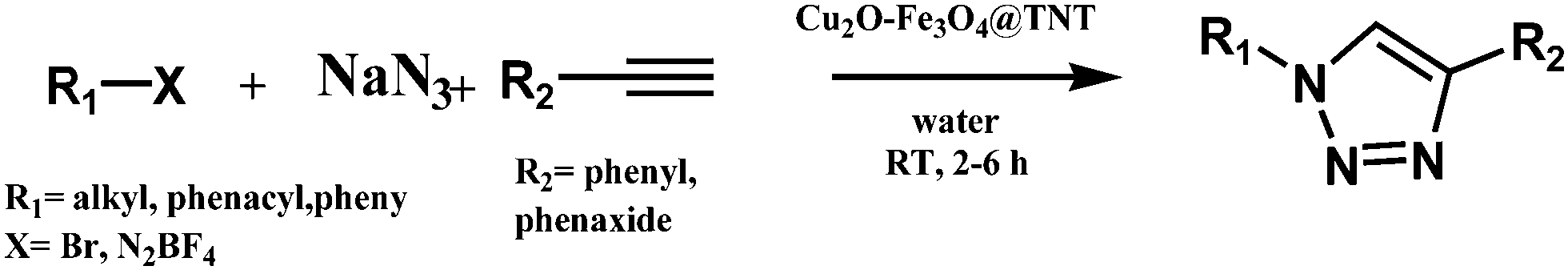DOI:
10.1039/D2MA00451H
(Paper)
Mater. Adv., 2022,
3, 7960-7965
Magnetically recoverable Cu2O–Fe3O4@TNT catalytic system for click chemistry in water: multi-component synthesis of 1,2,3-triazoles at room temperature†
Received
22nd April 2022
, Accepted 7th August 2022
First published on 9th August 2022
Abstract
The catalytic performance of a Cu2O–Fe3O4@TNT nanocatalyst derived from Fe(III) acetylacetonate, Cu(II) acetylacetonate and hydrogen titanate nanotubes (HTNTs) has been investigated for the first time for the click reaction in a water medium at room temperature. The nanocatalyst was characterized by performing XRD, HR-TEM, XPS, UV-DRS, H2-TPR, N2 adsorption-desorption, and ICP-OES analyses. The catalyst showed superior catalytic activity for the multicomponent synthesis of 1,2,3-triazoles in an aqueous medium. The nanocatalyst can be reused and recovered by using an external magnet.
Introduction
Click chemistry shortens the procedure for synthesizing 1,4-disubstituted 1,2,3-triazoles, providing quicker detection and optimization.1 Various triazoles have found clinical applications, such as in anti-bacterial agents2 and those displaying anti-HIV activity1a,3 as well as for β3-adrenergic receptor agonism.4 Also, triazoles are incorporated in dyes, herbicides, and fungicides.5 Cu-Catalyzed construction of triazole derivatives has been carried out by using a convenient scheme involving N3−-alkyne cycloaddition. Many homogeneous and heterogeneous catalysts have been developed for this (CuAAC) reaction. Included among the several heterogeneous catalytic systems that have been developed recently are Cu1-zeolite,6 a complex of NHC and Cu(I),7,8 Cu nanoparticles,9,10 a Cu2O catalyst,11,12 and magnetically separable catalysts.13–15
The ability to remove as much Cu as possible from a solution is a priority, particularly for biological applications.16 For pharmaceuticals, the permeability limits for metal content are <10 ppm.17 In this context, nano-catalysis has been developed as a modest and sustainable alternative to traditional catalysis because of the large surface-to-volume ratios of the nanoparticles, which increase their activity and selectivity, preserving the inherent characteristics of a heterogeneous catalyst.18
Inorganic supports having large surface areas can be used to capture metal nanoparticles and form especially active and recyclable catalysts as a result of the relatively high stability and dispersion of the particles. Various strategies involving centrifugation or filtration, often used for isolating catalysts in general, are unsustainable, noneconomic, outdated and particularly unsuitable for isolating nanosized catalysts. These strategies can be avoided when using magnetic nanoparticles (MNPs), which can be isolated by using an external magnet in a simple and efficient method to remove such nanocatalysts from reaction mixtures, and such retrieved MNPs have been shown to be ideal catalysts or supports. This alternative strategy has received excellent reviews.19
Hydrogen titanate nanotubes (HTNTs) have been intensively studied, attracting much attention in the past decade due to their unique textural and physicochemical properties, including versatile applications, particularly in catalysis and photocatalysis. Titanate NTs that are synthesized from TiO2 nanoparticles (particle size < 25 nm) using the hydrothermal method display high specific surface areas (≈200–400 m2 g−1), and are widely used because of their high amenability to being modified as desired. While the preparation of titanate NTs from TiO2 nanoparticles is well established, procedures for preparing them from commercial TiO2 powder are limited. The most important and attractive material for magnetic applications is nanostructured magnetite (Fe3O4), due to its biocompatibility, high magnetic susceptibility, chemical stability, innocuousness, high saturation magnetization, and inexpensiveness.22–25
As part of our ongoing interest in developing metal nanocatalysts for organic transformations in aqueous media,26–28 in our current work we developed a design and synthesis of the magnetically active nanocatalyst Cu2O–Fe3O4 supported on hydrogenated titanium NTs (Cu2O–Fe3O4@TNT) and effectively used it to catalyze the synthesis of 1,4-disubstituted 1,2,3-triazoles at room temperature in an eco-friendly medium involving the use of water as the solvent. The details of the preparation, characterization, and applications of the catalyst are described.
Experimental
All chemicals were received from commercial sources and used as received.
Preparation of catalysts
HTNT synthesis.
Hydrogen titanate NTs were prepared via a hydrothermal method. Typically, 3.5 g of TiO2 were dispersed in an aqueous solution of NaOH (10 M, 200 ml), and the resulting mixture was sonicated for 30 minutes and then transferred into a 300 ml Teflon-lined stainless-steel autoclave and kept there at 130 °C for 20 h (for NTs) and into a Teflon-lined autoclave at 180 °C per 20 h (for nanorods). The white precipitate in the autoclave was washed repeatedly with deionized water followed by dispersing the residues in 0.1 N HCl (100 ml) under stirring for 24 h.
Synthesis of Cu2O and Fe3O4 nanoparticles.
A mass of 3.5 g of iron(III) acetylacetonate and 1.4 g of Cu(III) acetylacetonate were together dissolved in 100 ml of oleylamine and then subjected to hydrothermal treatment in a 250 ml autoclave under an N2 atmosphere at 300 °C for 12 h. The resultant solution was poured into 500 ml of ethanol and the formed precipitate was collected and subjected to centrifugation after being redispersed in 100 ml of petroleum ether. Again the precipitate was dispersed in 100 ml of ethanol and100 ml of petroleum ether solution collected the sediment, and this process was repeated five times. The final obtained nanoparticles were allowed to dry at RT for one day.
Preparation of Cu2O–Fe3O4@TNT catalyst.
A mass of 0.8 g of HTNT was dispersed into 50 ml of petroleum ether and a mass of 0.2 g of Cu2O–Fe3O4 nanoparticles was dispersed into a separate 50 ml sample of petroleum ether, and each resulting dispersion was subjected to sonication for 30 minutes. The resulting latter solution was slowly added to the former under sonication, which was then continued for another 15 minutes. Catalyst was collected from the resultant mixture by using an external magnet and dried at 80 °C for 24 h. The amounts of Cu and Fe present in the collected catalyst were determined using ICP-OES.
Results and discussion
XRD studies
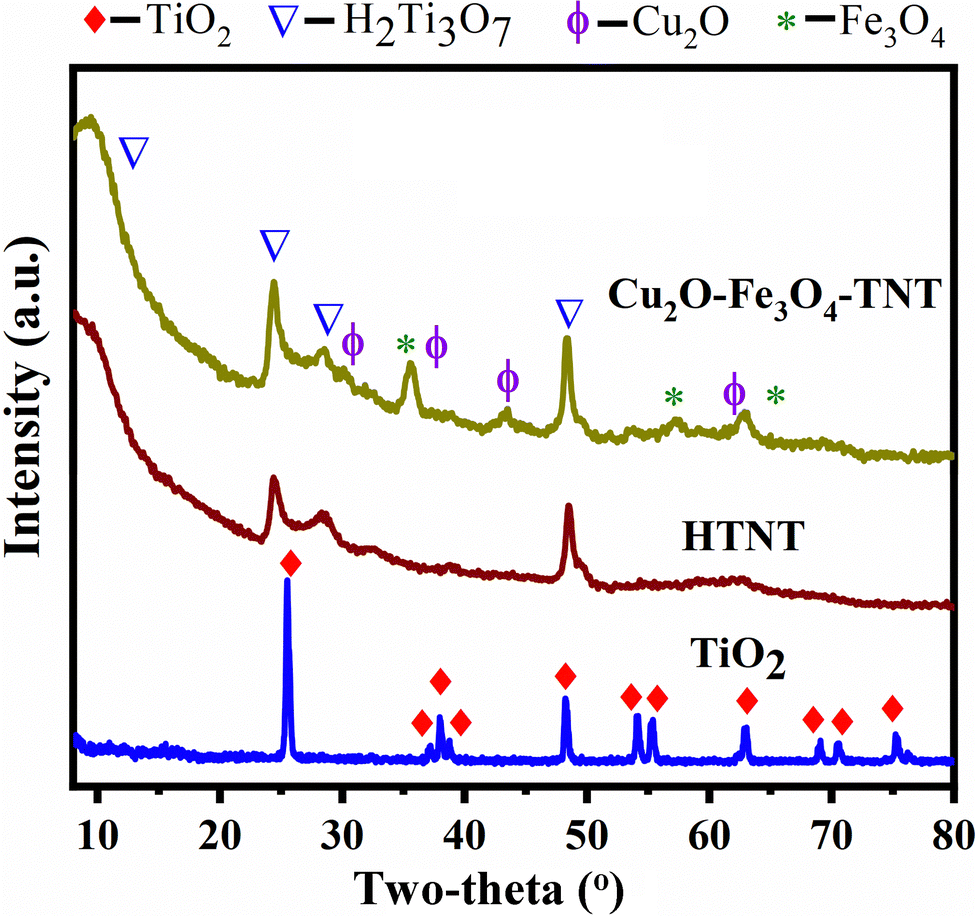
The XRD pattern of commercially purchased TiO2 powder shown in Fig. 1 indicated that it was in the anatase form in accordance with the diffraction peak positions at 2θ = 25.4, 37.9, 48.0, 54.0, 55.1, 62.7, 68.8, 70.4 and 75.1° and the corresponding (101), (004), (200), (105), (211), (204), (116), (200) and (215) crystal planes (JCPDS: 01-07-2486C). The XRD pattern of H2Ti3O7 was poorly resolved, confirming the transformation of highly crystalline anatase TiO2 into a nanoscopic material. Nevertheless, in this pattern, four diffraction peaks at 2θ = 11.2, 24.1, 28.5, and 48.5 and assigned to H2Ti3O7 were discerned, and the low-angle peaks indicated the layered structure of HTNT. The intensities at 2θ = 35.5, 57.5, and 62.8° originated from the magnetite Fe3O4 nanoparticles; note, in comparison, the standard XRD data for Fe3O4 (JCPDS No. 19-0629). The diffraction peaks at 29.6, 35.7, 43.52, and 62.84° derived from Cu2O (JCPDS No. 05-0667). The presence of Cu2O and Fe3O4 was confirmed using other analytical techniques, namely XPS, UV-DRS, and H2-TPR.
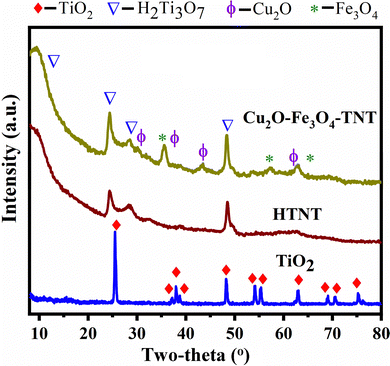 |
| | Fig. 1 XRD patterns of TiO2, TNT, and Cu2O–Fe3O4@TNT. | |
TPR studies for Cu2O–Fe3O4@HTNT
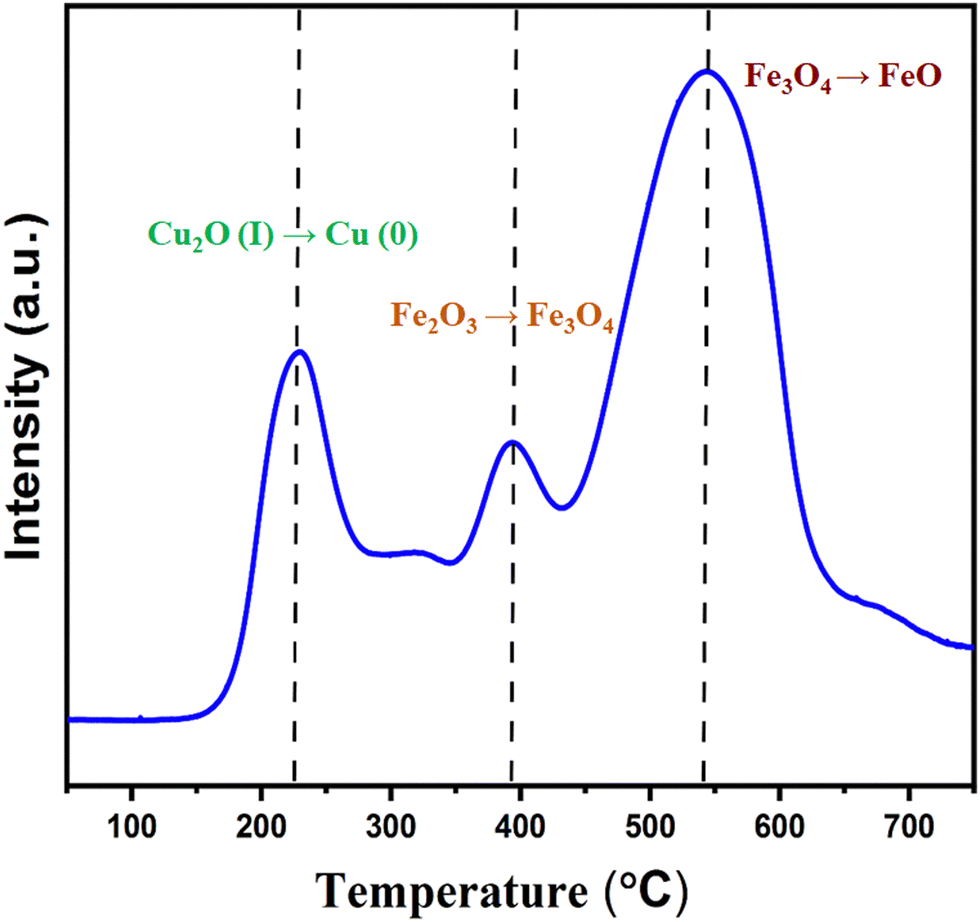
The TPR profile of Cu2O–Fe3O4@HTNT (Fig. 2) showed a reduction temperature region from 160 to 220 °C, attributed to the transformation Cu2O(I) → Cu (0). Note that the presence of Cu2O was also confirmed from XPS data.20 A small sharp peak was also observed at 400 °C, and corresponded to H2 consumption responsible for the transformation of Fe2O3 to Fe3O4, (maghemite → magnetite). In addition, a broad peak from 450 to 650 °C was observed, and was attributed to the conversion of Fe3O4 to FeO (magnetite → wustite).21
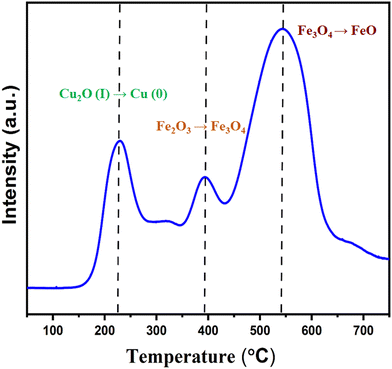 |
| | Fig. 2 H2-TPR of Cu2O–Fe3O4@TNT. | |
XPS studies
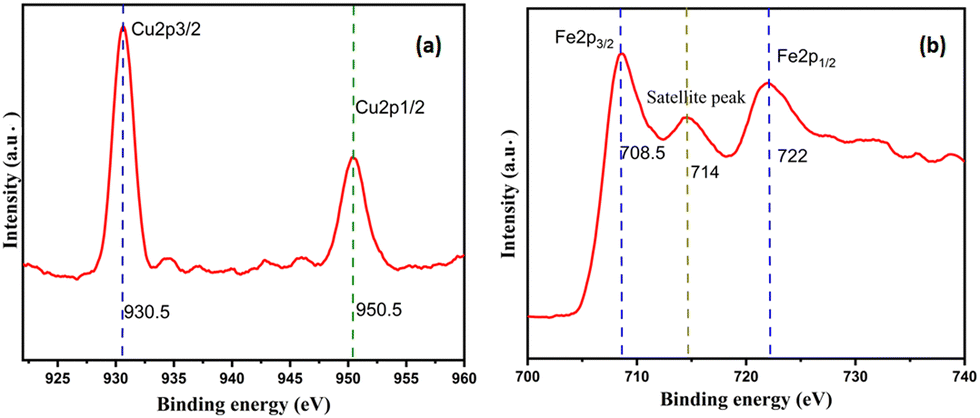
As shown in Fig. 3(b), the high-resolution XPS data for Cu2O–Fe3O4@HTNT showed two prominent peaks and one satellite peak in the Fe 2p region, specifically one peak at 722 eV and corresponding to Fe 2p1/2, the other peak at 708.5 eV and corresponding to Fe 2p3/2, and the satellite peak at 714 eV. The two spin–orbit couplings at Fe 2p1/2 and Fe 2p3/2 were attributed to Fe2+ and Fe3+. These results for Fe3O4 agreed with the literature.32 As shown in Fig. 3(a), the Cu 2p region showed two prominent peaks, at 930.5 eV and 950.5 eV and corresponding to Cu 2p3/2 and Cu 2p1/2, respectively, and indicating the presence of Cu2O.
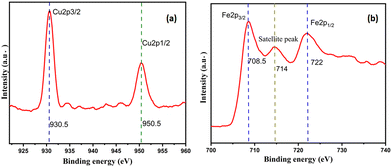 |
| | Fig. 3 XPS patterns of Cu2O–Fe3O4@HTNT in the (a) Cu and (b) Fe regions. | |
UV-visible studies
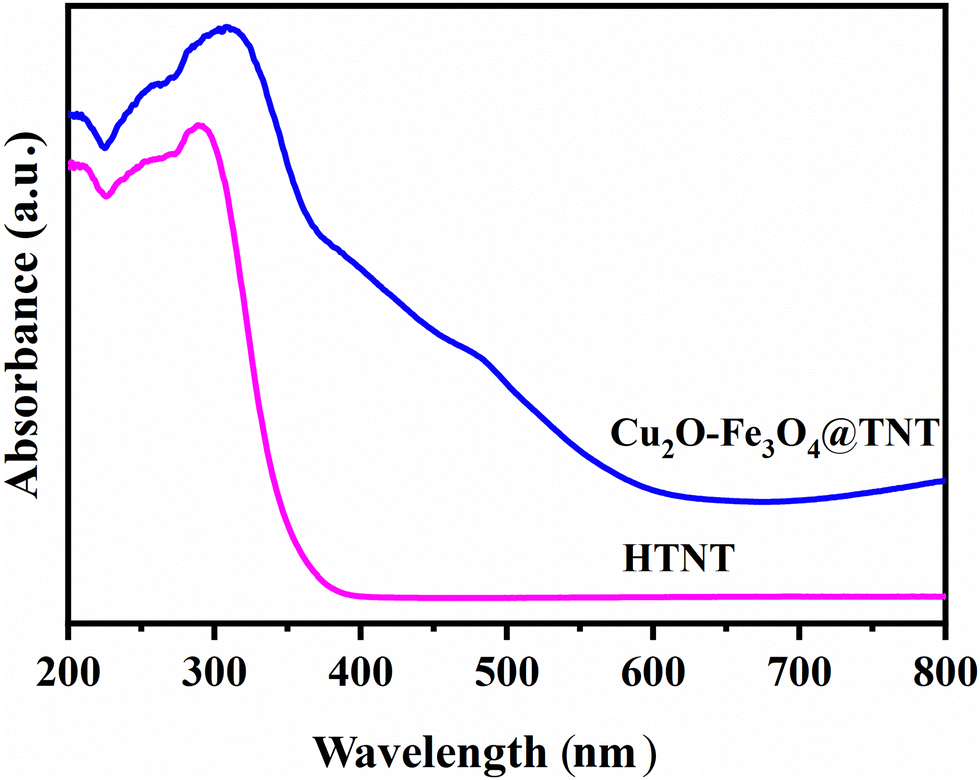
Optical properties of the catalysts were investigated by performing UV-Vis absorption studies. As shown in Fig. 4, the Cu2O–Fe3O4@TNT catalyst showed a broad absorption with the characteristic peak of Cu2O at a wavelength of 490 nm and the free excitation peak at 421 nm.29–31
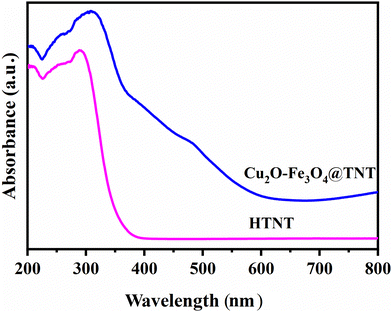 |
| | Fig. 4 UV-DRS spectrum of HTNT and Cu2O–Fe3O4@TNT. | |
TEM analysis
TEM images of our samples are shown in Fig. 5. Inspection of these images indicated that the titanate NTs produced from titanium powder retained their tubular structure with an internal diameter of 4.834 nm and interlayer distance of 0.35 nm—and that when Cu2O and Fe3O4 were introduced onto the HTNTs, the surfaces of the NTs became entirely shielded with Cu2O, Fe3O4 nanoparticles. Here, an average particle size of 5.5 nm was calculated from the latter TEM image.
 |
| | Fig. 5 TEM images of (a) HTNT and (b) the Cu2O–Fe3O4@TNT catalyst. | |
Activity test
The newly amassed catalyst (Cu2O–Fe3O4@TNT) was then tested for its ability to promote click addition through the reaction of terminal alkynes with in situ-generated organic azide. This one-pot procedure offered the benefit of not needing to pre-synthesize the azido partner, in contrast to the case for most nanoparticle-catalyzed click reactions. The reaction was carried out at room temperature, under air, using 50 mg of the catalyst, 7 mmol of phenyl acetylene, 7.5 mmol of NaN3 and 5 mmol of alkyl or benzyl halide in 5 ml of water. After completion of the reaction, ethyl acetate solvent was used to dissolve the product formed and the catalyst was recovered using an external magnet; the products were collected by concentrating the residual solution.
The results of various test reactions are listed in Table 1. The reaction neither occurred without catalyst nor with HTNT. The reaction progressed with Cu2O–Fe3O4@TNT catalyst, indicating the need of having active metal to perform the reaction (Scheme 1).
Table 1 Reaction optimization conditions
| Entry |
Catalyst |
Amount of catalyst (mg) |
Time (h) |
Yield (%) |
| Benzyl bromide (5 mmol), NaN3 (7.5 mmol), alkyne (7 mmol), and Cu2O–Fe3O4@TNT (50 mg), solvent = water (5 ml), stirred at RT for 4 h Reaction at 60 °C. Reaction at 100 °C. Neat at RT. |
| 1 |
HTNT |
20 |
12 |
0 |
| 2 |
No catalyst |
— |
12 |
0 |
| 3 |
Cu2O–Fe3O4@TNT |
20 |
4 |
54 |
| 4 |
Cu2O–Fe3O4@TNT |
30 |
4 |
82 |
| 5 |
Cu2O–Fe3O4@TNT |
50 |
4 |
98 |
| 6 |
Cu2O–Fe3O4@TNT |
70 |
4 |
86 |
| 7a |
Cu2O–Fe3O4@TNT |
50 |
4 |
93 |
| 8b |
Cu2O–Fe3O4@TNT |
50 |
4 |
98 |
| 9c |
Cu2O–Fe3O4@TNT |
50 |
4 |
67 |
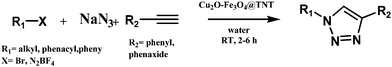 |
| | Scheme 1 Click reaction over Cu2O–Fe3O4@TNT catalyst. | |
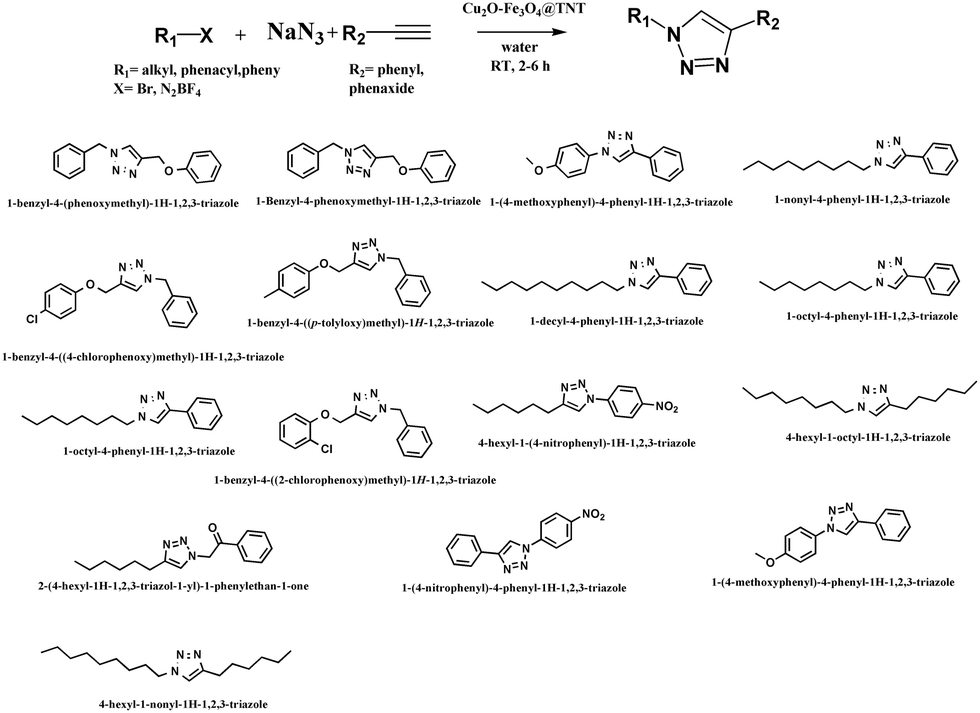
The catalytic system proved efficient on a variety of substrates (Table 2) and was active on various terminal alkynes such as phenyl acetylene derivatives. The structures of all of the isolated compounds (yields ranging from 95 to 99%) were confirmed from 1H-NMR data.
Table 2 Scope of the reaction
The most important advantage of carrying out the CuAAC reaction in an aqueous solvent was the easy isolation of analytically clean products, that is by removing the catalyst with an external magnet. The solid resulting from the reaction was then dissolved in an ethyl acetate solution; concentrating the residual solution gave clean products without the need for any tedious column isolation. The magnetically separated catalyst was washed with ethyl acetate twice and dried at 100 °C in a hot air oven for 12 h and used for the next run.
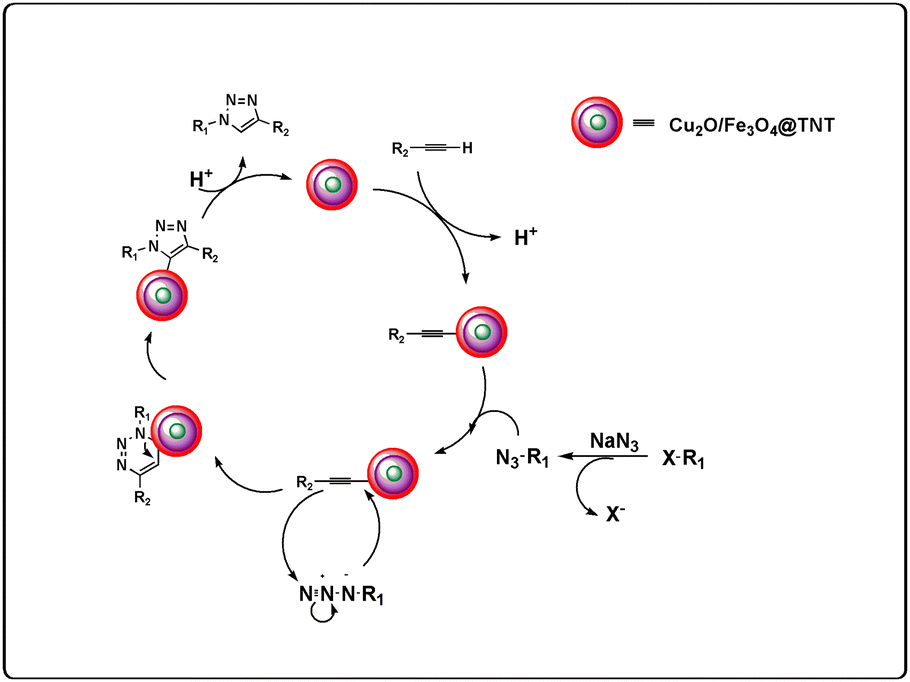
A mechanism has been proposed based on our experimental results and previous reports,33 and the details of this mechanism are shown in Fig. 6.
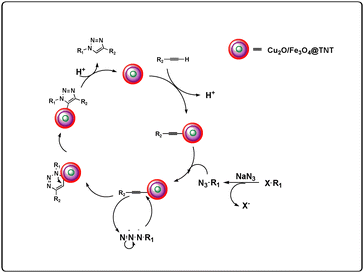 |
| | Fig. 6 Proposed mechanism. | |
Recyclability test
The catalyst was re-used four times and still led to a decent product yield, as depicted in Fig. 7. At the end of each cycle, the catalyst was collected by using an external magnet, washed with ethyl acetate thrice, and dried in a hot air oven at 100 °C for 12 h to be used for the next cycle.
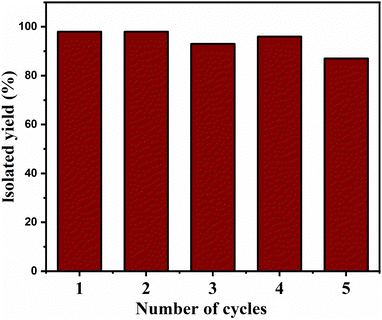 |
| | Fig. 7 Recyclability of the Cu2O–Fe3O4@TNT catalyst. | |
Conclusions
A novel magnetically recoverable Cu2O–Fe3O4@TNT catalyst was synthesized. The performance of the catalyst towards the click reaction for the synthesis of 1,4-disubstituted 1,2,3-triazoles via in situ-liberated organic azide with alkynes was established. The very high activity of catalyst was concluded to be due to the particles of Cu in the Cu1+ state. The catalyst can be used for the synthesis of a wide variety of substrates.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
V. R. K. Velpula and T. Ketike are thankful to Council of Scientific and Industrial Research (CSIR) and UGC, New Delhi, India, for research fellowships.
Notes and references
-
(a) P. W. Baures, Org. Lett., 1999, 1, 249 CrossRef CAS PubMed;
(b) A. K. Jordão, V. F. Ferreira, T. M. L. Souza, G. G. D. S. Faria, V. Machado, J. L. Abrantes, M. C. B. V. de Souza and A. C. Cunha, Bioorg. Med. Chem., 2011, 19, 1860 CrossRef PubMed;
(c) L. S. Kallander, Q. Lu, W. Chen, T. Tomaszek, G. Yang, D. Tew, T. D. Meek, G. A. Hofmann, C. K. S. Pritchard, W. W. Smith, C. A. Janson, M. D. Ryan, G. F. Zhang, K. O. Johanson, R. B. Kirkpatrick, T. F. Ho, P. W. Fisher, M. R. Mattern, R. K. Johnson, M. J. Hansbury, J. D. Winkler, K. W. Ward, D. F. Veber and S. K. Thompson, J. Med. Chem., 2005, 48, 5644 CrossRef CAS PubMed;
(d) S. G. Agalave, S. R. Maujan and D. V. S. Pore, Chem. – Asian J., 2011, 6, 2696 CrossRef CAS PubMed;
(e) A. Lauria, R. Delisi, F. Mingoia, A. Terenzi, A. Martorana, G. Barone and A. M. Almerico, Eur. J. Org. Chem., 2014, 3289–3306 CrossRef CAS;
(f) R. Aggarwal and G. Sumran, Eur. J. Med. Chem., 2020, 205, 112652 CrossRef CAS PubMed;
(g) P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905–4979 CrossRef CAS PubMed;
(h) D. Dheer, V. Singh and R. Shankar, Bioorg. Chem., 2017, 71, 30–54 CrossRef CAS PubMed;
(i) G. Mohammad and J. Neda, ACS Sustainable Chem. Eng., 2014, 2(12), 2658–2665 CrossRef;
(j) G. Mohammad, O. Erfan and M. S. Jose, ChemistrySelect, 2019, 4, 3151–3160 CrossRef.
- M. J. Genin, D. K. Hutchinson, D. A. Allwine, J. B. Hester, D. E. Emmert, S. A. Garmon, C. W. Ford, G. E. Zurenko, J. C. Hamel, R. D. Schaadt, D. Stapert, B. H. Yagi, J. M. Friis, E. M. Shobe and W. J. Adams, J. Med. Chem., 2000, 43, 953 CrossRef CAS PubMed.
- R. Alvarez, S. Velazquez, A. San-Felix, S. Aquaro, E. De Clercq, C. F. Perno, A. Karlsson, J. Balzarini and M. J. Camarasa, J. Med. Chem., 1994, 37, 4185 CrossRef CAS PubMed.
- L. L. Brockunier, E. R. Parmee, H. O. Ok, M. R. Candelore, M. A. Cascieri, L. F. Colwell, L. Deng, W. P. Feeney, M. J. Forrest, G. J. Hom, D. E. MacIntyre, L. Tota, M. J. Wyvratt, M. H. Fisher and A. E. Weber, Bioorg. Med. Chem. Lett., 2000, 10, 2111 CrossRef CAS PubMed.
-
H. Wamhoff, Comprehensive Heterocyclic Chemistry, Pergamon, Oxford, 1984, vol. 5, p. 669 Search PubMed.
- V. Beneteau, A. Olmos, T. Boningari, J. Sommer and P. Pale, Tetrahedron Lett., 2010, 51, 3673–3677 CrossRef CAS.
- M. A. Topchiy, A. A. Ageshina, P. S. Gribanov, S. M. Masoud, T. R. Akmalov, S. E. Nefedov, S. N. Osipov, T. R. Akmalov and A. F. Asachenko, Eur. J. Org. Chem., 2019, 1016–1020 CrossRef CAS.
- A. Mishra, P. Rai, M. Srivastava, B. P. Tripathi, S. Yadav, J. Singh and J. Singh, Catal. Lett., 2017, 147, 2600–2611 CrossRef CAS.
- M. Nasrollahzadeh, B. Jaleh, P. Fakhri, A. Zahraei and E. Ghadery, RSC Adv., 2015, 5, 2785 RSC.
- W. B. Yu, L. X. Jiang, C. Shen, W. M. Xu and P. F. Zhang, Catal. Commun., 2016, 79, 11–16 CrossRef CAS.
- Y. H. Tsai, K. Chanda, Y. T. Chu, C. Y. Chiu and M. H. Huang, Nanoscale, 2014, 6, 8704–8709 RSC.
- R. P. R. Bhoomireddy, L. G. B. Narla and V. G. R. Peddiahgari, Appl. Organomet. Chem., 2019, 33, e4752 CrossRef.
- A. Salamatmanesh, M. K. Miraki, E. Yazdani and A. Heydari, Catal. Lett., 2018, 148, 3257–3268 CrossRef CAS.
- Y. Jain, M. Kumari, R. P. Singh, D. Kumar and R. Gupta, Catal. Lett., 2020, 150, 1142–1154 CrossRef CAS.
- R. Bonyasi, M. Gholinejad, F. Saadati and C. Najera, New J. Chem., 2018, 42, 3078–3086 RSC.
-
(a) R. Wang, X. T. Wang, L. Wu, M. A. Mateescu and J. Toxicol, Environ. Health Part A, 1999, 57, 507 CrossRef CAS PubMed;
(b) M. K. Rauf, I. Din, A. Badshah, M. Gielen, M. Ebihara, D. de Vos and S. Ahmed, J. Inorg. Biochem., 2009, 103, 1135 CrossRef CAS PubMed;
(c) M. L. Teyssot, A. S. Jarrousse, A. Chevry, A. De Haze, C. Beaudoin, M. Manin, S. P. Nolan, S. Díez-González, L. Morel and A. Gautier, Chem. – Eur. J., 2009, 15, 314 CrossRef CAS PubMed.
- M. Crevoisier, E. L. Barle, A. Flueckiger, D. G. Dolan, M. Ovais and A. Walsh, Pharm. Dev. Technol., 2016, 1, 52–56 Search PubMed.
-
(a)
Nanoparticles and Catalysis, ed. D. Astruc, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed;
(b)
Selective Nanocatalysts and Nanoscience: Concepts for Heterogeneous and Homogeneous Catalysis, ed. A. Zecchina, S. Bordiga and E. Groppo, Wiley-VCH, Weinheim, Germany, 2011 Search PubMed;
(c)
Nanomaterials in Catalysis, ed. P. Serp and K. Philippot, Wiley-VCH, Weinheim, Germany, 2013 CrossRef;
(d) Y. Xia, H. Yang and C. T. Campbell, Acc. Chem. Res., 2013, 46, 1671–1910 CrossRef CAS PubMed.
-
(a) A.-H. Lu, E. L. Salabas and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 1222 CrossRef CAS PubMed;
(b) S. Shylesh, V. Schünemann and W. R. Thiel, Angew. Chem., Int. Ed., 2010, 49, 3428 CrossRef CAS PubMed;
(c) Y. Zhu, L. P. Stubbs, F. Ho, R. Liu, C. P. Ship, J. A. Maguire and N. S. Hosmane, ChemCatChem, 2010, 2, 365 CrossRef CAS;
(d) V. Polshettiwar, R. Luque, A. Fihri, H. Zhu, M. Bouhrara and J. M. Basset, Chem. Rev., 2011, 111, 3036 CrossRef CAS PubMed;
(e) L. M. Rossi, M. A. S. Garcia and L. L. R. J. Braz. Vono, Chem. Soc., 2012, 23, 1959 CAS;
(f) R. B. N. Baig and R. S. Varma, Chem. Commun., 2013, 49, 752 RSC;
(g) B. Karimi, F. Mansouri and H. M. Mirzaei, Chem. Rev., 2014, 114, 6949–6985 CrossRef PubMed.
- Y. Zhai, Y. Ji, G. Wang, Y. Zhu, H. Liu, Z. Zhong and F. Su, RSC Adv., 2015, 5, 73011 RSC.
- S. S. Kim, S. M. Lee and S. C. Hong, J. Ind. Eng. Chem., 2012, 2, 860–864 CrossRef.
- Y. M. Huh, Y. Jun, H. T. Song, S. Kim, J. Choi, J. H. Lee, S. Yoon, K. S. Kim, J. S. Shin, J. S. Suh and J. Cheon, J. Am. Chem. Soc., 2005, 127, 12387 CrossRef CAS PubMed.
- J. M. Perez, L. Josephson, T. O’Loughlin, D. Hogemann and R. Weissleder, Nat. Biotechnol., 2002, 20, 816 CrossRef CAS PubMed.
- S. Sun and H. Zeng, J. Am. Chem. Soc., 2002, 124, 8204 CrossRef CAS PubMed.
- Z. Xu, C. Shen, Y. Hou, H. Gao and S. Sun, Chem. Mater., 2009, 21, 1778–1780 CrossRef CAS.
- S. Ganji, S. Mutyala, C. K. P. Neeli, K. S. Rama Rao and D. R. Burri, RSC Adv., 2013, 3, 11533 RSC.
- S. Ganji, S. S. Enumula, R. K. Marella, K. S. Rama Rao and D. R. Burri, Catal. Sci. Technol., 2014, 4, 1813 RSC.
- S. Ganji, P. Bukya, Z. W. Liu, K. S. Rama Rao and D. R. Burri, New J. Chem., 2019, 43, 11871 RSC.
- Y. Pan, S. Deng, L. Polavarapu, N. Gao, P. Yuan, C. H. Sow and Q. Xu, Langmuir, 2012, 28, 12304–12310 CrossRef CAS PubMed.
- J. Kou, A. Saha, C. B. Stamper and R. S. Varma, Chem. Commun., 2012, 48, 5862–5864 RSC.
- L. Liu, S. Lin, J. Hu, Y. Liang and W. Cui, Appl. Surf. Sci., 2015, 330, 94–103 CrossRef CAS.
- C. L. C. Carvalho, A. T. B. Silva, R. A. S. Luz, G. M. B. Castro, C. L. Lima, V. R. Mastelaro, R. R. D. Silva, O. N. Oliveira and W. C. Silva, ACS Appl. Nano Mater., 2018, 1(8), 4283–4293 CrossRef.
-
(a) H. B. E. Ayouchia, L. Bahsis, H. Anane, L. R. Domingo and S.-E. Stiriba, RSC Adv., 2018, 8, 7670–7678 RSC;
(b) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS;
(c) C. W. Tornøe, C. Christensen and M. Melda, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *e,
David Raju
Burri
ab and
Krishna Mohan
Surapaneni
c
*e,
David Raju
Burri
ab and
Krishna Mohan
Surapaneni
c