
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydroxyapatite coatings: a critical review on electrodeposition parametric variations influencing crystal facet orientation towards enhanced electrochemical sensing
Priya Ranjan
Dev
 a,
Chekkottu Parambil
Anand†
ab,
David Samuvel
Michael†
a and
P.
Wilson
*a
a,
Chekkottu Parambil
Anand†
ab,
David Samuvel
Michael†
a and
P.
Wilson
*a
aDepartment of Chemistry, Madras Christian College (Autonomous), University of Madras, Chennai-600 059, India. E-mail: wilson@mcc.edu.in
bNMR Research Unit, University of Oulu, Oulu-90014, Finland
First published on 6th September 2022
Abstract
Electrochemical deposition presents a promising route toward the fabrication of hydroxyapatite (HAp) coatings attributed to its simplistic approach in the preparation of coatings over complex substrates with control over coating thickness and crystal orientation through variations in electrodeposition parameters. This article aims to consolidate and review the influence of reported parametric determinants associated with electrodeposition thereby providing insight into the outcome of HAp microstructures in terms of Ca/P ratio, crystal growth, and facet orientation of the HAp hexagonal crystal system. Additionally, a discussion on HAp and its composites as viable sensor materials for a myriad of aqueous-based analytes and gaseous molecules is presented. A survey into the application of HAp as a sensor is elaborated, emphasizing the level of sensitivity, selectivity, and sensor response characteristics regarding optimum performance at maximum % sensitivity. Mechanistic insights into the interaction of HAp with analyte species are discussed. Finally, the parameters that include both current density and pH are correlated to determine their synergistic effect as a deciding factor in the growth and preferential orientation of aligned hexagonal HAp crystal domains.
 Priya Ranjan Dev | Priya Ranjan Dev has completed his Masters in Chemistry and Bachelors in Education and is currently pursuing PhD at Madras Christian College, India. He has worked as a research fellow under the SERB-EMR project funded by DST, India (2016–2019). His research interests include the experimental synthesis of metal oxide based microstructures, their functionalization and the determination of the role of defects towards applications relating to energy materials, sensors, photocatalysis, and electrochemical applications. His research interests further extend to DFT studies and materials modelling. |
 Chekkottu Parambil Anand | Chekkottu Parambil Anand is presently a first year doctoral student at the NMR Research Unit of University of Oulu, Finland. He completed graduation from the University of Eastern Finland in 2020 and undergraduate degree from Madras Christian College, India, in 2018, both majoring in Chemistry. Currently he is interested in conducting theoretical and quantum-chemical studies on organometallic complexes of lanthanide and actinide ions which can be constructed as potential Single Molecular Magnets (SMM). This makes it possible to code information into individual molecules and has possible applications in quantum information processing. |
 David Samuvel Michael | David Samuvel Michael completed his graduate studies in Chemistry from Madras Christian College, India, in 2021. His research interests include computational chemistry and quantum mechanics. Presently he is working in the field of theoretical spectroscopy and fundamental quantum mechanics to understand electronic spectroscopy using TDDFT calculations and basic quantum mechanical models. |
 P. Wilson | P. Wilson received his PhD from Indian Institute of Technology, Madras, in 2000 and later worked as a postdoctoral fellow at Ben-Gurion University of the Negev in 2003. He worked as a lecturer in chemistry in Hindustan University, Chennai (1992–93), Bishop Heber College, Trichy (1993–94), and St. Joseph's College of Engineering, Chennai (2000–2002) prior to serving as an associate professor (2004–2015) and head (2015–2020) of Department of Chemistry, Madras Christian College, India. He currently serves as the Principal and Secretary, Madras Christian College. He has supervised research projects funded under DST, India, UGC, India and UBCHEA, USA. Moreover, he is a member of various research and academic bodies. His research publications and interests encompass the fabrication of biomaterials and metal oxide microstructural nanocomposites and their application in fields relating to photocatalysis towards water splitting and dye degradation, coating and corrosion science, biosensors, gas sensors and energy materials. Additionally, he has authored book chapters and continues to serve as a reviewer in ACS, Elsevier and Springer publications. He continues to publish in esteemed international publications. |
1. Introduction
Hydroxyapatite (HAp), a biomaterial composed of hydroxylated calcium phosphate (Ca10(PO4)6(OH)2), is an extensively studied calcium phosphate polymorph whose excellent biocompatibility finds predominant application in bone cement, implants with antibacterial properties,1,2 as well as in the fields of drug delivery3,4 and catalytic applications.5 Additionally, HAp exhibits ferro, pyro, and piezoelectric properties with practical application in bone regeneration and growth and as an immunosensor.6–8 Extensive applicatory studies have been carried out concerning its inherent physical and chemical properties. The versatility of HAp is mainly due to the possibilities arising from the modification of stoichiometric composition as a result of substitution of Ca2+ with divalent cations and replacement of the PO43− anionic tetrahedron and the OH− group with carbonate and halide anions. As a standalone material, HAp micro and macrostructures are inherently brittle with low orders of tensile strength making them impractical in load-bearing applications. Furthermore, they are incapable of exhibiting long-range electric properties. To mitigate these mechanical and electrical restrictions, HAp is generally coupled as a composite with metallic and metal oxide substrates of Ti, Mg, and carbon-based composites to act as a backbone of sorts in enhancing its mechanical properties9 and in providing a point of electrical contact and conduction. Nanocomposite coatings are considered an effective strategy in the fabrication of heterogeneous materials10,11 resulting in improvement in physico-chemical properties,12–18 mitigating detrimental degradation,19–21 and in enhancing its applicative aspects towards increased efficacy,22–25 in reinforcement,26–29 and sustainability.30The fabrication of these nanocomposites offers a certain degree of flexibility in their respective field of application. Among both wet and dry preparatory methods, HAp-based composites are preferably fabricated via the thermal spray deposition technique owing to its convenient industrial scalability.31,32 However, this technique does not necessarily yield robust HAp/metal coated biocomposites as the applied thermal effects result in the formation of cracks, defects, and mixed phases over the superficial macrostructure. This may result in increased susceptibility towards varying rates of biocorrosion. Other techniques include sol–gel33,34 and hydrothermal35,36 processes with each technique entailing its limitations concerning scalability, tunability of coating characteristics, and phase purity. However, the technique of electrochemical deposition presents a viable possibility of adjustment of coating thickness with control over the degree of crystallinity, grain size, and morphology. Additionally, electrochemical deposition offers growth on complex shaped substrates with relative ease and topographical uniformity.37 Furthermore, the coating characteristics can be modified by alteration of deposition parameters such as current density, applied potential, loading time, electrolytic pH, presence of additives, and external treatments like ultrasonication among many other factors. These variations significantly reflect in coating microstructure, porosity, and adhesion to the substrate.38 It is well known that control over crystal orientation influences the applicability of the material. This specificity in HAp crystal orientation may be a deciding factor in enhancing selectivity towards ionic and neutral species. Therefore, optimization of parametric variations encompassing electrochemical deposition and controlling preferentiality in HAp crystal orientation can lead to enhancement in properties.
Relevant HAp reviews have been published discussing the myriad synthetic and coating routes,9,39,40 and the fabrication of deformable ultralong HAp nanowires,41 along the line of application in the biomedical42–44 and pharmaceutical fields,4 and in the adsorptive removal of lead45 and dyes.46 Additionally, comprehensive reviews have been reported on adhesion quality towards bio-metallic substrates,47 electrical nature of HAp,48–50 heterogeneous organocatalytic aspects of HAp with emphasis on stability and reusability,51 and specifically on cationic, anionic, and multi-ionic substitutions of the lattice atoms of HAp.52 In the field of electrodeposition, a general review of electrodeposited biocoatings53 and a review focusing on the influence of applied current have been reported.54 However, there has not been any comprehensive publication exploring the myriad of driving and assisting electrochemical deposition parameters influencing nucleation and crystal orientation with a specific applicative focus on the influence of hexagonal HAp crystal systems towards sensing of various electrolytic and gaseous species.
This review aims to consolidate and disseminate the technique of electrochemical deposition of HAp with a focus on the classification of deposition parameters that determine and influence the dynamics of HAp coating performance reported in line with this preparatory route. This review will strive to hopefully enable any newcomer to grasp the nuances involved in the fabrication of specifically tuned HAp crystals via parametric modification during electrochemical deposition. Therefore, this review will begin firstly by elucidating the crystal facets of HAp to comprehend the significance of the surface properties of each facet. Secondly, a discussion entailing the effects of all significantly reported parametric modifications on nucleation, crystal growth, and microstructure of HAp coatings is presented. Next, a survey into the application of HAp and its composites towards electrochemical sensing is discussed emphasizing the level of sensitivity and selectivity of the crystal towards the sensing of gaseous and electrolytic species. Finally, a correlation between applied current density and pH is discussed, modeling the possible microstructural outcomes of HAp electrodeposits via variations of these two electrolytic parameters.
2. Crystal structure of HAp
The potency of HAp in fields such as ion exchange resin, drug/protein carriers, catalysts, and sensing can be related to the anisotropic properties exhibited by the crystal. HAp exists in two crystalline structures: monoclinic and hexagonal. Monoclinic HAp is thermodynamically stable in relation to the hexagonal system wherein the major difference between the two lies in the orientation of the columnar hydroxyl groups present within the crystal lattice.55 The hexagonal crystal system, illustrated in Fig. 1, belongs to the P63/m space group while maintaining a stoichiometric Ca/P ratio at 1.67. The framework of hexagonal HAp conforms to two crystal facets: the a,b-plane and c-plane.56 The c-plane (or P site) exposes both PO43− and OH− ions predominantly whereas a higher density of surface Ca2+ occupies the a,b-plane (or C site). Consequently, a net negative charge on the c-plane and a net positive charge on the a,b-plane persist due to the system being anionically and cationically richer respectively.57 This enables the system to exhibit anisotropic properties due to its differently charged facets making HAp an ideal candidate for the aforementioned applications. For instance, the possibility of exchange of Ca2+ and PO43− lattice ions makes HAp a versatile material for the selective removal of anionic and cationic species from the external environment.58 The proper utilization of this aspect can be related to the controlled growth of crystals along a specific orientation. The key to bringing out the efficacy of the hexagonal crystal system lies in controlling growth along either the c-plane or a,b-plane, which may be witnessed in terms of the overall Ca/P ratio and microstructural variations. The increase in exposed surface area of a target plane maximizes the capability of the crystal towards adsorption and exchange of ionic species. | ||
| Fig. 1 Schematic illustration of the hexagonal HAp crystal system with the PO43− and OH− groups (in red and blue respectively) occupying the c-plane and Ca2+ (green sphere) populating the a,b-plane. | ||
The a,b-plane and c-plane can be matched with the XRD peak profile of phase-pure HAp. The diffraction peak identified at ∼26° belongs to the (002) plane, i.e., the c-plane.59 The peaks profiled at 21.8° and 32.9° correspond to the (200) and (300) planes respectively, i.e., the a,b-plane.60 When the nucleation and growth predominate along the a,b-axis, the resultant microstructure is flake-like, whereas crystal growth along the c-axis witnesses the formation of hexagonal rods and acicular fibers. Additionally, during electrochemical deposition, the directional growth of crystals is generally perpendicular to the electrode surface. However, the variation in deposition parameters may better align the growth orientation either along the a,b-axes or along the c-axis as the predominantly exposed facet that can be typically identified from the XRD profiles.
3. Electrochemical deposition of HAp
3.1. Deposition techniques of HAp coatings
The control of microstructure, coating thickness and nucleation rate of HAp can be related to the migration and ionic concentration of Ca2+, PO43− and OH− near the vicinity of the cathode, which is dependent on the applied potential and current density over the working electrode. HAp can be electrochemically deposited via electrocathodic and electrophoretic deposition techniques. Both techniques can be performed in a two- or three-electrode electrolytic cell. The substrates employed in both electrochemical techniques are generally conductive metals and alloys, with a few exceptions like bioglass and conductive glass.61,62 Electrocathodic deposition is a popular coating technique comprising ionic species as the electrolyte, wherein fabrication of multi-component and multi-layered coatings can be achieved. Conversely, electrophoretic deposition is mainly a colloidal approach to electrodeposition as it involves employing non-dissociable electrolytic species, such as polymers61 and dispersed HAp nanoparticulate powders in the form of suspended colloids.Electrophoresis boasts of its capability as a technique with a high deposition rate, producing coating thickness ranging from 0.1 to 2.0 mm.63 Electrophoretic deposition emphasizes employing high voltages with very short deposition time. Additional factors influencing coating characteristics under electrophoretic deposition are suspension related factors such as particle size distribution, suspension viscosity and particle charging.64 To minimize viscosity effects, HAp powders are usually dispersed in organic solvents such as ethanol, isopropanol or LiCl-organic electrolytic HAp suspensions.64–66 Furthermore, the dispersed particles must remain in a colloidal suspension comprised of a liquid medium with an effective dielectric constant. Based on a relative comparison between isopropyl alcohol and ethanol, an effective electrophoretic efficiency was witnessed in the case of ethanol. Additionally, a high charge to mass ratio favors rapid deposition.67 The difference between electrocathodic and electrophoretic deposition is that the latter employs a low ionic strength solvent whereas the former employs a solution of ionic salts with high ionic strength. Additionally, electrophoretic deposition requires high electric fields to facilitate the migration of low ionic strength species from the bulk towards the electrode surface.
Irrespective of the technique employed, the coatings fabricated via both techniques are predetermined by a common primary set of deposition parameters such as current, potential, pH, loading time, and deposition temperature. The dissemination within this review will focus on elucidating the parameters reported under electrocathodic deposition, hereafter termed electrodeposition, and their effects on the growth and morphological outcome of HAp crystals along either the a,b-axes or c-axis.
3.2. Electrolytic cell setup and mode of fabrication
4. Mechanism of electrodeposition of HAp
The evolution of the phases of calcium phosphate is mainly driven by cathodic pH, primarily depending on the concentration of liberated OH− ions.74,75 The formation of H2PO4−, HPO42− and PO43− is dependent on the relative evolution of OH− ions. As a result, the concentration of OH− dictates the distribution of phosphate species near the cathode, thus influencing the phase transformation of the deposited polymorph and can be expressed as| 10Ca2+ + 6PO43− + 2OH− → Ca10(PO4)6(OH)2 (HAp phase) | (1) |
| 4Ca2+ + HPO42− + 2PO43− → Ca4H(PO4)3 (OCP phase) | (2) |
| Ca2+ + HPO42− + 2H2O → CaHPO4·2H2O (DCDP phase) | (3) |
| O2 + 2H2O + 4e− → 4(OH)−ads | (4) |
| 2H+ + 2e− → H2 | (5) |
| 2H2O + 2e− → H2 + 2(OH)−ads | (6) |
| 2TiO2 + 2H2O + e− → Ti(OH)3 + (OH)−ads | (7) |
| H2PO4− + H2O + 2e− → H2PO3− + 2(OH)−ads | (8) |
5. Parameters concerning electrochemical deposition of HAp
This section of the review involves the dissemination of electrodeposition parameters reported to control the nucleation rate, crystal growth, orientation of crystal facets and coating characteristics of HAp composites. The various reported electrochemical parameters were classified on the basis of driving and assisting parameters. Driving parameters encompass those factors that initiate HAp formation and influence the rate of nucleation while assisting parameters are segregated based on their influence towards tuning and enhancing the coating characteristics upon their introduction to the electrolytic system. The myriad of parameters reported in relation to HAp electrodeposition and their classification can be seen depicted in Fig. 2.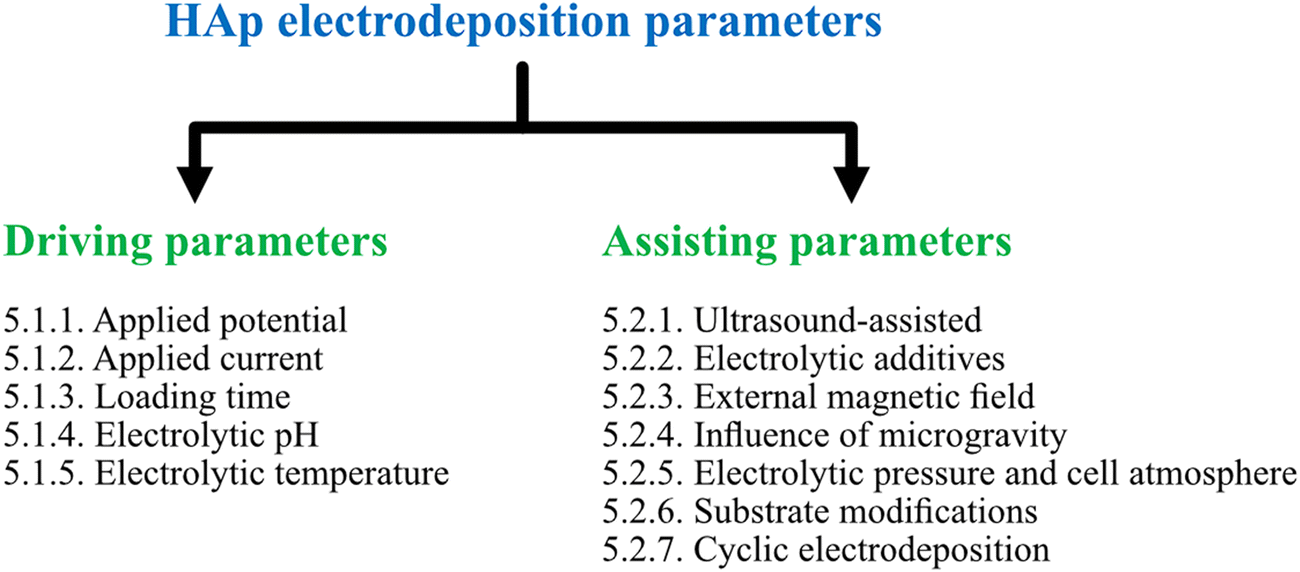 | ||
| Fig. 2 Classification of various applied parameters, as driving and assisting parameters, reported under the technique of electrodeposition of HAp. | ||
5.1. Driving parameters
| S. No. | Cathode | Electrolyte | Potential (V) | Loading time (min) | pH | Temp (°C) | Ca/P ratio | Comments | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Ti | 0.042 M Ca(NO3)2 + 0.025 M NH4H2PO4 + 0.1–1.5 wt% H2O2 | −1.4, −1.6, −1.8 | 1, 2, 3, 5, 30 | 4.3 | 65 | 1.67 | Two-electrode potentiostatic mode | 71 |
| 2 | CoNiCrMo alloy | 0.06 mM Ca(NO3)2 + 0.36 mM NH4H2PO4 + 0.1 M NaNO3 | −1.4, −1.6, −1.8 | 10, 20, 60 | 6.0 | 80 | 1.67 | Three-electrode configuration | 77 |
| 3 | Ti | 0.167 M CaCl2 + 0.1 M NH4H2PO4 | −1.5 | 30 | 6.0 | 80 | — | Needle-like HAp of varying sizes | 68 |
| 4 | Ti6Al4V rod | 0.61 mM Ca(NO3)2 + 0.38 mM NH4H2PO4 | −1.4 | 120 | — | 85 | 1.67 | Plate-like HAp using three-electrode setup | 253 |
| 5 | Ti mesh | 0.167 M CaCl2 + 0.1 M NH4H2PO4 + 0.1 M NaCl | −0.8 | 30 | 6.0 | 80 | 1.57 | Flower-like acicular HAp | 254 |
| −1.5 | 2.54 | ||||||||
| 6 | TiN/316L SS | 0.03 M Ca(NO3)2 + 0.018 M NH4H2PO4 + 0.15 M NaNO3 + 0–6% H2O2 | −1.5 to −1.8 | 30, 45, 60, 75, 90 | 4.0–5.5 | 25, 40, 50, 60, 70, 80 | — | Change from flakes to spherical particles with applied potential | 91 |
| 7 | Ti6Al4V alloy | Ca(NO3)2 + NH4H2PO4 | −1.4 to 0 (scan rate = 0.2 mV s−1) | 120 | 5.0 and 6.0 | 75 | 1.32 @ pH 5, 1.51 @ pH 6 | Potentiodynamic electrodeposition. Flakes (pH 5) and needle-like HAp (pH 6) | 76 |
| 8 | Ti6Al4V alloy | 0.08 mol L−1 Ca(NO3)2 + 0.05 mol L−1 K2HPO4 | 2.5, 3.5, 4.5, 5.5 | 5–45 | 4.6 | 67 | — | Phase change with variation in applied potential | 255 |
| 9 | Ti disc | 0.875–3.5 mM Ca(NO3)2 + 0.5–2.1 mM (NH4)2HPO4 | −1.5 | 60 | 8.3–8.4 | 80 | 1.67 | Three-electrode three-step potentiostatic mode | 79 |
5.1.1.1. Potentiostatic electrodeposition. T. Mokabber et al. investigated a two-electrode approach to electrodeposit HAp wherein a potentiostatic voltage between −1.8 V and −1.4 V was applied with loading times of 1, 2, 3, 5, and 30 min.71 At an applied potential of −1.4 V, a uniform dense coating with a plate-like morphology is observed while deposition at −1.6 V and −1.8 V resulted in the coating to suffer deterioration with a significant amount of cracks and pore formation due to increased H2 evolution. It was also determined that at −1.4 V, variation in loading time significantly brought about changes in crystal growth. For a deposition period of 1 to 3 min, the crystal nucleation dominated along the (010) and (030) planes indicating the initial growth being favored along the a,b-plane resulting in a flake- and plate-like microstructure. However, increased time resulted in the predominance of the acicular morphology with the XRD profile revealing increased intensity for the (002) c-plane.
Conversely, a three-electrode potentiostatic mode reported in ref. 77 addresses the growth and electrocrystallization of HAp in dilute solutions under experimental potentials ranging from −1.4 V to −2.2 V. At a lower potential of −1.4 V, it was observed that the coating predominantly comprised of OCP. A higher applied potential of −1.6 V, −1.8 V, −2.0 V, and −2.2 V favors the conversion of OCP to HAp. The XRD profile indicated the preferential crystal growth along the (002) plane with increasing applied potential. Additionally, SEM revealed the formation of vertically oriented hexagonal crystals with the progress in deposition time (shown in Fig. 3). At −1.8 V, an initial deposition time of 10 min resulted in the formation of flake-like crystals, whereas after 20 min, the microstructure was rod-like (Fig. 3a and b). The initially formed flakes, closest to the substrate, were denser with interspersed pores due to the effects of H2 bubble formation. Additionally, at the onset of deposition, the nucleation of HAp is horizontal (i.e. lateral) to the substrate. An increase in loading time witnessed that the nucleation and growth predominated along the direction perpendicular to the substrate resulting in the gradual formation of hexagonal rods. Furthermore, it was noted that the coatings electrodeposited at −1.8 V displayed enhanced adhesion to the substrate when subjected to sonofragmentation.78
 | ||
| Fig. 3 Microstructural modifications of the coating electrodeposited at –1.8 V over various time intervals of (a) 10 min, (b) 20 min and (c) 60 min. Adapted from ref. 77 with permission from Elsevier. | ||
A comparative analysis of the potentiostatic mode of electrodeposition of HAp under the two-electrode and three-electrode configuration can be made from ref. 71 and 77 respectively. In a two-electrode arrangement, the maximum working potential for uniform dense growth of HAp was reported as −1.4 V. An increase in applied potential above −1.5 V resulted in the generation of H2 gas bubbles leading to severely cracked coating with larger pores. Conversely, the three-electrode potentiostatic mode facilitated a maximum working potential of −1.8 V beyond which the coating stability deteriorates. In terms of crystal growth and orientation, the three-electrode arrangement yields hexagonal crystals at −1.8 V (shown in Fig. 3c).
5.1.1.2. Three-step potentiostatic electrodeposition. B.-O. Taranu et al. investigated a three-step potentiostatic HAp electrodeposition strategy wherein the procedure comprises the creation of an initial deposit of HAp, followed by a second step of thermal calcination and then completion by performing an additional electrodeposition onto the HAp crystals deposited in the first step.79 The process was performed under a three-electrode potentiostatic mode at −1.5 V with a loading time of 60 min. This three-step technique proved beneficial in lowering the rate of corrosion. The microstructure revealed spherical clusters of needle-like crystals with each successive deposition step increasing the orientation of growth along the c-plane. The overall effect of this technique resulted in the densification of the first layer of coating over the Ti substrate while the additional electrodeposition generated a secondary porous layer of shorter and thicker acicular clusters. An intermediate thermal sintering step facilitated the creation of HAp coating with a variation in porosity gradient. Additionally, the three-step treatment involving electrodeposition, calcination, and electrodeposition showed a reduction in the rate of corrosion to ∼7.7 μm year−1 when compared to the single-step electrodeposition.
