
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA detailed investigation of the core@shell structure of exchanged coupled magnetic nanoparticles after performing solvent annealing†
Kevin
Sartori
 abc,
Dris
Ihiawakrim
a,
Christophe
Lefèvre
a,
Solenn
Reguer
b,
Cristian
Mocuta
b,
Sylvie
Bégin-Colin
a,
Fadi
Choueikani
b and
Benoit P.
Pichon
*ac
abc,
Dris
Ihiawakrim
a,
Christophe
Lefèvre
a,
Solenn
Reguer
b,
Cristian
Mocuta
b,
Sylvie
Bégin-Colin
a,
Fadi
Choueikani
b and
Benoit P.
Pichon
*ac
aUniversité de Strasbourg, CNRS, Institut de Physique et Chimie des Matériaux de Strasbourg, UMR 7504, F-67000 Strasbourg, France. E-mail: Benoit.Pichon@unistra.fr; Fax: +33 03 88 10 72 47; Tel: +33 03 88 10 71 33
bSynchrotron SOLEIL, L’Orme des Merisiers, Saint Aubin – BP48, 91192 Gif-sur-Yvette, France
cInstitut Universitaire de France, 1 rue Descartes, 75231 Paris Cedex 05, France
First published on 29th September 2022
Abstract
Thanks to important advances in synthesis techniques, a wide collection of bimagnetic core–shell nanoparticles with tunable properties was reported in the literature. Such nanoparticles may combine two phases with different intrinsic magnetic properties (magnetization, anisotropy, coercive field, etc.). Core–shell structures with large interfaces usually favor efficient exchange coupling between both phases that may result in the enhancement of the effective magnetic anisotropy energy and of the coercive field. In this context, the chemical composition and the crystal structure of the core/shell interface in nanoparticles are crucial parameters to modulate efficiently their magnetic properties. Here, we report on the solvent mediated thermal annealing of Fe3−δO4@CoO nanoparticles in a high boiling point solvent. The structure of nanoparticles was investigated before and after thermal annealing by advanced characterization techniques such as high resolution transmission electron microscopy (HR-TEM), X-ray absorption spectroscopy (XAS), X-ray magnetic circular dichroism (XMCD) and diffraction anomalous near edge structure (DANES). The combination of DANES and XAS/XMCD allowed studying the cationic distribution in spinel and wüstite crystal structures as a function of site occupancy and oxidation state. To the best of our knowledge, it is the first time that DANES is performed to quantitatively investigate the chemical composition of biphasic metal oxide nanoparticles. Hence, we have investigated precisely the chemical composition of the spinel phase and that of the wüstite phase. Besides the partial solubilisation of the CoO shell observed by HR-TEM, thermal annealing favors the formation of a thicker intermediate Co-doped ferrite layer at the spinel/wüstite interface. Such significant modifications of the core@shell structure markedly influence interfacial coupling phenomena between the core and the shell, hence offering wide perspectives towards nanoparticles with tunable magnetic properties for a variety of applications.
Introduction
The design of complex nanostructures combining different crystal phases opens wide potential for advanced applications.1 Besides the combination of the properties of each phase into a single nano-object, the fine control of the large interface at the nanoscale usually affects intimately the physical properties of each phase by the other one and vice versa. Bimagnetic nanoparticles remarkably illustrate this purpose.2 The combination of two phases with different magnetic characteristics (anisotropy, magnetization, etc.) in a core@shell structure results in highly synergistic magnetic properties. The most interesting feature of this system is certainly the enhancement of the magnetic anisotropy energy of a soft magnetic nanoparticle after the growth of a harder magnetic phase onto its surface.3 The combination of both soft and hard magnetic phases results in the exchange coupling phenomenon at the core/shell interface. The pinning of soft spins by hard spins of the shell increases the magnetic anisotropy energy of the core, i.e. enhances its thermal stability. Therefore, such an approach is an efficient way to push back the superparamagnetic limit of most of the metal oxide nanoparticles by using few amounts of hard magnetic components. Nanoparticle sizes below 20 nm can be expected, thus preserving advantages resulting from a high surface-to-volume ratio such as the fine tuning of magnetic properties as a function of the size and shape.4 Furthermore, rare earth components which are critical raw materials usually involved in permanent magnets could be avoided.5The thermal decomposition of metal complexes at high temperatures (about 300 °C) became very popular during the last decade to generate metal oxide nanoparticles.6,7 Although it may apparently be rather simple to perform, the La Mer theory describes the mechanism pathway as a short burst of nucleation followed by a slow growth of nanoparticles and8 it is difficult to control nucleation and growth steps separately. Therefore, the fine control of extrinsic properties such as the narrow size distribution and a well-defined shape is still highly challenging, even when the operating conditions are slightly modulated. Large iron oxide nanoparticles (>20 nm) were produced through a multi-step seed-mediated growth process by performing successively the thermal decomposition of the same metal precursor.9–11 Although this approach allowed the narrow size distribution and the well-defined shape of nanoparticles, the fine tuning of magnetic properties is restricted to the size evolution.
The seed-mediated growth process is well adapted for the design of a wide panel of core–shell nanoparticles combining two different magnetic phases thanks to the very low lattice mismatch of crystal phases of a variety of metal oxides. For this purpose, Fe3−δO4 (partially oxidized magnetite) was repeatedly combined with CoxFe3−xO4 (0 ≤ x ≤ 1) or CoO phases in the last decade. Either direct12–,17 or reverse18–22 core–shell structures were investigated. The fine setting of synthesis parameters is particularly crucial to grow such phases at the surface of Fe3−δO4 nanoparticles. The concentration of the Co precursor influences the growth kinetics significantly. Low concentrations result in the diffusion of Co in vacancies or cation-exchange at the surface of the Fe3−δO4 nanoparticles,14,15,23–25 thus generating a CoxFe3−xO4 phase with a graded chemical composition from the surface which is a self-limited process when the stoichiometry is reached.25 In contrast, high concentrations favor faster decomposition kinetics resulting in the growth of CoO at the surface of crystal seeds.14–16,26 The surface of Fe3−δO4 nanoparticles consists of multiple crystal planes – the spherical shape being pure approximation – and it may favor the multiple growth of CoO guided by specific crystal orientations.14 Indeed, each crystal plane displays a specific surface energy which controls the growth kinetics differently from each facet.27
Besides the core–shell structures, pioneering studies push back the synthesis limits to grow a second shell onto the surface of the first shell, thus resulting in core@shell@shell nanoparticles.28–32 The creation of two different interfaces – core/shell and shell/shell – widely provided the possibility to modify the magnetic properties of nanoparticles. However, the exact chemical composition and the structure of these nanoparticles remain difficult to assess precisely. Indeed, the synthesis occurs at high temperatures (>300 °C), and the similarity of crystal phases and defects favors cationic diffusion at interfaces and/or partial solubilisation/recristalization.29,32 Recently, we showed that the CoO shell grown at the surface of Fe3−δO4 nanoparticles turned into cobalt ferrite when performing again the decomposition of the Fe precursor, leading to the growth of a second CoxFe3−xO4 shell.32 Fe3−δO4@CoO core@shell nanoparticles were also reported to contain an intermediate CoxFe3−xO4 layer. It was ascribed to the diffusion of Co2+ in vacancies at the surface layer of the Fe3−δO4 nanoparticles before the growth of the CoO shell.15,33 The repeated conduction of the thermal decomposition steps stimulates the formation of complex interfaces within nanoparticles. Therefore, the high temperature that is required to synthesize nanoparticles through thermal decomposition results in some instability of the core@shell structure.
Although such complex mechanisms result in unpredicted crystal phases (non-stoichiometric and defects), they are of high potential to generate unexpected magnetic properties. Therefore, understanding precisely the effect of the synthesis parameters on the crystal structure and the chemical composition of bimagnetic core@shell nanoparticles is a crucial purpose. Considering the high temperature (>300 °C) used for the thermal decomposition of metal complexes, we investigated the thermal stability of the core@shell structure and the potential ability of Fe and Co cations for further mobility across the core/shell interface. Fe3−δO4@CoO nanoparticles were annealed after dispersion in a high temperature boiling point solvent to study the core@shell structure including the size, the shape and the chemical composition of the core/shell interface. Besides usual characterization techniques (TEM, XRD and FTIR), we performed X-ray absorption spectroscopy (XAS) and X-ray magnetic circular dichroism (XMCD) in order to investigate the distribution of Fe and Co cations in the spinel structure (MFe2O4) by considering oxidation states. The structure of nanoparticles was fully described by performing diffraction anomalous near edge structure (DANES) in order to quantify precisely the distribution of Fe and Co in both the spinel and wüstite structures. These results were confronted to the magnetic properties and, more precisely, exchange interactions at the core/shell interface which were investigated by magnetometry.
