
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIodine-induced electrical conductivity of novel columnar lanthanide metal–organic frameworks based on a butterfly-shaped π-extended tetrathiafulvalene ligand†
Monica A.
Gordillo
,
Paola A.
Benavides
,
Colin
McMillen
 and
Sourav
Saha
*
and
Sourav
Saha
*
Department of Chemistry, Clemson University, 211 S. Palmetto Blvd., Clemson, SC 29634, USA. E-mail: souravs@clemson.edu
First published on 7th July 2022
Abstract
Novel columnar lanthanide metal–organic frameworks (Ln-MOFs) based on a butterfly-shaped electron-rich π-extended tetrathia-fulvalene ligand (ExTTFTB) were synthesized and their electronic properties were investigated. Upon iodine-induced ligand oxidation, the Tb-MOF displayed ca. 100-fold higher electrical conductivity (5 × 10−7 S m−1) than the neutral pristine MOF.
Metal–organic frameworks (MOFs)1 have emerged as one of the most attractive functional materials due to their diverse structures, properties, and functions,2–11 which can be easily fine-tuned by introducing different organic ligands and metal ions. The introduction of redox-active ligands12,13 and/or metal ions14–18 endows MOFs with mobile charge-carriers and helps create charge transport pathways, rendering them electrically conducting7,8 and expanding their utility in various energy production, transport, and storage systems, such as batteries, transistors, capacitors, photovoltaics and fuel cells, and so on.7,8,19–21 Among various redox-active building blocks, electron-rich tetrathiafulvalene (TTF) ligands have been used extensively to construct electrically conducting MOFs22–25 because (i) the TTF cores can be easily functionalized with different coordinating groups, such as carboxylic acids and pyridines, to enable MOF formation, (ii) they can be oxidized easily and reversibly to stable TTF˙+ radical cations and TTF2+ dications to enhance the charge-carrier concentration of the corresponding MOFs,26–45 and (iii) TTF ligands can form extended π-stacks that can facilitate long-range through-space charge transport within the MOFs.38,39
Aiming to construct new MOF architectures that would not only possess adequate charge-carrier concentration but also efficient charge transport pathways, recently, we introduced40 a butterfly-shaped electron-rich π-extended tetrathiafulvalene ligand containing four benzoate groups (ExTTFTB) that can coordinate metal ions. The ExTTFTB ligand has a convex π-surface consisting of a boat-shaped anthracene core attached to two 1,3-dithiolene rings at the 9- and 10-positions, which create a dihedral angle of ∼81°. This unique shape of the ExTTFTB ligand led to the formation of a novel double-helical Zn-EXTTFTB MOF in which, the convex ExTTF cores formed extended π–π-interactions along the seams of the neighboring strands. Upon iodine-mediated partial oxidation of about half of the ExTTFTB ligands to ExTTFTB˙+ radical cations in each double-helical strand, extended ExTTFTB/ExTTFTB˙+ π-donor/acceptor chains were formed along the seams of adjacent strands, which facilitated long-range charge movement and engendered 104-fold higher electrical conductivity (∼10−4 S m−1) than that of the pristine MOF. However, in the 2D double-helical Zn-EXTTFTB MOF, not all carboxylate arms of the ExTTFTB ligand were coordinated extensively with the Zn2+ ions, which prevented the formation a 3D architecture and led us to envision that if all carboxylate groups of the ExTTFTB coordinated extensively with the metal cluster nodes, then they could potentially form 3D MOFs. Based on this design principle, we have employed Ln3+ ions (Eu3+, Tb3+, and Er3+), postulating that the larger ionic radii and higher coordination number of Ln3+ ions should allow more extensive coordination of multiple ExTTFTB ligands at the same time, which should lead to the formation of a novel 3D columnar MOF architecture defined by the curvature of convex ExTTFTB ligands. Herein, we report the synthesis, structural characterization, and electronic and optical properties of a new 3D columnar Ln-ExTTFTB MOF architecture, which displayed almost two orders of magnitude higher electrical conductivity (10−7 S m−1) upon iodine-mediated oxidation of the ExTTFTB ligands to ExTTFTB˙+ radical cations possibly due to increased charge-carrier concentration.
