
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly active PdSb catalysts on porous carbon for electrochemical oxidation reactions of biomass-derived C1–C3 alcohols†
Daehee
Jang‡
,
Hyunsu
Han‡
,
Junbeom
Maeng
,
Wongeun
Yoon
,
Minseon
Park
and
Won Bae
Kim
 *
*
Department of Chemical Engineering, Pohang University of Science and Technology (POSTECH), 77 Cheongam-ro, Nam-gu, Pohang, Gyeongbuk, 37673, Republic of Korea. E-mail: kimwb@postech.ac.kr; Fax: +82-54-279-5528; Tel: +82-54-279-2397
First published on 9th June 2022
Abstract
The design of efficient and durable electrocatalysts is a challenge for the development of direct alcohol fuel cells (DAFCs). Herein, highly active and stable PdSb nanoparticle catalysts that are supported on three-dimensional (3D) porous carbon materials (PdxSby/PC) are prepared and investigated for electrooxidation reactions of various C1–C3 alcohols under alkaline conditions. From electrochemical tests, it is found that the optimized Pd0.90Sb0.10/PC catalysts exhibit the highest mass activities for various alcohols such as ethanol, ethylene glycol and glycerol, especially with 2.6 times higher activity than the commercial Pd/C catalysts in the ethanol oxidation reaction (EOR). Furthermore, it shows no significant decay in catalytic activity for over 6 hours of continuous operation. Detailed experimental activity studies and characterization analyses reveal that synergistic effects on the electrocatalytic performance are attributed to the electronic modification of the Pd catalyst by incorporating Sb and the structural feature of 3D porous carbon materials. These findings suggest a novel guidance to the catalyst fields for the direct alcohol-fed fuel cells which require great improvements in terms of catalytic performances and cost.
1. Introduction
Direct alcohol fuel cells (DAFCs) have drawn much attention due to their significant potential in green energy technology among several types of fuel cells. Alcohols, such as methanol, ethanol, ethylene glycol and glycerol, have been investigated as fuels due to their easy storage and transportation compared to pure dihydrogen.1,2 In particular, ethanol, ethylene glycol and glycerol are promising C2–C3 alcohol fuels because they can be largely produced from biomass and have a relatively low toxicity with a high energy density.3–6 Anion exchange membrane DAFCs (AEM-DAFCs) working in alkaline media have been attractive due to facilitated kinetics for the alcohol oxidation reaction (AOR) and the oxygen reduction reaction (ORR) compared to those performed in acidic media.7 Furthermore, the employment of low cost electrocatalysts instead of Pt-based ones is possible for the AOR and ORR in alkaline media. However, low poisoning resistances against AOR intermediates such as CO and difficulties involved in the C–C cleavage of alcohols remain to be alleviated for high performance catalysts. In this sense, highly active materials must be developed as anode catalysts along with a high CO tolerance for the practical application of DAFCs.8Pt-based catalysts have been studied widely for low temperature AORs due to their excellent catalytic activity for small organic molecules with low overpotential.9 Also, Pd-based catalysts have been investigated to compete with Pt-based catalysts in various reactions by improving the surface area, controlling the morphology, and alloying with other elements.10–14 In particular, Pd-based catalysts show excellent catalytic activity compared to other metals for the ethanol oxidation reaction (EOR) in alkaline media.15–17 The structure and composition of Pd-based metal catalysts have been systematically manipulated to further enhance the catalytic performance toward complete ethanol electrooxidation. It has been reported that an addition of a second metal to Pd catalysts has been known as an effective strategy to increase the rate of ethanol oxidation and CO tolerance induced by steric-, bifunctional-, or electronic effects.18–20 For instance, various bimetallic PdM (M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ni,21 Co,22 Cu,23etc.) catalysts have been studied and have shown enhanced catalytic performances in the EOR. However, bimetallic PdSb catalysts have been very rarely studied for electrooxidation of alcohols including ethanol, even though antimony (Sb) was reported to be effective as a second metal of Pd catalysts for direct formic acid fuel cells.24,25 In addition, choosing an appropriate support material is highly desirable to achieve high catalytic performance and good stability. For performance improvement, the introduction of 3D porous support materials with large surface area can promote rapid transports of electrons and AOR-related species and can also make active catalysts be highly dispersed with a strong interaction.26–29 Therefore, it is speculated that the synergistic effect over the 3D porous carbon supports and highly active bimetallic PdSb catalysts should lead to significant enhancement of catalytic performance toward AORs.
:Ni,21 Co,22 Cu,23etc.) catalysts have been studied and have shown enhanced catalytic performances in the EOR. However, bimetallic PdSb catalysts have been very rarely studied for electrooxidation of alcohols including ethanol, even though antimony (Sb) was reported to be effective as a second metal of Pd catalysts for direct formic acid fuel cells.24,25 In addition, choosing an appropriate support material is highly desirable to achieve high catalytic performance and good stability. For performance improvement, the introduction of 3D porous support materials with large surface area can promote rapid transports of electrons and AOR-related species and can also make active catalysts be highly dispersed with a strong interaction.26–29 Therefore, it is speculated that the synergistic effect over the 3D porous carbon supports and highly active bimetallic PdSb catalysts should lead to significant enhancement of catalytic performance toward AORs.
In this work, we developed a highly efficient electrocatalyst consisting of PdxSby alloy nanoparticles decorated on 3D porous carbon supports (PdxSby/PC) for the electrochemical AOR in an alkaline medium. The resulting PdxSby/PC catalysts showed better electrocatalytic properties for the electrooxidation reactions of methanol, ethanol, ethylene glycol and glycerol in terms of activity and stability compared to Pd/C. The results indicate that the AOR catalytic activity can be improved by modifying the support material structure and alloying a second metal to the Pd catalyst, and the Pd0.90Sb0.10/PC catalyst revealed remarkable EOR activity compared with the state-of-the-art catalysts. Herein, the structural advantages of porous carbon support materials and the effects of Sb addition into Pd metals were systemically demonstrated for the greatly improved electrocatalytic performance toward the AOR for a direct alcohol fuel cell.
2. Experimental section
2.1 Catalyst preparation
Sodium citrate tribasic dihydrate (C6H5Na3O7·2H2O, Sigma-Aldrich), tetraethylene glycol (C8H18O5, Sigma-Aldrich), palladium chloride (PdCl2, Sigma-Aldrich), antimony chloride (SbCl3, Sigma-Aldrich), Vulcan XC-72 (C, Cabot), sulfuric acid (H2SO4, Sigma-Aldrich), potassium hydroxide (KOH, Sigma-Aldrich), methanol (CH4O, Sigma-Aldrich), ethanol (C2H6O, Sigma-Aldrich), ethylene glycol (C2H6O2, Sigma-Aldrich) and glycerol (C3H8O3, Sigma-Aldrich) were purchased from the specified suppliers. Deionized water (DI, 18 MΩ cm, Millipore) was used for entire experimental steps.Porous carbon (PC) was prepared through direct carbonization of sodium citrate tribasic dihydrate.29,30 The carbonization takes place in a quartz tube under an inert Ar gas atmosphere for 1 h at 800 °C with a temperature increase rate of 3 °C min−1 from room temperature. The acid treatment with 0.5 M H2SO4 aqueous solution was carried out for the resultant black powder in order to eliminate remaining inorganic impurities such as sodium ions. The final product was washed with DI water several times, collected by centrifugation and dried in a freezing dryer.