Experimental section
Synthesis of the Fe(II) stearate precursor
Iron(II) stearate was synthesized following an already published protocol34,35 by precipitation of sodium stearate (98.8%, TCI) and ferrous chloride (99% Acros Organic) salt in distilled water. Typically, 9.8 g (32 mmol) of sodium stearate was dissolved in 320 mL of distilled water. The aqueous solution was stirred at reflux for 30 min until the complete dissolution of the stearate. Then, 3.16 g of iron(II) chloride tetra hydrate (16 mmol) was dissolved in 160 mL of distilled water and was added to the previous mixture under vigorous stirring. A light orange precipitate immediately appears and the solution was kept at reflux and under stirring for 15 min. The mixture was then cooled down to room temperature and the light orange precipitate was separated by centrifugation (14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 10 min), followed by a filtration using a Buchner funnel. The obtained powder was washed 3 times with distilled water before being dried for 14 h in an oven at 65 °C.
000 rpm, 10 min), followed by a filtration using a Buchner funnel. The obtained powder was washed 3 times with distilled water before being dried for 14 h in an oven at 65 °C.
Synthesis of the Co(II) stearate precursor
Cobalt stearate was synthesized through the adaptation of the iron(II) stearate protocol:A 1 L two-necked round bottom flask was charged with 9.8 g (32 mmol) of sodium stearate (98.8%, TCI) and 320 mL of distilled water. The mixture was heated at reflux under magnetic stirring until all the stearate was dissolved. Afterwards, 3.16 g (16 mmol) of cobalt(II) chloride hexahydrated dissolved in 160 mL of distilled water was poured in the round bottom flask. The mixture was kept another 15 min at reflux and under magnetic stirring. Then, the solution was allowed to cool down. The orange precipitate was collected by centrifugation (15000 rpm, 5 min) and washed by filtration using a Buchner funnel. Finally, the powder was dried in an oven at 65 °C for one night.
Synthesis of the pristine iron oxide core
The synthesis of Fe3−δO4@CoO core@shell (CS) nanoparticles was adapted from our previous work.13,36 In a first step, iron oxide Fe3−δO4 (C) nanoparticles were synthesized by performing the thermal decomposition of a homemade iron(II) stearate in the presence of oleic acid (OA, 99%, Alfa Aesar) in dioctylether (DOE, 99%, Sigma). The iron stearate (1.38 g for FeSt2, 2.2 mmol) was mixed with OA (1.24 g, 4.4 mmol) in 20 mL of DOE in a two-necked round-bottom flask. The mixture was stirred and heated at 120 °C for 30 min without a reflux condenser in order to remove volatile molecule residues during the solubilisation process. The reflux condenser was then connected to the flask and the solution was heated under air to reflux for 2 h (boiling point (Bp ≈ 290 °C)) at a heating rate of 5 °C min−1. Afterwards, the black solution was cooled down to room temperature and washed four times by centrifugation at 16000 rpm for 5 min by adding acetone and chloroform. The final suspension of nanoparticles was stored in chloroform. In a second step, Fe3−δO4@CoO core@shell (CS) nanoparticles were synthesized through a seed-mediated growth approach. 90% of the Fe3−δO4 (C) nanoparticles were poured in a two-necked round bottom flask. After chloroform was evaporated under vacuum, 10 mL of ether dioctyl was added. Then, 1.246 g of cobalt(II) stearate (2 mmol), 20 mL of 1-octadecene (BP = 320 °C) and 1.130 g of oleic acid (4 mmol) were poured in the flask. The solution was then sonicated for another 5 min and heated to 120 °C for 30 min under magnetic stirring to remove water and solvent residues. Then, the reaction medium was brought to reflux for 120 min under air at a heating ramp of 1 °C min−1. After cooling down, the nanoparticles were washed by centrifugation (14000 rpm, 5 min) using a chloroform: acetone (1:7) mixture. The CS nanoparticles were finally stored as a colloidal suspension in chloroform.
Thermal annealing of CS nanoparticles
The CS colloidal suspension in chloroform was added in a two-necked round bottom flask in order to evaporate the solvent under vacuum. The nanoparticles were then dispersed in 20 mL of dioctylether (BP = 290 °C) and 13.98 mg (5 × 10−5 mol) of oleic acid was added. The mixture was ultra-sonicated for 2 min and was then heated to 100 °C under magnetic stirring for 30 min. The mixture was then heated to reflux at a heating rate of 5 °C min−1. After 2 h, the reaction medium was cooled down to 100 °C. The nanoparticles were precipitated by the addition of an excess of acetone followed by centrifugation (14000 rpm, 5 min). A chloroform:acetone (1:5) mixture was added twice before centrifugation in order to remove an excess of oleic acid and possible side products. Finally, the nanoparticles were stored as a black suspension in chloroform.
Characterization techniques
High-resolution transmission electron microscopy (HR-TEM) was performed using a JEOL 2100 LaB6 TEM with a 0.2 nm point to point resolution. EDX spectroscopy was performed using a JEOL Si(Li) detector. The nanoparticle suspension was drop casted onto a TEM grid. The nanoparticle size was calculated by measuring at least 300 nanoparticles from TEM micrographs by using the Image J software. The mean size of nanoparticles and deviation were calculated by fitting the size distribution using a Gaussian function. The mean shell thickness corresponds to half of the difference between nanoparticle sizes which were measured before and after the second and third decomposition steps. Granulometry measurements based on dynamic light scattering (DLS) were performed on the suspension of nanoparticles in chloroform using a nanosize MALVERN (nano ZS) apparatus.Fourier transform infrared (FTIR) spectroscopy was performed using a PerkinElmer Spectrum spectrometer in the energy range of 4000–400 cm−1 on samples diluted in KBr pellets.
X-Ray diffraction (XRD) measurements were carried out using a Bruker D8 Advance diffractometer equipped with a monochromatic copper radiation source (Kα = 0.154056 nm) and a Sol-X detector in the 2θ range of 20–70° with a scan step of 0.02°. High purity silicon powder (a = 0.543082 nm) was mixed with nanoparticles in order to be systematically used as an internal standard. Crystal sizes were calculated by using the Debye formula.