5.1.1.3. Potentiodynamic electrodeposition. The electrodeposition of HAp under potentiodynamic conditions, as reported by A. Vladescu et al.,76 involved HAp formation wherein deposition was initiated by sweeping the potential from an initial −1.4 V to a final 0 V potential at a sweep rate of 0.2 mV s−1. A three-electrode system setup with Ti6Al4V alloy as the working electrode was employed. Further experiments were conducted with variations of pH under acidic conditions of 5 and 6. The morphological observations indicate the formation of HAp as flakes and needles at pH 5 and 6 respectively. The potential sweep ensured the formation of thick coatings with thickness reaching ∼455 nm for flakes and ∼2.27 μm for needles with crystal growth perpendicular to the substrate. In this scenario, the HAp flakes and needles had a Ca/P ratio of 1.32 and 1.51 respectively with the biocorrosion resistance being higher in the case of needle-like HAp when studied in a SBF electrolyte.
| S. No. | Cathode | Electrolyte | Current density (mA cm−2) | Loading time (min) | pH | Temp (°C) | Ca/P ratio | Comments | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Ti6Al4V disc | 0.61 mol L−1 Ca(NO3)2 + 0.36 mol L−1 NH4H2PO4 | 0.318 | 15, 30, 60, 180 | 4.5 | 90 | 1.67 | Galvanostatic electrodeposition with acicular HAp | 80 |
| 2 | Ti | 0.42 M CaCl2 + 0.25 M K2HPO4 + 2000 ppm H2O2 | Pulsed between 1.0 and 0 | 60 | — | — | — | Acicular HAp arranged in lotus-like fashion | 155 |
| 3 | Ti6Al4V | Ca(NO3)2 + NH4H2PO4 | Pulsed between (−0.25) and (−1.25) | 30 | 4–5 | 37 | — | Pulsed electrodeposition with a focus on the duty cycle | 86 |
| 4 | TiO2 NTs | 2.5 mM CaCl2 + 1.67 mM K2HPO4 + 0.15 M NaCl | Pulsed between −10 and 0.01 for 100 cycles | ∼77 | 7.2 | 80 | 1.43 | Thick acicular HAp | 238 |
| 5 | NiTi alloy | 0.0167 M Ca(NO3)2 + 0.01 M NH4H2PO4 + 0.1 M NaNO3 + 10 mL L−1 H2O2 | 0.5 | 40 | 6.0 | 65 | — | Flake-like HAp | 256 |
| 6 | Ti | 1 g/L SiO2 + 0.042 M Ca(NO3)2 + 0.025 M NH4H2PO4 | 0.8 | 30 | 4.2 | 65 | — | Needle-like HAp | 257 |
| 7 | AZ32B alloy | 0.042 mol L−1 Ca(NO3)2 + 0.025 mol L−1 NH4H2PO4 + 0.1 mol L−1 NaNO3 + 20 mL L−1 H2O2 | 3 | 120 | 5.0 | 23 | 1.98 | Plate-like HAp with variation in porosity | 258 |
| 6 | 2.28 | ||||||||
| 13 | 2.31 | ||||||||
| 8 | SS316 alloy | 0.15 M CaCl2 + 0.1 M NH4H2PO4 | 5 | 60 | — | 25 | 1.34 | Current pulsed between 0 and 5, 10, 20 | 235 |
| 10 | 1.35 | ||||||||
| 20 | 1.45 | ||||||||
| 9 | TiO2 NTs | 8 mM Ca(NO3)2 + 5 mM NH4H2PO4 + 30 mg L−1 GO | 15 (pulsed duty cycle: 0.1) | 65 | 6.0 | 65 | 1.57 (only CaP), 1.63 (CaP + GO) | Flake-like HAp/GO coatings with a,b-plane preferentiality | 87 |
| 10 | Ti | 1.3 mM CaCl2 + 0.84 mM K2HPO4 + 138 mM NaCl | 12.5 | 4 | 7.2 | 95 | — | Rod-like HAp coatings | 259 |
5.1.2.1 Galvanostatic electrodeposition. A report on preferentially oriented c-axis HAp crystals grown galvanostatically was presented by E. A. dos Santos et al.80 A three-electrode setup with Ti6Al4V disc as the working electrode and a galvanostatic current density of 0.318 mA cm−2 was employed. This study dictates the fact that the orientation of crystals along the a,b-axes or c-axis is time-dependent under galvanostatic conditions. A 15 min loading time resulted in a haphazard arrangement of plate-like HAp. However, as the loading time increased to 1 h, the plates converted to needle-like with the crystal orientation predominating along the (002) c-axis. Additionally, the conversion of the initial flake-like microstructure to an acicular one can be attributed to the lowering of surface free energy during crystallization by reorienting growth in a direction parallel to the electric field, vis-à-vis perpendicular to the substrate. This produced acicular microstructures favoring a c-axis orientation.
5.1.2.2. Pulse/reverse pulse electrodeposition. Generally, the incorporation of pulses in applied current is a manipulation reported to improve the growth and coating characteristics such as geometry, porosity, surface defects, and crystal orientation.81,82 Additionally, the inclusion of pulses has been reported to bring out uniformity and compactness with increased adhesive strength in comparison to coatings deposited under a direct continuous current.83 Pulses in current favor enhancement in nucleation rate over growth assimilation with refinement in grain size and roughness.84
The introduction of pulses in direct current can be applied in two ways: a pulse current method (viz. unipolar pulse current) wherein the current oscillates between zero and a forward bias, and a reverse pulse current method (viz. bipolar pulse current) wherein the current alternates between two non-zero bias voltages. The variations in pulse current modes are depicted in Fig. 4. The factors to be considered while pulsing are the duration of pulse on/off time and the amplitude range of the current density which proves to be crucial for nucleation and phase purity of HAp. Under a direct continuous current, a higher accumulation of H2 bubbles is witnessed. The consequences of these bubbles include detrimental effects on ion diffusion. However, by employing pulses, one essentially reduces the quantity of bubbles, thereby improving the mass transport between the diffusion layer and the substrate. This proves beneficial in terms of physico-chemical properties of the HAp coating with regard to crystallinity, adhesive strength, and biocorrosion.
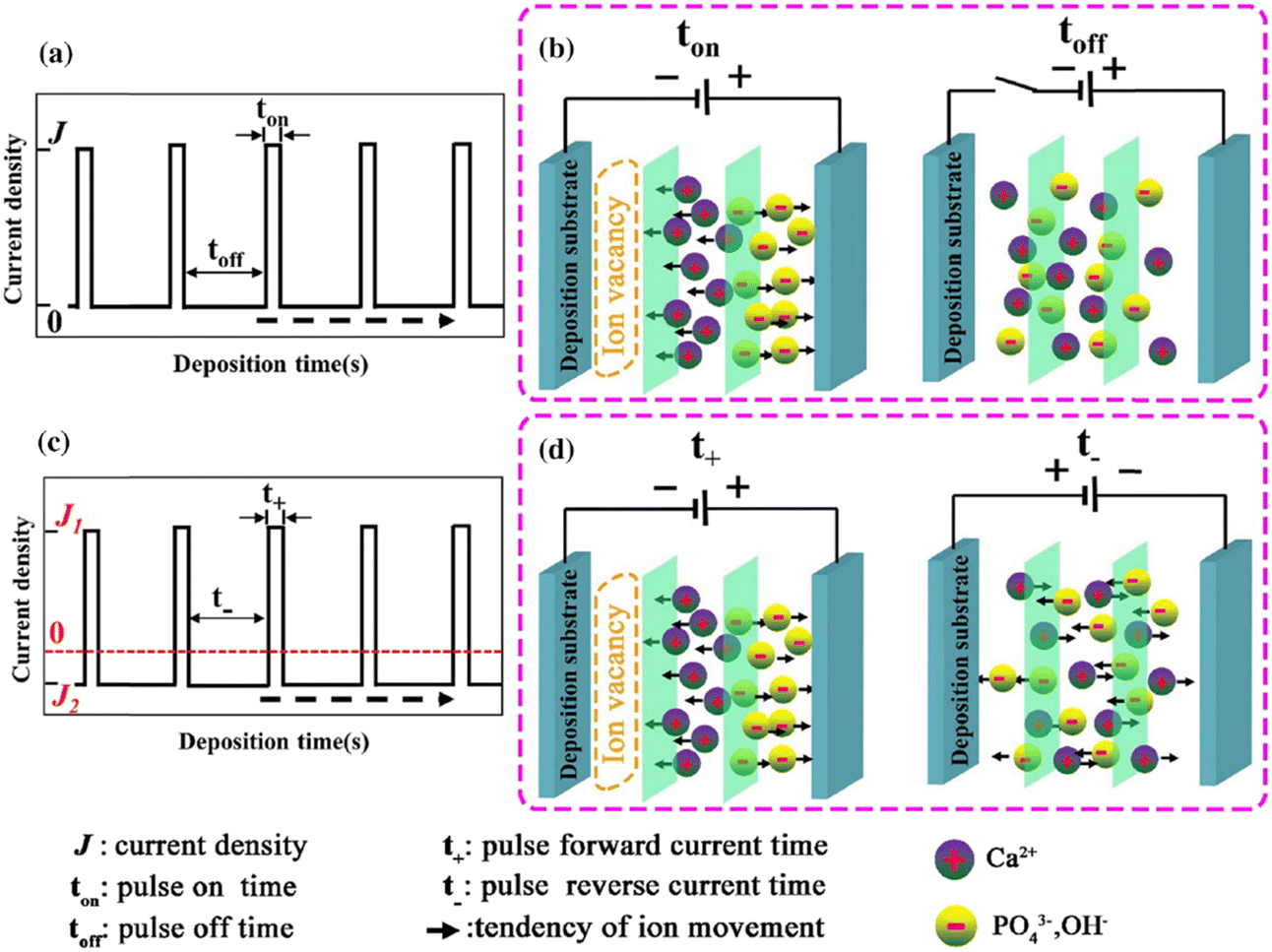 | ||
| Fig. 4 Schematic representing the (a) and (b) unipolar pulse current method and (c) and (d) the bipolar pulse current mode. Adapted from ref. 54 with permission from Springer. | ||
The difference between pulse and reverse pulse techniques is that unipolar pulses create an ion vacancy near the cathodic double layer as a result of depletion of ions during the ON cycle (Fig. 4a and b). During the OFF cycle, with applied current being zero, replenishment of ion vacancy takes longer. This can be mitigated by employing a bipolar pulse current wherein the vacancy of ions near the cathode is replenished during the reverse pulse current (i.e., a negative bias). This process ensures maintaining an ionic homogeneity in the diffusion layer (Fig. 4c and d). Furthermore, studies on the effect of pulse duration on homogeneity and microstructure with respect to duration of introduced pulses have been investigated.85 The manipulation in the duration of pulses is known as the duty cycle and is reported to be a ratio of the active duration to the total duration and is expressed as86
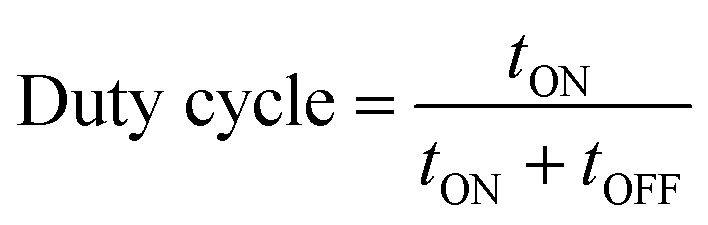 | (9) |
To summarize, the introduction of pulses in applied current witnessed an increase in the adhesive strength of the composite substrate compared to the galvanostatic electrodeposition. Furthermore, variation in duty cycle and magnitude of applied current significantly reduced the accumulation of H2 bubbles. The unipolar pulse electrodeposition incorporates a relaxation time during the pause (applied current being zero) resulting in a slow rate of recovery of depleted ions. In the case of bipolar pulse electrodeposition, the substrate may be coated with a multi-component layer assembled during each optimized cycle of applied forward or reverse bias. A comparative study of the continuous, pulse, and reverse pulse electrodeposition of HAp and its influence on morphology and electrochemical behavior has been elucidated.89
Another effect of pH variation can be witnessed in terms of the type of substrate employed during electrodeposition leading to an enhancement in nucleation, as investigated by employing a metal oxide cathode in ref. 93. This investigation revealed that at the TiO2/solution interface, charge polarization is observed between the positively charged Ti and negatively charged O2 sites. Due to this, the OH− groups are effectively adsorbed onto the TiO2 surface in comparison to the pure Ti metal. This isoelectric point for TiO2 is reached at an alkaline pH of approximately 5 and 6. Outside this isoelectric pH, if the electrolytic pH is acidic, the surface positive charge gets modified, whereas an alkaline pH alters the net negative charge on the Ti substrate. This variation in surface charge enhances the adsorption of Ca2+ and PO43− leading to the nucleation and growth of HAp. The progress of loading time significantly alters the Ca/P ratio on the adsorbed substrate. The pH near the vicinity of the cathode can be theoretically calculated from ref. 94 as
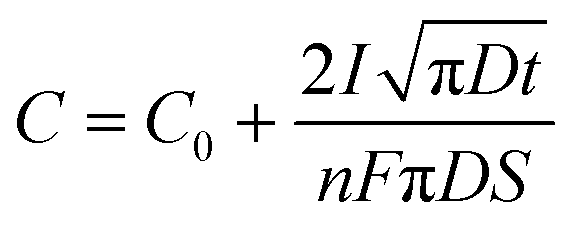 | (10) |
C. M. Cotrut et al. investigated the influence of deposition temperature on microstructural HAp coating wherein electrodeposition was carried out under a sweeping potential from 0 V to −1.4 V at a scan rate of 0.2 mV s−1.97 The Ti disc was immersed in a Ca2+ and PO43− precursor solution at pH 5 with a deposition temperature of 50 °C and 75 °C. The surface morphology of HAp deposited at both temperatures consists of plate-like microstructures with variations in coating density and porosity. However, coatings prepared at 50 °C were less crystalline with a Ca/P ratio of 1.39 as a result of ionic vacancies formed due to lower deposition rate. It can be concluded that electrodeposition temperature showed an influence over coating morphology and the Ca/P ratio. In an investigative work by Nam et al.,91 electrodeposition of HAp onto a TiN/316L SS substrate showed variation in the mass transfer and weight of HAp deposited upon temperature variation. The mass of deposited HAp increased up to a working temperature of 50 °C. However, temperatures nearing 80 °C resulted in diminished HAp deposits. This was witnessed as a flaking phenomenon wherein HAp coating shows decreased adhesion capacity with increased porosity. The microstructural changes observed at 25 °C were plate-like indicating an increase in the c-plane surface area. Above 50 °C, the morphology converts to acicular crystals with a dominant a,b-plane.
5.2. Assisting parameters
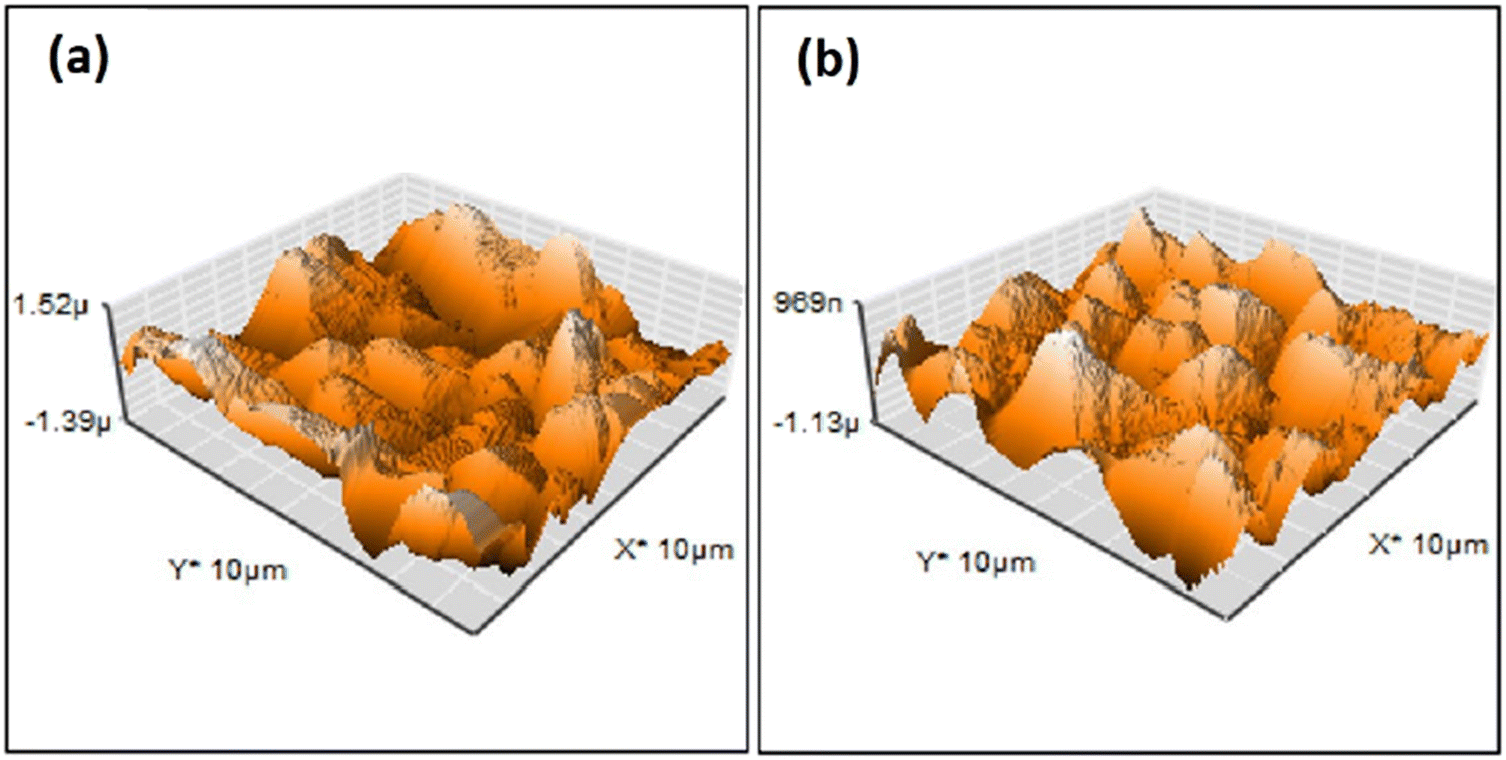 | ||
| Fig. 5 AFM surface profile of HAp deposited via pulse electrodeposition in (a) the absence of ultrasonic irradiation and (b) at 60 W ultrasonic power. Adapted from ref. 98 with permission from Elsevier. | ||
Another effect is a substantial decrease in porosity and an increase in surface smoothness.100 The microstructure resembled rounded needles in both cases with an increased density of needles with ultrasound exposure in relation to those without ultrasound (in Fig. 6). The effect of ultrasonic treatment and current density on the Ca/P ratio is significant, wherein an increase in current density from 2.5 mA cm−2 to 5.0 mA cm−2 changed the ratio from 1.53 to 1.58 but witnessed a decrease at higher current. The Ca/P ratios for ultrasonic assisted HAp at current densities of 2.5, 5.0 and 7.5 mA cm−2 were 1.69, 1.65 and 1.40 respectively. This may be attributed to the supporting effect of ultrasound facilitating the distribution of ions within the electrolyte. Its effects were noticeable on the crystallinity of deposited HAp. The surface roughness reduced from 7.04 μm to 0.76 μm at 7.5 mA cm−2 under 20 W ultrasonic power, indicating a control over surface roughness through control of current density and ultrasonic power. L. Fathyunes et al. assessed the effects of ultrasound during pulse electrodeposition of HAp/GO over anodized Ti.101 Ultrasound aided in the dispersion of GO with a refinement in overall coating. The formation and implosion of microbubbles near the cathode enhanced the migration and the subsequent nucleation process. Furthermore, this ultrasonic agitation contributes to the formation of OH radicals. In addition to the fracturing effects, a plate-like morphology with increased uniformity was witnessed. The effects of ultrasound were also investigated under coupled ultrasound-assisted pulse electrodeposition.102
 | ||
| Fig. 6 Variation in microstructure in the presence and absence of ultrasound at different applied current densities. Reproduced from ref. 100 with permission from Elsevier. | ||
The overall purview of implementing ultrasonics during the process of electrodeposition relates to the microstructural formation of uniform and dense coating with reduced roughness and pore size. Furthermore, the crystal orientation of HAp in an ultrasonic assisted electrodeposition favors horizontal growth of HAp crystals as a result of the fracturing capability of ultrasonic waves.
Other additives such as KNO3 and NaNO3 promote an increase in the ionic strength of electrolytic species hence increasing conductivity. The NO3− ion undergoes electrochemical reduction to NO2− with the harnessed generation of OH− ions.105,106 An applied potential ranging from −1 V to −1.5 V (vs. SCE) facilitates the electroreduction of NO3− ions which is expressed as
| NO3− + H2O + 2e− → NO2− + 2OH− | (11) |
The incorporation of CNTs in the electrolytic composition led to the formation of HAp crystals preferentially orienting along the a,b-plane as witnessed from the flake-like crystalline microstructures.107 The flakes grew perpendicular to the substrate with a thickness of up to 3 μm. Owing to the presence of CNTs, the composite displayed good corrosion resistance with reinforced mechanical strength.
5.2.2.1. Effect of H2O2. The reduction in interfacial adhesive strength between HAp and substrate is a cause for concern mainly attributed to the evolution of large amounts of H2 gas bubbles, a consequential product from the electrolytic dissociation reaction of H2O. This dissociation occurs at increased potentials beyond the range of −2 V.77 A higher potential of −5 V generates crater-like indents with diameters ranging from 40 μm to 250 μm.94 Furthermore, this disruption in mass transport affects the Ca/P ratio of the deposited crystals resulting in deposition of either phase-pure HAp or other polymorphs of calcium phosphate.73 The degradative effects of H2 bubbles on coating properties can be mitigated by altering certain parameters such as current density, modifying the cell atmosphere (later discussed in Section 5.2.5.3), introducing ultrasonic waves108 and application of pulses with addition of H2O2 to the electrolytic medium.109 Unlike H2O, the addition of H2O2 results in its electrolytic fragmentation generating OH− ions given as
| H2O2 + 2e− → 2OH− | (12) |
 | ||
| Fig. 7 SEM micrographs depicting electrodeposited HAp in a H2O2 electrolytic environment under the (a) direct current method and (b) pulsed current method. Adapted from ref. 111 with permission from Elsevier. | ||
D. Gopi et al. examined HAp deposition onto 316L SS at a lower current density of 0.5 mA cm−2 with the inclusion of unipolar pulses and H2O2 as an additive.112 The absence of H2O2 produced DCDP and OCP as the dominant phases. Lower H2O2 concentrations of 600 ppm and 1000 ppm resulted in DCDP crystals and trace amounts of HAp. However, H2O2 concentrations above 2000 ppm produced phase-pure HAp signifying the threshold levels of H2O2 needed to favor HAp crystal growth. This excess in OH− ions raises the pH to alkaline levels near the cathode thereby generating sufficient growth of HAp. The phase purity was also studied by varying current densities between 1, 2 and 3 mA cm−2 revealing that the HAp (002) intensity increased ever so slightly as the magnitude of current density increased at 1000 ppm H2O2. Higher current densities were avoided to prevent the coating to turn porous while low currents resulted in a non-uniform flake-like morphology. High concentrations of H2O2 (6 or 9 vol%) with 2 min pulse OFF time resulted in acicular crystals while lower concentration (600–2000 ppm) with 2 s pulse OFF time resulted in HAp flakes. The coupling of the H2O2 additive with pulse electrodeposition proved effective in improving the crystallinity and orientation along the c-plane.