The solvothermal reaction between ExTTFTB and Ln(NO3)3·5H2O (Ln3+ = Eu3+, Tb3+, and Er3+) in a mixture of dimethylformamide (DMF), H2O, and trifluoroacetic acid (TFA) at 65 °C yielded reddish orange crystals of the corresponding Ln-ExTTFTB MOFs (Fig. 1A, see ESI† for details). Single crystal X-ray diffraction (SXRD) analysis revealed that all three Ln-MOFs were isostructural and possessed a 3D columnar architecture (Fig. 1B) with a triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The asymmetric unit of representative Tb-MOF featured one Tb3+ ion, one ExTTFTB ligand, one Tb-coordinated H2O, and one free DMF molecule. In contrast to our previously reported 2D double-helical Zn-ExTTFTB MOF40 that contained two distinct Zn-nodes because only three carboxylate groups of each ExTTFTB were involved in Zn-coordination, in 3D Ln-ExTTFTB MOFs, all Ln3+ ions have the same coordination environment, and all four carboxylate groups of each ligand are coordinated to Ln3+ ions. For example, in Tb-MOF, the four carboxylate groups of each ExTTFTB ligand are coordinated to six Tb3+ ions: two trans-carboxylate groups coordinated four Tb3+ ions in a bridging bidentate mode with each binding two Tb3+ ions, the third carboxylate group chelated a Tb3+ ion in a bidentate mode, and the fourth one coordinated a Tb3+ ion in a monodentate fashion and remained in the acid form. The octa-coordinated Tb3+ ions of this MOF are coordinated by six carboxylate groups—four in a bridging bidentate mode, one in a chelating bidentate mode, and another in a monodentate mode—and one water molecule (Fig. S1, ESI†). The Ln–O bond lengths—2.352(4)–2.500(5) Å for Eu-MOF, 2.296(7)–2.507(4) Å for Tb-MOF, and 2.236(8)–2.462(4) Å for Er-MOF—roughly followed the ionic radius trend.30,43 The face-to-face oriented convex ExTTFTB ligands connected by Ln-cluster nodes formed large ovoid channels (ca. 18 × 12 Å in diameter) extended along the crystallographic a-axis and the Ln-nodes connected the parallel columns along the b-axis forming the 3D columnar arrays, which were packed along the c-axis (Fig. 1B). The Ln-coordination further augmented the curvature of the convex ExTTFTB ligands, which formed the walls of these columns. In Ln-MOFs, the dihedral angles between the two 1,3-dithiolene rings of the ExTTFTB ligand were 65–67° (vs. 81° in the free ligand). The columnar arrays of Ln-MOFs are packed in a similar fashion as double-helical strands of Zn-ExTTFTB MOF,40i.e., the convex ExTTFTB ligands of a given array of coordinatively linked columns protruded into the grooves of adjacent arrays (Fig. 1B). However, unlike the double-helical Zn-ExTTFTB MOF, in the Ln-EXTTFTB MOFs, the 1,3-dithiolene rings of the ExTTFTB ligands of neighboring columnar arrays were not aligned co-facially, which prevented meaningful intermolecular π–π stacking interactions (Fig. S2, ESI†) and efficient through-space charge movement (vide infra). The C
space group. The asymmetric unit of representative Tb-MOF featured one Tb3+ ion, one ExTTFTB ligand, one Tb-coordinated H2O, and one free DMF molecule. In contrast to our previously reported 2D double-helical Zn-ExTTFTB MOF40 that contained two distinct Zn-nodes because only three carboxylate groups of each ExTTFTB were involved in Zn-coordination, in 3D Ln-ExTTFTB MOFs, all Ln3+ ions have the same coordination environment, and all four carboxylate groups of each ligand are coordinated to Ln3+ ions. For example, in Tb-MOF, the four carboxylate groups of each ExTTFTB ligand are coordinated to six Tb3+ ions: two trans-carboxylate groups coordinated four Tb3+ ions in a bridging bidentate mode with each binding two Tb3+ ions, the third carboxylate group chelated a Tb3+ ion in a bidentate mode, and the fourth one coordinated a Tb3+ ion in a monodentate fashion and remained in the acid form. The octa-coordinated Tb3+ ions of this MOF are coordinated by six carboxylate groups—four in a bridging bidentate mode, one in a chelating bidentate mode, and another in a monodentate mode—and one water molecule (Fig. S1, ESI†). The Ln–O bond lengths—2.352(4)–2.500(5) Å for Eu-MOF, 2.296(7)–2.507(4) Å for Tb-MOF, and 2.236(8)–2.462(4) Å for Er-MOF—roughly followed the ionic radius