X-Ray absorption spectroscopy (XAS) and X-ray magnetic circular dichroism (XMCD) signals were recorded at both Fe L2,3 edges (700–735 eV) and Co L2,3 edges (770–805 eV) on the DEIMOS beamline at SOLEIL synchrotron.37 The measurement protocol was detailed earlier in ref. 38. The colloidal suspensions were drop-casted on silicon substrates and dried at room temperature. In contrast to other samples, CS_CoF1 was diluted in KBr powder to improve its stability during the measurement. The silicon substrates were affixed on a sample holder which was introduced into the cryomagnet.37 All spectra were recorded in total electron yields (TEYs) at 4.2 K, under UHV conditions (10−10 mbar) and under an applied magnetic field H (H+ = +6.5 Tesla and H− = −6.5 Tesla). The beam size was 800 × 800 μm2 and the photon energy resolution was 100 meV. XAS and XMCD spectra were plotted by considering the absorption cross-section measured with left (σL) and right (σR) circularly polarized X-rays. Isotropic XAS spectra were plotted as σXAS = (σ+ + σ−)/2 and XMCD spectra were plotted as σXMCD= (σ+ − σ−) where σ+ = [σL(H+) + σR(H−)]/2 and σ− = [σL(H−) + σR(H+)]/2. The circularly polarized X-rays are provided using an Apple-II HU-52 helical undulator for both XAS and XMCD measurements and by sweeping the magnetic field from +6.5 T to −6.5 T.37
Diffraction anomalous near edge structure (DANES) measurements were carried out on the Diffabs beamline at SOLEIL synchrotron using a fixed-exit double Si(111) monochromator and a macro-beam mode, with a beam spot size of about 300 μm2. Samples consist of drop-cast films on a silicon wafer which were mounted on the 6 + 2 circle diffractometer. The scans have been performed at both the Fe and Co K-edges (7.11 and 7.71 keV, respectively) in a range from −50 eV to +50 eV relative to the edges with a step of 1 eV. The intensity of the incident beam onto the sample was monitored using a thin Si photodiode, while the diffracted signals were acquired using a 2D XPAD detector.39,40 The different diffraction patterns associated with each energy step were obtained by the integration of the powder diffraction rings. The different Bragg peaks were then refined using a Pearson-VII function and the experimental DANES spectra were thus obtained by plotting the area of the peak vs. energy. Finally, refinements of spectra corresponding to the selected node were recorded by using the FDMNES code41 which led to the quantitative analysis of the distribution of Co and Fe cations in the Oh and Td sites of spinel and wüstite phases. The spinel phase was refined by considering the 220, 311 and 511 Bragg peaks which do not superimpose to the wüstite Bragg peaks ones. The common 222 (spinel) + 111 (wüstite) Bragg peaks were then refined considering a linear combination of previous results with the theoretical computation of the Co1−xFexO4 compound. More details are given in the ESI.†
Magnetic measurements were carried out using a Superconducting Quantum Interference Device (SQUID) magnetometer (Quantum Design MPMS-XL 5). Samples consist of a suspension of nanoparticles in chloroform that were evaporated in a small container which was sealed in order to avoid nanoparticles to move upon applying the magnetic field. Temperature dependent zero-field cooled (ZFC) and field cooled (FC) magnetization curves were recorded as follows: (i) the sample was introduced in the chamber at room temperature and cooled down to 5 K without an applied magnetic field and after applying a careful degaussing procedure; (ii) a magnetic field of 7.5 mT was applied, and the ZFC magnetization curve was recorded upon heating from 5 to 400 K; (iii) the sample was cooled down to 5 K under the same applied field; (iv) the FC magnetization curve was recorded upon heating from 5 to 400 K. Magnetization curves were also recorded as a function of the magnetic field (M(H)) at 5 and 300 K. The sample was also introduced in the SQUID at 300 K and cooled down to 5 K without an applied magnetic field (ZFC curve). The magnetization was then measured at a constant temperature by sweeping the magnetic field from +7 T to −7 T, and then from −7 T to +7 T. To confirm the exchange bias effect, FC M(H) curves were recorded further after heating up to 400 K and cooling down to 5 K under a magnetic field of 7 T. The FC hysteresis loop was then measured by applying the same field sweep as for the ZFC curve. The coercive field (HC) and MR/MS ratio were measured from ZFC M(H) curves. The exchange bias field (HE) was calculated as the HC difference of each branch in FC M(H) curves. The magnetization saturation (MS) was measured from hysteresis recorded at 5 K and was corrected by considering the mass of metal oxide vs. oleic acid. With this aim, thermogravimetry (TG) analysis was performed in order to discriminate the mass of metal oxide from the organic coating. A SDTQ600 apparatus from TA instrument was used to perform measurements on dried powders under air from 20 to 600 °C at a heating rate of 5 °C min−1.
Results and discussion
Fe3−δO4@CoO core@shell (CS) nanoparticles were synthesized in a similar way as we reported earlier (Fig. 1).13 Iron oxide (Fe3−δO4) nanoparticles were first synthesized by performing the thermal decomposition of iron stearate in dioctylether (BP = 290 °C). Then, cobalt stearate was decomposed in the presence of octadecene (BP = 310 °C) in order to grow a CoO shell at the surface of Fe3−δO4 nanoparticles (C). The side products resulting from the incomplete decomposition of reactants were removed by centrifugation. Finally, CS nanoparticles were heated again in dioctylether for 2 h without any addition of metal precursors in order to perform thermal annealing (CSR nanoparticles). Oleic acid was added in the reaction medium before annealing in order to avoid the aggregation of nanoparticles and to enhance the colloidal stability of the suspension. | ||
| Fig. 1 Synthesis pathway of CS nanoparticles corresponding to the seed-mediated growth process from C nanoparticles and their thermal annealing that led to CSR nanoparticles. | ||
TEM micrographs show that the shape of C nanoparticles is rather well-defined and may be assimilated to spheres that display a narrow size distribution centered at 8.3 ± 0.8 nm (Fig. 2). The size distribution of CS nanoparticles was increased to 10.8 ± 1.3 nm, thus corresponding to a size variation of 2.5 nm (Table 1). It can be roughly ascribed to the formation of a CoO shell with an average thickness of 1.2 nm. Nevertheless, the size distribution of CS is broader than that of C and the shape slightly deviates from spheres which may result from the inhomogeneous growth of the CoO shell at the surface of pristine iron oxide nanoparticles.14,27 CSR nanoparticles that were annealed at high temperatures are smaller than CS. Their mean size of 9.7 + 1.4 nm corresponds to a size reduction of 1.1 nm, suggesting the partial loss of the CoO shell.
 | ||
| Fig. 2 (a–c) TEM micrographs. (d–f) Size distributions. (a and d) C nanoparticles. (b and e) CS nanoparticles. (c and f) CSR nanoparticles. | ||
| C | CS | CSR | |
|---|---|---|---|
| TEM size (nm) | 8.3 ± 0.8 | 10.8 ± 1.3 | 9.7 ± 1.4 |
| Size variation from C (nm) | — | 1.3 | 0.7 |
| Fe:Co at ratio by EDX |
— | 43:57 |
58:42 |
| Hydrodynamic diameter (nm) | 9.9 | 13.7 | 18.1 |
| Cell parameter spinel (Å) | 8.37 ± 0.01 | 8.42 ± 0.01 | 8.42 ± 0.01 |
| Cell parameter wüstite (Å) | — | 4.24 ± 0.01 | n.c. |
| Crystal size (nm) | 8.3 ± 0.1 | 10.4 ± 0.1 | 10.6 ± 0.1 |
The increase of the Fe:Co atomic ratio measured by EDX from CS (43:57) to CSR (58:42) agrees with the partial removal of the CoO shell. However, considering the volume variations of CS and CSR with respect to C and considering a CoO phase grown onto a Fe3−δO4 core of 8.3 nm, the Fe:Co ratios should be 69:31 and 83:17 for CS and CSR, respectively. Therefore, the Co content measured by EDX is much higher than expected which may be ascribed to Co diffusion at the surface of nanoparticles upon the growth of the CoO shell.33
HR-TEM micrographs were recorded to further investigate the structure of the core–shell nanoparticles. In both samples, the shell could not be discriminated from the core (Fig. 3) because of the similar electronic densities. Continuous lattice fringes were observed across CS and CSR nanoparticles which look like single crystals. It shows the good epitaxial relationship between the core and the shell. Fast Fourier transforms (FFT) calculated from regions of interest (white squares) corresponding to the center of nanoparticles show very narrow spots which agree with the high crystal quality of the nanoparticles. Spots were indexed to hkl reflections of the spinel structure (either magnetite or maghemite or CoFe2O4) although those of the wüstite structure (CoO) could not be observed because the hkl reflections overlap.32 The calculation of the reverse FFT after applying a Bragg filter avoided the incoherent background and allowed displaying lattice fringes of the spinel phase separately that correspond to the specific hkl reflections. The reverse FFTs show some defects for both CS and CSR although those calculated for Fe3−δO4 pristine nanoparticles did not show any defects.42 Furthermore, closer analysis on the edge of nanoparticles shows that there are more defects in the shell of both CS and CSR (see the ESI†). It shows that the CoO shell either before or after annealing at high temperatures contains more defects than the core. Although the lattice mismatch (<1%) between the spinel and the wüstite phases is small, the growth of CoO from different facets of the Fe3−δO4 core seems to result in strains that propagates from the core–shell interface as we observe recently in Fe3−δO4@CoFe2O4@Fe3−δO4 nanoparticles.43
 | ||
| Fig. 3 (a–c) CS and (d–f) CSR. (a and d) HR-TEM micrographs. (b and e) FFT corresponding to the region of interest (white square) in HR-TEM micrographs. (c and f) Inverse FFT calculated from the FFT after applying a Bragg filter. hkl reflections (circles in the FFT) were indexed by measuring the distances between lattice fringes that were observed in reverse FFT. Pictures in ranks 2 and 4 correspond to reverse FFTs that were calculated separately for each (hkl) reflection shown in (b and e). The [uvw] indexes in the upper right corner in (c and f) correspond to the zone axis. | ||
Infrared spectroscopy showed typical bands of metal oxide nanoparticles coated with oleic acid (Fig. 4a). Bands at 2922 cm−1 (νasC–H) and 2952 cm−1 (νsC–H) correspond to the hydrocarbon chains of oleic acid. Bands at about 1630 cm−1 (νasC–O) and 1460 cm−1 (νsC–O) are ascribed to carboxylic acid groups which interact with the surface of metal oxide nanoparticles. Finally, the region corresponding to the lowest wavelengths (750–400 cm−1) corresponds to the bands of metal oxide bonds (Fig. 4b). The pristine Fe3−δO4 displays a broad band from 750 to 475 cm−1 which is typical of partially oxidized magnetite.26 It consists of a main contribution centered at 590 cm−1 and smaller contributions at higher wavelengths (see arrows). CS and CSR show narrower bands centered at 601 and 594 cm−1 which agree with a structure closer to magnetite (574 cm−1) with a higher content in Fe2+. It may also be ascribed to an increasing content in cobalt ferrite (591 cm−1).44 Finally, a second band centered at 512 cm−1 is ascribed to Co–O bonds in CoO.45 Its intensity decreases significantly from CS to CSR which agrees with the size reduction of nanoparticles, i.e. a thinner CoO shell.
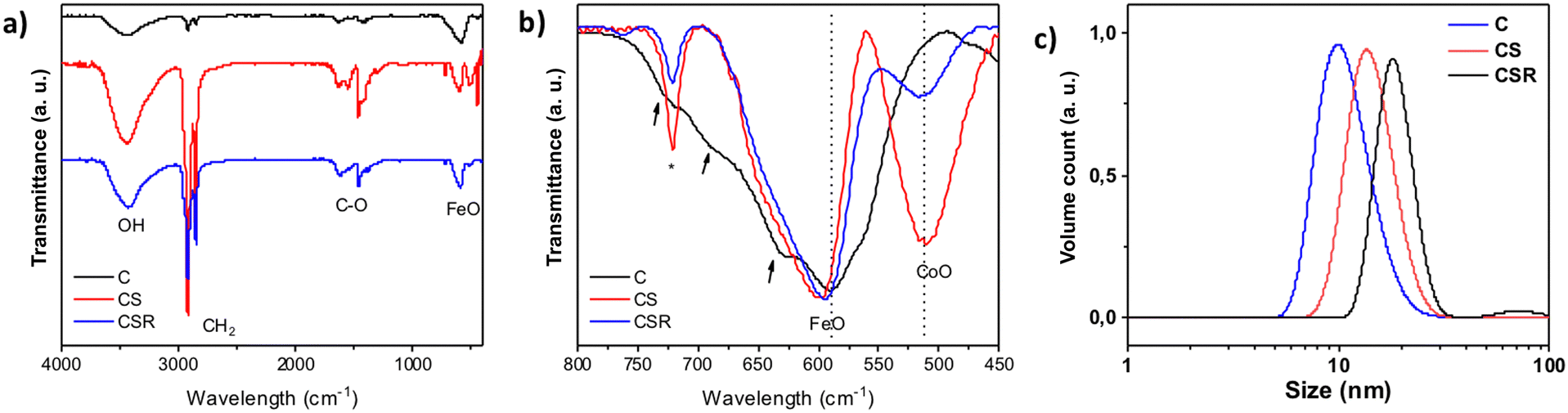 | ||
| Fig. 4 (a and b) Infrared spectra of C, CS and CSR. (c) Distributions of hydrodynamic diameters of nanoparticle suspensions in chloroform plotted as a function of volume counts. | ||
Granulometry measurements performed on colloidal suspensions in chloroform showed narrow size distributions of the hydrodynamic diameter which agree with the good colloidal stabilities of C, CS and CSR nanoparticles (Fig. 4c). The mean values of hydrodynamic diameters for C (9.9 nm) and CS (13.7 nm) are slightly larger than sizes measured from TEM micrographs because they include the oleic acid at the surface of nanoparticles. It is worthy of note that the hydrodynamic diameter of CSR (18.1 nm) is larger than the one of CS although it should be slightly smaller according to TEM micrographs. Such an increase can be correlated to the contribution centered at 80 nm observed in the case of intensity counts (see the ESI†) which is attributed to the partial aggregation of nanoparticles upon solvent-mediated annealing although most of nanoparticles remain non-aggregated.
XRD patterns recorded using a Cu cathode for each nanoparticle type displayed peaks that could be indexed to the Fd-3m spinel structure of iron oxide (Fe3O4, JCPDS card no. 19-062 and γ-Fe2O3, JCPDS card no. 39-1346) (Fig. 5). Additional peaks were attributed to the wüstite structure of CoO (JCPDS card no. 70-2856) in the patterns of CS and CSR. The intensity of the (222) and (400) reflections of the wüstite phase decreased from CS to CSR with respect to the ones of the spinel structure. These results agree with the size reduction of nanoparticles observed in TEM micrographs and with the lower intensity of the νCo–O band in the FTIR spectra which were ascribed to a thinner CoO shell in CSR than in CS. In contrast, peaks corresponding to the spinel structure become narrower from C, CS to CSR, agreeing with larger crystal domains which increased from 8.3 to 10.6 nm. Obviously, the crystal sizes of CS and CSR include Fe3−δO4 and the CoO shell that is supported by the crystal HR-TEM micrographs. The cell parameter of pristine Fe3−δO4 nanoparticles (8.37 ± 0.01 Å) is intermediate to those of magnetite (a = 8.396 Å) and maghemite (a = 8.338 Å). This feature agrees with the partial oxidation of Fe2+ at the surface of nanoparticles because they are exposed to air upon washing. Cell parameters calculated for CS and CSR are similar (8.42 ± 0.01 Å) and are larger than that of C and magnetite. It may be ascribed to the lattice mismatch between Fe3−δO4 and CoO phases because the cell parameter of CoO (a = 4.214 Å) is slightly larger than half of the one of magnetite (a/2= 4.198 Å). These results agree with strains resulting for the diffusion of cationic species at the spinel/wüstite interface as observed from TEM micrographs and as reported for cobalt ferrite nanoparticles.46
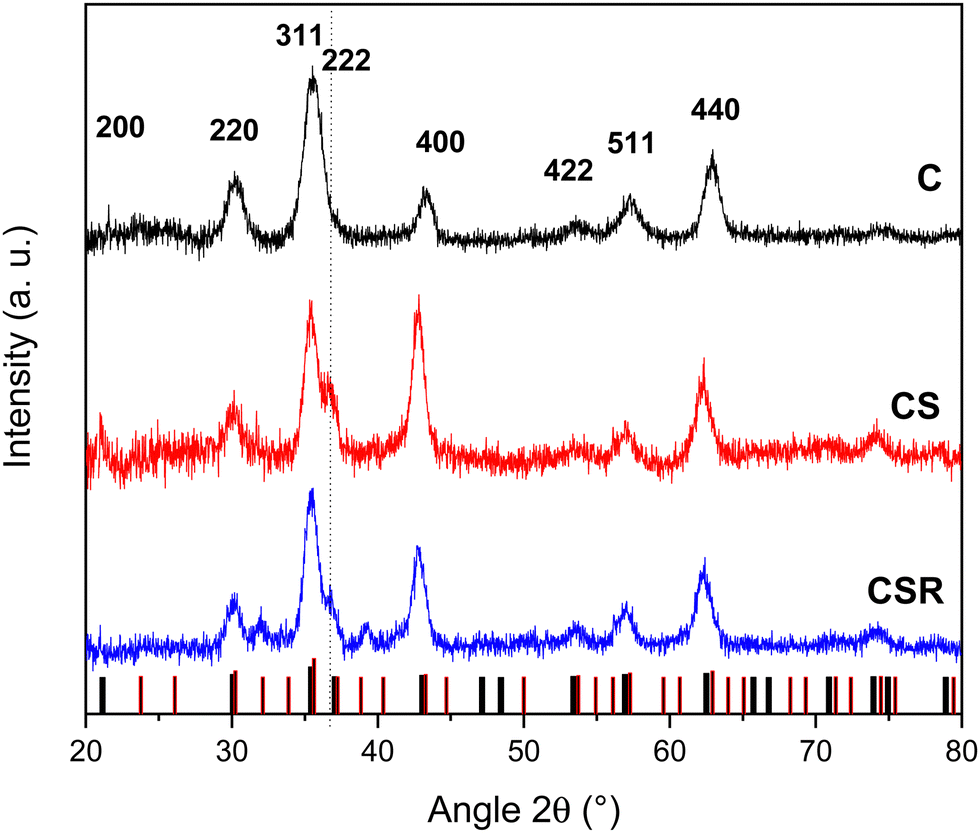 | ||
| Fig. 5 XRD patterns recorded with a monochromatic copper radiation (Kα = 0.154056 nm) for C, CS and CSR nanoparticles. Bar diagrams correspond to peak positions of the spinel (black) and wüstite (red) structures. | ||
The chemical compositions of CS and CSR nanoparticles were also investigated using soft X-ray absorption spectroscopy (XAS) and X-ray magnetic circular dichroism (XMCD) measurements (Fig. 6). XAS and XMCD spectra were recorded for C, CS and CSR to obtain complementary information on the oxidation state of cations as a function of their occupancy of octahedral (Oh) and tetrahedral (Td) sites. XAS spectra at the L2,3 Fe edge show two contributions that correspond to Fe2+ in Oh sites (I1) and Fe3+ in Oh and Td sites (I2) (Fig. 6a).47 Therefore, the increase of the intensity ratio I1/I2 from 0.57 (C) to 0.78 (CS) and further to 0.81 (CSR) agrees with a higher content of Fe2+.48 XMCD spectra recorded at the Fe edge are typical of ferrites (Fig. 6b).48 The negative contributions S1 and S3 correspond to the distribution of Fe2+/Fe3+ and Fe3+, respectively, in Oh sites. In contrast, the positive contribution S2 is ascribed to Fe3+ in Td sites. The intensity ratio S = (S1 + S2)/(S2 + S3) also increases from for C (0.75) to CS (0.77) and further to CSR (0.83). According to the theoretical values considered by Jungchaoren et al.49 for γ-Fe2O3 (0.69) and Fe3O4 (1.30) nanoparticles prepared by thermal decomposition, it corresponds to the gradual increase of Fe3O4 (10%, 13% and 23% for C, CS, CSR respectively) with respect to phases which do not contain Fe2+ (CoFe2O4 and γ-Fe2O3). Such a large increase between CS and CSR may result from the thinner wüstite shell since 80% of the XMCD signal arises from the first 2 nm deep from the nanoparticle surface.50 XAS spectra recorded at the Co L2,3 edges for CS and CSR correspond to similar chemical environments of Co2+ cations (Fig. 6c). The relative intensities of I3 and I4 peaks agree with the presence of CoO.20,51,66 XMCD spectra recorded at the Co L2,3 edges show an intense negative peak corresponding to Co2+ in Oh sites (Fig. 6d) of a spinel structure, i.e. cobalt ferrite.33,38,52 Such a contribution may also be partially ascribed to uncompensated spins at the surface of the CoO wüstite shell although it corresponds to a very weak signal.33 A small positive contribution corresponds to Co2+ in Td sites of the spinel structure. The magnitude of the XMCD signal with respect to that of XAS agrees with the coexistence of spinel cobalt ferrite and wüstite cobalt oxide (CoO).
 | ||
| Fig. 6 (a and c) XAS and (b and d) XMCD spectra at the (a and b) Fe L2,3 and (c and d) Co L2,3 edges of Fe3−δO4@CoO nanoparticles (CS) that have been subjected to reheating (CSR). | ||
The crystal structure and chemical composition of the spinel and the wüstite phases were investigated more deeply by performing DANES. Spectra were plotted from XRD patterns measured at different energy ranges according to Fe and Co-K edges with a high resolution of 1 eV. The integrated intensities of peaks corresponding to reflections of interest were plotted as a function of energy (Fig. 7, see the ESI† for more details). DANES spectra corresponding to the (220)s spinel node show a significant decrease of the integrated intensity for energy ranges corresponding to Fe and Co. The (220)s node being specific to the spinel structure, it shows that the Fe and Co atoms are present in Td sites. The investigation of the wüstite structure was highly challenging, because each node corresponding to the wüstite phase systematically overlap with those of the spinel phase. The DANES spectra corresponding to both (111)w and (222)s nodes of the wüstite and spinel phases, respectively, were refined by performing linear combinations on the basis of results obtained for (220)s, (311)s, and (511)s reflections which are specific to the spinel phase in order to estimate the contribution of the wüstite phase (see the ESI† for details). It is worth noting that the DANES spectra recorded at the Fe K edge also show a significant decrease of the integrated intensity which agree with the presence of Fe in the wüstite phase. The refinement of the DANES spectra corresponding to (220)s, (311)s, (511)s, (222)s and (111)w reflections led us to calculate the distribution of Fe and Co atoms in Td and Oh sites in the spinel and wüstite phases as follows:
| CS: (Fe0.89Co0.11)[Fe0.91Co0.09]2O4@Fe0.09Co0.91O |
| CSR: (Fe0.77Co0.23)[Fe0.80Co0.20]2O4@Fe0.03Co0.97O |
 | ||
| Fig. 7 DANES spectra recorded for (a and b) CS and (c and d) CSR. (a and c) (220) spinel nodes and (b and d) (222) spinel + (111) wüstite nodes at both Fe K-edge (left part) and Co K-edge (right part). Experimental (grey circles) and best fit (red curve). The low resolution of DANES spectra corresponding to wüstite in CSR agrees with the partial solubilisation of the shell. | ||
Discussion on the structure of nanoparticles
The contribution of Co2+ in the spinel phase increases from CS to CSR while the CoO shell is thinner. It unambiguously shows that heating to 290 °C for 2 h enhances the diffusion of cationic species at the spinel/wüstite interface. Such an increase of the Co content in the spinel phase of CSR can be assimilated to the presence of an interfacial cobalt ferrite layer between the Fe3−δO4 core and the wüstite shell.14,23,55,56 The diffusion of Co2+ is driven by vacancies in the Fe3−δO4 core and results in a gradient of concentration from the spinel/wüstite interface. We anticipate that such a mechanism is self-limited by the formation of stoichiometric cobalt ferrite which stops further cation exchange above a critical thickness.25,57 Therefore, we expect the chemical composition of the interfacial cobalt ferrite layer to be very close to stoichiometry. These results agree with the narrowing of XRD peaks from C to CS and further, to CSR and, with the slight shift of the M–O band in FTIR spectra to higher wavelengths. It is also confirmed by the Fe:Co ratios measured by EDX for CS and CSR which are lower than that of a core@shell structure with a well-defined interface (no cationic diffusion). In addition, DANES showed that the diffusion of Co2+ is concomitant to the partial solubilisation of the Fe3−δO4 core and to the subsequent recrystallization of Fe species upon the formation of the wüstite shell of CS, i.e. CoyFe1−yO. The significant decrease of Fe in the wüstite phase of CSR agrees with the transformation of unstable FeO into magnetite.54
According to the chemical compositions of the spinel and wüstite phases obtained by refining DANES patterns, we tentatively calculated the core size and shell thicknesses of CS and CSR. Considering the core@shell structure as spinel@wüstite and the original size of pristine Fe3−δO4 nanoparticles (8.3 nm), we expect the spinel core of CS to consist of a Fe3−δO4 core with a diameter of 7.8 nm and a Co-ferrite layer with a thickness of larger than 0.3 nm. For CSR, the Fe3−δO4 core diameter decreases down to 6.2 nm and the thickness of cobalt ferrite is 1.1 nm. Concerning the wüstite shell, we expect its thickness to correspond to the size variation observed in TEM micrographs with respect to pristine Fe3−δO4 nanoparticles C, e.g. 1.3 nm for CS and 0.7 nm for CSR.
The cationic distribution in such complex structures markedly influence the magnetic properties of CS and CSR nanoparticles which were investigated by SQUID magnetometry (Fig. 8 and Table 2). Temperature dependent magnetization curves recorded after zero field cooling (ZFC) all display a maximum magnetization that is shifted to higher temperatures from C, CS to CSR (Fig. 8a). It corresponds to the transition from blocked to unblocked magnetic moments when increasing the temperature. More precisely, the magnetic anisotropy energy of nanoparticles (KV, where K is the magnetic anisotropy constant and V is the volume of nanoparticles) becomes lower than thermal energy (kT, where k is the Boltzmann constant and T is the temperature). The derivative of the difference between the ZFC and FC M(T) curves (eqn (1)) describes more accurately the blocking temperature (TB) by considering a distribution of energy barriers.58
| f(TB) ≈ −(1/T)(d(MZFC–MFC)/dT) | (1) |
 | ||
| Fig. 8 Magnetic properties of C, CS and CSR nanoparticles. (a) Temperature dependent magnetization curves. (b) Distribution of blocking temperatures. Magnetization curves recorded against a magnetic field at (c) 300 K, (d) 10 K under zero field cooling (ZFC) and (e) 10 K after field cooling under 7 T. | ||
| C | CS | CSR | |
|---|---|---|---|
| H C 10 K ZFC (kOe) | 0.3 | 17.3 | 16.6 |
| H C 10 K FC (kOe) | 0.4 | 20.0 | 20.2 |
| H E 10 K FC (kOe) | — | 2.6 | 1.8 |
| T max (K) | 133 | 272 | 276 |
| T B (K) | 78 | 237 | 237 |
| M S at 10 K (emu g−1) | 56 | 38 | 45 |
M(H) curves recorded perfectly overlap at 300 K which correspond to unblocked magnetic moments, e.g. superparamagnetic behavior for each sample (Fig. 8c).4 The magnetization saturation (MS) of C (56 emu g−1) agrees with Fe3−δO4 nanoparticles which are partially oxidized.26,42 The lower MS for CS (38 emu g−1) results from the growth of antiferromagnetic wüstite which does not contribute to magnetization.33 In contrast, the increase of MS for CSR (45 emu g−1) perfectly agrees with the reduction of the volume of wüstite (40%) and the increase of the ferrimagnetic cobalt ferrite volume fraction.60 Although the volume of cobalt ferrite increases, it compensates the reduction of the Fe3−δO4 core by the same order of volume unit. Indeed, bulk CoFe2O4 displays similar MS (94 emu g−1) to that of magnetite (98 emu g−1).
M(H) curves recorded at 10 K after zero field cooling show an open-hysteresis which corresponds to blocked magnetic moments (Fig. 8d). Coercive fields (HC) measured for CS (17300 Oe) and CSR (16600 Oe) are much larger than that of pristine iron oxide nanoparticles (300 Oe) which is due to strong interfacial exchange coupling between the soft and hard magnetic phases. The soft spins pinned by hard spins at interfaces require a stronger magnetic field to reverse. Nevertheless, the decrease of HC from CS to CSR which is ascribed to slightly weaker interfacial exchange bias coupling. More details were obtained from the M(H) curves recorded after field cooling to 10 K under a field of 7 T (Fig. 8e). The hysteresis is shifted to lower magnetic fields which is typical of exchange bias effect resulting from exchange interactions at the interface between the ferrimagnetic (FiM) core and the antiferromagnetic (AFM) shell. Such a phenomenon can be quantified by calculating the exchange field (HE) which decreases from 2600 Oe (CS) to 1800 Oe (CSR). It is ascribed to the lower magnetic anisotropy energy of the thinner wüstite shell that reduces the pinning of soft spins by hard spins. Nevertheless, very similar HC (20000 Oe) measured from the FC M(H) curves of CS and CSR show that exchange bias coupling at the FiM/AFM interface is compensated by exchange coupling at the Fe3−δO4/CoFe2O4 interface.
Furthermore, some pinned spins in the AFM shell remained uncompensated as shown by the vertical shift of FC M(H) curves as usually observed for exchange bias coupled nanoparticles.61,62 It can be ascribed to the binary composition of the wüstite shell where Co spins are not fully compensated by Fe spins. The non-saturation of M(H) curves recorded at 10 K is ascribed to the high effective magnetic anisotropy energy of nanoparticles that is significantly enhanced by exchange coupling. One may notice the steep decrease of magnetization at zero field in both the ZFC and FC M(H) curves recorded for CSR which is attributed to the faster magnetic reversal of a population of nanoparticles. This behavior cannot be ascribed to small nanoparticles according to the monomodal size distribution of CSR. It may be attributed to a small fraction of nanoparticles that strongly interact through dipolar interactions which favor the magnetic moment reversal.63–65
As mentioned above, CSR consists of a Fe3−δO4 core and a FexCo1−xO shell with an intermediate layer of CoxFe3−xO4. According to the anisotropy constants (K(Fe3O4) ≈ 4.6 × 104 J m−3,66K(CoFe2O4) ≈ 3 × 105 J m−3)67,68 and K(CoO) ≈ 7 × 106 J m−369) and the volume of each phase in CS and CSR, the magnetic anisotropy energies (Table 3) agree with exchange coupling that occurs at both Fe3−δO4/CoxFe3−xO4 and CoxFe3−xO4/CoyFe1−yO interfaces. Therefore, Fe3−δO4 interfacial spins are pinned by those of CoxFe3−xO4 which are pinned by those of CoyFe1−yO. In CS, the intermediate shell is much thinner than its cell parameter, so it is better to consider a Co-doped ferrite with enhanced magnetic anisotropy energy (and intermediate anisotropy constant (K(Co0.29Fe2.72O4) = 1.2 × 105 J m−3). This consideration also agrees with a lower magnetic anisotropy energy of the spinel core (3.46 × 10−20 J m−3) than that of the wüstite shell (2.59 × 10−19 J m−3). It is worthy of note that the magnetic anisotropy of CoyFe1−yO is larger than the interfacial energy (Eint = HEMSD/6 where D is the diameter of FiM phases) of the spinel core in both CS (2.18 × 10−20 J) and CSR (1.19 × 10−20 J), thus consistent with exchange bias coupling.
| C | CS | CSR | |
|---|---|---|---|
| Fe3−δO4 | 1.33 × 10−20 | 9.41 × 10−21 | 4.90 × 10−21 |
| CoFe2O4 | — | 2.52 × 10−20 | 5.46 × 10−20 |
| CoyFe1−yO | — | 2.59 × 10−19 | 1.22 × 10−19 |
Although the Co-doped ferrite layer in CS contributes to increase the magnetic anisotropy energy of the spinel core of C through exchange coupling, it is much less efficient than the wüstite shell. In contrast, the enhanced diffusion of cationic species at the spinel/wüstite interface and the reduction of the wüstite shell by partial solubilisation from CS to CSR results in the increase of the magnetic anisotropy energy of the spinel core and in the decrease of that of the wüstite phase. Although it results in weaker exchange bias coupling at the spinel/wüstite interface in CSR, it is compensated by additional exchange coupling at the new Fe3−δO4/CoxFe3−xO4 soft/hard interface. Therefore, CS and CSR display similar effective magnetic anisotropy energies as shown by TB values.
Conclusion
Despite the effectiveness of the thermal decomposition method to synthesize exchange coupled nanoparticles with tunable magnetic properties, the thermal stability of the core@shell structure remained unexplored. We performed the solvent thermal annealing of Fe3−δO4@CoO nanoparticles in order to simulate the experimental conditions prior to the seed-mediated growth of an additional shell of a magnetic material as we reported recently.32,43 We studied the structure and magnetic properties of Fe3−δO4@CoO nanoparticles after thermal annealing at 290 °C in a high boiling temperature solvent. The morphology, size, chemical composition and crystal structure of nanoparticles were studied before and after thermal annealing by using advanced analysis techniques such as HR-TEM, XAS, XMCD and DANES. The magnetic properties were investigated by SQUID magnetometry.The size, chemical composition and structure were directly altered at high temperatures in a liquid medium. The size of nanoparticles was significantly reduced according to partial solubilisation of the CoO shell. Nevertheless, the size variation of nanoparticles measured from the HR-TEM micrographs after each synthesis step was not satisfactory to fully describe the nanoparticle structure. XAS/XMCD spectroscopy provided complementary information on the oxidation states of cations as a function of their site occupancy. The collected XAS and XMCD data demonstrated that Co cations diffuse in the spinel structure upon the formation of CS and the thermal annealing leading to CSR. Thanks to DANES measurements, the occupancy of Fe cations vs. Co cations in Td and Oh sites was quantified, separately in the spinel and wüstite phases, offering wide perspectives toward the fine understanding of the crystal structure and chemical composition of core@shell nanoparticles. Hence, it results in a reduced iron oxide core and in a thicker intermediate Co-doped ferrite layer. Fe cations are also present in the wüstite shell which consists of CoyFe1−yO. Indeed, the Fe3−δO4 pristine nanoparticles are certainly partially solubilized which leads to the subsequent crystallization of Fe species upon the decomposition of the Co precursor. Therefore, CS and CSR nanoparticles are better described as an onion structure that we tentatively assigned to Fe3−δO4@CoxFe3−xO4@CoyFe1−yO. Thermal annealing results in the reduction of both the Fe3−δO4 core and the CoyFe1−yO shell through partial solubilisation while the intermediate CoxFe3−xO4 shell expand through cation diffusion.