T. Mokabber et al. prepared HAp via a potentiodynamic process wherein H2O2 was added to lower the evolution of H2 gas bubbles.71 The presence of H2O2 leads to an increase in OH− formation, which in turn increases the concentrations of HPO42− and PO43−, according to the following reactions:
| H2PO4− + OH− → HPO42− + H2O | (13) |
| HPO42− + OH− → PO43− + H2O | (14) |
5.2.2.2. Effect of chelates. The influence of chelating agents on the crystallization of HAp has been reported by D. He et al., specifically the influence of EDTA on HAp deposition via the hydrothermal–electrochemical route.113 With varying concentrations of EDTA, it was determined that the percentage crystallinity and coating thickness can be altered. The coating thickness decreased from 21 μm to ∼13 μm as concentrations increased, while the adhesive strength towards the substrate increased. The topography of the composite displayed increased growth along the c-axis in the absence of EDTA whereas the addition of EDTA oriented the microstructure growth along the a,b-plane. This shift in microstructure is due to the effect of supersaturation of chelated Ca-EDTA. The binding affinity of EDTA towards the ions along the crystal plane is stronger with the (100) facet (viz. a,b-plane) than with the (001) facet (viz. c-plane) thereby hindering deposition along the c-axis.114 At reduced levels of EDTA, the supersaturation of the electrolyte is lowered with a consequential increased growth rate along the c-axis.113 The addition of EDTA as an additive takes advantage of the control of crystal orientation more towards the a,b-axes and can be related to control of the supersaturation of the chelate complex.
Trisodium citrate has been reportedly employed as an organic modifier and complexing additive in electrodeposition.115 After a 1 h loading time, a gradual increase in citrate ion concentration modified the hexagonal rod-like crystals (at [citrate] = 0) to a tapered acicular cone (at [citrate] > 5). The addition of citrate inhibits the early formation and nucleation of OCP, mainly attributed to an increase in pH near the vicinity of the electrode. Furthermore, the presence of citrate altered the crystal characteristics as a result of its adsorption and complexation with Ca2+ thereby arresting crystal growth along the a,b-axes while promoting c-axis growth during electrodeposition.
5.2.2.3. Simultaneous anodization–electrodeposition of HAp. Typically, electrodeposition of HAp is carried out employing ionic electrolytic precursors involving Ca2+ and PO43− salts. Furthermore, employing dispersed HAp nanoparticles within aqueous electrolytes is typically reported under the electrophoretic deposition technique.116 However, J.-M. Jang et al. investigated the electrodeposition of dispersed HAp nanoparticles onto the TiO2 NT substrate.117 The experimental protocol involved a unique simultaneous anodization–electrodeposition technique, shown in Table 3, involving the simultaneous growth of anodized TiO2 up to 800 nm in length while depositing a HAp coating with thickness up to 100 nm. The dispersed HAp nanoparticles formed a layer onto TiO2 and grew vertically with a particle size of 13–17 nm. The ultrafine deposited layer assumed a spherical microstructure (shown in Fig. 8) with crystallographic studies revealing the presence of phase-pure HAp. An XPS depth-profiling was carried out after employing an Ar+ ion sputtering surface treatment for 100, 200 and 300 s revealing varying content of Ca and P. The Ca and P content varied in the range of 0.61–0.90 and 0.58–0.24 respectively upon each successive Ar+ sputtering time. The reason for the decrease in the content of P with depth was attributed to the low capacity of adsorption and interaction between the nanotubular TiO2. However, as loading time increases, the quantity of P-sites increases due to the formation of hydrated Ti(OH)2. This facilitates the enhanced adsorption of PO43−, HPO42− and H2PO4− ions.
| S. No. | Cathode | Electrolyte | Potential (V) | Loading time (min) | Temp (°C) | Additive | Ca/P ratio | Comments | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Ti | 0.042 M Ca(NO3)2 + 0.025 M NH4H2PO4 + 0.1–1.5 wt% H2O2 | −1.4, −1.6, −1.8 | 1, 2, 3, 5, 30 | 65 | H2O2 | 1.67 | Reduction in cathodic H2 generation | 71 |
| 2 | Ti6Al4V | 0.025 mol L−1 Ca(NO3)2 + 0.015 mol L−1 NH4H2PO4 + (0.00025 to 0.001 mol L−1) EDTA-2Na. Current density = 1.25 mA cm−2 | — | 120 | 120 | EDTA-2Na | — | Chelate assisted electrodeposition of HAp | 113 |
| 3 | Ti | 0.042 mol L−1 Ca(NO3)2 + 0.025 mol L−1 K2HPO4 + 0–2 wt% CNTs (pH: 4.7) | −1.4 | 60 | — | CNTs | — | CNTs promote a,b-plane preferentiality | 107 |
| 4 | TiO2 NTs | 1.6 M NH4H2PO4 + 0.65 M NH4F + 0.8 wt% HAp | 21 | ∼47 | 28 | HAp nanoparticles | — | Simultaneous anodization–electrodeposition of ultrafine HAp | 117 |
| 5 | TiO2 NTs | 1.6 M NH4H2PO4 + 0.8 M NH4F + 0.1, 0.15, 0.25 wt% HAp | 28 | 120, 150 | — | HAp nanoparticles | 1.26, 1.53, 1.65 | HAp electrodeposition from dispersed HAp nanoparticles as the precursor | 118 |
| 6 | TiO2 NTs | 1.7 M NH4H2PO4 + 0.4 M NH4F + 0.1 wt% Pd + 0.2 wt% Ag + 0.3 wt% HAp | 28 | 10, 15, 30, 45, 90, 120 | — | HAp nanoparticles | 1.67 | Ultra-fine deposition of spherical HAp particles | 119 |
| 7 | Ti plate | HAp nanoslabs in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2O–ethanol mixture. Current density: 1, 3, 5 mA cm−2 :1 H2O–ethanol mixture. Current density: 1, 3, 5 mA cm−2 |
— | — | — | HAp nanoslabs | — | Dispersed HAp nanoslabs/nanoplates electrodeposited onto a Ti plate | 244 |
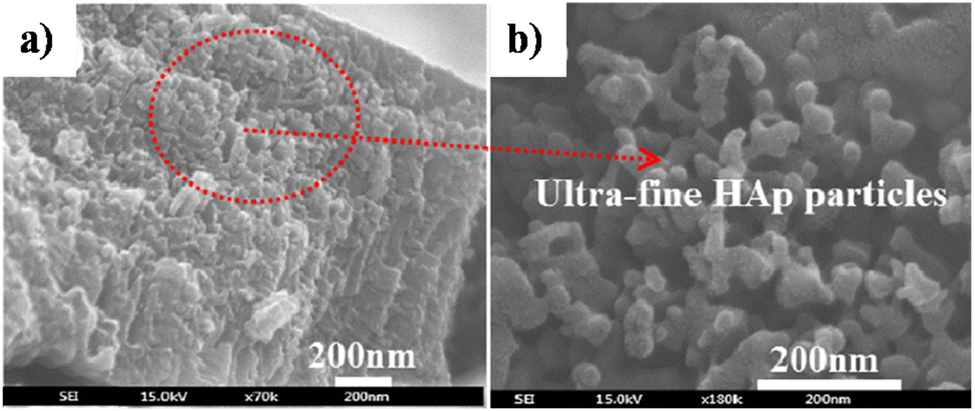 | ||
| Fig. 8 FESEM of (a) HAp electrodeposited over TiO2 NTs via the simultaneous anodization–electrodeposition technique and (b) the microstructure of the deposited HAp particles. Adapted from ref. 117 with permission from Elsevier. | ||
Similar reports on the simultaneous anodization–electrodeposition technique are reported in ref. 118 and 119 initiated by the anodization of the Ti substrate with simultaneous electrodeposition of HAp from a dispersion of ultra-fine HAp nanoparticles. The electrodeposited HAp nanoparticles displayed an increase in size in addition to an increase in the weight percentage of HAp. Furthermore, the technique of simultaneous anodization–electrodeposition proved effective in the modulation of Ca/P ratios of the deposits, wherein Ca/P ratios were 1.26, 1.53 and 1.65 for the mixed electrolyte containing 0.1, 0.15 and 0.25 wt% of dispersed HAp nanoparticles. The technique of simultaneous anodization–electrodeposition revealed that the incorporation of Ca2+ and PO43− ions is significantly affected by the composition of the substrate.
5.2.2.4. Simultaneous precipitation–electrodeposition of HAp. The technique of simultaneous precipitation–electrodeposition, unlike simultaneous anodization–electrodeposition, involves deposition of HAp crystals without electrochemical modification of the cathodic material like anodization.120 The method entails drop-wise addition of one precursor (either Ca or P) into an electrolytic cell containing the other precursor, while simultaneously initiating potentiostatic electrodeposition (experimental parameters in Table 3). XRD profiles reveal that when the Ca2+ precursor is added to the PO43− electrolyte, an enhanced preferential growth along the c-axis is witnessed over a loading time of 4 h. There is a significant improvement in the degree of crystallinity from this technique in lieu of conventional HAp electrodeposition. All samples had an initial uniform layer of submicron crystals with the increase in acicular crystals grouped as hemispherical clusters (shown in Fig. 9). The technique proved effective in improving the anti-corrosiveness and adhesion strength of the HAp coating.
 | ||
| Fig. 9 FESEM of HAp crystals deposited via the simultaneous precipitation–electrodeposition technique with dropwise addition of (a) P precursor, (b) Ca precursor and (c) Ca + P precursor. Reproduced from ref. 120 with permission from Elsevier. | ||
H. E. Lundager Madsen reported that phosphates of diamagnetic ions are affected under a magnetic field resulting in variation of phases.122 The precipitation resulted in the formation of large brushite crystals in the absence of B, whereas at 0.27 T, stable HAp crystals persisted. Although the crystal size reduced, the rate of crystallization increased with new nucleation sites leading to smaller but higher number of crystals. The precipitative crystallization of HAp under the influence of a magnetic field was understood via a proposed mechanism which includes a proton transfer from a weak acid to H2O. The rate of transfer of protons is slow in the case of a weak acid. Additionally, the rate of crystallization of HAp increases as the pH of the medium shifts towards alkaline.123 Although the magnetic field had no effect on the mass transport of the diamagnetic species, the relative increase in the rate of crystal formation was correlated to the rate of proton transfer from the weak acid to water (since proton spin is influenced by B) thereby revealing that the precipitation of crystals in a magnetic field can be related to the rate of proton transfer.
Unlike the previous study conducted at a low magnetic field of 0.27 T, slipcasting was carried out in the range of 10–12 T field strength wherein a slurry of HAp was allowed to set in a crucible.124,125 The magnetic field applied was parallel to the direction of the gravitational field. XRD profiles (shown in Fig. 10) revealed a remarkable effect of the high magnetic field on the orientation of HAp. Horizontal cut cross-section displayed a dominant peak intensity for the exposed a,b-plane. This is an indication that when a slurry of HAp is slipcast while exposed to an external high magnetic field, the nucleation and the subsequent crystal orientation can be controlled to a great extent. Similar experiments were carried out by employing a surfactant and performing milling before slipcasting126,127 to primarily prevent the aggregation of HAp particles during slipcasting.
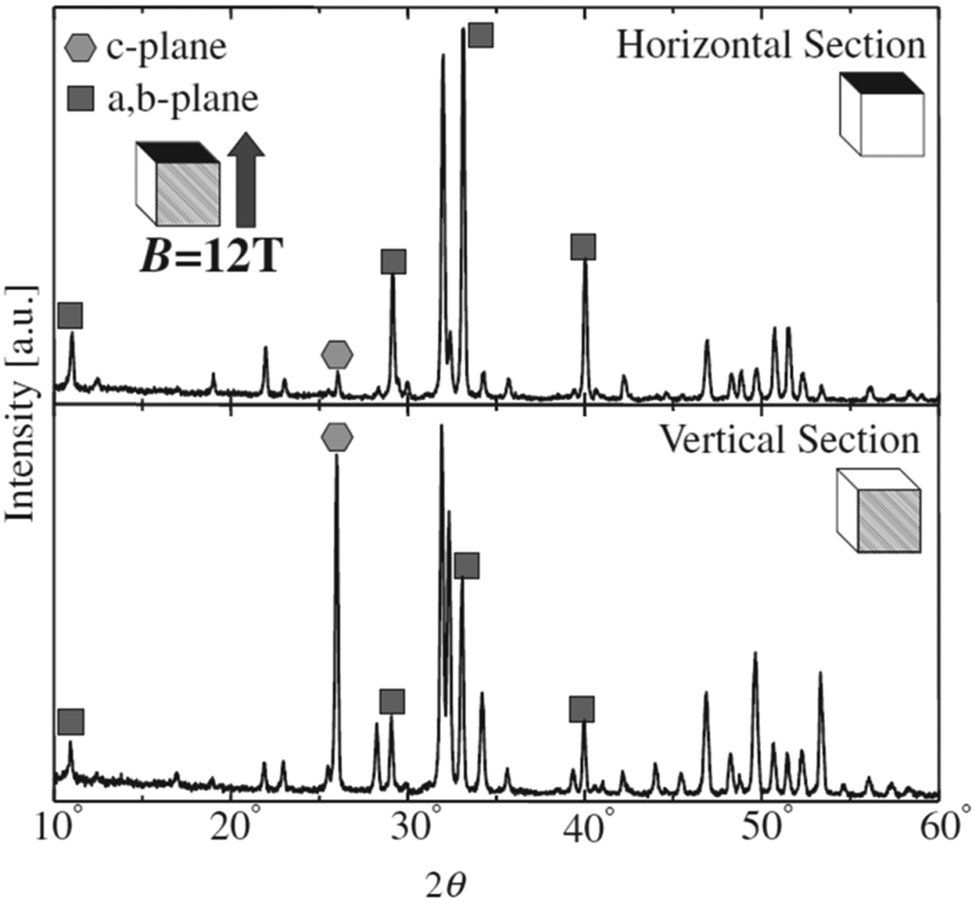 | ||
| Fig. 10 XRD diffraction indicating the orientation of the crystal facets of slipcast HAp at B = 12 T. Adapted from ref. 124 with permission from The Japan Institute of Metals and Materials. | ||
Okido et al. reported a magnetic field assisted deposition method, termed the heat substrate method, wherein a similar control over crystal orientation was accomplished.128 The experiment involved heating the substrate (a strip of Ti foil) with the aid of an electric current to precipitate HAp in the presence of a magnetic flux of 12 T. Note that this technique is not magnetoelectrodeposition since the applied electric current is for heating of the mounted substrate and there is an absence of electric flux (i.e., no cathode and anode). The foil was mounted both parallel and perpendicular to B. In the absence of B = 12 T, the HAp crystals were unoriented (Fig. 11a). At an applied field of 12 T, the resultant HAp crystals were hexagonal pillars when the foil was oriented parallel to B (Fig. 11b) and thin hexagonal flakes when the foil was perpendicular to B (Fig. 11c). For HAp, this would be the a,b-plane aligning to the direction of B. Additionally, it must be noted that both slipcasting and the heat substrate method, when performed at B = 12 T, resulted in predominant exposure of the c-plane when B was parallel to the slurry and Ti foil respectively.
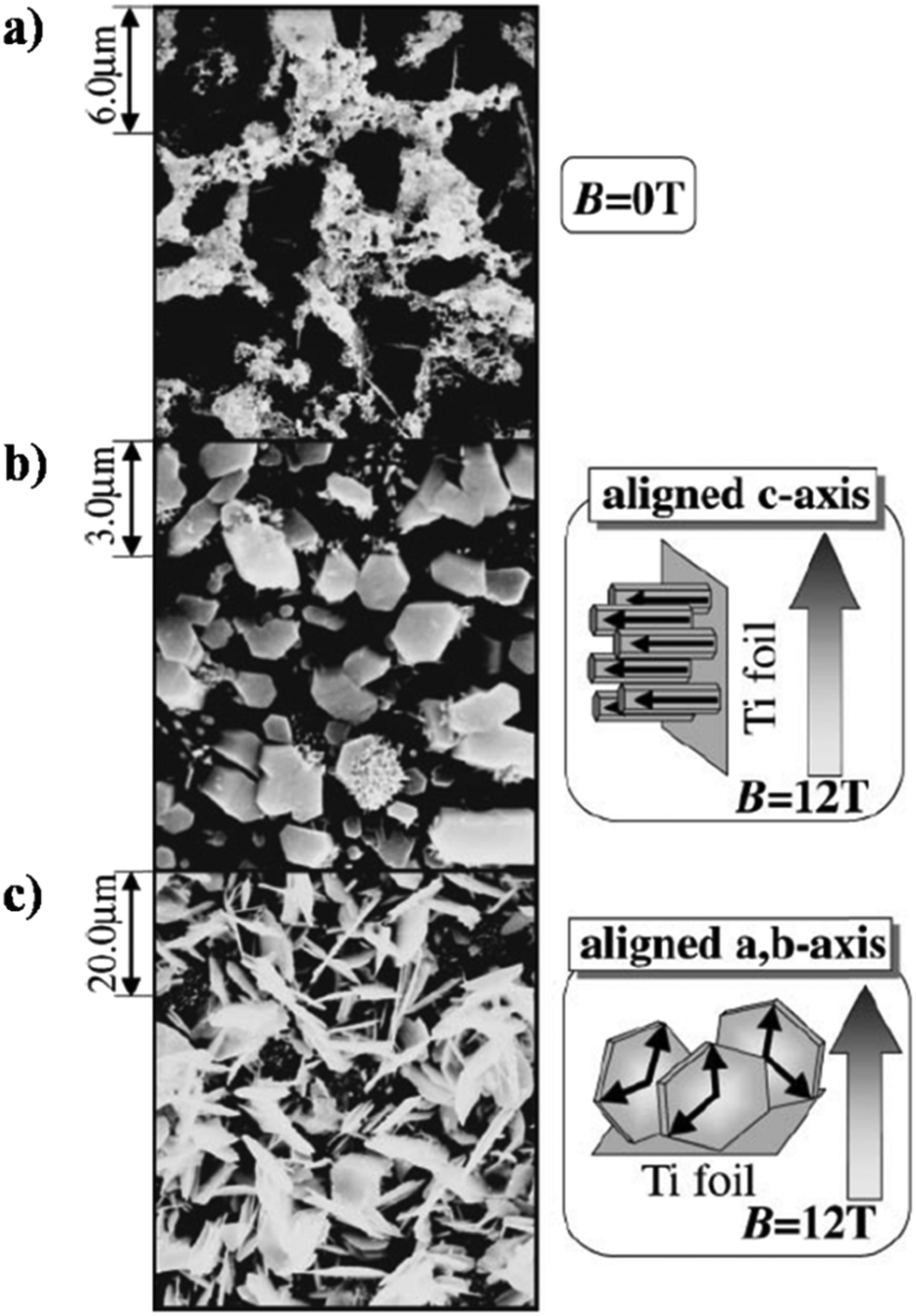 | ||
| Fig. 11 SEM images of HAp electrodeposited via the heat substrate method over Ti foil under a magnetic field strength of (a) B = 0, (b) B = 12 T (B ⊥ j) and (c) B = 12 T (B ‖ j). Reproduced from ref. 124 with permission from The Japan Institute of Metals and Materials. | ||
The variation between the heat substrate method and magnetoelectrodeposition is that the latter involves creation of MHD and mass transport of species between two electrodes. In order to determine the sphere of influence of magnetic flux in an electric field, a comprehensive account of its effect over mass transport, electrode kinetics and electrochemical equilibria has been reported by T. Z. Fahidy.129,130 The phenomenon of magnetoelectrodeposition involves correlating the effects of solution dynamics, termed magnetohydrodynamics (MHD), resulting from the Lorentz force and Kelvin force on the enhancement or suppression of migratory nature of the ionic species. The Lorentz force is the magnetohydrodynamic force (FB), mathematically expressed as the cross-product of magnetic field strength (B) and current density (j) vectors (FB = j × B). The vector cross-product is zero when both are parallel or antiparallel in their directional nature and maximum when they are perpendicular to each other. This maximum generates a convective flow within the electrolyte when (B ⊥ j). However, electrodeposition can occur when FB equals zero which is attributed to the concentration gradient of paramagnetic and diamagnetic species, i.e., the Kelvin force.131 As electrodeposition transpires in the presence of a magnetic field, the Lorentz force induces a convective flow which is speculated to alter the ionic concentration at the electrode–electrolytic diffusion layer.132 When the Lorentz force acts in the direction similar to the direction of conventional forces, an increase in current density is witnessed. This enhances the mass transport and size of the crystals.133
The effects of a superimposed external magnetic field with electric field (j) on the electrolyte on electrodeposition can be determined by recognizing the various possible combinations wherein magnetic flux (B) can align with respect to j. A schematic depicted in Fig. 12 illustrates the assembly of the electrodes between the poles of the electromagnet. The pole pieces of the electromagnet are kept horizontal to the electrodes. The magnets may be placed vertical to the electrodes; however, the outcome of the vector cross-product yields similar results. From the figure, it may be deduced that there may be five possibilities of interaction between B and j among the three possible electrode orientations while keeping the magnet positions fixed. The alignment in Fig. 12a and b represents B parallel to j (B ‖ j) and B perpendicular to j (B ⊥ j) respectively. Fig. 12d has an electrode setup similar to Fig. 12a but the vector alignment of B with j is anti-parallel in nature. Fig. 12c and e exhibits conditions wherein the alignment of B with j is perpendicular in nature but culminates in migration of ions working against the gravitational field (Fig. 12c) and vector forces working along the lines of the gravitational field (Fig. 12e) respectively.
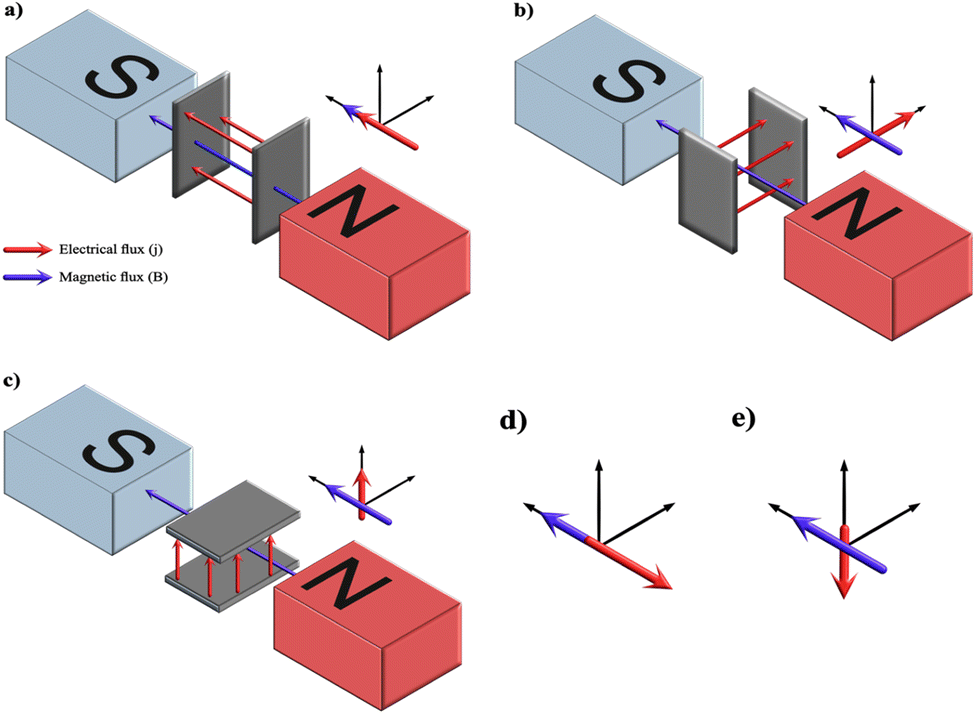 | ||
| Fig. 12 A schematic of the possibilities of orientation of electrode configuration with respect to the electromagnets and the consequent electrical and magnetic flux directions with its representation indicated as (a) parallel, (b) perpendicular, (c) perpendicular aligned against gravity, (d) antiparallel and (e) perpendicular aligned with gravity. | ||
For diamagnetic species, in the case of parallel and antiparallel orientation of B and j (cell configuration analogous to Fig. 12a and d respectively) the magnitude of j increases with B. This is attributed to the Kelvin force, i.e., concentration gradient force. The diamagnetic concentration gradient force can be up to three orders of magnitude smaller than the paramagnetic counterpart, yet it is sufficient to induce convection within the electrolyte.134 In the case of perpendicularity between j and B (Fig. 12b, c and e) the vector magnitude of j is maximum resulting in electrodeposition due to Lorentz force.