trend.30,43 The face-to-face oriented convex ExTTFTB ligands connected by Ln-cluster nodes formed large ovoid channels (ca. 18 × 12 Å in diameter) extended along the crystallographic a-axis and the Ln-nodes connected the parallel columns along the b-axis forming the 3D columnar arrays, which were packed along the c-axis (Fig. 1B). The Ln-coordination further augmented the curvature of the convex ExTTFTB ligands, which formed the walls of these columns. In Ln-MOFs, the dihedral angles between the two 1,3-dithiolene rings of the ExTTFTB ligand were 65–67° (vs. 81° in the free ligand). The columnar arrays of Ln-MOFs are packed in a similar fashion as double-helical strands of Zn-ExTTFTB MOF,40i.e., the convex ExTTFTB ligands of a given array of coordinatively linked columns protruded into the grooves of adjacent arrays (Fig. 1B). However, unlike the double-helical Zn-ExTTFTB MOF, in the Ln-EXTTFTB MOFs, the 1,3-dithiolene rings of the ExTTFTB ligands of neighboring columnar arrays were not aligned co-facially, which prevented meaningful intermolecular π–π stacking interactions (Fig. S2, ESI†) and efficient through-space charge movement (vide infra). The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond lengths between the central anthracene and 1,3-dithiolene rings are ca. 1.34–1.35 Å, which is consistent with a double-bond character, indicating that the ExTTFTB ligands in Ln-MOFs are mostly neutral.40,41 Since all three Ln-ExTTFTB MOFs are isostructural, herein, we used the representative Tb-ExTTFTB MOF for further studies of electronic and optical properties, which, in principle, can be extrapolated to all three MOFs.
C bond lengths between the central anthracene and 1,3-dithiolene rings are ca. 1.34–1.35 Å, which is consistent with a double-bond character, indicating that the ExTTFTB ligands in Ln-MOFs are mostly neutral.40,41 Since all three Ln-ExTTFTB MOFs are isostructural, herein, we used the representative Tb-ExTTFTB MOF for further studies of electronic and optical properties, which, in principle, can be extrapolated to all three MOFs.
 | ||
| Fig. 1 (A) Synthetic scheme and (B) single crystal X-ray (SXRD) structure of Ex-TTFTB-based Ln-MOFs (C, gray; O, red; S, yellow; and Ln, cyan; H-atoms and solvent molecules are omitted for clarity). | ||
The experimental and simulated powder X-ray diffraction (PXRD) patterns of pristine Tb-MOF are in good agreement (Fig. S3, ESI†). The higher intensity of some peaks in the experimental PXRD profile and some discrepancies between experimental and simulated patterns could be attributed to the preferred orientation of MOF crystallites (extensive grinding of MOF powders to disrupt such orientation diminished crystallinity).
To oxidize the electron-rich ExTTFTB ligands of the Tb-MOF, we exposed it to I2 vapor.40 After I2-treatment, the orange Tb-MOF turned into a brown powder, which was washed thoroughly to remove any physisorbed I2. The PXRD pattern of the I2-treated Tb-MOF was in good agreement with that of the pristine material (Fig. S3, ESI†), suggesting that it remained mostly crystalline after I2-induced partial oxidation of the ExTTFTB ligands to ExTTFTB˙+ radical cations (vide infra) although some signals became slightly broader and weaker, which is not uncommon in other I2-treated TTF-based MOFs.40 Thermogravimetric analysis revealed that both pristine and I2-treated Tb-MOFs (Fig. S4, ESI†) lost ca. 9% weight before 150 °C, which corresponded to the loss of residual solvent, followed by a plateau until ca. 320 °C, suggesting that no physisorbed I2 was left in the latter.
The I2-mediated oxidation of the Tb-ExTTFTB MOF was confirmed by quantitative electron paramagnetic resonance (EPR) spectroscopy (Fig. 2).38,39 Whereas the pristine Tb-MOF displayed a small EPR signal indicating the presence of a small fraction of aerobically oxidized paramagnetic ExTTFTB˙+ radical cations (1.7 × 1019 spins mol−1), the I2-treated Tb-MOF showed a significantly stronger EPR signal, revealing that I2 produced a much greater population of ExTTFTB˙+ radical cations (7.7 × 1020 spins mol−1). Thus, the latter contained a significantly higher charge carrier concentration, which led to enhanced framework conductivity.
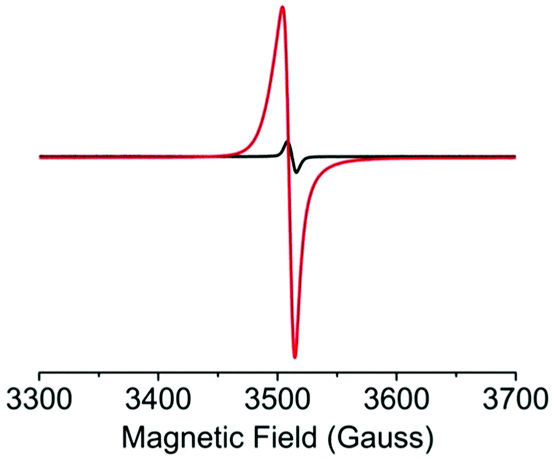 | ||
| Fig. 2 Solid-state EPR spectra of the pristine (black) and iodine-treated (red) Tb-MOF show I2-induced oxidation of ExTTFTB ligands to paramagnetic ExTTFTB˙+ radical cations. | ||
The diffuse reflectance spectrum (DRS) of the pristine Tb-MOF displayed (Fig. 3A) an absorption band with an onset at 618 nm, which corresponded to an optical band gap of ca. 2 eV. In contrast, the I2-treated Tb-MOF displayed a broader absorption band through ca. 760 nm, which corresponded to a narrower optical band gap of 1.6 eV.40 These optical band gaps of the pristine and I2-treated Tb-MOFs are in good agreement with those calculated from their respective Tauc plots (2.1 and 1.7 eV) (Fig. 3B). The narrower band gap of the latter is attributed to I2-mediated oxidation of the ExTTFTB to an ExTTFTB˙+ radical cation, which absorbs at a longer wavelength.
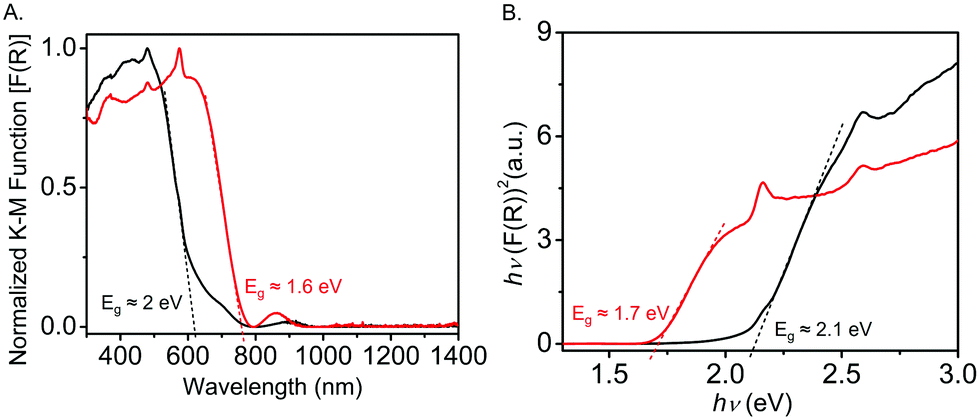 | ||
| Fig. 3 (A) Diffusion reflectance spectra and (B) Tauc plots of the pristine (black) and iodine-treated (red) Tb-MOF. | ||
Finally, to determine the effects of the I2-induced oxidation of the ExTTFTB ligands of Tb-MOF on its electrical conductivity, we measured the current–voltage (I–V) relationships (Fig. 4) of pressed pellets of the pristine and I2-treated Tb-MOF by the two-probe method using pellets placed between two stainless steel electrodes surrounded by a snug-fit Teflon tube.38–40 Both materials displayed linear I–V plots, albeit with different slopes, indicating ohmic conduction. The electrical conductivity of the I2-treated Tb-MOF (5 × 10−7 S m−1) was two orders of magnitude higher than that of the pristine material (7 × 10−9 S m−1), which can be attributed to its enhanced charge carrier concentration provided by a large ExTTFTB˙+ population. However, the conductivity of the I2-treated columnar Tb-MOF was lower than that of the previously reported I2-treated double-helical Zn-ExTTFTB MOF (∼10−4 S m−1).40 This difference can be attributed to the lack of π-donor/acceptor interactions between the spatially separated ExTTFTB ligands of the I2-treated columnar Tb-MOF, which hindered through-space charge movement, whereas the extended ExTTFTB/ExTTFTB˙+ π-donor/acceptor chains located along the seams of adjacent double helical strands of the I2-treated Zn-ExTTFTB MOF facilitated long-range charge movement. As often found in other electrically conducting Ln-MOFs,28,29,43–45 the ionic Tb–O bonds cannot support through-bond charge movement, leaving less efficient redox-hopping as the predominant charge transport mechanism in the Tb-ExTTFTB MOF. Due to grain-boundary and contact resistances, bulk conductivities measured with MOF pellets consisting of microcrystallites are usually 2–3 orders of magnitude smaller than the corresponding single crystal conductivity7,8,46 (in rare events suitable crystals are available for such measurements).
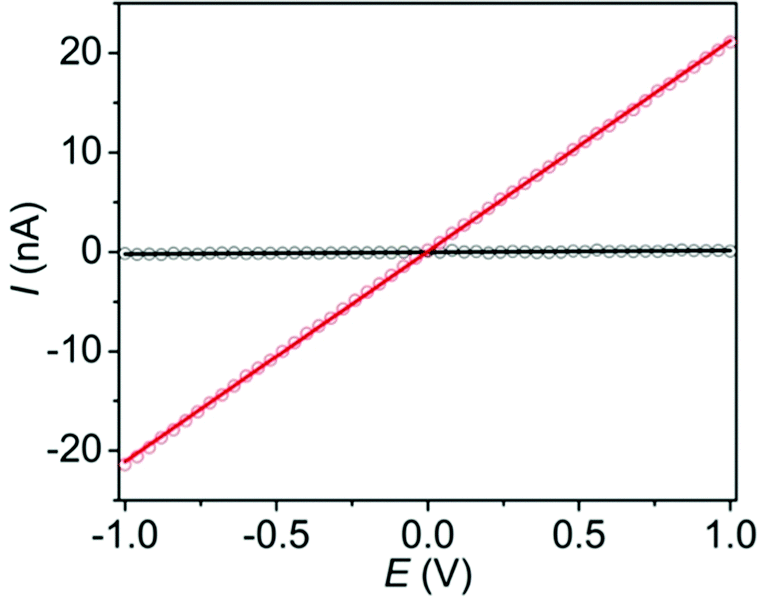 | ||
| Fig. 4 The I–V plots of pristine (black) and iodine-treated (red) Tb-MOF pellets show higher conductivity of the latter. | ||
In summary, we have synthesized new isostructural 3D columnar Ln-MOFs based on a butterfly-shaped electron-rich ExTTFTB ligand. While ExTTFTB formed a 2D double helical MOF with Zn2+, the introduction of larger Ln3+ ions with higher coordination number led to the formation of 3D columnar MOFs containing coordinatively linked parallel columns made of face-to-face oriented convex ExTTFTB ligands. I2-mediated oxidation of the Tb-MOF enhanced its electrical conductivity from ca. 10−9 to 10−7 S m−1 due to the formation of EXTTFTB˙+ radical cations, which increased the charge-carrier concentration, although the MOF lacked well-defined through-bond or through-space charge-transport pathways. Thus, these studies yielded novel 3D columnar Ln-MOFs and demonstrated that while improved charge-carrier concentration can modestly enhance the electrical conductivity of redox-active MOFs, efficient charge movement is vital for attaining very high electrical conductivity.
We acknowledge the US National Science Foundation (award no. NSF-1809092) for financial support for this work.
Conflicts of interest
There are no conflicts to declare.References
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444-1–12 CrossRef PubMed.
- J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- V. Stavila, A. A. Talin and M. D. Allendorf, Chem. Soc. Rev., 2014, 43, 5994–6010 RSC.
- L. Sun, M. G. Campbell and M. Dincă, Angew. Chem., Int. Ed., 2016, 55, 3566–3579 CrossRef CAS PubMed.
- L. S. Xie, G. Skorupskii and M. Dincǎ, Chem. Rev., 2020, 120, 8536–8580 CrossRef CAS PubMed.
- H. Wang, Q.-L. Zhu, R. Zou and Q. Xu, Chemistry, 2017, 2, 52–80 CrossRef CAS.
- M. A. Gordillo, D. K. Panda and S. Saha, ACS Appl. Mater. Interfaces, 2019, 11, 3196–3206 CrossRef CAS.
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC.
- E. A. Dolgopolova and N. B. Shustova, MRS Bull., 2016, 41, 890–896 CrossRef CAS.
- B. Ding, M. B. Solomon, C. F. Leong and D. M. D’Alessandro, Coord. Chem. Rev., 2021, 439, 213891 CrossRef CAS.
- D. M. D’Alessandro, Chem. Commun., 2016, 52, 8957–8971 RSC.
- D. Sheberla, L. Sun, M. A. Blood-Forsythe, S. Er, C. R. Wade, C. K. Brozek, A. Aspuru-Guzik and M. Dincă, J. Am. Chem. Soc., 2014, 136, 8859–8862 CrossRef CAS.
- J.-H. Dou, L. Sun, Y. Ge, W. Li, C. H. Hendon, J. Li, S. Gul, J. Yano, E. A. Stach and M. Dincă, J. Am. Chem. Soc., 2017, 139, 13608–13611 CrossRef CAS PubMed.
- J. Clough, N. M. Orchanian, J. M. Skelton, A. J. Neer, S. A. Howard, C. A. Downes, L. F. J. Piper, A. Walsh, B. C. Melot and S. C. Marinescu, J. Am. Chem. Soc., 2019, 141, 16323–16330 CrossRef PubMed.
- G. Chen, L. B. Gee, W. Xu, Y. Zhu, J. S. Lezama-Pacheco, Z. Huang, Z. Li, J. T. Babicz, S. Choudhury, T. H. Chang, E. Reed, E. I. Solomon and Z. Bao, J. Am. Chem. Soc., 2020, 142, 21243–21248 CrossRef CAS PubMed.
- L. S. Xie, L. Sun, R. Wan, S. S. Park, J. A. Degayner, C. H. Hendon and M. Dincǎ, J. Am. Chem. Soc., 2018, 140, 7411–7414 CrossRef CAS PubMed.
- W.-H. Li, K. Ding, H.-R. Tian, M.-S. Yao, B. Nath, W.-H. Deng, Y. Wang and G. Xu, Adv. Funct. Mater., 2017, 27, 1702067 CrossRef.
- D. Sheberla, J. C. Bachman, J. S. Elias, C. J. Sun, Y. Shao-Horn and M. Dincǎ, Nat. Mater., 2016, 16, 220–224 CrossRef PubMed.
- L. Wang, Y. Han, X. Feng, J. Zhou, P. Qi and B. Wang, Coord. Chem. Rev., 2016, 307, 361–381 CrossRef CAS.
- J. L. Segura and N. Martin, Angew. Chem., Int. Ed., 2001, 40, 1372–1409 CrossRef CAS.
- D. Canevet, M. Sallé, G. Zhang, D. Zhang and D. Zhu, Chem. Commun., 2009, 2245–2269 RSC.
- J. Liu, W. Zhou, J. Liu, I. Howard, G. Kilibarda, S. Schlabach, D. Coupry, M. Addicoat, S. Yoneda, Y. Tsutsui, T. Sakurai, S. Seki, Z. Wang, P. Lindemann, E. Redel, T. Heine and C. Wöll, Angew. Chem., Int. Ed., 2015, 54, 7441–7445 CrossRef CAS PubMed.
- H. Y. Wang, L. Cui, J. Z. Xie, C. F. Leong, D. M. D’Alessandro and J. L. Zuo, Coord. Chem. Rev., 2017, 345, 342–361 CrossRef CAS.
- T. C. Narayan, T. Miyakai, S. Seki and M. Dincă, J. Am. Chem. Soc., 2012, 134, 12932–12935 CrossRef CAS PubMed.
- H.-Y. Wang, J.-Y. Ge, C. Hua, C.-Q. Jiao, Y. Wu, C. F. Leong, D. M. D’Alessandro, T. Liu and J.-L. Zuo, Angew. Chem., Int. Ed., 2017, 56, 5465–5470 CrossRef CAS.
- L. S. Xie, E. V. Alexandrov, G. Skorupskii, D. M. Proserpio and M. Dincǎ, Chem. Sci., 2019, 10, 8558–8565 RSC.
- J. Su, S. Yuan, J. Li, H. Y. Wang, J. Y. Ge, H. F. Drake, C. F. Leong, F. Yu, D. M. D’Alessandro, M. Kurmoo, J. L. Zuo and H. C. Zhou, Chem. – Eur. J., 2021, 27, 622–627 CrossRef CAS PubMed.
- F. Leong, C.-H. Wang, C. D. Ling and D. M. D’Alessandro, Polyhedron, 2018, 154, 334–342 CrossRef.
- S. S. Park, E. R. Hontz, L. Sun, C. H. Hendon, A. Walsh, T. Van Voorhis and M. Dincă, J. Am. Chem. Soc., 2015, 137, 1774–1777 CrossRef CAS PubMed.
- J. Su, S. Yuan, H.-Y. Wang, L. Huang, J.-Y. Ge, E. Joseph, J. Qin, T. Cagin, J.-L. Zuo and H.-C. Zhou, Nat. Commun., 2017, 8, 2008 CrossRef PubMed.
- H. Y. Wang, Y. Wu, C. F. Leong, D. M. D’Alessandro and J. L. Zuo, Inorg. Chem., 2015, 54, 10766–10775 CrossRef CAS PubMed.
- H. Y. Wang, J. Su, J. P. Ma, F. Yu, C. F. Leong, D. M. D’Alessandro, M. Kurmoo and J. L. Zuo, Inorg. Chem., 2019, 58, 8657–8664 CrossRef CAS PubMed.
- Y. G. Weng, W. Y. Yin, M. Jiang, J. Le Hou, J. Shao, Q. Y. Zhu and J. Dai, ACS Appl. Mater. Interfaces, 2020, 12, 52615–52623 CrossRef CAS PubMed.
- Z. N. Yin, Y. H. Li, Y. G. Sun, T. Chen, J. Xu, Q. Y. Zhu and J. Dai, Inorg. Chem., 2016, 55, 9154–9157 CrossRef CAS PubMed.
- D. Bechu, L. S. Xie, N. Le Breton, S. Choua, M. Dincǎ, M. W. Hosseini and S. A. Baudron, Chem. Commun., 2020, 56, 2407–2410 RSC.
- S. Zhang, D. K. Panda, A. Yadav, W. Zhou and S. Saha, Chem. Sci., 2021, 12, 13379–13391 RSC.
- M. A. Gordillo, P. A. Benavides, K. Spalding and S. Saha, Front. Chem., 2021, 9, 792 Search PubMed.
- M. A. Gordillo, P. A. Benavides, D. K. Panda and S. Saha, ACS Appl. Mater. Interfaces, 2020, 12, 12955–12961 CrossRef CAS PubMed.
- J. Su, N. Xu, R. Murase, Z.-M. Yang, D. M. D’Alessandro, J.-L. Zuo and J. Zhu, Angew. Chem., Int. Ed., 2021, 60, 4789 CrossRef CAS PubMed.
- F. Gao, F. F. Zhu, X. Y. Wang, Y. Xu, X. P. Wang and J. L. Zuo, Inorg. Chem., 2014, 53, 5321–5327 CrossRef CAS PubMed.
- J. Su, T.-H. Hu, R. Murase, H.-Y. Wang, D. M. D’Alessandro, M. Kurmoo and J.-L. Zuo, Inorg. Chem., 2019, 58, 3698–3706 CrossRef CAS PubMed.
- J.-J. Hu, Y.-G. Li, H.-R. Wen, S.-J. Liu, Y. Peng and C.-M. Liu, Dalton Trans., 2021, 50, 14714–14723 RSC.
- G. Skorupskii, B. A. Trump, T. W. Kasel, C. M. Brown, C. H. Hendon and M. Dincă, Nat. Chem., 2020, 12, 131–136 CrossRef CAS PubMed.
- L. Sun, S. S. Park, D. Sheberla and M. Dincă, J. Am. Chem. Soc., 2016, 138, 14772–14782 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, characterization, and additional experimental data of Ln-MOFs, crystallographic data CCDC 2156276–2156278. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ma00606e |
| This journal is © The Royal Society of Chemistry 2022 |