
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLanthanoid coordination compounds as diverse self-templating agents towards hierarchically porous Fe–N–C electrocatalysts†
Itamar
Salton‡
,
Karina
Ioffe‡
,
Tomer Y.
Burshtein
,
Eliyahu M.
Farber
,
Nicola M.
Seraphim
,
Nofit
Segal
and
David
Eisenberg
 *
*
Schulich Faculty of Chemistry and The Grand Technion Energy Program Technion – Israel Institute of Technology Technion City, Haifa, 3200003, Israel. E-mail: eisenberg@technion.ac.il
First published on 23rd August 2022
Abstract
Pore structure is a critical material property of carbon materials, determining their surface area, active site accessibility, wettability, and efficiency of bubble removal. In the self-templating approach to pore design, inorganic particles form inside a carbon during pyrolysis, templating meso- and macropores. This strategy is simple and economic, yet limited by choice of templating elements and by incomplete understanding of carbon-template interactions. We followed the self-templating process of eight lanthanoid coordination compounds (the iminodiacetates of La3+, Nd3+, Sm3+, Eu3+, Gd3+, Tb3+, Er3+, and Yb3+), shedding light on the pore structure and the processes that form it. The resulting carbons showed high BET specific surface areas (up to 2700 m2 g−1), hierarchical micro-, meso- and macro-porosity, and lanthanoid-imprinted nitrogen moieties that could be transmetalated to yield atomically dispersed Fe–Nx sites. Some of the resulting Fe–N–C materials showed excellent activity towards hydrazine oxidation electrocatalysis, helping to identify several key links between porosity and electrocatalysis, especially the removal of electrode-blocking N2(g) bubbles. Overall, this detailed investigation expands the toolbox of rational design methods towards rich and useful electrocatalyst porosities.
Introduction
Carbon materials are key players in energy conversion and storage, environmental remediation, and catalysis.1–4 Carbons can store heat5 and fuels,6 harvest sunlight7,8 and water,9 and are key in electrochemical devices such as supercapacitors,10–12 batteries,13–15 and fuel cells.16–18 Carbons offer high specific surface areas, electronic conductivity, low cost, and low density. Their seemingly infinite structural tuneability is particularly important in electrocatalysis, where high power applications call for high volumetric surface area, a flow-enabling pore network, graphitic domains for electronic conductivity, and fine-tuned pore sizes for directing reaction selectivity through intermediate confinement.19–30 Thus, the structural design of carbons is a central challenge in electrocatalysis research.Highly precise carbon structures can be designed by ingenious multi-stage syntheses,31–35 yet complex methodologies often lead to low yields. At the other extreme, the pyrolysis of abundant biomass wastes is a popular route to useful carbons, but with little control over structure or composition.36,37 A promising golden path is the method of self-templating (Fig. 1). In this approach, pyrolysis of well-defined metal–organic coordination compounds – ideally composed of earth-abundant elements – yields carbon matrices with embedded inorganic particles of precise dimensions and compositions. Subsequent acid leaching of the templating particles produces hierarchically porous carbons. The self-templating method was pioneered by Inagaki et al. for magnesium compounds, and was explored for other alkaline earth metals.24,38–45 Surprisingly, few elements are known to be self-templating in carbons: the alkaline earths,24,38–45 a handful of post-transition metals,46,47 lanthanum,48,49 and several transition metals (which are often catalytic, thus convoluting structural and catalytic effects).50–54 To expand the utility of the self-templating pore design strategy, the range of templating elements must be extended, and the templating process must be better understood.
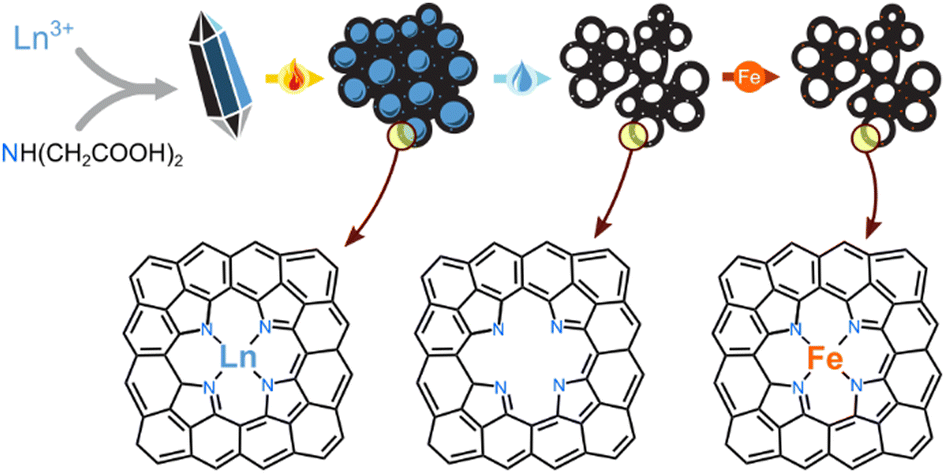 | ||
| Fig. 1 Simultaneous self-templating of pores and imprinting of active sites, to yield hierarchically porous Fe–N–C electrocatalysts: (1) precipitation of a lanthanoid salt, (2) pyrolysis at 800 °C, (3) acid leaching, (4) boiling with an iron salt solution. The bottom row shows a possible structure of an imprinted active site. | ||
The self-templating strategy can be enhanced even further with the simultaneous imprinting of catalytic sites, following the Fellinger method (Fig. 1).55–58 Metal-nonmetal moieties that form during pyrolysis (e.g. Zn2+–N4 or Mg2+–N4), can be transmetalated by catalytic elements such as Fe2+. The resulting materials belong to the highly important class of atomically-dispersed Fe–N–C electrocatalysts, which show great promise in the oxygen,50,59–62 nitrogen,50,63–68 and carbon cycles.69,70 This method is particularly useful for avoiding the formation of surface-blocking FeX particles during pyrolysis with iron precursors.50 The resulting Fe–N–C are particularly active in the hydrazine oxidation reaction (HzOR, N2H4 + 4OH− → N2 + 4H2O), an important reaction for alkaline fuel cells. Hydrazine hydrate (N2H4·H2O) is a promising alternative fuel: it is easily liquifiable, energy-dense, and thus a promising alternative to H2 in alkaline fuel cells.63,71–76 Fast HzOR requires active catalytic sites and fast mass transfer, but also efficient removal of electrode-blocking N2(g) bubbles.77,78
We now report the coordination compounds of eight lanthanoids (La, Nd, Sm, Eu, Gd, Tb, Er, Yb) as excellent and highly diverse precursors for the simultaneous self-templating and metal-imprinting of hierarchically porous carbons. The lanthanoids – historically part of the ‘rare earth metals’ – are more common in the earth's crust than many well-known elements (Fig. S1, ESI†).79 The lanthanoid-templated carbons were found to exhibit diverse and rich porous structures, created by a range of growth pathways during pyrolysis of the inorganic–carbon composite. Moreover, post-pyrolytic transmetalation of the lanthanoid-nitrogen moieties with Fe2+ yielded excellent electrocatalysts for hydrazine oxidation. The rich, fully hierarchical micro-, meso-, macro-porosity resulted in fast mass transfer during electrocatalysis, and the efficient removal of electrode-blocking N2 bubbles.