The crystal growth of HAp varies significantly as a result of the influence of Lorentz force and Kelvin force. Experimental reports by A. Tian et al.135 and C. Liu et al.136 discuss electrodeposition under a static magnetic field with magnitudes of 0.25 T, 0.5 T and 1.0 T in perpendicular (B ⊥ j) and parallel (B ‖ j) orientations. The experimental parameters can be seen in Table 4. The former study employed Ti foil as a substrate whereas the latter employed TiO2 nanotubular arrays. A zero magnetic flux environment was taken as control, which is analogous to bare electrodeposition. The control yielded a coating of thin flakes of HAp. However, the orientation (B ‖ j) produced spherical HAp while the (B ⊥ j) orientation generated needle-like crystals. The formation of spherical and acicular HAp can be explained due to the additional micro-MHD vortices occurring on the cathode surface.137,138 When (B ‖ j), the cathode is oriented perpendicular to both B and j (interpreted from Fig. 12a). In this scenario, the cathode experiences a primary MHD flow along the periphery of the electrode, resulting in additional micro-MHD flow. These micro-MHD forces are reported to suppress the 3D nucleation thereby promoting the lateral growth of spherical particles. However, when (B ⊥ j), the cathode is parallel to B (Fig. 12a and c), thereby resulting in nucleation and crystal growth along the vertical direction consequently yielding acicular HAp.
| S. No. | Cathode | Electrolyte | Current density (mA cm−2) | Loading time (min) | pH | Temp (°C) | Magnetic field (T) | Comments | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Ti foil | 0.3 mM Ca(H2PO4)2 + 0.7 mM CaCl2 | — | 15 | 7.0 | ∼140 | 12 | Heat substrate method of Ti foil favors a,b-axes growth along the direction of B | 128 |
| 2 | Ti | 0.042 M Ca(NO3)2 + 0.025 M NH4H2PO4 | 10, 15 | 40 | 4.5–5.5 | 85 | 0.5, 1.0 | (B ⊥ j) and (B ‖ j) result in c-plane and a,b-plane crystal orientation respectively | 135 |
| 3 | TiO2 NTs | 0.042 M Ca(NO3)2 + 0.025 M NH4H2PO4 | 10 | 60 | 4.5–5.5 | 85 | 0.25, 0.5, 1.0 | Plate like when B = 0, acicular when B ⊥ j and granular when B ‖ j | 136 |
| 4 | Micro-arc oxidized Mg alloy | 0.1 mol L−1 Ca(NO3)2 + 0.04 mol L−1 NaNO3 + 0.06 mol L−1 NH4H2PO4 | 10 | 60 | 6.0 | 85 | 1.0 | Spherical HAp at 1 T when B ‖ j | 140 |
| 5 | NiTi rod | 0.0084 M Ca(NO3)2 + 0.005 M NH4H2PO4 + 0.1 M NaNO3 + 0.0588 H2O2 | 3, 5 | 3, 5, 7, 10, 15 | 6.0 | 65 | 0.08, 0.101, 0.12, 0.25 | Variation in the Ca/P ratio depending on applied B | 139 |
The control of orientation and morphology of HAp crystals can be achieved through manipulation of mass transfer via controlled electric and magnetic fields. P. M. S. Tohidi et al. performed electromagnetic assisted deposition of HAp onto a NiTi substrate in a pulsed electrodeposition environment.139 In addition to variation of vector orientation, the experiments included the study of position of the cathode with respect to the magnetic pole pieces. The cathode may be placed near the north or south pole pieces of the electromagnet. An optimized experimental condition (i3-B||J-m101-t5) resulted in a plate-like microstructure. However, increasing the field strength from 101 mT to 250 mT increased the presence of micro-voids and porosity. Under a constant loading time, current density and field strength, the orientations of (B ⊥ j) and (B ‖ j) resulted in acicular and spherical morphology respectively. Furthermore, under (B ‖ j) orientation, the positioning of the working electrode closer to either the north or south pole of the electromagnet resulted in the formation of spherical or flake-like crystals respectively.
Likewise, HAp particles electromagnetically deposited over Mg alloy were spherical with a diameter of ∼50 nm.140 An improvement in the anchoring capability of HAp was witnessed when a micro-arc oxidized substrate was employed. A lowered (002) plane intensity can be ascribed to the formation of spherical particles. The control experiment carried out in the absence of a magnetic field indicated a crystal growth along the (300) plane.
To summarize, the heat substrate method resulted in hexagonal pillars at 12 T, whereas magnetoelectrodeposition produced needle-like crystals when (B ⊥ j) at a lower B of 0.25–1.0 T. Additionally, it was revealed that irrespective of the deposition method, the application of a magnetic field predominantly brought about crystal orientation along the (002) c-plane (2θ ≈ 26°) when (B ⊥ j). Similarly, the absence of a magnetic field favored crystal orientation along the a,b-axes. This leads to the revelation that the control and refinement of the crystal size, orientation of specific planes of HAp and control of coating roughness can be engineered by the application of a magnetic field during electrodeposition.
Y. Ya-Jing et al. reported on the microgravity effects over HAp electrodeposition onto Ti6Al4V substrates. The substrate was functionalized with negatively charged self-assembled mono layers (SAMs) comprised of –COOH and –PO3H in order to better facilitate induced growth by modification of substrate surface energy.142 Microgravity was simulated via a high magnetic field inducing superconducting magnet. The applied field strength had a maximum of 16 T. A near 0 G environment arose when the magnetization force Fm balanced the gravitational force FG in a vertical arrangement wherein the two forces were anti-parallel to each other.143 The HAp particles took on a spherical microstructure with not much agglomeration due to reduced collision kinetics in this microgravity environment. With HAp having a diamagnetic susceptibility, the ionic species Ca2+ and PO43− would remain suspended in the microgravity environment due to the drop in convection and solute mass transport. However, the effect of an applied electric field on nucleation (i.e. an orientation wherein j and B are anti-parallel and magnetic flux B aligns against gravitational field G) in this simulated microgravity environment has not yet been fully explored. By understanding the mechanics of the piezoelectric nature of HAp acting in synergy with MHD forces in a microgravity environment, one may effectively overcome the predicament of osteoporosis in space.
| S. No. | Cathode | Electrolyte | Potential (V) | Current density (mA cm−2) | Loading time (min) | pH | Temp (°C) | Pressure (Torr) | Ca/P ratio | Comments | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Ti plate | 2.5 mM CaCl2 + 1.67 mM K2HPO4 + 137.8 mM NaCl | — | 12.5 | 60 | 7.2 | 100, 150, 200 | ∼12000 @ 200 °C | 1.47–1.65 | Dissolution of metal resulting in mixed phase of calcium phosphate | 147,260 |
| Zn plate | 1.59–1.03 | ||||||||||
| Ni plate | 1.49–1.45 | ||||||||||
| Fe plate | 1.58–1.39 | ||||||||||
| SS plate | 1.45–1.51 | ||||||||||
| 2 | Ti plate | 2.5 mM CaCl2 + 1.67 mM K2HPO4 + 137.8 mM NaCl | — | 5–25 | 10–120 | 7.2 | 90–200 | — | — | High pressure electrodeposition | 146 |
| 3 | Ti mesh | 2.5 mM CaCl2 + 1.67 mM K2HPO4 + 137.8 mM NaCl | — | 12 | 60 | 7.2 | 90, 100, 150 | — | 1.38 @ 90 °C, 1.40 @ 100 °C, 1.54 @ 150 °C | Microstructure variation from plate-like to needles with the change in autoclave temperature | 148 |
| 4 | Anodized Ti6Al4V | 0.138 mol L−1 NaCl + 0.016 mol L−1 K2HPO4 + 0.02 mol L−1 CaCl2 | — | 1.25 | 120 | — | 120 | — | — | Reduction in the (002) plane | 261 |
| 5 | Ti6Al4V | 0.04 M MCPM | 4–10 | — | 60 | ∼3.0 | 0–60 | 80 | 1.05–1.69 | Transition from plate to needle to granular HAp | 150 |
| 6 | Ti6Al4V | 0.04 M MCPM | 4–10 | — | 20–120 | 3.0 | 60 | 80 | 1.54 | Low pressure electrodeposition | 262 |
| 7 | Ti6Al4V | 0.04 M MCPM | 4–10 | — | 20–120 | ∼3.0 | 0–60 | 80 | 1.09–1.83 | HAp content increases with potential and temperature at 80 Torr | 151 |
| 8 | Ti plate | 2.5 mM Ca(NO3)2 + 1.5 mM NH4H2PO4 | — | 1.0 | 30, 60 | 4.2–5.1 | 80 | Ambient pressure | — | Atmosphere controlled deposition | 152 |
5.2.5.1. Effect of high pressure. S. Ban and J. Hasegawa studied the effect of high pressure on HAp crystal growth by employing a combined hydrothermal-electrodeposition method.146 A quartz beaker electrochemical cell was sealed in a high pressure autoclave, thereby creating a closed system, wherein morphological modifications were studied with variation of temperature, current density and time. The crystals grown in this high pressure environment were hexagonal needles. A decrease in aspect ratio was observed with increasing temperature and loading time attributed to the decreased nucleation sites at high pressure. Furthermore, a reduction in solubility of crystals was witnessed. This resulted in a subsequent growth on existing crystals thereby increasing the crystal size. During electrodeposition, an increase in liberation of H2 gas at the cathode was seen due to the increase in current density. However, due to the closed environment, this created a saturation of overall electrolytic pH thereby enabling modulation of Ca2+ and PO43− sites.
An earlier study by S. Ban et al. reported a similar experimental work while varying the cathodic substrates.147 The growth of HAp on various metal substrates, herein Ti, Zn, Ni, Fe and stainless steel plates, was studied exemplifying the relation between formation of phase-pure HAp crystals and ionic dissolution of the cathode. The study was carried out under temperatures of 100, 150 and 200 °C. This comparative study noted the formation of phase-pure HAp on all substrates only at 100 °C. Temperatures exceeding 100 °C resulted in mixed phases for all substrates with the exception of Ti. Vertically oriented hexagonal HAp crystallized at all temperatures on the Ti plate. An increase in temperature resulted in an increased deposition over the already nucleated crystals thereby making the hexagonal rods thicker. Consequently, an increase in the Ca/P ratio was witnessed in the case of Ti, signifying the preferentiality of (002) crystal orientation with the exposed c-plane.
Hydrothermally assisted electrodeposition of HAp over a Ti mesh substrate was carried out by A. Yuda et al.148 The results presented indicate the formation of a homogeneous coating with variation in morphology as a function of hydrothermal temperature. At 90 °C, plate-like deposits and at 100 °C, a mixture of plate- and needle-like morphology were obtained. A further increase in temperature to 150 °C witnessed a complete shift in morphology to acicular. The dimensions of the needles grown at 100 °C were 7 μm in length and 0.5–1.0 μm in width while those grown at 150 °C were 60–90 μm in length and 2–7 μm in width. The assimilation of the needles was hexagonal with an increase in length dictating an increase in the Ca/P ratio of 1.38 (90 °C), 1.40 (100 °C) and 1.54 (150 °C). This electrodeposition technique of high pressure coupled with high temperature facilitates reduction in the viscosity of the electrolyte.149 The low viscosity enhances the rate of diffusion of ions, rate of crystal growth and orientation of the HAp crystal along the c-axis.
5.2.5.2. Effect of low pressure. Alternatively, the effect of sub-atmospheric pressure on morphology of HAp crystals grown on the electrode is quite a unique one elaborated in two literature reports. The experiments, both carried out by M.-C. Wang et al., were carried out in a reduced pressure environment, specifically 80 Torr. This reduced pressure environment was achieved by placing the electrolytic cell in a vacuum chamber. The first and second study involved the morphological study of the coatings on Ti6Al4V alloy with variations in deposition potential150 and deposition time respectively.151 With the precursor to be monocalcium phosphate monohydrate (MCPM) in both studies, the initial stages of electrodeposition generate both MCPM and DCDP as plates. However, variation of potential over a range from 4 V to 10 V resulted in an increase of HAp as the predominant phase. The morphology transitioned from plate-like at 4 V to acicular at 7 V and finally granular at 10 V.
This sub-atmospheric pressure condition proved to be advantageous in terms of mechanical strength of coating. This can be elucidated from the illustration depicted in Fig. 13. It is well known that any electrochemical process carried out in aqueous medium results in the generation of H2 gas bubbles at the cathode. The interspersed gas bubbles tend to distort the double layer between the electrode and the electrolyte (dotted line shown in Fig. 13a). The bubbles hinder the migration of solvated ions thereby obstructing crystal nucleation and growth sites. Consequently, this results in an uneven non-compact coating with porous volcano-like microstructures (illustrated in Fig. 13b). Upon lowering of electrolytic cell pressure to sub-atmospheric pressure, the gas bubbles were swiftly removed, accounting for the reduction in volcano-like structures.151 As a consequence, the minimization of double layer distortion resulted in an improved coating with an enhanced adhesive strength as shown in Fig. 14. The increase in deposition potential coupled with the reduction in pressure enhances mass transport of Ca2+ and PO43− ions thereby improving crystallinity and phase purity. Additionally, unlike the high pressure environment, this experimental sub-atmospheric pressure resulted in enhanced crystal nucleation and growth ensuing uniform HAp coating over the electrodeposited substrate. The tunability of ambient pressure in an electrolytic system noticeably improves the nature of deposition in terms of mechanical stability and bonding strength to the substrate.
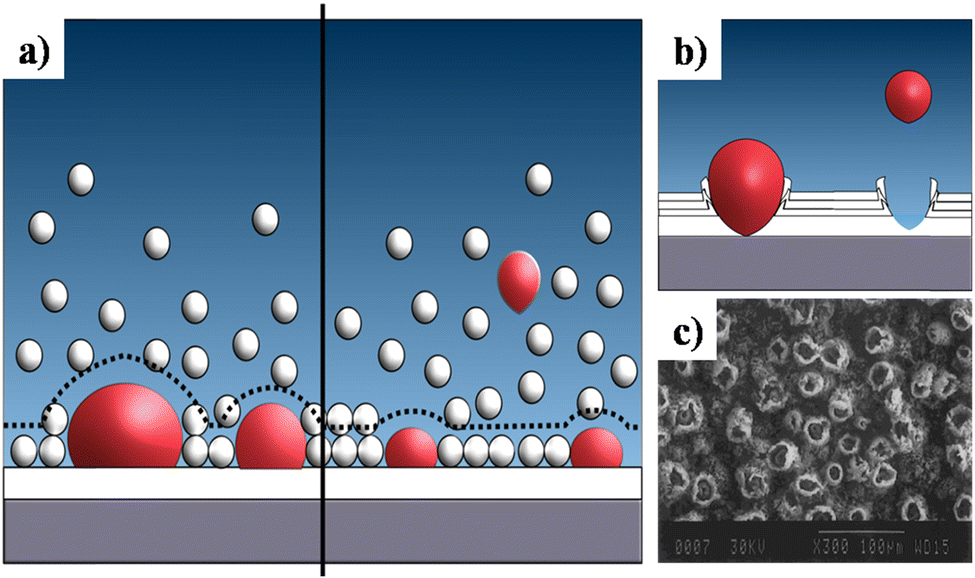 | ||
| Fig. 13 (a) A schematic representation of the double layer distortion (represented with dotted lines) at ambient pressure (left) and reduced pressure (right) as a result of adhering gas bubbles (in red), (b) an illustration of the formation of volcano-like structures and (c) SEM of DCDP coatings with volcano-like growth under ambient pressure. Image (c) reprinted from ref. 266 with permission from Wiley. | ||
 | ||
| Fig. 14 The enhanced adhesive strength displayed by electrodeposited HAp specimens onto Ti6Al4V at (a) ambient pressure and (b) 80 Torr reduced pressure. Adapted from ref. 150 with permission from Elsevier. | ||
5.2.5.3. Effect of controlled atmosphere-assisted electrodeposition. Unlike the previous studies of employing low and high pressure in a closed electrolytic cell, the manipulation of crystal orientation via the control of the cell atmosphere has been reported by X. Lu and group.152 Galvanostatic electrodeposition of HAp was performed on a Ti plate and the electrolytic setup was sealed and equipped for dynamic gas flow. The ambient atmospheres selected for this study were CO2, N2 and compressed air (comprising 78.08% N2, 20.95% O2 and 0.03% CO2) with a dynamic flow rate of 1 L min−1.
The controlled atmosphere facilitated manipulation of crystal orientation and phase purity. As shown in Fig. 15, the SEM micrographs indicate the formation of layered deposits of needle-like and plate-like coatings electrodeposited under N2 and CO2 atmospheres respectively. In a compressed air environment, comprising N2 and CO2, microstructural variations were observed with the progress in loading time with the coatings assuming a needle-like microstructure under a 30 min loading time with a gradual shift to a plate-like appearance beyond 30 min. The compressed air atmosphere resulted in the initial formation of phase-pure HAp needles. However, with the progress in loading time, the HAp gave way to the formation of OCP plates. This resulted in the coatings to be a HAp + OCP mixed phase with a mixed needle + flake microstructure under the controlled atmosphere of compressed air.
 | ||
| Fig. 15 SEM depicting the formation of (a) needle-like HAp in a N2 atmosphere and (b) plate-like HAp in a CO2 atmosphere electrodeposited under galvanostatic conditions. Adapted from ref. 152 with permission from Elsevier. | ||
The results indicate that the crystal size and growth can be controlled by the ambient atmosphere within the electrolyte cell. Under a constant deposition rate, HAp crystals are much larger when electrodeposited in a CO2 atmosphere than in a N2 environment. The role of the cell atmosphere also accounts for pH variation within the electrolyte. The saturation of CO2 provides for an acidic environment while N2 gas shifts the pH towards alkalinity. The control over the ambient atmosphere serves as a modulating factor for pH and growth rate along with a consequential influence on the Ca/P ratio, rate of nucleation and topography. A CO2 atmosphere resulted in crystal orientation along a,b-axes while a N2 atmosphere oriented the crystal along the c-axis.
5.2.6.1. Crystallinity of Ti and TiO2 NT substrates. Electrodeposition of HAp onto Ti alters the growth rate based on the morphology and crystallinity of TiO2 as investigated by Y. Parcharoen et al.153 Herein, Ti foils were anodized to synthesize TiO2 NTs of varying tube length. The electrodeposited HAp adhered with different capacities depending on the length of the NTs while larger diameters enhanced the formation and adhesion of the grown crystals. Another investigation by H. Tsuchiya et al.154 considered the influence that the crystalline phase of TiO2 NTs had on nucleation. An interesting observation is the indication of increased nucleation and formation of HAp over crystalline samples in comparison to amorphous TiO2 NTs. Furthermore, a reduction in growth was seen for phase-pure anatase TiO2 NTs in comparison to mixed anatase–rutile phase. This enhancement in nucleation rate for mixed anatase–rutile crystalline TiO2 NTs may be due to variation in thermodynamic variables.
D. Gopi et al. performed a pulse electrodeposition of HAp onto HELCDEB treated Ti in a H2O2 environment.155 The HAp crystals grown on the bare Ti substrate were flake-like while those on the HELCDEB surface treated Ti substrate nucleated as rods in a lotus-like fashion with an indicative lowered (002) plane. F. S. Utku et al. studied the effects of pulses and electrolytic temperature on the Ca/P chemistry of HAp deposits grown over a normal Ti plate and TiO2 substrates anodized in the presence and absence of F− ions.156 The investigation revealed that the deposits were plate-like in the case of the Ti plate with preferred crystal orientation along the (300) plane. However, the anodized TiO2 NTs both had varying sizes of needle-like crystals depending on the electrolyte temperature with varying magnitudes of the Ca/P ratio. The use of anodized and non-anodized Ti substrates indicated the control over crystal growth and orientation in a bipolar pulsed galvanodynamic mode.
5.2.6.2. Functionalized carbon substrates. HAp has been reported to have been coupled successfully to MWCNTs, graphene and reduced graphene-metallic substrates.157–159 These serve the purpose of improving mechanical and adhesive strength, as biocompatible fillers with anti-microbial properties and acting as a selective gateway for various ionic species. Additionally, their influence on growth of electrodeposited HAp in terms of porosity, hydrophilicity and morphology is enhanced.
The morphological improvement of electrodeposited HAp, reported in ref. 160, is carried out potentiostatically over anodized TiO2 NTs (Fig. 16a) with the resulting microstructure resembling flakes with a Ca/P ratio of 1.5 (Fig. 16b). However, the formation of HAp carried out in the presence of –COOH functionalized MWCNTs resulted in a homogeneous foam-like appearance with a Ca/P ratio of 1.6 (shown in Fig. 16c). This foam-like HAp coating grown over the substrate witnessed an increase in porosity which facilitates an enhanced migratory transport of ions within the composite coating via its micro- and macro-porous channels. Another relatable improvement can be witnessed in micro-hardness upon incorporation of MWCNTs with a decrease in contact angle signifying the hydrophilic nature of the coatings.
 | ||
| Fig. 16 SEM images depicting the microstructural variation in HAp coating electrodeposited over TiO2 NT and –COOH functionalized MWCNT/TiO2 NT substrates: (a) TiO2 NTs, (b) HAp/TiO2 NT nanocomposite and (c) HAp/MWCNT/TiO2 NT nanocomposite. Adapted and rearranged from ref. 160 with permission from Elsevier. | ||
Z. Leilei investigated the galvanostatic electrodeposition of HAp onto a carbon nanotube/nanofiber hybrid template with ultrasound assistance.161 The carbon nanotubes grew radially to the carbon nanofiber template. Subsequently, this was employed as a template for the deposition process. The carbon nanotubes maintained a straight and porous network (Fig. 17a) with gaps ranging from 1 to 10 μm. The HAp deposited network over carbon nanotubes and carbonated carbon nanofibers can be seen in Fig. 17b and c respectively. The deposits over carbon nanotubes have evolved into a particle shaped HAp while the carbon fibers are coated with a plate-like HAp with size ranging from 20 to 50 μm. The control over microstructure can prove effective in the tunability of the crystal orientation and the microstructure, depending on the electrode modifications employed.
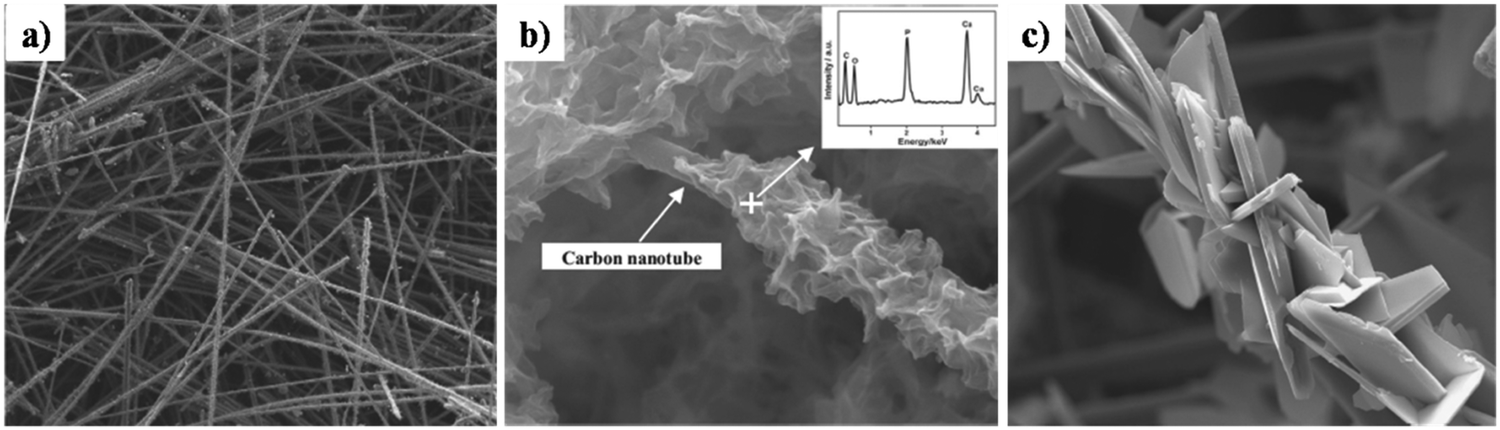 | ||
| Fig. 17 SEM of (a) carbon nanotubes, (b) carbonated HAp electrodeposited over carbon nanotubes and (c) carbonated HAp deposited over carbon fibers. Adapted from ref. 161 with permission from Elsevier. | ||
5.2.6.3. Nafion-Ni foam substrate. Q. Zhang et al. fabricated a Ni foam/HAp composite by employing Nafion-assisted electrodeposition.162 Herein, Nafion plays the role of an electrolytic membrane facilitating high proton conductivity and no anion uptake and exhibits excellent physico-chemical durability. The experimental protocol reported involves Nafion being used as a template for electrodeposition of HAp crystals. As shown in Fig. 18a, a bare Ni foam resulted in hexagonal needle-like HAp microstructures with coating thickness ranging from 10 to 15 μm. However, the incorporation of Nafion to the Ni foam substrate stimulated the growth of a 3D morphology comprised of a mixture of needles with agglomerates of spherical HAp (Fig. 18b). Every individual sphere comprised of needle-like nanocrystals 100 nm in length with an overall spherical diameter of 300 nm. An intense (002) peak signifies the favored directional growth along the c-axis. The composite witnessed an increase in the degree of crystallinity from 85.27% to 91.57% upon inclusion of Nafion onto the substrate. The Nafion membrane is speculated to facilitate an optimized distribution of OH− ions near the cathode surface thereby being conducive to the formation of a layered topography with enhanced crystallinity.
 | ||
| Fig. 18 Cross-sectional SEM of HAp electrodeposited over (a) Ni foam and (b) Nafion coated Ni foam. Adapted from ref. 162 with permission from Elsevier. | ||
5.2.6.4. Cation exchange membrane-assisted electrodeposition. A unique tubular shaped HAp was synthesized via the method of cation exchange membrane-assisted electrodeposition.163,164 The conceptual design of the experiment involves two glass containers separated by a Nafion N-117 membrane, with the Ca2+ and PO43− precursor in each container. Electrical contact is supplied through two electrodes, one placed in each electrolytic cell. When an applied potential is introduced, the Ca2+ ions penetrate through the exchange membrane towards the PO43− rich side. The formation of HAp occurs directly on the Nafion membrane. Upon completion, the Nafion membrane is carefully rinsed and dried and the tubular HAp powder is collected from the membrane. The peculiarity of this technique is that the HAp electrodeposition does not occur on any metallic working electrode but instead on the Nafion membrane. Since the driving force for the ions through the membrane is the applied potential, it is coined as cation exchange membrane-assisted electrodeposition. With a phase constitution of HAp, the preferred crystal orientation was along the (002) plane, i.e. the exposed c-axis. The growth mechanism of the tubular structures is reported to occur at the bottom, closer to the exchange membrane, and in some ways, it resembles an extrusion process. The tubular HAp microstructures and schematic regarding the growth of hollow tubular shaped HAp are depicted in Fig. 19. The formation of tubular HAp supports the hypothesis of growth occurring near the Nafion membrane as the Ca2+ ions diffuse through. It is speculated that the HAp crystals grow preferentially orienting along the c-axis to form nanosheets and then roll up to form tubular HAp to reduce surface energy.
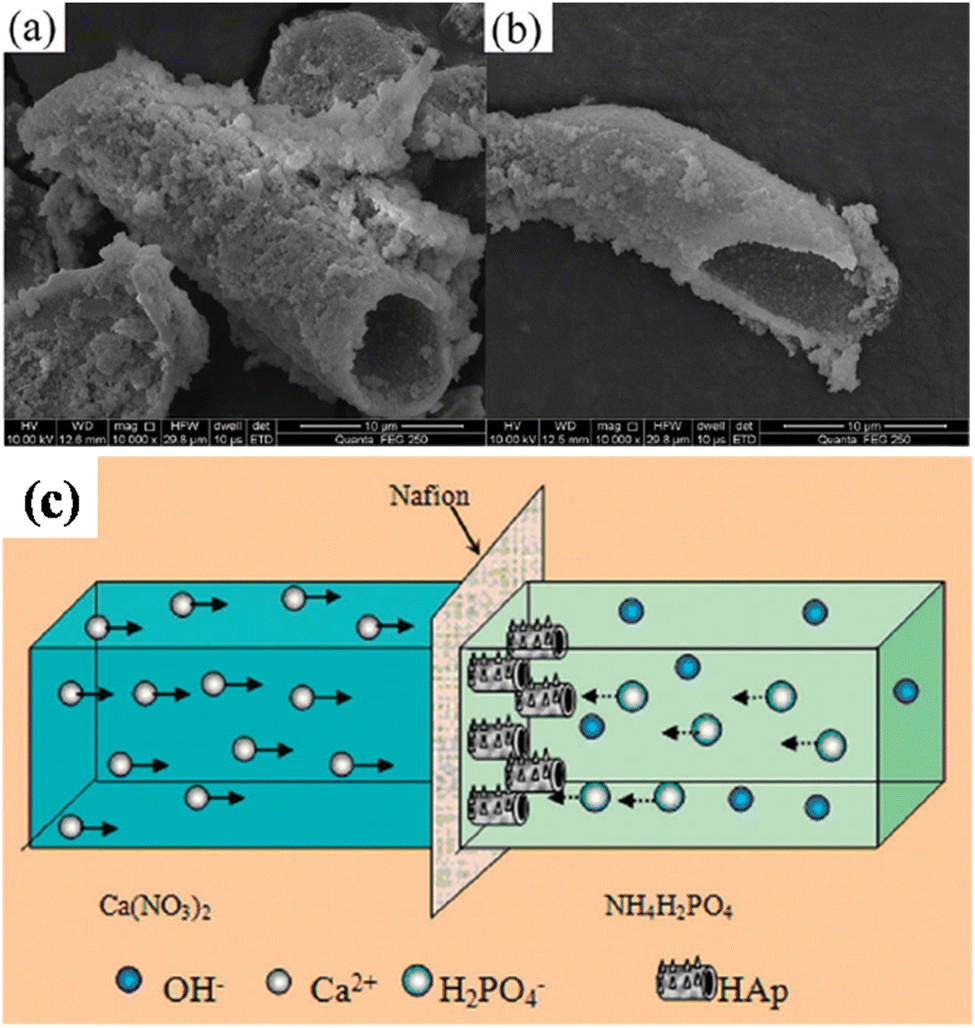 | ||
| Fig. 19 (a and b) SEM of tubular shaped HAp synthesized via the cation exchange membrane-assisted electrodeposition technique and (c) the schematic illustration of the formation of tubular HAp. Images (a) and (b) adapted from ref. 163 and image (c) reproduced from ref. 164 with permission from Royal Society of Chemistry. | ||
5.2.6.5. Template-assisted electrodeposition. S. Beaufils et al. employed a template-assisted electrodeposition approach towards the synthesis of HAp nanotubes and nanowires as powders.165 Deposition of HAp was performed in a three-electrode setup wherein the working electrode was a track-etched polycarbonate membrane with one side sputtered with 300 nm gold film for conductivity (shown in Fig. 20a). The morphology of the nanotubes and nanowires grown via this method can be seen in Fig. 20b and c respectively. The electrodeposition of HAp was examined via both potentiostatic and applied pulsed modes. H2O2 was added to minimize the formation of H2 bubbles which may otherwise disrupt the ionic diffusion process and block the pores of the etched polycarbonate template. The template was dissolved in dichloromethane upon completion of electrodeposition. By varying the membrane pore diameter and controlling the deposition parameters, the thickness of the needles and nanotubes can be controlled thereby altering the aspect ratio between 19 and 71 for nanowires. The aspect ratio for HAp nanotubes varied between 9 and 25 with high specific surface area. The Ca/P ratio of the HAp crystals fabricated via pulsed template-assisted electrodeposition was ∼1.5. The growth orientation of the nanowires and nanoneedles was along the c-axis while the predominantly exposed HAp facet indicated an increase in the a,b-plane during the application of pulses. Compared to the growth of HAp nanotubes via the cation exchange membrane-assisted electrodeposition, the approach of template-assisted electrodeposition facilitates the fabrication of HAp nanotubes and nanowires of controlled morphological homogeneity.
 | ||
| Fig. 20 FESEM of (a) the 100 nm porous polycarbonate membrane template and template-assisted electrodeposited microstructures of (b) HAp nanotubes and nanowires deposited under constant potential and (c) HAp nanowires deposited under pulsed mode. (d) SEM of HAp nanowire nanoarrays electrodeposited onto GCE via template-assisted electrodeposition. Images (a), (b), and (c) adapted from ref. 165 and image (d) adapted from ref. 229 with permission from Elsevier. | ||
:EtOH (2:1) at pH 5.5 and Na3PO4:EtOH (2:1) at pH 7.2 respectively. The working electrode was polarized at 1 mA cm−2 of cathodic current density when immersed in the Ca2+ electrolytic precursor followed by immersion in PO43− solution with an anodic current density of 1 mA cm−2 for a total of 20 cycles, 30 s each. Note that this technique does not incorporate a counter electrode. Instead, the polarity of the working electrode is reversed alternatively, wherein the porous silicon substrate is negatively charged when immersed in the Ca2+ solution and positively charged when immersed in the PO43− precursor (Fig. 21a and b). This alternating cyclic immersion incorporates a cyclic change in overall charge density over the substrate resulting in a heterogeneous coating of electrocrystallized HAp with isolated agglomerates (Fig. 21c and d). The density and size of the HAp clusters over the substrate witnessed an enhancement with the increase in the number of immersion cycles. The agglomerated HAp comprised spherical nanoparticles with a predominant crystal facet along the a,b-plane.
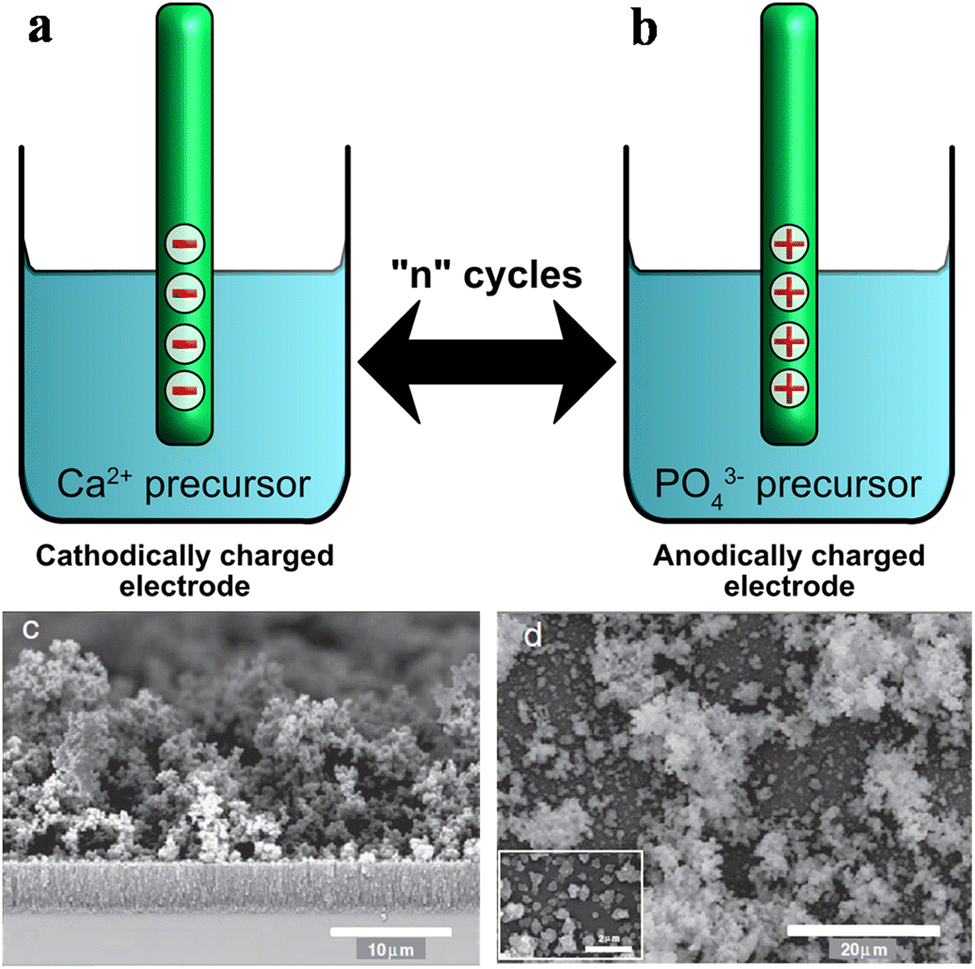 | ||
| Fig. 21 Schematic representation of the technique of cyclic electrodeposition where the electrode is alternatively immersed in precursor solution under galvanostatic conditions. (c) Cross-sectional and (d) topographical FESEM of agglomerated HAp spheres synthesized via cyclic electrodeposition. Images (c) and (d) adapted from ref. 167 with permission from Elsevier. | ||
6. Sensing applications of HAp
Sensors enable the quantification of physical and chemical changes occurring in our environment, ranging from biological and biomedical changes for human health and patient care,168–170 to chemical quantification of environmental pollutants and their remediation.171,172 The detection of gaseous components from industrial and manufacturing processes,173,174 and use as biosensors for glucose, peroxidase and protein estimation175–178 are some of the incredible applications of sensors. The materials constituting a sensor can include semiconductor metal oxides179–182 and carbon-based materials183,184 with functionality ranging from mechanical stimuli185–191 to environmental monitoring.192,193The quantitative detection and sensing of gaseous pollutants is vital in the industrial and manufacturing arena to safeguard human health. Emission control of hazardous substances can be realized via sensors. The detection of gaseous components generally occurs either by chemical interaction with surface active sites or through physisorption onto the sensing material. This interaction results in variations over surface charge distribution that can be measured electrochemically. Depending on the technique, the detection of the analyte can be resolved via change in resistance, impedance, capacitance and amperometry among other measurement systems. Among the myriad detection methods for chemical sensing, resistance-based sensing is the simplest. The common environmental gaseous pollutants that require rapid detection and response include CO, CO2, SO2, NH3, water vapor, NOx and VOCs, to name a few.
6.1. HAp composites towards detection of gaseous analytes
The unique property of the presence of surface active sites of crystalline HAp, viz. a,b-plane and c-plane, facilitates its sensing behavior towards gases and solvated ions. The crystal facets of hexagonal HAp comprise of acidic and basic sites thereby ensuring versatility towards the detection of these gases. The application of HAp in the field of gas sensing can attributed to its porosity, surface groups along the a,b-plane and c-plane and its ion exchange capability. The drawback of phase-pure HAp as a sensing material encompasses a low response time, high operating temperatures and selectivity towards acidic and basic gas molecules. To mitigate this, a sensor is generally activated through the incorporation of dopants resulting in binary and ternary composites so as to provide a framework and serve as a means for recording the variation in charge carrier density occurring from the analyte interaction over the active material. By modifying the physico-chemical properties of HAp with additives, dopants and microstructural and crystalline modifications, one may enhance the interaction of HAp towards ionic species.The technical detection of any analyte species can be performed in either static or dynamic environments. A dynamic environment takes into consideration the gas flow rate while ensuring the safety protocol involved in venting out the gas after measurement. However, we will limit our discussion to the static mode of gas sensing. The percentage sensitivity of a sensor towards the target gas can be determined via resistance-based measurements and is calculated from the relation
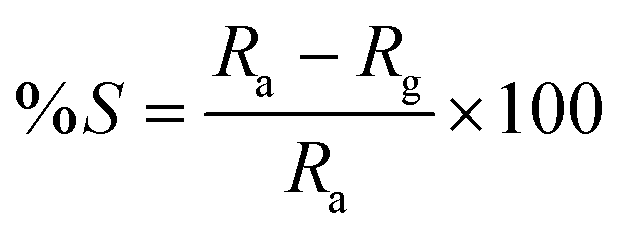 | (15) |
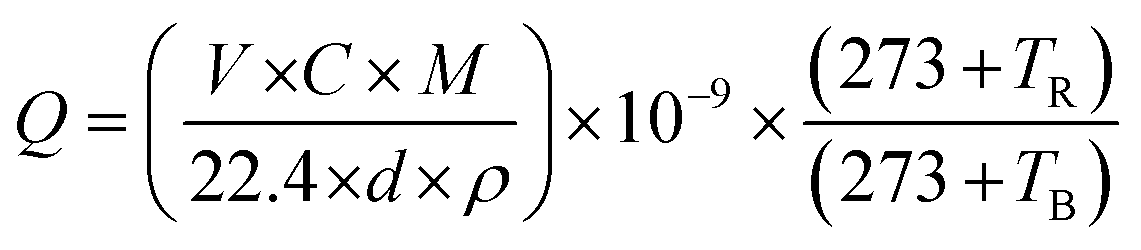 | (16) |
Gas sensing depends on the change in resistance induced during adsorption and desorption of the analyte species. Gases with functional groups (–NH2, –C6H5, –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) exhibit higher response upon interaction with HAp. The mechanism of detection based on HAp can be related to the density and migration of the charge carriers across the surface crystal lattice. At room temperature, it is reported that protons (H+) are the major charge carriers on HAp.163 The proton may migrate from one OH− to the adjacent OH− or PO43− groups on the P-site. If the growth orientation is predominant along the c-plane, the amount of ionic channels is enhanced thereby facilitating rapid adsorption and improved conductivity. With H+ being the charge carrier on HAp at room temperature, an increase in H+ concentration witnesses an increase in resistance. For a migrating proton between two adjacent OH− groups, the interaction may be related as
O) exhibit higher response upon interaction with HAp. The mechanism of detection based on HAp can be related to the density and migration of the charge carriers across the surface crystal lattice. At room temperature, it is reported that protons (H+) are the major charge carriers on HAp.163 The proton may migrate from one OH− to the adjacent OH− or PO43− groups on the P-site. If the growth orientation is predominant along the c-plane, the amount of ionic channels is enhanced thereby facilitating rapid adsorption and improved conductivity. With H+ being the charge carrier on HAp at room temperature, an increase in H+ concentration witnesses an increase in resistance. For a migrating proton between two adjacent OH− groups, the interaction may be related as
| 2OH− → O2− + H2O | (17) |
This section will focus on the aspect of sensing behavior in relation to the exposed and active crystal facet of hexagonal HAp nanocomposites. Additionally, the mechanism involved in the detection of each analyte is presented. The added benefits of porosity and surface acidity/basicity facilitate detection through increased density of active sites. Table 6 outlines the sensor material, its working temperature at maximum % sensitivity, response–recovery time and the predominantly exposed crystal facet.
| S. No. | Sensor | Analyte gas | Working temperature (°C) | Optimum concentration (ppm) | Sensitivity (%) | Response/recovery time (s) | Dominant HAp crystal facet | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | Tubular HAp | NH3 | RT | 2000 | 84.58 | 10/180 | c-Plane | 163 |
| 2 | PANI/tubular HAp | NH3 | RT | 500 | 85.0 | —/— | c-Plane | 194 |
| 3 | PPy/tubular HAp | NH3 | RT | 1000 | 90.7 | —/— | c-Plane | 194 |
| 4 | Au/tubular HAp | NH3 | RT | 200 | 86.7 | 20/25 | c-Plane | 196 |
| 5 | Graphene doped HAp | NH3 | RT | 50 | 54.0 | —/— | a,b-Plane | 195 |
| 6 | HAp/cellulose nanofiber | NH3 | 25 | 5 | 575 | 210/30 | — | 197 |
| 7 | Tubular HAp | H2S | RT | 2000 | 31.2 | —/— | a,b-Plane | 200 |
| Acidithiobacillus ferrooxidans incorporated tubular HAp | H2S | RT | 2000 | 76.0 | —/— | a,b-Plane | ||
| 8 | Tubular HAp | Acetone | RT | 1000 | ∼60 | 63/31 | c-Plane | 163 |
| n-Hexane | ∼46 | 25/40 | ||||||
| Styrene | ∼50 | 40/139 | ||||||
| 9 | Graphene doped HAp | Acetone | RT | 500 | 35.7 | —/— | a,b-Plane | 195 |
| Methanol | 41.6 | —/— | ||||||
| THF | 35.5 | —/— | ||||||
| 10 | CNT doped HAp | Ethanol | RT | 100 | 600 | 90/120 | c-Plane | 207 |
| 11 | CNT doped HAp | Methanol | 30 | 100 | ∼600 | 60/— | c-Plane | 263 |
| 12 | HAp-TiO2 | Methanol | 33 | 10 | 225 | 50/20 | a,b-Plane | 204 |
| Ethanol | 35 | 190 | 170/20 | |||||
| Propanol | 55 | 192 | 100/70 | |||||
| 13 | Graphite doped HAp | Methanol | 30 | 100 | 465 | 90/180 | c-Plane | 205 |
| Ethanol | 30 | 333 | 110/200 | |||||
| Propanol | 60 | 352 | 60/— | |||||
| 14 | HAp | CO | 195 | 1000 | 45 | 35/25 | c-Plane | 219 |
| Co-HAp | 150 | 135 | ∼25/∼20 | |||||
| Fe-HAp | 140 | 91 | 30/40 | |||||
| 15 | SWI HAp | CO | 195 | 1000 | 210 | 20/25 | c-Plane | 264 |
| 16 | SWI Co-HAp | CO | 150 | 1000 | 220 | 20/∼20 | c-Plane | 218 |
| 17 | SWI Fe-HAp | CO | 140 | 1000 | 250 | ∼20/25 | c-Plane | 217 |
| 18 | HAp-BaTiO3 | CO | 175 | 1000 | 78 | —/— | c-Plane | 219 |
| 19 | HAp-TiO2 | CO | 185 | 1000 | 60 | —/— | c-Plane | 219 |
| 20 | 0.05 wt% CNT-HAp | CO | 105 | 20 | 130 | 48/62 | a,b-Plane | 221 |
| 21 | HAp | CO2 | 165 | 1000 | 31 | 20/15 | a,b-Plane | 216 |
| Co-HAp | 135 | 75 | 35/45 | |||||
| 22 | SWI Co-HAp | CO2 | 135 | 1000 | 156 | 25/30 | c-Plane | 218 |
| 23 | Fe-HAp | CO2 | 155 | 1000 | 112 | 30/40 | c-Plane | 215 |
| 24 | SWI Fe-HAp | CO2 | 155 | 1000 | 290 | 15/20 | c-Plane | 217 |
| 25 | 0.1 wt% CNT-HAp | CO2 | 70 | 20 | 200 | 70/35 | a,b-Plane | 220 |
L. Huixia et al. reported the development of an effective NH3 sensor based on HAp composited with conducting polymers such as PANI and PPy.194 PANI and PPy polymers are p-type semiconductors. Tubular HAp microstructures were fabricated via the cation exchange membrane-assisted electrodeposition technique. The composites were prepared by mechanical mixing, and electrical measurements for gas sensing efficacy were recorded at room temperature under static conditions. As previously discussed (in Section 5.2.6.4), tubular HAp synthesized on the exchange membrane exhibited preferential crystal orientation along the c-plane with high specific surface area. The maximum sensitivity was displayed by 5% PPy/HAp and 20% PANI/HAp composites at NH3 concentrations of 1000 ppm and 500 ppm respectively. When compared to pure HAp, the response/recovery time of PANI and PPy nanocomposites was lower. However, lower detectable limits improved in conducting polymer composites. The selectivity towards NH3 was determined along with four interfering VOCs and the selectivity coefficient was calculated as
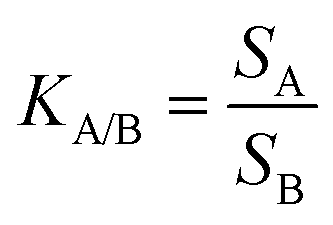 | (18) |
Additional reports involving HAp doped with graphene,195 Au196 and cellulose nanofibers197,198 revealed varying levels of sensitivity towards NH3 with the measured values tabulated in Table 6. Interestingly, the predominant crystal orientation of HAp in most reports was along the c-plane with the exception of the report in ref. 195 wherein the graphene doped HAp has its HAp crystals oriented along the a,b-plane (based on XRD interpretation) resulting in a reduced sensitivity of 54%. The HAp blended cellulose nanofibers exhibited 575% sensitivity towards 5 ppm NH3, mainly attributed to the content, distribution, specific surface area and pore density of the HAp particles blended with cellulose. The HAp/cellulose nanofibrils formed a dense net-like structure with a HAp Ca/P ratio for all nanocomposites ranging from ∼2.35 and 2.66. NH3, a polar molecule, has a likelihood of interacting with the physisorbed surface H2O molecules. The cellulose-based composites exhibited enhanced conductivity and sensitivity that can be attributed to the H-bonding affinity between NH3 and the –OH groups present on the six-membered polysaccharide.
The sensing mechanism of HAp towards NH3 can be explained in terms of proton exchange and conductivity occurring during the adsorptive interaction between the analyte gas and the adsorbed H2O molecules. HAp behaves as an n-type semiconductor whose resistance decreases upon exposure to ammonia.194 Furthermore, HAp is a well-known hydrophilic biopolymer with the H2O molecules being adsorbed onto the –OH groups present along the c-axis. The adsorbed H2O assists in the formation of a hydrogen bond with the NH3 molecule.199 This interaction results in the possible conversion of adsorbed NH3 to NH4+, expressed as
NH3(g) + H2O(ads) ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) NH4+(ads) + OH−(ads) NH4+(ads) + OH−(ads) | (19) |
| NH3 NH2− + H+ | (20) |
| NH2− + O2− NOxn− + H2O | (21) |
| H2S(g) + H2O(ads) → HS− + H3O+ | (22) |
| O2 + e− → O2˙− | (23) |
| NO + O2˙− → NO2 + O− | (24) |
| NO2 + O− → NO3− | (25) |
As per reports in ref. 163, tubular HAp, synthesized via cation exchange membrane-assisted electrodeposition, was employed in VOC sensing. The tubular HAp showed a larger response towards VOCs than hydrothermally prepared HAp. The maximum % sensitivity at room temperature towards 1000 ppm of acetone, n-hexane and styrene was 60%, 46% and 50% respectively. As already discussed, the tubular HAp prepared via the cation membrane-assisted electrodeposition technique had maximum crystal orientation predominating along the c-plane. However, compared to pure HAp, the presence of additives significantly altered the sensitivity levels of the composite towards VOC detection. S. R. Anjum et al. developed a graphite doped HAp selective alcohol sensor.205 The choice of graphite as a dopant lies in its capability of facile sorption/desorption of incoming gas molecules. The ideal adsorption/desorption behavior can be attributed to the weak dispersive forces towards gas molecules. XRD profiles reveal that the HAp crystals were hexagonal both before and after graphitic doping with a maximum intensity along the (002) plane, indicating predominantly c-plane oriented crystals. The total pore volume increased by a factor of ∼2 upon addition of graphite, from 0.087 cc g−1 (pristine HAp) to 0.201 cc g−1 (after doping). The analyte VOCs detected were 100 ppm methanol, ethanol and propanol. The sensitivity of pure HAp was 100%, 92% and 180% towards methanol, ethanol and propanol respectively, whereas 0.5 wt% graphite doped HAp possessed a sensing efficacy up to 465%, 333% and 352% respectively. It was witnessed that as the carbon chain length in the alcohol moiety increased, the adsorptive interaction and subsequent sensing capabilities of the sensor improved with the increase in thermal energy. As reported in ref. 205, sensing of propanol improves at an operating temperature of 60 °C, while for methanol and ethanol, the optimum temperature for maximum % sensitivity is 30 °C (shown in Table 6). Alcohols being reducing species with electron donating properties tend to increase the number of liberated electrons depending on the carbon chain length.205
The mechanism describing the interaction of HAp towards VOCs has been detailed in ref. 163 and 205. The exposure of HAp crystals to VOCs results in the adsorption and dissociation of the molecule via the loss of H+.
| CxHyOz CxHy−nOn− + nH+ | (26) |
| CxHy−nOn− + O2− CO2 + H2O + e− | (27) |
| CH3OH + 3O2− → CO2 + 2H2O + e− | (28) |
Furthermore, there exists the possibility of interaction of the CO2 gas with the OH− surface species along the c-plane. This may result in the formation of CO32− and its substitution within the lattice groups thus forming carbonated HAp.
| CO2 + 2OH− CO32− + H2O | (29) |
The mechanism involved in the detection of LPG is reported as follows:
| CnH2n+2 + O2− → CnH2nO + H2O + e− | (30) |
A significantly low response–recovery time towards CO2 gas was characteristic for pristine HAp with the predominant crystal orientation being the a,b-plane. The % sensitivity was indicated to be ∼31% due to reduced interaction along the exposed (300) facet.214 In order to mitigate the low levels of sensitivity, metallic dopants such as Fe and Co were added to HAp thereby witnessing a substantial increase in the response of the sensor film.215,216 In the case of Fe-doped HAp, it was determined that the microstructure of the composite varied from granular to plate-like up to an Fe concentration of 0.01 M. The plate-like composite reverted back to granular at 0.05 M Fe-doped HAp. An increase in Fe within the lattice resulted in an enhancement in surface area and microporous volume resulting in a spike in gas uptake capacity towards CO and CO2 gas. Co-Doped HAp thick film composites were explored as a viable sensor by M. P. Mahabole et al. towards detection of CO and CO2. The percentage of variation in the Co dopant ranged between 0.008 M and 0.05 M. This variation in the ion-exchanged composite altered the crystal size and porosity. The maximum response to CO2 was exhibited by the 0.01 M Co-doped HAp composite at 75% at an operating temperature of 135 °C. This is attributed to the reduction in (201) oriented crystal planes as the concentration of Co within the lattice increases.216
An extended study on these doped composites was carried out by modifying the surface of the sensor material through the process of swift heavy ion irradiation (SWI). Here, both the Fe-doped and Co-doped HAp screen printed sensor substrates were subjected to Ag7+ ion irradiation with an ion fluence ranging between 3 × 1011 ions cm−2 and 3 × 1013 ions cm−2, respectively. This process modified the surface by enhancing the pore size and the density of active adsorption sites.217,218 The response of the ceramic was tested with CO and CO2 gas at an optimum operating temperature of 140 °C and 155 °C respectively. The response–recovery time significantly improved with CO and CO2 gas response witnessing a ∼2.5 times increase in comparison to pristine HAp crystals. The orientation of HAp crystals in ref. 217–219 was along a predominantly exposed c-plane. The variation in % sensitivity between pristine HAp, Fe-doped HAp, Co-doped HAp, SWI-Fe-doped HAp and SWI-Co-doped HAp towards detection of CO gas can be seen in Table 6. With each modification, the response–recovery time of the composite sensor witnesses an improvement. The sensitivity for pure HAp is 45% while Fe-doped HAp and Co-doped HAp are enhanced by a factor of 2 and 3 respectively. On the other hand, SWI-HAp indicated a sensitivity of 210% (4.6 times increase from pure HAp). SWI improved the efficacy of Fe- and Co-doped HAp towards detection of CO gas. Similar trends were observed in the sensing of CO2 gas.
A carbon-based sensor material was reportedly synthesized by R. S. Khairnar et al. by mechanically grinding 0.1 wt% CNTs with 99.9 wt% HAp prepared via chemical precipitation.220,221 The addition of CNTs as a dopant over spherical HAp nanoparticles witnessed an improvement in the gas uptake capacity. Furthermore, the inherent electrical conductivity of CNTs facilitated the detection of 20 ppm of CO and CO2 gas at a reduced operating temperature of 105 °C and 70 °C respectively. However, the operating temperature for maximum % sensitivity for pure HAp was 175 °C. While the % sensitivity of pure HAp towards CO and CO2 was 100%, the detection level for CNT doped HAp towards CO and CO2 was 130% and 200% respectively. Although the spherical CNT doped HAp had a crystal preferentially along the (300) plane rather than (002), the sensitivity was higher due to the increased surface area for adsorption of the analyte gas.
The mechanism of adsorption and quantification of CO and CO2 gas can be related either through their interaction with the surface OH− group along the c-plane or through their reaction with O2− species.215 CO, a reducing gas, gets oxidized to CO2 and subsequently into the CO32− anion.222
| 2OH− + CO → CO2 + H2O | (31) |
| O2− + CO → CO2 + 2e− | (32) |
| O2− + CO2 → CO32− | (33) |
6.2. HAp nanocomposites towards detection of electrolytic analytes (biosensors)
Most electrolytic biosensors aid in the determination of dissolved analytes via employing enzymatic and non-enzymatic substrates. The ease of loading and binding of enzymes to HAp biomaterials makes it feasible towards electrochemical sensing of target molecules. The response characteristics of select HAp biosensors are tabulated in Table 7.| S. No. | HAp modified composite | Substrate electrode | Analyte species | Linear range (μM) | Limit of detection (μM) | Sensitivity (μA mM−1 cm−2) | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | HAp/GOx/Nafion | GCE | Glucose | 120–2160 | 20 | 6.75 mA M−1 | 224 |
| 2 | GOx/HAp/TiO2/MWCNT | GCE | Glucose | 10–15200 |
2.0 | 57.0 | 225 |
| 3 | PoPD/GA-GOx/HAp/Au | Au foil | Glucose | 3–1700 | 0.34 | 32.2 | 226 |
| 4 | RGO/HAp/GOx | GCE | Glucose | 100–11500 |
30 | 16.9 | 227 |
| 5 | PANI/HAp | Ni foam | Glucose | 1–30000 |
0.32 | 1704.53 | 228 |
| 6 | Cyt c/FCNFs/HAp | Au | H2O2 | 2–8700 | 0.3 | — | 265 |
| 7 | HAp/chitosan/tyrosinase | Au | Catechol | 0.01–7 | 0.005 | 2110 | 230 |
| Phenol | 0.07–5 | 0.01 | 1770 | ||||
| m-Cresol | 0.01–5 | 0.005 | 2050 | ||||
| 8 | Fe-HAp/tyrosinase | GCE | L-Tyrosine | 0.1–11 | 0.245 | — | 231 |
| 9 | Ce-HAp | GCE | NE | 0.1–200 | 0.058 | — | 232 |
| UA | 0.5–200 | 0.39 | — | ||||
| Tyr | 0.1–200 | 0.072 | — | ||||
| 10 | HAp | m-CPE | UA | 0.068–50 | 0.05 | — | 233 |
| GOx (FAD) + 2e− + 2H+ → GOx (FADH2) (in N2 saturated PBS) | (34) |
| GOx (FADH2) + glucose + O2 → GOx (FAD) + gluconolactone + H2O2 (in O2 saturated PBS) | (35) |
A non-enzymatic glucose biosensor was fabricated, reported in ref. 228, wherein the sensor composite comprised of HAp/PANI deposited onto a Ni foam substrate. The choice of Ni foam over Ni plate lies in its increased specific surface area. The nucleation and crystal orientation of the HAp crystals were along the c-axis resulting in needle-like crystals. The incorporation of PANI facilitated improvement in the conductivity of the biosensor while enhancing the current density required for electrocatalytic oxidation of glucose. The oxidation of glucose over a non-enzymatic HAp-Ni sensor is as follows:
| NiOOH + glucose → Ni(OH)2 + gluconolactone | (36) |
| Catechol + tyrosinase (O2) → o-quinone + H2O | (37) |
7. Discussion
Some of the general advantages in the modification of growth alignment in crystal orientation of hexagonal HAp lie in the fact that (a) the density of acidic and basic sites can be modulated thereby enhancing its applicatory nature, (b) an improvement in proton conductivity with respect to the predominantly exposed crystal facet can be achieved and (c) the physico-chemical variables such as adhesion, corrosion resistance and adsorption dynamics can be significantly enhanced with manipulation of HAp crystal growth. In general, the rate of nucleation, growth rate, crystal alignment and Ca/P ratio can be related to parametric modifications encompassing the electrochemical deposition technique. The parameters discussed in the review are categorized as driving parameters that initiate and control the deposition rate of HAp and assisting parameters that work synergistically with the driving parameters so as to enhance the nucleation rate, refinement in crystal orientation and improved coating characteristics. Furthermore, the preference of either two- or three-electrode electrochemical setup has been highlighted. The advantage of the three-electrode setup over the two-electrode counterpart can be attributed to circumventing the IR drop and maintaining a stable potential throughout electrodeposition despite variations in cathode charge density with progressive HAp growth.The role of applied potential and applied current density, under either static or dynamic conditions, in the microstructural outcome of HAp coating has been elaborated.80,234 The pulsing of potential or current has a significant effect in ensuring control over dissociation of H2O and the liberation of H2 gas at the cathode via employing a duty cycle with a shorter ON pulse duration.235,236 Additionally, the presence of pulses ensures a minimum shift in cathodic pH.237 The diffusion of phosphate species into a porous TiO2 surface can be controlled by tuning the pulse duration thereby resulting in variation in the Ca/P ratio238 and control over HAp crystal orientation.239 Modifications of the Ca/P ratio, crystal size and coating stability were controlled through electrodeposition parameters such as loading time, electrolyte temperature and pH.
In addition to these driving parameters, variations in assisting parameters reported under electrodeposition of HAp were elaborated in this review. The implementation of ultrasonics during electrodeposition witnesses a reduction in coating roughness and pore size, enhancement in dispersion of ionic species, rapid removal of H2 bubbles and removal of weakly adhered coatings. Furthermore, the crystal orientation of ultrasonically grown HAp may result in the formation of either plate- or needle-like microstructures depending on the applied current density.240 The effects of electrolytic additives like KOH and NaOH augment the formation of HAp while addition of NaNO3 witnesses an enhancement in the ionic conductivity of the electrolyte.241 The presence of chelating agents such as EDTA results in crystal orientation along the a,b-axes via the control of supersaturation of the Ca-EDTA chelate complex. The addition of H2O2 to the electrolytic system witnesses an increase in cathodic pH resulting in increased PO43− formation. The added advantage of H2O2 significantly improves the coating properties as evidenced by the decreased formation of H2 bubbles. As an alternative to solvated ionic Ca2+ and PO43− precursors, the possibility of employing dispersed HAp nanoparticles was explored under the simultaneous anodization–electrodeposition technique resulting in nanospherical deposits.242,243 Furthermore, the deposition of HAp nanoslabs/nanoplates onto a Ti plate is reported wherein HAp nanoslabs were synthesized prior to dispersal in a deposition electrolyte.244 The technique of simultaneous precipitation–electrodeposition proved fruitful in the preferential c-axis growth of acicular HAp with improved crystallinity and uniformity.120,245 An external applied magnetic field facilitates the growth of hexagonal rods or flakes depending on the orientation of the magnetic flux (B) with respect to electrical flux (j), either in a parallel or in a perpendicular fashion. The morphology, crystal size and orientation witnessed a variation, either as needles or as flakes, under a N2 or CO2 cell atmosphere.152 Employing high pressure during electrodeposition increased the size of deposited crystals. A reduced pressure facilitated removal of H2 bubbles from the cathode thus circumventing the formation of weak coatings.246 Substrate modification and pretreatment improved the overall coating characteristics.247 The rate of growth and adhesion to the substrate increased when TiO2 NTs were employed.248 The ease of electrochemically coating HAp over complex substrates such as TiO2 nanofibers,249 carbon fiber cloth,240 transparent MEA chip,250 foam,162 mesh241,251 and Nafion substrates163 is highlighted. Employing the cation exchange membrane-assisted electrodeposition technique significantly alters HAp formation as hollow tubular microstructures163,164,196 and the phase evolution of the coating.252 The technique of cyclic electrodeposition involving the alternative immersion of the substrate in Ca and P precursors under galvanostatic conditions resulted in the formation of HAp crystals with a predominantly exposed a,b-facet.166,167
These driving and assisting parameters, either acting alone or in synergy within a multitude of combinations, are shown to contribute to the morphological outcome of electrodeposited HAp in terms of preferential crystal orientation. The importance of oriented HAp crystals, facilitating the optimized sensing of gaseous and electrolytic analytes, has been indicated through the adsorptive interaction of the analyte species with HAp crystal facets.
8. Correlation in electrodeposited HAp architecture with respect to current density and pH
Among the discussed driving parametric determinants, the influence of both current density and pH on HAp microstructure formation is significant under the galvanostatic electrodeposition method. Relatively, other driving parameters such as loading time and electrolytic temperature serve in enhancing the crystal size and growth of HAp. Additionally, the applied potential plays a role in topography within the limits of under −1.4 V and −1.8 V for the two- and three-electrode system respectively. Beyond this range in applied potential, the coating characteristics and microstructure deteriorate due to electrolytic dissociation of H2O and the evolution of H2 gas bubbles on the cathode surface. Additionally, the electrolytic pH is an important parameter in crystal growth due to the consequential solubility of HAp deposits in a highly acidic environment and its formation beyond a particular pH range. It was understood that at acidic pH (<4), DCDP and OCP phases predominate over HAp formation. However, a conversion to the HAp phase is witnessed beyond pH 4 or when the deposits are immersed in an alkaline bath. This preliminary inference reveals that pH plays a role in phase change while applied current density initiates and influences HAp growth and morphology under the galvanostatic mode.Based on this hypothesis, an analysis has been made in this section, correlating the premise of control in both electrolytic pH and current density and its effects over the microstructural outcome and preferential crystal orientation of the HAp deposits. For our analysis, we collected data on pH and current density from reported literature while maintaining constancy of other experimental parameters. It was ensured that the substrate was Ti-based (like metallic Ti, TiO2 NTs and Ti6Al4V alloys) whereas the electrolyte comprised of Ca(NO3)2 and NH4H2PO4 as precursors. By maintaining this parametric constancy, the collected data were plotted (shown in Fig. 22) while maintaining the pH range between 4.0 and 7.5 and the galvanostatic current density between 0 and 20 mA cm−2. From the figure, it is inferred that within this pH range, the formation of needle-like or flake-like HAp can be controlled with modulation of applied current density. As discussed in this section, pH was selected from above 4.0 due to the formation of HAp via phase conversion from DCDP and OCP phases. The formation of needles at pH ∼ 4.5 up to 10 mA cm−2 indicates the control of these parameters resulting in preferential c-axis growth. However, for an increase above 10 mA cm−2 (at pH ∼ 4.5), the microstructure is witnessed to convert to a flake-like a,b-plane orientation. At pH ≥ 7, the probability of formation of a mixture of rod-like and flake-like HAp is higher. Furthermore, this analysis reveals the correlation of applied current density with pH and the subsequent selective morphological variation resulting in the controlled alignment of the electrochemically deposited HAp crystal facet, i.e., preferential growth either along the a,b-plane or along the c-plane. Current densities higher than the plate-like region (indicated in stripped red in Fig. 22) are speculated to result in the formation of spherical HAp nanoparticles, although data regarding higher current densities are limited for the prerequisites put forth in this correlation.
 | ||
| Fig. 22 Correlation of applied current density and pH and the probability range of their influence on the microstructural outcome of needle/flake type HAp crystals. | ||
9. Summary and future perspective
This review encompasses a discussion on the crystal structure of hexagonal HAp, focusing on the two important crystal facets of HAp, namely, the a,b-facet and the basal c-facet. The technique of electrodeposition of HAp and nucleation and crystal growth of HAp are presented. Notably, we highlighted the possibility of controlling the HAp crystal orientation through the variations in each parametric determinant involved under the technique of electrodeposition. Furthermore, by categorization of reported electrochemical parameters under driving and assisting parameters, their influence on the morphology was reviewed. The parameters of applied potential and current density, time, temperature, and pH, the influence of ultrasound assistance and additives, the effect of an external magnetic field and microgravity, electrolytic pressure and controlled atmosphere, and substrate-based electrodeposition of HAp were briefly discussed. Furthermore, this review presents a brief purview on the efficacy of phase pure HAp and its composites as sensor materials towards electrochemical detection of gaseous molecules and electrolytic analytes. The mechanistic interactions of HAp with gases such as NH3, NOx, H2S, CO, CO2 and VOCs and with the electrolytic sensing of glucose, CN− and solvated organics have been discussed. This review highlighted the importance of crystal facet exposure by correlating the predominant crystal orientation to sensitivity. The presence of hexagonal rods or flakes determines the efficacy of the HAp nanocomposite. The correlation of current density and pH indicates the possibility to tune the morphology thereby controlling the density of active sites. The conceptual elucidation discussed in this manuscript can be extrapolated to various substituted HAp composites for furthering the possibility of effective sensors. Additionally, the concept of exposing the favourable crystal facet will prove advantageous while employing HAp in the field of catalysis, ion exchange, and adsorption phenomena. Therefore, extending subsequent research along these lines will encourage future development of environmentally friendly and improved HAp biomaterials.Author contributions
Priya Ranjan Dev – conceptualization, supervision, formal analysis, writing – original draft, writing – review & editing; Chekkottu Parambil Anand – data curation, writing – editing & proofreading; David Samuvel Michael – data curation, writing – editing & proofreading; and P. Wilson – guidance and supervision.List of abbreviations
Abbreviations
| AFM | Atomic force microscopy |
| β-TCP | Beta-tricalcium phosphate |
| CNTs | Carbon nanotubes |
| Cyt c | Cytochrome c |
| DCDP | Dicalciumdiphosphate |
| j | Electrical flux |
| EDS | Energy dispersive spectroscopy |
| FESEM | Field emission scanning electron microscopy |
| FCNFs | Functionalized carbon nanofibers |
| GCE | Glassy carbon electrode |
| F G | Gravitational force |
| HELCDEB | High energy low current electron beam |
| HRP | Horseradish peroxidase |
| LPG | Liquified petroleum gas |
| LEL | Lower explosive limit |
| B | Magnetic flux |
| F m | Magnetization force |
| F B | Magnetohydrodynamic force |
| MHD | Magnetohydrodynamics |
| MEA | Microelectrode array |
| m-CPE | Modified carbon paste electrode |
| MCPM | Monocalcium phosphate monohydrate |
| MWCNT | Multi-walled carbon nanotubes |
| NTs | Nanotubes |
| OSHA | Occupational Safety and Health Administration |
| OCP | Octacalcium phosphate |
| PBS | Phosphate buffer solution |
| RT | Room temperature |
| SCE | Saturated calomel electrode |
| SBF | Simulated body fluid |
| SS | Stainless steel |
| SWI | Swift heavy ion irradiation |
| UV | Ultraviolet |
| VOCs | Volatile organic compounds |
| XRD | X-ray diffraction |
| XPS | X-ray photoelectron spectroscopy |
Chemical name
| NH3 | Ammonia |
| Ar+ | Argon cation |
| Ca2+ | Calcium cation |
| CO2 | Carbon dioxide |
| CO | Carbon monoxide |
| Cl− | Chloride anion |
| Co2+ | Cobalt cation |
| CN− | Cyanide anion |
| H2PO4− | Dihydrogen phosphate anion |
| EtOH | Ethanol |
| EDTA | Ethylenediaminetetraacetic acid |
| FAD | Flavin adenine dinucleotide |
| F− | Fluoride anion |
| GOx | Glucose oxidase |
| GA | Glutaraldehyde |
| Au | Gold |
| GO | Graphene oxide |
| H2S | Hydrogen sulfide |
| H2 | Hydrogen gas |
| H2O2 | Hydrogen peroxide |
| H3O+ | Hydronium ion |
| OH− | Hydroxide ion |
| Fe2+ | Iron cation |
| Pb2+ | Lead cation |
| LiCl | Lithium chloride |
| Mg | Magnesium |
| Mn2+ | Manganese cation |
| HPO42− | Monohydrogen phosphate anion |
| Ni | Nickel |
| NiOOH | Nickel oxide hydroxide |
| NO3− | Nitrate anion |
| NO | Nitric oxide |
| NO2− | Nitrite anion |
| NE | Norepinephrine |
| PO43− | Phosphate anion |
| Pt | Platinum |
| PoPD | Poly(o-phenylenediamine) |
| PANI | Polyaniline |
| PPy | Polypyrrolidine |
| H+ | Proton |
| RGO | Reduced graphene oxide |
| Na+ | Sodium cation |
| SO2 | Sulfur dioxide |
| THF | Tetrahydrofuran |
| TiO2 | Titania |
| Ti | Titanium |
| Tyr | Tyrosine |
| UA | Uric acid |
Units and symbols
| V | Applied potential |
| °C | Degree Celsius |
| cc g−1 | Cubic centimeter per gram |
| e− | Electron |
| h | Hour |
| K | Kelvin |
| L min−1 | Liter per minute |
| MPa | Megapascal |
| m2 g−1 | Square meter per gram |
| μA mM−1 cm−2 | Microampere per millimolar per square centimeter |
| μm | Micrometer |
| μm year−1 | Micrometer per year |
| μM | Micromolar |
| μmol dm−3 | Micromolar per cubic decimeter |
| mA cm−2 | Milliampere per square centimeter |
| mg L−1 | Milligram per liter |
| mm | Millimeter |
| mM | Millimolar |
| mT | Millitesla |
| mV s−1 | Millivolt per second |
| min | Minute |
| mol | Mole |
| mol L−1 | Mole per liter |
| nm | Nanometer |
| nM | Nanomolar |
| ppm | Parts per million |
| s | Second |
| T | Tesla |
| Torr | Torricelli |
| vol% | Volume percentage |
| W | Watt |
| wt% | Weight percentage |
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to acknowledge the Science and Engineering Research Board (SERB), India, under the Department of Science and Technology (DST) (Grant No.: EMR/2015/001406). Priya Ranjan Dev would like to thank Pavithra Dev and M. Lilly Mary for their support and motivation while consolidating the present review.References
- L. Yan, Y. Xiang, J. Yu, Y. Wang and W. Cui, ACS Appl. Mater. Interfaces, 2017, 9, 5023–5030 CrossRef CAS.
- H. R. Bakhsheshi-Rad, E. Hamzah, A. F. Ismail, M. Aziz, E. Karamian and N. Iqbal, Trans. Nonferrous Met. Soc. China, 2018, 28, 1553–1562 CrossRef CAS.
- E. Vidal, J. Buxadera-Palomero, C. Pierre, J. M. Manero, M.-P. Ginebra, S. Cazalbou, C. Combes, E. Rupérez and D. Rodríguez, Surf. Coat. Technol., 2019, 358, 266–275 CrossRef CAS.
- J. Kolmas, S. Krukowski, A. Laskus and M. Jurkitewicz, Ceram. Int., 2016, 42, 2472–2487 CrossRef CAS.
- H. Wang, C. Wang, B. Xiao, L. Zhao, J. Zhang, Y. Zhu and X. Guo, Catal. Today, 2016, 259, 340–346 CrossRef CAS.
- A. A. Gandhi, M. Wojtas, B. L. Sidney, A. L. Kholkin and S. A. M. Tofail, J. Am. Ceram. Soc., 2014, 97, 2867–2872 CrossRef CAS.
- R. Rodriguez, D. Rangel, G. Fonseca, M. Gonzalez and S. Vargas, Results Phys., 2016, 6, 925–932 CrossRef.
- Y. Ding, J. Liu, H. Wang, G. Shen and R. Yu, Biomaterials, 2007, 28, 2147–2154 CrossRef CAS.
- A. Dehghanghadikolaei and B. Fotovvati, Materials, 2019, 12, 1795 CrossRef CAS PubMed.
- P. Xie, Z. Shi, M. Feng, K. Sun, Y. Liu, K. Yan, C. Liu, T. A. A. Moussa, M. Huang, S. Meng, G. Liang, H. Hou, R. Fan and Z. Guo, Adv. Compos. Hybrid Mater., 2022, 5, 679–695 CrossRef.
- P. E. Lokh, e e, V. Kadam, C. Jagatap, U. S. Chavan, R. Udayabhaskar and H. M. Pathan, ES Mater. Manuf., 2022, 17, 53–56 Search PubMed.
- M. Lian, Y. Huang, Y. Liu, D. Jiang, Z. Wu, B. Li, Q. Xu, V. Murugadoss, Q. Jiang, M. Huang and Z. Guo, Adv. Compos. Hybrid Mater., 2022 DOI:10.1007/s42114-022-00475-6.
- D. K. Shah, D. KC, T.-G. Kim, M. S. Akhtar, C. Y. Kim and O.-B. Yang, Eng. Sci., 2022, 18, 243–252 CAS.
- Y. Cheng and G. Zhang, ES Energy Environ., 2022, 17, 1–10 CAS.
- J. Cai, V. Murugadoss, J. Jiang, X. Gao, Z. Lin, M. Huang, J. Guo, S. A. Alsareii, H. Algadi and M. Kathiresan, Adv. Compos. Hybrid Mater., 2022, 5, 641–650 CrossRef CAS.
- J. Guo, Z. Chen, Z. M. El-Bahy, H. Liu, H. M. Abo-Dief, W. Abdul, K. M. Abualnaja, A. K. Alanazi, P. Zhang, M. Huang, G. Hu and J. Zhu, Adv. Compos. Hybrid Mater., 2022, 5, 899–906 CrossRef CAS.
- J. Chen, Y. Zhu, Z. Guo and A. G. Nasibulin, Eng. Sci., 2020, 12, 13–22 CAS.
- B. Dai, Y. Ma, F. Dong, J. Yu, M. Ma, H. K. Thabet, S. M. El-Bahy, M. M. Ibrahim, M. Huang, I. Seok, G. Roymahapatra, N. Naik, B. B. Xu, J. Ding and T. Li, Adv. Compos. Hybrid Mater., 2022, 5, 704–754 CrossRef.
- X. Sun, D. Zhu, M. Liu, Y. Song and J. Wang, Eng. Sci., 2022, 20 DOI:10.30919/es8d715.
- S. E. Chavez, H. Ding, B. L. Williams, S. Nam, Z. Hou, D. Zhang and L. Sun, ES Mater. Manuf., 2022, 15, 72–77 CAS.
- D. Pan, G. Yang, H. M. Abo-Dief, J. Dong, F. Su, C. Liu, Y. Li, B. Bin Xu, V. Murugadoss, N. Naik, S. M. El-Bahy, Z. M. El-Bahy, M. Huang and Z. Guo, Nano-Micro Lett., 2022, 14, 118 CrossRef CAS.
- Y. Ma, X. Xie, W. Yang, Z. Yu, X. Sun, Y. Zhang, X. Yang, H. Kimura, C. Hou, Z. Guo and W. Du, Adv. Compos. Hybrid Mater., 2021, 4, 906–924 CrossRef CAS.
- C. Dang, Q. Mu, X. Xie, X. Sun, X. Yang, Y. Zhang, S. Maganti, M. Huang, Q. Jiang, I. Seok, W. Du and C. Hou, Adv. Compos. Hybrid Mater., 2022, 5, 606–626 CrossRef.
- R. Ma, B. Cui, D. Hu, S. M. El-Bahy, Y. Wang, I. H. E. Azab, A. Y. Elnaggar, H. Gu, G. A. M. Mersal, M. Huang and V. Murugadoss, Adv. Compos. Hybrid Mater., 2022, 5, 1477–1489 CrossRef CAS.
- Z. Wang, X. Li, L. Wang, Y. Li, J. Qin, P. Xie, Y. Qu, K. Sun and R. Fan, Adv. Compos. Hybrid Mater., 2020, 3, 1–7 CrossRef.
- C. Stephen, B. Shivamurthy, M. Mohan, A.-H. I. Mourad, R. Selvam and B. H. S. Thimmappa, Eng. Sci., 2022, 18, 75–97 CAS.
- S. M. Shahabaz, S. Sharma, N. Shetty, S. D. Shetty and G. M. C., Eng. Sci., 2021, 16, 26–46 CAS.
- B. Vinod, S. Suresh and D. Sudhakara, Adv. Compos. Hybrid Mater., 2020, 3, 194–204 CrossRef.
- J. C. D. M. Neto, N. R. do Nascimento, R. H. Bello, L. A. de, V. Verçosa, J. E. Neto, J. C. M. da Costa, F. Rol and O. V. Diaz, Eng. Sci., 2022, 17, 28–44 Search PubMed.
- N. Naik, B. Shivamurthy, B. H. S. Thimmappa, A. Gupta, J. Z. Guo and I. Seok, Eng. Sci., 2022, 20 DOI:10.30919/es8d713.
- R. S. Pillai, M. Frasnelli and V. M. Sglavo, Ceram. Int., 2018, 44, 1328–1333 CrossRef CAS.
- X. Zhang, Y. Wu, Y. Lv, Y. Yu and Z. Dong, Surf. Coat. Technol., 2020, 386, 125483 CrossRef CAS.
- T. V. Khamova, O. V. Frank-Kamenetskaya, O. A. Shilova, V. P. Chelibanov, A. M. Marugin, E. A. Yasenko, M. A. Kuz’mina, A. E. Baranchikov and V. K. Ivanov, Crystallogr. Rep., 2018, 63, 254–260 CrossRef CAS.
- M. Catauro, F. Papale, L. Sapio and S. Naviglio, Mater. Sci. Eng., C, 2016, 65, 188–193 CrossRef CAS PubMed.
- R. Chen and J. Shen, Mater. Lett., 2020, 259, 126881 CrossRef CAS.
- O. Yigit, B. Dikici, T. C. Senocak and N. Ozdemir, Surf. Coat. Technol., 2020, 394, 125858 CrossRef CAS.
- X. Sun, H. Lin, C. Zhang, J. Jin and S. Di, Coatings, 2019, 9, 667 CrossRef CAS.
- R. I. M. Asri, W. S. W. Harun, M. A. Hassan, S. A. C. Ghani and Z. Buyong, J. Mech. Behav. Biomed. Mater., 2016, 57, 95–108 CrossRef CAS PubMed.
- M. Sadat-Shojai, M.-T. Khorasani, E. Dinpanah-Khoshdargi and A. Jamshidi, Acta Biomater., 2013, 9, 7591–7621 CrossRef CAS PubMed.
- Y. Lu, W. Dong, J. Ding, W. Wang and A. Wang, Nanomaterials from Clay Minerals, Elsevier, 2019, pp. 485–536 Search PubMed.
- Y.-J. Zhu and B.-Q. Lu, ACS Biomater. Sci. Eng., 2019, 5, 4951–4961 CrossRef CAS.
- S. Awasthi, S. K. Pandey, E. Arunan and C. Srivastava, J. Mater. Chem. B, 2021, 9, 228–249 RSC.
- M. Prakasam, J. Locs, K. Salma-Ancane, D. Loca, A. Largeteau and L. Berzina-Cimdina, JFB, 2015, 6, 1099–1140 CrossRef CAS PubMed.
- F. Anene, J. Aiza, I. Zainol, A. Hanim and M. T. Suraya, Proc. Inst. Mech. Eng., Part L, 2020, 234, 1450–1460 CAS.
- S. Omar, M. S. Muhamad, L. Te Chuan, T. Hadibarata and Z. C. Teh, Water, Air, Soil Pollut., 2019, 230, 275 CrossRef CAS.
- S. Pai, M. S. Kini and R. Selvaraj, Environ. Sci. Pollut. Res., 2021, 28, 11835–11849 CrossRef CAS.
- W. S. W. Harun, R. I. M. Asri, J. Alias, F. H. Zulkifli, K. Kadirgama, S. A. C. Ghani and J. H. M. Shariffuddin, Ceram. Int., 2018, 44, 1250–1268 CrossRef CAS.
- A. Saxena, M. Pandey and A. K. Dubey, J. Indian Inst. Sci., 2019, 99, 339–359 CrossRef.
- A. Das and D. Pamu, Mater. Sci. Eng., C, 2019, 101, 539–563 CrossRef CAS PubMed.
- F. R. Baxter, C. R. Bowen, I. G. Turner and A. C. E. Dent, Ann. Biomed. Eng., 2010, 38, 2079–2092 CrossRef CAS.
- A. Fihri, C. Len, R. S. Varma and A. Solhy, Coord. Chem. Rev., 2017, 347, 48–76 CrossRef CAS.
- M. Šupová, Ceram. Int., 2015, 41, 9203–9231 CrossRef.
- A. Zielinski and M. Bartmanski, Coatings, 2020, 10, 782 CrossRef CAS.
- T.-T. Li, L. Ling, M.-C. Lin, H.-K. Peng, H.-T. Ren, C.-W. Lou and J.-H. Lin, J. Mater. Sci., 2020, 55, 6352–6374 CrossRef CAS.
- G. Ma and X. Y. Liu, Cryst. Growth Des., 2009, 9, 2991–2994 CrossRef CAS.
- M. Okada and T. Matsumoto, Jpn. Dent. Sci. Rev., 2015, 51, 85–95 CrossRef.
- Z. Zhuang, T. J. Fujimi, M. Nakamura, T. Konishi, H. Yoshimura and M. Aizawa, Acta Biomater., 2013, 9, 6732–6740 CrossRef CAS.
- M. Ibrahim, M. Labaki, J.-M. Giraudon and J.-F. Lamonier, J. Hazard. Mater., 2020, 383, 121139 CrossRef CAS PubMed.
- Z. S. Stojanović, N. Ignjatović, V. Wu, V. Žunič, L. Veselinović, S. Škapin, M. Miljković, V. Uskoković and D. Uskoković, Mater. Sci. Eng., C, 2016, 68, 746–757 CrossRef PubMed.
- N. Mohan, R. Palangadan and H. Varma, Ceram. Int., 2016, 42, 17259–17268 CrossRef CAS.
- S. Khanmohammadi, M. Ojaghi-Ilkhchi and M. Farrokhi-Rad, Ceram. Int., 2021, 47, 1333–1343 CrossRef CAS.
- A. Janković, S. Eraković, M. Vukašinović-Sekulić, V. Mišković-Stanković, S. J. Park and K. Y. Rhee, Prog. Org. Coat., 2015, 83, 1–10 CrossRef.
- S. V. Dorozhkin, JFB, 2010, 1, 22–107 CrossRef PubMed.
- A. Janković, S. Eraković, M. Mitrić, I. Z. Matić, Z. D. Juranić, G. C. P. Tsui, C. Tang, V. Mišković-Stanković, K. Y. Rhee and S. J. Park, J. Alloys Compd., 2015, 624, 148–157 CrossRef.
- M. Farrokhi-Rad, J. Alloys Compd., 2018, 741, 211–222 CrossRef CAS.
- D. Drdlik, M. Slama, H. Hadraba, K. Drdlikova and J. Cihlar, Ceram. Int., 2016, 42, 16529–16534 CrossRef CAS.
- A. Stoch, A. Brożek, G. Kmita, J. Stoch, W. Jastrzębski and A. Rakowska, J. Mol. Struct., 2001, 596, 191–200 CrossRef CAS.
- N. N. C. Isa, Y. Mohd and N. Yury, APCBEE Proc., 2012, 3, 46–52 CrossRef CAS.
- T. Mokabber, Q. Zhou, A. I. Vakis, P. van Rijn and Y. T. Pei, Mater. Sci. Eng., C, 2019, 100, 475–484 CrossRef CAS.
- S. Cometa, M. A. Bonifacio, M. Mattioli-Belmonte, L. Sabbatini and E. De Giglio, Coatings, 2019, 9, 268 CrossRef CAS.
- T. Mokabber, L. Q. Lu, P. van Rijn, A. I. Vakis and Y. T. Pei, Surf. Coat. Technol., 2018, 334, 526–535 CrossRef CAS.
- Y. Han, T. Fu, J. Lu and K. Xu, J. Biomed. Mater. Res., 2001, 54, 96–101 CrossRef CAS PubMed.
- D. J. Blackwood and K. H. W. Seah, Mater. Sci. Eng., C, 2010, 30, 561–565 CrossRef CAS.
- R. Hu, C. Lin, H. Shi and H. Wang, Mater. Chem. Phys., 2009, 115, 718–723 CrossRef CAS.
- S. K. Yen and C. M. Lin, Mater. Chem. Phys., 2003, 77, 70–76 CrossRef CAS.
- A. Vladescu, D. M. Vranceanu, S. Kulesza, A. N. Ivanov, M. Bramowicz, A. S. Fedonnikov, M. Braic, I. A. Norkin, A. Koptyug, M. O. Kurtukova, M. Dinu, I. Pana, M. A. Surmeneva, R. A. Surmenev and C. M. Cotrut, Sci Rep., 2017, 7, 16819 CrossRef PubMed.
- D.-Y. Lin and X.-X. Wang, Surf. Coat. Technol., 2010, 204, 3205–3213 CrossRef CAS.
- D.-Y. Lin and X.-X. Wang, Ceram. Int., 2011, 37, 403–406 CrossRef CAS.
- B.-O. Taranu, A. I. Bucur and I. Sebarchievici, J. Coat. Technol. Res., 2020, 17, 1075–1082 CrossRef CAS.
- E. A. dos Santos, M. S. Moldovan, L. Jacomine, M. Mateescu, J. Werckmann, K. Anselme, P. Mille and H. Pelletier, Mater. Sci. Eng., B, 2010, 169, 138–144 CrossRef CAS.
- M. Heydari Gharahcheshmeh and M. Heydarzadeh Sohi, Mater. Chem. Phys., 2012, 134, 1146–1152 CrossRef CAS.
- P. R. Dev, T. M. David, S. J. Samuel Justin, P. Wilson and A. Palaniappan, J. Nanopart. Res., 2020, 22, 56 CrossRef CAS.
- P. Peng, S. Kumar, N. H. Voelcker, E. Szili, R. S. C. Smart and H. J. Griesser, J. Biomed. Mater. Res., 2006, 76A, 347–355 CrossRef CAS.
- E. Tóth-Kádár, I. Bakonyi, L. Pogány and Á. Cziráki, Surf. Coat. Technol., 1997, 88, 57–65 CrossRef.
- D. Gopi, A. Karthika, S. Nithiya and L. Kavitha, Mater. Chem. Phys., 2014, 144, 75–85 CrossRef CAS.
- F. A. Azem, T. K. Delice, G. Ungan and A. Cakir, Mater. Sci. Eng., C, 2016, 68, 681–686 CrossRef CAS.
- L. Fathyunes, J. Khalil-Allafi and M. Moosavifar, J. Mech. Behav. Biomed. Mater., 2019, 90, 575–586 CrossRef CAS PubMed.
- T. Zhou, L. Yan, C. Xie, P. Li, L. Jiang, J. Fang, C. Zhao, F. Ren, K. Wang, Y. Wang, H. Zhang, T. Guo and X. Lu, Small, 2019, 15, 1805440 CrossRef PubMed.
- R. Chakraborty and P. Saha, Surf. Interfaces, 2018, 12, 160–167 CrossRef CAS.
- Z. Zhao, G. Zhang and H. Li, J. Cent. South Univ. Technol., 2004, 11, 147–151 CrossRef CAS.
- P. T. Nam, T. D. Lam, H. T. Huong, N. T. Phuong, N. T. Thu Trang, T. Hoang, N. T. Thanh Huong, L. B. Thang, C. Drouet, D. Grossin, E. Kergourlay, G. Bertrand, D. Devilliers and D. T. Mai Thanh, J. Nanosci. Nanotechnol., 2015, 15, 9991–10001 CrossRef CAS.
- N. Eliaz and T. M. Sridhar, Cryst. Growth Des., 2008, 8, 3965–3977 CrossRef CAS.
- J. Kunze, L. Müller, J. M. Macak, P. Greil, P. Schmuki and F. A. Müller, Electrochim. Acta, 2008, 53, 6995–7003 CrossRef CAS.
- M. Horynová, M. Remešová, L. Klakurková, K. Dvořák, I. Ročňáková, S. Yan, L. Čelko and G.-L. Song, J. Am. Ceram. Soc., 2019, 102, 123–135 CrossRef.
- H. R. Le, K. Y. Chen and C. A. Wang, J. Sol–Gel Sci. Technol., 2012, 61, 592–599 CrossRef CAS.
- M. C. Kuo and S. K. Yen, Mater. Sci. Eng., C, 2002, 20, 153–160 CrossRef.
- C. M. Cotrut, A. Vladescu, M. Dinu and D. M. Vranceanu, Ceram. Int., 2018, 44, 669–677 CrossRef CAS.
- L. Fathyunes and J. Khalil-Allafi, Ultrason. Sonochem., 2018, 42, 293–302 CrossRef CAS PubMed.
- N. Edwin and P. Wilson, Ultrason. Sonochem., 2019, 50, 188–199 CrossRef.
- T.-T. Li, L. Ling, M.-C. Lin, Q. Jiang, Q. Lin, C.-W. Lou and J.-H. Lin, Mater. Sci., 2019, 105, 13 Search PubMed.
- L. Fathyunes, J. Khalil-Allafi, S. O. R. Sheykholeslami and M. Moosavifar, Mater. Sci. Eng., C, 2018, 87, 10–21 CrossRef CAS PubMed.
- L. Fathyunes and J. Khalil-Allafi, Ceram. Int., 2017, 43, 13885–13894 CrossRef CAS.
- S. Oh, R. Finones, C. Daraio, L. Chen and S. Jin, Biomaterials, 2005, 26, 4938–4943 CrossRef CAS PubMed.
- T. Kokubo, H.-M. Kim, M. Kawashita and T. Nakamura, J. Mater. Sci.: Mater. Med., 2004, 15, 99–107 CrossRef CAS.
- M. Nobial, O. Devos, O. R. Mattos and B. Tribollet, J. Electroanal. Chem., 2007, 600, 87–94 CrossRef CAS.
- Y. Yajing, D. Qiongqiong, H. Yong, S. Han and X. Pang, Appl. Surf. Sci., 2014, 305, 77–85 CrossRef.
- D. Gopi, E. Shinyjoy, M. Sekar, M. Surendiran, L. Kavitha and T. S. Sampath Kumar, Corros. Sci., 2013, 73, 321–330 CrossRef CAS.
- L. Ling, T.-T. Li, M.-C. Lin, Q. Jiang, H.-T. Ren, C.-W. Lou and J.-H. Lin, Mater. Lett., 2020, 261, 126989 CrossRef CAS.
- X. Chen, Z. Zhao, A. Chen and H. Li, Trans. Nonferrous Met. Soc. China, 2007, 17, 617–621 CrossRef.
- D. J. Blackwood and K. H. W. Seah, Mater. Sci. Eng., C, 2009, 29, 1233–1238 CrossRef CAS.
- R. Drevet, H. Benhayoune, L. Wortham, S. Potiron, J. Douglade and D. Laurent-Maquin, Mater. Charact., 2010, 61, 786–795 CrossRef CAS.
- D. Gopi, J. Indira and L. Kavitha, Surf. Coat. Technol., 2012, 206, 2859–2869 CrossRef CAS.
- D. He, J. Du, P. Liu, X. Liu, X. Chen, W. Li, K. Zhang and F. Ma, Surf. Coat. Technol., 2019, 365, 242–247 CrossRef CAS.
- D. He, X. Zhang, P. Liu, X. Liu, X. Chen, F. Ma, W. Li, K. Zhang and H. Zhou, Surf. Coat. Technol., 2021, 406, 126656 CrossRef CAS.
- D.-Y. Lin, X.-X. Wang and Y. Jiang, J. Biomed. Mater. Res., 2011, 96B, 1–8 CrossRef CAS PubMed.
- N. Karimi, M. Kharaziha and K. Raeissi, Mater. Sci. Eng., C, 2019, 98, 140–152 CrossRef CAS PubMed.
- J.-M. Jang, H.-C. Choe and W. A. Brantley, Appl. Surf. Sci., 2019, 483, 76–84 CrossRef CAS.
- S.-J. Park and J.-M. Jang, J. Nanosci. Nanotechnol., 2011, 11, 7167–7171 CrossRef CAS PubMed.
- J.-M. Jang, S.-W. Chung, H.-C. Choe and W. A. Brantley, Surf. Coat. Technol., 2017, 320, 383–390 CrossRef CAS.
- A. I. Bucur, E. Linul and B.-O. Taranu, Appl. Surf. Sci., 2020, 527, 146820 CrossRef CAS.
- M. E. Zilm, L. Chen, V. Sharma, A. McDannald, M. Jain, R. Ramprasad and M. Wei, Phys. Chem. Chem. Phys., 2016, 18, 16457–16465 RSC.
- H. E. L. Madsen, J. Cryst. Growth, 1995, 152, 94–100 CrossRef.
- J. S. Sørensen and H. E. Lundager Madsen, J. Cryst. Growth, 2000, 216, 399–406 CrossRef.
- K. Inoue, K. Sassa, Y. Yokogawa, Y. Sakka, M. Okido and S. Asai, Mater. Trans., 2003, 44, 1133–1137 CrossRef CAS.
- S. Asai, Modell. Simul. Mater. Sci. Eng, 2004, 12, R1–R12 CrossRef CAS.
- Y. Sakka, K. Takahashi, T. S. Suzuki, S. Ito and N. Matsuda, Mater. Sci. Eng., A, 2008, 475, 27–33 CrossRef.
- S. Asai, K. Sassa and M. Tahashi, Sci. Technol. Adv. Mater., 2003, 4(5), 455–460 CrossRef CAS.
- M. Okido, K. Kuroda, M. Ishikawa, R. Ichino and O. Takai, Solid State Ionics, 2002, 151, 47–52 CrossRef CAS.
- T. Z. Fahidy, J. Appl. Electrochem., 1983, 13, 553–563 CrossRef CAS.
- T. Z. Fahidy, Prog. Surf. Sci., 2001, 68, 155–188 CrossRef CAS.
- L. M. A. Monzon and J. M. D. Coey, Electrochem. Commun., 2014, 42, 42–45 CrossRef CAS.
- P. Dunne and J. M. D. Coey, J. Phys. Chem. C, 2019, 123, 24181–24192 CrossRef CAS.
- A. Bund, A. Ispas and G. Mutschke, Sci. Technol. Adv. Mater., 2008, 9, 024208 CrossRef.