The modification of the nanoparticle structure results in similar HC and TB. However, it is not representative of the complexity of the magnetic structure of CS and CSR. Indeed, the variation of core sizes and shell thicknesses led to the reduction of the magnetic anisotropy energy of the FiM core and of the AFM shell which were compensated by that of the thicker CoxFe3−xO4 intermediate layer. The lower volume of the AFM phase from CS to CSR resulted in a significantly weaker exchange bias coupling between the FiM core and the AFM shell (HE is 40% lower). In contrast, the thicker cobalt ferrite intermediate hard magnetic layer resulted in exchange coupling with the FiM core. Although exchange bias coupling was reduced at the FiM/AFM interface, it was efficiently compensated by exchange coupling at the FiM/FiM interface. These results show that core@shell structures and the arising magnetic properties at the nanoscale can be easily modulated by thermal annealing in high boiling temperature solvents which is very appealing for the design of nanomagnets for advanced applications in areas such as biomedicine.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
K. S. was supported by a PhD grant from the French Agence Nationale de la Recherche (ANR) under the reference ANR-11-LABX-0058-NIE within the Investissement d'Avenir program ANR-10-IDEX-0002-02 and the SOLEIL synchrotron/Laboratoire Léon Brillouin fellowship. The authors are grateful to the SOLEIL synchrotron for providing the access to DEIMOS and DiffAbs beamlines and they thank the respective staff for support during experiments.References
- C.-W. Nan and Q. Jia, MRS Bull., 2015, 40, 719–724 CrossRef.
- J. Nogues, J. Sort, V. Langlais, V. Skumryev, S. Surinach, J. S. Munoz and M. D. Baro, Phys. Rep., 2005, 422, 65–117 CrossRef.
- A. López-Ortega, M. Estrader, G. Salazar-Alvarez, A. G. Roca and J. Nogués, Phys. Rep., 2015, 553, 1–32 CrossRef.
- S. Bedanta and W. Kleemann, J. Phys. D: Appl. Phys., 2008, 42, 013001 CrossRef.
- O. Gutfleisch, M. A. Willard, E. Brück, C. H. Chen, S. G. Sankar and J. P. Liu, Adv. Mater., 2011, 23, 821–842 CrossRef CAS PubMed.
- T. Hyeon, S. S. Lee, J. Park, Y. Chung and H. B. Na, J. Am. Chem. Soc., 2001, 123, 12798–12801 CrossRef CAS.
- A.-H. Lu, E. L. Salabas and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 1222 CrossRef CAS.
- V. K. La Mer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847–4854 CrossRef CAS.
- S. Sun and H. Zeng, J. Am. Chem. Soc., 2002, 124, 8204–8205 CrossRef CAS PubMed.
- J. Park, E. Lee, N.-M. Hwang, M. Kang, S. C. Kim, Y. Hwang, J.-G. Park, H.-J. Noh, J.-Y. Kim, J.-H. Park and T. Hyeon, Angew. Chem., Int. Ed., 2005, 44, 2872–2877 CrossRef CAS.
- M. Levy, A. Quarta, A. Espinosa, A. Figuerola, C. Wilhelm, M. García-Hernández, A. Genovese, A. Falqui, D. Alloyeau, R. Buonsanti, P. D. Cozzoli, M. A. García, F. Gazeau and T. Pellegrino, Chem. Mater., 2011, 23, 4170–4180 CrossRef CAS.
- I. Panagiotopoulos, G. Basina, V. Alexandrakis, E. Devlin, G. Hadjipanayis, L. Colak, D. Niarchos and V. Tzitzios, J. Phys. Chem. C, 2009, 113, 14609 CrossRef CAS.
- W. Baaziz, B. P. Pichon, C. Lefevre, C. Ulhaq-Bouillet, J.-M. Greneche, M. Toumi, T. Mhiri and S. Begin-Colin, J. Phys. Chem. C, 2013, 117, 11436–11443 CrossRef CAS.
- X. Liu, B. P. Pichon, C. Ulhaq, C. Lefèvre, J.-M. Grenèche, D. Bégin and S. Bégin-Colin, Chem. Mater., 2015, 27, 4073–4081 CrossRef CAS.
- T. Gaudisson, R. Sayed-Hassan, N. Yaacoub, G. Franceschin, S. Nowak, J.-M. Grenèche, N. Menguy, Ph Sainctavit and S. Ammar, CrystEngComm, 2016, 18, 3799–3807 RSC.
- G. Franceschin, T. Gaudisson, N. Menguy, B. C. Dodrill, N. Yaacoub, J.-M. Grenèche, R. Valenzuela and S. Ammar, Part. Part. Syst. Charact., 2018, 35, 1800104 CrossRef.
- N. Flores-Martinez, G. Franceschin, T. Gaudisson, P. Beaunier, N. Yaacoub, J.-M. Grenèche, R. Valenzuela and S. Ammar, Part. Part. Syst. Charact., 2018, 35, 1800290 CrossRef.
- E. Lima, E. L. Winkler, D. Tobia, H. E. Troiani, R. D. Zysler, E. Agostinelli and D. Fiorani, Chem. Mater., 2012, 24, 512–516 CrossRef CAS.
- E. L. Winkler, E. Lima, D. Tobia, M. E. Saleta, H. E. Troiani, E. Agostinelli, D. Fiorani and R. D. Zysler, Appl. Phys. Lett., 2012, 101, 252405 CrossRef.
- W. Baaziz, B. P. Pichon, Y. Liu, J.-M. Grenèche, C. Ulhaq-Bouillet, E. Terrier, N. Bergeard, V. Halté, C. Boeglin, F. Choueikani, M. Toumi, T. Mhiri and S. Begin-Colin, Chem. Mater., 2014, 26, 5063–5073 CrossRef CAS.
- W. Baaziz, B. P. Pichon, J.-M. Grenèche and S. Begin-Colin, CrystEngComm, 2018, 20, 7206–7220 RSC.
- G. C. Lavorato, E. L. Jr, D. Tobia, D. Fiorani, H. E. Troiani, R. D. Zysler and E. L. Winkler, Nanotechnology, 2014, 25, 355704 CrossRef PubMed.
- A. Uheida, G. Salazar-Alvarez, E. Björkman, Z. Yu and M. Muhammed, J. Colloid Interface Sci., 2006, 298, 501–507 CrossRef CAS PubMed.
- M. Sytnyk, R. Kirchschlager, M. I. Bodnarchuk, D. Primetzhofer, D. Kriegner, H. Enser, J. Stangl, P. Bauer, M. Voith, A. W. Hassel, F. Krumeich, F. Ludwig, A. Meingast, G. Kothleitner, M. V. Kovalenko and W. Heiss, Nano Lett., 2013, 13, 586–593 CrossRef CAS PubMed.
- S. Lentijo-Mozo, D. Deiana, E. Sogne, A. Casu and A. Falqui, Chem. Mater., 2018, 30, 8099–8112 CrossRef CAS.
- W. Baaziz, B. P. Pichon, S. Fleutot, Y. Liu, C. Lefevre, J.-M. Greneche, M. Toumi, T. Mhiri and S. Begin-Colin, J. Phys. Chem. C, 2014, 118, 3795–3810 CrossRef CAS.
- Y. Xia, X. Xia and H.-C. Peng, J. Am. Chem. Soc., 2015, 137, 7947–7966 CrossRef CAS PubMed.
- L. Catala, D. Brinzei, Y. Prado, A. Gloter, O. Stéphan, G. Rogez and T. Mallah, Angew. Chem., Int. Ed., 2009, 48, 183 CrossRef.
- G. Salazar-Alvarez, H. Lidbaum, A. López-Ortega, M. Estrader, K. Leifer, J. Sort, S. Suriñach, M. D. Baró and J. Nogués, J. Am. Chem. Soc., 2011, 133, 16738–16741 CrossRef CAS PubMed.
- K. L. Krycka, J. A. Borchers, M. Laver, G. Salazar-Alvarez, A. Lopez-Ortega, M. Estrader, S. Surinach, M. D. Baro, J. Sort and J. Nogués, J. Appl. Phys., 2013, 113 Search PubMed.
- V. Gavrilov-Isaac, S. Neveu, V. Dupuis, D. Taverna, A. Gloter and V. Cabuil, Small, 2015, 11, 2614–2618 CrossRef CAS PubMed.
- K. Sartori, F. Choueikani, A. Gloter, S. Begin-Colin, D. Taverna and B. P. Pichon, J. Am. Chem. Soc., 2019, 141, 9783–9787 CrossRef CAS.
- K. Sartori, G. Cotin, C. Bouillet, V. Halte, S. Begin-Colin, F. Choueikani and B. P. Pichon, Nanoscale, 2019, 11, 12946–12958 RSC.
- G. Cotin, C. Kiefer, F. Perton, M. Boero, B. Özdamar, A. Bouzid, G. Ori, C. Massobrio, D. Begin, B. Pichon, D. Mertz and S. Begin-Colin, ACS Appl. Nano Mater., 2018, 1, 4306–4316 CrossRef CAS.
- Z. Xu, C. Shen, Y. Tian, X. Shi and H.-J. Gao, Nanoscale, 2010, 2, 1027–1032 RSC.
- X. Liu, B. P. Pichon, C. Ulhaq, C. Lefevre, J.-M. Greneche, D. Begin and S. Begin-Colin, Chem. Mater., 2015, 27, 4073 CrossRef CAS.
- P. Ohresser, E. Otero, F. Choueikani, K. Chen, S. Stanescu, F. Deschamps, T. Moreno, F. Polack, B. Lagarde, J.-P. Daguerre, F. Marteau, F. Scheurer, L. Joly, J.-P. Kappler, B. Muller, O. Bunau and Ph Sainctavit, Rev. Sci. Instrum., 2014, 85, 013106 CrossRef CAS PubMed.
- N. Daffé, F. Choueikani, S. Neveu, M.-A. Arrio, A. Juhin, P. Ohresser, V. Dupuis and P. Sainctavit, J. Magn. Magn. Mater., 2018, 460, 243–252 CrossRef.
- P. Pangaud, S. Basolo, N. Boudet, J.-F. Berar, B. Chantepie, P. Delpierre, B. Dinkespiler, S. Hustache, M. Menouni and C. Morel, Nucl. Instrum. Methods Phys. Res., Sect. A, 2007, 571, 321–324 CrossRef CAS.
- K. Medjoubi, T. Bucaille, S. Hustache, J. F. Bérar, N. Boudet, J. C. Clemens, P. Delpierre and B. Dinkespiler, J. Synchrotron Radiat., 2010, 17, 486–495 CrossRef CAS PubMed.
- O. Bunău and Y. Joly, J. Phys.: Condens. Matter, 2009, 21, 345501 CrossRef PubMed.
- K. Sartori, D. Gailly, C. Bouillet, J.-M. Grenèche, P. Dueñas-Ramirez, S. Begin-Colin, F. Choueikani and B. P. Pichon, Cryst. Growth Des., 2020, 20, 1572–1582 CrossRef CAS.
- K. Sartori, A. Musat, F. Choueikani, J.-M. Grenèche, S. Hettler, P. Bencok, S. Begin-Colin, P. Steadman, R. Arenal and B. P. Pichon, ACS Appl. Mater. Interfaces, 2021, 13, 16784–16800 CrossRef CAS PubMed.
- G. V. M. Jacintho, A. G. Brolo, P. Corio, P. A. Z. Suarez and J. C. Rubim, J. Phys. Chem. C, 2009, 113, 7684–7691 CrossRef CAS.
- C.-W. Tang, C.-B. Wang and S.-H. Chien, Thermochim. Acta, 2008, 473, 68–73 CrossRef CAS.
- A. López-Ortega, E. Lottini, C. D. J. Fernández and C. Sangregorio, Chem. Mater., 2015, 27, 4048–4056 CrossRef.
- H. J. Lee, G. Kim, D. H. Kim, J.-S. Kang, C. L. Zhang, S.-W. Cheong, J. H. Shim, S. Lee, H. Lee, J.-Y. Kim, B. H. Kim and B. I. Min, J. Phys.: Condens. Matter, 2008, 20, 295203 CrossRef.
- E. Pellegrin, M. Hagelstein, S. Doyle, H. O. Moser, J. Fuchs, D. Vollath, S. Schuppler, M. A. James, S. S. Saxena, L. Niesen, O. Rogojanu, G. A. Sawatzky, C. Ferrero, M. Borowski, O. Tjernberg and N. B. Brookes, Phys. Status Solidi B, 1999, 215, 797–801 CrossRef CAS.
- P. Jungcharoen, M. Pédrot, F. Choueikani, M. Pasturel, K. Hanna, F. Heberling, M. Tesfa and R. Marsac, Environ. Sci.: Nano, 2021, 8, 2098–2107 RSC.
- G. van der Laan and A. I. Figueroa, Coord. Chem. Rev., 2014, 277–278, 95–129 CrossRef CAS.
- S. Y. Istomin, O. A. Tyablikov, S. M. Kazakov, E. V. Antipov, A. I. Kurbakov, A. A. Tsirlin, N. Hollmann, Y. Y. Chin, H.-J. Lin, C. T. Chen, A. Tanaka, L. H. Tjeng and Z. Hu, Dalton Trans., 2015, 44, 10708–10713 RSC.
- J. Li, N. Menguy, M.-A. Arrio, P. Sainctavit, A. Juhin, Y. Wang, H. Chen, O. Bunau, E. Otero, P. Ohresser and Y. Pan, J. R. Soc., Interface, 2016, 13, 20160355 CrossRef PubMed.
- D. Peddis, N. Yaacoub, M. Ferretti, A. Martinelli, G. Piccaluga, A. Musinu, C. Cannas, G. Navarra, J. M. Greneche and D. Fiorani, J. Phys.: Condens. Matter, 2011, 23, 426004 CrossRef CAS PubMed.
- B. P. Pichon, O. Gerber, C. Lefevre, I. Florea, S. Fleutot, W. Baaziz, M. Pauly, M. Ohlmann, C. Ulhaq, O. Ersen, V. Pierron-Bohnes, P. Panissod, M. Drillon and S. Begin-Colin, Chem. Mater., 2011, 23, 2886–2900 CrossRef CAS.
- E. Skoropata, R. D. Desautels, C.-C. Chi, H. Ouyang, J. W. Freeland and J. van Lierop, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 024410, DOI:10.1103/PhysRevB.89.024410.
- E. Tronc, J.-P. Jolivet, J. Lefebvre and R. Massart, J. Chem. Soc., Faraday Trans. 1, 1984, 80, 2619 RSC.
- Z. Fan, L.-C. Lin, W. Buijs, T. J. H. Vlugt and M. A. van Huis, Nat. Commun., 2016, 7, 1–8 Search PubMed.
- I. J. Bruvera, P. Mendoza Zélis, M. Pilar Calatayud, G. F. Goya and F. H. Sánchez, J. Appl. Phys., 2015, 118, 184304 CrossRef.
- A. Demortiere, P. Panissod, B. P. Pichon, G. Pourroy, D. Guillon, B. Donnio and S. Begin-Colin, Nanoscale, 2011, 3, 225–232 RSC.
- Q. Song and Z. J. Zhang, J. Am. Chem. Soc., 2004, 126, 6164–6168 CrossRef CAS PubMed.
- E. Arenholz, K. Liu, Z. Li and I. K. Schuller, Appl. Phys. Lett., 2006, 88, 072503 CrossRef.
- O. Iglesias, A. Labarta and X. Batlle, J. Nanosci. Nanotechnol., 2008, 8, 2761 CrossRef CAS.
- B. P. Pichon, M. Pauly, P. Marie, C. Leuvrey and S. Begin-Colin, Langmuir, 2011, 27, 6235–6243 CrossRef CAS PubMed.
- S. Ammar, A. Helfen, N. Jouini, F. Fiévet, I. Rosenman, F. Villain, P. Molinié and M. Danot, J. Mater. Chem., 2001, 11, 186–192 RSC.
- H. Sharifi Dehsari and K. Asadi, J. Phys. Chem. C, 2018, 122, 29106–29121 CrossRef CAS.
- M. Pauly, B. P. Pichon, P. Panissod, S. Fleutot, P. Rodriguez, M. Drillon and S. Begin-Colin, J. Mater. Chem., 2012, 22, 6343–6350 RSC.
- Y. Suzuki, R. B. van Dover, E. M. Gyorgy, J. M. Phillips and R. J. Felder, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 14016–14019 CrossRef CAS PubMed.
- R. Chen, M. G. Christiansen and P. Anikeeva, ACS Nano, 2013, 7, 8990–9000 CrossRef CAS PubMed.
- W. H. Meiklejohn and C. P. Bean, Phys. Rev., 1957, 105, 904–913 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: HRTEM micrographs and calculated FFTs on the edge of CS and CSR nanoparticles, the hydrodynamic diameter plotted as a function of intensity counts, additional DANES results and corresponding data analysis. See DOI: https://doi.org/10.1039/d2ma00629d |
| This journal is © The Royal Society of Chemistry 2022 |