
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePNIPAAm microgels with defined network architecture as temperature sensors in optical stretchers†
Nicolas
Hauck
a,
Timon
Beck
bc,
Gheorghe
Cojoc
b,
Raimund
Schlüßler
b,
Saeed
Ahmed
b,
Ivan
Raguzin
a,
Martin
Mayer
 a,
Jonas
Schubert
a,
Paul
Müller
c,
Jochen
Guck
bc and
Julian
Thiele
*a
a,
Jonas
Schubert
a,
Paul
Müller
c,
Jochen
Guck
bc and
Julian
Thiele
*a
aLeibniz-Institut für Polymerforschung Dresden e.V., Institute of Physical Chemistry and Polymer Physics, D-01069 Dresden, Germany. E-mail: thiele@ipfdd.de
bCenter for Molecular and Cellular Bioengineering, Biotechnology Center, Technische Universität Dresden, D-01307 Dresden, Germany
cMax Planck Institute for the Science of Light, Staudtstraße 2, D-91058 Erlangen, Germany
First published on 5th July 2022
Abstract
Stretching individual living cells with light is a standard method to assess their mechanical properties. Yet, heat introduced by the laser light of optical stretchers may unwittingly change the mechanical properties of cells therein. To estimate the temperature induced by an optical trap, we introduce cell-sized, elastic poly(N-isopropylacrylamide) (PNIPAAm) microgels that relate temperature changes to hydrogel swelling. For their usage as a standardized calibration tool, we analyze the effect of free-radical chain-growth gelation (FCG) and polymer-analogous photogelation (PAG) on hydrogel network heterogeneity, micromechanics, and temperature response by Brillouin microscopy and optical diffraction tomography. Using a combination of tailor-made PNIPAAm macromers, PAG, and microfluidic processing, we obtain microgels with homogeneous network architecture. With that, we expand the capability of standardized microgels in calibrating and validating cell mechanics analysis, not only considering cell and microgel elasticity but also providing stimuli-responsiveness to consider dynamic changes that cells may undergo during characterization.
Introduction
Hydrogels are three-dimensional, crosslinked polymer networks capable of retaining large amounts of water.1 Due to the ability of many hydrogels to swell and de-swell upon environmental changes, this sensitivity is attractive for advanced applications in the fields of drug delivery, photonics, actuation, and sensing.2 Poly(N-isopropylacrylamide) (PNIPAAm) is one such hydrogel material reacting to a thermal stimulus at a volume phase transition temperature (VPTT) of around 32 °C in aqueous media.3 As the transition temperature is close to the human body temperature, PNIPAAm has been widely investigated for biomedical applications,4,5 and various physicochemical as well as mechanical properties of PNIPAAm hydrogels have been tailored depending on the hydrogels' network architecture.6,7An idealized image of a hydrogel network commonly consists of a completely regular array of meshes. This regularity depends mainly on whether monomer- or polymer-based building blocks are used, their architecture (e.g., linear or multi-arm), and the synthesis conditions during the gelation process.8 Focusing on the synthesis of PNIPAAm hydrogels, we can primarily distinguish between two key gelation mechanisms, namely free-radical chain-growth gelation (FCG) and polymer-analogous photogelation (PAG) (Fig. 1). In the former case, N-isopropylacrylamide monomers crosslinked by N,N′-methylenebisacrylamide (BIS) commonly leads to spatial inhomogeneities of the hydrogel crosslinking density. It has been reported that high initiator concentrations as well as a polymerization temperature close to the lower critical solution temperature (LCST) lead to a fast and exothermic free-radical polymerization and pronounced structural inhomogeneity on the micro- and nanometer scale.9,10 This is due to reaction heat that cannot be efficiently transferred away, leading to an increase of the reaction temperature close to or eventually above the LCST of PNIPAAm chains in water.8 On the nanometer scale, network inhomogeneities in the form of broad mesh size distribution accompanied by structural imperfections, such as loops and dangling chains, can be found because of random incorporation of monomer and crosslinker during chain growth. On the micrometer scale, domains form due to temperature-driven microphase separation.11 These domains or nanogel clusters arise during the early phase of network formation and become only loosely interconnected during further hydrogel growth.12 Eventually, micrometer-sized inhomogeneities can be observed by optical microscopy. To improve PNIPAAm hydrogel formation via FCG, decreasing the amount of initiator led to lower reaction temperatures below the LCST and to a gel architecture being uniform on the micrometer scale, but still bearing the same structural inhomogeneities on the nanometer-scale.10
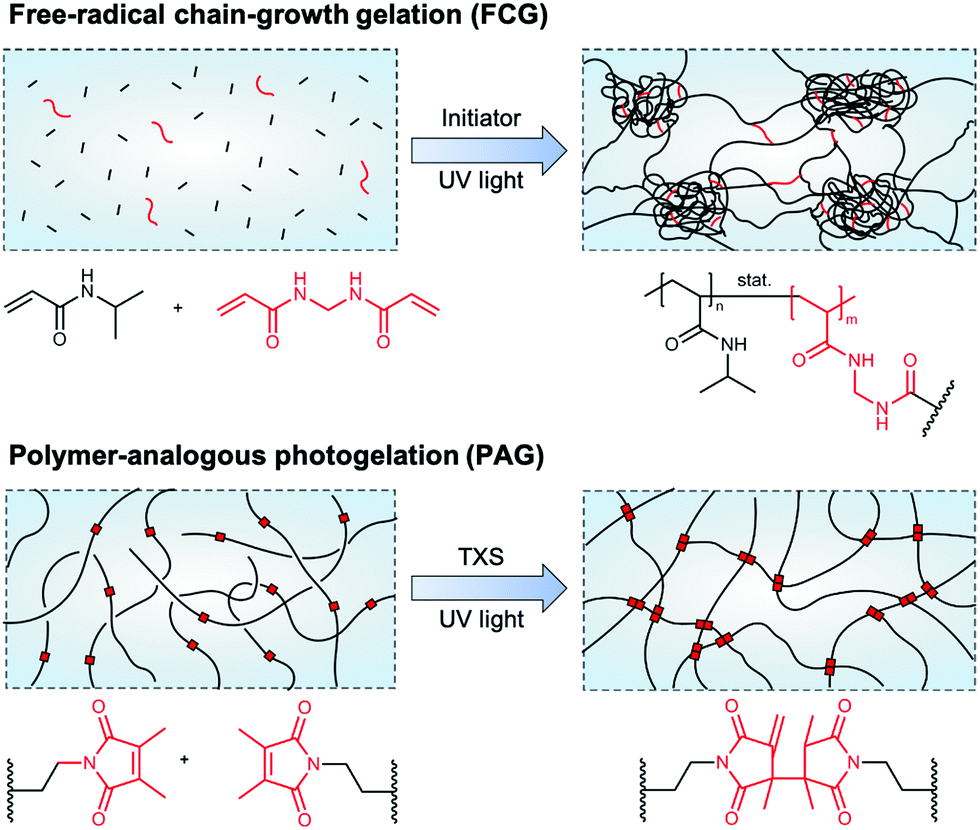 | ||
| Fig. 1 Schematics of NIPAAm-based hydrogel network architectures formed via different synthesis approaches. (top) Hydrogel network formation by free-radical chain-growth gelation of NIPAAm and BIS leads to pronounced network inhomogeneities on the nano- and, in case of reaction temperatures close to or above the LCST of PNIPAAm in water, also micrometer scale. The exothermic process of initiator decomposition and chain-growth polymerization leads to the formation of nanogel clusters that are loosely interconnected. (bottom) Hydrogel network formation via polymer-analogous photogelation leads to homogeneous networks on the nano- and micrometer scale. Pre-synthesized polymeric precursors (macromers) are interconnected by UV light-induced, TXS-catalyzed dimerization of incorporated DMMI moieties. Schematic adapted with permission from ref. 12. | ||
In contrast to FCG, PAG provides polymer networks with a high homogeneity independent of the rate of crosslinking, with macromolecular precursor chains functionalized with crosslinkable side groups being the basis for hydrogel formation.8 A well-suited crosslinkable side group for this approach is dimethylmaleimide (DMMI) that undergoes a UV light-induced dimerization, catalyzed by the addition of the triplet sensitizer thioxanthone disulfonate (TXS).13,14 In molar concentrations up to 4.5 mol%, DMMI has been shown to have only a minor influence on the LCST of a PNIPAAm-based copolymer.15 By interconnecting these hydrogel network precursors, gels form that appear clear and translucid in optical microscopy, exhibiting network homogeneity on the micro-scale. This is because no exothermic processes, such as initiator decomposition and chain-growth propagation, are involved in network formation. Beyond visual observations, static light scattering according to the Debye–Bueche method reveals that this type of hydrogel only shows small root-mean-square concentration fluctuations, indicating nanoscale homogeneity.8 This proves that the polymer-analogous reaction strategy is suitable for synthesizing well-defined PNIPAAm polymer networks with a high level of control over the homogeneity on the micro- and nanoscale, and thus reproducible temperature response.
For applications, which require a fast and reproducible response of a PNIPAAm hydrogel toward heat as an external stimulus, we do not only require a defined network structure but also the object size is likewise crucial. Here, hydrogel particles in the micrometer range – so-called microgels according to the IUPAC definition16 – exhibit a quicker feedback than their macroscopic analogs.17 For fabricating microgels in the range from 10 to 1000 μm with narrow size distribution (<5%) and controlled network homogeneity,8,18 droplet microfluidics is the method of choice.19–21 Here, an aqueous stream of hydrogel precursor solution is periodically broken up into surfactant-stabilized droplets with a reproducible size by a flow of a second immiscible fluid. The gelation of droplets containing PNIPAAm hydrogel precursors can be triggered by means of heat,22 light,23 or addition of a reaction accelerator,24 giving rise to micrometer-sized hydrogel particles. For actual usage, as-formed microgels have to be transferred into an aqueous environment by breaking the emulsion, which has developed beyond chemically induced surfactant exchange in recent years.25,26
The responsiveness of PNIPAAm microgels towards external stimuli renders them well-suited as temperature sensors, which can be easily calibrated by recording the microgel diameter as a function of environmental temperature with optical microscopy. PNIPAAm microgels can provide reliable and quantitative information on the heat input into microscopic volumes, e.g., inside optical traps. This is particularly important when optical traps are used to trap and manipulate living cells via laser light directly,27 such as in optical stretchers,28 and optical cell rotators.29,30 In optical stretchers (OS), two coaxially aligned, non-focused laser beams with a Gaussian intensity profile are integrated into a microfluidic device to trap cells passing through a microchannel.31 By increasing the laser power, trapped objects are stretched by optically induced surface forces along the beams' axis. This allows for measuring the cells' viscoelastic properties, which in turn permits insights into physiological and pathological functional changes in cells in situ.32–37 While, conventionally, a good sensor and analysis tool should not alter the properties of the analyte during characterization, the viscoelastic properties of trapped cells depend on the temperature during the measurement38–41 wherefore the quantification of the lasers' heat input is crucial. While the temperature in the trapping volume of optical stretchers was quantified by fluorescence ratio thermometry42 and the impact of laser-induced heating on the cell viability after single-cell trapping analyzed,43 we are reporting on a method that employs a cell-sized, standardized calibration tool to directly derive the temperature in the trap.
In this work, we utilize microfluidically produced PNIPAAm microgels as cell-sized sensor particles and calibration tool in optical stretchers. For that, we compare microgels emerging from free-radical chain-growth gelation (FCG) and polymer-analogous photogelation (PAG) and provide an in-depth analysis of polymer network homogeneity on the microscale as well as temperature-dependent mechanics by utilizing advanced optical microscopy based on optical diffraction tomography and Brillouin microscopy that has previously been used to provide a detailed view on cellular structure and composition.44,45 Microgels in the size range of living cells made from macromolecular precursors and crosslinked by PAG are then utilized as temperature sensors to quantify the amount of heat introduced to living cells by laser-induced heating in OS setups. To determine the absorbed energy, we record the absorbance of PNIPAAm hydrogel in the range of the two laser wavelengths at 780 and 1064 nm. By introducing PNIPAAm microgels with well-defined physicochemical and mechanical properties as a cell-like replacement into OS setups, we quantify the amount of heat introduced under the same experimental conditions as in real-life cell stretching experiments. With that, we provide the basis to describe the extent of undesired heating in the theoretical description of viscoelastic properties of living cells extracted from cell-stretching experiments.
Results and discussion
Microgel preparation by droplet microfluidics
Droplet microfluidics is employed to fabricate PNIPAAm-based microgels with a diameter lower than 20 μm to match the size of eukaryotic cells, e.g., BALB 3T3 fibroblasts and breast epithelial cells,28,32 whose mechanical properties are commonly analyzed by optical stretching. In addition, droplet microfluidics provides microgels with reproducible and uniform size via fast mixing of hydrogel precursors prior to droplet solidification, which is a prerequisite for homogeneous hydrogel network formation. As we require reproducible temperature response and volume phase transition (VPTT) behavior, respectively, actual knowledge of hydrogel composition and structure is crucial.18 On this account, we compare hydrogel network formation via PAG and FCG. In the former case, a pre-polymerized statistical copolymer of N-isopropylacrylamide (NIPAAm) and dimerizable dimethylmaleimide-functionalized acrylamide (DMMIAAm), PNIPAAm-co-DMMIAAm, is utilized for UV light-induced crosslinking of the hydrogel network. PNIPAAm-co-DMMIAAm is synthesized based on a protocol from Adler and coworkers.46 To improve reaction kinetics, the triplet sensitizer TXS is added, synthesized based on a protocol from Kronfeld and Timpe.47 In the case of FCG, hydrogel networks inside microfluidically prepared droplets are formed by statistical chain-growth copolymerization of NIPAAm and BIS initiated by UV light-induced cleavage of the radical initiator Irgacure® 2959.For droplet generation from both PAG and FCG precursor solutions, polydimethylsiloxane (PDMS)-on-glass microflow cells fabricated by combined photo- and soft lithography are utilized. Emulsion droplets are pinched off from an aqueous stream of hydrogel precursor solutions by geometrically confined breakup from a flow-focusing junction with a nozzle diameter of 15 μm (Fig. 2). As-formed water-in-oil (W/O) emulsion droplets are stabilized against coalescence in the surrounding continuous fluorocarbon oil phase, 3M™ Novec™ 7500, by a non-ionic home-made ABA triblock copolymer surfactant,48 set to a concentration of 2% (w/w).
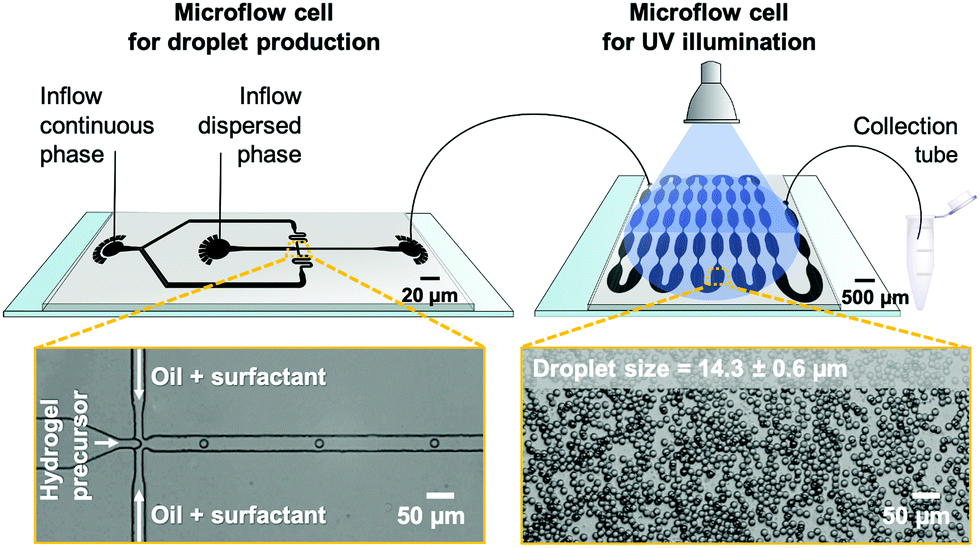 | ||
| Fig. 2 (top) Experimental setup for microgel fabrication by polymer-analogous gelation (PAG). AutoCAD®-designed microflow cells for droplet formation and UV illumination connected via tubing. After illumination, the emulsion is collected in an Eppendorf tube. (bottom) Bright-field microscopy images of emulsion droplets inside the droplet-generating flow cell (left) and during UV illumination (right). The mean size of emulsion droplets (N = 100) with their standard deviation is indicated. | ||
Since the properties of a good sensor should not have an impact on its readout, it is important to render the microgel probes as invisible as possible to the IR lasers of the stretcher setup or at least comparable to the cells commonly analyzed in optical stretchers. To ensure this, bulk gelation tests are carried out based on PAG before performing the more elaborate microfluidic emulsion formation of PNIPAAm-co-DMMIAAm to enable microgel particle fabrication with the highest possible transparency and the lowest possible absorption. Here, the lowest concentration for forming mechanically stable PAG hydrogels from dimerizing PNIPAAm-co-DMMIAAm is found to be 30 mg mL−1 in deionized (DI) water. Therefore, this concentration is used for subsequent microfluidic experiments. As-formed emulsion droplets are transferred to a second microflow cell for solidification by UV illumination. Its key design feature is a sequence of tapered microchannels that ensures equal residence and illumination time for all droplets passing through the flow cell by repeatedly repositioning droplets from the microchannel walls with more friction and thus lower velocity to the center of the emulsion flow and vice versa.48,49 From our experience, utilizing on-chip UV illumination should enable uniform and complete crosslinking of all droplets as compared to conventional off-chip solidification in collection vials containing multi-layered emulsion droplets that would scatter and dim UV light penetrating from top-to-bottom through the collected emulsion. The setup for on-chip illumination, which also allows for precisely adjusting the UV beam size, has been previously introduced by Heida et al.50
The illumination time for PAG experiments in the second flow cell is approximately 4 min with a UV-light intensity of 160 mW cm−2 on the surface of the microflow cell. Bright-field microscopy images of precursor microdroplets indicate the formation of uniform emulsion droplets with an average diameter of 14.3 ± 0.6 μm. However, the surfactant shell of the gelled emulsion droplets cannot be broken by conventional chemically induced coalescence through the addition of a weakly stabilizing surfactant such as 1H,1H,2H,2H-perfluoro-1-octanol (PFO) that would enable the microgels to transfer into water. PNIPAAm microgels exhibit an amphiphilic character and stabilize the water-fluorocarbon oil mixture that forms during PFO-induced droplet coalescence. By adsorbing at the interface of water and oil droplets, these remain stable even after surfactant removal. Therefore, an alternative purification process is employed. After removing excess oil by repeated centrifugation and syringe aspiration, an excess amount of isopropyl alcohol (IPA) is added to the solidified emulsion droplets, causing droplet coalescence and yielding a dispersion of PNIPAAm microgels in IPA. Due to the known effect of co-non-solvency of IPA and water,51 microgels cannot directly transfer to water by a stepwise exchange of the surrounding media. Instead, we use 1,4-dioxane as an auxiliary solvent being miscible with both IPA and water. That way, our microgels first transfer from IPA to dioxane, and then from dioxane to water. It should also be noted that due to the microgels' small diameter and low material content, a separation from the supernatant by centrifugation before changing from one eluent to the next is not feasible. Therefore, the solvents IPA and dioxane are gradually exchanged by consecutive steps of evaporation before the microgels’ final transition into water. Despite swelling and collapsing during the process of solvent change, PAG-made microgels maintain their low size distribution with 18.4 ± 0.6 μm in IPA, 19.6 ± 0.6 μm in dioxane, and 16.2 ± 0.5 μm in water. After considerable swelling in IPA (2.1-fold in volume) and dioxane (2.6-fold in volume), these microgels reveal a minor 1.5-fold increase in volume compared to the original droplet template size of 14.3 ± 0.6 μm (cf. above).
To compare the effect of different gelation mechanisms on PNIPAAm polymer network architecture, we investigate a second type of microgels synthesized via FCG. In contrast to PAG microgels formed from PNIPAAm-co-DMMIAAm with an overall material content of 30 mg mL−1 in the precursor solution and 3 mol% dimerizable DMMI groups, corresponding to 1.5 mol% crosslinks, mechanically stable microgels cannot be fabricated by FCG using 1.5 mol% BIS as crosslinker. Therefore, the molar percentage of BIS crosslinker is increased stepwise from 1.5 mol% to 6 mol% relative to the NIPAAm content. By this, we confirm the finding by Seiffert et al. that PAG is more efficient than FCG, enabling hydrogel network formation at significantly lower amounts of crosslinker.8 Using the same setup and oil/surfactant system as for the PAG-reacted microgels, no stable FCG microgels are initially formed, presumably due to insufficient illumination energy inside the second flow cell previously utilized for on-chip solidification of emulsion droplets. By changing the UV illumination from on-chip to in-tubing (Fig. S1, ESI†), a higher local UV light intensity is realized, leading to sufficient cleavage of initiator and, eventually, hydrogelation via FCG. The intensity at the PE tubing connecting the outflow port of the microfluidic droplet generator and the collection vial is 360 mW cm−2 with a droplet residence time in the UV-irradiated tubing volume of 20 s compared to 4 min at 160 mW cm−2 for the on-chip illumination setup used for PAG.
After UV irradiation and utilizing the protocol for emulsion decomposition and microgel transfer introduced for the PAG microgel purification above, PNIPAAm precursor droplets with a size of 14.6 ± 0.7 μm yield microgels with a size of 17.0 ± 0.9 μm (1.6-fold swelling in volume) in IPA. However, these cannot be transferred to water with the support of dioxane. Instead, our FCG-prepared microgels exhibit a brittle appearance as shown by bright-field microscopy and experience a swelling-related shock leading to their fragmentation. We account this behavior to the microgels' marked porosity visible on the microscale (discussed in more detail in the following section) with micropore sizes of approximately 1 μm, which are only one magnitude smaller than the microgel diameter and could, therefore, lead to the observed instability during swelling. To deepen the insights on the network homogeneity, PNIPAAm microgels formed by PAG and FCG will be compared by optical techniques being swollen in IPA. Since pure IPA is, like water, a good solvent for PNIPAAm and the swelling behavior of the microgels in these solvents rather similar,52 our findings on the PNIPAAm hydrogel network architecture derived from investigations in IPA can be transferred to microgels swollen in water used for the stretching experiments.
Analysis of microgel homogeneity by optical techniques
The homogeneity of the microgel network architecture created either via PAG and FCG is investigated on the microscale by optical methods utilizing phase-contrast microscopy, optical diffraction tomography, and Brillouin microscopy. After purification, PNIPAAm microgels swell in IPA and reduce their already low material content of 3% (w/v, 30 mg mL−1) in the precursor solutions to approximately 1.4% (w/v) for PAG and 1.9% (w/v) for FCG in the microgel state. This low material content is the reason why the refractive index (RI) of these microgels, which consist to more than 98% out of solvent accordingly, only differs slightly from the RI of the surrounding IPA medium. Therefore, phase-contrast microscopy is utilized for a more detailed analysis. The corresponding image in Fig. 3 (top) shows that PAG microgels do not exhibit visible inhomogeneities on the microscale and a high optical transparency. Opposed to that, phase-contrast microscopy analysis of FCG microgels in Fig. 3 (bottom) reveals pronounced inhomogeneities visible in the form of brighter and darker domains, which can be assigned to microscopic pores formed during the gelation due to heat generated by the exothermic free-radical network formation. As the temperature inside the droplets increases towards or above the LCST of the as-formed PNIPAAm chains in water, a microphase separation occurs that is permanently locked by further crosslinking. The combination of these heterogeneities of approximately 1 μm at an overall microgel size of approximately 18 μm (cf. above) with a low material content of only 1.9% (w/v) explains the insufficient stability of microgels obtained via FCG during solvent exchange from IPA over dioxane to water.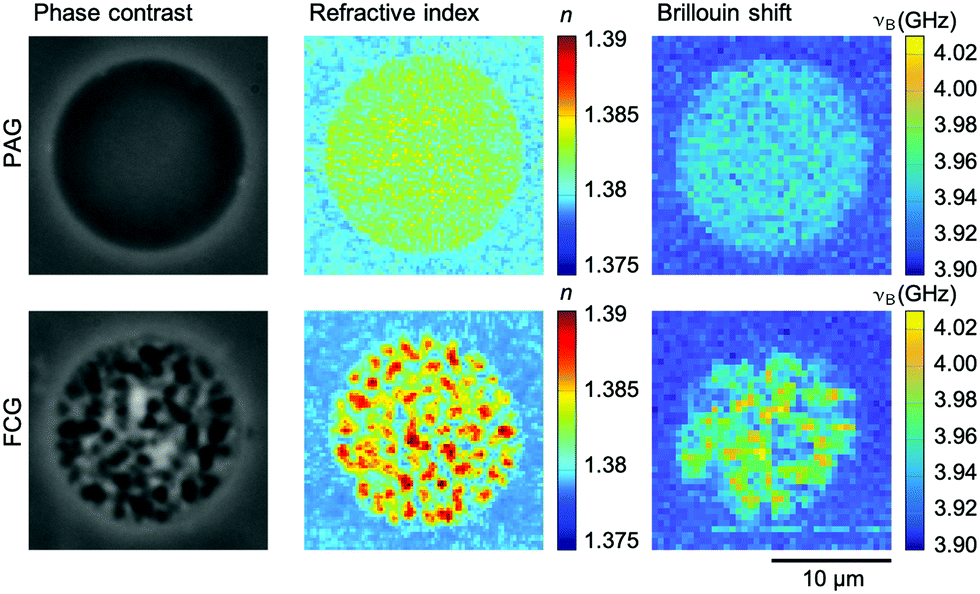 | ||
| Fig. 3 Phase-contrast microscopy, optical diffraction tomography, and Brillouin microscopy images of PNIPAAm microgels in IPA fabricated via polymer-analogous photogelation (PAG) (top) and free-radical chain-growth gelation (FCG) (bottom). All three methods reveal a high homogeneity of the polymer network architecture on the microscale for microgels synthesized via PAG and visible heterogeneities of microgels synthesized via FCG. | ||
Next, we employ optical diffraction tomography (ODT) as an imaging technique to obtain a quantitative RI map of the microgels in 3D. For ODT reconstructions, spatially modulated holograms of PAG and FCG microgels are recorded from multiple angles from which the complex optical field is obtained by a Fourier-based field retrieval algorithm.45 Utilizing the Fourier diffraction theorem, the RI tomogram is then reconstructed from the complex optical fields. Fig. 3 shows corresponding quantitative RI images from the center plane of both microgel types recorded by ODT. Both are in good agreement with the results of phase-contrast microscopy. The PAG microgels reveal a homogenous RI distribution, which points toward a homogeneous hydrogel network structure on the microscale. For FCG microgels, refractive indices obtained within the pores are equal to the RI in the surrounding medium, indicating the formation of interconnected pores in the micrometer range within the PNIPAAm microgels.
The ODT findings are supported by confocal Brillouin microscopy. Brillouin microscopy exploits the inelastic scattering of incident photons by acoustic phonons within the sample. It measures the Brillouin shift νB, which is related to the longitudinal modulus, the RI, and the density of the sample. For recording spatially resolved maps of microgels made via PAG and FCG, the microscope stage is moved automatically and confocal measurements of the Brillouin shift are recorded. The detectable variations of the Brillouin shift within the PAG microgel are equal to the random error of the measurement system, whereas the mechanical heterogeneity in the FCG microgels is evident (Fig. 3).
Homogeneity of the hydrogel network and its mechanics on the microscale and excellent transparency presumably render PAG-made microgels more suitable for application as reproducible temperature sensors in OS than FCG-made microgels. Thus, our subsequent investigations focus on the characterization of PAG-based microgels in water as the target medium in conventional OS characterization of living cells. For calibration purposes, we investigate microgel swelling as a function of temperature, microgel mechanics, and absorbance of the hydrogel network.
Swelling behavior of microgels fabricated by PAG
The size of PNIPAAm microgels in water prepared by PAG is recorded as a function of temperature to determine their swelling behavior. While the amount of DMMI moieties in the microgel precursors should only have little effect on the VPTT,15 we investigate the continuity of the volume phase transition behavior. For that, we fill PNIPAAm microgels into a fluid cell on a temperature-controlled stage to precisely control and increase the medium's temperature in a stepwise fashion from 20 to 33 °C.The upper part of Fig. 4A shows the change in microgel diameter with temperature. At low temperatures of approximately 20 °C, the microgels are fully swollen in water. By increasing the temperature, the hydrogen bonds between the protons of water and the polymer chains become less preferred than intra- or intermolecular interactions. Thus, the hydrogel network releases water causing the microgel collapse at higher temperatures. To determine the VPTT, we fit the experimental data with a sigmoidal BiDoseResp function (Origin) and calculate the first derivate of this fit based on a procedure from Cors et al.53 In the lower part of Fig. 4A, the derivative is plotted as a function of the temperature. The inflection point of the fit lies at the minimum of the first derivative, which is indicated by a vertical line at a VPTT of 30.5 °C. Beyond the VPTT, the sigmoidal fit reveals that microgels undergo a phase transition in a continuous fashion, which is consistent with the predictions for a homogeneous hydrogel network from Li and Tanaka.54 The change of RI derived from quantitative phase images is plotted as a function of the temperature in Fig. 4B. Here, the increasing RI with increasing temperature is in good accordance with the temperature-dependent swelling behavior of PAG microgels, displayed in Fig. 4A.
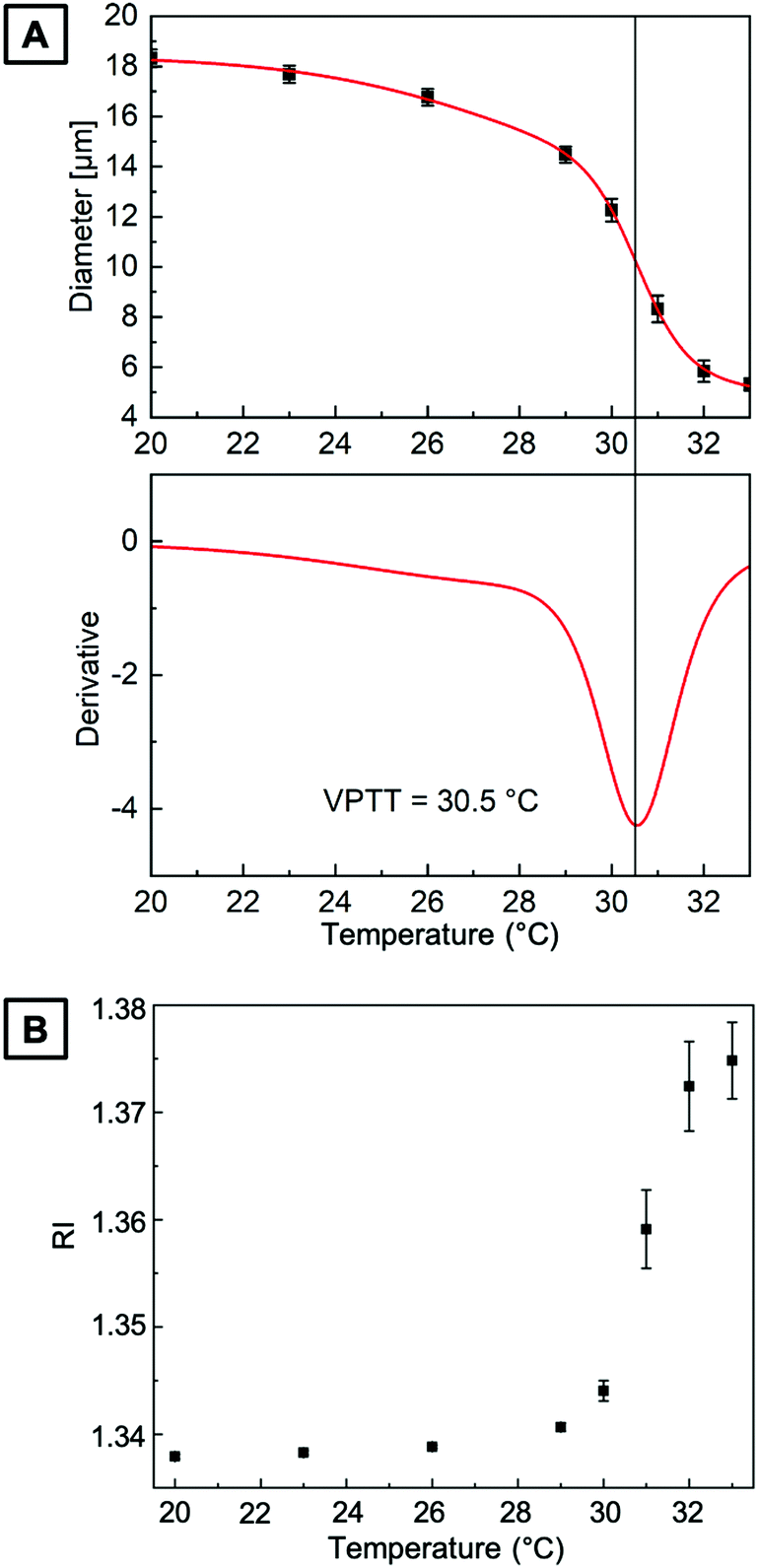 | ||
| Fig. 4 (A) (top) Swelling of PNIPAAm microgels in water prepared by PAG as a function of temperature. Red line is a fit of a sigmoidal function to the experimental data (black squares). (bottom) First derivative of the fitted experimental data. The vertical line indicates the VPTT to be 30.5 °C. (B) RI derived from quantitative phase images of microgels in water as a function of temperature. | ||
Temperature-dependent micromechanics of PNIPAAm microgels
To further investigate the mechanical properties of the PNIPAAm microgels made by PAG, we use Brillouin microscopy and determine the longitudinal modulus at different temperatures (Fig. 5). At room temperature, the longitudinal modulus of the microgels is mainly determined by the bulk modulus of water, as the polymer content is in the range of only a few percent. With increasing temperature, the longitudinal modulus of the surrounding water shows a well-known, weak temperature dependence, which is approximated by a linear fit. Hydrogels with a high water content can be typically described by a binary mixture model approximating the mechanical properties as the weighted sum of solute and solvent.56,57 This approximation also works for other solute molecules, not exclusively for polymers.58 Yet, the general temperature dependence of the longitudinal modulus is not covered by a binary mixture model. We thus use a simplified binary mixture model and approximate the general temperature dependence of M by| ΔM(T)microgel ≈ ΔM(T)water ≈ b × T | (1) |
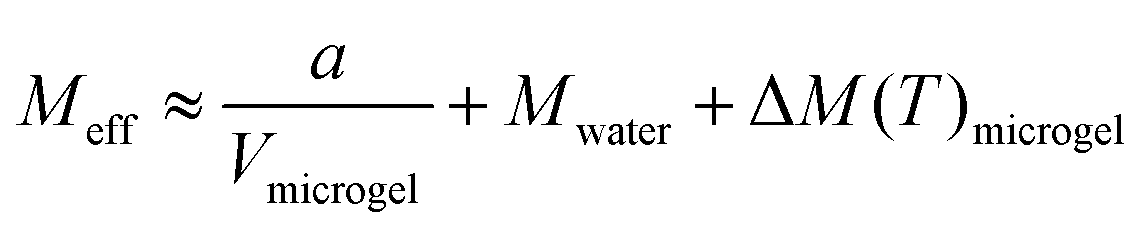 | (2) |
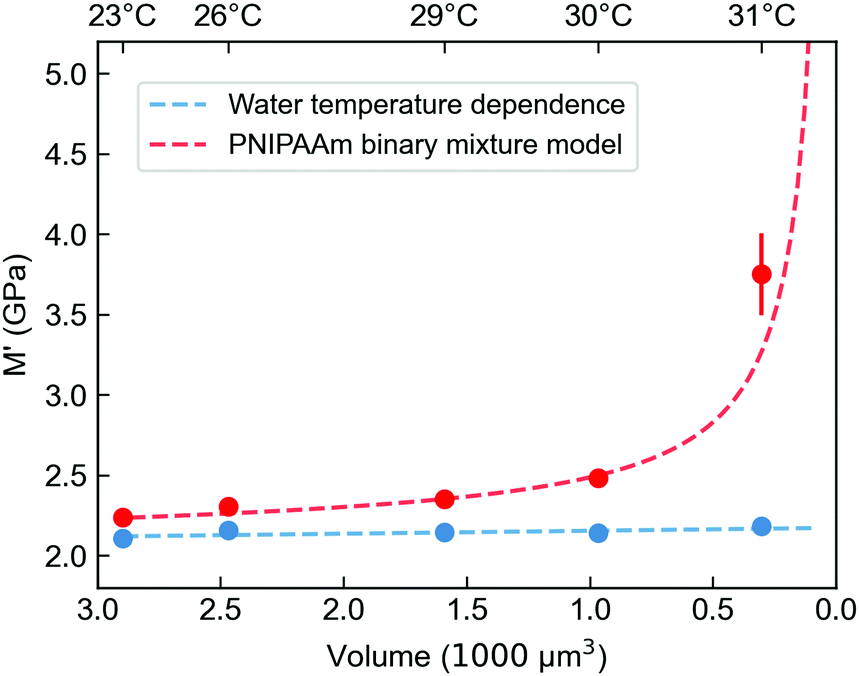 | ||
| Fig. 5 Longitudinal storage modulus M′ of PNIPAAm microgels prepared by PAG derived from Brillouin microscopy measurements at different temperatures with corresponding volumes of the microgels. The red line shows the fit for an adapted binary mixture model (see ESI†). The blue curve is a linear approximation for the temperature-dependent longitudinal modulus of the surrounding water. The volume is calculated from a series of quantitative phase imaging (QPI) and extracted with an algorithm as described in Müller et al.55 | ||
PNIPAAm microgels as temperature sensors in optical stretchers
As introduced above, a common challenge when measuring mechanical properties of biological cells by means of light is the amount of heat introduced due to the light absorption of the water and the cellular material. At this point, it is crucial to understand the increase in temperature due to the increase in laser power and decouple the mechanical response of the cell from its thermal response.To characterize the temperature increase in the OS in a typical creep compliance measurement protocol, we employ our optically characterized PNIPAAm microgels as thermoresponsive probes. The corresponding OS setup is shown in Fig. 6A and schematically explained in Fig. 6B. One of the most important features of an OS is the ability of sudden change of the optical stress applied on the sample. This is achieved by precisely tuning the optical output power of the lasers, usually in the range of ms, with three distinct phases (cf.Fig. 6C): trapping of the particles at lower laser power, exposure at high laser power phase and a recurrent phase of lower laser power exposure. The behavior of microgels in these steps is recorded with a camera, and the size and shape of a particle can be extracted for each frame. For our comparative analysis, we quantify the size of the PNIPAAm microgels as the average of the collected data in the last second of each phase. PNIPAAm microgels are trapped at low total power (P = 0.1 W with 0.05 W per optical fiber), before a higher pulse is applied identically as in experiments with living cells. The first set of experiments is carried out at room temperature (T = 23 °C). We use the highest available total power of P = 1.5 W at 780 nm. The increase in laser power induces a noticeable, but minimal size reduction of the microgel (Fig. 6C and D). Even at powers as high as 1.5 W, the microgels do not fully collapse. This means that the temperature increase induced by this light source is in the range of few K W−1, which is in line with previous estimates.40 In contrast, using a 1064 nm laser, a total power of P = 0.6 W is sufficient to observe a full collapse of the microgels resulting in a diameter reduction of 40%. With the VPTT established as T = 30.5 °C, the diameter reduction corresponds to a temperature difference of approximately 8 K induced by the 0.6 W power of the laser, which translates to a temperature increase of 13.3 K W−1 within the OS. This result is in good agreement with a previous study using fluorescence ratio thermometry without a trapped object.42
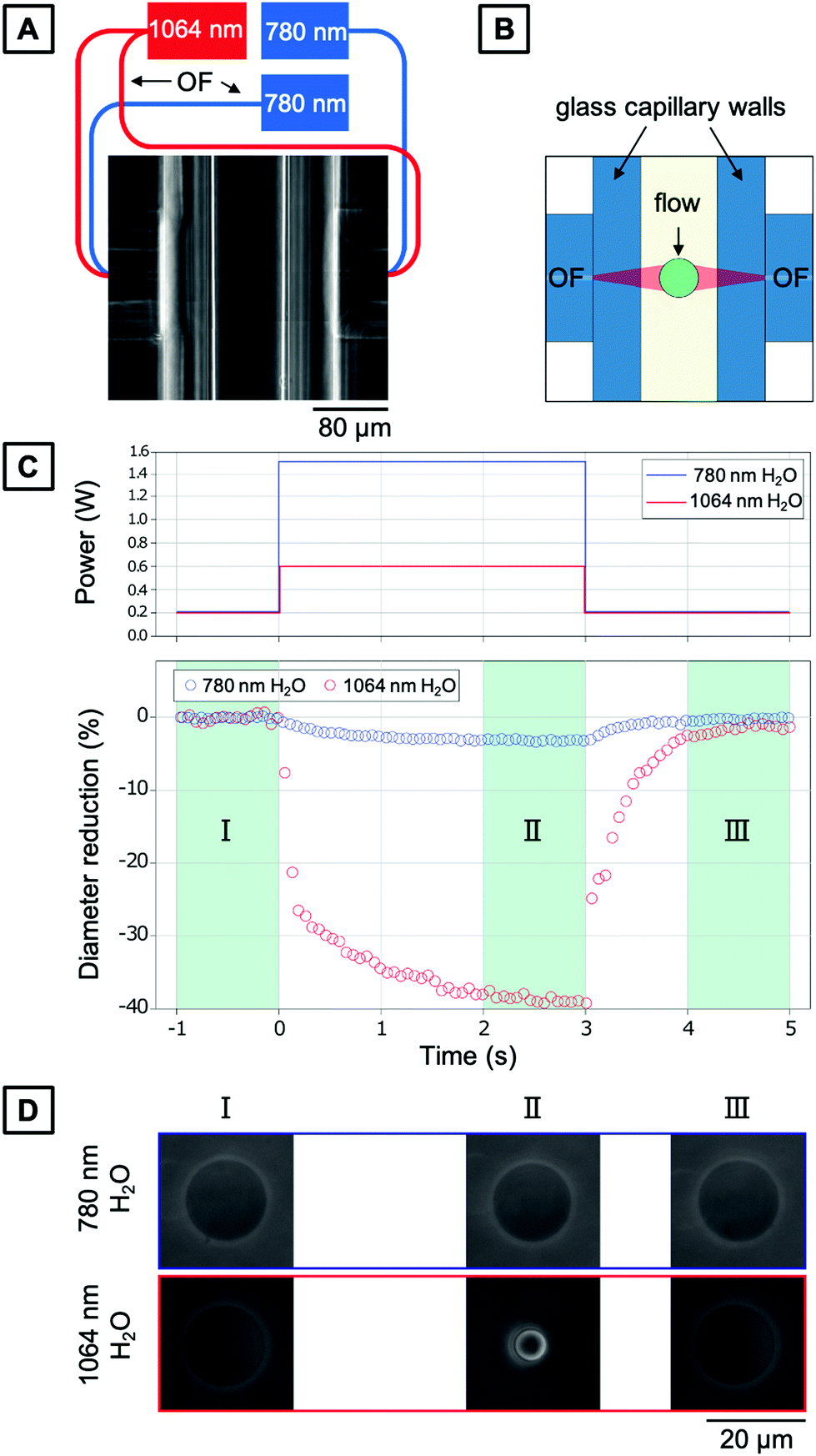 | ||
| Fig. 6 Optical stretcher setup and experimental procedure for trapping PNIPAAm microgels. (A) Top view of the trapping region. Two opposing optical fibers are mounted perpendicular to a glass capillary acting as a microfluidic channel. Note: each pair of fibers is connected to a different optical stretcher setup, but for simplicity integrated in the same sketch in here. (B) Schematic of the top view of the trapping region. Optical fibers and the glass capillary are depicted in light blue, and the core of the fibers is depicted in dark blue. Two divergent beams (red) are creating the trap. A cell is depicted in green. The flow (arrow) is adjusted by means of gravity. (C) Top: Typical optical step stress applied. Bottom: Corresponding diameter reduction of the PNIPAAm microgels during the three phases. (D) Size of PNIPAAm microgels as function of the wavelength and phase of the optical stress. | ||
Absorbance of PNIPAAm hydrogels
To provide an explanation of the differences of thermal energy introduced by the 780 nm 1064 nm lasers leading to the observed temperature increase in the OS setup, the absorbance of PNIPAAm hydrogels synthesized via PAG is investigated. Absorbance describes the intensity loss of light for a given light path length through a sample. The relevant processes contributing to the absorbance are elastic scattering in the sample and light absorption that causes sample heating. Due to the emission wavelengths of the utilized lasers at 780 nm and 1064 nm, a region of interest from 700 to 1100 nm is analyzed. To receive information on the absorbance of the hydrogel in H2O, bulk gels are prepared directly inside cuvettes tailored for absorbance measurements, and compared to pure H2O. Recorded spectra are displayed in Fig. 7. Below 900 nm, our PNIPAAm hydrogels show a higher absorbance than pure water, whereas the offset gradually increases for decreasing wavelength. To investigate this behavior more closely, the decoupled contributions of scattering and absorption are calculated by measuring the complete scattering response and the scattering response without zero-order transmission. D2O is used as a solvent and reference to avoid distortion of the weak scattering signal in the IR by the scattering of the stretching vibrations of H2O (cf. Fig. S2, ESI†). The detected scattering signal of the PNIPAAm hydrogel network in D2O increases with decreasing wavelengths, which is also the reason for the increasing absorbance with decreasing wavelength in Fig. 7. As the pore size of the polymer network is much smaller than the laser wavelength, the elastic scattering processes in the gel can be described by Rayleigh scattering. The cross section of Rayleigh scattering scales with λ−4, and therefore elastic scattering contributes much more to the absorbance at shorter wavelength. Above 900 nm, the absorbance of the microgels in H2O drastically increases. As this increase is not present in the gels suspended in D2O, it can be attributed to the absorption of H2O giving rise to a stronger heating effect. These findings explain the much more pronounced temperature increase of PNIPAAm hydrogels illuminated with a wavelength of 1064 nm compared to 780 nm.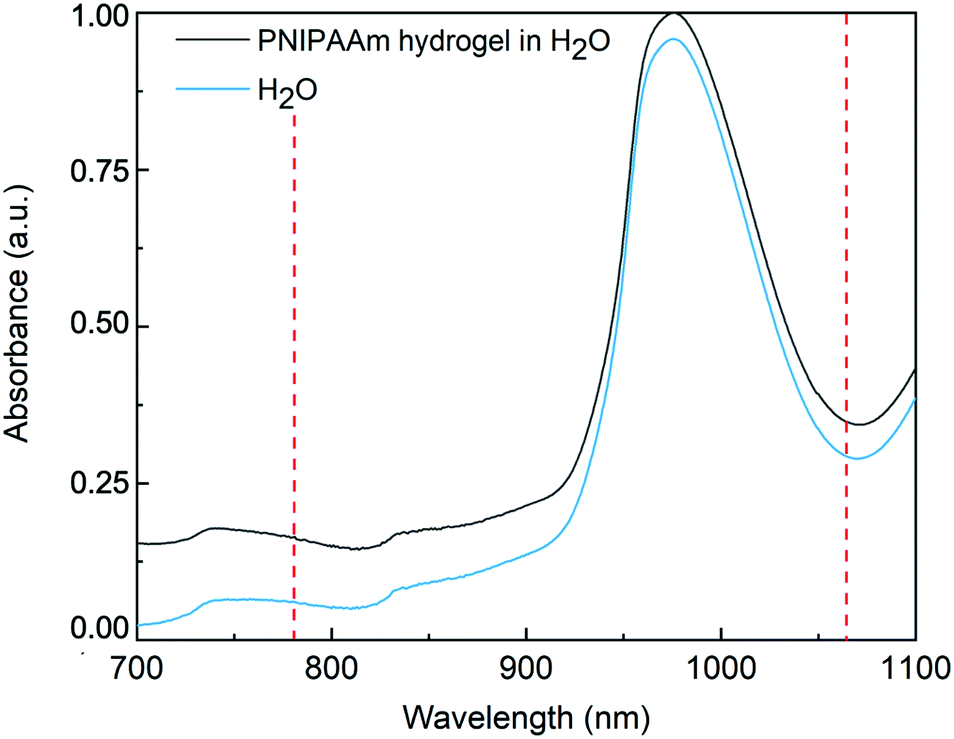 | ||
| Fig. 7 Normalized absorbance spectrum of PNIPAAm bulk hydrogel made by PAG, swollen in H2O, and the associated spectrum of pure H2O. Dashed red lines indicate the wavelengths of the lasers of the optical stretcher setup at 780 nm and 1064 nm used in this study. | ||
Conclusion
In summary, polymer-analogous photogelation of tailor-made crosslinkable PNIPAAm macromers combined with microfluidic emulsion formation provides the means to reproducibly form sub-20 μm microgels as cell-sized temperature sensors in optical stretchers. Optical analysis of these microgels by phase-contrast microscopy, optical diffraction tomography, and confocal Brillouin microscopy reveals a homogeneous hydrogel network structure on the microscale. Investigating the swelling behavior, we found that our PAG microgels show a VPTT at 30.5 °C undergoing a phase transition in a continuous fashion, which is in consistency with the predictions for a homogeneous hydrogel network from Li and Tanaka.54 Investigating the temperature-dependent micro-mechanics, we determined the longitudinal modulus of our PAG microgels at different temperatures. We found an average longitudinal modulus close to water at room temperature, followed by a step increase when the VPTT was approached. The observed elevation of the longitudinal modulus for higher temperatures could be addressed to a larger polymer volume fraction, according to previous studies that investigated gelatin gels57 and hydrated collagen fibers.59In addition, we established relationships between temperature, microgel diameter and refractive index to quantify the heating effect of an optical stretcher at its operating wavelengths of 780 and 1064 nm. While values derived from microgel swelling for a local temperature increase in the trapping region are shown to be in the range of a few K W−1 for the 780 nm laser, measurements utilizing the 1064 nm laser exhibit a significantly higher increase of the local temperature of 13.3 K W−1. The observed temperature increase utilizing the 1064 nm laser is in good agreement with previous investigations that were performed utilizing thermosensitive fluorescent dyes.42 Absorbance measurements carried out on bulk PNIPAAm hydrogels and pure H2O reveal a significantly higher absorbance in the region above 900 nm, which can be attributed to the absorption of H2O leading to the observed differences of the temperature increase for the different lasers.
For calibrating optical stretcher setups, the very first trapped hydrogel particle taken from a microfluidically prepared, homogeneous microgel population will already provide information on the heat input by the lasers in the trapping volume by its temperature-induced collapse. To guarantee a statistically reliable calibration prior to stretching experiments of living cells, a microgel quantity of N ≥ 20 would be utilized.
Materials and methods
Materials and instrumentation for microgels synthesis
All chemicals were used without further purification unless stated otherwise. Anhydrous sodium carbonate, acryloyl chloride, magnesium sulfate, dimethylsulfoxide-d6, chloroform-d3, deuterium oxide, N-isopropylacrylamide, N,N′-methylenebis(acrylamide), 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone (Irgacure® 2959), azobisisobutyronitrile, thioxanthen-9-one, sulfuric acid fuming (20% SO3), calcium carbonate, barium chloride dihydrate, sodium carbonate, and phenolphthalein were purchased from Sigma Aldrich/Merck (Germany). N-tert-Butyloxycarbonyl-1,2-ethylenediamine was purchased from Carbolution Chemicals (Germany). Dichloromethane, chloroform, N,N-dimethylformamide, acetone, and isopropanol were purchased from Fisher Scientific (USA). Sodium chloride was purchased from Carl Roth (Germany). Trifluoroacetic acid, triethylamine, and 2,3-dimethylmaleic anhydride were purchased from TCI Deutschland (Germany). Diethyl ether was purchased from VWR Chemicals (USA). Deionized (DI) water with a resistance of 18.2 MΩ cm was prepared in a Milli-Q Direct 8 water purification system (Merck Millipore, USA).NMR spectra were recorded on a Bruker Avance III 500 (USA). Chemical shifts are reported in ppm units relative to tetramethylsilane (δ = 0.00) as internal standard. SEC measurements were performed on an Agilent LC 1100 (USA) utilizing Zorbax PSM Trimodal-S columns with an eluent composed of dimethylacetamide (DMAc), 2% (v/v) water, and 3 g L−1 lithium chloride operated at a flow rate of 0.5 mL min−1. Samples were dissolved at a concentration of 2 mg mL−1 and passed through a 0.2 μm syringe filter. Calibration was performed with poly(2-vinylpyridine) (P2VP) standards. Hence, molecular weights are obtained as apparent P2VP equivalent values.
Microfluidic device fabrication and general microfluidic setup
Microflow cells were fabricated by combined photo- and soft lithography. Briefly, the negative photoresist SU-8 2015 (Micro Resist Technology, Germany) was spin-coated onto the polished side of a 3-inch silicon wafer (Siegert Wafer, Germany). Using a mask aligner (MJB3, Süss MicroTec, Germany), the desired microchannel structure, created by employing a computer-aided design software (AutoCAD®, Autodesk, USA) and beforehand printed on a photomask, was transferred into the photoresist. After that, the non-illuminated parts of the photoresist were removed with the developer mr-Dev 600 (Micro Resist Technology, Germany). The desired channel height and width of 15 μm at the droplet-forming nozzle of the master structure was verified by confocal microscopy (μsurf expert, NanoFocus AG, Germany). After that, polydimethylsiloxane (PDMS) base material and crosslinker (Sylgard 184 silicone elastomer kit, Dow Corning, USA) were mixed in a ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (w/w) and degassed in a planetary centrifugal mixer (ARE-250, Thinky, USA), then poured onto the master structure, and cured for at least 2 hours at 65 °C. The microstructures were cut out of the PDMS layer, and access ports for the tubing were stamped with a biopsy punch (diameter: 1.0 mm, KAI Medical, Germany). Finally, the PDMS-based microchannel replica was bonded to a glass slide (76 mm × 26 mm) after surface treatment by oxygen plasma (100 W, 15 s, MiniFlecto 10, Plasma Technology, Germany). To render the surface of the microchannels hydrophobic, a solution of (tridecafluoro-1,1,2,2-tetrahydrooctyl)trichlorosilane (1% v/v) (Gelest, USA) in 3M™ Novec™ 7500 (IoLiTec, Germany) was injected into the channels approximately 15 min before usage.
:1 (w/w) and degassed in a planetary centrifugal mixer (ARE-250, Thinky, USA), then poured onto the master structure, and cured for at least 2 hours at 65 °C. The microstructures were cut out of the PDMS layer, and access ports for the tubing were stamped with a biopsy punch (diameter: 1.0 mm, KAI Medical, Germany). Finally, the PDMS-based microchannel replica was bonded to a glass slide (76 mm × 26 mm) after surface treatment by oxygen plasma (100 W, 15 s, MiniFlecto 10, Plasma Technology, Germany). To render the surface of the microchannels hydrophobic, a solution of (tridecafluoro-1,1,2,2-tetrahydrooctyl)trichlorosilane (1% v/v) (Gelest, USA) in 3M™ Novec™ 7500 (IoLiTec, Germany) was injected into the channels approximately 15 min before usage.
The setup for microfluidics experiments included two high-precision syringe pumps (Harvard Apparatus 11 Pico Plus Elite, Harvard Apparatus, USA), to which the microflow cell was connected via PE tubing (inner diameter: 0.38 mm, outer diameter: 1.09 mm, Hartenstein, Germany). A 500 μL gastight syringe (1750 TLL SYR, Hamilton, USA) was used for the dispersed phase, and a 3 mL disposable syringe (BD Luer-Lok tip, Becton Dickinson, USA) was used for the continuous phase. Microfluidic droplet formation was followed on an inverted bright-field microscope (Axio Vert.A1, Carl Zeiss, Germany) equipped with a high-speed digital camera (Miro eX4, Vision Research Inc., USA).
Microgel synthesis
The outflow of the on-chip illumination flow cell was connected to an Eppendorf tube (1.5 mL, Eppendorf, Germany), which was sealed with Parafilm (Bemis Company, Inc., USA). The emulsion was collected in batches at intervals of 30 min. For microgel purification, the fluorinated oil phase with surfactant was removed using a syringe. Isopropanol (IPA) was added to break the emulsion and transfer the microgels into the organic phase. Due to the co-non-solvency of IPA and water, 1,4-dioxane (Fisher Scientific, USA) had to be used as an auxiliary solvent to support the transition of the microgels from IPA to water. For that, IPA was evaporated from the microgel dispersion to 10% of its volume and repeatedly refilled with either the same or the successive solvent within the solvent exchange series. During the evaporation procedure, 1,4-dioxane was refilled five times, and water as well as deuterium oxide four times, depending on the desired target medium.
Emulsion droplets were collected in batches every 20 min. For microgel purification, the CP was removed using a syringe, and IPA was added to break the emulsion and transfer the microgels into the organic phase.
Bright-field and phase-contrast microscopy
Bright-field and phase-contrast microscopy were conducted on a DMi8 (Leica, Germany) equipped with 10× and 40× objective lenses (both air) or on a Axio Observer (Zeiss, Germany) equipped with a 63× objective. Microgel samples were loaded into the cavities created by an imaging spacer with a diameter of 9 mm and a thickness of 0.12 mm (Grace Bio-Labs SecureSeal, Sigma Aldrich, USA) between two glass slides. For evaluating the emulsion droplet and corresponding microgel size in different solvents (IPA, 1,4-dioxane, water, deuterium oxide), at least 50 droplets and microgels, respectively, were measured manually using ImageJ. From this data, mean diameters and standard deviations were calculated. | ||
| Fig. 8 Quantitative phase imaging (QPI) and data evaluation. (A) PAG microgel samples are loaded onto a dynamic thermo-stage on an inverted microscope, and a commercial QPI camera is used for data acquisition (cf. text for details). (B) Bright-field and (C) quantitative phase images of a PAG PNIPAAm microgel at 23 °C (below its LCST). The white dashed circle indicates the microgel perimeter obtained with the analysis software DryMass. (D) Bright-field and (E) quantitative phase images of a PAG PNIPAAm microgel at 32 °C (above its LCST). | ||
Optical diffraction tomography
Optical diffraction tomography was performed using a custom-built optical setup employing Mach-Zehnder interferometry.65 Individual microgel samples were illuminated under different angles by a collimated laser beam at a wavelength of 532 nm. A dual-axis galvanometer mirror was utilized to diffract the illumination beam and generate different illumination angles. After passing through the sample, the beam was interfered with a reference beam at the image plane of a CCD camera that recorded the spatially modulated hologram.Confocal Brillouin microscopy
Brillouin measurements were performed on a custom-built confocal Brillouin microscope based on a Zeiss Axiovert 200 M microscope (Zeiss, Oberkochen, Germany) and a two-stage VIPA spectrometer, as described in detail elsewhere.44,66 Briefly, a microgel sample was illuminated by a frequency-modulated diode laser (DLC TA PRO 780; Toptica, Gräfelfing, Germany) with a laser power of 10 mW at a wavelength of 780.24 nm. Brillouin images were acquired at room temperature with a Zeiss water-immersion C-Apochromat 40/1.2 NA objective. For bulk measurements at different temperatures, a Zeiss C-Apochromat 40/0.95 NA objective was used. The temperature of the microgel sample was controlled either with a Petri dish heater and a temperature probe in the buffer (23 °C–30 °C) or with a heating stage (31 °C).63 Brillouin spectra were evaluated with a self-written MATLAB (The MathWorks, Natick, MA) code, which can be downloaded from https://github.com/BrillouinMicroscopy/BrillouinEvaluation.Optical stretcher setup
To investigate the influence of transient heat on the PNIPAAm microgels prepared via PAG, two OS setups with different laser wavelengths (1064 nm and 780 nm) were employed to trap individual microgels at low power (P = 0.1 W, 0.05 W per fiber) and heat them up at various laser powers (Fig. 6A). The general setup has been described in detail elsewhere.31 The first OS device was mounted on an inverted microscope (Zeiss Axiovert 200M) equipped with a LD Plan-NEOFLUAR Ph2 40×/0.60 NA objective and a single-mode, continuous-wave fiber laser operating at λ = 1064 nm (YLM-5-1070-LP; IPG Photonics, Oxford, MA, USA). This setup not only allows for applying mechanical stress, but also inducing a concomitant temperature increase of 13 K W−1 in a comparable OS setup.42 The second OS device was mounted on an inverted microscope (IX71; Olympus, Melville, NY, USA) equipped with a Plan Fluor 40×/NA 0.75 objective (Olympus) and a single-mode, continuous-wave fiber laser at λ = 780 nm (Eylsa 780; Quantel, Les Ulis, France) with an estimated temperature increase of only 1 K W−1.39 A CCD camera (AVT MARLIN F-146B; Allied Vision, CA) was attached to both microscopes for image acquisition.Vis-IR absorbance measurements
Vis-IR spectra were recorded on a Cary 5000 spectrophotometer (Agilent, USA). Scattering losses were determined with the external diffuse reflectance accessory 2500 (integrating sphere) as described by Steiner et al.67 The sample and reference were positioned in the center of the integrating sphere to collect forward and backward scattering of the sample.Author contributions
N. H., G. C, J. G., and J. T. conceived and designed the experiments; N. H., T. B., G. C., R. S., M. M., and J. S. performed the experiments and analyzed the data; G. C. and S. A. developed the OS setup; R. S. and P. M. developed the QPI and ODT setups; I. R. performed initial experiments on the material basis; N. H. wrote the original draft; all authors reviewed the final version of the submitted manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (Grant agreement No. 852065). J. T. acknowledges funding by the Federal Ministry of Education and Research (BMBF, Biotechnology 2020+: Leibniz Research Cluster, 031A360C), the Volkswagen Foundation (“Experiment!”), and the German Research Foundation (DFG, Research Training Group 1865: Hydrogel-based Microsystems). N. H. thanks Thomas Heida and Niclas Weigel for providing surfactants, Max J. Männel for designing the microfluidic devices (all Leibniz IPF Dresden), and Martin Schumann (TU Dresden) for designing the table of contents graphic.References
- E. M. Ahmed, J. Adv. Res., 2015, 6, 105–121 CrossRef CAS PubMed.
- M. C. Koetting, J. T. Peters, S. D. Steichen and N. A. Peppas, Mater. Sci. Eng., R, 2015, 93, 1–49 CrossRef PubMed.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- Y. Guan and Y. Zhang, Soft Matter, 2011, 7, 6375–6384 RSC.
- F. Doberenz, K. Zeng, C. Willems, K. Zhang and T. Groth, J. Mater. Chem. B, 2020, 8, 607–628 RSC.
- M. A. Haq, Y. Su and D. Wang, Mater. Sci. Eng., C, 2017, 70, 842–855 CrossRef PubMed.
- S. Schmidt, M. Zeiser, T. Hellweg, C. Duschl, A. Fery and H. Möhwald, Adv. Funct. Mater., 2010, 20, 3235–3243 CrossRef CAS.
- S. Seiffert and D. A. Weitz, Soft Matter, 2010, 6, 3184–3190 RSC.
- J. Nie, B. Du and W. Oppermann, Macromolecules, 2004, 37, 6558–6564 CrossRef CAS.
- C. Erbil, Y. Yıldız and N. Uyanık, Polym. Int., 2000, 49, 795–800 CrossRef CAS.
- E. S. Matsuo, M. Orkisz, S.-T. Sun, Y. Li and T. Tanaka, Macromolecules, 1994, 27, 6791–6796 CrossRef CAS.
- A. Habicht, W. Schmolke, F. Lange, K. Saalwächter and S. Seiffert, Macromol. Chem. Phys., 2014, 215, 1116–1133 CrossRef CAS.
- S. Seiffert, W. Oppermann and K. Saalwächter, Polymer, 2007, 48, 5599–5611 CrossRef CAS.
- S. Seiffert, J. Thiele, A. R. Abate and D. A. Weitz, J. Am. Chem. Soc., 2010, 132, 6606–6609 CrossRef CAS PubMed.
- D. Kuckling, J. Hoffmann, M. Plötner, D. Ferse, K. Kretschmer, H.-J. P. Adler, K.-F. Arndt and R. Reichelt, Polymer, 2003, 44, 4455–4462 CrossRef CAS.
- J. V. Alemán, A. V. Chadwick, J. He, M. Hess, K. Horie, R. G. Jones, P. Kratochvíl, I. Meisel, I. Mita, G. Moad, S. Penczek and R. F. T. Stepto, Pure Appl. Chem., 2007, 79, 1801–1829 CrossRef.
- T. Tanaka and D. J. Fillmore, J. Chem. Phys., 1979, 70, 1214–1218 CrossRef CAS.
- S. Seiffert, Macromol. Rapid Commun., 2012, 33, 1135–1142 CrossRef CAS PubMed.
- T. Heida, J. W. Neubauer, M. Seuss, N. Hauck, J. Thiele and A. Fery, Macromol. Chem. Phys., 2017, 218, 1600418 CrossRef.
- N. Hauck, N. Seixas, S. P. Centeno, R. Schlüßler, G. Cojoc, P. Müller, J. Guck, D. Wöll, L. A. Wessjohann and J. Thiele, Polymers, 2018, 10, 1055 CrossRef PubMed.
- S. Girardo, N. Träber, K. Wagner, G. Cojoc, C. Herold, R. Goswami, R. Schlüßler, S. Abuhattum, A. Taubenberger, F. Reichel, D. Mokbel, M. Herbig, M. Schürmann, P. Müller, T. Heida, A. Jacobi, E. Ulbricht, J. Thiele, C. Werner and J. Guck, J. Mater. Chem. B, 2018, 6, 6245–6261 RSC.
- M. S. Boybay, A. Jiao, T. Glawdel and C. L. Ren, Lab Chip, 2013, 13, 3840–3846 RSC.
- T. Kanai, K. Ohtani, M. Fukuyama, T. Katakura and M. Hayakawa, Polym. J., 2011, 43, 987–990 CrossRef CAS.
- R. K. Shah, J.-W. Kim, J. J. Agresti, D. A. Weitz and L.-Y. Chu, Soft Matter, 2008, 4, 2303–2309 RSC.
- I. Akartuna, D. M. Aubrecht, T. E. Kodger and D. A. Weitz, Lab Chip, 2015, 15, 1140–1144 RSC.
- V. Chokkalingam, Y. Ma, J. Thiele, W. Schalk, J. Tel and W. T. S. Huck, Lab Chip, 2014, 14, 2398–2402 RSC.
- A. Ashkin and J. M. Dziedzic, Ber. Bunseng. Phys. Chem., 1989, 93, 254–260 CrossRef.
- J. Guck, R. Ananthakrishnan, H. Mahmood, T. J. Moon, C. C. Cunningham and J. Käs, Biophys. J., 2001, 81, 767–784 CrossRef CAS PubMed.
- M. K. Kreysing, T. Kießling, A. Fritsch, C. Dietrich, J. R. Guck and J. A. Käs, Opt. Express, 2008, 16, 16984–16992 CrossRef CAS PubMed.
- M. Kreysing, D. Ott, M. J. Schmidberger, O. Otto, M. Schürmann, E. Martin-Badosa, G. Whyte and J. Guck, Nat. Commun., 2014, 5, 1–6 Search PubMed.
- B. Lincoln, S. Schinkinger, K. Travis, F. Wottawah, S. Ebert, F. Sauer and J. Guck, Biomed. Microdevices, 2007, 9, 703–710 CrossRef PubMed.
- J. Guck, S. Schinkinger, B. Lincoln, F. Wottawah, S. Ebert, M. Romeyke, D. Lenz, H. M. Erickson, R. Ananthakrishnan, D. Mitchell, J. Käs, S. Ulvick and C. Bilby, Biophys. J., 2005, 88, 3689–3698 CrossRef CAS PubMed.
- F. Lautenschläger, S. Paschke, S. Schinkinger, A. Bruel, M. Beil and J. Guck, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15696–15701 CrossRef PubMed.
- H. K. Matthews, U. Delabre, J. L. Rohn, J. Guck, P. Kunda and B. Baum, Dev. Cell, 2012, 23, 371–383 CrossRef CAS PubMed.
- K. J. Chalut, M. Höpfler, F. Lautenschläger, L. Boyde, C. J. Chan, A. Ekpenyong, A. Martinez-Arias and J. Guck, Biophys. J., 2012, 103, 2060–2070 CrossRef CAS PubMed.
- S. M. Man, A. Ekpenyong, P. Tourlomousis, S. Achouri, E. Cammarota, K. Hughes, A. Rizzo, G. Ng, J. A. Wright and P. Cicuta, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 17588–17593 CrossRef CAS PubMed.
- A. E. Ekpenyong, N. Toepfner, C. Fiddler, M. Herbig, W. Li, G. Cojoc, C. Summers, J. Guck and E. R. Chilvers, Sci. Adv., 2017, 3, e1602536 CrossRef PubMed.
- T. R. Kießling, R. Stange, J. A. Käs and A. W. Fritsch, New J. Phys., 2013, 15, 045026 CrossRef.
- C. J. Chan, G. Whyte, L. Boyde, G. Salbreux and J. Guck, Interface Focus, 2014, 4, 20130069 CrossRef CAS PubMed.
- C. J. Chan, W. Li, G. Cojoc and J. Guck, Biophys. J., 2017, 112, 1063–1076 CrossRef CAS PubMed.
- C. Huster, D. Rekhade, A. Hausch, S. Ahmed, N. Hauck, J. Thiele, J. Guck, K. Kroy and G. Cojoc, New J. Phys., 2020, 22, 085003 CrossRef CAS.
- S. Ebert, K. Travis, B. Lincoln and J. Guck, Opt. Express, 2007, 15, 15493 CrossRef CAS PubMed.
- F. Wetzel, S. Rönicke, K. Müller, M. Gyger, D. Rose, M. Zink and J. Käs, Eur. Biophys. J., 2011, 40, 1109–1114 CrossRef PubMed.
- R. Schlüßler, S. Möllmert, S. Abuhattum, G. Cojoc, P. Müller, K. Kim, C. Möckel, C. Zimmermann, J. Czarske and J. Guck, Biophys. J., 2018, 115, 911–923 CrossRef PubMed.
- R. Schlüßler, K. Kim, M. Nötzel, A. Taubenberger, S. Abuhattum, T. Beck, P. Müller, S. Maharana, G. Cojoc, S. Girardo, A. Hermann, S. Alberti and J. Guck, bioRxiv, 2020, 2020.10.30.361808.
- L. Ling, W. D. Habicher, D. Kuckling and H.-J. Adler, Des. Monomers Polym., 1999, 2, 351–358 CrossRef CAS.
- K.-P. Kronfeld and H.-J. Timpe, J. Prakt. Chem., 1988, 330, 571–584 CrossRef CAS.
- T. Heida, T. Köhler, A. Kaufmann, M. J. Männel and J. Thiele, ChemSystemsChem, 2020, 2, e1900058 CAS.
- L. Frenz, K. Blank, E. Brouzes and A. D. Griffiths, Lab Chip, 2009, 9, 1344–1348 RSC.
- T. Heida, O. Otto, D. Biedenweg, N. Hauck and J. Thiele, Polymers, 2020, 12, 1760 CrossRef CAS PubMed.
- C. Scherzinger, A. Schwarz, A. Bardow, K. Leonhard and W. Richtering, Curr. Opin. Colloid Interface Sci., 2014, 19, 84–94 CrossRef CAS.
- S. Y. Oh, H. J. Kim and Y. C. Bae, Polymer, 2013, 54, 6776–6784 CrossRef CAS.
- M. Cors, L. Wiehemeier, J. Oberdisse and T. Hellweg, Polymers, 2019, 11, 620 CrossRef PubMed.
- Y. Li and T. Tanaka, Annu. Rev. Mater. Sci., 1992, 22, 243–277 CrossRef CAS.
- P. Müller, G. Cojoc and J. Guck, BMC Bioinf., 2020, 21, 1–8 CrossRef PubMed.
- P.-J. Wu, I. V. Kabakova, J. W. Ruberti, J. M. Sherwood, I. E. Dunlop, C. Paterson, P. Török and D. R. Overby, Nat. Methods, 2018, 15, 561–562 CrossRef CAS PubMed.
- M. Bailey, M. Alunni-Cardinali, N. Correa, S. Caponi, T. Holsgrove, H. Barr, N. Stone, C. P. Winlove, D. Fioretto and F. Palombo, 2019, arXiv:1912.08292.
- S. V. Adichtchev, Yu. A. Karpegina, K. A. Okotrub, M. A. Surovtseva, V. A. Zykova and N. V. Surovtsev, Phys. Rev. E, 2019, 99, 062410 CrossRef CAS PubMed.
- S. Cusack and S. Lees, Biopolymers, 1984, 23, 337–351 CrossRef CAS PubMed.
- L. Comez, L. Lupi, M. Paolantoni, F. Picchiò and D. Fioretto, J. Chem. Phys., 2012, 137, 114509 CrossRef CAS PubMed.
- R. Prevedel, A. Diz-Muñoz, G. Ruocco and G. Antonacci, Nat. Methods, 2019, 16, 969–977 CrossRef CAS PubMed.
- C. Holtze, A. C. Rowat, J. J. Agresti, J. B. Hutchison, F. E. Angilè, C. H. J. Schmitz, S. Köster, H. Duan, K. J. Humphry, R. A. Scanga, J. S. Johnson, D. Pisignano and D. A. Weitz, Lab Chip, 2008, 8, 1632–1639 RSC.
- M. Mittasch, P. Gross, M. Nestler, A. W. Fritsch, C. Iserman, M. Kar, M. Munder, A. Voigt, S. Alberti and S. W. Grill, Nat. Cell Biol., 2018, 20, 344–351 CrossRef CAS PubMed.
- P. Müller, M. Schürmann, S. Girardo, G. Cojoc and J. Guck, Opt. Express, 2018, 26, 10729–10743 CrossRef PubMed.
- S. Abuhattum, K. Kim, T. M. Franzmann, A. Eßlinger, D. Midtvedt, R. Schlüßler, S. Möllmert, H.-S. Kuan, S. Alberti and V. Zaburdaev, Front. Phys., 2018, 6, 131 CrossRef.
- G. Scarcelli and S. H. Yun, Opt. Express, 2011, 19, 10913–10922 CrossRef PubMed.
- A. M. Steiner, M. Mayer, D. Schletz, D. Wolf, P. Formanek, R. Hübner, M. Dulle, S. Förster, T. A. König and A. Fery, Chem. Mater., 2019, 31, 2822–2827 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00296e |
| This journal is © The Royal Society of Chemistry 2022 |