
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImplementation of heteroatom-doped nanomaterial/core–shell nanostructure based electrocatalysts for fuel cells and metal-ion/air/sulfur batteries
Saravanan
Nagappan
*a,
Malarkodi
Duraivel
b,
NaHyun
Park
a,
Kandasamy
Prabakar
 b and
Kang Hyun
Park
*a
b and
Kang Hyun
Park
*a
aDepartment of Chemistry, Chemistry Institute for Functional Materials, Pusan National University, Busan, 46241, Republic of Korea. E-mail: chemistry@pusan.ac.kr; saravananagappan@gmail.com
bDepartment of Electrical Engineering, Pusan National University, 2 Busandaehak-ro 63beon-gil, Geumjeong-Gu, Busan 46241, Republic of Korea
First published on 30th June 2022
Abstract
Core–shell nanostructures (CSNs) have been extensively used in energy conversion and storage (ECS) applications. CSNs feature abundant active sites and large surface areas. In addition, their size, shape, and morphology can be tailored to be uniform and controlled. The catalytic activity and performance of CSNs can be significantly improved by heteroatom doping to CSNs, which makes them even more advantageous for ECS applications. In addition to various noble metals, transition metals have been used in the cores and shells of nanostructures featuring heteroatoms. Such materials are inexpensive and exhibit excellent catalytic activity and stability. Likewise, heteroatom-doped nanomaterial and core–shell nanostructure (HCSN) based electrocatalysts serve as bifunctional electrocatalysts for the oxygen reduction reaction (ORR) and hydrogen evolution reaction (HER) as well as the ORR and oxygen evolution reaction (OER). These bifunctional electrodes are highly suitable for fuel cell and battery applications. In this review article, we exclusively focus on the important role of HCSNs in various types of fuel cells and batteries. In particular, we discuss the important roles and applications of HCSNs in proton exchange membranes (PEMs), anion exchange membranes (AEMs), direct methanol, microbial, and other types of fuel cells as well as metal ion batteries such as lithium (Li), sodium (Na), and potassium (K)-ion batteries. The applications of HCSNs in Li–sulfur (S) batteries and metal–air batteries, such as Li, zinc (Zn), and Na–air batteries, have also been addressed. Finally, we have discussed the technical advancements related to the applications of HCSNs in fuel cells and batteries along with their disadvantages, scope, and future prospects. This review will benefit researchers working in the ECS field as well as those investigating various other applications, such as drug delivery, sensors, and adsorption.
 Saravanan Nagappan | Saravanan Nagappan is working as a Post-Doc Fellow under Professor Kang-Hyun Park in the Department of Chemistry and Chemistry Institute for Functional Materials, Pusan National University, Korea. His research interests include the synthesis and fabrication of various organic–inorganic hybrid nanomaterials, MXenes, graphene, carbon, and nanoclay based materials for coatings, catalysis, environmental remediation, biomedicine, electrocatalysis, and energy storage and conversion applications. |
 Malarkodi Duraivel | Malarkodi Duraivel will obtain her PhD, in Electrical and Electronics Engineering (August 2022) under the supervision of Professor Kandasamy Prabakar at Pusan National University, Korea. Her current research focuses on the development of low-cost and efficient electrocatalysts for energy conversion applications including water splitting and oxygen reduction reactions. |
 NaHyun Park | NaHyun Park is currently pursuing a Master's in the Department of Chemistry at Pusan National University, Korea. Her research interests focus on the development of nanomaterials for battery applications. |
 Kandasamy Prabakar | Kandasamy Prabakar is a Professor at the Department of Electrical Engineering, Pusan National University, Korea. His current research is focussed on the development of various nanohybrid electrodes for energy conversion and storage applications. In particular, he is working on the development of novel electrodes for hybrid capacitors, photo-electrocatalytic hydrogen and oxygen evolution reactions for total water-splitting, and reduction reactions for metal–air batteries. |
 Kang Hyun Park | Kang Hyun Park is a Professor at the Department of Chemistry and Chemistry Institute for Functional Materials, Pusan National University, Korea. His research interests include the development of new transition metal-nanoparticle catalyzed reactions. Currently, his research is focused on the development of multifunctional nanohybrid materials for electrocatalysis, especially on the synthesis of multifunctional nanohybrid electrodes for fuel cell and battery applications. |
1. Introduction
Research on core–shell nanostructures (CSNs) has attracted considerable interest due to the continuous demand for electrocatalysts with improved performance and stability that are suitable for energy conversion and storage (ECS) applications.1–5 Various transition metals, metal oxides, and noble metals are used to construct the cores and shells of CSNs which are used in drug delivery, energy and environmental applications, and catalysis, among others.6–11 Numerous methods have been employed to synthesize CSNs based on top-down and bottom-up approaches.12 Micro-fabrication methods or various electrical, mechanical, and optical tools (e.g., lithographic, laser treatment, mechanical mixing, and polishing techniques) are used to develop CSNs via top-down approaches.12 In contrast, bottom-up approaches involve the alteration of the chemical properties using self-assembly and seed-growth techniques, sol–gel processes involving the Stöber method, film deposition and growth, colloidal aggregation, and chemical vapor deposition (CVD), among others.12 These methods have unique advantages and disadvantages based on surface finishing and metal compositions. Compared to top-down approaches, bottom-up approaches are simpler, cost-effective, and considerably easier to reproduce without using expensive instrumental set-ups. They also ensure control of the nanoparticle size, structure, and morphology. Top-down and bottom-up approaches can be combined by first constructing a uniform core via a top-down approach, followed by a thin shell layer by a bottom-up approach to afford significantly improved and controlled nanoarchitectures, which are extremely useful for electrocatalytic applications.12Platinum (Pt), palladium (Pd), iridium (Ir), ruthenium (Ru), rhodium (Rh), gold (Au), and other expensive noble metals were widely used in ECS owing to their superior electrochemical properties and stability compared to transition metals.7,13,14 The high cost and insufficient availability of Pt and other noble metals limit their use in practical and commercial applications, such as fuel cells and batteries. Several attempts have been made to minimize noble metal consumption by alloying them with inexpensive and earth-abundant transition metals (M = cobalt (Co), nickel (Ni), manganese (Mn), iron (Fe), aluminum (Al), magnesium (Mg), and tin (Sn), among others).13–20 Among various structurally and morphologically distinct nanomaterials, CSNs have been widely used in ECS applications due to their advanced properties and precisely controlled morphologies and chemical compositions (core or shell). In addition, their core and shell structures can be conveniently controlled using single or multiple layers.4,6 Long et al. studied the effective role of Pt and Pt–Pd core–shell nanoparticles in electrocatalysts.16 Pt–Pd core–shell nanoparticles were synthesized by a modified polyol method using silver nitrate (AgNO3) as a support in the presence of ethylene glycol.16 Jian et al. developed efficient CSNs (CNT@RuO2) via the sol–gel method using carbon nanotubes (CNTs) and ruthenium oxide (RuO2).21 These CSNs served as bifunctional catalysts, with superior performances in both the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER), and also delivered appreciable results in rechargeable Li–air batteries. The CNT core (15 nm in diameter) and RuO2 shell (thickness of approximately 3.5 nm) demonstrated outstanding catalytic activity and usability in Li–air batteries.21 The Li–air battery cell fabricated using the CNT@RuO2 nanocomposite exhibits a high round-trip efficiency (ca. 79%) at a current of 100 mA gtotal−1 with superior cycling stability and high rate performance.21
Heteroatom doped nanomaterials and core–shell nanostructures (HCSNs) are highly decorated with heteroatoms on the surface or in the chemical structures, and the presence of low-cost and earth-abundant metals either in the core or in the shell or noble metal deposits (a few layers thick) on the shells can enhance their electrocatalytic properties, stability, and tolerance against severe environmental conditions.22,23 Single heteroatoms, such as N, S, P, and boron (B), or combinations of two or more such heteroatoms are suitable for various applications due to the enhanced electrochemical activity which results from the formation of oxygen vacancies and defects in metal-based catalysts.24,25 Furthermore, doping heteroatoms with earth-abundant transition metals or conjugated polymers leads to improved properties and stability concerning different electrochemical applications.26,27 Heteroatom doping on CSNs has many unique advantages. It increases the number of active sites on the CSN surface, enhances the electrochemical activity and conductivity, facilitates charge transfer from the CSNs and heteroatoms, and improves the stability of the catalysts.28 The particle size, shape, morphology, and chemical compositions of the core and shell are amenable to precise control, which facilitates the development of CSNs and HCSNs tailored for various ECS applications. Despite the difficulties related to dissolution under acidic conditions, these nanostructures offer the major advantages of high surface area and pore volume, more active sites and defects, and reduced use of noble metals. The disadvantages can be overcome through the appropriate selection of materials to maximize production and minimize the cost of the electrocatalysts. Typically, heteroatom doping is performed either using a direct in situ doping technique, such as CVD, bottom-up synthesis, and ball-milling, or post-synthetic treatments, such as wet chemical methods and thermal annealing.28 The valence electron numbers and sizes of N and B are almost identical to those of carbon (C). Therefore, it is easier to dope graphene-based C materials with N and B than with other heteroatoms.28 Various N-, B-, S-, and P-based compounds have been used for heteroatom doping on CSNs using different in situ doping techniques or post-synthetic treatments.12,28,29 HCSNs feature highly oriented structures with large surface areas, porosity, and large numbers of active sites as well as defects. Recent attempts have focused on studying the role and theoretical background of multi-heteroatoms in electrocatalysis, particularly for the ORR owing to the increasing demand for highly stable ORR electrodes for numerous end-use applications.23,24,30 In addition, continuous attention has been given to the use of HCSNs for various fuel cells and battery applications.31–34
The various aspects of CSNs related to ECS applications have been discussed in a few reviews.4,35,36 Feng et al. briefly reviewed the important aspects and applications of CSNs with concern for storage systems including various types of supercapacitors and Li-based batteries, energy conversion systems, such as solar cells and fuel cells, and photocatalytic hydrogen production.35 CSNs synthesized using transition metals and encapsulated in C materials were discussed thoroughly and the key advantages of CSNs for electrochemical applications were summarized.36 The use of C-based CSNs is prevalent in various ECS applications due to their enhanced conductivity and catalytic activity compared to other materials.4 Combinations of the various properties of the mentioned earth-abundant transition metals and C structures with those of the heteroatoms significantly improve the catalytic properties of CSNs.
Bai et al. synthesized N and S co-doped mesoporous C, followed by the deposition of manganese oxide (MnO2) nanoflakes via a solvothermal reaction to obtain an excellent supercapacitor behavior of CSNs with a maximum capacitance of 310 F g−1, energy and power densities of 44 W h kg−1 and 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 W h kg−1, respectively, and a 90% retention of capacitance after 5000 cycles.37 Zhao et al. developed bifunctional heteroatom doped core–shell C fibers by first preparing composite nanofibers by an electrospinning technique using a mixture of polyacrylonitrile (PAN, Mw = 230000), polyvinylpyrrolidone (PVP, Mw = 1300000), and cobalt acetylacetonate (Co(acac)2), dissolved in N,N-dimethylformamide (DMF).38 The composite nanofibers were reacted with cobalt nitrate hexahydrate (Co(NO3)2·6H2O) and zinc nitrate hexahydrate (Zn(NO3)2·6H2O) in the presence of 2-methylimidazole (2-MeIM) via immersion in methanol to form metal–organic framework (MOF) based zeolitic imidazolate frameworks (ZIFs). The PAN@ZIFs were subjected to pyrolysis at 800 °C (at a heating rate of 2 °C min−1) for 2 h in an N2 atmosphere to obtain heteroatom doped core–shell C fibers. The as-prepared catalyst delivered excellent bifunctional catalytic activity for both the ORR and OER. The catalyst also functions as a primary rechargeable Zn–air battery with power and energy densities of 140.1 mW cm−2 and 878.9 W h kgzn−1, respectively, and excellent long-term cycling stability (constant voltage gap after 150 h).38
000 W h kg−1, respectively, and a 90% retention of capacitance after 5000 cycles.37 Zhao et al. developed bifunctional heteroatom doped core–shell C fibers by first preparing composite nanofibers by an electrospinning technique using a mixture of polyacrylonitrile (PAN, Mw = 230000), polyvinylpyrrolidone (PVP, Mw = 1300000), and cobalt acetylacetonate (Co(acac)2), dissolved in N,N-dimethylformamide (DMF).38 The composite nanofibers were reacted with cobalt nitrate hexahydrate (Co(NO3)2·6H2O) and zinc nitrate hexahydrate (Zn(NO3)2·6H2O) in the presence of 2-methylimidazole (2-MeIM) via immersion in methanol to form metal–organic framework (MOF) based zeolitic imidazolate frameworks (ZIFs). The PAN@ZIFs were subjected to pyrolysis at 800 °C (at a heating rate of 2 °C min−1) for 2 h in an N2 atmosphere to obtain heteroatom doped core–shell C fibers. The as-prepared catalyst delivered excellent bifunctional catalytic activity for both the ORR and OER. The catalyst also functions as a primary rechargeable Zn–air battery with power and energy densities of 140.1 mW cm−2 and 878.9 W h kgzn−1, respectively, and excellent long-term cycling stability (constant voltage gap after 150 h).38
Fig. 1 clearly illustrates the continuous growth of CSNs, heteroatom doping, and the utility of CSNs in fuel cells and battery applications. Recently, there has been an increase in the demand for CSNs for ECS applications due to their advanced properties and highly controlled size and morphology. Heteroatom doping of CSNs has been utilized for developing improved electrocatalysts for wider applications. We briefly reviewed the various aspects of synthesis and application of HCSN based electrocatalysts for the ORR,1 whereas a few other review articles have focused on the role of heteroatom-doping of CSNs in ECS-related applications. However, none of them has discussed the relationship between the structural properties of HCSNs and their utility in various types of fuel cells and batteries. Their pertinence to future ECS demands has also not been commented on. Therefore, in this review, we exclusively cover the recent advances in HCSNs and their emerging applications in different fuel cells and batteries. A special emphasis has been placed on the role of HCSNs in ECS applications and suggestions have been provided for improving the properties and mitigating the disadvantages of HCSNs. This review is expected to provide constructive guidance to those working in related fields. Fig. 2 also illustrates the effective approaches of using heteroatom doped nanomaterials and core–shell nanostructures for various fuel cells and batteries. These approaches summarize that HCSNs play an effective role in various electrochemical ECS applications.
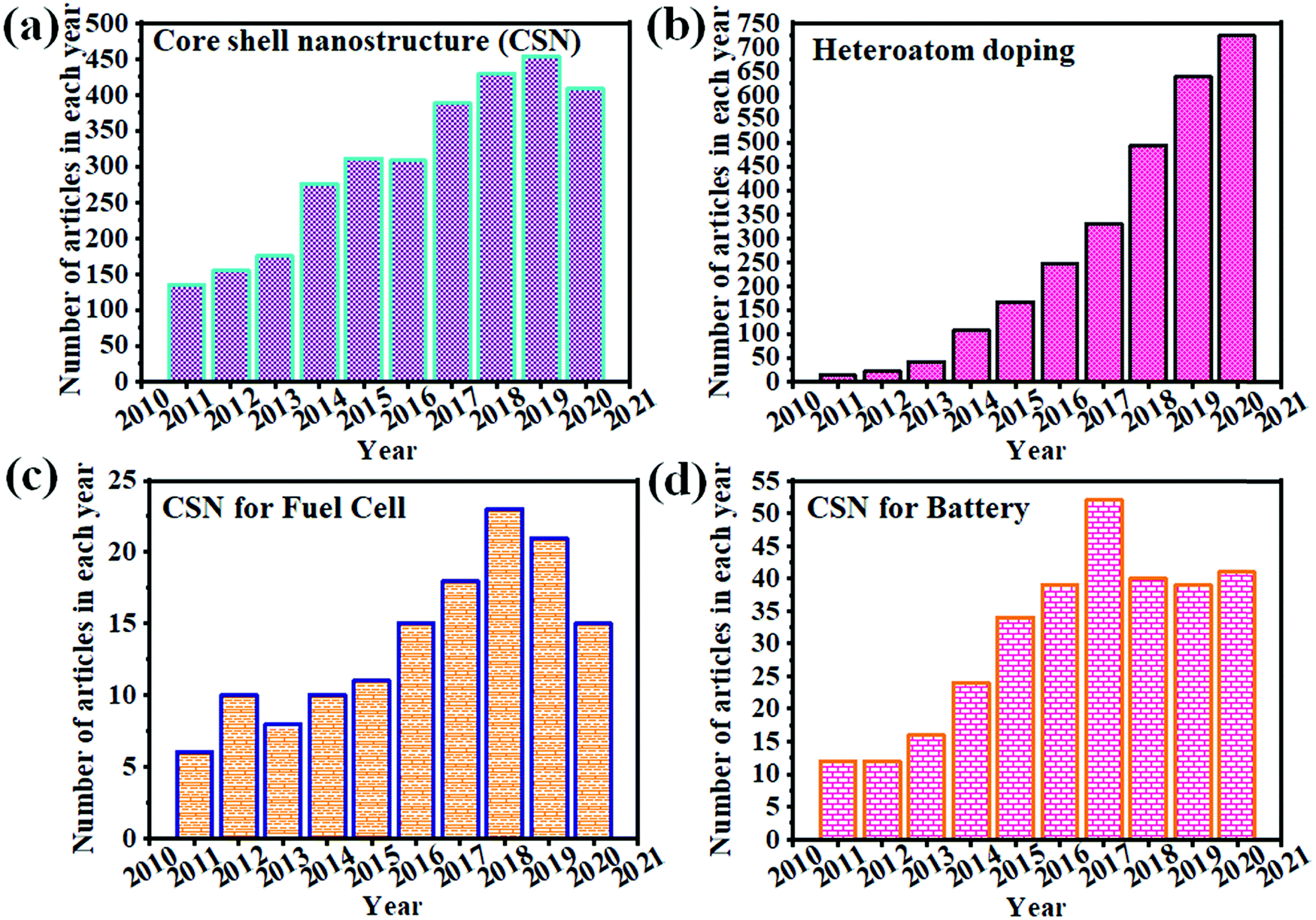 | ||
| Fig. 1 Number of papers published under the topic of (a) core–shell nanostructures, (b) heteroatom doping, (c) CSNs for fuel cells, and (d) CSNs for batteries from 2011 to 2020 (source: https://apps.webofknowledge.com/). | ||
 | ||
| Fig. 2 The schematic illustration and applications of heteroatom doped nanomaterials and core–shell nanostructures for various fuel cells and batteries. | ||
2. Implementation of heteroatom doped nanomaterials/core–shell nanostructures (HCSNs)
CSNs and HCSNs have considerable utility in biomedical, energy, and environmental applications. They are also used in catalysis, water treatment, and sensing applications.12,24 Recently, CSNs and HCSNs have been used in ECS applications because of their improved properties, minimal use of noble metals, and reduced cost. The important applications of HCSNs in PEM, AEM, direct methanol, microbial, and other types of fuel cells have been discussed. In addition, HCSN applications in metal-ion batteries, such as Li-ion and Na-ion batteries, metal–air batteries, such as Li, Zn, and Na–air batteries, and metal–S batteries (MSBs) have also been addressed.2.1. HCSNs for fuel cells
The combustion of fossil fuels produces electrical and thermal energies that are required for ECS applications.39 However, the high consumption of fossil fuel generates various environmental issues that can be mitigated by using alternative sources of electricity. Fuel cells are electrochemical devices that generate electricity by converting chemical energy to electrical energy.40 A typical fuel cell consists of an anode, a cathode, and an electrolyte membrane.41,42 Hydrogen is passed through the anode and oxygen is evolved at the anode of a fuel cell. At the anode side, hydrogen molecules are split into electrons and protons in the presence of a catalyst and transferred to the PEM. Electrons are simultaneously transferred to the circuit and produce electric current and heat. The protons, electrons, and oxygen combine at the cathode site to generate water.41 Fuel cells are used widely to supply electricity to hospitals, homes, grocery stores, and business offices. They also provide energy to buses, trucks, cars, trains, and other vehicles.41 Fuel cells are low-maintenance devices capable of continuous electricity generation.41 Noble metals have been extensively used for high-performance fuel cell applications because they impart improved stability and longer working cycles compared to other materials.43–45 On the other hand, noble metal catalysts are expensive and have poor tolerance to alcohol. Functional nanomaterials with enhanced properties have thus been developed as a viable replacement for noble metals. Various types of fuel cells fabricated using HCSN electrocatalysts have been discussed in the following sub-sections.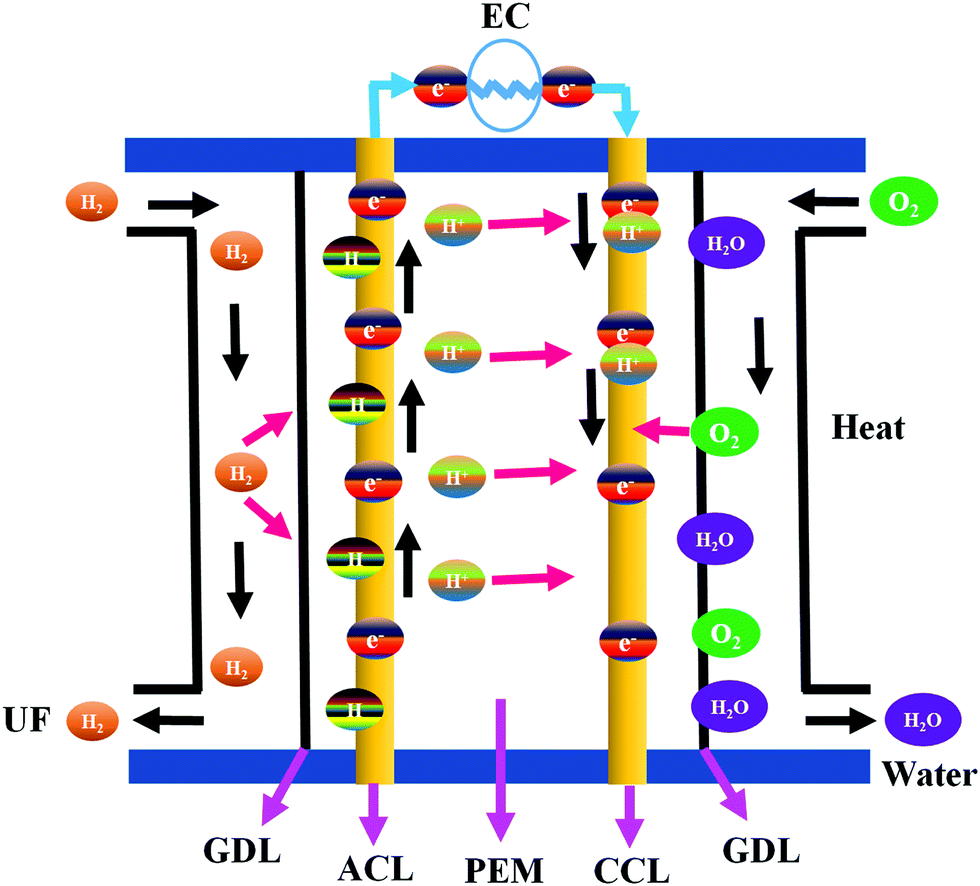 | ||
| Fig. 3 The working principle of PEMFCs (note: H2 – hydrogen, O2 – oxygen, H2O – water, UF – unused fuel, GDL – gas diffusion layer, ACL – anode catalyst layer, PEM – proton exchange membrane, CCL – cathode catalyst layer, H+ – proton, e− – electron). | ||
Recently, Jiang et al. briefly reviewed the role of electrocatalysts used to develop CSNs for the ORR. In particular, the authors discussed the important aspects of the obtained ORR electrocatalyst related to PEMFCs along with the approaches currently adopted to enhance the activity of CSN based electrocatalysts used in PEMFCs.54 In this section, we specifically discuss the fabrication of HCSN electrocatalysts for fuel cell applications with a detailed analysis of the reported literature.55–58
The PEMFC reactions are as follows:46
| Anode: 2H2 → 4H+ + 4e− E° = 0 V | (1) |
| Cathode: O2 + 4H+ + 4e− → 2H2O E° = 1.23 V | (2) |
| Overall: 2H2 + O2 → 2H2O E° = 1.23 V | (3) |
An ultra-thin Pt monolayer (0.025 mg cm−2) was deposited on a Pd/C substrate to produce a core–shell catalyst which delivered high performance in PEMFCs due to the higher specific surface area (SA) of Pt (SA, 205–240 m2g Pt−1) and reduced the cost of the fuel cells.52 In addition, the costs and stack sizes of the fuel cells were further reduced by increasing the voltage loss at a high current density (HCD).52 Karuppannan et al. studied the importance of depositing ultra-thin Pt–C layers on C nanofiber (CNF) surfaces by coating with a wet Pt–aniline complex for optimizing the performance of PEMFCs.59 The carbonization of the prepared material at 900 °C under an N2 atmosphere led to the uniform deposition of 3–4 nm-sized Pt nanoparticles with a thin layer of C on the CNF surface. The Pt@CS-CNF900 electrode had an E1/2 value that was 10 mV higher than that of the Pt/C electrode.59 The electrochemically active surface area (ECSA) values observed for the samples carbonized at 600 (95.6 m2g Pt−1), 700 (100.5 m2g Pt−1), and 900 °C (99.6 m2g Pt−1) indicated the presence of more active sites than in Pt/C. A higher mass activity was observed for the sample that was carbonized at 900 °C (116.7 ± 1 m2g Pt−1) compared to those carbonized at 600 (97.8 ± 1 m2g Pt−1) and 700 °C (90.8 ± 1 m2g Pt−1). The cell voltage decreased from 0.796 to 0.794 V for Pt@CS-CNF900, while that for Pt@CS-CNF600 decreased from 0.799 to 0.754 V.59 These results proved that the stability and performance were improved by carbonizing the sample at 900 °C.
Lin et al. studied the PEMFC application of the synthesized core–shell Co@Pt (1:3)–C electrocatalyst and the heat-treated sample (Co@Pt (1:3)–C (reduced)) obtained at 500 °C under a 5 vol% H2/N2 gas flow and compared their activities with that of a commercial Johnson Matthey (JM) Pt/C catalyst.55 The synthesized Co@Pt (1:3)–C electrocatalyst had an average particle size of approximately 3 nm and excellent dispersibility on the electrode. According to the experimental results, the heat-treatment of Co@Pt (1:3)–C that resulted in the formation of Co@Pt (1:3)–C (reduced) significantly enhanced the half-wave potential values (0.803 V and 0.842 V for the former and latter electrocatalysts, respectively) which were comparable to that of the commercial Pt/C catalyst. The Tafel slopes of the (JM) Pt/C, Co@Pt (1:3)–C (reduced), and Co@Pt (1:3)–C electrocatalysts were 74, 78, and 85 mV decade−1, respectively. Furthermore, the PEMFC performance of the Co@Pt (1:3)–C (reduced) electrocatalyst was studied in a single cell set-up using the Co@Pt (1:3)–C (reduced) catalyst as the cathode and Pt/C as the anode. Electrochemical impedance spectroscopy (EIS) was performed using (JM) Pt/C and Co@Pt (1:3)–C (reduced) as the working electrodes and Pt/C as the counter electrode under H2 saturation conditions. The ohmic resistances of the (JM) Pt/C and Co@Pt (1:3)–C (reduced) catalysts were 10.1 mΩ and 13.9 mΩ, respectively. The diameters of the kinetic loops for Pt/C (JM) and Co@Pt (1:3)–C (reduced) were determined to be 12 and 12.2 mΩ, respectively.55 Additionally, the Co@Pt (1:3)–C (reduced) electrocatalyst imparted exceptional stability and durability to the PEMFCs without losing its characteristic properties.
Various CSNs were studied exclusively concerning PEMFC applications.45,55,60–65 Ahn et al. discussed the importance of the Nafion ionomer content for the preparation of Pt/ordered mesoporous C (OMC) electrocatalyst slurries used in PEMFCs.61 The optimum ionomer contents were found to be ∼20 and 30 wt% when carbon black was used as the catalyst support in the HCD region, whereas the use of the Pt/OMC catalyst support required only 10 wt% of ionomer for optimized PEMFC performance (Fig. 4).61 The gram-scale synthesis of the core–shell structure was performed using a microwave-assisted polyol method on a carbon black support using a CuPt alloy as the core which was encased in a Pt shell. The obtained catalyst delivered high electrochemical activity toward the ORR and demonstrated a high potential for use in PEMFCs.66 The pyrolysis of Pt@Zn-based ZIF-8 at 800 °C under a mixed gaseous atmosphere of H2 and Ar (1:20) for 5 h afforded N-doped C decorated PtZn intermetallic PtZn@NC.67 The 10%-PtZn@NC-800 electrocatalyst displayed an increased E1/2 value of 0.912 V vs. reversible hydrogen electrode (RHE) for the cathodic ORR in an acidic medium compared to that of Pt/C (0.886 V). In addition, the 10%-PtZn@NC-800 electrocatalyst exhibited 3 to 5 times higher mass and specific activities than the commercial Pt/C electrocatalyst with excellent stability after 5000 cycles. Moreover, the electrocatalyst exhibited superior activity with respect to methanol and formic acid oxidation reactions. These results suggest the wide applicability of the 10%-PtZn@NC-800 electrocatalyst in PEMFCs.
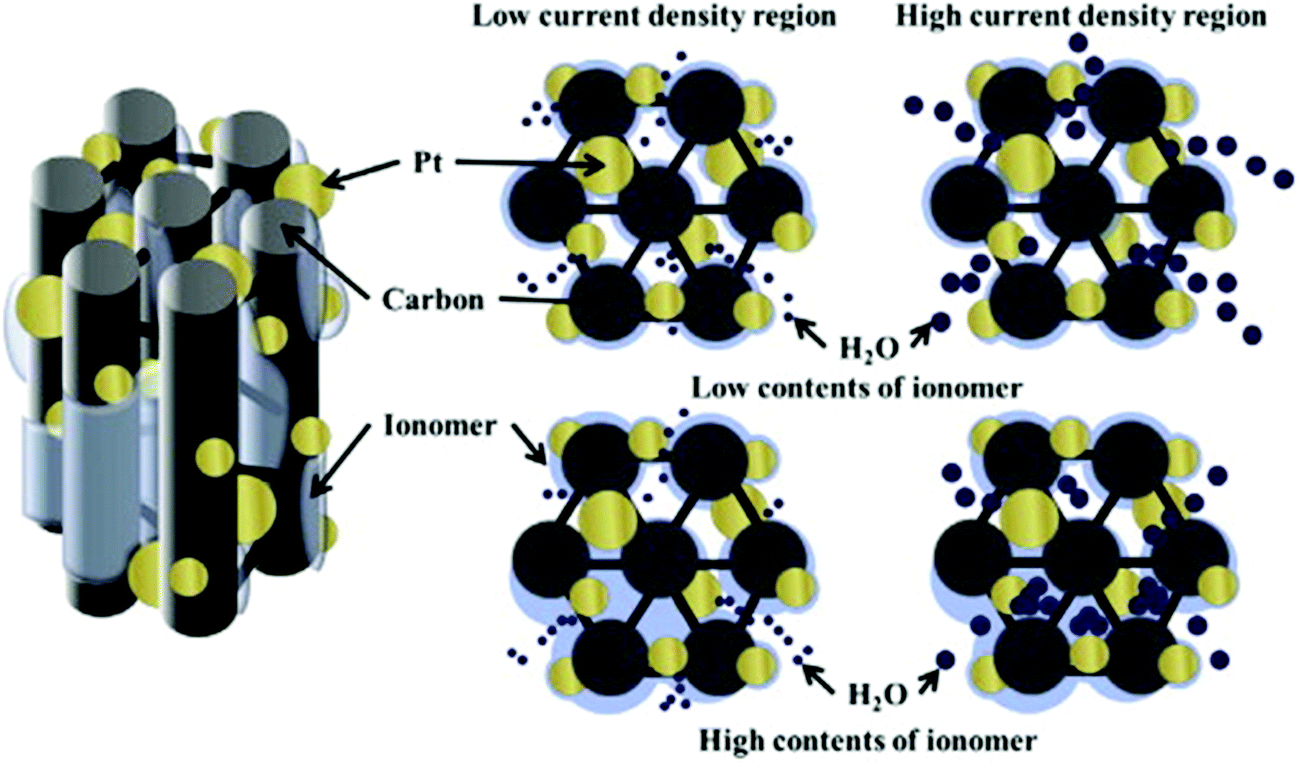 | ||
| Fig. 4 Schematic illustration of the effect of ionomer contents in the low and high current density regions. Reproduced from ref. 61 with permission from Elsevier B.V. | ||
Several studies were performed on the various aspects of CSNs using different nanomaterial architectures in AEMFCs.74–77 Recently, highly conducting hydroxide AEM electrolytes have been employed in an alkaline electrolyte-based fuel cell.74,78–80 AAEMFCs undergo the HOR at the anode, which generates hydroxide anions based on the ORR at the cathode and produces water as the final product at the anode.79 Extensive separation of electrodes from the AAEM polymeric electrolyte is observed and the particular transport of hydroxide anions from the cathode to the anode facilitates the migration of water from the anode to the cathode without reactant/oxidant gas crossover.79 Sa et al. synthesized CNTs with heteroatom-doped C (HDC) derived from ionic liquids (ILs), such as [1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide (BMITFSI)], in the presence of tetraethyl orthosilicate (TEOS).74 The ILs containing N, S, and fluorine (F) in their structures were uniformly deposited on the CNTs through acid reflux, followed by thermal annealing at 800–1000 °C. The CNT/HDC prepared at 1000 °C demonstrated excellent ORR activity with superior kinetic current density and half-wave potential in alkaline media compared to those subjected to other carbonization temperatures and its performance was almost identical to that of commercial Pt/C.74 In alkaline fuel cells, the CNT/HDC-1000 based MEA exhibited a very high onset potential of 0.85 V, which was superior to that observed in the case of a CNT-based MEA. The power (221 mW cm−2) and current density (368 mA cm−2) of the CNT/HDC-1000 cathode at 0.6 V in an H2/O2 atmosphere in an alkaline fuel cell were 23.3 times higher than the corresponding values for the CNT-based cathode.74 Lee et al. prepared cobalt-supported HDC from natural bean sprouts as a biosource that absorbed the metal precursor. The material was subsequently carbonized at 900 °C for 2 h at a heating rate of 10 °C min−1 under an Ar atmosphere.75 The prepared electrocatalyst delivered outstanding ORR activity in alkaline media due to the presence of heteroatoms and a large surface area. The heteroatom-doped electrocatalyst had a higher positive half-wave potential value (0.836 V) than a metal-supported C electrocatalyst devoid of heteroatoms (0.767 V), C derived from bean sprouts (0.683 V), and a commercial Pt/C electrocatalyst (0.825 V). Furthermore, the cobalt supported HDC electrocatalyst exhibited a high power density of 172.2 mW cm−2 at 0.373 V in AEMFCs.75
Woo et al. synthesized P- and S-doped Fe–N/C electrocatalysts by a silica coating mediated method, which delivered an excellent ORR performance with a half-wave potential of 0.91 V vs. RHE in alkaline media.76 The P- and S-doped Fe–N/C electrocatalysts also exhibited higher current densities of 904 and 977 mA cm−2 at 0.6 V compared to the undoped Fe–N/C electrocatalyst which showed a current density of 669 mA cm−2 at 0.6 V (Fig. 5(a)).76 The introduction of heteroatoms to the undoped Fe–N/C electrocatalyst enhanced the half-cell activity in both the rotating disc electrode and single-cell performance tests (Fig. 5(b)).76 The S-doped Fe–N/C electrocatalyst exhibited a power density of 635 mW cm−2 in AEMFC applications with a markedly improved MEA performance in both acidic and alkaline electrolytes.
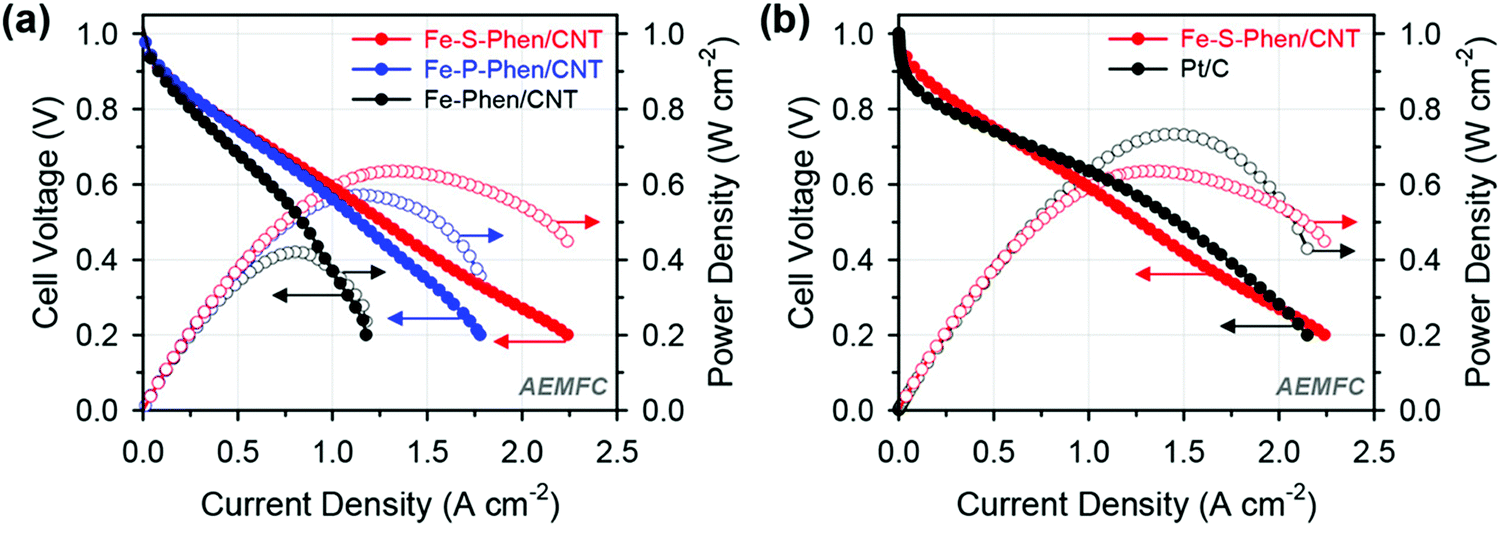 | ||
| Fig. 5 Alkaline AEMFC performance of MEAs using (a) Fe-Phen/CNT, Fe-P-Phen/CNT, and Fe-S-Phen/CNT as cathode catalysts and (b) Fe-S-Phen/CNT and Pt/C as cathode catalysts. Reproduced from ref. 76 with permission from the American Chemical Society. | ||
A carbide-derived C material co-doped with a transition metal and N (M/N/CDC) was prepared by reacting titanium carbide-derived C with dicyandiamide in the presence of ferric chloride (FeCl3) or cobalt chloride (CoCl2) as the dopant.81 The MEA of the obtained M/N/CDC cathode delivered an excellent power density of 80 mW cm−2 compared to the Pt/C cathode (90 mW cm−2). Dekel et al. briefly reviewed the various aspects of hydrogen-fueled AEMFCs, Pt-free hydrogen AEMFCs, and the stability of hydrogen AEMFCs.82 AEMFCs are used under alkaline conditions, whereas acidic conditions are applied in PEMFCs. The hydroxide anions migrate from the cathode to the anode via the AEMs in AEMFCs which is analogous to the H+ conduction pathway in PEMFCs.82 AEMs function as solid electrolytes which conduct hydroxide and carbonate anions owing to the presence of positively charged cationic groups on their polymer backbone.82 An Fe–tin (Sn) C nitride graphene core–shell electrocatalyst prepared at 900 °C delivered an ORR overpotential of ∼70 mV, thus demonstrating potential utility in AEMFCs.83
Recently, AAEMFCs have been applied more extensively in fuel cell technology compared with AEMFCs and PEMFCs because they are capable of performing the selective transport of hydroxide ions from the cathode to the anode via a membrane.79,84–86 Thin membranes derived from solid polymer electrolytes are used in AAEMFCs. However, the presence of carbonate anions diminishes the ionic conductivity of the membranes over time, which is a major issue.84 Polymer electrolytes also suffer from low ion exchange conductivity (IEC). Polymer electrolytes with improved IEC and stable mechanical properties are necessary to achieve efficient AAEMFC activity. Various AEMs have been prepared using different types of polymer backbones and functional ionic groups for successful use in AAEMFCs. We have listed some of the important CSN and HCSN based catalysts and their results using AEMFCs and AAEMFCs (Table 1).
| Catalyst | Membrane | Loading density (mg cm−2) | OCV (V) | Maximum power density (mW cm−2) | Ref. |
|---|---|---|---|---|---|
| OCV: open-circuit voltage, CNT-HDC-1000: carbon nanotube/heteroatom-doped carbon, NBSCP: Co2P nanoparticles supported on heteroatom-doped carbon catalysts, CoMn-pNGr: CoMn alloy oxide nanoparticles on N-doped porous graphene, Fe–N-comp-0.5: nitrogen doped iron containing carbide-derived carbon (Fe–N-CDC) and multi-walled carbon nanotubes (MWCNTs), Fe-M-La-C-700: pyrolyzed iron melamine lipoic acid carbon composite at 700 °C, Fe-NMG: iron containing nicarbazin, methylimidazole, and glucoril, Fe–N–C (1000 °C): Fe-containing N-doped carbon, AT-FeNC: aminothiazole derived Fe-containing N-doped carbon, CoFeN–C-HLH: cobalt and iron containing N-doped carbon [HLH: (pyrolyzed:acidic treatment:pyrolyzed)], NCNHs: nitrogen-doped single-walled carbon nanohorns, Fe-NCNH-900: single-walled carbon nanohorns by simultaneous doping with Fe and N at 900 °C. | |||||
| CNT-HDC-1000 | — | 2.0 | — | 270 | 74 |
| NBSCP | FAA-3 | 3.0 | 1.01 | 172 | 75 |
| CoMn-pNGr | FAA-3 | 2.0 | 0.92 | 35.20 | 87 |
| Fe–Co-NpGr | FAA-3 | 2.5 | 0.85 | 35 | 88 |
| Fe–N-comp-0.5 | A201 | 2.6 ± 0.2 | 0.92 | 160 | 89 |
| Fe-M-La-C-700 | A201 | 4.0 | 0.64 | 137 | 90 |
| Fe-NMG | A201 | 3.5 | 1.05 | 218 | 91 |
| Fe-NMG-35 wt% | A201 | 3.5 | — | 218 | 91 |
| Fe–N–C (1000 °C) | aQAPS-S8 | 2.0 | — | 485 | 92 |
| Fe–N–C (800 °C) | aQAPS-S8 | 2.0 | — | 450 | 92 |
| AT-FeNC | aQAPS-S8 | 4.0 | — | 164 | 93 |
| FeCoN-C-HLH | Tokuyama A-201 | 4.0 | — | 177 | 94 |
| N-CNT | — | 5.0 | — | 37 | 95 |
| NCNHs | Fumapem FAA | 3.0 | 0.75 | 30 | 96 |
| Fe-NCNH-900 | Fumapem FAA | 4.0 | 0.83 | 35 | 97 |
The Pt electrodes in DMFCs also degrade the catalyst through the formation of carbon monoxide (CO) by the electro-oxidation of methanol, which diminishes the performance.98 These disadvantages are addressed by mixing Pt with other noble metals, transition metals, or metal oxides which help maintain the stability and tolerance of the electrodes against alcohol while mitigating the effects of CO poisoning.102 The performance and stability of the electrocatalysts in DMFCs are also enhanced by using porous materials to support the catalytic materials.103,104 The porous materials are required to possess high surface areas and appreciable electrical conductivity. Additionally, they should facilitate catalyst recovery with repeated cycles, interact strongly with the catalysts, and have superior corrosion resistivity.105 Yuda et al. briefly reviewed the different synthetic pathways for preparing graphitic carbon nitrides (g-C3N4) and their structural characteristics. The effects of incorporating non-metals and noble and non-noble metals in g-C3N4 structures on the methanol oxidation reaction (MOR) were discussed along with the applications of these materials in DMFCs.99 The doping of g-C3N4 with heteroatoms or metals improved their electrocatalytic performance with respect to DMFC applications.99,106–108
Various metals and metal–C based hybrid CSNs, such as Ni–Pd, Ru–Pt, Se–Ru, Au–Pd, Au–PdAg, Au–PtCu, PdCuM (M = Ru, Rh, and Ir), PdCu@Pt, Pt3Co–Pt, PtCuCo, NiCoO4-MWCNTs, and CeO2–rGO–Pt, were used for DMFC applications.109–120 Jin et al. designed trimetallic alloy nanosheet (PdCuRu, PdCuRh, and PdCuIr) based electrocatalysts and studied their electrocatalytic behavior toward the MOR. The synthesized Pd59Cu33Ru8 nanosheets exhibited outstanding mass (1660.8 mA mg−1) and specific activities (4.7 mA cm−2), which were 4.9- and 2.8-fold higher than those of the other trimetallic alloys and the Pd/C catalyst, respectively.115 Parthiban et al. studied the role of sulfonic acid-functionalized graphene (SFG) in conjunction with proton-conducting Nafion in controlling the methanol crossover effect in DMFCs.121 The material delivered superior electrocatalytic activity and the combination of SFG and Nafion reduced the extent of methanol crossover compared to that observed in the presence of pristine Nafion (Fig. 6(a)).121 In addition, the functionalized material retained its stability after 50 h of operation owing to its considerably low methanol permeability (Fig. 6(b)).121 The obstruction to the methanol pathway caused by the binding of S-graphene on the polymer membrane is responsible for the lower methanol diffusivity of the material.
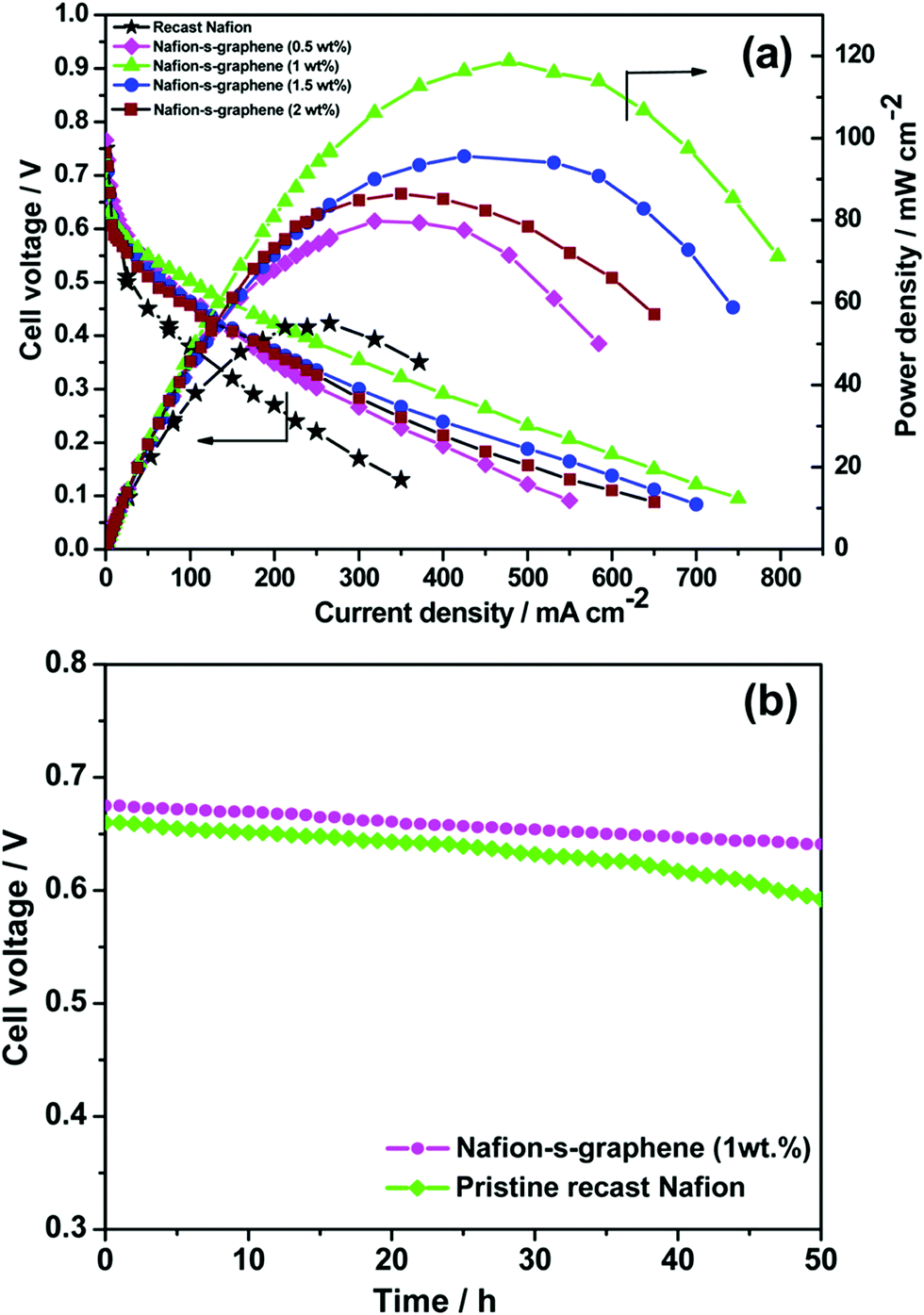 | ||
| Fig. 6 (a) DMFC performance of pristine Nafion and Nafion–S-graphene hybrid membrane at 70 °C under ambient pressure. (b) Stability test of the membranes for 50 h under fuel cell configuration at OCV. Reproduced from ref. 121 with permission from the American Chemical Society. | ||
The DMFC polarization and performance curves indicate that the hybrid membranes have considerably higher open-circuit voltages (OCVs) at 70 °C under ambient pressure than the pristine Nafion membrane, which indicates a lower extent of methanol crossover in the presence of the hybrid membrane (Fig. 6(a)).121 The hybrid membrane prepared using 1 wt% S-graphene exhibited an outstanding power density of 118 mW cm−2 which was considerably higher than those recorded for the pristine Nafion membrane (54 mW cm−2) and the other hybrid membranes containing different proportions of S-graphene. The sulfonic acid functional group present in the hybrid membrane has excellent proton-conducting characteristics and enhances the power stability, making the hybrid membrane highly suitable for DMFC applications (Fig. 6(b)).121 The conductivity of the membrane was partially reduced upon increasing the S-graphene content as the ionic conducting pathways were hindered.121 Moreover, a reduction in the extent of methanol crossover was reported upon using the sodium dodecyl sulfate (SDS) surfactant as a template to develop a porous C-Nafion membrane.122
Çögenli et al. synthesized an N and B doped 3D porous graphene aerogel, followed by Pt loading on the material by a microwave heating method.123 The obtained heteroatom-doped materials demonstrated superior catalytic activity in both formic acid and methanol oxidations.123 A highly stable and methanol tolerant electrocatalyst was also developed by applying the core–shell technique to C nanofibers using Pt cores and C shells in the presence of aniline. The electrocatalyst was found to be highly effective in DMFC applications.124 In Table 2, we outlined the important results of DMFCs such as methanol concentration with respect to a power density of the CSN based catalyst. To the best of our knowledge, there are very few studies on the role of heteroatom doping and CSNs in DMFC applications. Detailed studies are extremely necessary to understand the importance of CSNs and HCSNs in enhancing the electrocatalytic performance of DMFCs.
| Catalyst | Methanol concentration (M) | Power density (mW cm−2) | Ref. |
|---|---|---|---|
| Au–Ag2S–Pt: gold and silver sulfide core–shell decorated with platinum, modified-anode MPLs: the combination of commercial carbon black (CB, (50 vol%)) and platelet carbon nanofibers (PCNFs, (50 vol%)) with a micro-porous layer, poly(SHS-ddm): cross-linked sulfonic acid-containing polybenzoxazine, PMFSP: porous metal fiber sintered plate. | |||
| Au–Ag2S–Pt nanocomposites | 0.5–15 | 89.7 | 125 |
| Hydrophobic anode MPL | 8 | 78 | 126 |
| Modified-anode MPLs | 5 | 77.9 | 127 |
| Modified-anode MPLs | 7 | 67.7 | 127 |
| Poly(SHS-ddm) | 7 | 66.5 | 128 |
| GO nanoplatelets | 5–10 | 50 | 129 |
| PMFSP | 2–4 | 7 | 130 |
Yang et al. prepared a bio-waste derived catalytic material by carbonizing the anaerobic digestion products of Broussonetia papyrifera at 800 °C for 2 h under an N2 atmosphere at a heating rate of 5 °C min−1.133 N doped C (N–C) and Fe and N co-doped C (Fe–N–C) were also produced in the absence and presence of ferric chloride (FeCl3) at 800 °C under an N2 atmosphere for 2 h (Fig. 7(A)).133 The prepared Fe–N–C electrode displayed excellent ORR activity due to the formation of active Fe–N–C sites which facilitated the transfer of electrons and protons. In addition, the enhanced formation of graphitic structures in the catalyst increased its conductivity. The Fe–N–C catalyst exhibited appropriate adsorption of oxygenates (*O, *O2, *OH, and *OOH), which led to the enhancement of the ORR activity.133 Furthermore, the Fe–N–C electrode, when used in MFCs, dispatched a maximum power density of 1308 mW m−2 which was superior to that recorded in the case of the N–C electrode (638 mW m−2). The open-circuit potentials of the Fe–N–C, Pt/C, and N–C catalysts in the MFCs were found to be 0.63, 0.62, and 0.47 V, respectively (Fig. 7(B)).133 In addition, the Fe–N–C catalyst exhibited outstanding activity in the MFC after 500 cycles of operation due to the presence of a large specific surface area and a high number of active sites, which were responsible for the excellent stability of the catalyst.133 Luo et al. prepared an ordered mesoporous C (Fe–N–C) material containing atomically dispersed Fe–Nx active sites derived from MOFs.134 The developed material had excellent properties, such as high values of maximum power density (1232.9 mW m−2), constant output voltage (0.46 V), and open-circuit voltage (0.644 V). These values established that the developed materials had considerably superior catalytic properties to 20% Pt/C in MFCs.134 The presence of active Fe–Nx sites and pyridine- and graphite-like N structures is responsible for enhancing the catalytic activity in MFCs. Various types of Fe–N–C based materials have been widely used in MFCs.135–139 In addition, various core–shell-based electrocatalysts fabricated using heteroatom-doped transition metals have been studied with respect to MFCs.25,140–144 The catalytic activity of different HCSNs in MFCs is also presented in Table 3.
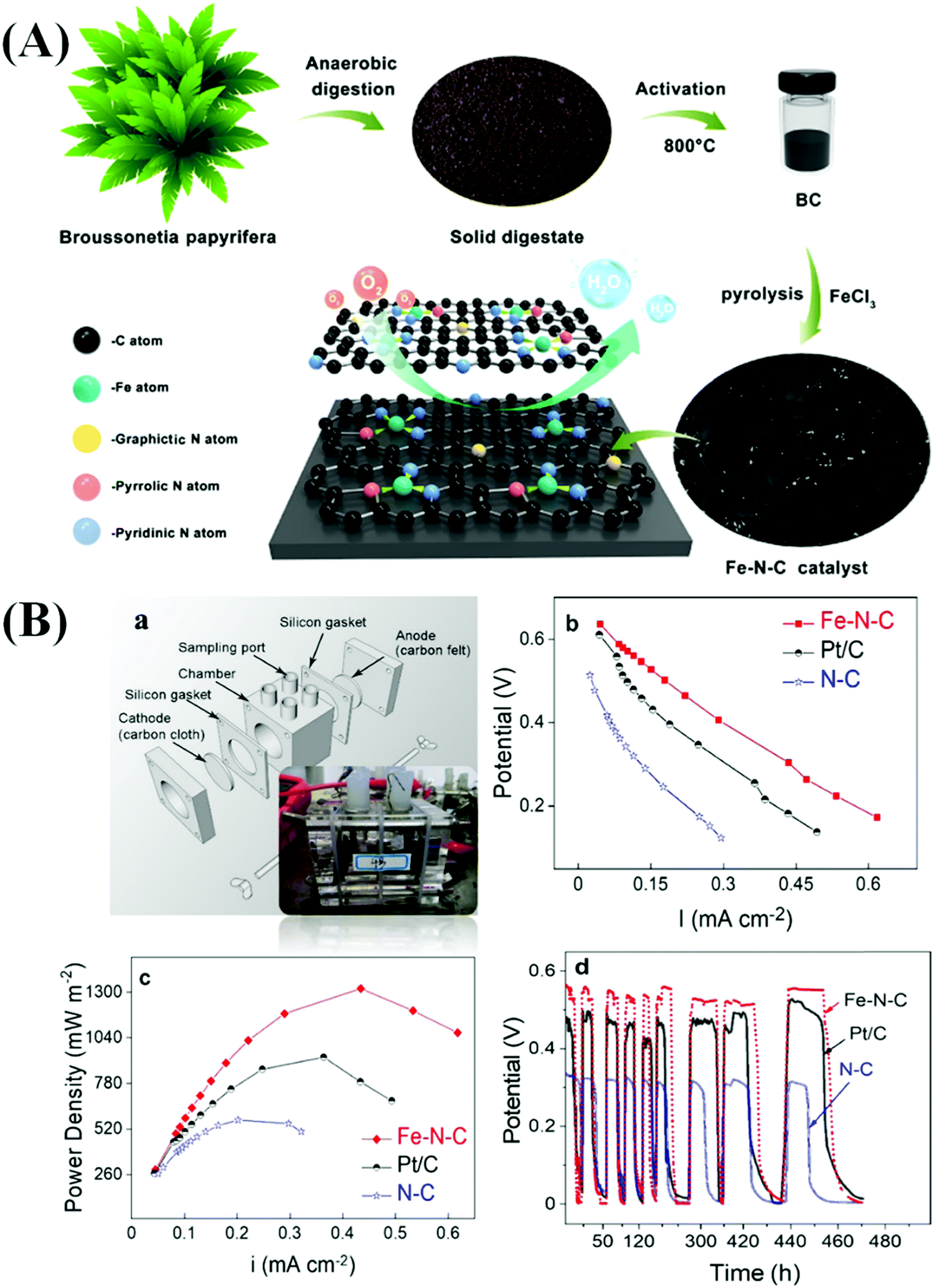 | ||
| Fig. 7 (A) Schematic illustration of the fabrication of hierarchical Fe–N–C catalysts derived from the solid digestate. (B) Schematic illustration of a single-chamber MFC (a); power density (b) and electrode polarization curves (c), and the periodic variation of single-chamber MFCs coupled with the Fe–N–C, Pt/C, and N–C catalysts (d). Reproduced from ref. 133 with permission from the American Chemical Society. | ||
| Catalyst | Maximum power density (mW m−2) | Ref. |
|---|---|---|
| Note: NP-C: N and P dual-doped carbon from cellulose, CP-M-Z: magnesium and zinc containing pyrolyzed carbon, FePc-CS500: iron(II)-phthalocyanine containing macroporous hollow nanocarbon shells, Fe–N–C-G: iron–nitrogen–carbon nanorod network-anchored graphene nanohybrid, Fe–N–C: Fe–N-doped carbon, Fe/N-HCN: Fe–N-doped porous hollow carbon nanospheres (HCNs), Co–Co9S8-NPGC: nitrogen-doped Co–Co9S8-partly-graphitized carbon, Fe–N-AC: iron–nitrogen/activated carbon, N-CA: N-doped carbon aerogel, Fe–C-Ns-900: Fe-containing N-doped carbon, NFe-CNS: nitrogen and iron co-doped carbon nanospheres, Fe–N co-doped GNR-CNT: iron, nitrogen co-doped graphene nanoribbon-carbon nanotube, FePc-PID-CNTs: iron phthalocyanine and polyindole on carbon nanotubes/carbon Vulcan, NB-CPs: nitrogen and boron-co-doped core–shell carbon nanoparticles, NPOMC: nitrogen- and phosphorus-doped ordered mesoporous carbon. | ||
| CoN–C-5 | 1202.3 | 25 |
| Fe–N co-doped GNR-CNT | 801 | 136 |
| Fe–N–C-50 | 1166 | 137 |
| NB-CPs | 642 | 141 |
| CP-M-Z | 2288 ± 30 | 142 |
| NP-C | 2293 ± 50 | 145 |
| FePc-CS500 | 2160 ± 20 | 146 |
| Fe–N–C-G | 1601 | 147 |
| Fe–N–C | 1620 ± 30 | 148 |
| Fe–N-HCN | 1300 ± 64 | 149 |
| Co-Co9S8-NPGC | 1156 ± 18 | 150 |
| Fe–N-AC | 1092 | 151 |
| N-CA | 967 ± 34 | 152 |
| Fe–C-Ns-900 | 900 | 153 |
| NFe-CNS | 866.5 ± 7 | 154 |
| FePc-PID-CNTs | 799 | 155 |
| NPOMC | 245.8 | 156 |
Cao et al. investigated the applications of ultra-small Ag@Pd CSNs coated on Vulcan XC72R carbon support in DFAFCs. Ag/C nanocrystals were prepared by mixing silver nitrate and Vulcan XC72R carbon with ammonium hydroxide and formaldehyde.159 The synthesized Ag/C nanocrystals were subsequently modified by doping with palladium chloride in the presence of ethylenediaminetetraacetic acid (EDTA) disodium salt at 160 °C. Ag–Pd–C CSNs of three different compositions were prepared using different Ag/Pd atomic ratios: Ag–Pd–C-1 (63:37), Ag–Pd–C-2 (44:56), and Ag–Pd–C-3 (19:81). The formic acid oxidation reaction (FAOR) was performed using the prepared Ag–Pd–C CSNs. The mass-specific activities (MSA) at a potential of 0.345 V of the three samples were 1.06, 1.47, and 0.84 mA μg−1, respectively.159 The MSA of Pd–C and commercial Pd black were found to be 1.04 mA μg−1 and 0.49 mA μg−1, respectively. According to these results, of all the materials tested, the Ag–Pd–C-2 CSNs exhibited the highest electrocatalytic activity. In addition, the Ag–Pd–C-2 CSNs exhibited a superior current density (0.124 mA μg−1) to Pd–C (0.084 mA μg−1) and Pd black (0.083 mA μg−1) after 2000 s.159 The multi-walled carbon nanotube (MWCNT)-supported Ru–Pd CSNs synthesized by Zhang et al. also manifested superior FAOR and ethanol oxidation reaction (EOR) properties.160 The FAOR activities of the synthesized Ru–Pd-MWCNT and Pd-MWCNT materials corresponded to forward peak current densities of 1757 and 976 mA mg−1Pd, respectively. Likewise, on examining the EOR activities of the Ru–Pd-MWCNT, Pd-MWCNT, and Pd–C samples, the peak current densities were found to be 2507.5, 1378.2, and 943.5 mA mg−1Pd, respectively.160 The obtained results demonstrate the superior electrochemical activity of the Ru–Pd-MWCNT material toward the FAOR and EOR compared to Pd-MWCNT and Pd–C.160 Pt3Pb nanocrystals encased in Pt CSNs also exhibit a higher FAOR activity compared to the nanocrystals devoid of Pt.161
2.2. HCSNs for metal-ion batteries
Metal-ion batteries are a promising technology related to ECS systems.34 Metal-ion batteries principally require high specific power and energy densities along with sufficient operational stability. Metal-ion batteries employ polymeric cathodes, such as N,N′-substituted dihydrophenazine and hexaazatrinaphthylene, and NaK alloy anodes impregnated in C cloth.163 PAN equipped with organic cyclic chains is used as an effective anode material for metal-ion batteries in alkaline media, exhibiting a reversible capacitance of 1238 mA h g−1 under a current density of 50 mA g−1.164 Likewise, organic electrodes featuring carbonyl and disulfide linkages, and those obtained via superlithiation, azo and anion insertion, and imine reactions have been used in metal-ion batteries.165 Various HCSNs have been used for improving the performances of metal-ion batteries due to their efficient electrocatalytic properties and highly controlled architectures. Herein, we briefly describe the different HCSNs used for the development of metal-ion batteries such as Li, sodium (Na), and K-ion batteries.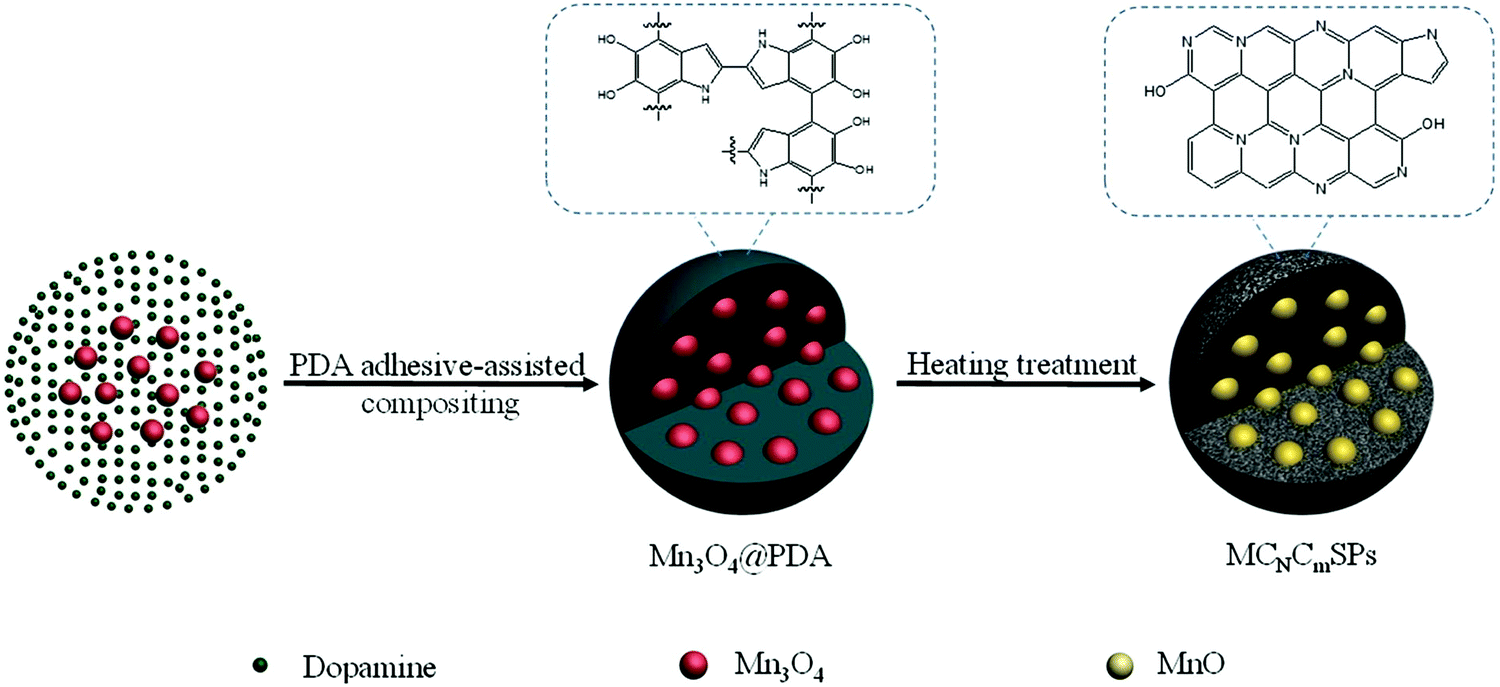 | ||
| Fig. 8 Schematic illustration of the fabrication of MCNCmSPs by a PDA adhesive-assisted compositing procedure. Reproduced from ref. 177 with permission from Elsevier Ltd. | ||
Zhou et al. also studied the LIB behavior of heteroatom-doped core–shell carbonaceous framework materials derived from ZIF-8 and cyclomatrix poly(organophosphazenes) (POPs).22 Li-ions and atoms have apertures sizes of ∼1.5 Å and 3.0 Å, respectively, whereas ZIF-8 has apertures measuring ∼3.4 Å. These values demonstrate the excellent potential for interaction and migration of Li-ions and atoms in ZIF-8 networks and the feasibility of their application in LIBs. POPs are composed of organic–inorganic hybrid frameworks with intrinsic heteroatoms such as P, S, N, and O and covalently cross-linked polymer backbones.22 The size of the synthesized POP nanosphere core is around 200 nm and that of the ZIF-8 shell is in the range of 50 to 300 nm. The POP-ZIF-8 material has an approximate surface area of 1557 m2 g−1. The POP-ZIF-8 materials contain transition metals, such as Zn, and heteroatoms such as P, N, and S. In addition, the carbonization of the POP-ZIF-8 material leads to the formation of core@shell heteroatom-doped carbonaceous structures. The LIB efficiency of the material was examined using the coin cell approach over a series of charge and discharge cycles at cell voltages of 0.01 and 2 V vs. Li/Li+ and a current density of 74 mA g−1. The POP-ZIF-8 synthesized using 1:2.5 and 1:8 weight ratios have initial charge capacities of 493 and 580 mA h g−1, and discharge capacities of 538 and 647 mA h g−1, respectively.22 An increase in the capacity was observed upon increasing the POP-ZIF-8 weight ratio due to the abundance of heteroatoms and metals, which generated more reactive sites for the pseudo-redox reactions. Moreover, the obtained results suggest that an increase in the proportion of ZIF-8 results in an increase in the anode capacity and stability, which enhances the anode reactivity toward the lithiation and de-lithiation steps in LIBs.
Hua et al. explained the mechanism of the (de)lithiation process in the core–shell NCM electrode during the first cycle (Fig. 9).179 Lithium ions are first taken from the Ni-rich core phase, which has a higher concentration of reactive Ni, due to the microscopic open pores of the core–shell NCM material. Because the initial charge of the Mn-rich phase is smaller than that of the Ni-rich phase, the state of charge of the shell controls the electrochemical properties of the core–shell NCM.
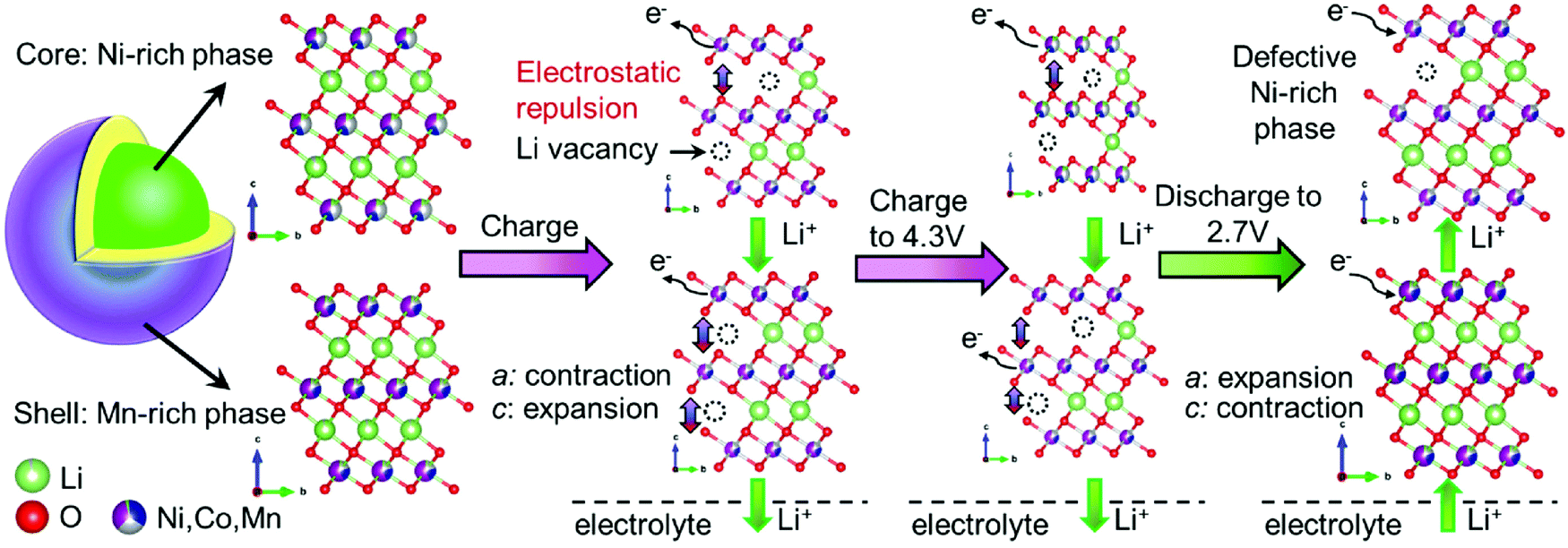 | ||
| Fig. 9 Schematic illustration of a subsequent (de)lithiation mechanism in the core–shell NCM cathode materials during the first cycle, showing an obvious change in the unit-cell volume of the Ni-rich phase within the interior region of a secondary particle with respect to the Mn-rich phase. TM: Ni – whitish; Mn – magenta; Co – blue. Reproduced from ref. 179 with permission from the Authors, Elsevier Ltd. | ||
Zhang et al. synthesized low-cost K-PB nanoparticles (KPBNPs, K0.220Fe[Fe(CN)6]0.805·4.01H2O) as potential cathode materials for KIBs. The cathode prepared using KPBNPs exhibits a high discharge voltage (3.1–3.4 V) and reversible capacity (73.2 mA h g−1) with excellent stability.196 The full-cell battery set-up constructed using KPBNPs exhibits an excellent capacity of 68.5 mA h g−1 at 100 mA g−1 with a capacity retention of approximately 93.4% after 50 cycles. The cyclic voltammetry (CV) curve of the KPBNP based electrode indicates the occurrence of a reversible redox reaction at a high potential interval due to the reversible K+ intercalation–deintercalation mechanism involving C–FeIII–FeII.196 Binder-free cathodes have been developed by combining rusty stainless steel meshes (RSSM) with PB nanocubes and RGO. These cathode materials are highly stable, inexpensive, and exhibit superior electrical conductivity. Use of these cathode materials in KIBs was associated with a high capacity (96.8 mA h g−1), rate capability (1000 mA g−1, 42% capacity retention), and discharge voltage (3.3 V) along with superior cycle stability (305 cycles, 75.1% capacity retention).197 Moreover, high-performance cathode materials developed via the intercalation of non-flammable electrolytes such as ethylene carbonate (EC) with depotassiated K0.5MnO2 significantly enhance the electrochemical performance of KIBs, delivering a high reversible capacity of 120 mA h g−1 and 84% capacity retention after 400 cycles.199
Wang et al. synthesized molybdenum selenide (MoSe2)–C CSNs having pistachio shuck-like structures by mixing sodium molybdate, oleylamine, and dibenzyl diselenide and degassing at 90 °C for 30 min, followed by two successive pyrolysis steps at 250 °C for 30 min (heating rate: 8 °C min−1) and 550 °C for 2 h (heating rate: 8 °C min−1) in an inert gas atmosphere.200 The synthesized unique structures facilitate the diffusion of the K+ ions. Moreover, they maintain their structural stability and exhibit excellent cycle stability during the charge and discharge processes. The electrode fabricated using the (MoSe2)–C material exhibits charge–discharge capacities of 402 and 635 mA h g−1 in the first cycle with 63.4% coulombic retention. In addition, it maintains a capacity of 322 mA h g−1 at 0.2 A g−1 for over 100 cycles and a discharging capacity of 226 mA h g−1 at a maximum current density of 1 A g−1 after 1000 cycles.200 According to the results of density functional theory (DFT) studies on (MoSe2)–C CSNs, the accelerated diffusion of K+ ions occurred due to the higher energy barrier of bulk MoSe2 than that of the surface of expanded MoSe2 nanosheets. In addition, the presence of C on the surface facilitates the electron transfer between the CSNs and ensures appreciable structural stability and cycling performance.200 On the other hand, when the hollow mesoporous C structure obtained by combining MOFs with CoSe2–N-doped C was used as an anode, initial charging–discharging capacities of 448 and 675 mA h g−1 were observed along with a coulombic efficiency of 66%, a rate capability of 263 mA h g−1 at 2 A g−1 and a stable cycle performance of 442 mA h g−1 at 0.1 A g−1 after 120 cycles.201 It is plausible that the large volume expansion of the active material caused by pulverization was responsible for its outstanding performance in KIBs.
Chu et al. synthesized a bimetallic Fe–Mo selenide-N-doped C (FMSC) based CSN as an active anode for KIBs, which exhibited a high reversible capacity of 298 mA h g−1 and maintained a capacity of up to 200 mA h g−1 over 100 cycles.202 The presence of a few layers of bimetallic Se at the core facilitates the transfer of electrons and K+ ions due to the expanded interlayer spacing (0.74 nm) and high intrinsic conductivity. In addition, the presence of a shell layer of flexible N-doped C facilitates the enhanced transfer of electrons and improves the electrocatalytic activity due to the volume expansion of the active materials induced by pulverization. Luo et al. prepared PB nanocubes and subjected them to direct sulfurization at 400 °C for 3 h under an Ar atmosphere to synthesize improved FeS2–C nanoparticles.203 The prepared core–shell nanoparticles exhibit nanocube shapes and deliver an exceptional electrochemical rate capability of 664 mA h g−1 at a current density of 0.1 A g−1 due to the enhanced electrical conductivity due to volume expansion. Furthermore, the performance of HCSN based catalysts for different MIBs such as LIBs, SIBs, and KIBs was compared and the results are shown in Table 4.
| Catalyst | Reversible capacity (mA h g−1) | Cycle time | Current density (A g−1) | Ref. |
|---|---|---|---|---|
| Co9S8–MoS2–rGO: cobalt and molybdenum sulphide core–shell nanostructures functionalized with RGO, FeOx-N-doped GC: iron oxide and nitrogen-doped graphitic carbon, GaP: gallium phosphide, GaP-C: pyrolyzed GaP, SnS: tin sulfide, Sn4P3-graphite: tin phosphide graphite, Mn1−xFexP: phosphorus-containing manganese and iron, Ge–N-doped graphene: germanium–nitrogen-doped graphene yolk–shell nanoarchitectures, Fe2P-GC: iron and phosphide doped graphitized carbon, Ni2P-pGN: nickel phosphide on a porous graphene network, PNAF-NP: poriferous nanoflake-assembled flower-like nickel tetraphosphide, CoP-C-RGO-NF: cobalt phosphide with carbon and RGO on nickel foam, FeS2–C: pyrite based carbon, SnS2-NGS: tin sulfide on nitrogen-doped graphene sheets, MoSe2–C: carbonized molybdenum selenide, FexO-NFLG-240: iron oxide on N-doped few-layer graphene framework, Sb-NPMC: antimony-nanoparticles encapsulated in nitrogen and phosphorus co-doped mesoporous carbon nanofibers, Co9S8-N-C-MoS2 DHNCs: N,S doped cobalt and molybdenum carbon. | ||||
| LIBs | ||||
| POP-ZIF-8 | 538 | 250 | — | 22 |
| MCNCmSPs | 578 | 60 | 0.1 | 177 |
| Co9S8–MoS2–rGO | 2014.5 | 200 | 0.3 | 204 |
| Ge–N-doped graphene | 1220 | 1000 | 1 C | 205 |
| FeOx–N-doped GC | 1071 | 1000 | 1.0 | 206 |
| MoS2 graphene quantum dots | 1031 | 80 | 0.1 | 207 |
| GaP | 305 | 60 | 0.5 | 208 |
| GaP–C | 832 | 100 | 0.5 | 208 |
| Co9S8–graphene nanocomposites | 754 | 100 | 0.1 | 209 |
| Sn4P3-graphite | 651 | 100 | 0.1 | 210 |
| Mn1−xFexP | 506 | 40 | 0.1 | 211 |
| Fe2P-GC | 592 | 200 | 0.1 | 212 |
| Ni2P-pGN | 457 | 500 | 0.3 | 213 |
| SIBs | ||||
| FeS2–FeSe2 | 395 | 270 | 1 | 189 |
| TiO2−x-NC | 245.6 | 200 | 0.1 | 191 |
| Ni2P-pGN | 181 | 100 | 0.2 | 213 |
| NiP3 | 980 | 15 | 0.1 | 214 |
| PNAF-NP | 456.34 | 300 | 0.2 | 215 |
| Sn4P3 | 718 | 100 | 0.1 | 216 |
| Sn4P3–C | 701 | 50 | 0.1 | 217 |
| CoP-C-RGO-NF | 473 | 100 | 0.1 | 218 |
| Ni2P–C | 433 | 100 | 0.05 | 219 |
| FeS2–C | 551 | 100 | 0.1 | 220 |
| FeS2–C | 330 | 800 | 2.0 | 220 |
| SnS2-NGS | 450 | 100 | 0.2 | 221 |
| Co3S4-polyaniline | 253 | 100 | 0.2 | 222 |
| FeS-rGO | 547 | 50 | 0.5 | 223 |
| KIBs | ||||
| KFeC2O4F | 112 | 2000 | 0.2 | 192 |
| Fe7S8-MCC | 336 | 100 | 0.2 | 193 |
| Fe7S8-MCC | 267 | 500 | 1.0 | 193 |
| KPBNPs | 68.5 | 50 | 0.1 | 196 |
| PB-SSM | 96.8 | 305 | 1 | 197 |
| K0.5MnO2 | 120 | 400 | 0.4 | 199 |
| MoSe2–C | 266 | 1000 | 1 | 200 |
| CoSe2–N-doped C | 442 | 120 | 0.1 | 201 |
| FMSC | 298 | 100 | 0.2 | 202 |
| FeS2-C | 664 | 100 | 0.1 | 203 |
| S,N co-doped thin carbon | 320 | 100 | 0.05 | 224 |
| FexO-NFLG-240 | 423 | 100 | 0.05 | 225 |
| Sb-NPMC | 130 | 100 | 1 | 226 |
| Co9S8-N-C-MoS2 | 100 | 100 | 1 | 227 |
| N-doped porous carbon | 144.4 | 5000 | 1 | 228 |
2.3. HCSNs for metal–air-batteries (MABs)
Similar to LIBs, MABs have received considerable attention from the research community. Although LIBs are advantageous for energy storage applications, their construction entails the use of expensive materials. They also suffer from poor recyclability, electrolyte degradation, blockage of cathodic reaction sites by the electrolyte, solid electrolyte interface formation, and large overpotentials.229,230 In addition, the availability of Li in the earth's crust is limited. MABs are widely used to overcome some of the disadvantages associated with LIBs.231,232 Many types of MABs have been reported, and they are differentiated based on the metal used to construct the anode. Important examples include Li, Zn, Na, Al, Mg, Fe, and Sn–air batteries. Among the various MABs, Li, Zn, and Na–air batteries have been widely investigated in energy storage applications.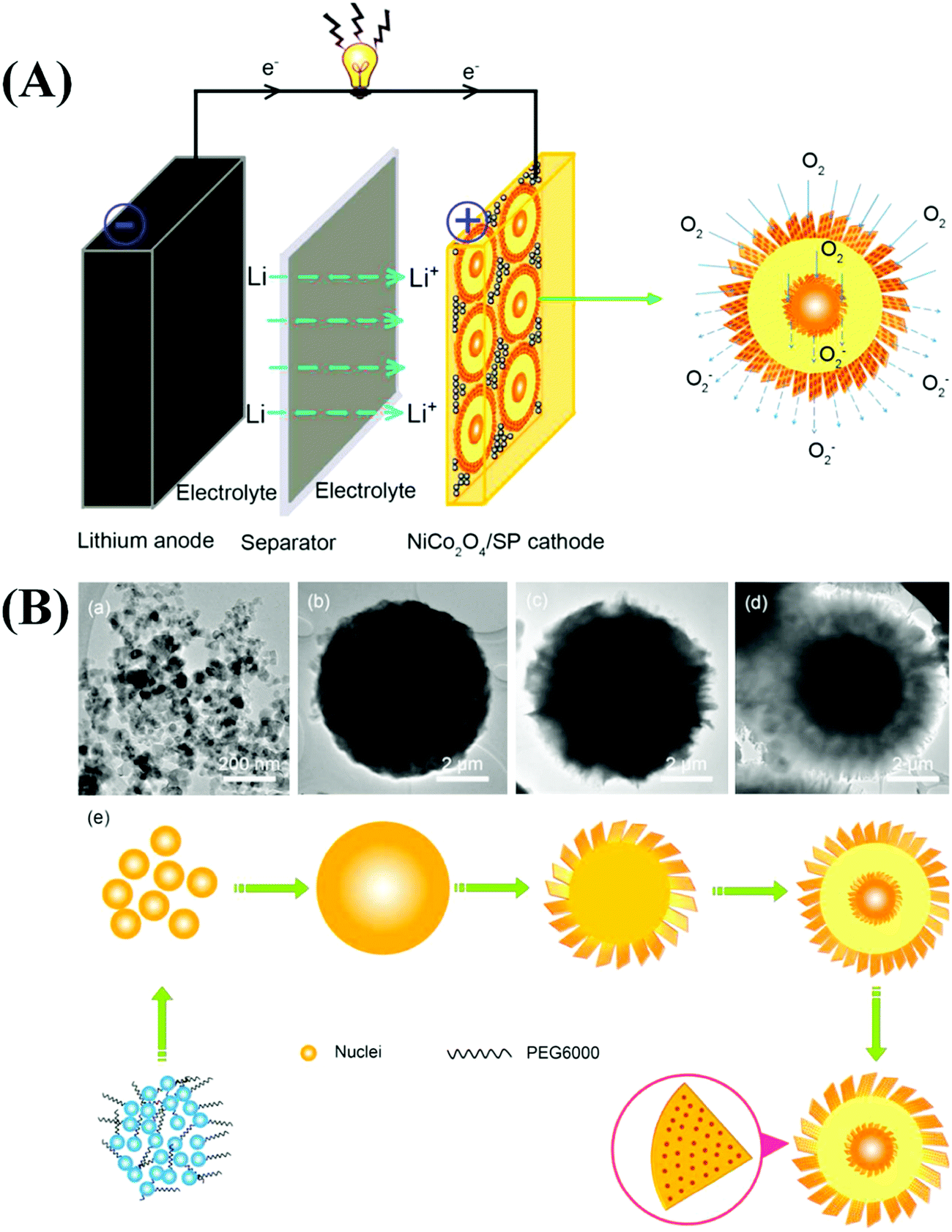 | ||
| Fig. 10 (A) Representative schematic illustration of the Li–O2 battery catalyzed by 3D porous NiCo2O4 core–shell microspheres. (B) Time-dependent experiment of NiCo2O4 at different reaction times: (a) 1 h, (b) 3 h, (c) 5 h, and (d) 10 h, and (e) the corresponding scheme of the reaction process. Reproduced from ref. 239 with permission from Elsevier Ltd. | ||
Konderi et al. synthesized inexpensive trimolybdenum phosphide (Mo3P) nanoparticles with exceptional bifunctional activities toward the OER and ORR and demonstrated their application in LABs.240 The fabricated Mo3P electrode had a long cycle life and a low overpotential and achieved current densities of 7.21 and 6.85 mA cm−2 at 2 and 4.2 V, respectively, vs. Li/Li+ in the presence of an O2-saturated non-aqueous electrolyte. Moreover, the catalyst showed low charge and discharge overpotentials of 270 and 80 mV, respectively, with low ORR and OER overpotentials of 4 and 5.1 mV, respectively. The corresponding Tafel slopes were 35 and 38 mV dec−1, respectively. MOF-based FeCo alloy cores containing N-doped shell nanoparticles encapsulated in CNTs were prepared via pyrolysis at 600 °C. These materials demonstrated superior electrocatalytic performance in LIBs.241 Exemplary bifunctional catalytic activity was observed in an alkaline electrolyte with a ΔE (EOER@10 mA cm−2 − EORR@-1 mA cm−2) value of 0.79 V. Moreover, the catalyst exhibited remarkable Li–O2 charge/discharge cycling stability (up to 40 cycles) and retained its catalytic activity after several cycles. The strong chemical/electrical coupling of the FeCo alloy core to the NC shell facilitated electron transfer and protected the alloy from agglomeration or dissolution.241
To date, a diverse range of electrocatalysts with superior ORR and OER activities have been developed using perovskite oxides, heteroatom-doped C, and transition metal nitrides/oxides/sulfides, which are applicable in ZABs.242–245 HCSNs with bifunctional properties have been extensively used in ZABs.246–258 An MCO/CNF@NC catalyst has demonstrated outstanding catalytic performance in ZABs with a discharge capacity of 478 F g−1 at a discharge current density of 1 A g−1.259 The OER activity of the catalyst was associated with a low overpotential (0.41 V) at a current density of 10 mA cm−2 and matching onset (1 V vs. RHE) and half-wave potential (0.76 V vs. RHE) values were obtained with an ORR activity that was approximately 40 mV lower than that of a Pt–C catalyst. The catalyst also exhibited a lower ORR/OER potential gap (0.88 V), which indicated the excellent bifunctional behavior of the catalyst. Furthermore, the specific capacitance and energy density values of the synthesized catalyst were determined to be 695 mA h g−1zn and 778 W h kg−1zn, respectively, at 20 mA cm−2.
Chen et al. synthesized an N-doped C material containing Fe-enriched FeNi3 intermetallic nanoparticles by plasma engineering using a maximum amount of iron(II) chloride tetrahydrate and adjustable amounts of nickel(II) acetate tetrahydrate in the presence of pyridine, and the obtained material was further pyrolyzed at 800 °C for 1 h under an N2 atmosphere (1.5 cc min−1).260 The synthesized Fe-enriched FeNi3/NC electrocatalyst had excellent bifunctional catalytic activities toward the ORR and OER. The schematic of the reaction pathway of the corresponding ZAB is shown in Fig. 11(a).260 The open circuit potential of the Fe-enriched FeNi3/NC electrocatalyst was 1.43 V (Fig. 11(b)).260 In addition, the synthesized material delivered a low charge/discharge voltage gap (0.89 V) with a peak power density, current density, and specific capacity of 89 mW cm−2, 200 mA cm−2, and 734 mA h g−1, respectively, for 1 mg cm−2 of catalyst loading. The corresponding ZAB exhibited appreciable stability, cycle durability, and rechargeability (Fig. 11(c)–(e)).260 On the other hand, the corresponding values for the electrode prepared using 20 wt% Pt–C and Ir–C were 74 mW cm−2, 160 mA cm−2, and 707 mA h g−1, respectively. When the Fe-enriched FeNi3–NC electrocatalyst was utilized for optical imaging by connecting a red light-emitting diode (LED) to two liquid ZABs (in series), continuous illumination was observed for over 10 days (Fig. 11(f)).260
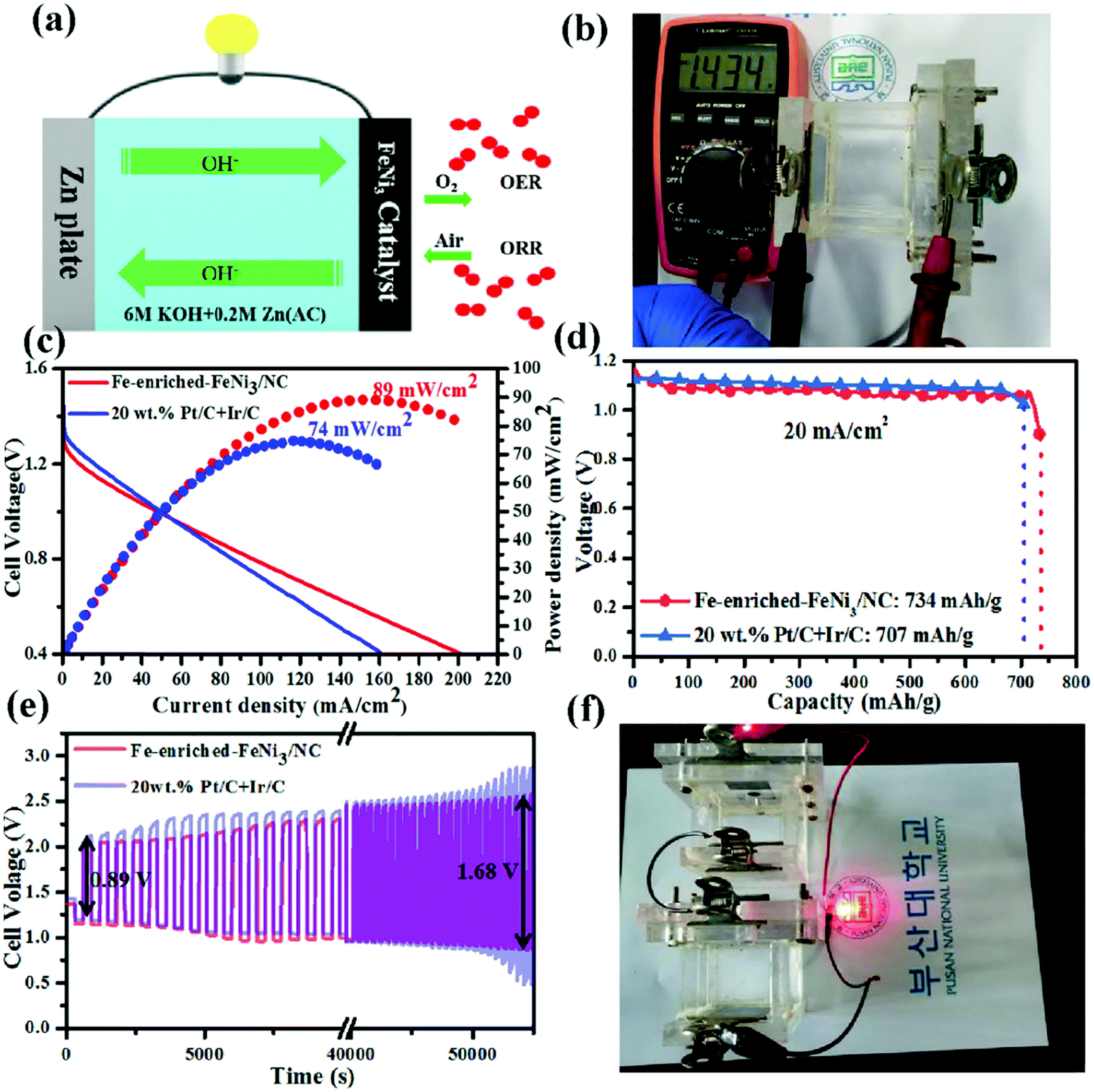 | ||
| Fig. 11 (a) A schematic illustration of the home-made Zn–air battery. (b) OCV of the as-assembled Zn–air battery with an Fe-enriched-FeNi3/NC air–cathode. (c) The discharge polarization curves and the corresponding power density curves of the battery with Fe-enriched-FeNi3/NC and 20 wt% Pt/C + Ir/C air-electrodes, respectively. (d) Specific capacity plots of Fe-enriched-FeNi3/NC and 20 wt% Pt/C + Ir/C based Zn–air batteries tested at 20 mA cm−2. (e) Galvanostatic discharge–charge cycling curves of Fe-enriched-FeNi3/NC and 20 wt% Pt/C + Ir/C based batteries at a current density of 10 mA cm−2, respectively. (f) Photograph of a red LED lightened by two series-connected liquid Zn–air batteries with an Fe-rich-FeNi3/NC air–cathode. Reproduced from ref. 260 with permission from Elsevier Inc. | ||
A Co@CNT based CSN bifunctional electrocatalyst obtained from ZIF-67 and melamine via a simple surface treatment demonstrated exceptional stability, low charge/discharge voltage gap, and larger power density in ZABs.257Fig. 12(a) shows a schematic illustration of the synthesis of Co@CNT from ZIF-67.257 ZIF-67 was modified by mixing with melamine which acted as a carbon and nitrogen source. The sample was subsequently pyrolyzed at 700 °C for 3 h under an N2 gas flow at a rate of 2 °C min−1. The electrode fabricated from the synthesized Co@CNT CSNs delivered a superior bifunctional ORR and OER electrocatalytic activity (Fig. 12(a)).257 DFT studies were conducted to predict the free energy of the bifunctional electrocatalyst in an alkaline (pH = 13) solution and a probable mechanism was investigated (Fig. 12(b)).257 The ORR catalytic reaction mediated by Co@CNT occurred via four-electron transfer pathways with the adsorption of three active species (OOH*, OH*, and O*) on the catalytic surface. The Gibbs free energies (ΔG values) corresponding to the adsorption of OOH*, OH*, and O* were −0.44, −0.56, and −0.41 eV, respectively, while that corresponding to the desorption of OH* was −0.41 eV. The highest ΔG value observed corresponded to the adsorption of OH*, which indicated that the adsorption of OH* constituted the rate-determining step (RDS) of the catalyst. In addition, the low overpotential value of 0.56 eV indicates the excellent ORR performance of the catalyst. The ORR catalytic behavior of the catalyst was investigated in alkaline and acidic environments using 0.1 M KOH and 0.5 M H2SO4, respectively, and the obtained results were correlated with K–L plots acquired at different rotating speeds of the catalyst electrodes in the respective electrolytes (Fig. 12(c)–(h)).257 The Co–CNT electrocatalyst exhibited appreciable ORR stability and OER performance with a low overpotential and high catalytic activity. These results demonstrate the potential utility of the bifunctional catalyst in ZABs. Here, Zn foil acts as the anode and the Co–CNT catalyst (0.5 mg cm−2) loaded on carbon cloth (CC)-GDL acts as the cathode, while zinc chloride (ZnCl2, 0.2 M) containing 6 M KOH behaves as the electrolyte. The Co–CNT (1:1) catalyst delivers superior battery performance at a high current density with a reduced charge/discharge voltage gap (a current density of 50 mA cm−2 necessitated a voltage gap as low as 1 V) compared with that observed in the case of the Pt–C + Ir–C catalyst. Additionally, Co–CNT (1:1) delivered a peak power density of 149.2 mW cm−2 which was higher than that offered by the Pt–C–Ir–C catalyst (90 mW cm−2). These results demonstrate the outstanding rechargeability and reactivity of the Co–CNT (1:1) catalyst.257 According to the galvanostatic charge/discharge cycling curve of the Co–CNT (1:1) catalyst determined at 2 mA cm−2, a discharge voltage as high as 1.2 V was observed with a modest charge voltage of 2 V, which corresponded to a charge–discharge gap of 0.8 V and an energy efficiency of over 60%. These results, which were maintained for over 40 h (120 cycles), indicated the superior stability of the catalyst due to its efficient electronic conductivity, controlled surface texture with a high surface area, interfacial physicochemical properties, and high intrinsic activity.257 Zhao et al. developed a B and N heteroatom doped ultrathin carbon nanosheet via a simple and effective sodium chloride (NaCl)-assisted pyrolysis method.261 The BN–C produced is made up of ultrathin nanosheets as thin as 5 nm and has a large surface area of 1085 m2 g−1 with both micro- and mesopores. BN–C has strong ORR activity, with a half-wave potential of 0.8 V. Furthermore, in comparison to the commercial Pt–C + RuO2 based battery, the BN–C + RuO2 based Zn–air battery with a liquid electrolyte demonstrates improved performance and cycle stability after 1000 continuous discharge–charge cycles (for 14 days of operation).261 The specific capacity of the BN–C + RuO2 catalyst is determined to be 802 W h kgZn−1 at 5 mA cm−2, which is higher than the capacity of the Pt/C + RuO2 based Zn–air battery, which was only 774 W h kgZn−1 based on the amount of Zn that was consumed.261 The author has generated atomically distributed Co sites that are anchored on interconnecting B,N-doped carbon nanotubes (B, N, Co–C nanotubes).262 These nanotubes are prepared using a simple molten-salt-assisted pyrolysis of B–N–Co precursors which comes after chemical etching. Various other HCSN based materials have demonstrated efficient catalytic activity in ZABs. We also compared and summarized the results of some of the HCSN catalysts used in ZIBs (Table 5).
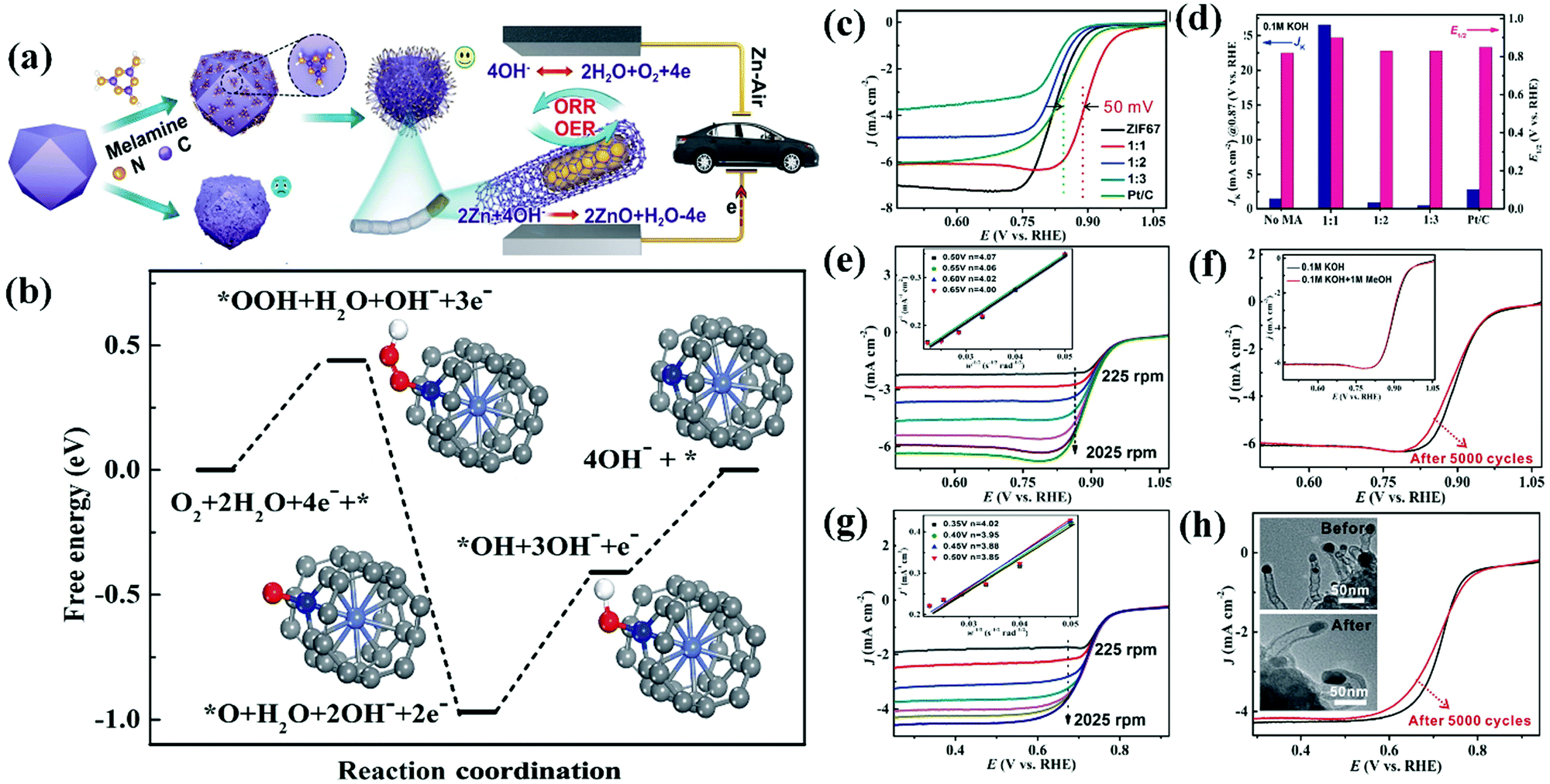 | ||
| Fig. 12 (a) Schematic illustration of the synthetic strategy for Co@CNTs. (b) Free-energy diagram for the ORR on Co@CNTs in alkaline (pH = 13) media. ORR activities of Co@CNTs in both alkaline and acidic media. (c) LSV curves and (d) JK and E1/2 of Co@CNTs and other comparative catalysts in 0.1 M KOH. (e) LSV curves of the Co@CNTs at different rotating rates. Inset : the K–L plot at various potentials. (f) LSV curves of Co@CNTs and the sample after 5000 cycles (the inset shows 0.1 M KOH solution with and without 1 M methanol). (g) LSV curves of Co@CNTs at different rotating rates in 0.5 M H2SO4. Inset: the K–L plot at various potentials. (h) LSV curves of Co@CNTs and the sample after 5000 cycles (inset: TEM images before and after 5000 cycles). MA:melamine; 1:1, 1:2 and 1:3 are the ratios of weightZIF-67:weightmelamine, respectively. Reproduced from ref. 257 with permission from Science Press and Dalian Institute of Chemical Physics, Chinese Academy of Sciences. Published by Elsevier B.V. and Science Press. | ||
| Catalyst | Specific capacity (mA h gzn−1)@j (mA cm−2) | Energy density (Wh kgzn g−1)@j (mA cm−2) | Power density (mW cm−2) | Current density (mA cm−2) | Ref. |
|---|---|---|---|---|---|
| CMT–CNT: carbon microtube@nanotube core–shell, Co4N-CNW-CC: metallic Co4N with carbon fiber network on carbon cloth, Co-SAs-NC: Co single atoms on N-doped carbon, N-GCNT-FeCo-3: bimetal FeCo nanoparticles enveloped by N-doped graphitic carbon nanotubes, CoNi-NHCS-TUC-3: cobalt–nickel supported on nitrogen-doped hollow spherical carbon and tubular carbon, GNCNTs-4: nitrogen-doped carbon nanotubes-graphene, ZOMC: pyrolysis of ZIF-67 to form ordered macroporous carbon, CoNi-BCF: CoNi alloy nanoparticles supported by a butterfly wing-derived carbon framework, NDGs-800: pyridinic-N-dominated doped defective graphene, FeNC–S–FexC–Fe: sulfuration of an Fe–N–C catalyst containing FexC–Fe species, FeS–Fe3C-NS-C-900: FeS–Fe3C nanoparticles embedded in a porous N,S-dual doped carbon, NCN-1000-5: nitrogen doped ultrathin carbon nanosheets, NPCS-900: nitrogen and phosphorous co-doped carbon spheres, SCoNC: monodisperse Co single atoms on a nitrogen-doped 2D carbon nanosheet, CoZn-NC-700: bimetallic nitrogen-doped carbon, NiCo2S4-gC3N4-CNT: NiCo2S4-graphitic carbon nitride/carbon nanotube, zinc plate-Pt–SCFP-CC: platinum with a perovskite on carbon cloth, zinc plate-Co–Co3O4-NAC-NF: Co single-atom anchored on Co3O4 and nitrogen-doped active carbon on nickel foam, Fe–Co4N–N–C: co-doped Fe–Co4N–N–C nanosheet array. | |||||
| CMT–CNT | 781.7@10 | 930.2@10 | 160.6 | 10 | 255 |
| Co4N-CNW-CC | 774@10 | — | 174 | 10 | 263 |
| Co-SAs-NC | 897.1@20 | — | 105 | 20 | 264 |
| N-GCNT-FeCo-3 | 872.2@100 | 653.2@100 | 89.3 | 100 | 265 |
| CoNi-NHCS-TUC-3 | 756.5 | 957 | 255.9 | 361.8 | 266 |
| GNCNTs-4 | 728 | — | 253 | 383 | 267 |
| ZOMC | 795.3@5 | 969.4@5 | 221.1 | 5 | 268 |
| ZOMC | 697.9@10 | 772@10 | 221.1 | 10 | 268 |
| CoNi-BCF | 710.9@10 | 853.1@10 | 155.1 | 10 | 269 |
| NDGs-800 | 750.8@10 | 872.3 | 115.2 | 10 | 270 |
| FeNC–S–FexC–Fe | 663@10 | 795@10 | 149.4 | 10 | 271 |
| FeS–Fe3C-NS-C-900 | 750 | — | 90.9 | 20 | 272 |
| NCN-1000-5 | 672@10 | 805@10 | 207 | 10 | 273 |
| NPCS-900 | 625@20 | 656.25@20 | 79 | 20 | 274 |
| SCoNC | 690@5 | 945@5 | 194 | 5 | 275 |
| CoZn-NC-700 | 578@10 | 694@10 | 152 | 10 | 276 |
| NiCo2S4-gC3N4-CNT | 485.7@10 | 530.5@10 | 142 | 10 | 277 |
| Zinc plate-Pt–SCFP-CC | 790.4@5 | — | 122 | 10 | 278 |
| Zinc plate-Co–Co3O4-NAC-NF | 721@10 | — | 164 | 10 | 279 |
| Fe–Co4N–N–C | 806@5 | 934 | 105 | 5 | 280 |
An et al. used DFT simulations to assess the surface energetics of NiFe2O4 and FeNi2S4 clusters connected via their (311) or (100) planes and projected the link between the interface and catalysis of these clusters for ZIBs (Fig. 13).281 The increased electrical affinity of oxygen causes the electron to move from the FeNi2S4 domain to the NiFe2O4 domain, according to DFT investigations, which also demonstrate that oxygen prefers to adsorb on surface Ni near the interface. These also suggest that NiFe2O4 and FeNi2S4 domains have a significant electrical connection. The results of their calculations showed that the intermediates *O, *OH, and *OOH scaled linearly, allowing the use of oxygen adsorption energy (E0) as an universal descriptor to forecast and assess the ORR and OER activities.281 It is well known that metal oxide catalysts experience excessively high oxygen adsorption, which slows down the reaction kinetics. Therefore, improving the oxygen electrocatalytic characteristics of the catalysts requires significantly reduced E0 at the NiFe2O4/FeNi2S4 interface.281 Wang et al. also proposed the mechanism of vertically-aligned porous nanoarrays made of ultrafine nitrogen-doped cobalt oxide (NP-Co3O4) nanoparticles (4–5 nm) prepared in situ on carbon cloth (CC) by mildly oxidizing Co-based zeolitic-imidazolate-framework (Co-ZIF) nanoarrays (Fig. 14).282
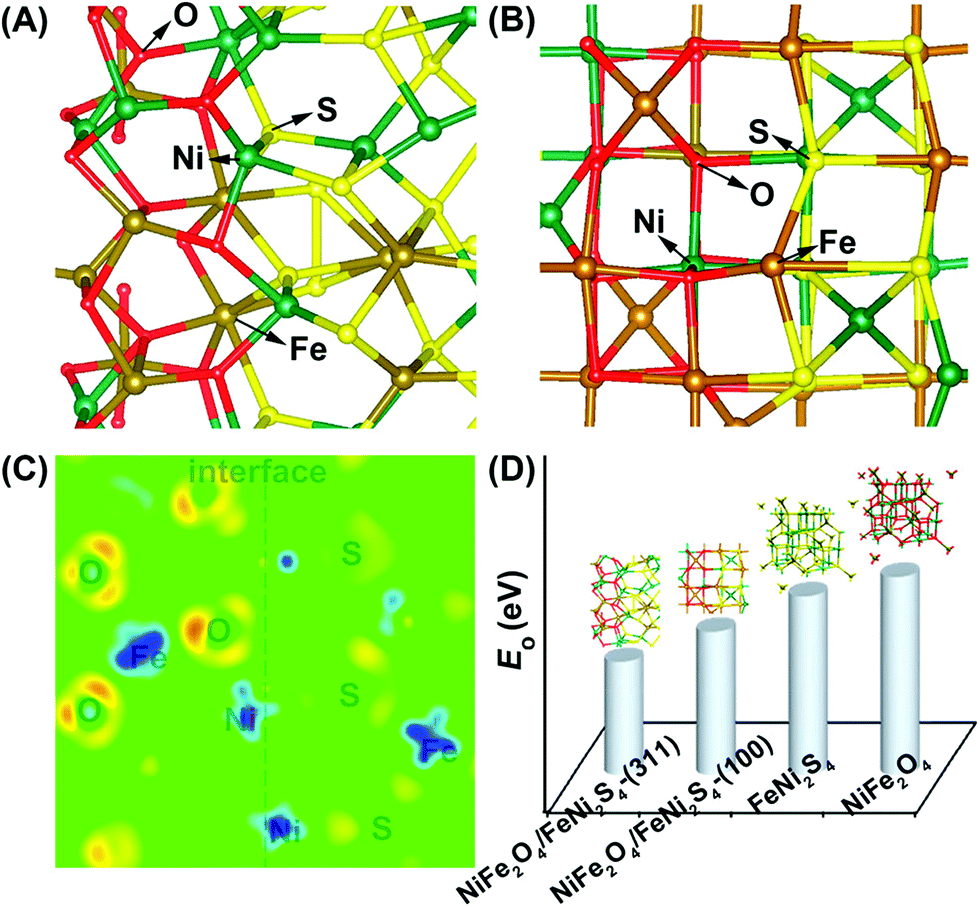 | ||
| Fig. 13 (A), (B) Optimized geometry of oxygen adsorption over the NiFe2O4/FeNi2S4(311) interface model (A) and the NiFe2O4/FeNi2S4(100) interface model (B). (C) Differential charge density of the cross-sectional view of the NiFe2O4/FeNi2S4(311) interface model. The red/light (blue/dark) areas mark an increase (decrease) of the electron density. (D) Computational surface EO comparison of NiFe2O4/FeNi2S4-(311), NiFe2O4/FeNi2S4-(100), FeNi2S4, and NiFe2O4. Reproduced from ref. 281 with permission from the American Chemical Society. | ||
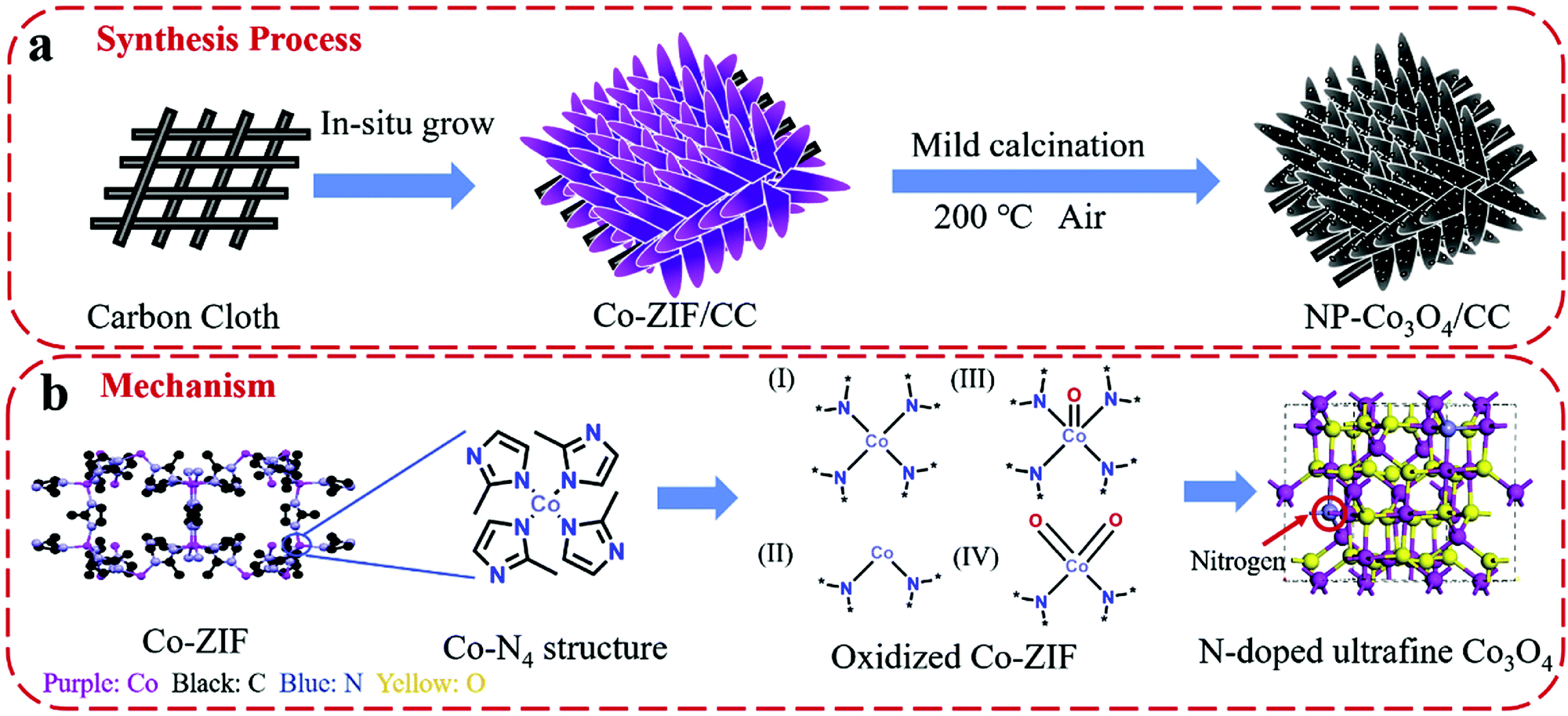 | ||
| Fig. 14 Schematic demonstration of (a) the fabrication process of NP-Co3O4/CC and (b) the proposed mechanism of nitrogen doping in cobalt oxide lattices. Reproduced from ref. 282 with permission from Elsevier B.V. | ||
| Anode: Na ↔ Na+ + e− E° = +2.71 V | (4) |
| Cathode: O2 + 2H2O + 4e− ↔ 4OH− E° = +0.40 V | (5) |
| Overall: 4Na + O2 + 2H2O ↔ 4NaOH E° = 3.11 V | (6) |
The charge–discharge behavior of the GNS-MC catalyst in an aqueous SAB was characterized by a lower ΔV (ΔV denotes the voltage differences between charge and discharge voltages) (115 mV) compared with the values observed for the Pt–C (179 mV), Ir–C (364 mV), and C paper (698 mV) catalysts, which was indicative of the outstanding performance of the catalyst. The charge–discharge cycle stability of the GNS-MC electrocatalyst was examined for 10 cycles, and the results indicated that the electrocatalytic properties of the material were sustained.289 Recent research has focused on improving the performance of the electrocatalysts used in SABs.290–293 The reported results demonstrate the outstanding activity of the electrocatalysts used in SABs and establish the excellent utility of SABs in ECS applications.294,295
2.4. HCSNs for Li–sulfur batteries (LSBs)
Similar to the metal-ion and metal–air batteries discussed in previous sections, LSBs have attracted considerable attention from the research community.296 LSBs are noted for being cost-effective and demonstrating enhanced gravimetric energy density and safety.297–301 The theoretical energy density of Li–S rechargeable batteries can be as high as 2600 W h kg−1, which makes them highly suitable for application in various energy storage systems.299,302 S is highly inexpensive compared to transition metals (e.g., Co and Mn) and phosphates, which are used to develop Li-based batteries.297 Moreover, the synthesis of LSB electrode materials is considerably easier and safer compared to those of Li-ion and Li–air battery electrode materials. Therefore, S can be widely used to develop rechargeable batteries with high energy densities.297,303 However, LSBs suffer from the incomplete utilization of S due to the insulating nature and low conductivity of S. The instability of the Li metal surface is another disadvantage. The electrode material experiences a considerable volumetric expansion of ∼80% during lithiation which involves pulverization of the active material. These factors result in decreased coulombic efficiency and capacity decay, sluggish redox kinetics, and shuttle effects.297,299 These disadvantages can be addressed by the incorporation of various functional materials in the S cathodes, such as metal compounds, porous carbon, graphene, RGO, and CNTs.297 Enhanced accumulation of S can be achieved in LSBs using a C support. The presence of a C support improves S loading and results in a high surface area and large pore volume. The formation of hierarchical meso–microporous structures with robust mechanical properties is also observed. C supported S delivers superior electrochemical performance due to its improved conductivity.302 Gai et al. recently built a high performance LSB using multifunctional 3D-mesoporous carbon nanocomposites such as RGO with embedded Co nanoparticles and elemental N (Co–NrGO) followed by ground mixture with powdered S at a mass ratio of 1:6 (Co–NrGO–S) used as cathode-separator-coated interlayers and working electrodes in assembled Li–S cells, respectively.296 The LSB cells that were developed exhibited outstanding endurance (905 mA h g−1 in the 250th cycle at 0.5 or 0.2 C), high-rate capabilities (835 mA h g−1, 2.0 C), and superior cycling stability (1070 mA h g−1 in the 100th cycle at 0.2 C). These results also further proved that the lack of Co–NrGO coatings and/or Co nanoparticles in the constructed LSB may be insufficient to ensure enhanced S availability which leads to a poorer performance in the LSB. However, the better performance of Co–NrGO–S is mostly due to the catalytic capacity of the inserted metallic Co nanoparticles for the reversible transformation of S8 into Li2S.296
Zhen et al. prepared S-doped ordered meso@microporous core–shell (S/MMCS) C nanoparticles and compared their electrochemical performance with those of S doped mesoporous C (S/mesoC) and S doped microporous C (S/microC) materials in LSBs.302 The cores of the developed structures facilitate increased S loading due to their ordered porous structures with high surface areas and pore volumes, whereas the shells having microporous C structures accommodate lower amounts of S. The overall loading of S in the meso–microporous structure is thus optimized (Fig. 15(A)).302 The prepared S/mesoC, S/microC, and S/MMCS electrodes exhibit excellent specific capacities of 1123 (739), 1071 (322), and 1212 (734), respectively, after 2 charge/discharge cycles (Fig. 15(B)(a) and (b)).302 The S/MMCS electrodes exhibit appreciable capacity retention, which is evidenced by the capacity values of 1014 (615) and 837 (506) mA h g−1 observed after 100 and 200 cycles, respectively. They also exhibit a high current density of 0.5 C (here C refers to the capacity of a battery), capacity retention exceeding 80%, and 95% coulombic efficiency (Fig. 15(c)).302 The LSB cell prepared using S/MMCS exhibits discharge capacities of 1182 (715), 1038 (628), 930 (563), 840 (508), and 605 (366) mA h g−1 at 0.1, 0.2, 0.5, 1, and 2 C, respectively (Fig. 15B(d)).302 According to these results, the superior electrical conductivity, large pore volume, and high S loading of S-MMCS ensure its utility in LSBs. Similarly, various S-doped C-containing CSNs were synthesized by various techniques and applied successfully in LSBs.304–307 Two dimensional S (5 mg cm−2)-doped C yolk–shell nanosheets were prepared, which displayed volumetric and areal capacitance values of 1330 mA h cm−3 and 5.7 mA h cm−2, respectively. The areal capacitance was enhanced (up to 11.4 mA h cm−2) upon increasing the S doping from 5 to 10 mg cm−2.304 Tao et al. synthesized an efficient anode material for Li–Na-ion batteries using multi-heteroatom (N, P, and S) decorated C containing magnetite-based iron oxide (Fe3O4) nanospheres which exhibited excellent electrochemical performance, as evidenced by the high specific capacity and cycle stability.305 Fe3O4@doped C@graphene (Fe3O4@C@G) 3D structured anode materials were synthesized, which delivered a reversible capacity of 919 mA h g−1 at 0.1 A g−1 in LIBs. The corresponding value observed for an S-ion battery was 180 mA h g−1 at 0.1 A g−1 after 600 cycles.305
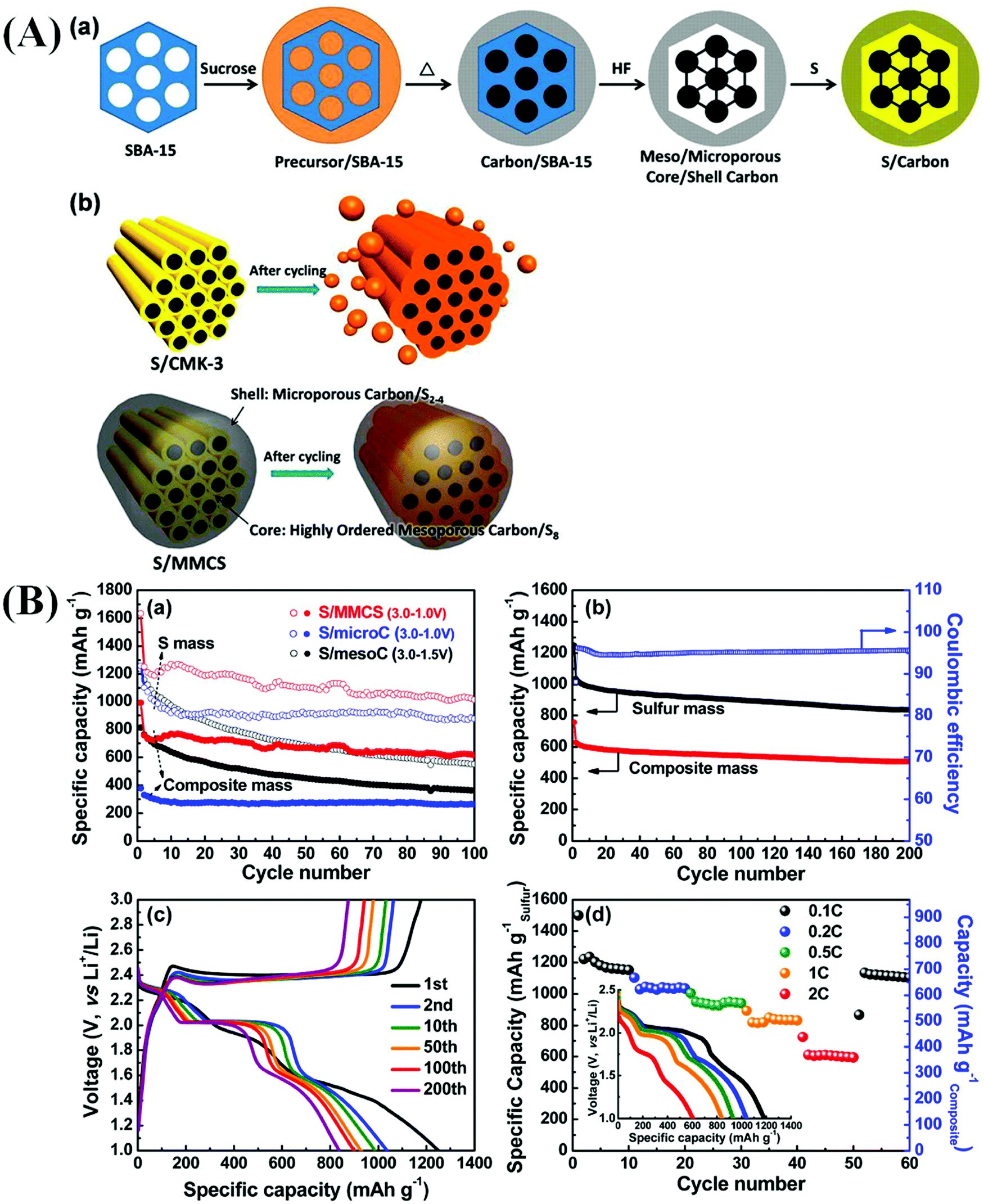 | ||
| Fig. 15 (A) Schematic of preparation of the highly ordered meso–microporous core–shell (MMCS) carbon and sulfur/carbon composite (a). Schematic of the mesoC/S8-microC/S2–4 core–shell structure (b). (B) Cycle performances of S/MMCS, S/mesoC, and S/microC at 0.1C (a and b), voltage profiles during cycling at 0.5C (c), and rate capacities at various C-rates of S/MMCS in the voltage range of 3.0–1.0 V vs. Li+/Li (d). The inset of (d) shows the discharge voltage profiles at current densities from 0.1 to 2C. Reproduced from ref. 302 with permission from the American Chemical Society. | ||
3. Conclusions
In this review, we have briefly described the various properties of HCSNs and the recent advances made in their applications to fuel cell and battery technologies. The fuel cell activity of HCSNs has been improved by optimizing the synthetic processes and adopting different approaches, such as the insertion of dissimilar metals in the HCSN cores, formation of alloys with unassociated metals, controlling the size, shape (or facet), and morphology, and application of sacrificial materials as supports.54,308 The main advantage of CSNs is the tunability of their core and shell compositions. Tuning is performed by incorporating one or more low-cost transition metals or metal oxides either in the core or in the shell to reduce the proportion of Pt, which significantly improves the catalytic activity and tolerance against different environments. Some of the important challenges encountered during the application of CSNs in fuel cells include insufficient catalytic stability, unoptimized catalyst designs, and insufficient understanding of ORR activity and mechanisms.54The catalytic activity of CSNs in fuel cells is enhanced by doping them with heteroatoms, which increases the number of active sites and defects while modifying the facets of the nanoparticles on the shell. Rigorous studies are necessary on the structural modifications of the materials induced by catalytic applications along with theoretical modeling and experimental measurements. In addition, optimization of the MEA fabrication process is required by the respective catalyst inks/layers and the associated testing parameters.54 The use of Pt in electrode fabrication should be minimized to reduce costs and enhance the stability and tolerance against the external environment. Earth-abundant elements can be successfully used to reduce the proportion of Pt in the CSN cores.309 However, the 3d transition metals used in CSN cores suffer from electrochemical corrosion. This disadvantage can be addressed by using carbides, nitrides, and oxides to construct the cores of CSNs featuring noble metal shells.309
Bifunctional ORR/OER catalysts have excellent electrochemical properties, which make them highly useful for various types of battery applications. The performances of bifunctional electrocatalysts are improved by controlling the crystal facets, defects, morphologies, and phases through strain engineering and the introduction of heteroatoms and the formation of M–N–C structures. Highly active electrocatalysts for fuel cells and Li-ion–air–S batteries are developed by doping C nanomaterials with heteroatoms. Research aimed at the continuous improvement of performance and stability, and reduction of the final cost of the electrocatalysts is highly important for addressing the future energy demands. More studies are necessary to develop improved catalysts for use in fuel cell and battery applications.
Author contributions
S. N. contributed to the conceptualization, writing the original draft, and review and editing. M. D. and N. P. contributed to the literature review, methodology, and summary. K. P. contributed to the outline of the review article as well as review and editing. K. H. P. contributed to the conceptualization, supervision, review and editing, and funding acquisition. All the authors discussed and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) grant funded by the Korea Government (MSIP) (NRF-2020R1I1A3067208).Notes and references
- S. Nagappan, M. Duraivel, S. A. Hira, K. Prabakar, C.-S. Ha, S. H. Joo, K. M. Nam and K. H. Park, J. Mater. Chem. A, 2022, 10, 987–1021 RSC.
- S. W. Kim, J. Ryu, C. B. Park and K. Kang, Chem. Commun., 2010, 46, 7409–7411 RSC.
- E. Lim, J. Changshin, M. S. Kim, M. H. Kim, J. Chun, H. Kim, J. Park, K. C. Roh, K. Kang, S. Yoon and J. Lee, Adv. Funct. Mater., 2016, 26, 3711–3719 CrossRef CAS.
- H. P. Feng, L. Tang, G. M. Zeng, J. Tang, Y. C. Deng, M. Yan, Y. N. Liu, Y. Y. Zhou, X. Y. Ren and S. Chen, J. Mater. Chem. A, 2018, 6, 7310–7337 RSC.
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. Yu, Z. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. F. Toney and A. Nilsson, Nat. Chem., 2010, 2, 454–460 CrossRef CAS PubMed.
- A. M. El-Toni, M. A. Habila, J. P. Labis, Z. A. Alothman, M. Alhoshan, A. A. Elzatahry and F. Zhang, Nanoscale, 2016, 8, 2510–2531 RSC.
- G. Li and Z. Tang, Nanoscale, 2014, 6, 3995–4011 RSC.
- K. Mondal and A. Sharma, RSC Adv., 2016, 6, 83589–83612 RSC.
- K. C. Ho and L. Y. Lin, J. Mater. Chem. A, 2019, 7, 3516–3530 RSC.
- M. Yusuf, S. Song, S. Park and K. H. Park, Appl. Catal., A, 2021, 613, 118025 CrossRef CAS.
- A. Sivanantham, P. Ganesan, L. Estevez, B. P. McGrail, R. K. Motkuri and S. Shanmugam, Adv. Energy Mater., 2018, 8, 1702838 CrossRef.
- R. G. Chaudhuri and S. Paria, Chem. Rev., 2012, 112, 2373–2433 CrossRef PubMed.
- C. Wang, N. M. Markovic and V. R. Stamenkovic, ACS Catal., 2012, 2, 891–898 CrossRef CAS.
- N. V. Long, M. Ohtaki, T. D. Hien, J. Randy and M. Nogami, Electrochim. Acta, 2011, 56, 9133–9143 CrossRef.
- M. Sun, H. Liu, J. Qu and J. Li, Adv. Energy Mater., 2016, 6, 1600087 CrossRef.
- Y. Shabangoli, M. S. Rahmanifar, M. F. El-Kady, A. Noori, M. F. Mousavi and R. B. Kaner, Energy Storage Mater., 2018, 11, 282–293 CrossRef.
- Q. Lu, J. Yu, X. Zou, K. Liao, P. Tan, W. Zhou, M. Ni and Z. Shao, Adv. Funct. Mater., 2019, 29, 1904481 CrossRef.
- L. Wang, Y. Sun, S. Zhang, H. Di, Q. Liu, X. Du, Z. Li, K. Yang and S. Chen, ChemElectroChem, 2021, 8, 4286–4295 CrossRef CAS.
- Z. Zhou, Y. Kong, H. Tan, Q. Huang, C. Wang, Z. Pei, H. Wang, Y. Liu, Y. Wang, S. Li, X. Liao, W. Yan and S. Zhao, Adv. Mater., 2022, 34, 2106541 CrossRef CAS PubMed.
- K. H. Wu, Y. Liu, X. Tan, Y. Liu, Y. Lin, X. Huang, Y. Ding, B. J. Su, B. Zhang, J. M. Chen, W. Yan, S. C. Smith, I. R. Gentle and S. Zhao, Chem Catal., 2022, 2, 372–385 CrossRef.
- Z. Jian, P. Liu, F. Li, P. He, X. Guo, M. Chen and H. Zhou, Angew. Chem., Int. Ed., 2014, 53, 442–446 CrossRef CAS PubMed.
- Y. Zhou, Q. Huang, C. T. J. Low, R. I. Walton, T. McNally and C. Wan, New J. Chem., 2019, 43, 5632–5641 RSC.
- X. Huang, X. Zou, Y. Meng, E. Mikmeková, H. Chen, D. Voiry, A. Goswami, M. Chhowalla and T. Asefa, ACS Appl. Mater. Interfaces, 2015, 7, 1978–1986 CrossRef CAS PubMed.
- J.-C. Li, P.-X. Hou and C. Liu, Small, 2017, 13, 1702002 CrossRef PubMed.
- M. Li, J. Zhou, Y. G. Bi, S. Q. Zhou and C. H. Mo, Chem. Eng. J., 2020, 383, 123152 CrossRef CAS.
- Q. Abbas, R. Raza, I. Shabbir and A. G. Olabi, J. Sci.: Adv. Mater. Devices, 2019, 4, 341–352 Search PubMed.
- Y. He, X. Han, Y. Du, B. Zhang and P. Xu, Polymers, 2016, 8, 366 CrossRef PubMed.
- X. Wang, G. Sun, P. Routh, D. H. Kim, W. Huang and P. Chen, Chem. Soc. Rev., 2014, 43, 7067–7098 RSC.
- G. L. Chai, K. Qiu, M. Qiao, M. M. Titirici, C. Shang and Z. Guo, Energy Environ. Sci., 2017, 10, 1186–1195 RSC.
- A. Sarapuu, E. Kibena-Põldsepp, M. Borghei and K. Tammeveski, J. Mater. Chem. A, 2018, 6, 776–804 RSC.
- H. Cui, Z. Zhou and D. Jia, Mater. Horiz., 2017, 4, 7–19 RSC.
- N. Mansor, T. S. Miller, I. Dedigama, A. B. Jorge, J. Jia, V. Brázdová, C. Mattevi, C. Gibbs, D. Hodgson, P. R. Shearing, C. A. Howard, F. Corà, M. Shaffer, D. J. L. Brett and P. F. McMillan, Electrochim. Acta, 2016, 222, 44–57 CrossRef CAS.
- Z. Song, N. Cheng, A. Lushington and X. Sun, Catalysts, 2016, 6, 116 CrossRef.
- Z. Cui, G. Fu, Y. Li and J. B. Goodenough, Angew. Chem., Int. Ed., 2017, 56, 9901–9905 CrossRef CAS PubMed.
- H. P. Feng, L. Tang, G. M. Zeng, Y. Zhou, Y. C. Deng, X. Ren, B. Song, C. Liang, M. Y. Wei and J. F. Yu, Adv. Colloid Interface Sci., 2019, 267, 26–46 CrossRef CAS PubMed.
- D. T. Tran, T. Kshetri, D. C. Nguyen, J. Gautam, V. H. Hoa, H. T. Le, N. H. Kim and J. H. Lee, Nano Today, 2018, 22, 100–131 CrossRef.
- X. Bai, D. Cao and H. Zhang, Inorg. Chem. Front., 2020, 7, 411–420 RSC.
- Y. Zhao, Q. Lai, J. Zhu, J. Zhong, Z. Tang, Y. Luo and Y. Liang, Small, 2018, 14, 1704207 CrossRef PubMed.
- J. Zhang, Z. Xia and L. Dai, Sci. Adv., 2015, 1, e1500564 CrossRef PubMed.
- I. Staffell, D. Scamman, A. Velazquez Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC.
- L. Carrette, K. A. Friedrich and U. Stimming, ChemPhysChem, 2000, 1, 162–193 CrossRef CAS PubMed.
- G. J. K. Acres, J. Power Sources, 2001, 100, 60–66 CrossRef CAS.
- S. Guo and E. Wang, Nano Today, 2011, 6, 240–264 CrossRef CAS.
- B. Cai, S. Henning, J. Herranz, T. J. Schmidt and A. Eychmüller, Adv. Energy Mater., 2017, 7, 1700548 CrossRef.
- L. Shi, X. Lin, F. Liu, Y. Long, R. Cheng, C. Tan, L. Yang, C. Hu, S. Zhao and D. Liu, ACS Catal., 2022, 12, 5397–5406 CrossRef CAS.
- M. Yurukcu, E. O. Badradeen, S. Bilnoski, F. M. Yurtsever and M. Begum, Mater. Sci. Eng. Int. J., 2018, 2, 58–64 Search PubMed.
- K. Jiao and M. Ni, Int. J. Energy Res., 2017, 41, 1793–1797 CrossRef CAS.
- X. Wang, Z. Li, Y. Qu, T. Yuan, W. Wang, Y. Wu and Y. Li, Chem, 2019, 5, 1486–1511 CAS.
- X. Ren, Y. Wang, A. Liu, Z. Zhang, Q. Lv and B. Liu, J. Mater. Chem. A, 2020, 8, 24284–24306 RSC.
- M. S. Garapati and R. Sundara, Int. J. Hydrogen Energy, 2019, 44, 10951–10963 CrossRef CAS.
- B. Fang, M.-S. Kim, J. H. Kim, M. Y. Song, Y.-J. Wang, H. Wang, D. P. Wilkinson and J.-S. Yu, J. Mater. Chem., 2011, 21, 8066–8073 RSC.
- A. Kongkanand, N. P. Subramanian, Y. Yu, Z. Liu, H. Igarashi and D. A. Muller, ACS Catal., 2016, 6, 1578–1583 CrossRef CAS.
- K. Jiao, J. Xuan, Q. Du, Z. Bao, B. Xie, B. Wang, Y. Zhao, L. Fan, H. Wang, Z. Hou, S. Huo, N. P. Brandon, Y. Yin and M. D. Guiver, Nature, 2021, 595, 361–369 CrossRef CAS PubMed.
- R. Jiang, S. on Tung, Z. Tang, L. Li, L. Ding, X. Xi, Y. Liu, L. Zhang and J. Zhang, Energy Storage Mater., 2018, 12, 260–276 CrossRef.
- R. Lin, C. Cao, T. Zhao, Z. Huang, B. Li, A. Wieckowski and J. Ma, J. Power Sources, 2013, 223, 190–198 CrossRef CAS.
- Y. J. Sa, C. Park, H. Y. Jeong, S. H. Park, Z. Lee, K. T. Kim, G. G. Park and S. H. Joo, Angew. Chem., Int. Ed., 2014, 126, 4186–4190 CrossRef.
- J. S. Walker, N. V. Rees and P. M. Mendes, J. Exp. Nanosci., 2018, 13, 258–271 CrossRef CAS.
- S. J. Hwang, S. J. Yoo, J. Shin, Y. H. Cho, J. H. Jang, E. Cho, Y. E. Sung, S. W. Nam, T. H. Lim, S. C. Lee and S. K. Kim, Sci. Rep., 2013, 3, 1309 CrossRef PubMed.
- M. Karuppannan, Y. Kim, S. Gok, E. Lee, J. Y. Hwang, J.-H. Jang, Y.-H. Cho, T. Lim, Y.-E. Sung and O. J. Kwon, Energy Environ. Sci., 2019, 12, 2820–2829 RSC.
- H. Wu, D. Wexler, G. Wang and H. Liu, J. Solid State Electrochem., 2012, 16, 1105–1110 CrossRef CAS.
- C. Y. Ahn, J. Y. Cheon, S. H. Joo and J. Kim, J. Power Sources, 2013, 222, 477–482 CrossRef CAS.
- K. Mohanraju, G. Kousik and L. Cindrella, New J. Chem., 2016, 40, 8681–8695 RSC.
- D. J. You, D. H. Kim, J. R. De Lile, C. Li, S. G. Lee, J. M. Kim and C. Pak, Appl. Catal., A, 2018, 562, 250–257 CrossRef CAS.
- G. Samjeské, S. Nagamatsu, S. Takao, K. Nagasawa, Y. Imaizumi, O. Sekizawa, T. Yamamoto, Y. Uemura, T. Uruga and Y. Iwasawa, Phys. Chem. Chem. Phys., 2013, 15, 17208–17218 RSC.
- W.-Y. Huang, M.-Y. Chang, Y.-Z. Wang, Y.-C. Huang, K.-S. Ho, T.-H. Hsieh and Y.-C. Kuo, Polymers, 2020, 12, 617 CrossRef CAS PubMed.
- M. Luo, L. Wei, F. Wang, K. Han and H. Zhu, J. Power Sources, 2014, 270, 34–41 CrossRef CAS.
- Y. Xue, H. Li, X. Ye, S. Yang, Z. Zheng, X. Han, X. Zhang, L. Chen, Z. Xie, Q. Kuang and L. Zheng, Nano Res., 2019, 12, 2490–2497 CrossRef CAS.
- J. C. Douglin, J. R. Varcoe and D. R. Dekel, J. Power Sources Adv., 2020, 5, 100023 CrossRef.
- S. Gottesfeld, D. R. Dekel, M. Page, C. Bae, Y. Yan, P. Zelenay and Y. S. Kim, J. Power Sources, 2018, 375, 170–184 CrossRef CAS.
- W. E. Mustain, M. Chatenet, M. Page and Y. S. Kim, Energy Environ. Sci., 2020, 13, 2805–2838 RSC.
- D. Henkensmeier, M. Najibah, C. Harms, J. Žitka, J. Hnát and K. Bouzek, J. Electrochem. Energy Convers. Storage, 2021, 18, 024001 CrossRef CAS.
- H. S. Dang, E. A. Weiber and P. Jannasch, J. Mater. Chem. A, 2015, 3, 5280–5284 RSC.
- J. Ponce-González, D. K. Whelligan, L. Wang, R. Bance-Soualhi, Y. Wang, Y. Peng, H. Peng, D. C. Apperley, H. N. Sarode, T. P. Pandey, A. G. Divekar, S. Seifert, A. M. Herring, L. Zhuang and J. R. Varcoe, Energy Environ. Sci., 2016, 9, 3724–3735 RSC.
- Y. J. Sa, C. Park, H. Y. Jeong, S. H. Park, Z. Lee, K. T. Kim, G. G. Park and S. H. Joo, Angew. Chem., Int. Ed., 2014, 53, 4102–4106 CrossRef CAS PubMed.
- D. W. Lee, J. H. Jang, I. Jang, Y. S. Kang, S. Jang, K. Y. Lee, J. H. Jang, H. J. Kim and S. J. Yoo, Small, 2019, 15, 1902090 CrossRef PubMed.
- J. Woo, S. Y. Yang, Y. J. Sa, W. Y. Choi, M. H. Lee, H. W. Lee, T. J. Shin, T. Y. Kim and S. H. Joo, Chem. Mater., 2018, 30, 6684–6701 CrossRef CAS.
- D. S. Baek, K. A. Lee, J. Park, J. H. Kim, J. Lee, J. S. Lim, S. Y. Lee, T. J. Shin, H. Y. Jeong, J. S. Son, S. J. Kang, J. Y. Kim and S. H. Joo, Angew. Chem., Int. Ed., 2021, 60, 1441–1449 CrossRef CAS PubMed.
- V. Vijayakumar and S. Y. Nam, J. Ind. Eng. Chem., 2019, 70, 70–86 CrossRef CAS.
- N. Ramaswamy and S. Mukerjee, Chem. Rev., 2019, 119, 11945–11979 CrossRef CAS PubMed.
- F. Xu, Y. Su and B. Lin, Front. Mater., 2020, 4 CrossRef.
- S. Ratso, I. Kruusenberg, M. Käärik, M. Kook, L. Puust, R. Saar, J. Leis and K. Tammeveski, J. Power Sources, 2018, 375, 233–243 CrossRef CAS.
- D. R. Dekel, J. Power Sources, 2018, 375, 158–169 CrossRef CAS.
- E. Negro, A. Bach Delpeuch, K. Vezzù, G. Nawn, F. Bertasi, A. Ansaldo, V. Pellegrini, B. Dembinska, S. Zoladek, K. Miecznikowski, I. A. Rutkowska, M. Skunik-Nuckowska, P. J. Kulesza, F. Bonaccorso and V. Di Noto, Chem. Mater., 2018, 30, 2651–2659 CrossRef CAS.
- Z. F. Pan, L. An, T. S. Zhao and Z. K. Tang, Prog. Energy Combust. Sci., 2018, 66, 141–175 CrossRef.
- M. Hren, M. Božič, D. Fakin, K. S. Kleinschek and S. Gorgieva, Sustainable Energy Fuels, 2021, 5, 604–637 RSC.
- M. C. L. Santos, L. C. Nunes, L. M. G. Silva, A. S. Ramos, F. C. Fonseca, R. F. B. de Souza and A. O. Neto, ChemistrySelect, 2019, 4, 11430–11434 CrossRef CAS.
- S. K. Singh, V. Kashyap, N. Manna, S. N. Bhange, R. Soni, R. Boukherroub, S. Szunerits and S. Kurungot, ACS Catal., 2017, 7, 6700–6710 CrossRef CAS.
- T. Palaniselvam, V. Kashyap, S. N. Bhange, J.-B. Baek and S. Kurungot, Adv. Funct. Mater., 2016, 26, 2150–2162 CrossRef CAS.
- S. Ratso, M. Käärik, M. Kook, P. Paiste, V. Kisand, S. Vlassov, J. Leis and K. Tammeveski, ChemElectroChem, 2018, 5, 1827–1836 CrossRef CAS.
- H.-C. Huang, Y.-C. Lin, S.-T. Chang, C.-C. Liu, K.-C. Wang, H.-P. Jhong, J.-F. Lee and C.-H. Wang, J. Mater. Chem. A, 2017, 5, 19790–19799 RSC.
- M. M. Hossen, K. Artyushkova, P. Atanassov and A. Serov, J. Power Sources, 2018, 375, 214–221 CrossRef CAS.
- H. Ren, Y. Wang, Y. Yang, X. Tang, Y. Peng, H. Peng, L. Xiao, J. Lu, H. D. Abruña and L. Zhuang, ACS Catal., 2017, 7, 6485–6492 CrossRef CAS.
- C. Chen, X.-D. Yang, Z.-Y. Zhou, Y.-J. Lai, M. Rauf, Y. Wang, J. Pan, L. Zhuang, Q. Wang, Y.-C. Wang, N. Tian, X.-S. Zhang and S.-G. Sun, Chem. Commun., 2015, 51, 17092–17095 RSC.
- X. Li, B. N. Popov, T. Kawahara and H. Yanagi, J. Power Sources, 2011, 196, 1717–1722 CrossRef CAS.
- C. V. Rao and Y. Ishikawa, J. Phys. Chem. C, 2012, 116, 4340–4346 CrossRef.
- S. M. Unni, S. N. Bhange, R. Illathvalappil, N. Mutneja, K. R. Patil and S. Kurungot, Small, 2015, 11, 352–360 CrossRef CAS PubMed.
- S. M. Unni, S. Ramadas, R. Illathvalappil, S. N. Bhange and S. Kurungot, J. Mater. Chem. A, 2015, 3, 4361–4367 RSC.
- A. Yuda, A. Ashok and A. Kumar, Catal. Rev.: Sci. Eng., 2020, 64, 126–228 CrossRef.
- A. Yuda and A. Kumar, Int. J. Hydrogen Energy, 2022, 47, 3371–3395 CrossRef CAS.
- Y. Tong, X. Yan, J. Liang and S. X. Dou, Small, 2021, 17, 1904126 CrossRef CAS PubMed.
- D. J. You, K. Kwon, S. H. Joo, J. H. Kim, J. M. Kim, C. Pak and H. Chang, Int. J. Hydrogen Energy, 2012, 37, 6880–6885 CrossRef CAS.
- A. Glüsen, F. Dionigi, P. Paciok, M. Heggen, M. Müller, L. Gan, P. Strasser, R. E. Dunin-Borkowski and D. Stolten, ACS Catal., 2019, 9, 3764–3772 CrossRef.
- Z. Wen, J. Liu and J. Li, Adv. Mater., 2008, 20, 743–747 CrossRef CAS.
- S. H. Joo, K. Kwon, D. J. You, C. Pak, H. Chang and J. M. Kim, Electrochim. Acta, 2009, 54, 5746–5753 CrossRef CAS.
- N. Jha, A. Leela Mohana Reddy, M. M. Shaijumon, N. Rajalakshmi and S. Ramaprabhu, Int. J. Hydrogen Energy, 2008, 33, 427–433 CrossRef CAS.
- Y. Zhang, X. Zhang, X. Ma, W. Guo, C. Wang, T. Asefa and X. He, Sci. Rep., 2017, 7, 43366 CrossRef PubMed.
- N. Bhandary, P. P. Ingole and S. Basu, ChemistrySelect, 2017, 2, 8151–8157 CrossRef CAS.
- Q. Liu and J. Zhang, Langmuir, 2013, 29, 3821–3828 CrossRef CAS PubMed.
- M. G. Hosseini, R. Mahmoodi and V. Daneshvari-Esfahlan, Energy, 2018, 161, 1074–1084 CrossRef CAS.
- N. Muthuswamy, J. L. G. De La Fuente, D. T. Tran, J. Walmsley, M. Tsypkin, S. Raaen, S. Sunde, M. Rønning and D. Chen, Int. J. Hydrogen Energy, 2013, 38, 16631–16641 CrossRef CAS.
- Z. Y. Shih, Z. Yang, Z. H. Lin and H. T. Chang, Int. J. Hydrogen Energy, 2011, 36, 7303–7309 CrossRef CAS.
- Q. Tan, C. Du, G. Yin, P. Zuo, X. Cheng and M. Chen, J. Catal., 2012, 295, 217–222 CrossRef CAS.
- W. Yang, Q. Zhang, C. Peng, E. Wu, S. Chen, Y. Ma, J. Hou, Y. He, B. Zhang and L. Deng, RSC Adv., 2019, 9, 931–939 RSC.
- Z. L. Zhao, L. Y. Zhang, S. J. Bao and C. M. Li, Appl. Catal., B, 2015, 174–175, 361–366 CrossRef CAS.
- L. Jin, H. Xu, C. Chen, H. Shang, Y. Wang, C. Wang and Y. Du, ACS Appl. Mater. Interfaces, 2019, 11, 42123–42130 CrossRef CAS PubMed.
- H. Wang, R. Wang, H. Li, Q. Wang, J. Kang and Z. Lei, Int. J. Hydrogen Energy, 2011, 36, 839–848 CrossRef CAS.
- Y. Yang, C. Tan, Y. Yang, L. Zhang, B. Zhang, K. Wu and S. Zhao, ChemCatChem, 2021, 13, 1587–1594 CrossRef CAS.
- T. Kwon, M. Jun, H. Y. Kim, A. Oh, J. Park, H. Baik, S. H. Joo and K. Lee, Adv. Funct. Mater., 2018, 28, 1706440 CrossRef.
- T. H. Ko, K. Devarayan, M. K. Seo, H. Y. Kim and B. S. Kim, Sci. Rep., 2016, 6, 20313 CrossRef CAS PubMed.
- X. Yu, L. Kuai and B. Geng, Nanoscale, 2012, 4, 5738–5743 RSC.
- V. Parthiban, S. Akula, S. G. Peera, N. Islam and A. K. Sahu, Energy Fuels, 2015, 30, 725–734 CrossRef.
- V. Parthiban, S. Akula and A. K. Sahu, J. Membr. Sci., 2017, 541, 127–136 CrossRef CAS.
- M. S. Çögenli and A. Bayrakçeken Yurtcan, Int. J. Hydrogen Energy, 2020, 45, 650–666 CrossRef.
- D. Lee, S. Gok, Y. Kim, Y. E. Sung, E. Lee, J. H. Jang, J. Y. Hwang, O. J. Kwon and T. Lim, ACS Appl. Mater. Interfaces, 2020, 12, 44588–44596 CrossRef CAS PubMed.
- Y. Feng, H. Liu and J. Yang, Sci. Adv., 2017, 3, e170058 Search PubMed.
- K. Kang, G. Lee, G. Gwak, Y. Choi and H. Ju, Int. J. Hydrogen Energy, 2012, 37, 6285–6291 CrossRef CAS.
- Y. S. Kim, D. H. Peck, S. K. Kim, D. H. Jung, S. Lim and S. H. Kim, Int. J. Hydrogen Energy, 2013, 38, 7159–7168 CrossRef CAS.
- B. Yao, X. Yan, Y. Ding, Z. Lu, D. Dong, H. Ishida, M. Litt and L. Zhu, Macromolecules, 2014, 47, 1039–1045 CrossRef CAS.
- A. Paneri, Y. Heo, G. Ehlert, A. Cottrill, H. Sodano, P. Pintauro and S. Moghaddam, J. Membr. Sci., 2014, 467, 217–225 CrossRef CAS.
- W. Yuan, J. Deng, Z. Zhang, X. Yang and Y. Tang, Renewable Energy, 2014, 62, 640–648 CrossRef CAS.
- J. Liu, Y. Zheng, Z. Hong, K. Cai, F. Zhao and H. Han, Sci. Adv., 2016, 2, e1600858 CrossRef PubMed.
- N. Tian, Z. Y. Zhou, S. G. Sun, Y. Ding and L. W. Zhong, Science, 2007, 316, 732–735 CrossRef CAS PubMed.
- G. Yang, Z. Zhang, X. Kang, L. Li, Y. Li and Y. Sun, ACS Appl. Energy Mater., 2020, 3, 11929–11938 CrossRef CAS.
- X. Luo, W. Han, W. Du, Z. Huang, Y. Jiang and Y. Zhang, J. Power Sources, 2020, 469, 228184 CrossRef CAS.
- J. Liu, L. Wei, C. Cao, F. Zhang, F. Lang, H. Wang, H. Yang and J. Shen, Nanoscale, 2019, 11, 13431–13439 RSC.
- M. Zhang, Z. Ma and H. Song, Nanomaterials, 2021, 11, 377 CrossRef CAS PubMed.
- D. Wang, J. Hu, J. Yang, K. Xiao, S. Liang, J. Xu, B. Liu and H. Hou, Int. J. Hydrogen Energy, 2020, 45, 3163–3175 CrossRef CAS.
- W. Han, C. Li, Y. Jiang, Z. Ma, Y. Zhang, X. Yan and X. Zheng, J. Alloys Compd., 2021, 852, 156994 CrossRef CAS.
- J. Ji, J. Woo, Y. Chung, S. H. Joo and Y. Kwon, Chem. Eng. J., 2020, 381, 122679 CrossRef CAS.
- G. Yang, D. Chen, P. Lv, X. Kong, Y. Sun, Z. Wang, Z. Yuan, H. Liu and J. Yang, Sci. Rep., 2016, 6, 35252 CrossRef CAS PubMed.
- S. Zhong, L. Zhou, L. Wu, L. Tang, Q. He and J. Ahmed, J. Power Sources, 2014, 272, 344–350 CrossRef CAS.
- W. Yang, Y. Dong, J. Li, Q. Fu and L. Zhang, Int. J. Hydrogen Energy, 2021, 46, 2530–2542 CrossRef CAS.
- L. Jiang, J. Chen, D. Han, S. Chang, R. Yang, Y. An, Y. Liu and F. Chen, J. Power Sources, 2020, 453, 227877 CrossRef CAS.
- Y. Xu, S. Zhou and M. Li, Int. J. Hydrogen Energy, 2019, 44, 13875–13884 CrossRef CAS.
- Q. Liu, Y. Zhou, S. Chen, Z. Wang, H. Hou and F. Zhao, J. Power Sources, 2015, 273, 1189–1193 CrossRef CAS.
- Y. Zou, J. Li, Q. Fu, L. Zhang, Q. Liao and X. Zhu, Electrochim. Acta, 2019, 320, 134590 CrossRef CAS.
- C. Cao, L. Wei, G. Wang, J. Liu, Q. Zhai and J. Shen, Inorg. Chem. Front., 2017, 4, 1930–1938 RSC.
- B. Mecheri, R. Gokhale, C. Santoro, M. A. C. de Oliveira, A. D’Epifanio, S. Licoccia, A. Serov, K. Artyushkova and P. Atanassov, ACS Appl. Energy Mater., 2018, 1, 5755–5765 CAS.
- L. Zhou, C. Yang, J. Wen, P. Fu, Y. Zhang, J. Sun, H. Wang and Y. Yuan, J. Mater. Chem. A, 2017, 5, 19343–19350 RSC.
- R. Li, Y. Dai, B. Chen, J. Zou, B. J. Jiang and H. Fu, J. Power Sources, 2016, 307, 1–10 CrossRef CAS.
- Y. Liu, Y. S. Fan and Z. M. Liu, Chem. Eng. J., 2019, 361, 416–427 CrossRef CAS.
- X. Tian, M. Zhou, C. Tan, M. Li, L. Liang, K. Li and P. Su, Chem. Eng. J., 2018, 348, 775–785 CrossRef CAS.
- P. Ren, S. Ci, Y. Ding and Z. Wen, Appl. Surf. Sci., 2019, 481, 1206–1212 CrossRef CAS.
- X. Shi, J. Zhang and T. Huang, Energy Technol., 2017, 5, 1712–1719 CrossRef CAS.
- M. T. Nguyen, B. Mecheri, A. Iannaci, A. D’Epifanio and S. Licoccia, Electrochim. Acta, 2016, 190, 388–395 CrossRef CAS.
- Y. E. Song, S. Lee, M. Kim, J. G. Na, J. Lee, J. Lee and J. R. Kim, J. Power Sources, 2020, 451, 227816 CrossRef CAS.
- N. T. Q. Tran, H. S. Gil, G. Das, B. H. Kim and H. H. Yoon, Korean Chem. Eng. Res., 2019, 57, 387–391 CAS.
- N. Senthilkumar, G. G. Kumar and A. Manthiram, Adv. Energy Mater., 2018, 8, 1702207 CrossRef.
- J. Cao, Z. Zhu, W. Zhao, J. Xu and Z. Chen, Chin. J. Chem., 2016, 34, 1086–1092 CrossRef CAS.
- X. J. Zhang, J. M. Zhang, P. Y. Zhang, Y. Li, S. Xiang, H. G. Tang and Y. J. Fan, Mol. Catal., 2017, 436, 138–144 CrossRef CAS.
- Y. Kang, L. Qi, M. Li, R. E. Diaz, D. Su, R. R. Adzic, E. Stach, J. Li and C. B. Murray, ACS Nano, 2012, 6, 2818–2825 CrossRef CAS PubMed.
- S. S. Hossain, Int. J. Electrochem. Sci., 2021, 16, 150926 CrossRef CAS.
- R. R. Kapaev, F. A. Obrezkov, K. J. Stevenson and P. A. Troshin, Chem. Commun., 2019, 55, 11758–11761 RSC.
- W. Zhang, M. Sun, J. Yin, E. Abou-Hamad, U. Schwingenschlögl, P. M. F. J. Costa and H. N. Alshareef, Angew. Chem., Int. Ed., 2021, 60, 1355–1363 CrossRef PubMed.
- J. J. Shea and C. Luo, ACS Appl. Mater. Interfaces, 2020, 12, 5361–5380 CrossRef CAS PubMed.
- W. Lu, X. Guo, Y. Luo, Q. Li, R. Zhu and H. Pang, Chem. Eng. J., 2019, 355, 208–237 CrossRef CAS.
- L. Trahey, F. R. Brushett, N. P. Balsara, G. Ceder, L. Cheng, Y. M. Chiang, N. T. Hahn, B. J. Ingram, S. D. Minteer, J. S. Moore, K. T. Mueller, L. F. Nazar, K. A. Persson, D. J. Siegel, K. Xu, K. R. Zavadil, V. Srinivasan and G. W. Crabtree, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12550–12557 CrossRef CAS.
- D. Ganguly, A. P. Ajay, A. Ghosh and S. Ramaprabhu, Sci. Rep., 2020, 10, 9945 CrossRef CAS.
- L. Su, Y. Jing and Z. Zhou, Nanoscale, 2011, 3, 3967–3983 RSC.
- L. Shen, H. Li, E. Uchaker, X. Zhang and G. Cao, Nano Lett., 2012, 12, 5673–5678 CrossRef CAS PubMed.
- W. Zhou, J. Zhu, C. Cheng, J. Liu, H. Yang, C. Cong, C. Guan, X. Jia, H. J. Fan, Q. Yan, C. M. Li and T. Yu, Energy Environ. Sci., 2011, 4, 4954–4961 RSC.
- G. Tan, F. Wu, Y. Yuan, R. Chen, T. Zhao, Y. Yao, J. Qian, J. Liu, Y. Ye, R. Shahbazian-Yassar, J. Lu and K. Amine, Nat. Commun., 2016, 7, 11774 CrossRef CAS PubMed.
- R. Rajagopal and K. S. Ryu, J. Electroanal. Chem., 2020, 856, 113669 CrossRef CAS.
- G. Jeong, J. G. Kim, M. S. Park, M. Seo, S. M. Hwang, Y. U. Kim, Y. J. Kim, J. H. Kim and S. X. Dou, ACS Nano, 2014, 8, 2977–2985 CrossRef CAS PubMed.
- L. Gao, F. Qu and X. Wu, J. Mater. Chem. A, 2014, 2, 7367–7372 RSC.
- S. Lee, J. Ha, H. Cheng, J. W. Lee, T. S. Jang, Y. G. Jung, Y. Huang, J. A. Rogers and U. Paik, Adv. Energy Mater., 2014, 4, 1300472 CrossRef.
- H. Liu, Z. Li, Y. Liang, R. Fu and D. Wu, Carbon, 2015, 84, 419–425 CrossRef CAS.
- L. Thirugnanam, M. Palanisamy, S. Kaveri, S. Ramaprabhu, V. G. Pol and M. Dutta, Int. J. Hydrogen Energy, 2021, 46, 2464–2478 CrossRef CAS.
- W. Hua, B. Schwarz, R. Azmi, M. Müller, M. S. Dewi Darma, M. Knapp, A. Senyshyn, M. Heere, A. Missyul, L. Simonelli, J. R. Binder, S. Indris and H. Ehrenberg, Nano Energy, 2020, 78, 105231 CrossRef CAS.
- L. Liang, Y. Xu, L. Wen, Y. Li, M. Zhou, C. Wang, H. Zhao, U. Kaiser and Y. Lei, Nano Res., 2017, 10, 3189–3201 CrossRef CAS.
- C. Wu, X. Tong, Y. Ai, D. S. Liu, P. Yu, J. Wu and Z. M. Wang, Nano-Micro Lett., 2018, 10, 40 CrossRef PubMed.
- R. Chandrabhan Shende, P. Chandran and S. Ramaprabhu, Nanotechnology, 2020, 31, 235403 CrossRef PubMed.
- Z. Xiong, D. Sun, X. Jia and J. Zhou, Carbon, 2020, 166, 339–349 CrossRef CAS.
- S.-Y. Duan, J.-Y. Piao, T.-Q. Zhang, Y.-G. Sun, X.-C. Liu, A.-M. Cao and L.-J. Wan, NPG Asia Mater., 2017, 9, e414 CrossRef CAS.
- A. P. Vijaya Kumar Saroja, M. S. Garapati, R. ShyiamalaDevi, M. Kamaraj and S. Ramaprabhu, Appl. Surf. Sci., 2020, 504, 144430 CrossRef CAS.
- F. Kong, Z. Han, S. Tao and B. Qian, J. Energy Chem., 2021, 55, 256–264 CrossRef.
- C. Chen, Z. Han, S. Chen, S. Qi, X. Lan, C. Zhang, L. Chen, P. Wang and W. Wei, ACS Appl. Mater. Interfaces, 2020, 12, 7144–7152 CrossRef CAS PubMed.
- J. Yin, Y. Shen, C. Li, C. Fan, S. Sun, Y. Liu, J. Peng, L. Qing and J. Han, ChemSusChem, 2019, 12, 4786–4790 CrossRef CAS PubMed.
- W. Zhao, C. Guo and C. M. Li, J. Mater. Chem. A, 2017, 5, 19195–19202 RSC.
- Y. Mei, Y. Huang and X. Hu, J. Mater. Chem. A, 2016, 4, 12001–12013 RSC.
- Y. L. Bai, R. Xarapatgvl, X. Y. Wu, X. Liu, Y. S. Liu, K. X. Wang and J. S. Chen, Nanoscale, 2019, 11, 17860–17868 RSC.
- B. Ji, W. Yao, Y. Zheng, P. Kidkhunthod, X. Zhou, S. Tunmee, S. Sattayaporn, H.-M. Cheng, H. He and Y. Tang, Nat. Commun., 2020, 11, 1225 CrossRef CAS PubMed.
- Y. He, Y. Xu, J. Li, Z. Xu, Z. Zhang, J. Sun, M. Zhang, X. Zhu and X. Zhou, Energy Fuels, 2021, 35, 3490–3496 CrossRef CAS.
- J. Bao, W. Deng, J. Liu and C.-F. Sun, Energy Storage Mater., 2021, 42, 806–814 CrossRef.
- H. Sun, P. Liang, G. Zhu, W. H. Hung, Y.-Y. Li, H.-C. Tai, C.-L. Huang, J. Li, Y. Meng, M. Angell, C.-A. Wang and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 27847–27853 CrossRef CAS PubMed.
- C. Zhang, Y. Xu, M. Zhou, L. Liang, H. Dong, M. Wu, Y. Yang and Y. Lei, Adv. Funct. Mater., 2017, 27, 1604307 CrossRef.
- Y. Zhu, Y. Yin, X. Yang, T. Sun, S. Wang, Y. Jiang, J. Yan and X. Zhang, Angew. Chem., Int. Ed., 2017, 56, 7881–7885 CrossRef CAS PubMed.
- Y. Xu, F. Bahmani, M. Zhou, Y. Li, C. Zhang, F. Liang, S. H. Kazemi, U. Kaiser, G. Meng and Y. Lei, Nanoscale Horiz., 2018, 4, 202–207 RSC.
- L. Deng, T. Wang, Y. Hong, M. Feng, R. Wang, J. Zhang, Q. Zhang, J. Wang, L. Zeng, Y. Zhu and L. Guo, ACS Energy Lett., 2020, 5, 1916–1922 CrossRef CAS.
- W. Wang, B. Jiang, C. Qian, F. Lv, J. Feng, J. Zhou, K. Wang, C. Yang, Y. Yang and S. Guo, Adv. Mater., 2018, 30, 1801812 CrossRef PubMed.
- S. H. Yang, S.-K. Park and Y. C. Kang, Nano-Micro Lett., 2021, 13, 9 CrossRef PubMed.
- J. Chu, Q. Yu, D. Yang, L. Xing, C. Y. Lao, M. Wang, K. Han, Z. Liu, L. Zhang, W. Du, K. Xi, Y. Bao and W. (Alex) Wang, Appl. Mater. Today, 2018, 13, 344–351 CrossRef.
- Y. Luo, M. Tao, J. Deng, R. Zhan, B. Guo, Q. Ma, M. K. Aslam, Y. Qi and M. Xu, Inorg. Chem. Front., 2020, 7, 394–401 RSC.
- H. Ren, C. Gu, J. Zhao, S. W. Joo and J. Huang, Appl. Surf. Sci., 2019, 473, 918–927 CrossRef CAS.
- R. Mo, D. Rooney, K. Sun and H. Y. Yang, Nat. Commun., 2017, 8, 13949 CrossRef CAS PubMed.
- J. H. Kim and Y. C. Kang, Small, 2017, 13, 1701585 CrossRef PubMed.
- J. Guo, H. Zhu, Y. Sun, L. Tang and X. Zhang, J. Mater. Chem. A, 2016, 4, 4783–4789 RSC.
- J. Ma, H. Zhang, Y. Xin, S. Liu, Y. Li, L. Yang, G. Xu, T. Lou, H. Niu and S. Yang, Dalton Trans., 2021, 50, 1703–1711 RSC.
- S. Liu, J. Wang, J. Wang, F. Zhang and L. Wang, Appl. Surf. Sci., 2016, 390, 86–91 CrossRef CAS.
- Y. Ding, Z.-F. Li, E. V. Timofeeva and C. U. Segre, Adv. Energy Mater., 2018, 8, 1702134 CrossRef.
- K.-H. Kim, W.-S. Kim and S.-H. Hong, Nanoscale, 2019, 11, 13494–13501 RSC.
- J. Yang, Y. Ouyang, H. Zhang, H. Xu, Y. Zhang and Y. Wang, J. Mater. Chem. A, 2016, 4, 9923–9930 RSC.
- C. Wu, P. Kopold, P. A. van Aken, J. Maier and Y. Yu, Adv. Mater., 2017, 29, 1604015 CrossRef PubMed.
- J. Fullenwarth, A. Darwiche, A. Soares, B. Donnadieu and L. Monconduit, J. Mater. Chem. A, 2014, 2, 2050–2059 RSC.
- R. Zhang, V. Raveendran, Y. He, A. Yau, A. Chang, C. Chi, S. Bong, F. Cheng, W. Ma and J. Chen, Energy Mater. Adv., 2021, 2021, 2124862 Search PubMed.
- Y. Kim, Y. Kim, A. Choi, S. Woo, D. Mok, N.-S. Choi, Y. S. Jung, J. H. Ryu, S. M. Oh and K. T. Lee, Adv. Mater., 2014, 26, 4139–4144 CrossRef CAS PubMed.
- L. Ma, P. Yan, S. Wu, G. Zhu and Y. Shen, J. Mater. Chem. A, 2017, 5, 16994–17000 RSC.
- X. Ge, Z. Li and L. Yin, Nano Energy, 2017, 32, 117–124 CrossRef CAS.
- J. Wang, B. Wang, X. Liu, G. Wang, H. Wang and J. Bai, J. Colloid Interface Sci., 2019, 538, 187–198 CrossRef CAS PubMed.
- Z. Liu, T. Lu, T. Song, X.-Y. Yu, X. W. (David) Lou and U. Paik, Energy Environ. Sci., 2017, 10, 1576–1580 RSC.
- Y. Jiang, Y. Feng, B. Xi, S. Kai, K. Mi, J. Feng, J. Zhang and S. Xiong, J. Mater. Chem. A, 2016, 4, 10719–10726 RSC.
- Q. Zhou, L. Liu, Z. Huang, L. Yi, X. Wang and G. Cao, J. Mater. Chem. A, 2016, 4, 5505–5516 RSC.
- S. Y. Lee and Y. C. Kang, Chem. – Eur. J., 2016, 22, 2769–2774 CrossRef CAS PubMed.
- A. Mahmood, S. Li, Z. Ali, H. Tabassum, B. Zhu, Z. Liang, W. Meng, W. Aftab, W. Guo, H. Zhang, M. Yousaf, S. Gao, R. Zou and Y. Zhao, Adv. Mater., 2019, 31, 1805430 CrossRef PubMed.
- Q. Tan, P. Li, K. Han, Z. Liu, Y. Li, W. Zhao, D. He, F. An, M. Qin and X. Qu, J. Mater. Chem. A, 2019, 7, 744–754 RSC.
- W. Zhang, W. Miao, X. Liu, L. Li, Z. Yu and Q. Zhang, J. Alloys Compd., 2018, 769, 141–148 CrossRef CAS.
- Y. Han, W. Li, K. Zhou, X. Wu, H. Wu, X. Wu, Q. Shi, G. Diao and M. Chen, ChemNanoMat, 2020, 6, 132–138 CrossRef CAS.
- D. Li, X. Ren, Q. Ai, Q. Sun, L. Zhu, Y. Liu, Z. Liang, R. Peng, P. Si, J. Lou, J. Feng and L. Ci, Adv. Energy Mater., 2018, 8, 1802386 CrossRef.
- F. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172–2192 RSC.
- C. Wang, Y. Yu, J. Niu, Y. Liu, D. Bridges, X. Liu, J. Pooran, Y. Zhang and A. Hu, Appl. Sci., 2019, 9, 2787 CrossRef CAS.
- Y. Li and J. Lu, ACS Energy Lett., 2017, 2, 1370–1377 CrossRef CAS.
- A. Kraytsberg and Y. Ein-Eli, Nano Energy, 2013, 2, 468–480 CrossRef CAS.
- K. Song, J. Jung, M. Park, H. Park, H. J. Kim, S. Il Choi, J. Yang, K. Kang, Y. K. Han and Y. M. Kang, ACS Catal., 2018, 10, 9006–9015 CrossRef.
- Z. Ma, X. Yuan, L. Li, Z. F. Ma, D. P. Wilkinson, L. Zhang and J. Zhang, Energy Environ. Sci., 2015, 8, 2144–2198 RSC.
- S. Y. Kim, H. T. Lee and K. B. Kim, Phys. Chem. Chem. Phys., 2013, 15, 20262–20271 RSC.
- D. U. Lee, M. G. Park, Z. P. Cano, W. Ahn and Z. Chen, ChemSusChem, 2018, 11, 406–414 CrossRef CAS PubMed.
- M. J. Song and M. W. Shin, Appl. Surf. Sci., 2014, 320, 435–440 CrossRef CAS.
- C. Tang, Y. Mao, J. Xie, Z. Chen, J. Tu, G. Cao and X. Zhao, Inorg. Chem. Front., 2018, 5, 1707–1713 RSC.
- S. Peng, Y. Hu, L. Li, X. Han, F. Cheng, M. Srinivasan, Q. Yan, S. Ramakrishna and J. Chen, Nano Energy, 2015, 13, 718–726 CrossRef CAS.
- A. Kondori, Z. Jiang, M. Esmaeilirad, M. Tamadoni Saray, A. Kakekhani, K. Kucuk, P. Navarro Munoz Delgado, S. Maghsoudipour, J. Hayes, C. S. Johnson, C. U. Segre, R. Shahbazian-Yassar, A. M. Rappe and M. Asadi, Adv. Mater., 2020, 32, 2004028 CrossRef CAS PubMed.
- H. Wang, F. Yin, P. Lv, T. Fan, X. He and B. Chen, Int. J. Hydrogen Energy, 2017, 42, 2127–2133 CrossRef CAS.
- P. Gu, M. Zheng, Q. Zhao, X. Xiao, H. Xue and H. Pang, J. Mater. Chem. A, 2017, 5, 7651–7666 RSC.
- W. Sun, F. Wang, B. Zhang, M. Zhang, V. Küpers, X. Ji, C. Theile, P. Bieker, K. Xu, C. Wang and M. Winter, Science, 2021, 371, 46–51 CrossRef CAS PubMed.
- J. S. Lee, S. T. Kim, R. Cao, N. S. Choi, M. Liu, K. T. Lee and J. Cho, Adv. Energy Mater., 2011, 1, 34–50 CrossRef CAS.
- Z. Wang, J. Huang, L. Wang, Y. Liu, W. Liu, S. Zhao and Z. Q. Liu, Angew. Chem., Int. Ed., 2022, 61, e202114696 CAS.
- X. Lu, H. M. Chan, C. L. Sun, C. M. Tseng and C. Zhao, J. Mater. Chem. A, 2015, 3, 13371–13376 RSC.
- Z. Wang, Y. Lu, Y. Yan, T. Y. P. Larissa, X. Zhang, D. Wuu, H. Zhang, Y. Yang and X. Wang, Nano Energy, 2016, 30, 368–378 CrossRef CAS.
- M. Wang, Y. Liu, K. Zhang, F. Yu, F. Qin, J. Fang, Y. Lai and J. Li, RSC Adv., 2016, 6, 83386–83392 RSC.
- Y. Cheng, S. Dou, J. P. Veder, S. Wang, M. Saunders and S. P. Jiang, ACS Appl. Mater. Interfaces, 2017, 9, 8121–8133 CrossRef CAS PubMed.
- Z. Guo, F. Wang, Y. Xia, J. Li, A. G. Tamirat, Y. Liu, L. Wang, Y. Wang and Y. Xia, J. Mater. Chem. A, 2018, 6, 1443–1453 RSC.
- X. Liu, L. Wang, P. Yu, C. Tian, F. Sun, J. Ma, W. Li and H. Fu, Angew. Chem., Int. Ed., 2018, 57, 16166–16170 CrossRef CAS PubMed.
- W. Fang, H. Hu, T. Jiang, G. Li and M. Wu, Carbon, 2019, 146, 476–485 CrossRef CAS.
- S. Ibraheem, S. Chen, J. Li, Q. Wang and Z. Wei, J. Mater. Chem. A, 2019, 7, 9497–9502 RSC.
- J. Zhang, M. Zhang, L. Qiu, Y. Zeng, J. Chen, C. Zhu, Y. Yu and Z. Zhu, J. Mater. Chem. A, 2019, 7, 19045–19059 RSC.
- W. Xie, J. Li, Y. Song, S. Li, J. Li and M. Shao, Nano-Micro Lett., 2020, 12, 97 CrossRef CAS PubMed.
- F. Wang, H. Zhao, Y. Ma, Y. Yang, B. Li, Y. Cui, Z. Guo and L. Wang, J. Energy Chem., 2020, 50, 52–62 CrossRef.
- J. Jiao, Y. Pan, B. Wang, W. Yang, S. Liu and C. Zhang, J. Energy Chem., 2020, 53, 364–371 CrossRef.
- Q. Zhou, Z. Zhang, J. Cai, B. Liu, Y. Zhang, X. Gong, X. Sui, A. Yu, L. Zhao, Z. Wang and Z. Chen, Nano Energy, 2020, 71, 104592 CrossRef CAS.
- T. T. Gebremariam, F. Chen, Q. Wang, J. Wang, Y. Liu, X. Wang and A. Qaseem, ACS Appl. Energy Mater., 2018, 1, 1612–1625 CrossRef CAS.
- K. Chen, S. Kim, R. Rajendiran, K. Prabakar, G. Li, Z. Shi, C. Jeong, J. Kang and O. L. Li, J. Colloid Interface Sci., 2021, 582, 977–990 CrossRef CAS PubMed.
- R. Zhao, Q. Li, Z. Chen, V. Jose, X. Jiang, G. Fu, J.-M. Lee and S. Huang, Carbon, 2020, 164, 398–406 CrossRef CAS.
- R. Zhao, J. Chen, Z. Chen, X. Jiang, G. Fu, Y. Tang, W. Jin, J.-M. Lee and S. Huang, ACS Appl. Energy Mater., 2020, 3, 4539–4548 CrossRef CAS.
- F. Meng, H. Zhong, D. Bao, J. Yan and X. Zhang, J. Am. Chem. Soc., 2016, 138, 10226–10231 CrossRef CAS PubMed.
- X. Han, X. Ling, Y. Wang, T. Ma, C. Zhong, W. Hu and Y. Deng, Angew. Chem., Int. Ed., 2019, 58, 5359–5364 CrossRef CAS PubMed.
- C.-Y. Su, H. Cheng, W. Li, Z.-Q. Liu, N. Li, Z. Hou, F.-Q. Bai, H.-X. Zhang and T.-Y. Ma, Adv. Energy Mater., 2017, 7, 1602420 CrossRef.
- K. Sheng, Q. Yi, A.-L. Chen, Y. Wang, Y. Yan, H. Nie and X. Zhou, ACS Appl. Mater. Interfaces, 2021, 13, 45394–45405 CrossRef CAS PubMed.
- Y. Xu, P. Deng, G. Chen, J. Chen, Y. Yan, K. Qi, H. Liu and B. Y. Xia, Adv. Funct. Mater., 2020, 30, 1906081 CrossRef CAS.
- A. I. Douka, Y. Xu, H. Yang, S. Zaman, Y. Yan, H. Liu, M. A. Salam and B. Y. Xia, Adv. Mater., 2020, 32, 2002170 CrossRef CAS PubMed.
- W. Wan, X. Liu, H. Li, X. Peng, D. Xi and J. Luo, Appl. Catal., B, 2019, 240, 193–200 CrossRef CAS.
- Q. Wang, Y. Ji, Y. Lei, Y. Wang, Y. Wang, Y. Li and S. Wang, ACS Energy Lett., 2018, 3, 1183–1191 CrossRef CAS.
- Y. Qiao, P. Yuan, Y. Hu, J. Zhang, S. Mu, J. Zhou, H. Li, H. Xia, J. He and Q. Xu, Adv. Mater., 2018, 30, 1804504 CrossRef PubMed.
- Y.-W. Li, W.-J. Zhang, J. Li, H.-Y. Ma, H.-M. Du, D.-C. Li, S.-N. Wang, J.-S. Zhao, J.-M. Dou and L. Xu, ACS Appl. Mater. Interfaces, 2020, 12, 44710–44719 CrossRef CAS PubMed.
- H. Jiang, J. Gu, X. Zheng, M. Liu, X. Qiu, L. Wang, W. Li, Z. Chen, X. Ji and J. Li, Energy Environ. Sci., 2019, 12, 322–333 RSC.
- S. Chen, L. Zhao, J. Ma, Y. Wang, L. Dai and J. Zhang, Nano Energy, 2019, 60, 536–544 CrossRef CAS.
- J. Wu, H. Zhou, Q. Li, M. Chen, J. Wan, N. Zhang, L. Xiong, S. Li, B. Y. Xia, G. Feng, M. Liu and L. Huang, Adv. Energy Mater., 2019, 9, 1900149 CrossRef.
- B. Chen, X. He, F. Yin, H. Wang, D.-J. Liu, R. Shi, J. Chen and H. Yin, Adv. Funct. Mater., 2017, 27, 1700795 CrossRef.
- X. Han, W. Zhang, X. Ma, C. Zhong, N. Zhao, W. Hu and Y. Deng, Adv. Mater., 2019, 31, 1808281 CrossRef PubMed.
- X. Wang, J. Sunarso, Q. Lu, Z. Zhou, J. Dai, D. Guan, W. Zhou and Z. Shao, Adv. Energy Mater., 2020, 10, 1903271 CrossRef CAS.
- X. Zhong, W. Yi, Y. Qu, L. Zhang, H. Bai, Y. Zhu, J. Wan, S. Chen, M. Yang, L. Huang, M. Gu, H. Pan and B. Xu, Appl. Catal., B, 2020, 260, 118188 CrossRef CAS.
- Q. Xu, H. Jiang, Y. Li, D. Liang, Y. Hu and C. Li, Appl. Catal., B, 2019, 256, 117893 CrossRef CAS.
- L. An, Z. Zhang, J. Feng, F. Lv, Y. Li, R. Wang, M. Lu, R. B. Gupta, P. Xi and S. Zhang, J. Am. Chem. Soc., 2018, 140, 17624–17631 CrossRef CAS PubMed.
- X. Wang, Z. Liao, Y. Fu, C. Neumann, A. Turchanin, G. Nam, E. Zschech, J. Cho, J. Zhang and X. Feng, Energy Storage Mater., 2020, 26, 157–164 CrossRef.
- X. Yu, Y. Kang, S. Wang, K. S. Hui, K. N. Hui, H. Zhao, J. Li, B. Li, J. Xu, L. Chen and H. Shao, J. Mater. Chem. A, 2020, 8, 16355–16365 RSC.
- W. W. Yin and Z. W. Fu, ChemCatChem, 2017, 9, 1545–1553 CrossRef CAS.
- H. Yadegari and X. Sun, Trends Chem., 2020, 2, 241–253 CrossRef CAS.
- X. Xu, K. S. Hui, D. A. Dinh, K. N. Hui and H. Wang, Mater. Horiz., 2019, 6, 1306–1335 RSC.
- Q. Sun, J. Liu, B. Xiao, B. Wang, M. Banis, H. Yadegari, K. R. Adair, R. Li and X. Sun, Adv. Funct. Mater., 2019, 29, 1808332 CrossRef.
- C. Xia, R. Black, R. Fernandes, B. Adams and L. F. Nazar, Nat. Chem., 2015, 7, 496–501 CrossRef CAS PubMed.
- J. Y. Cheon, K. Kim, Y. J. Sa, S. H. Sahgong, Y. Hong, J. Woo, S. D. Yim, H. Y. Jeong, Y. Kim and S. H. Joo, Adv. Energy Mater., 2016, 6, 1501794 CrossRef.
- Q. Sun, H. Yadegari, M. N. Banis, J. Liu, B. Xiao, X. Li, C. Langford, R. Li and X. Sun, J. Phys. Chem. C, 2015, 119, 13433–13441 CrossRef CAS.
- H. Yadegari, Y. Li, M. N. Banis, X. Li, B. Wang, Q. Sun, R. Li, T. K. Sham, X. Cui and X. Sun, Energy Environ. Sci., 2014, 7, 3747–3757 RSC.
- P. Hartmann, C. L. Bender, M. Vračar, A. K. Dürr, A. Garsuch, J. Janek and P. Adelhelm, Nat. Mater., 2013, 12, 228–232 CrossRef CAS PubMed.
- X. Bi, R. Wang, Y. Yuan, Y. Yuan, D. Zhang, T. Zhang, L. Ma, T. Wu, R. Shahbazian-Yassar, K. Amine, K. Amine and J. Lu, Nano Lett., 2020, 20, 4681–4686 CrossRef CAS PubMed.
- F. Liang, X. Qiu, Q. Zhang, Y. Kang, A. Koo, K. Hayashi, K. Chen, D. Xue, K. N. Hui, H. Yadegari and X. Sun, Nano Energy, 2018, 49, 574–579 CrossRef CAS.
- Z. Khan, M. Vagin and X. Crispin, Adv. Sci., 2020, 7, 1902866 CrossRef CAS PubMed.
- L. Gai, C. Zhao, Y. Zhang, Z. Hu and Q. Shen, Carbon Energy, 2022, 4, 142–154 CrossRef CAS.
- J. B. Robinson, K. Xi, R. V. Kumar, A. C. Ferrari, H. Au, M.-M. Titirici, A. Parra-Puerto, A. Kucernak, S. D. S. Fitch, N. Garcia-Araez, Z. L. Brown, M. Pasta, L. Furness, A. J. Kibler, D. A. Walsh, L. R. Johnson, C. Holc, G. N. Newton, N. R. Champness, F. Markoulidis, C. Crean, R. C. T. Slade, E. I. Andritsos, Q. Cai, S. Babar, T. Zhang, C. Lekakou, N. Kulkarni, A. J. E. Rettie, R. Jervis, M. Cornish, M. Marinescu, G. Offer, Z. Li, L. Bird, C. P. Grey, M. Chhowalla, D. Di Lecce, R. E. Owen, T. S. Miller, D. J. L. Brett, S. Liatard, D. Ainsworth and P. R. Shearing, J. Phys. Energy, 2021, 3, 031501 CrossRef CAS.
- J. Tian, F. Xing and Q. Gao, Molecules, 2021, 26, 2507 CrossRef CAS PubMed.
- R. Yan, T. Ma, M. Cheng, X. Tao, Z. Yang, F. Ran, S. Li, B. Yin, C. Cheng and W. Yang, Adv. Mater., 2021, 33, 2008784 CrossRef CAS PubMed.
- S. Rehman, K. Khan, Y. Zhao and Y. Hou, J. Mater. Chem. A, 2017, 5, 3014–3038 RSC.
- M. R. Kaiser, Z. Han, J. Liang, S. X. Dou and J. Wang, Energy Storage Mater., 2019, 19, 1–15 CrossRef.
- Z. Li, Y. Jiang, L. Yuan, Z. Yi, C. Wu, Y. Liu, P. Strasser and Y. Huang, ACS Nano, 2014, 8, 9295–9303 CrossRef CAS PubMed.
- X. Long Huang, S. X. Dou, Z. M. Wang, H.-J. Peng, S. Xue Dou and Z. M. Wang His, Mater. Horiz., 2021, 8, 2870–2885 RSC.
- F. Pei, L. Lin, D. Ou, Z. Zheng, S. Mo, X. Fang and N. Zheng, Nat. Commun., 2017, 8, 482 CrossRef PubMed.
- X. Tao, Y. Li, H. guo Wang, X. Lv, Y. Li, D. Xu, Y. Jiang and Y. Meng, J. Colloid Interface Sci., 2020, 565, 494–502 CrossRef CAS PubMed.
- H. Gao, S. Ning, Y. Zhou, S. Men and X. Kang, Chem. Eng. J., 2021, 408, 127323 CrossRef CAS.
- W. Zhou, X. Xiao, M. Cai and L. Yang, Nano Lett., 2014, 14, 5250–5256 CrossRef CAS PubMed.
- S. Il Choi, M. Shao, N. Lu, A. Ruditskiy, H. C. Peng, J. Park, S. Guerrero, J. Wang, M. J. Kim and Y. Xia, ACS Nano, 2014, 8, 10363–10371 CrossRef PubMed.
- S. Gong, Y. X. Zhang and Z. Niu, ACS Catal., 2020, 10, 10886–10904 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |