and R. Strey, J. Chem. Phys., 1987, 87, 3195–3200 CrossRef CAS
- M. Kosmulski, J. Colloid Interface Sci., 2009, 337, 439–448 CrossRef CAS PubMed
- J.-M. Schweitzer, M. Servel, F. Salvatori, A. Dandeu, M. Minière, J.-F. Joly, Q. Gaubert, S. Barbosa and F. R. Onofri, Chem. Eng. Res. Des., 2021, 170, 224–238 CrossRef CAS
- A. Goeppert, S. Meth, G. S. Prakash and G. A. Olah, Energy Environ. Sci., 2010, 3, 1949–1960 RSC
- K. Li, J. Jiang, S. Tian, F. Yan and X. Chen, J. Mater. Chem. A, 2015, 3, 2166–2175 RSC
- R. Sanz, G. Calleja, A. Arencibia and E. Sanz-Perez, Appl. Surf. Sci., 2010, 256, 5323–5328 CrossRef CAS
- R. Sanz, G. Calleja, A. Arencibia and E. S. Sanz-Perez, Microporous Mesoporous Mater., 2012, 158, 309–317 CrossRef CAS
- X. Xu, C. Song, J. M. Andresen, B. G. Miller and A. W. Scaroni, Energy Fuels, 2002, 16, 1463–1469 CrossRef CAS
- S. J. Datta, C. Khumnoon, Z. H. Lee, W. K. Moon, S. Docao, T. H. Nguyen, I. C. Hwang, D. Moon, P. Oleynikov and O. Terasaki, Science, 2015, 350, 302–306 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00785a |
| This journal is © The Royal Society of Chemistry 2022 |