- A. Bund and H. H. Kuehnlein, J. Phys. Chem. B, 2005, 109, 19845–19850 CrossRef CAS PubMed.
- A. Tian, X. Xue, C. Liu, J. He and Z. Yang, Mater. Lett., 2010, 64, 1197–1199 CrossRef CAS.
- C. Liu, A. Tian, H. Yang, Q. Xu and X. Xue, Appl. Surf. Sci., 2013, 287, 218–222 CrossRef CAS.
- R. Morimoto, M. Miura, A. Sugiyama, M. Miura, Y. Oshikiri, I. Mogi, S. Takagi, Y. Yamauchi and R. Aogaki, J. Electroanal. Chem., 2019, 847, 113255 CrossRef CAS.
- L. M. A. Monzon and J. M. D. Coey, Electrochem. Commun., 2014, 42, 38–41 CrossRef CAS.
- P. Mohammad Salahi Tohidi, M. S. Safavi, M. Etminanfar and J. Khalil-Allafi, Mater. Chem. Phys., 2020, 254, 123511 CrossRef CAS.
- Q. Xu, Y. Liu, C. Liu, A. Tian, X. Shi, C. Dong, Y. Zhou and H. Zhou, RSC Adv., 2015, 5, 14458–14464 RSC.
- X. Y. Liu, Appl. Phys. Lett., 2001, 79, 3539–3541 CrossRef CAS.
- Y.-J. Ye, Y.-Y. Liu and D.-C. Yin, Appl. Surf. Sci., 2015, 351, 594–599 CrossRef CAS.
- Y. Ya-Jing, Y. Da-Chuan and S. Peng, Appl. Surf. Sci., 2010, 256, 7535–7539 CrossRef.
- P. Ming, W. Lv, Y. Li, J. Shang and J. Wang, Electrochim. Acta, 2014, 120, 6–15 CrossRef CAS.
- J. Du, X. Liu, D. He, P. Liu, F. Ma, Q. Li and N. Feng, Rare Met. Mater. Eng., 2014, 43, 830–835 CrossRef CAS.
- S. Ban and J. Hasegawa, Biomaterials, 2002, 23, 2965–2972 CrossRef CAS PubMed.
- S. Ban, K. Matsuo, N. Mizutani and J. Hasegawa, Dent. Mater. J., 1999, 18, 259–270 CrossRef CAS PubMed.
- A. Yuda, S. Ban and Y. Izumi, Dent. Mater. J., 2005, 24, 588–595 CrossRef CAS PubMed.
- D. He, P. Liu, X. Liu, F. Ma, X. Chen, W. Li, J. Du, P. Wang and J. Zhao, J. Alloys Compd., 2016, 672, 336–343 CrossRef CAS.
- S.-H. Wang, W.-J. Shih, W.-L. Li, M.-H. Hon and M.-C. Wang, J. Eur. Ceram. Soc., 2005, 25, 3287–3292 CrossRef CAS.
- M.-C. Wang, W.-J. Shih, K.-M. Chang, S.-H. Wang, W.-L. Li and H.-H. Huang, J. Non-Cryst. Solids, 2010, 356, 1546–1553 CrossRef CAS.
- X. Lu, Z. Zhao and Y. Leng, J. Cryst. Growth, 2005, 284, 506–516 CrossRef CAS.
- Y. Parcharoen, P. Kajitvichyanukul, S. Sirivisoot and P. Termsuksawad, Appl. Surf. Sci., 2014, 311, 54–61 CrossRef CAS.
- H. Tsuchiya, J. M. Macak, L. Müller, J. Kunze, F. Müller, P. Greil, S. Virtanen and P. Schmuki, J. Biomed. Mater. Res., 2006, 77A, 534–541 CrossRef CAS PubMed.
- D. Gopi, A. Karthika, M. Sekar, L. Kavitha, R. Pramod and J. Dwivedi, Mater. Lett., 2013, 105, 216–219 CrossRef CAS.
- F. S. Utku, E. Seckin, G. Goller, C. Tamerler and M. Urgen, Ceram. Int., 2014, 40, 15479–15487 CrossRef CAS.
- D. Lahiri, S. Ghosh and A. Agarwal, Mater. Sci. Eng., C, 2012, 32, 1727–1758 CrossRef CAS PubMed.
- A. Stoch, A. Brożek, S. Błażewicz, W. Jastrzębski, J. Stoch, A. Adamczyk and I. Rój, J. Mol. Struct., 2003, 651–653, 389–396 CrossRef CAS.
- M. Li, Q. Liu, Z. Jia, X. Xu, Y. Cheng, Y. Zheng, T. Xi and S. Wei, Carbon, 2014, 67, 185–197 CrossRef CAS.
- M. Prodana, M. Duta, D. Ionita, D. Bojin, M. S. Stan, A. Dinischiotu and I. Demetrescu, Ceram. Int., 2015, 41, 6318–6325 CrossRef CAS.
- Z. Leilei, L. Hejun, L. Kezhi, S. Qiang, F. Qiangang, Z. Yulei and L. Shoujie, Ceram. Int., 2015, 41, 4930–4935 CrossRef.
- Q. Zhang, Y. Liu, Y. Zhang, X. Ji, Y. Tan and Q. Liu, Mater. Lett., 2013, 107, 337–339 CrossRef CAS.
- L. Huixia, L. Yong, T. Yanni, L. Lanlan, Z. Qing, L. Kun and T. Hanchun, New J. Chem., 2015, 39, 3865–3874 RSC.
- Y. Zhang, Y. Liu, X. Ji, C. E. Banks and W. Zhang, Chem. Commun., 2011, 47, 4126 RSC.
- S. Beaufils, T. Rouillon, P. Millet, J. Le Bideau, P. Weiss, J.-P. Chopart and A.-L. Daltin, Mater. Sci. Eng., C, 2019, 98, 333–346 CrossRef CAS PubMed.
- J. Hernández-Montelongo, A. Muñoz-Noval, V. Torres-Costa, R. J. Martín-Palma and M. Manso-Silvan, Int. J. Electrochem. Sci., 2012, 7, 12 Search PubMed.
- J. Hernandez-Montelongo, D. Gallach, N. Naveas, V. Torres-Costa, A. Climent-Font, J. P. García-Ruiz and M. Manso-Silvan, Mater. Sci. Eng., C, 2014, 34, 245–251 CrossRef CAS PubMed.
- Z. Luo, J. Che, L. Sun, L. Yang, Y. Zu, H. Wang and Y. Zhao, Eng. Regener., 2021, 2, 257–262 Search PubMed.
- Y. Yu, Q. Wang, C. Wang and L. Shang, Eng. Regener., 2021, 2, 96–104 Search PubMed.
- A. Balakrishnan, J. Medikonda, P. K. Namboothiri, M. Manik and A. Natarajan, Eng. Sci., 2022, 19, 5–19 Search PubMed.
- J. Sun, Q. Mu, H. Kimura, V. Murugadoss, M. He, W. Du and C. Hou, Adv. Compos. Hybrid Mater., 2022, 5, 627–640 CrossRef.
- D. Rohilla, S. Chaudhary and A. Umar, Eng. Sci., 2021, 16, 47–70 CAS.
- T. M. David, K. I. Gnanasekar, P. Wilson, P. Sagayaraj and T. Mathews, ACS Omega, 2020, 5, 11352–11360 CrossRef CAS PubMed.
- T. M. David, P. Sagayaraj, P. Wilson, C. Ramesh, N. Murugesan, E. Prabhu, A. S. R. Murthy and S. Annapoorani, J. Electrochem. Soc., 2016, 163, B15–B18 CrossRef CAS.
- C. Tian, L. Wang, F. Luan and X. Zhuang, Talanta, 2019, 191, 103–108 CrossRef CAS PubMed.
- X. Huo, P. Liu, J. Zhu, X. Liu and H. Ju, Biosens. Bioelectron., 2016, 85, 698–706 CrossRef CAS PubMed.
- L. Cai, D. Xu, H. Chen, L. Wang and Y. Zhao, Eng. Regener., 2021, 2, 109–115 Search PubMed.
- X. Wang, X. Xia, X. Zhang, W. Meng, C. Yuan and M. Guo, Mater. Sci. Eng., C, 2017, 80, 174–179 CrossRef CAS PubMed.
- G. G. Mandayo, F. González, I. Rivas, I. Ayerdi and J. Herrán, Sens. Actuators, B, 2006, 118, 305–310 CrossRef CAS.
- T. M. David, P. Wilson, C. Ramesh, N. Murugesan and P. Sagayaraj, Mater. Res. Express, 2018, 6, 015006 CrossRef.
- T. M. David, P. Wilson, R. Mahesh, C. Ramesh and P. Sagayaraj, Mater. Res. Express, 2019, 6, 095065 CrossRef CAS.
- T. M. David, P. R. Dev, P. Wilson, P. Sagayaraj and T. Mathews, Electrochem. Sci. Adv., 2022, 2, e202100083 Search PubMed.
- U. Rajaji, R. Arumugam, S.-M. Chen, T.-W. Chen, T.-W. Tseng, S. Chinnapaiyan, S.-Y. Lee and W.-H. Chang, Int. J. Electrochem. Sci., 2018, 13, 6643–6654 CrossRef CAS.
- C. Feng, G. Xu, H. Liu, J. Lv, Z. Zheng and Y. Wu, J. Electrochem. Soc., 2014, 161, B1–B8 CrossRef CAS.
- Y. Wu, J. Liu, S. Lin, K. Huang, E. Chen, K. Huang and M. Lei, Eng. Sci., 2022, 18, 105–112 CAS.
- M. Fan, L. Wu, Y. Hu, M. Qu, S. Yang, P. Tang, L. Pan, H. Wang and Y. Bin, Adv. Compos. Hybrid Mater., 2021, 4, 1039–1047 CrossRef CAS.
- Y. He, M. Zhou, M. H. H. Mahmoud, X. Lu, G. He, L. Zhang, M. Huang, A. Y. Elnaggar, Q. Lei, H. Liu, C. Liu and I. H. E. Azab, Adv. Compos. Hybrid Mater., 2022 DOI:10.1007/s42114-022-00525-z.
- K. H. M. and R. Dilli, Eng. Sci., 2022, 19, 186–197 Search PubMed.
- D. Kong, Z. M. El-Bahy, H. Algadi, T. Li, S. M. El-Bahy, M. A. Nassan, J. Li, A. A. Faheim, A. Li, C. Xu, M. Huang, D. Cui and H. Wei, Adv. Compos. Hybrid Mater., 2022 DOI:10.1007/s42114-022-00531-1.
- Y. Wu, E. Chen, X. Weng, Z. He, G. Chang, X. Pan, J. Liu, K. Huang, K. Huang and M. Lei, Eng. Sci., 2022, 18, 113–120 CAS.
- M. Cao, D. D. An Sun, H. Li, D. Lou, F. Yang, P. Du, D. Xu and S. Wang, ES Mater. Manuf., 2022 DOI:10.30919/esmm5f727.
- R. Kumar, A. Umar, R. Kumar, M. S. Chauhan, G. Kumar and S. Chauhan, Eng. Sci., 2021, 16, 288–300 CAS.
- S. Mishra, P. Chaudhary, B. C. Yadav, A. Umar, P. Lohia and D. K. Dwivedi, Eng. Sci., 2021, 15, 138–147 CAS.
- L. Huixia, L. Yong, L. Lanlan, T. Yanni, Z. Qing and L. Kun, Mater. Sci. Eng., C, 2016, 59, 438–444 CrossRef PubMed.
- Q. Zhang, Y. Liu, Y. Zhang, H. Li, Y. Tan, L. Luo, J. Duan, K. Li and C. E. Banks, Analyst, 2015, 140, 5235–5242 RSC.
- L. Luo, Y. Liu, Y. Tan, H. Li, Q. Zhang and K. Li, J. Cent. South Univ, 2016, 23, 18–26 CrossRef CAS.
- V. N. Narwade, S. R. Anjum, V. Kokol and R. S. Khairnar, Cellulose, 2019, 26, 3325–3337 CrossRef CAS.
- V. N. Narwade, K. A. Bogle and V. Kokol, Emergent Mater., 2022, 5(2), 445–454 CrossRef CAS.
- M. Sun, Z. Li, Y. Gu, S. Wu and X. Wang, Mater. Res. Express, 2019, 6, 106401 CrossRef CAS.
- Y. Tan, H. Li, Y. Liu, J. Xie, J. He and J. Pan, RSC Adv., 2016, 6, 76874–76878 RSC.
- A. Mitsionis, T. Vaimakis, C. Trapalis, N. Todorova, D. Bahnemann and R. Dillert, Appl. Catal., B, 2011, 106, 398–404 CrossRef CAS.
- H. Tanaka, T. Watanabe and M. Chikazawa, Faraday Trans., 1997, 93, 4377–4381 RSC.
- A. G. Kontos, A. Katsanaki, T. Maggos, V. Likodimos, A. Ghicov, D. Kim, J. Kunze, C. Vasilakos, P. Schmuki and P. Falaras, Chem. Phys. Lett., 2010, 490, 58–62 CrossRef CAS.
- S. Taha, S. Begum, V. N. Narwade, D. I. Halge, J. W. Dadge, M. P. Mahabole, R. S. Khairnar and K. A. Bogle, Mater. Chem. Phys., 2020, 240, 122228 CrossRef CAS.
- S. R. Anjum, V. N. Narwade, K. A. Bogle and R. S. Khairnar, Nano-Struct. Nano-Objects, 2018, 14, 98–105 CrossRef CAS.
- S. Taha, S. Begum, V. N. Narwade, D. I. Halge, J. W. Dadge, M. P. Mahabole, R. S. Khairnar and K. A. Bogle, Appl. Phys. A, 2021, 127, 514 CrossRef CAS.
- S. R. Anjum and R. S. Khairnar, Sens Imaging, 2016, 17, 18 CrossRef.
- M. Yashima, N. Kubo, K. Omoto, H. Fujimori, K. Fujii and K. Ohoyama, J. Phys. Chem. C, 2014, 118, 5180–5187 CrossRef CAS.
- S. K. Avinashi, A. Hussain, K. Kumar, B. C. Yadav and C. Gautam, Oxford Open Mater. Sci., 2020, 1, itab006 CrossRef.
- A. Noori, F. Ziaie and M. Shafaei, J. Nanomed. Nanotechnol., 2016, 7(4) DOI:10.4172/2157-7439.1000389.
- T. Ishikawa, H. Saito, A. Yasukawa and K. Kandori, J. Chem. Soc., Faraday Trans., 1993, 89, 3821–3825 RSC.
- M. Nagai, T. Saeki and T. Nishino, J. Am. Ceram. Soc., 1990, 73, 1456–1460 CrossRef CAS.
- M. Nagai, T. Nishino and T. Saeki, Sens. Actuators, 1988, 15, 145–151 CrossRef CAS.
- R. U. Mene, M. P. Mahabole, R. C. Aiyer and R. S. Khairnar, Open Appl. Phys. J., 2010, 3, 10–16 CAS.
- R. U. Mene, M. P. Mahabole, K. C. Mohite and R. S. Khairnar, Mater. Res. Bull., 2014, 50, 227–234 CrossRef CAS.
- M. P. Mahabole, R. U. Mene and R. S. Khairnar, AML, 2013, 4, 46–52 CrossRef.
- R. U. Mene, M. P. Mahabole, K. C. Mohite and R. S. Khairnar, J. Alloys Compd., 2014, 584, 487–493 CrossRef CAS.
- R. U. Mene, M. P. Mahabole and R. S. Khairnar, Proceedings IMCS 2012, AMA Service GmbH, Von-Münchhausen-Str. 49, 31515 Wunstorf, Germany, Nürnberg/Nuremberg, Germany, 2012, pp. 1180–1183 Search PubMed.
- R. S. Khairnar, R. U. Mene, S. G. Munde and M. P. Mahabole, AIP Conference Proceedings, Bali, (Indonesia), 2011, pp. 189–192 Search PubMed.
- R. S. Khairnar, S. R. Anjum and M. P. Mahabole, Int. J. Eng. Sci. Innovative Technol., 2014, 3, 9 Search PubMed.
- R. S. Khairnar, S. R. Anjum, V. Kokol and M. P. Mahabole, Int. J. Mod. Commun. Technol. Res., 2014, 2, 7 Search PubMed.
- M. P. Mahabole, R. C. Aiyer, C. V. Ramakrishna, B. Sreedhar and R. S. Khairnar, Bull. Mater. Sci., 2005, 28, 535–545 CrossRef CAS.
- H. Y. Zhao, H. M. Zhou, J. X. Zhang, W. Zheng and Y. F. Zheng, Biosens. Bioelectron., 2009, 25, 463–468 CrossRef CAS PubMed.
- R. Ma, B. Wang, Y. Liu, J. Li, Q. Zhao, G. Wang, W. Jia and H. Wang, Sci. China, Ser. B: Chem., 2009, 52, 2013–2019 CrossRef CAS.
- J. Li, D. Kuang, Y. Feng, F. Zhang and M. Liu, Microchim. Acta, 2012, 176, 73–80 CrossRef CAS.
- Q. Zhang, C. Chen, Q. Xie and P. Liu, Microchim. Acta, 2009, 165, 223–229 CrossRef CAS.
- G. Bharath, R. Madhu, S.-M. Chen, V. Veeramani, A. Balamurugan, D. Mangalaraj, C. Viswanathan and N. Ponpandian, J. Mater. Chem. B, 2015, 3, 1360–1370 RSC.
- L. Jia, X. Wei, L. Lv, X. Zhang, X. Duan, Y. Xu, K. Liu and J. Wang, Electrochim. Acta, 2018, 280, 315–322 CrossRef CAS.
- S. Wang, Y. Lei, Y. Zhang, J. Tang, G. Shen and R. Yu, Anal. Biochem., 2010, 398, 191–197 CrossRef CAS PubMed.
- L. Lu, L. Zhang, X. Zhang, S. Huan, G. Shen and R. Yu, Anal. Chim. Acta, 2010, 665, 146–151 CrossRef CAS PubMed.
- P. Kanchana, N. Lavanya and C. Sekar, Mater. Sci. Eng., C, 2014, 35, 85–91 CrossRef CAS PubMed.
- P. Kanchana, M. Navaneethan and C. Sekar, Mater. Sci. Eng., B, 2017, 226, 132–140 CrossRef CAS.
- E. Iyyappan, S. J. Samuel Justin, P. Wilson and A. Palaniappan, ACS Appl. Nano Mater., 2020, 3, 7761–7773 CrossRef CAS.
- D. T. M. Thanh, P. T. Nam, N. T. Phuong, L. X. Que, N. V. Anh, T. Hoang and T. D. Lam, Mater. Sci. Eng., C, 2013, 33, 2037–2045 CrossRef CAS.
- R. Chakraborty, S. Sengupta, P. Saha, K. Das and S. Das, Mater. Sci. Eng., C, 2016, 69, 875–883 CrossRef CAS PubMed.
- M. R. Etminanfar, J. Khalil-Allafi and A. B. Parsa, J. Alloys Compd., 2016, 678, 549–555 CrossRef CAS.
- M. Furko, Z. May, V. Havasi, Z. Kónya, A. Grünewald, R. Detsch, A. R. Boccaccini and C. Balázsi, J. Solid State Electrochem., 2018, 22, 555–566 CrossRef CAS.
- F. S. Utku, E. Seckin, G. Goller, C. Tamerler and M. Urgen, Appl. Surf. Sci., 2015, 350, 62–68 CrossRef CAS.
- N. Aboudzadeh, C. Dehghanian and M. A. Shokrgozar, Surf. Coat. Technol., 2019, 375, 341–351 CrossRef CAS.
- F. Olivier, Q. Picard, S. Delpeux-Ouldriane, J. Chancolon, F. Warmont, V. Sarou-Kanian, F. Fayon and S. Bonnamy, Surf. Coat. Technol., 2020, 389, 125507 CrossRef CAS.
- J. Xue, A. Farris, Y. Wang, W. Yeh, C. Romany, J. K. Guest, W. L. Grayson, A. S. Hall and T. P. Weihs, Coatings, 2020, 10, 715 CrossRef CAS.
- J.-M. Jang, T.-E. Park, S.-D. Kim and H.-C. Choe, J. Nanosci. Nanotechnol., 2018, 18, 2058–2062 CrossRef CAS PubMed.
- J.-M. Jang and H.-C. Choe, Appl. Surf. Sci., 2019, 477, 271–279 CrossRef CAS.
- M. Sharma, R. Nagar, V. K. Meena and S. Singh, RSC Adv., 2019, 9, 11170–11178 RSC.
- B.-O. Taranu, P. Ianasi, S. F. Rus and A. I. Bucur, Coatings, 2022, 12, 288 CrossRef CAS.
- H.-T. Chen, M.-C. Wang, K.-M. Chang, S.-H. Wang, W.-J. Shih and W.-L. Li, Metall. Mater. Trans. A, 2014, 45, 2260–2269 CrossRef CAS.
- V. Huynh, N. K. Ngo and T. D. Golden, Int. J. Biomater., 2019, 1–21 CrossRef.
- Y. Wang, J. Tao, L. Wang, P. He and T. Wang, Trans. Nonferrous Met. Soc. China, 2008, 18, 631–635 CrossRef CAS.
- L. Chetibi, D. Hamana and S. Achour, Mater. Chem. Phys., 2014, 144, 301–309 CrossRef CAS.
- F. Zhang, L.-X. Lin, G.-W. Wang, R. Hu, C.-J. Lin and Y. Chen, Electrochim. Acta, 2012, 85, 152–161 CrossRef CAS.
- T.-T. Li, L. Ling, M.-C. Lin, Q. Jiang, Q. Lin, J.-H. Lin and C.-W. Lou, Nanomaterials, 2019, 9, 679 CrossRef CAS PubMed.
- S. Sun, Q. Chen and Q. Song, CrystEngComm, 2020, 22, 8243–8250 RSC.
- H. Wang, N. Eliaz and L. W. Hobbs, Mater. Lett., 2011, 65, 2455–2457 CrossRef CAS.
- N. N. C. Isa, Y. Mohd and N. Yury, 2012 IEEE Colloquium on Humanities, Science and Engineering (CHUSER), IEEE, Kota Kinabalu, Sabah, Malaysia, 2012, pp. 771–775 Search PubMed.
- H. San, J. Hu, Y. Zhang, J. Han and S. Tang, J. Appl. Electrochem., 2019, 49, 485–501 CrossRef CAS.
- D. Qiu, L. Yang, Y. Yin and A. Wang, Surf. Coat. Technol., 2011, 205, 3280–3284 CrossRef CAS.
- Y. Huang, S. Han, X. Pang, Q. Ding and Y. Yan, Appl. Surf. Sci., 2013, 271, 299–302 CrossRef CAS.
- M. Uddin, C. Hall and V. Santos, Surf. Coat. Technol., 2020, 385, 125363 CrossRef CAS.
- X. Zhang and M. Z. Yates, Surf. Coat. Technol., 2018, 351, 60–67 CrossRef CAS.
- S. Ban and S. Maruno, J. Biomed. Mater. Res., 1998, 42, 387–395 CrossRef CAS.
- D. He, P. Wang, P. Liu, X. Liu, F. Ma, W. Li, X. Chen, J. Zhao and H. Ye, J. Wuhan Univ. Technol., Mater. Sci. Ed., 2016, 31, 461–467 CrossRef CAS.
- W.-J. Shih, M.-C. Wang, K.-M. Chang, C.-L. Wang, S.-H. Wang, W.-L. Li and H.-H. Huang, Metall. Mater. Trans. A, 2010, 41, 3509–3516 CrossRef CAS.
- S. R. Anjum and R. S. Khairnar, SENSORDEVICES 2016: The Seventh International Conference on Sensor Device Technologies and Applications, 2016, pp. 48–52 Search PubMed.
- R. U. Mene, M. P. Mahabole, R. Sharma and R. S. Khairnar, Vacuum, 2011, 86, 66–71 CrossRef CAS.
- K. Cui, Y. Song, Q. Guo, F. Xu, Y. Zhang, Y. Shi, L. Wang, H. Hou and Z. Li, Sens. Actuators, B, 2011, 160, 435–440 CrossRef CAS.
- M. Kumar, H. Dasarathy and C. Riley, J. Biomed. Mater. Res., 1999, 45, 9 CrossRef.
Footnote |
| † Equal contributing authors. |
| This journal is © The Royal Society of Chemistry 2022 |