Results and discussion
Hierarchical porosity templating with lanthanoid inorganic phases
Eight lanthanoid metal ions (Ln3+) were precipitated with the same bidentate ligand, iminodiacetic acid (NH(CH2COOH)2, IDA), from aqueous solutions of LnCl3 and IDA (molar ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3). After drying, washing and grinding, the structures of the resulting compounds were solved by single crystal X-ray diffraction (to be reported in a separate study), yielding the compositions of La(IDA)3, Nd2(IDA)3(H2O)4, Sm2(IDA)3(H2O)4, Eu2(IDA)3(H2O)4, Gd2(IDA)3(H2O)4, Tb(IDA)(H2O), Er(IDA)(H2O)4, and Yb(IDA)3. Some of the structures contained additional weakly bound chlorides and water. The Ln-IDA salts were imaged by scanning electron microscopy (SEM, Fig. S2, ESI†), revealing layered macroscopic particles. Upon heat treatment of the metal–organic coordination compounds in argon at 800 °C, a variety of carbon–inorganic composites formed (dubbed LnX@NC). According to powder X-ray diffraction (XRD, Fig. 2), the materials contain either inorganic oxides (Eu, Tb, Er, Yb), oxycarbonates (Sm), both (La, Nd) or amorphous phases (Gd).
:3). After drying, washing and grinding, the structures of the resulting compounds were solved by single crystal X-ray diffraction (to be reported in a separate study), yielding the compositions of La(IDA)3, Nd2(IDA)3(H2O)4, Sm2(IDA)3(H2O)4, Eu2(IDA)3(H2O)4, Gd2(IDA)3(H2O)4, Tb(IDA)(H2O), Er(IDA)(H2O)4, and Yb(IDA)3. Some of the structures contained additional weakly bound chlorides and water. The Ln-IDA salts were imaged by scanning electron microscopy (SEM, Fig. S2, ESI†), revealing layered macroscopic particles. Upon heat treatment of the metal–organic coordination compounds in argon at 800 °C, a variety of carbon–inorganic composites formed (dubbed LnX@NC). According to powder X-ray diffraction (XRD, Fig. 2), the materials contain either inorganic oxides (Eu, Tb, Er, Yb), oxycarbonates (Sm), both (La, Nd) or amorphous phases (Gd).
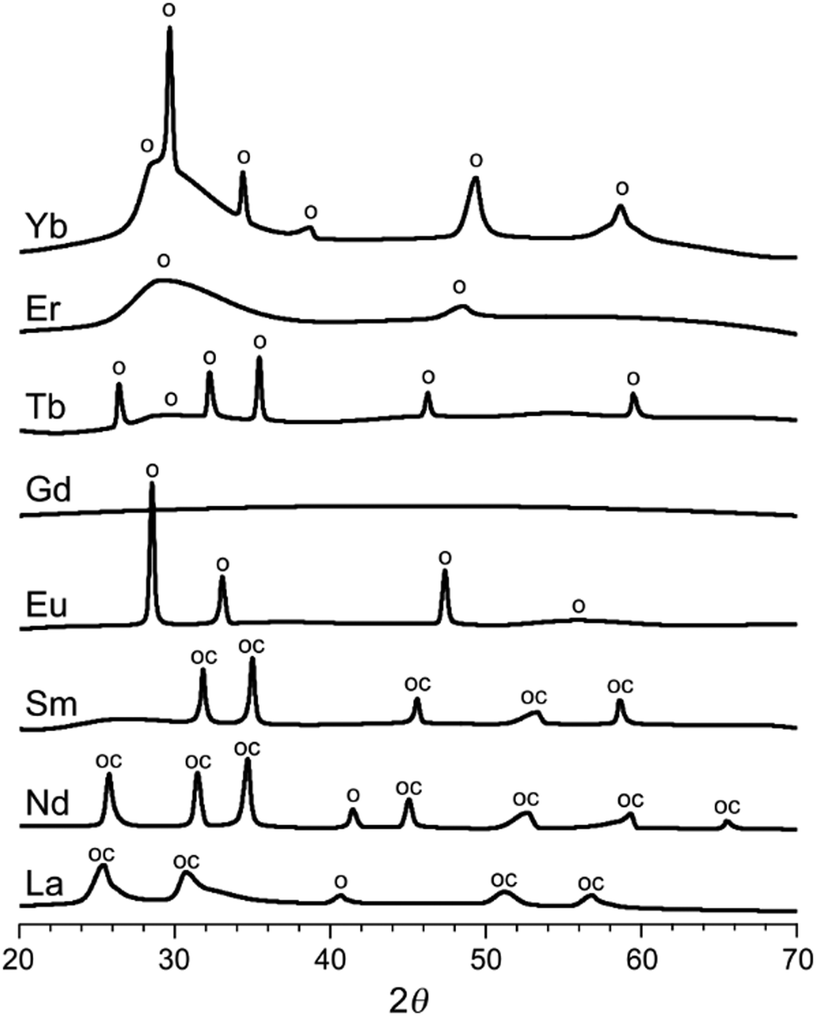 | ||
| Fig. 2 X-Ray diffractograms of the heat-treated carbon–inorganic composites (smoothed). The peaks are assigned to Ln2O3 oxides, marked o (JCPDS: La 05-0602, Nd 43-1023, Eu 83-7117, Tb 76-0156, Er 08-8239, Yb 41-1106) and to Ln2O2CO3 oxycarbonates, marked oc (JCPDS: La 37-0804, Nd 37-0806, Sm 37-0807). Visual peak matching in Fig. S4 (ESI†). | ||
The inorganic phases nucleate, crystallize and grow inside the carbon during the heat treatment. According to the thermogravimetric and calorimetric analysis, the last phase transformation occurs at roughly the same temperature for all materials (350–420 °C, Fig. S3, ESI†). Importantly, these inorganic phases can serve as templates for internal porosity only if they remain inside the carbon: if they are exsolved too early in the carbonization process, the templated voids collapse.24 The tendency for exsolution and carbon coating is determined by interplay between (1) the hydrophobicity of the inorganic phase, and (2) the crystallization energy, activation energy, and habit, which could favour the formation of compact particles. Such is the behaviour of GdX@NC and ErX@NC (as seen by scanning electron microscopy, SEM, Fig. 3a and b): the inorganic phases are exsolved from the carbon to form a skin-like layer at its surface. The GdX and ErX phases are also the most amorphous in the series (Fig. 2). Similarly, in LaX@NC, the inorganic phases (La2O3 and La2O2CO3) are limited to the carbon surface (Fig. 3c), despite being crystalline. Thus, these three lanthanoids (La, Gd, Er) are unlikely candidates for self-templating of internal porosity.
 | ||
| Fig. 3 (a–h) SEM micrographs of three types of LnX@NC composites (exsolved/embedded coating/particles), pyrolyzed at 800 °C. Scalebars are 1 μm. (i) Contact angle of ethylene glycol on Ln2O3 surfaces, as reported by Varanasi et al.80 and used here as a proxy for LnX hydrophobicity. | ||
Some exsolved phases, such as Nd2O3, Nd2O2CO3, Tb2O3 and Yb2O3 show up as well-defined crystalline platelets at the surface of their respective carbons (Fig. 3d–f). Such particles are more promising as templates for tuneable porosity than skin-like oxides because some particles may remain inside the carbon. Even more promising are Eu2O3 and Sm2O2CO3, which form as rounded particles, and are well-dispersed inside the carbon matrix (Fig. 3g, h, and Fig. S4, ESI†). In fact, the pore templating is directly seen in the micrographs: many inorganic particles appear alongside the pores from which they fell out. The degree of exsolution may vary slightly in different locations of the specimen, due to slight variations in the temperature experienced by that portion of the sample, but the micrographs presented here are the most representative.