Porous carbon supported 20 wt% PdxSby catalysts (PdxSby/PC) were synthesized using polyol synthesis. 100 mg of PC was ultrasonically dispersed in 30 mL of tetraethylene glycol and subsequently the pre-calculated amounts of PdCl2 and SbCl3 were added into the PC suspension. A potassium hydroxide solution (1 M solution in tetraethylene glycol) was added until the pH of the suspended solution reached approximately 10. The solution was heat treated at 180 °C for 1 h with stirring under reflux conditions followed by cooling to room temperature. The resulting black powder was washed multiple times with acetone and DI water to remove the residual tetraethylene glycol, and then dried in a freezing dryer. Finally, the dried PdxSby/PC catalysts were annealed at 120 °C for 1 h under a 4% H2/Ar atmosphere. The obtained catalysts are denoted as Pd/PC, Pd0.93Sb0.07/PC, Pd0.90Sb0.10/PC and Pd0.85Sb0.15/PC according to the weight ratios of Pd and Sb elements. In addition, Pd/AC catalysts were also prepared using the same process as the PdxSby/PC catalysts but by using activated carbon (AC) as the support material instead of the home-made PC.
2.2 Physicochemical characterization of catalysts
The morphology and metal particle size of the prepared catalysts were analyzed through scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images, using a JEOL-JSM7800F instrument operated at 15 kV and a JEOL-JEM2200FS instrument operated at 200 kV, respectively. Inductively coupled plasma atomic emission spectroscopy (ICP-AES, Optima 7300DV & Avio500) was performed to determine the actual compositions of the samples. The Brunauer–Emmett–Teller (BET) surface areas of samples were evaluated using the Quadrasorb evo using N2 adsorption/desorption isotherms at −196 °C. X-Ray diffraction (XRD) analysis data were obtained using a Bruker-Advanced D8 diffractometer using Cu Kα (λ = 1.5418 Å) radiation operated at 40 kV and 30 mA. X-ray photoelectron spectroscopy (XPS) was performed for the purpose of recording the XPS spectra of palladium using a NEXSA spectrometer. The XANES spectra of the Pd K-edge were recorded in the Pohang Accelerator Laboratory (PLS-II, 7D beamline), and the XAS data were analyzed using specific modules of the Demeter program.2.3 Electrochemical measurements
Electrochemical measurements were performed in a three-electrode batch cell connected to a potentiostat (ZIVE MP2A). A glassy carbon electrode of 3 mm diameter with a geometric area of 0.196 cm2 was used as the working electrode and the prepared catalyst loading on the working electrode was controlled to 0.1 mg cm−2. The Pt wire and saturated calomel electrode (SCE) were used as the counter electrode and the reference electrode, respectively. The electrochemical surface area (ECSA) was measured in 1 M KOH aqueous solution, and the ethanol oxidation reaction was performed in a 1 M KOH electrolyte containing 1 M ethanol at a scan rate of 50 mV s−1. Electrochemical impedance spectroscopy (EIS) was performed at –0.3 V (vs. SCE) in 1 M ethanol containing 1 M KOH aqueous solution in the frequency range from 105 Hz to 0.1 Hz. Electrochemical oxidation reactions of different alcohols and EIS measurements were carried out in the same procedure using methanol, ethylene glycol and glycerol. The reaction products after alcohol oxidation were investigated using high-performance liquid chromatography (HPLC, Waters e2695) using a UV-Vis detector. A Biorad Aminex HPX-87H column was used at 40 °C with 5 mM H2SO4 as the eluent at a flow rate of 0.5 mL min−1. For the CO stripping test, CO was pre-adsorbed on the catalyst by introducing the CO gas into 1 M KOH for 30 min. Then, the electrolyte is purged with a high purity N2 gas for 20 min and CO stripping voltammograms were recorded from −0.9 V to 0.3 V (vs. SCE) at a scan rate of 50 mV s−1.3. Results and discussion
Fig. 1a illustrates the preparation step and hypothetical structure of highly dispersed PdSb alloy nanoparticles on 3D porous carbon materials (PdxSby/PC). Briefly, 3D porous carbon (PC) was synthesized using sodium citrate via a simple pyrolysis process without involving various complicated steps or corrosive chemical agents.30 Over the PC, the polyol synthesis method using tetraethylene glycol (TEG) was employed for obtaining a series of PdxSby/PC catalysts with palladium and antimony precursors. Subsequently, the prepared catalysts were treated under a gas mixture of H2/Ar.31 The actual amounts of Pd and Sb on the PC were verified using inductively coupled plasma (ICP) spectrometry, as summarized in Table S1 (ESI†). The nominal metal loading was fixed at 20 wt% of total metals in the catalyst but the actual metal contents in the prepared samples were found to be around 15 wt%. The antimony contents in Pd0.93Sb0.07/PC, Pd0.90Sb0.10/PC and Pd0.85Sb0.15/PC were found to be approximately 8.0, 10.0 and 15.4 wt% of the total metal components, respectively.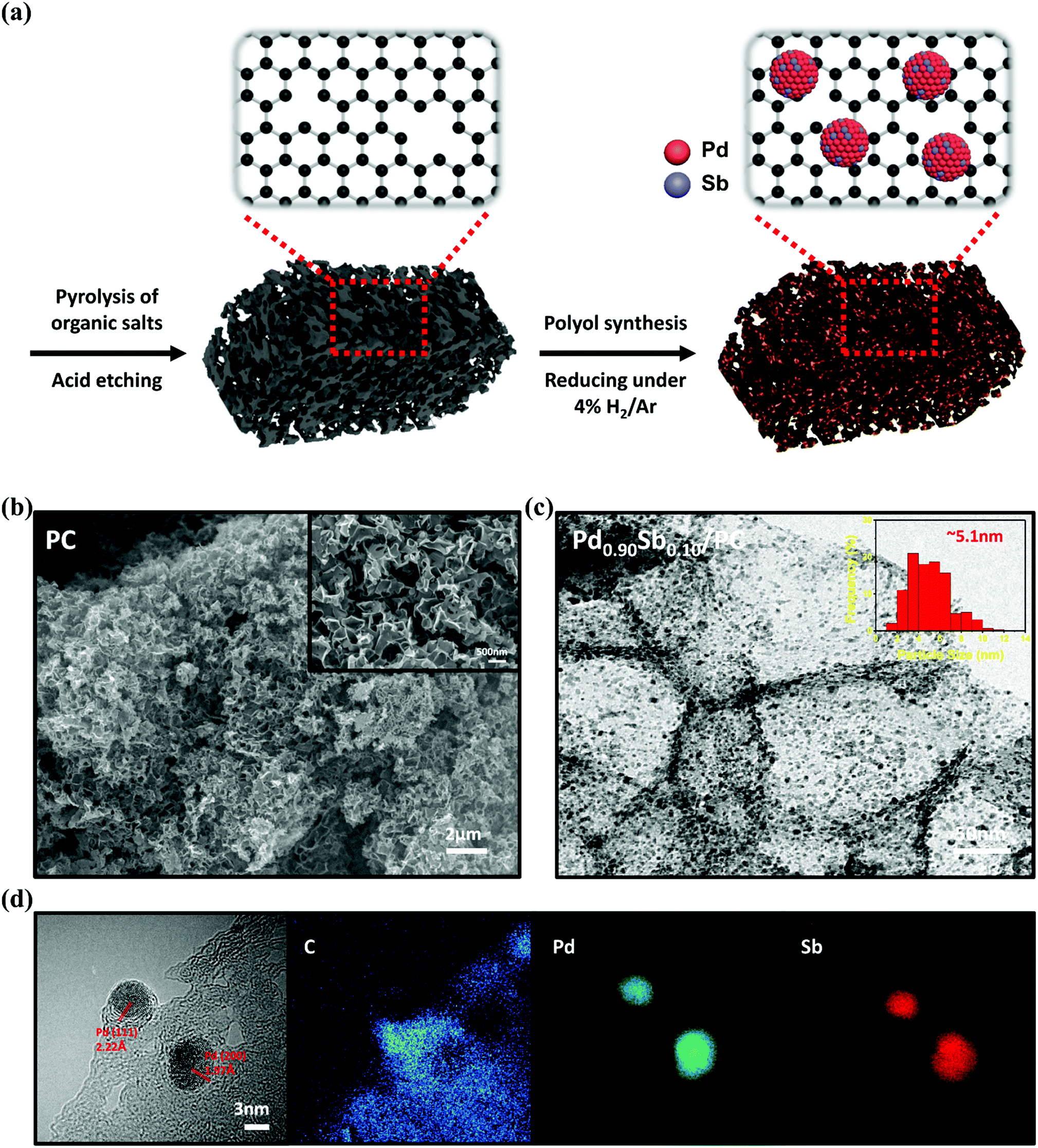 | ||
| Fig. 1 (a) Schematic illustration of the overall fabrication process of PdSb/PC catalysts. (b) SEM image of PC. (c) TEM image and (d) the EDS mapping of Pd0.90Sb0.10/PC catalysts. | ||
As shown in Fig. 1b, PC represented a well-organized 3D porous structure consisting of numerous macropores in 100–500 nm size with multidirectional channels. It was found that the morphology of PC was maintained even after the nanoparticles were loaded onto the PC (Fig. S1, ESI†). Textural properties were examined by N2 adsorption/desorption analysis, as demonstrated in Fig. S2 (ESI†) and listed in Table S2 (ESI†). The BET surface area and total pore volume of PC were 630 m2 g−1 and 1.21 cm3 g−1, respectively, which were much greater than those of AC (215 m2 g−1 and 0.63 cm3 g−1) and C (165 m2 g−1 and 0.52 cm3 g−1). From the SEM image and N2 sorption analysis, the specific structure with a high BET surface area and a large pore volume can be beneficial for the improved electrocatalysis as they can lead to highly dispersed active sites with facilitated mass transfer.32–34 The transmission electron microscopy (TEM) image with the PdSb metal alloy particle size distribution histogram of the Pd0.90Sb0.10/PC catalyst is presented in Fig. 1c. The TEM image indicated that the PdSb nanoparticles are well dispersed on the PC supports without significant agglomeration. The Pd/PC, Pd0.93Sb0.07/PC and Pd0.85Sb0.15/PC also showed the similar morphology, as revealed in Fig. S3 (ESI†). The size distribution of the PdSb particles of the Pd0.90Sb0.10/PC was estimated using randomly selected 300 particles seen in the TEM image and the average particle size was about 5.1 nm. This value is similar to that of commercial Pd/C and other PdxSby/PC catalysts, suggesting that a size effect of the nanoparticle should be negligible on the catalytic activity. Fig. 1d shows clear lattice fringes with ca. 2.22 and 1.97 Å, which are assigned to the (111) and (200) crystal planes of Pd, respectively (PDF No. 46-1043). In addition, according to energy dispersive spectroscopy (EDS) mapping, palladium and antimony appeared to be uniformly distributed in the PdSb nanoparticles.
The crystallographic data of Pd/PC and PdxSby/PC catalysts were obtained via X-ray diffraction (XRD) patterns shown in Fig. 2a. All samples display a broad diffraction peak at around 2θ = 23.4° which is ascribed to the (002) plane of graphitic carbons.35 The Pd/PC catalyst presents three diffraction peaks at approximately 40.0, 43.5 and 68.0, which correspond to the (111), (200) and (220) facets of the Pd face centered cubic (fcc) structure, respectively.36 The three main diffraction peaks of PdxSby/PC catalysts appeared to shift to lower values compared to those of Pd/PC catalysts, implying that the increase in the lattice constant was ascribed by the partial substitution of Pd atoms with Sb atoms which have a larger atomic size.37 Simultaneously, the full width at half maximum (FWHM) of the Pd peaks from the Pd/PC and PdxSby/PC catalysts was similar to each other, which is coincident with the particle size result measured from the TEM data. As the electronic properties of an alloy can be modified by charge transfer and lattice strain between different metal species, widening or narrowing of the d-band could take place along with the shifting of the d-band center (with regard to the Fermi level).38,39 In this sense, the electronic states of synthesized catalysts are determined by X-ray photoelectron spectroscopy (XPS) measurements (Fig. 2b). Compared to that of Pd/PC, the core-level binding energy (B.E.) of Pd0.90Sb0.10/PC moved to lower binding energies by 0.3 eV. This core-level B.E. shifting into lower energy indicated transfer of electrons in the Sb to the Pd element, leading to the modification of the electronic structures of Pd.
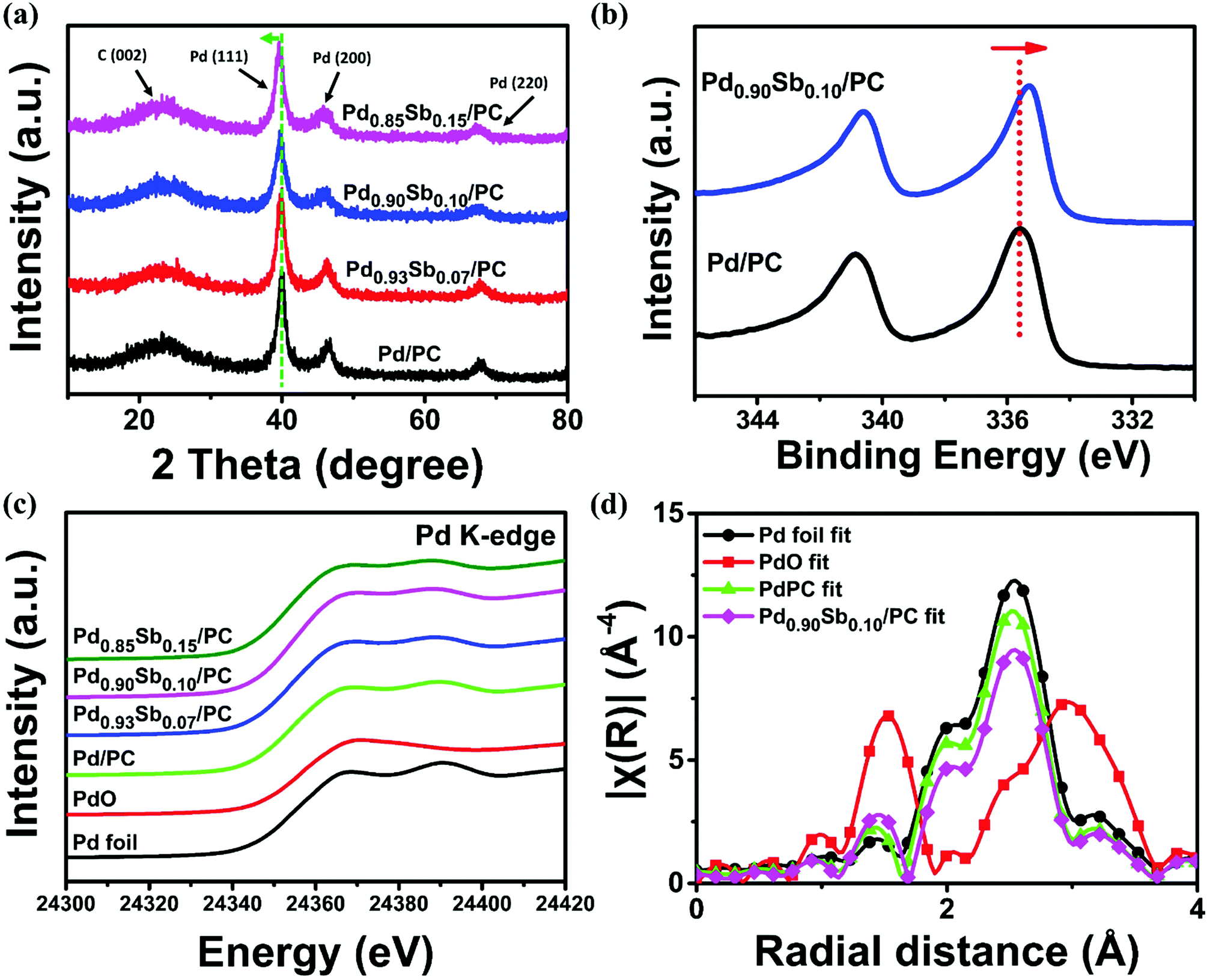 | ||
| Fig. 2 (a) XRD patterns of Pd/PC, Pd0.93Sb0.07/PC, Pd0.90Sb0.10/PC and Pd0.85Sb0.15/PC catalysts. (b) High resolution Pd 3d spectra of Pd/PC and Pd0.90Sb0.10/PC catalysts. (c) Pd K-edge XANES spectra of reference samples, Pd/PC and PdxSby/PC. (d) k3-Weighted Fourier transforms of Pd K-edge spectra. | ||
X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) were employed over the Pd K-edge spectra to additionally examine the local structural properties of the prepared catalysts. Fig. 2c shows the normalized XANES spectra of Pd foil, PdO, Pd/PC and PdxSby/PC catalysts. The XANES shapes of the PdxSby/PC catalysts are similar to that of Pd foil, implying that Pd nanoparticles primarily exist in a metallic state. Importantly, the local electronic structure changes of Pd can be investigated using the white line (WL) intensity.40 Compared to Pd/PC, an increased WL intensity of Pd0.90Sb0.10/PC was attributed to the 4d electron loss of Pd in the PdSb alloys (refer to Fig. S4 and Table S3, ESI†) due to oxidation and hybridization.31,41 Furthermore, the k3-weighted Fourier transformed EXAFS spectra of Pd0.90Sb0.10/PC and Pd/PC are reported in Fig. 2d, and the best fit results are shown in Table S4 (ESI†). As demonstrated by the EXAFS spectra of Pd foil reference and PdO reference, the dominant states of Pd in the Pd0.90Sb0.10/PC and Pd/PC catalysts were the metallic Pd phase with a partly oxidized one. The best-fitting analysis shows that Pd0.90Sb0.10/PC catalysts have a Pd–O coordination number of 1.04 which is a little bit bigger than that of Pd/PC (0.89). The Pd–Pd distance of Pd0.90Sb0.10/PC appears to increase as compared to that of Pd/PC, which indicates an expansion of the Pd lattice due to the incorporation of the Sb element, as shown in the XRD result. EXAFS analysis reveals that palladium species exists with an oxide layer which can lead to the AOR performance enhancement due to the lower adsorption energy of alcohols and intermediates such as acetaldehyde to the oxide layer than to the pure Pd metal surface.42 The expanded lattice (see the above XRD data) and the charge transfer effect induced by the introduction of Sb may tune the adsorption energy of reactive and intermediate species at the Pd site, leading to the improvement of electrocatalytic alcohol oxidation.43
The electrochemical active surface area (ECSA) of the prepared catalyst was evaluated by integrating the charge amount associated with the reducing monolayer PdO to Pd (405 μC cm−2) on the Pd surface in a 1.0 M KOH solution (Fig. 3a). The values of the ECSA for Pd/PC, Pd0.93Sb0.07/PC, Pd0.90Sb0.10/PC and Pd0.85Sb0.15/PC were 42.6, 54.2, 63.0 and 59.1 m2gPd−1, respectively. It could be found that the ECSA value was increased with the increasing Sb content and subsequently deceased with the excessive Sb addition, indicating that there is an optimized content for achieving a high ECSA. Pd0.95Sb0.05/PC and Pd0.80Sb0.20/PC catalysts were additionally examined and their ECSA values are summarized in Fig. S5 (ESI†). The amount of active sites appeared to decrease because the addition of the second metal should cover some parts of Pd surfaces.44 The higher ECSA value for the prepared catalysts implied the formation of a greater number of active sites where reactants can access at the catalyst surface, leading to a higher electrocatalytic activity for alcohol oxidation.45
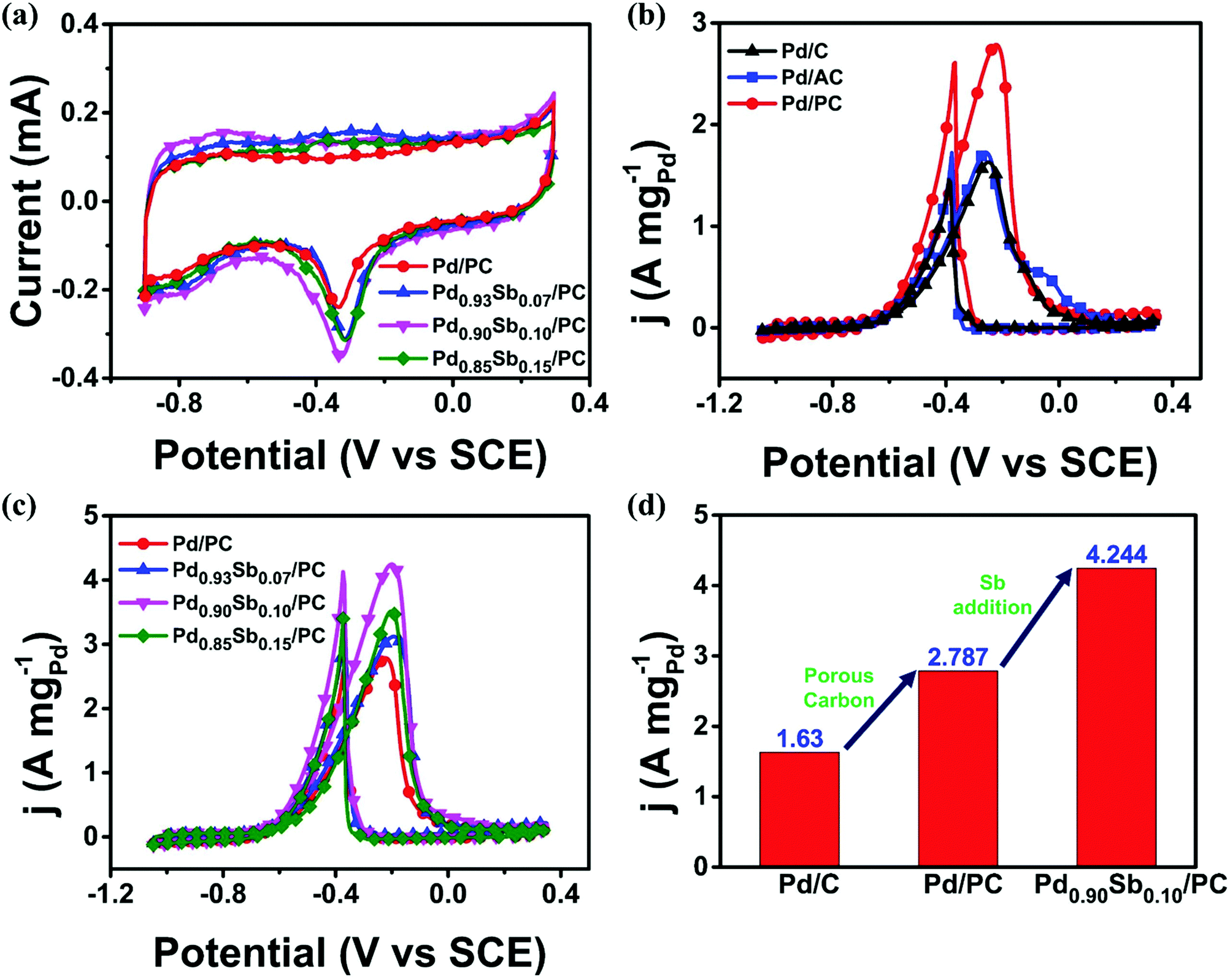 | ||
| Fig. 3 (a) Cyclic voltammetry of Pd/PC and PdxSby/PC in 1.0 M KOH solution. (b) Cyclic voltammetry of commercial Pd/C, Pd/AC and Pd/PC. (c) Cyclic voltammetry of Pd/PC and PdxSby/PC in 1.0 M KOH + 1.0 M EtOH solution. (d) Mass activities of Pd/C, Pd/PC and Pd0.90Sb0.10/PC. | ||
To investigate the electrocatalytic activity for alcohol oxidation over the prepared catalysts, cyclic voltammetry (CV) measurements were performed in the aqueous solutions of 1.0 M KOH containing 1.0 M alcohols at a scan rate of 50 mV s−1. The alcohol electrooxidation over Pd catalysts in alkaline media can take place according to the following steps:46
| Pd + RCH2O− ↔ Pd-RCH2O + e− | (1) |
| Pd-RCH2O + 2OH− ↔ Pd-RCO + 2H2O + 2e− | (2) |
| Pd + OH− ↔ Pd-OH + e− | (3) |
| Pd-RCO + Pd-OH ↔ Pd + Pd-RCOOH | (4) |
| Pd-RCOOH + OH− ↔ Pd + RCOO− + H2O | (5) |
Ethanol electrooxidation was first performed since Pd-based catalysts are known as highly effective electrocatalysts for the EOR and ethanol can be represented for biomass-derived alcohols. As shown in Fig. 3b and c, the CV curves for ethanol electrooxidation represent two well-defined peaks at around −0.25 V for the forward and at −0.4 V during the reverse scanning. The forward scan peak is ascribed to the oxidation of newly chemisorbed ethanol species according to eqn (1)–(4). The backward scan peak is generally related to the removal process of carbonaceous species that can be formed on the Pd catalyst surface but incompletely oxidized during the forward scan.47
Fig. 3b and c show the effects of the introduction of support materials and second metal on the electrocatalytic activities. The mass activities of Pd catalysts with different support materials showed the following order: Pd/PC (2.787 A mgPd−1) > Pd/AC (1.728 A mgPd−1) > commercial Pd/C (1.630 A mgPd−1) (Fig. 3b). This result could be attributed to different morphologies and textural properties of support materials. In terms of catalytic activity, types of support materials are considered to be one of the most important factors. As revealed by SEM images and N2 adsorption/desorption properties, the PC support material represents the 3D porous network with a higher surface area and total pore volume which could promote transportations of reactants and electron charges and lead to well dispersion of metal nanoparticles, improving the catalytic activity for ethanol electrooxidation. As illustrated in Fig. 3c, all the PdxSby/PC catalysts revealed better catalytic performances than Pd/PC, and their catalytic performance was maximized with the Pd0.90Sb0.10/PC catalyst, which is one of the best catalysts for the ethanol electrooxidation as summarized and compared with the formerly reported state-of-the-art catalysts (Table S6, ESI†). Sb addition to the Pd can modify the electronic structure and lattice constant of Pd, as previously revealed by XRD, XPS and XANES analyses. These electronic and steric effects could lead to the alteration of the adsorption energies of carbonaceous species associated with the oxidation process of ethanol molecules at the catalytic Pd sites, which could make the catalyst more appropriate to oxidize the ethanol and intermediate species with enhanced catalytic activity. The catalytic activity for the EOR was improved by the both effects of porous support materials and metal alloy catalysts as shown in Fig. 3d.
The Pd/C, Pd/PC and Pd0.90Sb0.10/PC catalysts were further applied in the electrooxidation reactions of methanol, ethylene glycol and glycerol (MOR, EGOR, and GOR) to demonstrate and compare the catalytic performances for the oxidation reaction of various alcohols (Fig. 4). As summarized in Table S7, (ESI†), Pd/PC and Pd0.90Sb0.10/PC catalysts appear to enhance the catalytic activities for the oxidation of all the studied alcohols. The Pd0.90Sb0.10/PC catalyst not only showed the greatest mass activity in the EOR under the alkaline conditions, but also performed 2.22, 2.54, and 2.32 times higher mass activity than the commercial Pd/C catalyst for the MOR, EGOR and GOR, respectively. The Pd0.90Sb0.10/PC catalyst also presented the highest specific activity even though the ECSA of Pd0.90Sb0.10/PC was much bigger than those of Pd/C and Pd/PC (Fig. S6, ESI†). Furthermore, the Pd0.90Sb0.10/PC catalyst exhibited negative shifts in the onset potential for each alcohol oxidation, which indicates substantial kinetic improvements by the catalyst studied in this work.
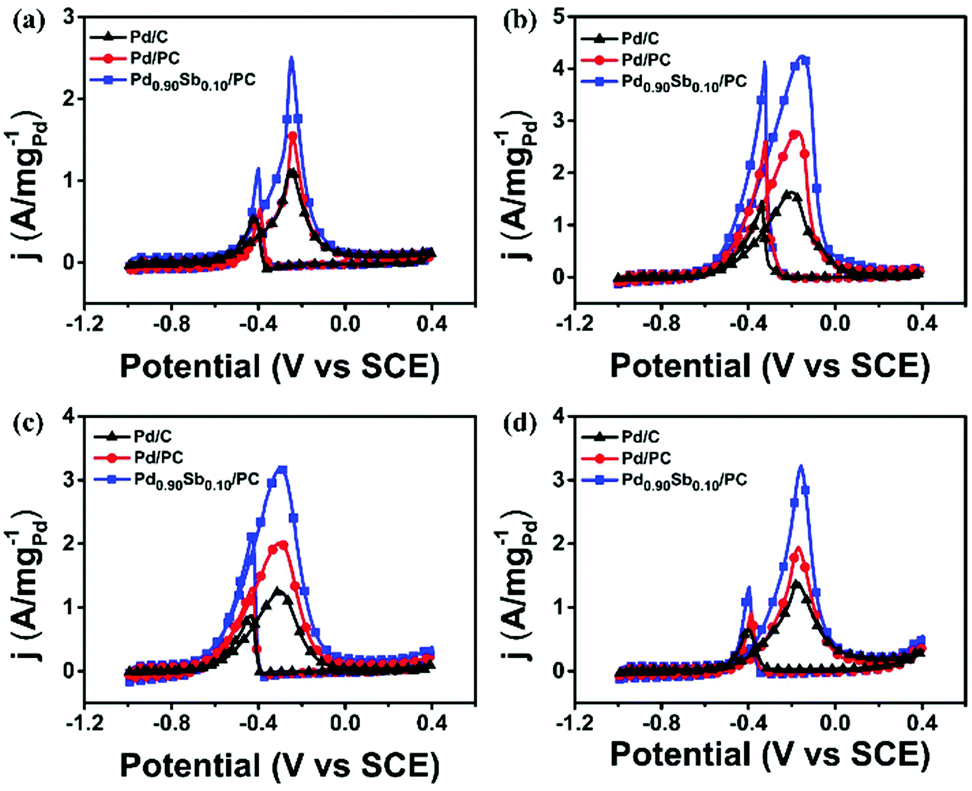 | ||
| Fig. 4 Cyclic voltammetry of commercial Pd/C, Pd/PC and Pd0.90Sb0.10/PC in 1.0 M KOH solution, containing (a) methanol, (b) ethanol, (c) ethylene glycol and (d) glycerol, respectively. | ||
According to eqn (1)–(5), various products can be formed from alcohol oxidation processes when they are not completely oxidized to CO2. To investigate oxidation products after alcohol oxidation, the solutions after the MOR, EOR, EGOR and GOR for 2 h using the Pd0.90Sb0.10/PC catalyst were analyzed by HPLC. Only formic acid and acetic acid, which are simple carboxylic acids, were produced after the MOR and EOR, but various organic acids were detected after the EGOR and GOR. C2 products, such as acetic acid, glycolic acid and oxalic acid, were dominantly produced during the EOR and EGOR. Likewise, C3 chemicals, such as lactic acid, pyruvic acid and glyceric acid, were major products from the GOR.
Electrochemical impedance spectroscopy (EIS) is an efficient method for investigating the dynamics of alcohol electrooxidation and is generally employed to study the kinetics in terms of charge transfer resistance throughout the electrocatalytic process. The electrochemical impedance parameters were evaluated using the equivalent circuit model shown in the inset of Fig. 5a, where Rs, Rct and CPE represent the electrolyte solution resistance, the charge transfer resistance and the constant phase element referring to the double layer capacitance, respectively.48 According to the fitted data in Table S8, (ESI†) the value of Rct is proportional to the semicircle radius in the Nyquist plots. As shown in Fig. 5a, the Nyquist plots indicated that the Pd0.90Sb0.10/PC catalysts showed the smallest semicircle radius among all the prepared catalysts, suggesting promoted charge transfer between electrolyte and catalysts. This improved charge transfer should be related to a reduced activation barrier for the electrooxidation of ethanol,49 resulting in the enhanced overall catalytic performance. The Pd0.90Sb0.10/PC catalyst also showed decreased semicircle radii in the Nyquist plots that were obtained in methanol, ethylene glycol and glycerol solutions as shown in Fig. S7 (ESI†). Furthermore, the Tafel plots are obtained by linear sweep voltammetry of the MOR, EOR, EGOR and GOR for the Pd/C, Pd/PC and Pd0.90Sb0.10/PC catalysts (Fig. S8, ESI†). The lower Tafel slope signifies the facilitated reaction kinetics of electrocatalysts. According to the plots, the Tafel slope of Pd0.90Sb0.10/PC is smaller than those of Pd/PC and Pd/C, indicating facilitated kinetics for the alcohols oxidation reaction by the incorporation of Sb.50 This result is in good agreement with the EIS analysis for the charge transfer resistance of the catalysts. Both EIS data and Tafel slopes suggest that the Pd0.90Sb0.10/PC catalyst shows better electron transfer kinetics for the MOR, EOR, EGOR and GOR than Pd/PC and Pd/C.
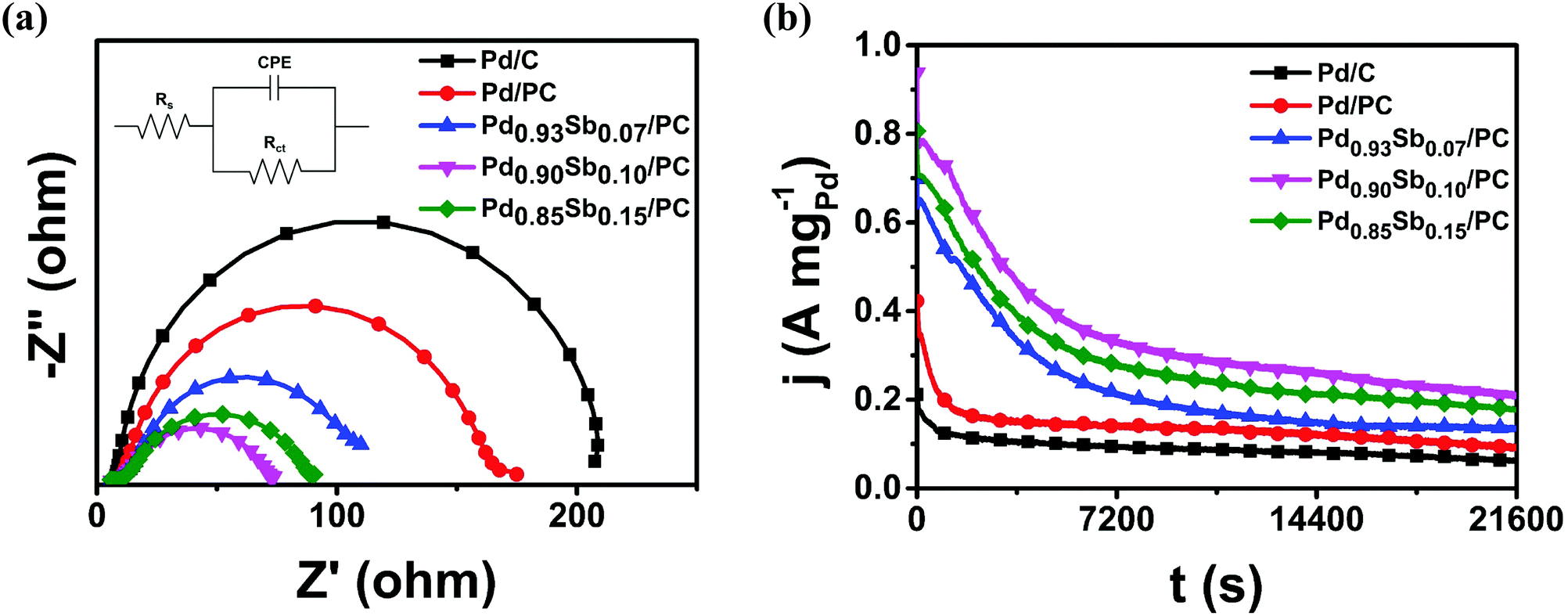 | ||
| Fig. 5 (a) Nyquist plots at −0.30 V (vs. SCE); inset shows the equivalent circuit model and (b) chronoamperometric measurements at −0.35 V (vs. SCE) in 1.0 M KOH + 1.0 M C2H5OH solution. | ||
For the design of the electrocatalyst, the stability issue is another crucial factor. Therefore, chronoamperometric measurements were performed for 6 hours in 1.0 M KOH containing 1.0 M alcohols to examine the stability of the catalysts in the AOR. As shown in Fig. 5b and Fig. S9 (ESI†), initial rapid decreases in the mass activity plots appeared for all the catalysts, which could be associated with the blocking active sites resulting from the strong adsorption of intermediates (e.g., CO). Afterwards, the activity decreased gradually and arrived at pseudo-steady states. Notably, a series of PdxSby/PC catalysts maintained quite higher activities in comparison to the commercial Pd/C and Pd/PC catalysts. In particular, the Pd0.90Sb0.10/PC catalysts showed 3.4 times higher mass activity than the commercial Pd/C catalysts after 6 h reaction, suggesting that the introduction of Sb metal could provide better tolerance of the intermediate species to the active Pd sites toward ethanol electrooxidation.
CO stripping voltammetry (Fig. 6) was performed to investigate the CO tolerance of catalysts, since the CO species are main poisoning intermediates during the electrooxidation of most alcohols. In comparison with the commercial Pd/C and Pd/PC catalysts, the CO stripping peak potential values of PdxSby/PC catalysts are negatively shifted by approximately 40 mV, indicating that the PdSb alloy catalysts can oxidize CO more easily at lower potentials. This enhancement of the oxidative removal of CO could be ascribed to the bifunctional effects of Sb addition.51 The presence of Sb promotes the formation of adsorbed oxygenated species, such as OHad, which can play a beneficial role in the catalytic activity since they participate in the oxidation reaction of ethanol for the removal of COad through equation (6), recovering the active sites of Pd.43,52,53
| Pd-OH + Pd-CO + OH− → 2Pd + CO2 + H2O + e− | (6) |
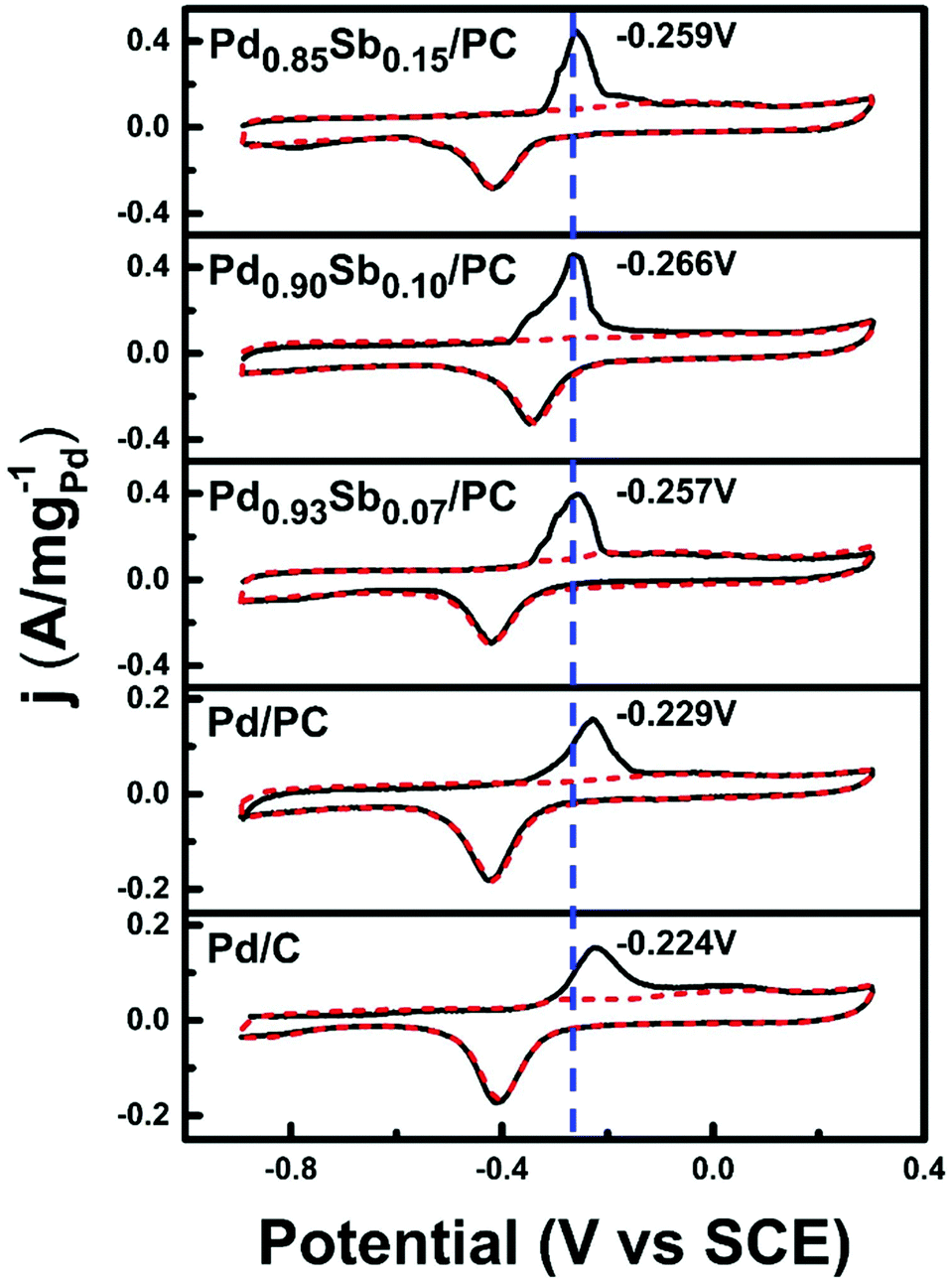 | ||
| Fig. 6 Cyclic voltammetry for CO stripping on PdxSby/PC, Pd/PC and commercial Pd/C in 1 M KOH solution. The black solid line shows the first scanning cycle, and the red dotted line shows the second scanning cycle. | ||
Conclusions
In summary, we have successfully synthesized 3D porous carbon network supported PdSb alloy nanoparticles as highly efficient electrocatalysts for electrochemical oxidation reactions of various alcohols. Through modifications of the electronic structure of Pd and the introduction of porous carbon supports, Pd0.90Sb0.10/PC achieved a maximum peak current density of 4.24 A mgPd−1 at a potential of −250 mV (vs. SCE), which is one of the best performing electrocatalysts for ethanol electrooxidation as reported in the literature. Moreover, this catalyst also demonstrated at least two times higher mass activities than the commercial Pd/C for the electrooxidation reactions of methanol, ethylene glycol, and glycerol. The results suggested that the activity improvements in alcohol electrooxidation could be attributed to the morphological benefit of 3D porous carbon supports and the modification of the electronic properties of Pd induced by Sb addition. PdxSby/PC catalysts have emerged as a suitable and promising anode material for direct alcohol fuel cells, wherein there is a great need for improving the catalytic performance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. Jang and H. Han contributed equally to this work. This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (NRF-2019R1A2C2088174) and by the “Human Resources Program in Energy Technology” of the Korea Institute of Energy Technology Evaluation and Planning (KETEP), granted financial resource from the Ministry of Trade, Industry & Energy, Republic of Korea (No. 20204010600100), and also by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (No. 2021R1A5A1084921).References
- K. Matsuoka, Y. Iriyama, T. Abe, M. Matsuoka and Z. Ogumi, J. Power Sources, 2005, 150, 27–31 CrossRef CAS.
- H. An, L. Pan, H. Cui, B. Li, D. Zhou, J. Zhai and Q. Li, Electrochim. Acta, 2013, 102, 79–87 CrossRef CAS.
- E. Antolini, J. Power Sources, 2007, 170, 1–12 CrossRef CAS.
- B. Katryniok, H. Kimura, E. Skrzynska, J. S. Giradon, P. Fongarland, M. Capron, R. Ducoulombier, N. Mimura, S. Paul and F. Dumeignil, Green Chem., 2011, 13, 1960–1979 RSC.
- N. Ji, T. Zhang, M. Zheng, A. Wang, H. Wang, X. Wang and J. G. Chen, Angew. Chem., Int. Ed., 2008, 47, 8510–8513 CrossRef CAS PubMed.
- H. Wan, C. Dai, L. Jin, S. Luo, F. Meng, G. Chen, Y. Duan, C. Liu, Q. Xu, J. Lu and Z. J. Xu, ACS Appl. Mater. Interfaces, 2022, 14, 14293–14301 CrossRef CAS PubMed.
- T. Jurzinsky, C. Cremers, F. Jung, K. Pinkwart and J. Tübke, Int. J. Hydrogen Energy, 2015, 40, 11569–11576 CrossRef CAS.
- F. Vigier, C. Coutanceau, A. Perrard, E. M. Belgsir and C. Lamy, J. Appl. Electrochem., 2004, 34, 439–446 CrossRef CAS.
- L. An, T. S. Zhao and Y. S. Li, Renewable Sustainable Energy Rev., 2015, 50, 1462–1468 CrossRef CAS.
- W. Li, Y. Huang, D. Tang, T. Zhang and Y. Wang, Electrochim. Acta, 2015, 174, 178–184 CrossRef CAS.
- O. O. Fashedemi, H. A. Miller, A. Marchionni, F. Vizza and K. I. Ozoemena, J. Mater. Chem. A, 2015, 13, 7145–7156 RSC.
- Y. Zhang, Q. Huang, G. Chang, Z. Zhang, T. Xia, H. Shu and Y. He, J. Power Sources, 2015, 280, 422–429 CrossRef CAS.
- X. Chang, A. F. Liu, B. Cai, J. Y. Luo, H. Pan and Y. B. Huang, ChemSusChem, 2016, 9, 3330–3337 CrossRef CAS PubMed.
- Y. Mun, S. Lee, A. Cho, S. Kim, J. W. Han and J. Lee, Appl. Catal., B, 2019, 246, 82–88 CrossRef CAS.
- Y. Y. Yang, J. Ren, Q. X. Li, Z. Y. Zhou, S. G. Sun and W. B. Cai, ACS Catal., 2014, 4, 798–803 CrossRef CAS.
- H. Han, Y. Noh, Y. Kim, V. S. K. Yadav, S. Park, W. Yoon, S. Lee and W. B. Kim, ChemistrySelect, 2017, 2, 6260–6268 CrossRef CAS.
- K. Kakaei and M. Dorraji, Electrochim. Acta, 2014, 143, 207–215 CrossRef CAS.
- H. Zhang, Y. Shang, J. Zhao and J. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 16635–16643 CrossRef CAS PubMed.
- Y. Wang, Z. M. Sheng, H. Yang, S. P. Jiang and C. M. Li, Int. J. Hydrogen Energy, 2010, 35, 10087–10093 CrossRef CAS.
- L. Jin, H. Xu, C. Chen, H. Shang, Y. Wang, C. Wang and Y. Du, ACS Appl. Mater. Interfaces, 2019, 11, 42123–42130 CrossRef CAS PubMed.
- S. Y. Shen, T. S. Zhao, J. B. Xu and Y. S. Li, J. Power Sources, 2010, 195, 1001–1006 CrossRef CAS.
- Z. Zhang, S. Liu, X. Tian, J. Wang, P. Xu, F. Xiao and S. Wang, J. Mater. Chem. A, 2017, 5, 10876–10884 RSC.
- A. Shafaei Douk, H. Saravani and M. Noroozifar, Int. J. Hydrogen Energy, 2017, 42, 15149–15159 CrossRef CAS.
- J. L. Haan, K. M. Stafford, R. D. Morgan and R. I. Masel, Electrochim. Acta, 2010, 55, 2477–2481 CrossRef CAS.
- X. Yu and P. G. Pickup, Electrochem. Commun., 2010, 12, 800–803 CrossRef CAS.
- H. Tan, J. Tang, J. Henzie, Y. Li, X. Xu, T. Chen, Z. Wang, J. Wang, Y. Ide, Y. Bando and Y. Yamauchi, ACS Nano, 2018, 12, 5674–5683 CrossRef CAS PubMed.
- L. Nan, W. Yue and Y. Jiang, J. Mater. Chem. A, 2015, 3, 22170–22175 RSC.
- L. Nan, Z. Fan, W. Yue, Q. Dong, L. Zhu, L. Yang and L. Fan, J. Mater. Chem. A, 2016, 4, 8898–8904 RSC.
- H. Han, Y. Noh, Y. Kim, S. Park, W. Yoon, D. Jang and W. B. Kim, Green Chem., 2020, 22, 71–84 RSC.
- H. Han, Y. Noh, Y. Kim, W. S. Jung, S. Park and W. B. Kim, Nanoscale, 2019, 11, 2423–2433 RSC.
- S. Lee, H. J. Kim, E. J. Lim, Y. Kim, Y. Noh, G. W. Huber and W. B. Kim, Green Chem., 2016, 18, 2877–2887 RSC.
- H. Han, S. Park, D. Jang, S. Lee and W. B. Kim, ChemSusChem, 2020, 13, 539–547 CrossRef CAS PubMed.
- T. Yoskamtorn, S. Yamazoe, R. Takahata, J.-I. Nishigaki, A. Thivasasith, J. Limtrakul and T. Tsukuda, ACS Catal., 2014, 4, 3696–3700 CrossRef CAS.
- H. Han, S. Park, D. Jang and W. B. Kim, J. Alloys Compd., 2021, 853, 157338 CrossRef CAS.
- H. Han, S. Jin, S. Park, M. H. Seo and W. B. Kim, Appl. Catal., B, 2021, 292, 120173 CrossRef CAS.
- Z. Liu, X. Zhang and L. Hong, Electrochem. Commun., 2009, 11, 925–928 CrossRef CAS.
- C. Hahn, D. N. Abram, H. A. Hansen, T. Hatsukade, A. Jackson, N. C. Johnson, T. R. Hellstern, K. P. Kuhl, E. R. Cave, J. T. Feaster and T. F. Jaramillo, J. Mater. Chem. A, 2015, 3, 20185–20194 RSC.
- W. Zhang, L. Wang, H. Liu, Y. Hao, H. Li, M. U. Khan and J. Zeng, Nano Lett., 2017, 17, 788–793 CrossRef CAS PubMed.
- Y. Wang, F.-F. Shi, Y.-Y. Yang and W.-B. Cai, J. Power Sources, 2013, 243, 369–373 CrossRef CAS.
- L. E. Betancourt, A. Rojas-Pérez, I. Orozco, A. I. Frenkel, Y. Li, K. Sasaki, S. D. Senanayake and C. R. Cabrera, ACS Appl. Energy Mater., 2020, 3, 2342–2349 CrossRef CAS.
- Y. Kim, H. W. Kim, S. Lee, J. Han, D. Lee, J.-R. Kim, T.-W. Kim, C.-U. Kim, S.-Y. Jeong, H.-J. Chae, B.-S. Kim, H. Chang, W. B. Kim, S. M. Choi and H. J. Kim, ChemCatChem, 2017, 9, 1683 CrossRef CAS.
- A. Krittayavathananon, S. Duangdangchote, P. Pannopard, N. Chanlek, S. Sathyamoorthi, J. Limtrakul and M. Sawangphruk, Sustainable Energy Fuels, 2020, 4, 1118–1125 RSC.
- J. L. Haan, K. M. Stafford and R. I. Masel, J. Phys. Chem. C, 2010, 114, 11665–11672 CrossRef CAS.
- Y. Huang, J. Cai and Y. Guo, Appl. Catal., B, 2013, 129, 549–555 CrossRef CAS.
- M. Zareie Yazdan-Abad, M. Noroozifar, A. R. Modaresi Alam and H. Saravani, J. Mater. Chem. A, 2017, 5, 10244–10249 RSC.
- Z. X. Liang, T. S. Zhao, J. B. Xu and L. D. Zhu, Electrochim. Acta, 2009, 54, 2203–2208 CrossRef CAS.
- M. S. Ahmed and S. Jeon, ACS Catal., 2014, 4, 1830–1837 CrossRef CAS.
- S. R. Chowdhury, T. Maiyalagan, S. K. Bhattachraya and A. Gayen, Electrochim. Acta, 2020, 342, 136028 CrossRef.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. D. Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T.-K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed.
- M. Yang, M. Pang, J. Chen, F. Gao, H. Li and P. Guo, ACS Appl. Mater. Interfaces, 2021, 13, 9830–9837 CrossRef CAS PubMed.
- S. Kwon, D. Ham, T. Kim, Y. Kwon, S. Lee and M. Cho, ACS Appl. Mater. Interfaces, 2018, 10, 39581–39589 CrossRef CAS PubMed.
- B. Łosiewicz, L. Birry and A. Lasia, J. Electroanal. Chem., 2007, 611, 26–34 CrossRef.
- T. P. Scachetti and A. C. D. Angelo, Electrocatalysis, 2015, 6, 472–480 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00277a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |