
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe extent of carbon surface oxygen affinity and its effects on the activity of metal-free carbon catalysts in the oxygen reduction reaction: the interplay of porosity and N-, O- and S-enriched surface chemistry†
Marc
Florent
,
Raabia
Hashmi
and
Teresa J.
Bandosz
 *
*
Department of Chemistry and Biochemistry, The City College of The City University of New York, 160 Convent Avenue, New York, NY 10031, USA. E-mail: tbandosz@ccny.cuny.edu
First published on 27th September 2022
Abstract
Highly porous carbon black, Black Pearl 2000, was modified with urea and thiourea to introduce only N- or N- and S-containing functional groups, affecting its activity in the oxygen reduction reaction. The samples were exposed to heating at 450 and 850 °C to vary surface features. Their chemistry and porosity were evaluated using various physical and chemical methods. An almost identical porosity of the urea-modified samples allowed for the evaluation of the effects of their surface chemistry on the ORR, assuming similar contributions of their micropores activity to an adsorption-induced oxygen reduction process. Even though the results suggested that pyridones were involved in the oxygen reduction activity, the number of electron transferred (∼4) and the onset potential were found to be directly dependent on the affinity of the surface to transport and adsorb dissolved oxygen, either into ultramicropores and onto N- and/or S- and N-based catalytic centers. The introduction of N and S led to a high catalytic activity. Sulfur, mainly in hydrophobic thiophenic species, compensated for the smaller absolute amounts of nitrogen. For the thiourea-modified series, the effect of ultramicropores in advancing the ORR was clearly indicated by the higher activity with fewer catalytic sites on the surface.
1. Introduction
The oxygen reduction reaction, ORR, is one of the processes that determine the efficiency of a fuel cell. A device is effective when 4 electrons are transferred and either H2O or OH− are formed in an acidic or basic electrolyte, respectively. Moreover, a high current density is desired and the onset potential should be as positive as possible. Even though these conditions are met with noble metal electrocatalysts, and especially on Pt,1 their high costs and scarcity may prove problematic to meeting the demand of the intended wide spread application of fuel cell technology and as such has prompted research into ORR electrocatalysts with transition metals,2,3 as a platinum replacement, with emphasis on their dispersion on a porous carbon phase rich in nitrogen heteroatoms.4 In particular, metals can coordinate with nitrogen,5 allowing the formation of highly efficient single atom electrocatalysts.6Another intensively explored line of research focuses on non-metal catalysts and here the interest of researchers is concentrated on carbon-based catalysts,7–16 graphitic carbon nitride17,18 or boron nitride.19 In this group, carbon materials are considered as attractive candidates to replace Pt/C owing to their conductivity and the possibility of modifying their electronic structure by the introduction of heteroatoms into a carbon matrix.20–30 The modifications, with a special emphasis on nitrogen20–23,31 provide basic sites to a carbon surface, which attract oxygen and then contribute to its reduction either to OH− or H2O, depending on the conditions. In a recent review by Cazorla-Amoros and co-workers, not only has the role of nitrogen species (such as pyridines and quaternary nitrogen) introduced to the carbon matrix been emphasized but also that of nitrogen–oxygen configurations such as pyridones.32 The stress was on the catalytic effects of the specific geometry of oxygen-active center interactions. Recently the role of sulfur in the carbon matrix on the ORR has been recognized24,25 and the results showed that it is especially beneficial for the ORR when located in the proximity of nitrogen-containing centers.33
For an efficient carbon-based electrocatalyst the number and distribution of active centers as well as their availability/accessibility are important, and the extent of porosity has also been targeted as a research topic.8,34–39 Besides carbons of naturally hierarchical porosity,36–39 3-D structures in the family of new nanocarbons have been built and explored.8 Not without importance is that recent studies have demonstrated that pores in highly porous carbons are built of distorted graphene layers9 and this finding could be regard as important when the applications of these materials in electrocatalysis and other “beyond adsorption” applications are considered.40,41 Here it is also worth mentioning that these distorted “graphene-like” pore walls have been presented as having many defects and defects have been also recently identified as active sites for the ORR.16,22,42
In our scientific explorations, and based on the literature data demonstrating the excellent oxygen reduction performance of nanoporous carbons either heteroatom-rich30 or heteroatom-free,43–47 we have proposed small micropores/ultramicropores as pseudocatalytic centers advancing the overall reduction process. This is due to the strong adsorption of dioxygen on their hydrophobic surfaces, followed by oxygen–oxygen bond breaking and the transfer of four electrons.43–47 This hypothesis has been independently supported by the results indicating the dependence of the catalytic efficiency descriptors, such as the kinetic current density or number of electron transfers, or even the onset potential, on a surface area or porosity.34,35 Moreover, theoretical modeling has shown an effect of carbon pore sizes on the oxygen reduction mechanism and on the number of electron transfers.36
Carbon blacks, owing to their conductivity, are commonly used as supports for the high dispersion of platinum, catalytically active in the ORR. Their application in the ORR was investigated from the viewpoints of corrosion48 or desired support features.49–51 Nevertheless, until now their direct usage as ORR catalysts has been reported rarely.52 Appleby and Marie53 evaluated the kinetics of the ORR on 19 carbon blacks, among 37 carbon-based materials, and they found a linear dependence of the measured current on the surface area, SBET. Recently, we tested the efficiency of oxygen reduction on a series of carbon blacks with the increasing surface area (from 110 m2 g−1 to 1600 m2 g−1) of a Black Pearl family.52 On highly porous carbon black, Black Pearl 2000, the number electron transferred reached 4 and a high kinetic current density was measured. Moreover, some dependence of the number of electron transferred and the onset potential on the volume of micropores or the surface area has been established. Regarding surface chemistry, its effect has been found to be similar to that found on carbon molecular sieves.47 Even though some oxygen was present, the electron-rich surface and a high volume of micropores provided sites attracting oxygen. Upon its strong adsorption, a bond splitting, electron transfer and hydrogenation took place. Moreover, the important role of the porosity of this series of carbon blacks for oxygen adsorption was demonstrated by the direct dependence of the extent of oxygen adsorption from O2 saturated water on the CB surface areas and micropore volumes.52
In recent reports, N-doped carbon blacks were used as catalysts themselves54 or as supports for other active phases.55–57 In the latter category, the performance of these supports is rarely reported in detail and specific information on the carbon black type is not always provided. Dong and Li55 introduced nitrogen under ammonia annealing to carbon black of a surface area of 250 m2 g−1. On the doped sample the number of electron transferred was 3.16 and the kinetic current density reached 2.2 mA cm−2 with an onset potential 0.79 V vs. RHE in an alkaline electrolyte. This relatively good performance was linked to nitrogen in pyrroles and graphitic configurations. Detailed results on N-doped Ketjen Black as an ORR catalyst were reported by Oh and co-workers.54 Their samples were treated with melamine and heated in two steps. The best sample was obtained with extensive energy consumption (preheated at 750 °C followed by treatment at 1000 °C). The resulting catalysts had 0.7 at% of N and 2.4 at% of O2 and a surface area of 838 m2 g−1. The onset potential was 0.97 V (on Pt/C 0.95 was reported), the number of electron transferred was 3.9 and the kinetic current density was 6.1 mA cm−2. The similarity to Pt/C in the performance was linked to N-doping and to the positive effect of the pretreatment at 750 °C on decreasing an electrical resistance at the interfaces.
Following the previously published encouraging results obtained on the series Black Pearl carbon blacks,52 our previous findings,30,43–47 and the well-established role of nitrogen and sulfur chemistry when incorporated into the carbon matrix,32 we have expanded our investigations of the suitability of the application of metal-free ORR porous carbon catalysts to carbon blacks whose surface was modified with nitrogen and with nitrogen and sulfur.58 For this purpose, we have chosen the best catalyst from the series investigated previously,52 highly porous Black Pearl 2000. Even though N-doped carbons blacks of different features have been studied as ORR catalysts54 or catalyst supports,55–57 the novelty of our approach lies in analyzing the effects of surface chemistry and porosity, either separately or combined, on oxygen adsorption in order to further advance understanding of the ORR on porous carbon catalysts. When evaluating of the performance, we counted on the effects of nitrogen and sulfur incorporation into carbon black and changes caused by them in the overall chemical and structural environment affecting the ORR performance. Contrary to other reports,54–57 our CB samples are rather rich in nitrogen, oxygen and sulfur, which, when combined with specific porosity, play an important role in catalyzing the oxygen reduction process. In the analysis, we focused on the affinity of surfaces, including textural and chemical features, to interact with O2 dissolved in an aqueous electrolyte.
2. Experimental
2.1. Materials
Commercial carbon black from Cabot Corporation, Black Pearls 2000 (named hereafter BP), was modified with urea (Amresco) or thiourea (99%, Alfa Aesar), in an as-received form or oxidized. Oxidation of BP was carried out by heating it in concentrated nitric acid (68–70%, BDH) at 80 °C for 30 minutes. The carbon was then filtered and rinsed with water before washing it in a Soxhlet apparatus until reaching a constant pH. After this, the sample was dried at 120 °C.Urea or thiourea were used as modifiers to introduce nitrogen and nitrogen and sulfur heteroatoms into the carbon matrix, respectively. First, initial or oxidized BP was impregnated with a solution of urea or thiourea (the mass ratio of carbon to an impregnant was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). After drying, the mixtures were heated under nitrogen (100 mL min−1) at 450 °C or 850 °C for 30 minutes with a heating rate of 10 °C min−1, and, after this step, washed in Soxhlet apparatus until reaching a constant pH. The materials were then dry at 120 °C. The oxidized, modified with urea or thiourea samples are designated with, –O, –U or –TU, respectively, added to their names. The last three digits represent the heat treatment temperatures. For instance, BP-U-450 represents BP used as-received and impregnated with urea and pyrolyzed at 450 °C, and BP-O-TU-850 represents BP first oxidized and then modified with thiourea and heat treated at 850 °C.
:1). After drying, the mixtures were heated under nitrogen (100 mL min−1) at 450 °C or 850 °C for 30 minutes with a heating rate of 10 °C min−1, and, after this step, washed in Soxhlet apparatus until reaching a constant pH. The materials were then dry at 120 °C. The oxidized, modified with urea or thiourea samples are designated with, –O, –U or –TU, respectively, added to their names. The last three digits represent the heat treatment temperatures. For instance, BP-U-450 represents BP used as-received and impregnated with urea and pyrolyzed at 450 °C, and BP-O-TU-850 represents BP first oxidized and then modified with thiourea and heat treated at 850 °C.
2.2. Methods
Thermogravimetric (TG) and differential TG (DTG) analyses were completed using an SDT Q600 (TA Instruments) by heating the samples up to 1000 °C at a rate of 10 °C min−1 in a 100 mL min−1 argon or air flow.
The surface group chemical character in a moist environment was evaluated via potentiometric titration, using an 888 Titrando (Methrom). 0.1 g of CBs were dispersed in 50 ml of a 0.01 M NaNO3 solution under a nitrogen flow. After acidification to pH ∼ 3.2 with 0.100 M HCl, the dispersion was titrated with 0.100 M NaOH. Proton binding curves were calculated from the titration curves, and from them the pKa distributions of surface groups59 were obtained using a SAIEUS approach.60
XPS spectra were recorded on a Physical Electronics PHI 5000 VersaProbe II spectrometer, using an Al Kα X-ray source (50 W, 15 kV, 1486.6 eV) at a take-off angle of 45° by using a concentric hemispherical analyzer operating in a constant-pass-energy mode, at 29.35 eV, with a 200 μm diameter analysis area. Multipack software was used to deconvolute the spectra.
Raman spectra were recorded on a WITec alpha300R confocal Raman microscope at 100× magnification and with a 5 mW laser at 532 nm.
Resistivity of the materials was measured using a Keithley 2400 Multimeter on a 0.5 × 0.45 cm gold interdigitated electrode covered with a thin film of carbon.
Water adsorption was estimated by measuring the weight gain of the dry samples left for four hours in a closed chamber saturated in water vapor.
Oxygen adsorption in solution was measured by recording the concentration of dissolved oxygen in 50 ml water using a Hach IntelliCAL LDO probe upon the addition 250 mg of the carbon sample.
Cyclic voltammetry (CV) curves were measured in either a nitrogen or oxygen-saturated electrolyte solution with a scan rate of 5 mV s−1.
An electrochemically active surface area (ECSA) was estimated by deriving a double layer capacitance, CDL, from CV curves measured in a non-faradaic region by varying the scan rate ν from 5 to 150 mV s−1 (Fig. S1 of the ESI†):61
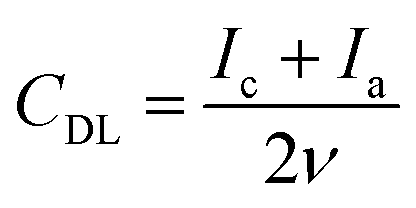 | (1) |
Assuming that all samples have a similar specific capacitance CS as carbon black, 27.5 μF cm−2, ECSA is given by:61
 | (2) |
The LSV curves were used to calculate the kinetic current using the Koutecky–Levich eqn (3):62
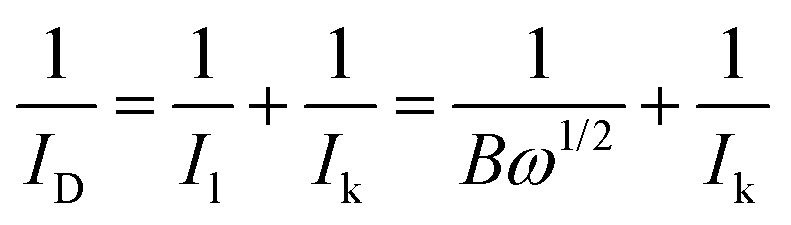 | (3) |
LSV curves measured at 2000 rpm were used to obtain the onset potential using the unbiased second derivative method,63 the electron transfer number n and the amount of H2O2 produced, using eqn (4) and (5) respectively:62
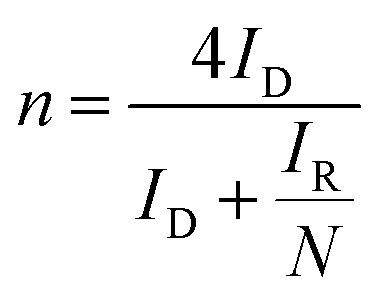 | (4) |
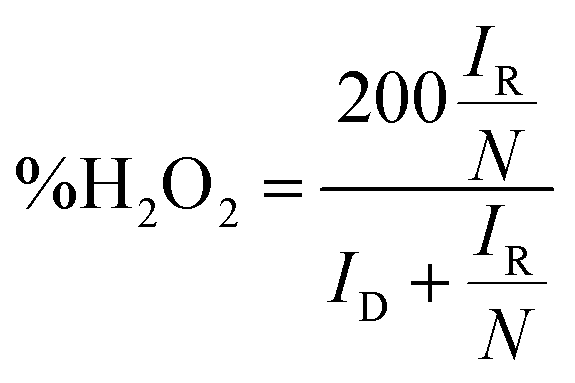 | (5) |
Tafel slopes were obtained by linear fit of the potential as a function of log(|Jk|) in a region close to the onset potential.
The stability of the samples was evaluated by measuring the current for 24 hours at the potential giving the maximum oxygen reduction current, with a constant O2 flow through the electrolyte.
Methanol resistance was tested by measuring a current at the potential giving the maximum oxygen reduction current by adding 2.5 ml of methanol into 60 ml of the electrolyte solution.
3. Results and discussion
All CV curves measured in a nitrogen saturated electrolyte (Fig. S2, ESI†) have similar quasi-rectangular shapes indicating some capacitance, which we link to the developed pore structure of the BP series.52 In the presence of dissolved O2, all samples show very clear humps on a cathodic current indicating their activity in oxygen reduction.The LSV curves (Fig. S3, ESI†) show that the measured current depends on the rotation rate, indicating a mixed kinetic-diffusion control process. Current densities at 2000 rpm for the urea and thiourea modified samples are compared in Fig. 1(A) and (B), respectively. Even though not all modifications led to an increase in the current compared to that for the initial BP, the shapes of the curves, reflecting the mechanism of the process, were markedly changed. While for BP two waves are clearly visible and linked to the two step oxygen reduction in pores of various sizes,36 the curves for the modified samples, with an increase in the treatment temperature, gradually resemble a typical shape of the LS curve of Pt/C carbon (included for comparison in Fig. 1(A) and (B)). This is especially pronounced for the samples modified with urea, and might suggest a predominance of one type of highly kinetically active catalytic centers. This trend after chemical/thermal modifications of Katjen Black was also reported by Oh and coworkers.54 Generally, for both series of modified carbons, preoxidation of the carbon surface increased the current density. BP-O-U-450 and BP-O-U-850 exhibit the highest current density (∼6.1–6.2 mA cm−2), and perform better than Pt/C (5.5 mA cm−2). The unoxidized samples and those heated at a low temperature appear to be inferior to BP and this is true for both series of the catalysts. The samples heated at 850 °C exhibited the higher current densities, regardless of the chemistry of the modifier.
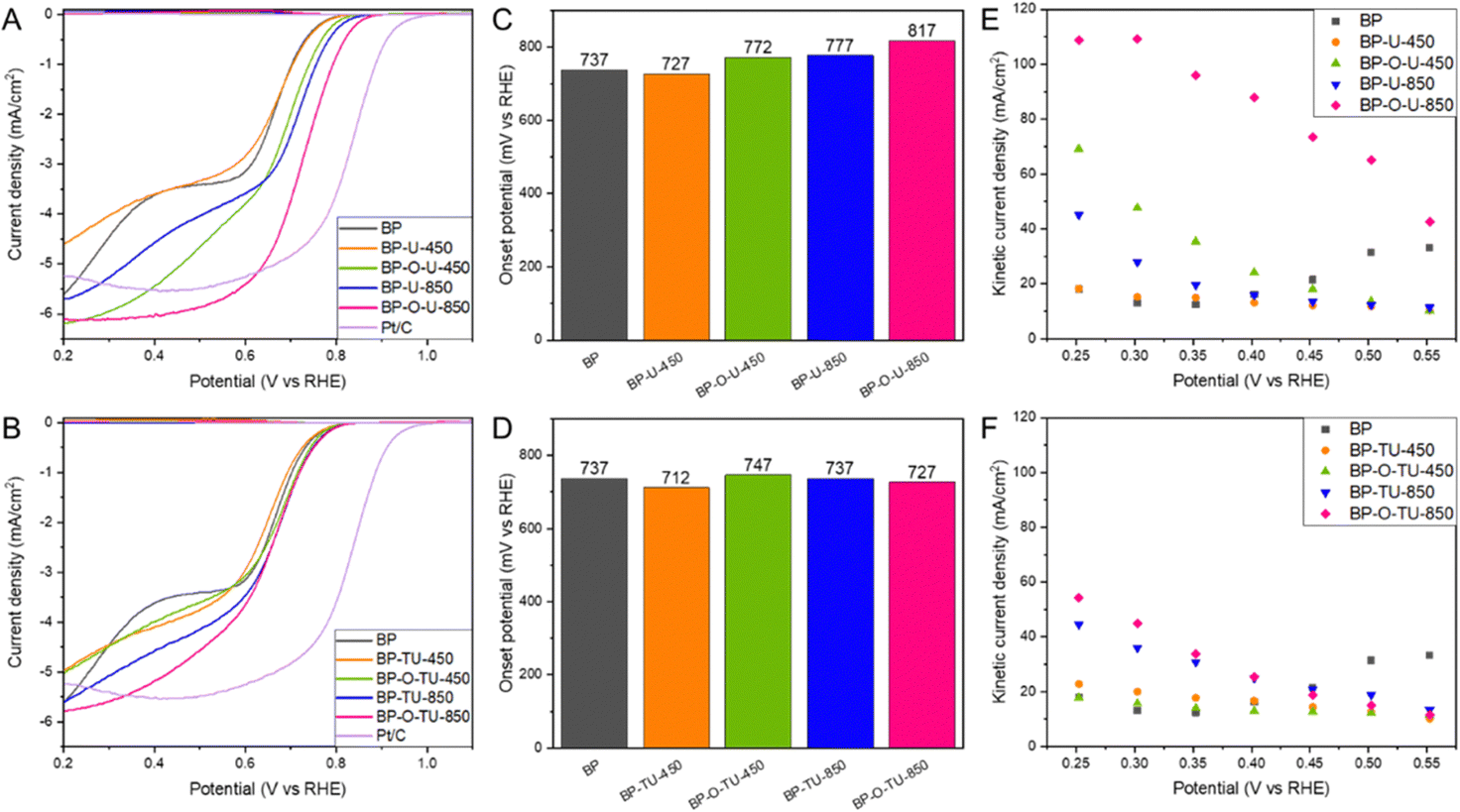 | ||
| Fig. 1 Current density for the urea modified samples (A) and thiourea modified ones (B); onset potentials for the urea modified samples (C) and thiourea modified ones (D). The kinetic current densities calculated using the Koutecky–Levich equation for the urea modified samples (E) and thiourea modified ones (F). | ||
Onset potentials calculated from the LSV curves using the unbiased second derivative method63 are shown in Fig. 1(C) and (D). While the modification with urea at 850 °C increased the onset potential by 40 mV compared to that of the initial BP, preoxidation had an even more positive effect with an 80 mV increase. By contrast, the modification at 450 °C led to a small decrease in the onset potential. However, here too, oxidation of BP before the modification resulted in the more positive onset potential, though not as high as that after the 850 °C treatment. The modification with thiourea had rather a small effect on the onset potential. It was not affected by the treatment at 850 °C and heating at 450 °C resulted in the lowest onset potential among all samples tested (25 mV decrease compared to that of BP).
The kinetic current densities calculated using the Koutecky–Levich equation are shown in Fig. 1(E) and (F). The samples modified with thiourea at 450 °C (Fig. 1(F)), regardless of the pretreatment, and BP-U-450, revealed the lowest kinetic currents, similar to that of pristine BP (∼20 mA cm−2). However, the samples modified at 850 °C with thiourea, and BP-U-850 show higher kinetic currents of ∼45–55 mA cm−2. And again, while carbon oxidation does not visibly affect the behavior of the samples modified with thiourea, for the samples modified with urea, prior oxidation of BP resulted in the highest kinetic current. For BP-O-U-450 it reached ∼70 mA cm−2 and for BP-O-U-850 a remarkable 110 mA cm−2 was measured. Thus, urea brought a more positive effect to the performance of the catalysts compared with that of thiourea.
Another important descriptor of the ORR efficiency is the number of electron transferred, n, as it indicates how complete the oxygen reduction is. The numbers of electron transferred for our samples are shown in Fig. 2(A) and (B). All samples show high n, above 3.8, and, at most, 12% of O2 is converted into H2O2 on the least effective sample, BP-TU-850 (Fig. 2(D)). On the best performing sample, BP-O-U-850 3.98 electrons were transferred and the trend in n with the potential is almost the same as that on Pt/C. On this sample only 2% of H2O2 (Fig. 2(C)) was formed, similarly to the amount detected on Pt/C. Even though all samples pass through a minimum in the n values, these minima are least pronounced for the oxidized samples heated at 850 °C from both series, indicating the steady mechanism of oxygen reduction. At 0.2 V, for all catalysts, n is above 3.9 and three samples are worse than pristine BP: BP-TU-850, BP-TU-450 and BP-U-450. For BP-O-U-450, n varies between 3.80 and 3.97. Four carbons exhibit higher n than does BP: BP-O-TU-450, BP-U-850, BP-O-TU-850 and BP-O-U-850. These trends suggest that modifying pristine BP with thiourea, and/or heating at a low temperature deteriorate the electrocatalysts in terms of the numbers of electrons transferred. However, using urea, and/or the high temperature and/or preoxidation significantly improve n. Interestingly, for the number of electron transferred, the positive effect of carbon oxidation is clearly visible, even when modifying with thiourea.
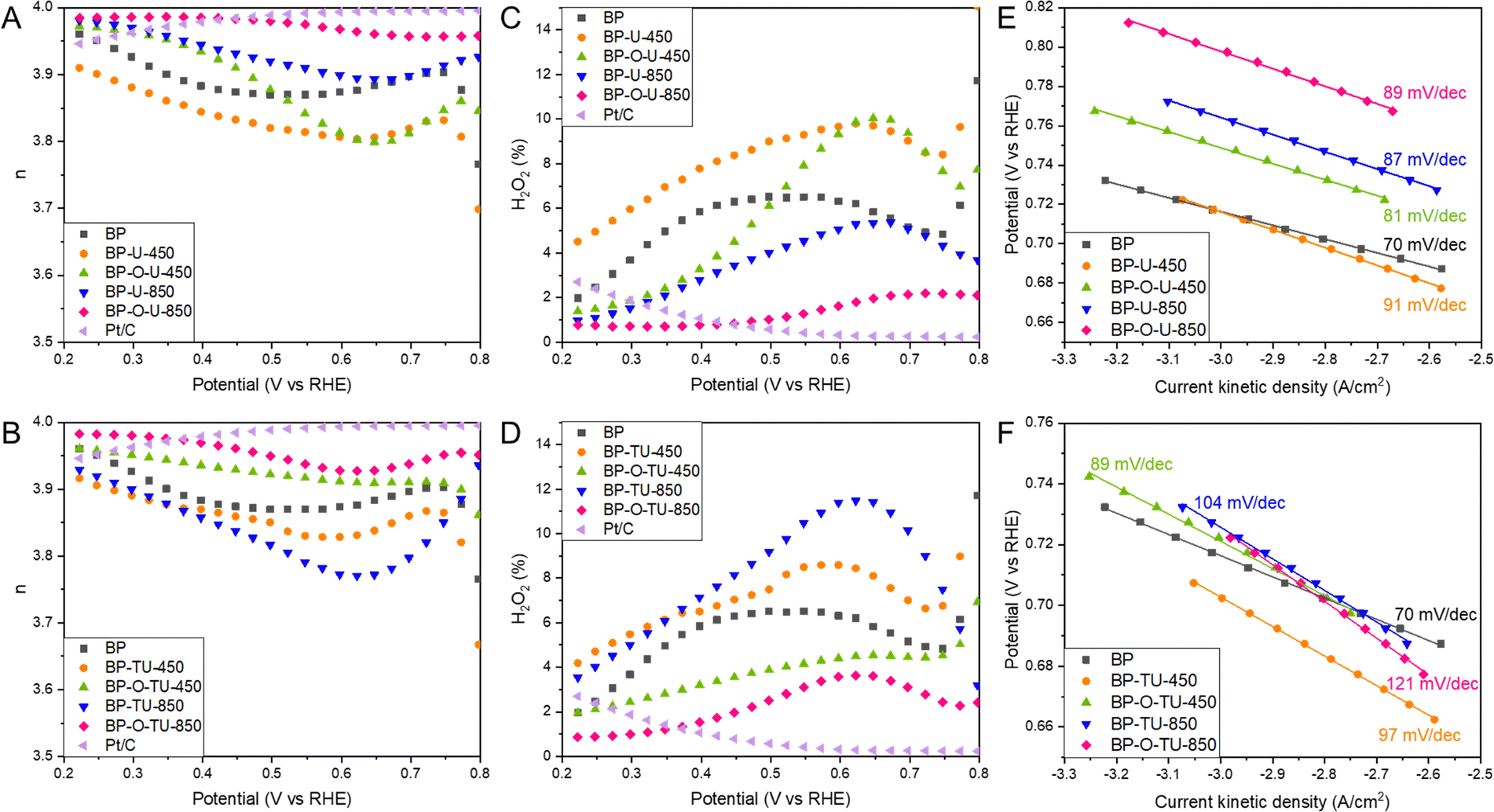 | ||
| Fig. 2 The number of electron transferred, n, for the urea modified samples (A) and thiourea modified ones. (B) Percent of H2O2 formed on the urea modified samples (C) and thiourea modified ones. (D) Tafel plot for the urea modified samples (E) and thiourea modified ones (F). | ||
Tafel plot slope values are linked to the kinetics of an electron transfer.27Fig. 2(E) and (F) show the Tafel plots with their calculated slopes. For Pt/C the slope of 0.81 mV dec−1 was found.52 The slopes for the urea modified series are almost the same for all samples, similar to that for Pt/C, and only slightly larger than that for BP, which indicate a fast electron transfer and likely some similarities in the mechanisms of the ORR process. For the thiourea modified samples, on the other hand, the differences in the slopes are more pronounced upon the specific modifications than those for the urea-modified catalysts. The slopes are larger than those for BP and Pt/C and the best performing sample- BP-O-TU-850, in terms of the onset potential, number of electron transfer, and kinetic current, has the largest slope, suggesting a complex mechanism of the ORR. All thiourea-modified catalysts exhibit a similar stability to that of Pt/C (Fig. S4, ESI†) but BP-O-TU-450 and BP-TU-850 are slightly more stable. Interestingly, the urea-modified samples show the same trend, with BP-O-U-450 and BP-U-850 being more stable, and BP-O-U-850 and BP-U-450 being less stable than Pt/C is. All samples show a much better tolerance to methanol cross-over than that of Pt/C (Fig. S4, ESI†). Pt/C loses about 30% of its efficiency after an injection of methanol, while the thiourea samples lose only 5–7.5% and the urea samples less than 5%.
To identify factors governing the difference in the performance of modified BP in the ORR, extensive surface characterization was performed. To account for the effect of defects the Raman spectra were obtained and deconvoluted using the Sadezky approach.64 For all materials the spectra are very similar – all have an ID/IG ratio between 1.1 and 1.2- and so are the contributions of specific defects in all samples (Fig. S5, ESI†). Resistivity measurements revealed the conductive nature of all catalysts with very small differences between samples (8–39 ohms). The results of these measurements allowed us to exclude the effects of defects and conductivity differences from the interpretation of the trends in the performance of our catalysts.
To expand our understanding of the differences caused by the specific modification method, a thermal analysis in argon was carried out (Fig. 3(A) and (B)). It revealed differences in the bulk chemistry of the samples, through differences in thermal decomposition patterns. The results for the urea-modified samples showed that the catalysts heated at 850 °C are thermally more stable than BP 2000 is (Fig. 3(A)), indicating changes in chemistry upon the applied treatment. BP-U-450 showed some weight loss starting at about 300 °C, and BP-O-U-450 – at about 500 °C, due to the decomposition of the thermally unstable surface groups.58 This indicates the marked differences in chemistry of these two samples caused by different modes of urea interactions with the as-received and oxidized surfaces, and thus differences in its thermal decomposition products (NH3, HNCO).65 Even though the oxidized sample is more stable at low temperatures, its weight loss over 600 °C exceeds that of the corresponding as-received sample. Generally, when oxygen groups on the carbon surface are considered, carboxylic acids decompose between 200 and 400 °C and then carbonyl does (∼600 °C) and phenols appear as the most stable one.66 The picture is much more complex when nitrogen and/or sulfur groups are also present, and it is an expectation in the case of our catalysts. In the former category, amines are considered as the least stable, followed by pyridinic and graphitic nitrogen.67 In the case of sulfur functional groups of a carbon surface, sulfonic acids decompose at about 200 °C, sulfones and sulfoxides are slightly more stable66,68 and sulfur in thiophenic configurations is considered as the most resistant to the thermal decomposition.68 Nevertheless, we do not attempt to make any precise assignment owing to the expected complex bonds with an involvement of oxygen. The thermally stable surface of the samples obtained at 850 °C suggests that they either have small amounts of groups on the surface, a significant number of heteroatoms are introduced to carbon rings and/or the bonds are thermally stable.
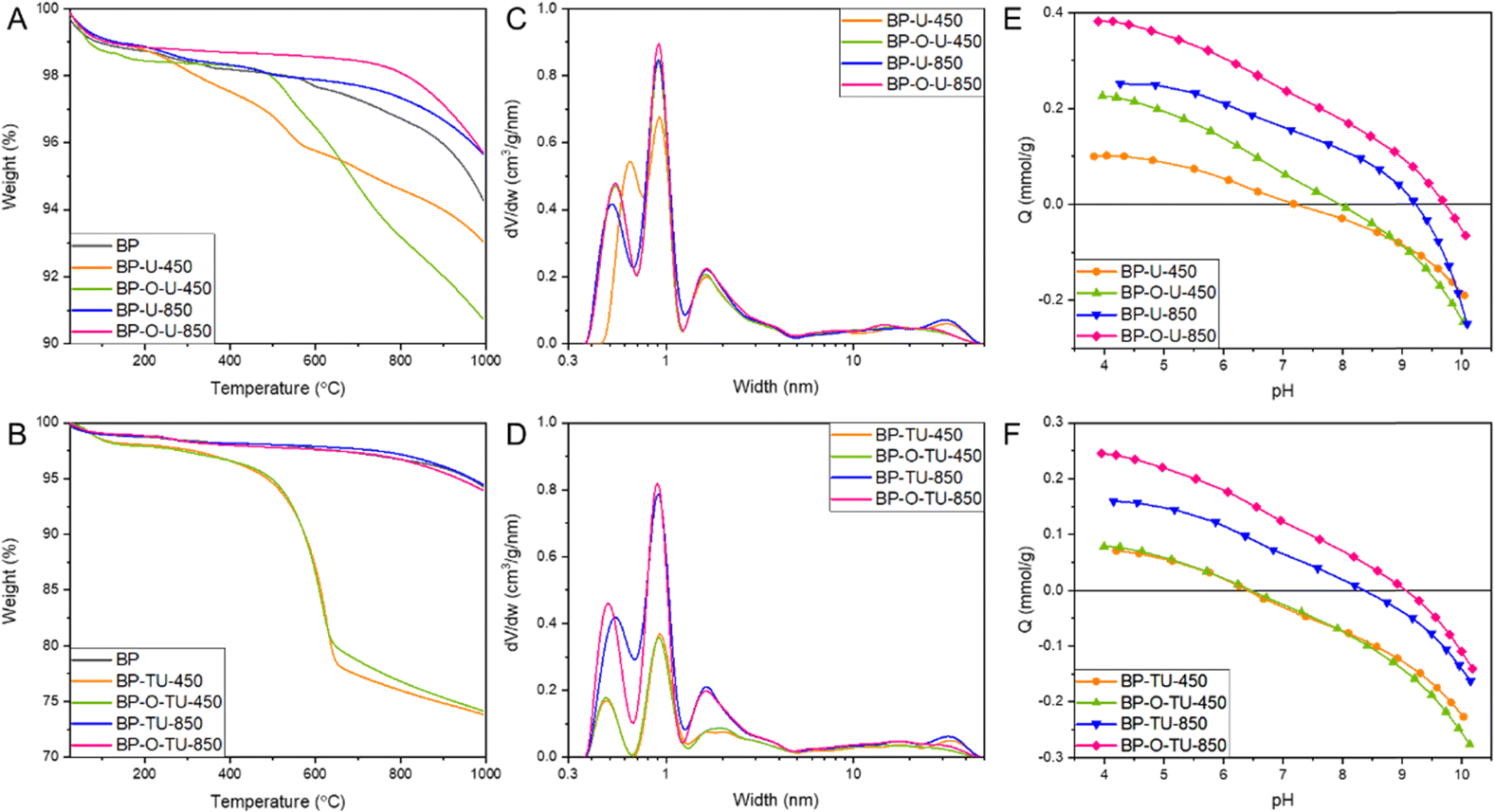 | ||
| Fig. 3 TG curves in argon for the urea modified samples (A) and thiourea modified ones. (B) Pore size distributions for the urea modified samples (C) and thiourea modified ones. (D) Proton binding curves for the urea modified samples (E) and thiourea modified ones (F). | ||
The samples exposed to thiourea and heated at 850 °C show exactly the same stability as BP2000, regardless of the pretreatment. On the other hand, treatment at 450 °C, apparently left a significant amount of a thermally unstable residue, about 25 wt% of both samples, which decomposed between 500 and 600 °C. Apparently, and also noticed when the performance was analyzed, thiourea interactions with the surface of our carbon blacks are less sensitive to the surface chemistry modification by oxidation than are those of urea. Moreover, the thermal behavior of thiourea differs from that of urea.69 As a pure compound, it is transformed into ammonium thiocyanite at 130 °C and at 200 °C decomposes into NH3, H2S and CS2 with a residual guanidinium thiocyanite. Its decomposition into NH3 and SC2 is expected to result in the incorporation of S and N heteroatoms to the carbon matrix. It is important to mention that the oxidized BP sample loses 11% weight almost linearly starting from 200 °C (Fig. S6, ESI†) and this pattern is not visible for the modified samples. This clearly indicates the surface chemical heterogeneity of the formed oxygen functional groups and these groups were apparently involved in interactions with the modifiers during the thermal treatment.70 To further support this hypothesis, we run the TG analysis of urea, thiourea and BP2000 impregnated with these two compounds (Fig. S7, ESI†). For the urea-modified samples a distinct decomposition of the modifier, only slightly influenced by the presence of carbon, takes place at 300 °C, thus already affecting the chemistry of the samples heated at 450 °C. On the other hand, thiourea stability when mixed with carbon totally differs from that of the pure compound. While the main decomposition of the latter takes place as a distinct event at about 200 °C, and with an increase in the temperature the small intensity events are visible at 250, 400 and about 600 °C, on carbon the first step is gradual and takes place between 200 and 400 °C, and the second one between 450 and 625 °C. The latter step is seen in Fig. 3(B). These results clearly indicate a reaction between thiourea and the carbon support.
Nitrogen adsorption isotherms were measured to obtain information on the extent of porosity, which we consider as important for the overall efficiency of the porous electrocatalysts30,39,43–47 (Fig. S8, ESI†). From them, pore size distributions (Fig. 3(C) and (D)) and pore structure parameters, including surface areas and pore volumes (Table 1) were calculated. The shapes of the isotherms are similar indicating a micro/mesoporous nature. Generally, a decrease in the surface area is observed upon the treatment, which we link to a pore blocking effect and thus inaccessibility of the N2 molecules to the pore system in the case of urea and all high temperature-treated samples and to the deposition of the significant amounts of nonporous thiourea residue on the samples modified with this compound and heated at 450 °C, as demonstrated from the TA analysis and discussed above. All urea-modified samples but BP-O-U-450 exhibit almost identical porosity with predominant pore sizes between 0.4–0.5 nm (0.53 nm), 0.9–1.0 nm, and 1.8 nm. Large mesopores important for mass transfer are also present. Thus, the contribution of the oxygen adsorption strength in ultramicropores to the efficiency of the ORR is expected to be similar and differences in the performance of these electrocatalysts could be mostly linked to the differences in surface chemistry and the volumes of ultramicropores, not their sizes.71 On the other hand, slightly larger ultramicropores in BP-U-450 (0.64 nm) than in the other samples of this series, with a similar volume, likely result in a decrease in the strength of oxygen adsorption, decreasing the efficiency of the contribution of the adsorption-mediated part of the ORR process. Indeed, the performance of this sample is worse among all urea modified samples. This increase in the pore size is likely caused by blocking of the smallest pores by the bulky functional groups, likely amine-type, formed at a low temperature range of the heat treatment. It is interesting that the ultramicropore blocking did not take place for BP-O-U-450, suggesting that a change in surface chemistry upon oxidation resulted in a change in the mode of urea interactions with the surface and, as a result, in its thermal decomposition pattern and in a final effect on carbon surface chemistry. Indeed, in this case the chemical interactions of the carboxylic and carbonyl surface groups with urea and the products of its decomposition (NH3, HNCO) were expected.72
| Sample | S BET (m2 g−1) | S >2nm (m2 g−1) | V mic (cm3 g−1) | V <0.7nm (cm3 g−1) | V tot (cm3 g−1) |
|---|---|---|---|---|---|
| BP | 1609 | 316 | 0.54 | 0.10 | 2.71 |
| BP-U-450 | 1346 | 280 | 0.44 | 0.08 | 2.29 |
| BP-O-U-450 | 1336 | 269 | 0.45 | 0.10 | 1.94 |
| BP-U-850 | 1472 | 305 | 0.49 | 0.09 | 2.53 |
| BP-O-U-850 | 1449 | 302 | 0.47 | 0.11 | 2.09 |
| BP-TU-450 | 704 | 209 | 0.19 | 0.03 | 1.63 |
| BP-O-TU-450 | 688 | 198 | 0.19 | 0.03 | 1.29 |
| BP-TU-850 | 1396 | 279 | 0.47 | 0.10 | 2.33 |
| BP-O-TU-850 | 1291 | 265 | 0.43 | 0.08 | 1.92 |
In the case of the thiourea-modified samples, the samples heated at 450 °C, BP-TU-450 and BP-O-TU-450, adsorbed small amounts of nitrogen, likely due to pore clogging by the partially decomposed thiourea residue, as seen in the TG curves (Fig. 3(B)). For these two samples, the effect of oxidation is only seen in the slightly smaller amount of nitrogen adsorbed at high p/po. The difference in the porosity between BP-O-U-450 and BP-O-TU-450 indicates the difference in the chemical interface formed upon the treatment applied. The pore size distributions show (Fig. 3(D)) that the smallest ultramicropores in the thiourea-modified samples are in BP-O-TU-850, and this factor might contribute to its good performance through an increase in the strength of oxygen adsorption. The worse performing samples in this series in terms of the current density, BP-TU-450 and BP-O-TU-450, have the smallest volume of ultramicropores and thus are expected to adsorb the least oxygen among all samples through a physical adsorption mechanism. It is interesting that BP-O-TU-450 shows a relatively good performance in terms of the number of electron transferred. This complex behavior of the thiourea modified samples suggests the various extents of the contributions of porosity and chemistry to the efficiency of the ORR process and it is also reflected in the differences in the Tafel plot for the samples in this series.
Potentiometric titration experiments were performed to evaluate the acid–base character of the carbon surface. This method was expected to bring a complementary view of surface chemistry to that of the XPS analysis,40 discussed below. The proton binding curves are presented in Fig. 3(E) and (F). Calculated from them the pKa distributions of the surface protonated/deprotonated groups and their amounts are shown in Fig. S9 and Table S1 (ESI†). For the urea-modified series, marked differences in an acid–base behavior are visible in the extent of the proton uptake (basic character – positive values) and proton release (acidic character – negative values).59 Based on the proton binding curves, among the urea modified samples only BP-O-U-850 (Fig. 3(E)) exhibits a basic character and the other three samples are amphiprotic, as is as-received BP.52 BP-U-450 has almost equal amounts of acidic and basic groups. The pH values, representing the average number and strength of the acidic groups for BP, BP-U-450, BP-O-U-450, BP-U-850, and BP-O-U-850 are 8.0, 6.9, 7.5 7.6, and 7.9, respectively. The samples also differ in the amounts of groups detected in our experimental window between pH 3–11. On BP, BP-U-450, BP-U-850, BP-O-U-450 and BP-O-U-850 0.41, 0.42, 0.85, 0.67, and 0.65 mmol g−1 were detected, respectively. Interestingly the number of dissociating groups on BP-U-850 is highest among all samples tested, and almost equal to that on BP-oxidized. Nevertheless, the groups on the surface of these two samples are of different strength/chemistry and those on the latter sample are much more acidic since its pH is 4.7. On the other hand, BP-U-850 and BP-O-U-450 have similar pH values but the latter has less groups and the marked difference in the amount of the groups with pKa 10–11 (0.67 mmol g−1 for BP-U-850 and 0.35 mmol g−1 for BP-O-U-450) (Fig. S8 and Table S1, ESI†). There is also a marked difference between the samples heated at 850 °C. The oxidized sample is more basic on average and has a smaller number of groups. This consistently shows the differences in the interactions of urea and its decomposition products with the as-received and oxidized samples. Differences in the quality and quantity of the protonated groups might affect the wettability73 and thus mass transfer to small pores of the catalysts. An increase in pH upon the modifications applied was expected, following the incorporation of the nitrogen groups to the carbon matrix.58 The trend in the acid–base behavior of the thiourea-modified samples with the heat treatment temperature is similar to that of their urea-treated counterparts. The average surface pH for BP-TU-450, BP-TU-850, BP-O-TU-450 and BP-O-TU-850 are 6.8, 7.5, 6.8 and 7.7, respectively. Preoxidation of the carbon did not result in significant differences in pH for the thiourea samples, contrarily to the temperature of the heat treatment. Even though the numbers of dissociating groups are very similar, treatment at 850 °C made the surface more basic and the proton binding curve of BP-O-TU-850 shows a predominant proton uptake process (Fig. 3(F)).
The surface chemistry was also evaluated in detail using XPS. The spectra of C 1s, O 1s, N 1s and S 2p with their deconvolutions are shown in Fig. S10, S11 and Table S2 of the ESI,† and the results are summarized in Fig. 4. All samples, even pristine BP, contain some nitrogen on their surface. Interestingly, since oxidation of BP removed all nitrogen (Fig. S8, ESI†), the nitrogen functionalities in the oxidized sample and urea- or thiourea-modified ones might be of different chemistry than those of the as-received carbon sample. This surface “cleaning” with nitric acid was not expected since, on the contrary, in some cases this oxidation method introduces nitrogen to a carbon surface.58 Thus, it is possible that nitrogen on the as-received sample was incorporated to some secondary carbon phase, which was oxidized upon the HNO3 treatment and removed as CO2/CO and NOx. The treatment with urea reintroduced nitrogen to the oxidized samples and slightly increased nitrogen in the as-received samples. BP-U-450 and BP-U-850 have very similar elemental contents, however the sample prepared at 850 °C has more nitrogen in a pyridone form. These species are important sites for oxygen reduction, as suggested by Cazorla-Amoros and coworkers74 and their presence might contribute to the much better performance of this catalyst. Thus, the highest contribution of these species along with quaternary nitrogen, only detected in BP-O-U-850, might explain its excellent catalytic activity in the ORR. This sample also has the smallest oxygen content and highest percentage of sp2 carbon which contributes to its hydrophobicity enhancing oxygen adsorption in small pores. Moreover, BP-O-U-850 is the only one in the urea-modified catalyst series that has the smallest ultramicropores and its surface is the most basic. Since free electrons can contribute to the basicity of the carbon matrix,75 their presence might also enhance the oxygen reduction process. BP-O-U-850 is also the only sample with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds and quinones were suggested as the catalytic centers for the ORR.76 Relatively high and similar oxygen contents in all urea-modified samples, but in BP-O-U-850, might be a consequence of the presence of oxygen in the modifier. The surface oxygen of the modified samples seems to differ from that on the BP sample and that in the oxidized one. In the latter sample, oxygen is mainly in phenols and carboxylic acids, as expected after HNO3 oxidation58 and this explains the acidic pH of this sample. On the other hand, the combined analysis of the C 1s, O 1s and N 1s suggests that oxygen in the urea-modified samples is in a close vicinity to nitrogen, in pyridones.
O bonds and quinones were suggested as the catalytic centers for the ORR.76 Relatively high and similar oxygen contents in all urea-modified samples, but in BP-O-U-850, might be a consequence of the presence of oxygen in the modifier. The surface oxygen of the modified samples seems to differ from that on the BP sample and that in the oxidized one. In the latter sample, oxygen is mainly in phenols and carboxylic acids, as expected after HNO3 oxidation58 and this explains the acidic pH of this sample. On the other hand, the combined analysis of the C 1s, O 1s and N 1s suggests that oxygen in the urea-modified samples is in a close vicinity to nitrogen, in pyridones.
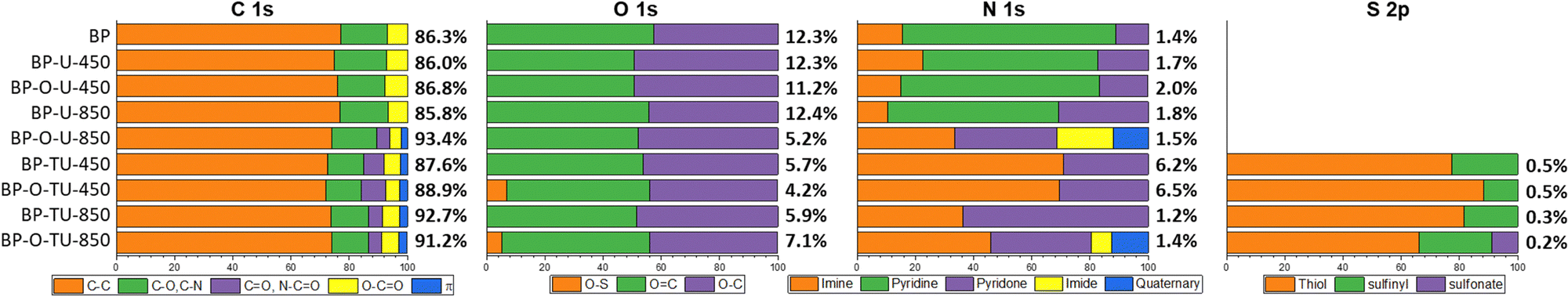 | ||
| Fig. 4 Atomic% of the element on the surface (in bold) and the results of the deconvolution of C 1s, O 1s, N 1s and S 2p core energy level spectra (in% contribution). | ||
The thiourea modified samples reveal a higher level of chemical heterogeneity than do their urea-modified-counterparts and, along with heterogeneity in the pore sizes, this factor is reflected in the complexity of the ORR on this series of catalysts. As in the case of urea, the high temperature of the heat treatment resulted in a high content of sp2 carbon, but in this case the oxygen content decreased dramatically for both samples. This might be a consequence of a lack of oxygen in thiourea and its different mode of interactions with the carbon surface, resulting in specific thermal decomposition patterns (Fig. S6B, ESI†). A reduction of the oxygen groups by the sulfur species (CS2) during the heating phase probably occurred. The lack of the significant effect on the surface pH, onset potential and reduction current of the thiourea modified samples, regardless of the pretreatment/oxidation might be caused by this process. A decrease in the contribution at about 288 eV (CO, C–NO) after heating at 850 °C can be only related to a decrease in the number of carbonyl groups, since the deconvolution of N 1s shows a dramatic increase in the contribution of pyridones, especially, for BP-TU-850. A decrease in the content of nitrogen from ∼6% to about 1% is associated with the decomposition of the partially pyrolyzed thiourea residue at ∼600 °C, as seen from the TA analysis (Fig. 3(B)). Between the two low-temperature-carbonized samples, BP-O-TU-450 has more pyridones than its as-received counterpart (Table S2, ESI†), which might be related to the difference in the oxygen speciation between BP and BP-O. The absolute content of nitrogen in BP-TU-450, BP-O-TU-450, BP-O-TU-800 and BP-O-TU-850 is 1.63, 1.98, 0.76, and 0.48 at%, respectively. The high number of pyridines acting as the ORR catalytic sites and their high densities on the surfaces of low temperature oxidized samples (owing to their low surface areas) might explain their relatively good performance in the number of transferred electrons, especially in the case of BP-O-TU-450. On the other hand, the residue (∼25%) of the not-fully-carbonized organic matter likely negatively affects the electron transfer through the carbon matrix resulting in the rather small current and kinetic current densities. Not without importance, is the quaternary nitrogen, which was detected only in BP-O-TU-850 and which might contribute to the good performance of this sample. Another marked difference between the urea- and thiourea-modified samples is the presence of sulfur in the latter. Even in a small amount (∼0.2%), that sulfur, when in the vicinity of nitrogen containing centers might be engaged in the oxygen reduction process.33 Based on the ratio of nitrogen to sulfur in the sample obtained at 450 °C (∼12), the residue existing in these samples is nitrogen-rich, implying the thermal instability of thiourea CS bonds. That ratio changes to 4 and 7 for BP-TU-850 and BP-O-TU-850, respectively, and the majority of sulfur in both samples is in thiophenic compounds, which might also contribute to the hydrophobicity and thus increase oxygen adsorption.
To probe the ability of our catalysts to adsorb/interact with oxygen, a change in the oxygen concentration in O2 saturated water was measured and the results are presented in Fig. 5(A) and (B). Since the porosities of the urea-modified samples are quite similar, as seen from nitrogen adsorption, the trends observed are expected to be mainly caused by the differences in the pore surface chemistry. The accessibility of oxygen to the hydrophobic pores is expected to be important and here the density of the dissociating groups in larger pores might play a marked role. Moreover, the electron rich-surface, even though it might attract oxygen, could also hinder its diffusion through the pore system when dissolved in water. Adsorption of oxygen on the urea-modified samples does not directly follow the trend in the total oxygen content measured by XPS, since, as discussed above, this method provides the content of both hydrophobic and hydrophilic oxygen groups. BP-U-450 adsorbs the least oxygen, followed by BP-O-U-450, BP-U-850 and BP-O-U-850, as the most oxygen attracting sample. This is the exact trend in the number of electron transferred and a high correlation between these two parameters is presented in Fig. 5(C). Surprisingly, for this series of samples a very similar dependence is found for the onset potential. Since the overall porosity of these series of samples is similar, the differences in oxygen adsorption are governed by surface chemistry. They must include both the affinity of oxygen to interact with various heteroatom-based groups, including pyridines, pyridones, quaternary nitrogen,32 quinones,76 and ethers47 and the efficiency of mass transfer of oxygen dissolved in water to enter deeply into the pore system and to be physically adsorbed in ultramicropores. Owing to the complexity of oxygen adsorption/interactions with heteroatom-based catalytic centers, and difficulties in calculating their combined amount, no sound dependence between the onset potential and that quantity could be established. The efficiency of mass transfer indeed affects the onset potential and when the porosity is similar, as that of the urea modified samples, the onset potential is dependent on the surface ability to transport oxygen. It is visible in a dependence (R2 = 0.95) of the onset potential on the parameter Φ expressing the contribution of dissociating groups (derived from potentiometric titration) in the total heteroatom content (oxygen + nitrogen, from XPS) (Fig. 5(D)). It assumes that dissociating groups, owing to geometrical constrains, exist in pores larger than 2 nm and the surface area in those pores are similar for this series of carbons. Indeed, those pores are the ones large enough to facilitate mass transfer and to accommodate the surface functionalities (Fig. 5(D)). Dissociating groups provide hydrophilicity and thus improve surface wetting, while other groups will repel water solutions.
 | ||
| Fig. 5 Change in oxygen concentration upon immersion (at time 0) of urea-modified samples (A) and thiourea modified samples (B) in oxygen saturated water. Dependence of the number of electron transfers for the urea-modified samples (C, left) and of the onset potential for the urea- and thiourea-modified samples (C, right) on the amount of adsorbed oxygen. Dependence of onset potential on Φ (the ratio of the number dissociating groups, in mmol g−1, derived from potentiometric titration, and the total heteroatom content, in at% (oxygen + nitrogen, from XPS), for the urea-modified samples) (D). ECSA of the different samples (E). | ||
In the case of the thiourea-modified samples the situation is much more complex since their chemistry is more convoluted and differences in porosity are also expected to affect the ORR performance. That complexity is seen in the results from oxygen adsorption tests (Fig. 5(B)), which show that the samples obtained at 450 °C, upon immersion in water, briefly increased the oxygen concentration, which is likely caused by their high affinity to chemisorb oxygen from the atmosphere on the outer surface. It is important to mention that only these two samples have the under pyrolyzed phase of the N-rich thiourea deposit. In the analysis of the oxygen affinity of these samples, a decrease in the initial oxygen concentration was taken into consideration. As seen in Fig. 5(C), for this group of catalysts a linear dependence of the onset potential on the extent of oxygen adsorbed was found with R2 = 0.87. However, no dependence of n on the amount of oxygen adsorbed could be established and no trend in the onset potential versus the fraction of dissociating groups were found, probably due to the complexity of the mechanism involving the effect of heteroatom based catalytic centers and that of the sizes of ultramicropores.
Interestingly, our evaluation of the affinity of the catalysts to adsorb water vapor indicated a similar behavior of all samples and a high affinity to attract water from its saturated vapors (Fig. S12, ESI†).
To complete the analysis of the electrochemical activity, we measured ECSA and the results are collected in Fig. 5(E). Even though this parameter is considered to reflect the specific activity of a catalyst, the collected results rather suggest that it is of a limited significance for highly porous electrocatalysts whose combined chemistry and porosity, which, through not-electrochemically-active components, play an important role in a catalytic process. The only general trend, which could be found in these values, are smaller ECSA for the preoxidized samples than for the as-received ones for the samples heated at 850 °C and, on the contrary, higher values for the preoxidized samples than for the received counterparts for the samples heated at 450 °C. The differences observed are larger, in general, for the urea-modified samples than for those treated with thiourea. This might suggest that surface chemistry is more important for the activity of the low temperature heated samples than for those obtained at 850 °C, where the changes in the carbon matrix are naturally more pronounced, and such factors as porosity and hydrophobicity play an important role in the observed activity.
4. Conclusions
The results collected and discussed in this paper showed both the complexity of the ORR process on porous and heteroatom N- and S-modified carbon materials and the direct effect of oxygen adsorption and its interactions with the carbon surface on this process. Even though N-, S-modified porous carbons have already been studied as ORR electrocatalysts, by investigating carbons with practically the same porosity in terms of pore sizes and volumes, as those CBs modified with urea, we have shown that both oxygen adsorption in pores and that governed by surface chemistry directly affect the number of electron transferred and the onset potential. With an assumed constant contribution of the former, we have clearly shown the advancing effect of the latter on the efficiency of the ORR. Here, we demonstrated that by combining the activities of both processes, although different in their principles, 4 electrons can be transferred and the reduction can initiate at favorable values of potentials, close to that on Pt/C. Moreover, highly porous carbon black, a commodity, can be converted into an efficient ORR catalyst by simple modification methods. For a good performance, not only surface hydrophobicity in ultramicropores is important, as promoting oxygen adsorption, but also hydrophilic surface in larger pores, as enhancing mass transfer of an aqueous electrolyte with dissolved oxygen to small pores, to take full advantage of such a dual system. Even though one might consider this kind of arrangement as hard to find in materials, and maybe difficult to synthesize, the unique nature of porous carbon surface makes such an arrangement feasible and relatively easy to achieve. In the carbon pores of small sizes is exactly where the adsorption of oxygen is the strongest (<0.7 nm), where dissociating groups have very small probability to exist, and where heteroatom are expected to exist only if introduced to carbon rings in specific synthesis efforts. The surface in larger pores, on the other hand, can easily be modified and accommodate not only electrochemically active heteroatom-based catalytic centers but also hydrophilic groups helping oxygen dissolved in an electrolyte to reach these centers. Along with ultramicropores withdrawing oxygen and, by its strong adsorption, participating in its reduction process, this configuration is a promising one leading to the development of efficient ORR catalysts competitive to those based on noble metals.Conflicts of interest
There are no conflicts to declare.References
- S. Sui, X. Y. Wang, X. T. Zhou, Y. H. Su, S. Riffatc and C. J. Liu, J. Mater. Chem. A, 2017, 5, 1808–1825 RSC.
- G. Wu and P. Zelenay, Acc. Chem. Res., 2013, 46, 1878–1889 CrossRef.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef.
- R. P. Jia, Z. Z. Gan, H. Huang and Z. M. Sheng, Electrochim. Acta, 2021, 368, 137617 CrossRef.
- Z. Z. Gan, Z. M. Sheng, H. Huang, X. Y. Dai, R. L. Niu and R. P. Jia, Sustainable Energy Fuels, 2019, 3, 3335–3343 RSC.
- J. Han, J. Bian and C. Sun, Research, 2020, 2020, 9512763 Search PubMed.
- X. Liu and L. Dai, Nat. Rev. Mater., 2016, 1, 16064 CrossRef.
- A. B. Jorge, R. Jervis, A. P. Periasamy, M. Qiao, J. Feng, L. N. Tran and M.-M. Titirici, Adv. Energy Mater., 2020, 10, 1902494 CrossRef CAS.
- J. Guo, J. R. Morris, Y. Ihm, C. I. Contescu, N. C. Gallego, G. Duscher, S. J. Pennycook and M. F. Chisholm, Small, 2012, 8, 3283–3288 CrossRef CAS PubMed.
- Z. Yang, H. Nie, X. Chen and S. Huang, J. Power Sources, 2013, 236, 238–249 CrossRef CAS.
- J. Zhang, Z. Xia and L. Dai, Sci. Adv., 2015, 1, e1500564 CrossRef PubMed.
- L. Yu, X. Pan, X. Cao, P. Hu and X. Bao, J. Catal., 2011, 282, 183–190 CrossRef CAS.
- M. Shao, Q. Chang, J. P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed.
- R. Ma, G. Lin, Y. Zhou, Q. Liu, T. Zhang, G. Shan, M. Yang and J. Wang, npj Comput. Mater., 2019, 5, 1–15 CrossRef CAS.
- L. Chen, X. Xu, W. Yang and J. Jia, Chin. Chem. Lett., 2020, 31, 626–634 CrossRef CAS.
- X. Yan, Y. Jia and X. Yao, Chem. Soc. Rev., 2018, 47, 7628–7658 RSC.
- K. Srinivasu, B. Modak and S. K. Ghosh, J. Phys. Chem. C, 2014, 118, 26479–26484 CrossRef CAS.
- Y. Gong, M. Li and Y. Wang, ChemSusChem, 2015, 8, 931–946 CrossRef CAS PubMed.
- C. Huang, C. Chen, M. Zhang, L. Lin, X. Ye, S. Lin, M. Antonietti and X. Wang, Nat. Commun., 2015, 6, 7698 CrossRef PubMed.
- H. Li, W. Kang, L. Wang, Q. Yue, S. Xu, H. Wang and J. Liu, Carbon, 2013, 54, 249–257 CrossRef CAS.
- J. Liang, Y. Zheng, J. Chen, J. Liu, D. Hulicova-Jurcakova, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 51, 3892–3896 CrossRef CAS PubMed.
- C. Z. Zhang, R. Hao, H. B. Liao and Y. L. Hou, Nano Energy, 2013, 2, 88–97 CrossRef CAS.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS.
- J. Liang, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 51, 11496–11500 CrossRef CAS.
- Z. Yang, Z. Yao, G. Li, G. Fang, H. Nie, Z. Liu, X. Zhou, X. a Chen and S. Huang, ACS Nano, 2012, 6, 205–211 CrossRef CAS.
- S. Yang, L. Zhi, K. Tang, X. Feng, J. Maier and K. Müllen, Adv. Funct. Mater., 2012, 22, 3634–3640 CrossRef CAS.
- C. H. Choi, S. H. Park and S. I. Woo, ACS Nano, 2012, 6, 7084–7091 CrossRef CAS.
- D. Von Deak, E. J. Biddinger, K. A. Luthman and U. S. Ozkan, Carbon, 2010, 48, 3637–3639 CrossRef CAS.
- S. Wang, L. Zhang, Z. Xia, A. Roy, D. W. Chang, J. B. Baek and L. Dai, Angew. Chem., Int. Ed., 2012, 51, 4209–4212 CrossRef CAS.
- M. Florent, R. Wallace and T. J. Bandosz, ChemCatChem, 2019, 11, 851–860 CrossRef CAS.
- Z. M. Sheng, C. Y. Hong, N. N. Li, Q. Z. Chen, R. P. Jia, D. Y. Zhang and S. Han, Electrochim. Acta, 2018, 259, 1104–1109 CrossRef CAS.
- J. Quilez-Bermejo, E. Morallon and D. Cazorla-Amoros, Carbon, 2020, 165, 434–454 CrossRef CAS.
- D. Li, Y. Jia, G. Chang, J. Chen, H. Liu, J. Wang, Y. Hu, Y. Xia, D. Yang and X. Yao, Chem, 2018, 4, 2345–2356 CAS.
- R. G. Morais, N. Rey-Raap, J. L. Figueiredo and M. F. R. Pereira, Beilstein J. Nanotechnol., 2019, 10, 1089–1102 CrossRef PubMed.
- K. M. Eblagon, N. Rey-Raap, J. L. Figueiredo and M. F. R. Pereira, Appl. Surf. Sci., 2021, 548, 149242 CrossRef.
- A. Gabe, R. Ruiz-Rosas, C. González-Gaitán, E. Morallón and D. Cazorla-Amorós, J. Power Sources, 2019, 412, 451–464 CrossRef.
- D. Eisenberg, P. Prinsen, N. J. Geels, W. Stroek, N. Yan, B. Hua, J.-L. Luo and G. Rothenberg, RSC Adv., 2016, 6, 80398–80407 RSC.
- T. J. Bandosz, Nanomaterials, 2021, 11, 407 CrossRef PubMed.
- T. J. Bandosz, Carbon, 2022, 188, 289–304 CrossRef.
- C. O. Ania, P. A. Armstrong, T. J. Bandosz, F. Beguin, A. P. Carvalho, A. Celzard, E. Frackowiak, M. A. Gilarranz, K. László, J. Matos and M. F. R. Pereira, Carbon, 2020, 164, 69–84 CrossRef.
- T. J. Bandosz, Chem. Rec., 2016, 16, 205–218 CrossRef CAS.
- Y. Jiang, L. Yang, T. Sun, J. Zhao, Z. Lyu, O. Zhuo, X. Wang, Q. Wu, J. Ma and Z. Hu, ACS Catal., 2015, 5, 6707–6712 CrossRef CAS.
- M. Seredych, A. Szczurek, V. Fierro, A. Celzard and T. J. Bandosz, ACS Catal., 2016, 6, 5618–5628 CrossRef CAS.
- J. Encalada, K. Savaram, N. A. Travlou, W. Li, Q. Li, C. Delgado-Sánchez, V. Fierro, A. Celzard, H. He and T. J. Bandosz, ACS Catal., 2017, 7, 7466–7478 CrossRef CAS.
- D. Barrera, M. Florent, K. Sapag and T. J. Bandosz, ACS Appl. Energy Mater., 2019, 2, 7412–7424 CrossRef CAS.
- D. Barrera, M. Florent, M. Kulko and T. J. Bandosz, J. Mater. Chem. A, 2019, 7, 27110–27123 RSC.
- G. de Falco, M. Florent, J. Jagiello, Y. Q. Cheng, L. L. Daemen, A. J. Ramirez-Cuesta and T. J. Bandosz, iScience, 2021, 24, 102216 CrossRef CAS PubMed.
- J. Wang, G. Yin, Y. Shao, S. Zhang, Z. Wang and Y. Gao, J. Power Sources, 2007, 171, 331–339 CrossRef CAS.
- H. M. Fruehwald, I. I. Ebralidze, P. D. Melino, O. V. Zenkina and E. B. Easton, J. Electrochem. Soc., 2020, 167, 084520 CrossRef CAS.
- M. Tarasevich, P. Mazin and N. Kapustina, Russ. J. Electrochem., 2011, 47, 923–932 CrossRef CAS.
- G. V. Zhutaeva, V. A. Bogdanovskaya, E. S. Davydova, L. P. Kazanskii and M. R. Tarasevich, J. Solid State Electrochem., 2014, 18, 1319–1334 CrossRef CAS.
- G. de Falco, M. Florent and T. J. Bandosz, Carbon, 2022, 189, 230–239 CrossRef CAS.
- A. Appleby and J. Marie, Electrochim. Acta, 1979, 24, 195–202 CrossRef CAS.
- J. Oh, S. Park, D. Jang, Y. Shin, D. Lim and S. Park, Carbon, 2019, 145, 481–487 CrossRef CAS.
- Y. Dong and J. Li, Chem. Commun., 2015, 51, 572–575 RSC.
- H. Yang, Y. Ko, W. Lee, A. Züttel and W. Kim, Mater. Today Energy, 2019, 13, 374–381 CrossRef.
- G. Cheng, G. Liu, P. Liu, L. Chen, S. Han, J. Han, F. Ye, W. Song, B. Lan and M. Sun, Front. Chem., 2019, 766 CrossRef CAS PubMed.
- T. J. Bandosz and C. O. Ania, in Interface Science and Technology, ed. T. J. Bandosz, Elsevier, 2006, vol. 7, pp. 159–229 Search PubMed.
- J. Jagiello, T. J. Bandosz and J. A. Schwarz, Carbon, 1994, 32, 1026–1028 CrossRef CAS.
- J. Jagiello, Langmuir, 1994, 10, 2778–2785 CrossRef CAS.
- B. H. Suryanto and C. Zhao, Chem. Commun., 2016, 52, 6439–6442 RSC.
- R. Zhou, Y. Zheng, M. Jaroniec and S.-Z. Qiao, ACS Catal., 2016, 6, 4720–4728 CrossRef CAS.
- G. de Falco, M. Florent, A. De Rosa and T. J. Bandosz, J. Colloid Interface Sci., 2021, 586, 597–600 CrossRef CAS PubMed.
- A. Sadezky, H. Muckenhuber, H. Grothe, R. Niessner and U. Pöschl, Carbon, 2005, 43, 1731–1742 CrossRef CAS.
- S. Tischer, M. Bornhorst, J. Amsler, G. Schoch and O. Deutschmann, Phys. Chem. Chem. Phys., 2019, 21, 16785–16797 RSC.
- J. L. Figueiredo, M. F. R. Pereira, M. M. A. Freitas and J. J. M. Órfão, Carbon, 1999, 37, 1379–1389 CrossRef.
- J. R. Pels, F. Kapteijn, J. A. Moulijn, Q. Zhu and K. M. Thomas, Carbon, 1995, 33, 1641–1653 CrossRef.
- O. L. Li, Z. Shi, H. Lee and T. Ishizaki, Sci. Rep., 2019, 9, 1–10 CrossRef CAS.
- Z. D. Wang, M. Yoshida and B. George, Comput. Theor. Chem., 2013, 1017, 91–98 CrossRef CAS.
- E. Deliyanni and T. J. Bandosz, Langmuir, 2011, 27, 1837–1843 CrossRef CAS.
- J.-Z. Liang, Polym. Test., 2017, 62, 219–224 CrossRef CAS.
- D. Hulicova-Jurcakova, M. Seredych, G. Q. Lu and T. J. Bandosz, Adv. Funct. Mater., 2009, 19, 438–447 CrossRef CAS.
- M. Florent, K. Rotnicki, N. Przybylska, M. Sliwinska-Bartkowiak and T. J. Bandosz, Carbon, 2021, 185, 252–263 CrossRef CAS.
- M. J. Mostazo-López, D. Salinas-Torres, R. Ruiz-Rosas, E. Morallón and D. Cazorla-Amorós, Materials, 2019, 12, 1346 CrossRef.
- C. A. Leon y Leon, J. M. Solar, V. Calemma and L. R. Radovic, Carbon, 1992, 30, 797–811 CrossRef CAS.
- H. Zhang, K. Lv, B. Fang, M. C. Forster, R. Dervişoğlu, L. B. Andreas, K. Zhang and S. Chen, Electrochim. Acta, 2018, 292, 942–950 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00762b |
| This journal is © The Royal Society of Chemistry 2022 |