To understand the three regimes of carbon|template interactions, highlighted in Fig. 3, we compared the hydrophobicity of lanthanoid oxides, reported in a systematic study by Varanasi et al.80 Specifically, we plotted the wetting angle of ethylene glycol on Ln2O3 materials, which is a useful proxy for the hydrophobicity of the LnX phases in this study. The most hydrophobic phases will tend to maximize their carbon interface area; indeed these are the phases that are exsolved to the surface and coat it (GdX, ErX, La2O3). The least hydrophobic phases will tend to minimize their interactions with the carbon, also leading to exsolution, and to ‘curling up’ into particles – these are the phases that form exsolved particles (Nd2O3, Nd2O2CO3, Tb2O3 and Yb2O3). Intermediate hydrophobicity, as in Eu2O3 and Sm2O2CO3, corresponds to the carbon|template interactions that favour the formation of embedded particles, which are optimal for pore templating. While crystallization thermodynamics and kinetics play a role in particle formation, self-templating proceeds mostly under kinetic control, impeding quantification of this aspect. Nevertheless, the hydrophobicity of the LnX phase is shown here to be a good indicator for carbon|template interactions during pyrolysis, and their effect on the ultimate porosity.
To follow the progress of exsolution along the pyrolysis, we selected two materials with partially embedded particles, YbX@NC and SmX@NC, different in composition and structure: Yb2O3 is a cubic oxide, while Sm2O2CO3 is a hexagonal oxycarbonate. The YbIDA and SmIDA salts were pyrolyzed at temperatures between 700–1000 °C, and followed by ex situ HRSEM (Fig. 4). At 700 °C, the inorganic particles are small (30–90 nm) and embedded in the carbon. The particles have roughly the same sizes in both materials, throughout the temperature range, indicating the central role of the carbon matrix in confining particle growth. At 800 °C, the particles begin falling out, leaving visible voids with sizes matching the template size. This is also the temperature where faceting starts, stressing the link between crystallization and exsolution. At 900 °C the voids begin to fill up, probably with amorphous pyrolysis product, while particle crystallization continues. Finally, at 1000 °C the inorganic phases reach their maximum size (150–250 nm) and are fully exsolved in the observed area of the carbon. In other areas of SmX@NC, however, many particles remain inside the carbon, as in Fig. 3 and Fig. S3 (ESI†). Thus, the temperature range of 800–900 °C is optimal for forming well-defined, easily removable templating particles.
 | ||
| Fig. 4 Following the progress of exsolution during pyrolysis of (a) SmX@NC, and (b) YbX@NC by HRSEM, along pyrolysis temperatures of 700–1000 °C. Scalebars are 100 nm. | ||
The pore templating was then completed by leaching out the inorganic particles by HCl 1 M for all eight LnX@NC materials. The porosity of the resulting carbons (dubbed NCLn) was quantified by N2 sorption porosimetry (Fig. 5). Complete leaching was confirmed by the disappearance of oxide and oxycarbonate peaks in the XRD (Fig. S6, ESI†), and by the trace amounts of lanthanoids measured by ICP-MS (0.01–0.04 at%). All N2 adsorption–desorption isotherms rise steeply at low p/p0 values (indicating significant microporosity), and all exhibit a brief and steep rise at p/p0 approaching 1 (due to condensation in macropores). The isotherms belong to either pure type I (microporous material) or have some type IV character (mesoporous material) mixed in, as observed in the hysteresis at intermediate-high p/p0 values.81
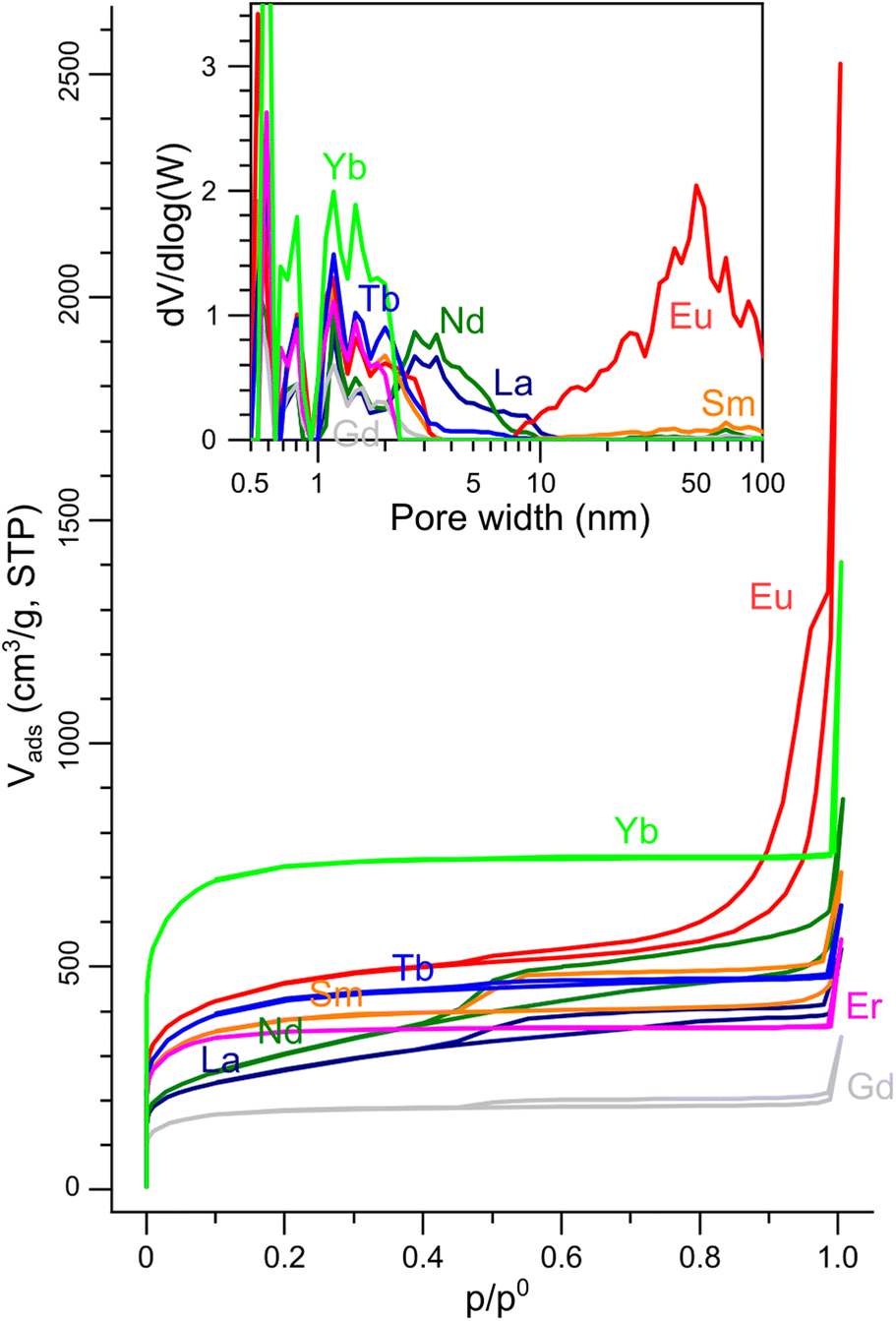 | ||
| Fig. 5 N2 sorption isotherms on the porous NCLn carbons (77 K). Inset: Pore size distributions calculated from the isotherms by the NLDFT model. | ||
The carbons in which the inorganic phase was exsolved to the surface during pyrolysis (NCGd, NCEr, NCLa, NCNd) had BET surface areas in the range of 660–1330 m2 g−1 (Table 1). These are high values, but are still the lowest in the series, where BET SSAs reached up to 1650 m2 g−1 in NCEu and even 2710 m2 g−1 in NCYb. These observations suggest that keeping the particles inside the carbon (by a proper tuning of the carbon|oxide interfacial energy) is crucial for exposing a high internal surface area in the carbon. NCYb is strictly microporous (type I isotherm), while NCEu has a full-house combination of micro-, meso- and macropores. Quantification of the pore size distribution by NLDFT analysis of the isotherms (Fig. 5, inset) revealed that all carbons have micropores (<2 nm), some have small mesopores (2–10 nm), but only NCEu also has a rich distribution of mesopores (10–50 nm) and macropores. Moreover, the total pore volume is highest in NCEu, reaching 1.91 mL g−1 – compared to 1.2 mL g−1 in strictly-microporous NCYb, and <0.8 mL g−1 in the others. The well-developed hierarchical porosity of NCEu is also visible at several magnifications by SEM and TEM (Fig. 6), as very fine wrinkles on a porous undulating surface. The richly porous morphology of NCEu and NCYb is promising for exposing active sites and enhancing flow during electrocatalysis. Moreover, the many sharp edges visible in the fine porous structure create low-area three-phase contact layers (bubble–electrolyte–solid),82 useful for preventing bubble pinning and enhancing gas evolution. The thin and structured carbon walls (Fig. 6d) help maximize the volumetric surface area of the electrode, while maintaining structural rigidity.
| Ln | C in LnX@NCa (wt%) | N in NCb (wt%) | Ln in NCb (wt%) | Fe in Fe–NCb (wt%) | BET SSAc (m2 g−1) | V pore (cm3 g−1) | I D/IGe | L a (nm) |
|---|---|---|---|---|---|---|---|---|
| a Carbon yield in thermogravimetric combustion (Fig. S7, ESI). b ICP-MS; the remainder is O and possibly Cl from the washing. c Isothermal N2 adsorption at 77 K. d Determined at p/p0 = 0.99. e Deconvoluted Raman spectra (Fig. S8, ESI). f Calculated from the ID/IG ratio. | ||||||||
| La | 27 | 13.5 | 0.49 | 0.31 | 930 | 0.61 | 1.76 | 10.9 |
| Nd | 21 | 10.2 | 0.36 | 0.22 | 1030 | 0.86 | 1.46 | 13.2 |
| Sm | 23 | 11.4 | 0.37 | 1.10 | 1390 | 0.71 | 1.79 | 10.7 |
| Eu | 18 | 11.3 | 0.11 | 2.56 | 1650 | 1.91 | 1.60 | 12.0 |
| Gd | 20 | 13.9 | 0.42 | 0.56 | 660 | 0.31 | 1.76 | 10.9 |
| Tb | 21 | 11.8 | 0.21 | 0.81 | 1530 | 0.74 | 1.59 | 12.1 |
| Er | 34 | 13.5 | 0.33 | 0.32 | 1330 | 0.57 | 1.54 | 12.5 |
| Yb | 39 | 13.5 | 0.48 | 2.07 | 2710 | 1.16 | 1.70 | 11.3 |
 | ||
| Fig. 6 SEM micrographs of hierarchically porous NCEu at four magnifications, obtained by (a and b) SEM, and (c and d) TEM. | ||
The high SSA and fine morphology of NCEu suggest that Eu3+ ions are not only excellent mesopore templates, but also particularly efficient in micropore etching during pyrolysis. When metal–organic compounds are heat treated and carbonized, metal ions can oxidize the carbon matrix, etching micropores into it and becoming easily washable reduced particles. Such processes have been observed in the pyrolysis of Group 1-containing precursors.40,43 Indeed, a careful examination of the carbon yield in each pyrolysis (Fig. 7a), as calculated from the thermogravimetric combustion of each of the NCLn carbons (Fig. S7, ESI†), reveals that most carbon is removed in NCEu. Moreover, the lanthanoid trace content is lowest in the case of Eu (Fig. 7b); this metal loss can be explained by efficient carbothermal reduction (i.e. micropore etching) and subsequent evaporation of the metal.40 Interestingly, Eu3+ is the strongest oxidant of all eight lanthanoids in this study, when comparing standard reduction potentials of the Ln3+/Ln couple (Fig. 7c). Although E° values were determined at conditions dissimilar to a hot carbon matrix, they do offer a qualitative trend to support the efficient etching of micropores by Eu3+.
 | ||
| Fig. 7 Indirect evidence for carbon etching by Eu3+ ions during pyrolysis: (a) efficient carbon etching, as seen by the thermogravimetrically determined carbon fraction in the LnX@NC composite; (b) remaining traces of Ln atoms after the acid wash (ICP-MS); (c) standard reduction potentials of the Ln3+/Ln couple,83 possibly correlated with the tendency to oxidize the carbon during pyrolysis. | ||
Formation of lanthanoid-imprinted Fe–N–C active sites
In the active site imprinting concept, proposed by Fellinger et al., the templating lanthanoid ions bind and retain the nitrogen atoms during the pyrolysis, and can later be replaced (transmetalated) with a catalytically active metal such as iron.55–58 We have performed such an iron transmetalation by refluxing the NCLn in a methanol solution of an iron salt, followed by washing and drying. The resulting materials (dubbed FeNCLn) contain very high nitrogen content (8.9–12.1 at%, Table 1). This is higher than the typical N content of Group 2-templated carbons (∼6 at%),24,43 and could arise from the stronger Ln–N coordination. The carbons also contain residual oxygen, hydrogen, and possibly chlorine from the acid wash. The oxygen phases are important for rendering the carbon surface hydrophilic enough for wetting by electrolyte, yet also serve as possible reaction sites for corrosion. Importantly for electronic conductivity, the carbons are quite graphitic, as measured by Raman spectroscopy (Fig. S8, ESI†). The intensity ratio between the Raman D band (associated with carbon defects) and the G band (graphitic band) varies slightly around 1.65 ± 0.12 across the series (Table 1). These values correspond to graphitic plane domain lengths (La) of 11.7 ± 0.9 nm. The similarity in degree of graphitization across the series reveals that IDA ligand, common to all eight salts, is solely responsible for directing the textural properties of the carbon. Thus, the role of the lanthanoid metal is only to direct the porous structure of the materials.The final iron content varied between 0.2–1.1 wt% for most carbons (ICP-MS, Table 1) but peaked in the highly porous carbons, reaching 2.1 wt% in Fe–NCYb and 2.6 wt% in Fe–NCEu. The nitrogen binding modes were analysed in two high-surface-area carbons, Fe–NCEu and Fe–NCSm, by X-ray photoelectron spectroscopy (XPS) in the N 1s region (Fig. 8). The spectra were deconvoluted into peaks of metal-bound, pyridinic, pyrrolic, quaternary, graphitic, and oxidized nitrogen positions.84 Both materials contain a significant fraction of metal-bound nitrogens (NMe), more in Fe–NCEu (33%) than in Fe–NCSm (25%). The difference is equally distributed across the other nitrogen positions (Table S1, ESI†). This suggests that the Eu3+ and Sm3+ ions were successful in imprinting Ln–Nx moieties that could be converted to Fe–Nx sites. Moreover, Eu3+ ions are somewhat better than Sm3+ ions in the imprinting process.
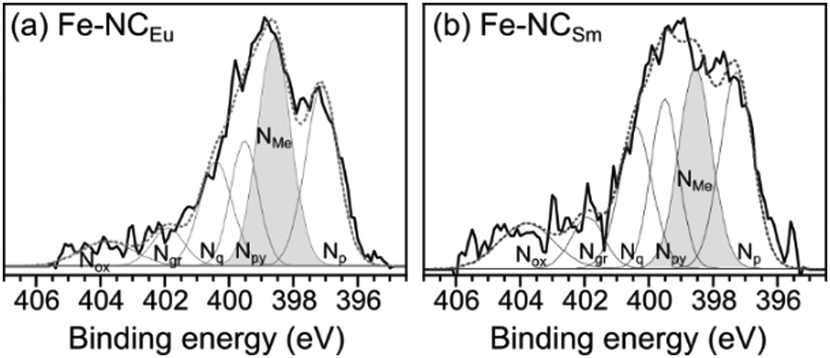 | ||
| Fig. 8 X-Ray photoelectron spectra in the N 1s region on Fe-transmetalated porous carbons (a) Fe–NCEu and (b) Fe–NCSm. The spectra are deconvoluted84 to identify different nitrogen moieties, including pyridinic (Np), metal-bound (NMe), pyrrolyic (Npy), quaternary (Nq), graphitic (Ng) and oxidized (Nox). | ||
All NCLn carbons and all transmetalated Fe–NCLn carbons were tested as hydrazine oxidation electrocatalysts in 1 M KOH solution, without electrode rotation (Fig. 9). Some of the iron-free NCLn carbons show reasonable HzOR activity, with onset potentials ranging 0.38–0.48 V vs. RHE at pH 14 (Fig. 9a). The oxidation occurs in two waves, assigned to two active sites: first, a small wave on the nitrogen dopants; second, a larger wave on carbon defects.85
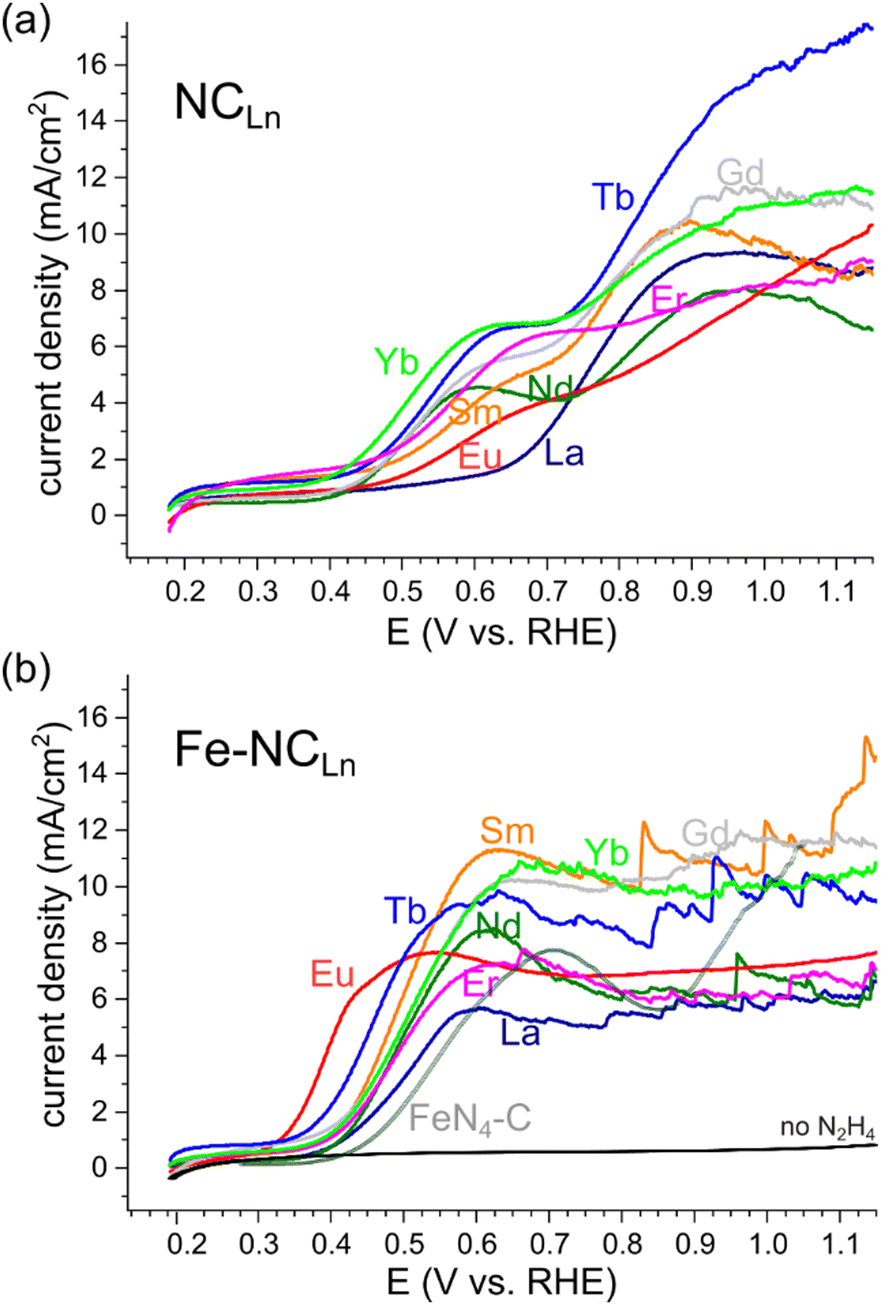 | ||
| Fig. 9 Hydrazine oxidation electrocatalysis on (a) metal-free porous NCLn carbons, and on (b) iron-transmetalated porous Fe–NCLn carbons, including a comparison to a recent FeN4-C reference material.64 LSVs taken without electrode rotation, in 1 M KOH, 0.1 M N2H4, scan rate 10 mV s−1. | ||
Fluctuations in the current, indicating irregular detachment of N2(g) bubbles produced by the HzOR, appear at high overpotentials (>0.9 V vs. RHE) for some of the materials. Fast bubble generation is encouraging for efficient HzOR but highlights the need for efficient bubble removal.
Upon transmetalation with iron, the HzOR activity improves drastically (Table S2 and Fig. S9, ESI†).56 The onset potentials are shifted negative, down to 0.37–0.38 V for most carbons, and even 0.32 V vs. RHE for Fe–NCEu. These onsets approach those of the best Fe–N–C electrocatalysts reported for the HzOR, to within 70 mV (see Table S3, ESI†).63 Importantly, the HzOR wave shows a constant oxidation current, indicating fast, mass-transfer limited electrocatalysis. This contrasts all previously reported Fe–N–C HzOR electrocatalysts, where Fe is typically deactivated, resulting in a peak-shaped voltametric wave.50,63,64 The atomical dispersion of the Fe–Nx moieties is further supported by cyanide and nitrite poisoning experiments (Fig. S10, ESI†), which show lower current densities upon selective poisoning of iron centers.66,86–88 Tafel analysis confirms a distinct change in electrocatalytic mechanism (Fig. S11, ESI†): the iron-free NC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) materials show Tafel slopes of 270 ± 70 mV dec−1, whereas the iron-imprinted Fe–NCLn catalysts exhibit steeper slopes of 140 ± 25 mV dec−1 (Table S4, ESI†). However, these values are hard to interpret on their own, since mass-transfer phenomena cloud the kinetic analysis.
materials show Tafel slopes of 270 ± 70 mV dec−1, whereas the iron-imprinted Fe–NCLn catalysts exhibit steeper slopes of 140 ± 25 mV dec−1 (Table S4, ESI†). However, these values are hard to interpret on their own, since mass-transfer phenomena cloud the kinetic analysis.
The Fe–NCLn catalysts start evolving N2 bubbles at early potentials (0.6 V vs. RHE). This is indicated by the current spikes, which correspond to single events of bubble release from the electrode surface. High power HzOR operation calls for fast removal of bubbles, which would result in small, frequent, and regular current spikes. This is the case for Fe–NCYb, where the spikes are regular and small. The efficient bubble removal is visible as continuous ‘frothing’ from the entire electrode surface (Fig. 10). Even more remarkable is Fe–NCEu, showing no current spikes at all – but rather a steady catalytic plateau from 0.5 V vs. RHE and on. Although N2 is still generated in large quantities (see also the Koutecký–Levich analysis below), the bubbles only grow at the perimeter of the electrode, indicating immediate removal to the sides (Fig. 10) and a complete lack of bubble pinning. This efficient bubble removal also explains the early HzOR onset potential on Fe–NCEu, since it allows the active site to start operating at maximum efficiency immediately; without removal of the N2 products, higher over-potentials are needed to make the reaction favorable. Overall, the differences in bubble removal efficiencies within the series are striking, and suggest the central role of the pore structure and fine surface texture in this important phenomenon.
 | ||
| Fig. 10 Photographs of two electrocatalysts showing the most efficient bubble removal, at E = 0.55 V, at the conditions of Fig. 9, rotating at 10 rpm. | ||
Koutecký–Levich analysis was further used to calculate the apparent number of electrons transferred per N2H4 molecule (Fig. S12, ESI†). The analysis revealed 2.1 ± 0.3 e− for Fe–NCYb and 3.2 ± 0.1 for Fe–NCEu. Complete oxidation to N2 would yield 4 e−/N2H4. The losses may result from chemical decomposition of the hydrazine.89 A more complete oxidation is linked with intermediate confinement in specific pore sizes. Thus, the higher value for the Fe–NCEu carbon further confirms its higher activity for the HzOR, which arises from its unique, hierarchically porous nanostructure.
Conclusions
Eight lanthanoid metal iminodiacetates (La, Nd, Sm, Eu, Gd, Tb, Er, Yb) are reported as diverse precursors for the simultaneous self-templating and metal-imprinting of hierarchically porous carbon electrocatalysts. The lanthanoid coordination compounds were pyrolyzed to yield a series of LnX@NC materials, exhibiting different types of carbon|template interactions, such as coating, exsolution, and embedding. The spatial evolution of template particles was shown to be critical for the exposure of internal microporosity: upon removal of the inorganic templates, a rich variety of porous carbons was obtained, exhibiting microporosity (correlated with BET SSAs from 660 to 2700 m2 g−1), some mesoporosity, and in some cases (Eu, Sm) complex porosity on multiple length-scales. Finally, post-pyrolytic transmetalation with Fe2+ yielded excellent electrocatalysts for the hydrazine oxidation reaction, with early onsets (down to Eonset = +0.32 V vs. RHE in 1 M KOH on NCEu) and stable catalytic currents without deactivation. Bubble removal, as assessed by current spikes, varied with the carbon texture, and very efficient bubble removal was observed on NCEu and NCYb. The outstanding HzOR activity of Fe–NCEu is no doubt related to its exceptional material properties: high specific surface area (1650 m2 g−1), high pore volume (1.91 mL g−1), hierarchical micro-, meso- and macroporosity, and higher Fe content. These properties, in turn, arise from the unique suitability of Eu ions for driving multi-level self-templating – both of nitrogen-based active sites, and of a flow-enabling pore network. These hierarchically carbons are expected to be useful supports for other electrocatalytic reactions. The systematic understanding of the self-templating process expands the toolbox of pore engineering strategies, to applications in catalysis, batteries, adsorption, and more.Author contributions
I. S. synthesized the carbons with the help of N. S. and coordinated all material characterizations, electrochemical measurements, and data analysis. K. I. performed transmetalation, electrochemical experiments, and helped coordinate the experiments. T. Y. B. performed Raman and XPS analysis. E. M. F. performed electron microscopy analysis. N. M. S. performed XRD analysis. D. E. acquired funds and helped coordinate the project. I. S., K. I., and D. E. wrote the manuscript. All authors edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Nancy & Stephen Grand Technion Energy Program (GTEP) and the Israel Ministry of Energy for partial financial support.Notes and references
- F. Rodríguez-Reinoso, Carbon, 1998, 36, 159–175 CrossRef.
- C. Zhang, W. Lv, Y. Tao and Q.-H. Yang, Energy Environ. Sci., 2015, 8, 1390–1403 RSC.
- M. Wu, J. Liao, L. Yu, R. Lv, P. Li, W. Sun, R. Tan, X. Duan, L. Zhang, F. Li, J. Kim, K. H. Shin, H. Seok Park, W. Zhang, Z. Guo, H. Wang, Y. Tang, G. Gorgolis, C. Galiotis and J. Ma, Chem. – Asian J., 2020, 15, 995–1013 CrossRef CAS PubMed.
- Q. Chen, X. Tan, Y. Liu, S. Liu, M. Li, Y. Gu, P. Zhang, S. Ye, Z. Yang and Y. Yang, J. Mater. Chem. A, 2020, 8, 5773–5811 RSC.
- X. Chen, P. Cheng, Z. Tang, X. Xu, H. Gao and G. Wang, Adv. Sci., 2021, 8, 2001274 CrossRef CAS PubMed.
- M. Mohan, V. K. Sharma, E. A. Kumar and V. Gayathri, Energy Storage, 2019, 1, e35 CrossRef CAS.
- M. Volokh, G. Peng, J. Barrio and M. Shalom, Angew. Chem., Int. Ed., 2019, 58, 6138–6151 CrossRef CAS PubMed.
- D. Xiao, M. Jiang, X. Luo, S. Liu, J. Li, Z. Chen and S. Li, ACS Sustainable Chem. Eng., 2021, 9, 4139–4145 CrossRef CAS.
- Z. Chen, S. Song, B. Ma, Y. Li, Y. Shao, J. Shi, M. Liu, H. Jin and D. Jing, Sol. Energy Mater. Sol. Cells, 2021, 230, 111233 CrossRef CAS.
- A. Borenstein, O. Hanna, R. Attias, S. Luski, T. Brousse and D. Aurbach, J. Mater. Chem. A, 2017, 5, 12653–12672 RSC.
- J. Biemolt, I. M. Denekamp, T. K. Slot, G. Rothenberg and D. Eisenberg, ChemSusChem, 2017, 10, 4018–4024 CrossRef CAS PubMed.
- P. Ratajczak, M. E. Suss, F. Kaasik and F. Béguin, Energy Storage Mater., 2019, 16, 126–145 CrossRef.
- Z.-L. Xu, J.-K. Kim and K. Kang, Nano Today, 2018, 19, 84–107 CrossRef CAS.
- J. Lach, K. Wróbel, J. Wróbel, P. Podsadni and A. Czerwiński, J. Solid State Electrochem., 2019, 23, 693–705 CrossRef CAS.
- L. Xie, C. Tang, Z. Bi, M. Song, Y. Fan, C. Yan, X. Li, F. Su, Q. Zhang and C. Chen, Adv. Energy Mater., 2021, 11, 2101650 CrossRef CAS.
- A. L. Dicks, J. Power Sources, 2006, 156, 128–141 CrossRef CAS.
- T. B. Ferriday and P. H. Middleton, Int. J. Hydrogen Energy, 2021, 46, 18489–18510 CrossRef CAS.
- Y. Yang, C. R. Peltier, R. Zeng, R. Schimmenti, Q. Li, X. Huang, Z. Yan, G. Potsi, R. Selhorst, X. Lu, W. Xu, M. Tader, A. V. Soudackov, H. Zhang, M. Krumov, E. Murray, P. Xu, J. Hitt, L. Xu, H.-Y. Ko, B. G. Ernst, C. Bundschu, A. Luo, D. Markovich, M. Hu, C. He, H. Wang, J. Fang, R. A. DiStasio, L. F. Kourkoutis, A. Singer, K. J. T. Noonan, L. Xiao, L. Zhuang, B. S. Pivovar, P. Zelenay, E. Herrero, J. M. Feliu, J. Suntivich, E. P. Giannelis, S. Hammes-Schiffer, T. Arias, M. Mavrikakis, T. E. Mallouk, J. D. Brock, D. A. Muller, F. J. DiSalvo, G. W. Coates and H. D. Abruña, Chem. Rev., 2022, 122, 6117–6321 CrossRef CAS PubMed.
- M. Ferrandon, A. J. Kropf, D. J. Myers, K. Artyushkova, U. Kramm, P. Bogdanoff, G. Wu, C. M. Johnston and P. Zelenay, J. Phys. Chem. C, 2012, 116, 16001–16013 CrossRef CAS.
- N. Daems, X. Sheng, I. F. J. Vankelecom and P. P. Pescarmona, J. Mater. Chem. A, 2014, 2, 4085–4110 RSC.
- A. S. Varela, N. Ranjbar Sahraie, J. Steinberg, W. Ju, H. S. Oh and P. Strasser, Angew. Chem., Int. Ed., 2015, 54, 10758–10762 CrossRef CAS.
- W. Zhou, C. Wang, Q. Zhang, H. D. Abruña, Y. He, J. Wang, S. X. Mao and X. Xiao, Adv. Energy Mater., 2015, 5, 1401752 CrossRef.
- D. Hursán, A. A. Samu, L. Janovák, K. Artyushkova, T. Asset, P. Atanassov and C. Janáky, Joule, 2019, 3, 1719–1733 CrossRef PubMed.
- E. M. Farber, K. Ojha, T. Y. Burshtein, L. Hasson and D. Eisenberg, Mater. Adv., 2020, 1, 20–33 RSC.
- A. B. Jorge, R. Jervis, A. P. Periasamy, M. Qiao, J. Feng, L. N. Tran and M.-M. Titirici, Adv. Energy Mater., 2020, 10, 1902494 CrossRef CAS.
- T. Asset and P. Atanassov, Joule, 2020, 4, 33–44 CrossRef CAS.
- F. Luo, A. Roy, L. Silvioli, D. A. Cullen, A. Zitolo, M. T. Sougrati, I. C. Oguz, T. Mineva, D. Teschner, S. Wagner, J. Wen, F. Dionigi, U. I. Kramm, J. Rossmeisl, F. Jaouen and P. Strasser, Nat. Mater., 2020, 19, 1215–1223 CrossRef CAS PubMed.
- S. Ratso, A. Zitolo, M. Käärik, M. Merisalu, A. Kikas, V. Kisand, M. Rähn, P. Paiste, J. Leis, V. Sammelselg, S. Holdcroft, F. Jaouen and K. Tammeveski, Renew. Energy, 2021, 167, 800–810 CrossRef CAS.
- Q. Han, H. Jiao, L. Xiong and J. Tang, Mater. Adv., 2021, 2, 564–581 RSC.
- X. Shu, M. Yang, D. Tan, K. San Hui, K. Nam Hui and J. Zhang, Mater. Adv., 2021, 2, 96–114 RSC.
- A. A. Zakhidov, R. H. Baughman, Z. Iqbal, C. Cui, I. Khayrullin, S. O. Dantas, J. Marti and V. G. Ralchenko, Science, 1998, 282, 897–901 CrossRef CAS PubMed.
- C. Liang, K. Hong, G. A. Guiochon, J. W. Mays and S. Dai, Angew. Chem., Int. Ed., 2004, 43, 5785–5789 CrossRef CAS PubMed.
- D.-Y. Kang and J. H. Moon, Sci. Rep., 2014, 4, 5392 CrossRef CAS PubMed.
- A. Guirguis, J. W. Maina, L. Kong, L. C. Henderson, A. Rana, L. H. Li, M. Majumder and L. F. Dumée, Carbon, 2019, 155, 660–673 CrossRef CAS.
- R. Wang, K. Lan, R. Lin, X. Jing, C.-T. Hung, X. Zhang, L. Liu, Y. Yang, G. Chen, X. Liu, C. Fan, A. M. El-Toni, A. Khan, Y. Tang and D. Zhao, ACS Nano, 2021, 15, 7713–7721 CrossRef CAS PubMed.
- S. Tabac and D. Eisenberg, Curr. Opin. Electrochem., 2021, 25, 100638 CrossRef CAS.
- Y. Wang, M. Zhang, X. Shen, H. Wang, H. Wang, K. Xia, Z. Yin and Y. Zhang, Small, 2021, 17, 2008079 CrossRef CAS.
- T. Morishita, K. Ishihara, M. Kato and M. Inagaki, Carbon, 2007, 45, 209–211 CrossRef CAS.
- M. Inagaki, H. Orikasa and T. Morishita, RSC Adv., 2011, 1, 1620–1640 RSC.
- M. Sevilla and A. B. Fuertes, J. Mater. Chem. A, 2013, 1, 13738–13741 RSC.
- N. Díez, M. Sevilla and A. B. Fuertes, Carbon, 2021, 178, 451–476 CrossRef.
- D. Eisenberg, W. Stroek, N. J. Geels, C. S. Sandu, A. Heller, N. Yan and G. Rothenberg, Chem. – Eur. J., 2016, 22, 501–505 CrossRef CAS PubMed.
- D. Eisenberg, W. Stroek, N. J. Geels, S. Tanase, M. Ferbinteanu, S. J. Teat, P. Mettraux, N. Yan and G. Rothenberg, Phys. Chem. Chem. Phys., 2016, 18, 20778–20783 RSC.
- D. Eisenberg, P. Prinsen, N. J. Geels, W. Stroek, N. Yan, B. Hua, J.-L. Luo and G. Rothenberg, RSC Adv., 2016, 6, 80398–80407 RSC.
- E. M. Farber, K. Ojha, T. Y. Burshtein and D. Eisenberg, J. Electrochem. Soc., 2020, 167, 064517 CrossRef CAS.
- Z. J. Zhang, P. Cui and X. Y. Chen, Ind. Eng. Chem. Res., 2013, 52, 16211–16219 CrossRef CAS.
- X. Y. Chen, Y. Y. He, H. Song and Z. J. Zhang, Carbon, 2014, 72, 410–420 CrossRef CAS.
- D. H. Youn, M. L. Meyerson, K. C. Klavetter, K. A. Friedman, S. S. Coffman, J.-W. Lee, A. Heller and C. B. Mullins, J. Electrochem. Soc., 2016, 163, A953–A957 CrossRef CAS.
- A. Heller, J.-W. Lee, D. Eisenberg, K. A. Friedman and S. Coffman, US Pat., US20200299136, 2020.
- T. Y. Burshtein, D. Aias, J. Wang, M. Sananis, E. M. Farber, O. M. Gazit, I. Grinberg and D. Eisenberg, Phys. Chem. Chem. Phys., 2021, 23, 26674–26679 RSC.
- R. Das, P. Pachfule, R. Banerjee and P. Poddar, Metal and metal oxide nanoparticle synthesis from metal organic frameworks (MOFs): Finding the border of metal and metal oxides, 2012, vol. 4 Search PubMed.
- W. Niu, L. Li, X. Liu, N. Wang, J. Liu, W. Zhou, Z. Tang and S. Chen, J. Am. Chem. Soc., 2015, 137, 5555–5562 CrossRef CAS PubMed.
- L. Wang, X. Jin, J. Fu, Q. Jiang, Y. Xie, J. Huang and L. Xu, J. Electroanal. Chem., 2018, 825, 65–72 CrossRef CAS.
- W. Yang, X. Li, Y. Li, R. Zhu and H. Pang, Adv. Mater., 2019, 31, 1804740 Search PubMed.
- A. Mehmood, J. Pampel, G. Ali, H. Y. Ha, F. Ruiz-Zepeda and T.-P. Fellinger, Adv. Energy Mater., 2018, 8, 1701771 CrossRef.
- D. Menga, F. Ruiz-Zepeda, L. Moriau, M. Šala, F. Wagner, B. Koyutürk, M. Bele, U. Petek, N. Hodnik, M. Gaberšček and T.-P. Fellinger, Adv. Energy Mater., 2019, 9, 1902412 CrossRef CAS.
- D. Menga, J. L. Low, Y.-S. Li, I. Arčon, B. Koyutürk, F. Wagner, F. Ruiz-Zepeda, M. Gaberšček, B. Paulus and T.-P. Fellinger, J. Am. Chem. Soc., 2021, 143, 18010–18019 CrossRef CAS PubMed.
- B. Koyutürk, E. M. Farber, F. E. Wagner, T.-P. Fellinger and D. Eisenberg, J. Mater. Chem. A, 2022 10.1039/D2TA00925K.
- H. Ren, Y. Wang, Y. Yang, X. Tang, Y. Peng, H. Peng, L. Xiao, J. Lu, H. D. Abruña and L. Zhuang, ACS Catal., 2017, 7, 6485–6492 CrossRef CAS.
- P. Chen, T. Zhou, L. Xing, K. Xu, Y. Tong, H. Xie, L. Zhang, W. Yan, W. Chu, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2017, 56, 610–614 CrossRef CAS PubMed.
- C. H. Choi, W. S. Choi, O. Kasian, A. K. Mechler, M. T. Sougrati, S. Brüller, K. Strickland, Q. Jia, S. Mukerjee, K. J. J. Mayrhofer and F. Jaouen, Angew. Chem., Int. Ed., 2017, 56, 8809–8812 CrossRef CAS PubMed.
- Y. Pan, S. Liu, K. Sun, X. Chen, B. Wang, K. Wu, X. Cao, W.-C. Cheong, R. Shen, A. Han, Z. Chen, L. Zheng, J. Luo, Y. Lin, Y. Liu, D. Wang, Q. Peng, Q. Zhang, C. Chen and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 8614–8618 CrossRef CAS PubMed.
- Y. Zheng, F. He, M. Chen, J. Zhang, G. Hu, D. Ma, J. Guo, H. Fan, W. Li and X. Hu, ACS Appl. Mater. Interfaces, 2020, 12, 38183–38191 CrossRef CAS PubMed.
- Y. Shahaf, A. Mahammed, A. Raslin, A. Kumar, E. M. Farber, Z. Gross and D. Eisenberg, ChemElectroChem, 2022, 9, e202200045 CrossRef CAS.
- C. Linares-Flores, J. Espinoza-Vergara, J. H. Zagal and R. Arratia-Perez, Chem. Phys. Lett., 2014, 614, 176–180 CrossRef CAS.
- D. Malko, A. Kucernak and T. Lopes, J. Am. Chem. Soc., 2016, 138, 16056–16068 CrossRef CAS PubMed.
- M. Wang, S. Liu, T. Qian, J. Liu, J. Zhou, H. Ji, J. Xiong, J. Zhong and C. Yan, Nat. Commun., 2019, 10, 341 CrossRef CAS PubMed.
- D. H. Kim, S. Ringe, H. Kim, S. Kim, B. Kim, G. Bae, H.-S. Oh, F. Jaouen, W. Kim, H. Kim and C. H. Choi, Nat. Commun., 2021, 12, 1856 CrossRef CAS PubMed.
- C. Genovese, M. E. Schuster, E. K. Gibson, D. Gianolio, V. Posligua, R. Grau-Crespo, G. Cibin, P. P. Wells, D. Garai, V. Solokha, S. Krick Calderon, J. J. Velasco-Velez, C. Ampelli, S. Perathoner, G. Held, G. Centi and R. Arrigo, Nat. Commun., 2018, 9, 935 CrossRef PubMed.
- J. Gu, C.-S. Hsu, L. Bai, H. M. Chen and X. Hu, Science, 2019, 364, 1091–1094 CrossRef CAS PubMed.
- A. Serov and C. Kwak, Appl. Catal., B, 2010, 98, 1–9 CrossRef CAS.
- N. V. Rees and R. G. Compton, Energy Environ. Sci., 2011, 4, 1255–1260 RSC.
- A. Serov, M. Padilla, A. J. Roy, P. Atanassov, T. Sakamoto, K. Asazawa and H. Tanaka, Angew. Chem., Int. Ed., 2014, 53, 10336–10339 CrossRef CAS PubMed.
- Y. Meng, X. Zou, X. Huang, A. Goswami, Z. Liu and T. Asefa, Adv. Mater., 2014, 26, 6510–6516 CrossRef CAS PubMed.
- K. Ojha, E. M. Farber, T. Y. Burshtein and D. Eisenberg, Angew. Chem., Int. Ed., 2018, 57, 17168–17172 CrossRef CAS PubMed.
- T. Zhang and T. Asefa, Adv. Mater., 2019, 31, 1804394 CrossRef PubMed.
- K. Akbar, J. H. Kim, Z. Lee, M. Kim, Y. Yi and S.-H. Chun, NPG Asia Mater., 2017, 9, e378–e378 CrossRef CAS.
- J. Jeong, M. Choun and J. Lee, Angew. Chem., Int. Ed., 2017, 56, 13513–13516 CrossRef CAS PubMed.
- W. M. Haynes, in CRC Handbook of Chemistry and Physics, CRC Press; Hoboken, 95th edn, 2014, pp. 14–19 Search PubMed.
- G. Azimi, R. Dhiman, H.-M. Kwon, A. T. Paxson and K. K. Varanasi, Nat. Mater., 2013, 12, 315–320 CrossRef CAS PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- H. Y. Erbil, Colloids Interfaces, 2021, 5, 8 CrossRef.
- P. Vanýsek, in CRC Handbook of Chemistry and Physics, ed. W. M. Haynes, CRC Press; Hoboken, 95th edn, 2014, pp. 5–80 Search PubMed.
- K. Artyushkova, A. Serov, S. Rojas-Carbonell and P. Atanassov, J. Phys. Chem. C, 2015, 119, 25917–25928 CrossRef CAS.
- T. Y. Burshtein, K. Tamakuwala, M. Sananis, I. Grinberg, N. R. Samala and D. Eisenberg, Phys. Chem. Chem. Phys., 2022, 24, 9897–9903 RSC.
- F. J. Pérez-Alonso, C. Domínguez, S. A. Al-Thabaiti, A. O. Al-Youbi, M. Abdel Salam, A. A. Alshehri, M. Retuerto, M. A. Peña and S. Rojas, J. Power Sources, 2016, 327, 204–211 CrossRef.
- M. Primbs, Y. Sun, A. Roy, D. Malko, A. Mehmood, M.-T. Sougrati, P.-Y. Blanchard, G. Granozzi, T. Kosmala, G. Daniel, P. Atanassov, J. Sharman, C. Durante, A. Kucernak, D. Jones, F. Jaouen and P. Strasser, Energy Environ. Sci., 2020, 13, 2480–2500 RSC.
- M. Mazzucato, G. Daniel, A. Mehmood, T. Kosmala, G. Granozzi, A. Kucernak and C. Durante, Appl. Catal., B, 2021, 291, 120068 CrossRef CAS.
- J. Sanabria-Chinchilla, K. Asazawa, T. Sakamoto, K. Yamada, H. Tanaka and P. Strasser, J. Am. Chem. Soc., 2011, 133, 5425–5431 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00596d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |