
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
2D material based heterostructures for solar light driven photocatalytic H2 production
Yingying
Wang
 ,
Zhu
Ding
,
Nayab
Arif
,
Wei-Chao
Jiang
* and
Yu-Jia
Zeng
*
,
Zhu
Ding
,
Nayab
Arif
,
Wei-Chao
Jiang
* and
Yu-Jia
Zeng
*
Key Laboratory of Optoelectronic Devices and Systems of Ministry of Education and Guangdong Province, College of Physics and Optoelectronic Engineering, Shenzhen University, Shenzhen 518052, Guangdong, China. E-mail: jiang.wei.chao@szu.edu.cn; yjzeng@szu.edu.cn
First published on 22nd March 2022
Abstract
Photocatalytic H2 production using solar energy has long been considered as a promising solution for renewable energy production to solve energy crisis and environmental issues. 2D materials with unique layered structures and electronic properties have led to considerable breakthroughs in energy fields. By constructing heterostructures to facilitate solar light absorption and charge transfer and separation, the photocatalytic activity of 2D materials can be highly boosted. In this review, we summarize recent progress in 2D material based heterostructures for solar light driven photocatalytic H2 production, including black phosphorus (BP), g-C3N4 (CN), transition metal dichalcogenides (TMDs), layered double hydroxides (LDHs), MXenes, and other emerging 2D materials. We provide a fundamental understanding of the functions of various heterojunctions due to their enhanced photocatalytic activity. The band alignments of different heterojunctions with enhanced visible light absorption and improved charge transfer in terms of water splitting reaction are specially discussed. Finally, the existing challenges and prospects of the practical application of 2D material-based heterostructures in photocatalytic H2 production are presented.
1 Introduction
Since solar energy is limitless, free and clean, it has become the key among renewable energy resources (biomass, wind, geothermal, marine, etc.) to alleviate the energy crisis and environmental issues.1,2 Meanwhile, as an abundant element on earth, hydrogen is also the most promising energy carrier, which releases only water after usage and results in truly zero emission. Owing to its cleanliness, plentiful quantity, high-energy efficiency, storability, and easy-transportation, hydrogen plays a crucial role in driving modern energy systems for the sustainable development of humans. Thus, the conversion of solar energy into usable and storable hydrogen in both an economically and environmentally friendly way is the most desirable solution to the energy crisis issue.3,4 The decomposition of water into hydrogen and oxygen using titania photoanodes was first reported by Honda and Fujishima in 1972;5 since then, photocatalytic water splitting using solar energy in the presence of semiconductor photocatalysts has been studied as the potential route for H2 production.6,7 Afterwards, a number of materials (e.g., ZnO, Fe2O3, WO3, BiVO4, CdSe) were developed successively with acceptable photocatalytic performance.8–10 However, further practical applications of traditional semiconductor photocatalysts have been restricted by their low solar-to-hydrogen (STH) conversion efficiency (approximately 1%), which is due to either their large band gap or the rapid recombination of photogenerated electron–hole pairs.11During the past decade, various strategies have been implemented to achieve a high efficiency of photocatalytic water splitting, including optimizing the available photocatalysts and exploring new types of photocatalysts and heterostructures.12,13 Two-dimensional (2D) materials with van der Waals (vdW) stacking of layers of atoms have been well recognized as rising stars following the successful isolation of graphene in 2004.14 2D materials include 2D allotropes of various elements (e.g., graphene, borophene, silicene, phosphorene and germanene) or compounds (e.g., h-BN, g-C3N4 (CN), transition metal dichalcogenides (TMDs), MXenes, layered double hydroxides (LDHs)), whose family is still growing.15–18 These 2D materials with unique structural and electronic properties result in extensive breakthroughs in the catalysis and energy conversion fields.19–21 It has been found that 2D semiconductors which have large specific surface areas with abundant surface-active sites could be promising photocatalysts providing superior electron mobility and short diffusion distance for efficient charge separation.22–24 Moreover, the planar structure of 2D materials with weak vdW force and dangling-bond-free surfaces allows the facile formation of heterostructures, which are integrated with not only 2D materials but also materials of different dimensionalities. The construction of heterostructures offers unique properties and promotes photocatalytic activity resulting from the synergies of their individual components.25–27 Various heterostructures of 2D materials have been synthesized successfully with optimized kinetics and energetics for photocatalytic water splitting.26,28,29 Previous reviews have been focused on individual 2D materials, such as BP based heterostructures,30–32 CN based heterostructures,33,34 and TMD based heterostructures,35–38 or focused on photocatalysis for environmental remediation,38,39 or on DFT calculation results.40 As the family of 2D materials is extending rapidly, an updated review of all-inclusive 2D material based heterostructures for photocatalytic H2 production is needed, in particular for their fundamental mechanisms.
Herein, we summarize recent advances of well functioned heterostructures based on various 2D materials, including BP, CN, TMDs, MXenes, and LDHs, and their applications in photocatalytic H2 production. Various 2D material-based heterojunctions are systematically discussed, such as type-I, type-II, Z-scheme, S-scheme and Schottky junctions. The band alignments in heterojunctions successfully enhance visible light absorption and suppress the recombination of photogenerated electron–hole pairs. Construction of vdW heterostructures can provide abundant surface-active sites for photocatalytic reactions. Moreover, the intimate interfacial contact between two components can also improve the charge transfer and separation of photogenerated electron–hole pairs. Owing to the synergy of components in the heterostructure, the efficiency of photocatalytic H2 production is highly improved. Many innovative studies on efficient visible light driven photocatalysts achieving remarkable H2 production results are particularly highlighted. This review provides comprehensive and critical information for developing vdW based heterostructures with excellent photocatalytic properties. Finally, our prospects on the explorations of vdW heterostructures in the field of renewable energy production are also presented.
2 Introduction to van der Waals (vdW) heterostructures
The family of 2D materials consisting of different elements is expanding dramatically, which is highlighted in Fig. 1. In addition to graphene, a series of layered materials were continuously exfoliated into their 2D forms, including graphene-like hexagonal boron nitride (h-BN),41,42 graphitic carbon nitride (CN),43–45 transition metal dichalcogenides (TMDs),46,47 group-III monochalcogenides,48–50 and layered double hydroxides (LDHs).51–54 Emerging members such as black phosphorus (BP),55–57 MXenes,24,58,59 silicene,60,61 metal–organic frameworks (MOFs),62–64 and polymers65–67 were further flourished in the energy-related areas. When 2D materials are applied in photocatalytic/photoelectrochemical water splitting, the photons are absorbed by photocatalysts and electron–hole pairs are generated for the subsequent redox reactions. The severe recombination of electron–hole pairs largely suppresses the solar-to-hydrogen conversion efficiency.68 By constructing heterostructures, the charge separation and transfer are highly improved and the recombination of electron–hole pairs are effectively prevented.69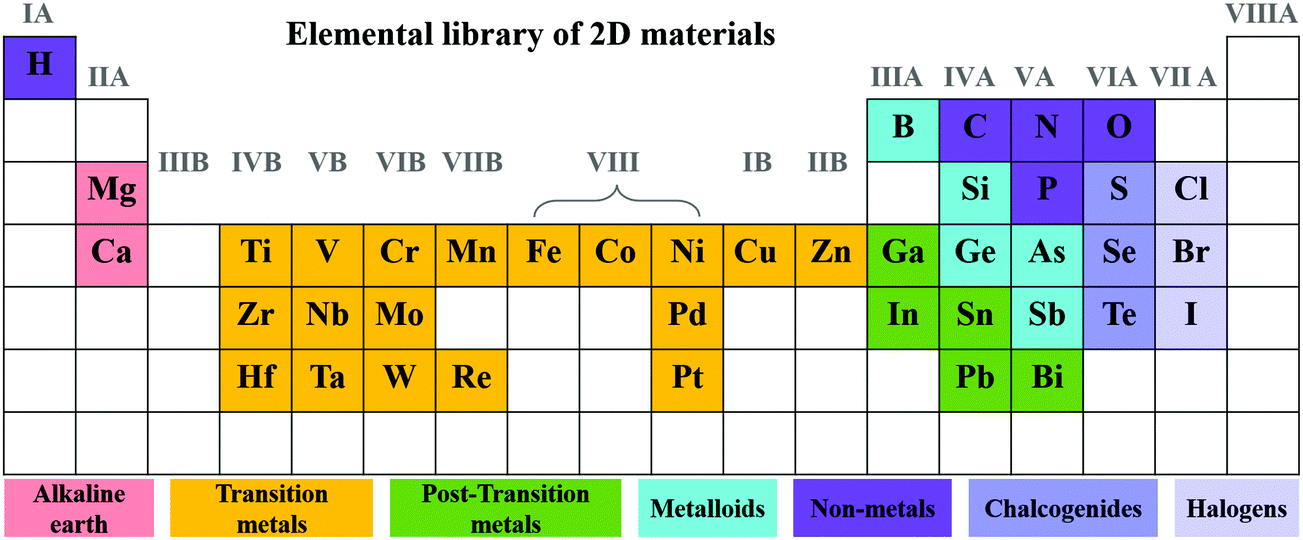 | ||
| Fig. 1 Element distribution of 2D materials in the periodic table. | ||
Avoiding direct chemical bonding between different layers and constraints of crystal lattice matching, 2D materials bring more possibilities for heterostructures with mixed dimensions. For example, 0D nanoparticles and quantum dots (QDs),70 1D nanowires and nanorods,71 and 1.5D nanosheets/nanoflakes (non-vdW structures but 2D morphologies)29 can be constructed with vdW heterostructures as shown in Fig. 2. A vdW hybrid (vdWH) not only combines the properties of the original component materials but also exhibits novel physicochemical properties and unique functions. For example, the MoS2/WSe2 heterostructure was reported to achieve ultrafast charge dynamics within a timescale of 100 fs.72,73 The ultrafast transfer of photo-excited electrons was also found in the BP/InSe heterostructure.74 The band alignment of the g-GaN/Mg(OH)2 vdWH boosted the oxidation and reduction reactions for water splitting. And the potential drop across the interface of the heterostructure could lead to a large built-in electric field which promotes charge separation.75 The electronic properties and band gap structure of the ZnO/GaN vdWH can be tuned using external biaxial strain for realizing high photocatalytic activity.76 Therefore, vdW heterostructures could be promising photocatalysts or co-catalysts for photocatalytic/photoelectrochemical water splitting.
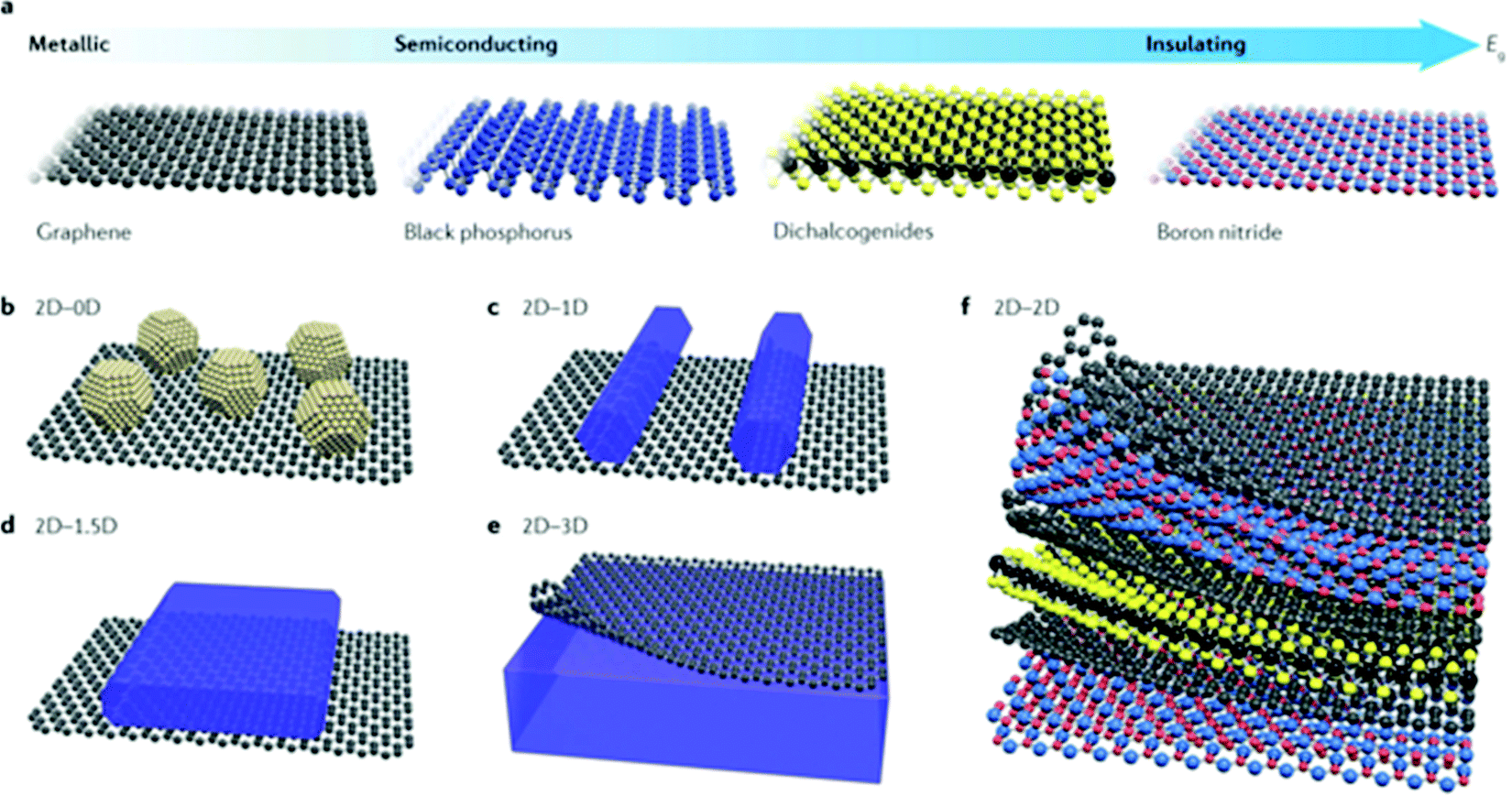 | ||
| Fig. 2 (a) Frontiers of some 2D materials and typical vdWHs: (b) 2D–0D, (c) 2D–1D, (d) 2D–1.5D, (e) 2D–3D, and (f) 2D–2D. Reprinted with permission.77 Copyright 2017 Nature. | ||
Depending on the band structures of 2D material-based heterostructures, there are mainly four types of heterojunctions (straddling alignment (type-I), staggered alignment (type-II), Z-scheme, and S-scheme heterojunctions), as illustrated in Fig. 3. In a type I heterojunction, the VB and CB of semiconductor 1 are lower and higher than those of semiconductor 2, respectively. Therefore, the photogenerated electrons and holes from semiconductor 1 can migrate to the CB and VB of semiconductor 2, respectively. However, the electrons in the CB of semiconductor 2 still easily recombine with holes in the VB of semiconductor 2, resulting in limited improvement in charge carrier separation. It is worth noting that a work function difference between two semiconductor photocatalysts is the pre-requisite for inducing charge redistribution and the formation of an internal electric field, which significantly affect the photogenerated charge carrier separation and transfer processes. In a type II heterojunction, both the CB and VB of semiconductor 2 are lower than those of semiconductor 1, forming a stable heterostructure. The photoinduced electrons and holes can migrate to the CB of semiconductor 2 and the VB of semiconductor 1 and the steps in the CB and VB go in the same direction. Importantly, the difference of chemical potential between semiconductors 1 and 2 causes band bending at the interface of the junction. The band bending induces a built-in field, which drives the photogenerated electrons and holes to move in opposite directions, leading to a spatial separation of the electrons and holes on the different sides of the heterojunction. Therefore, the efficient spatial separation of charge carriers and a prolonged electron lifetime are achieved. Despite the competency of the heterojunction-type of 2D-materials in facilitating charge separation, the redox ability of the photocatalytic system is weakened due to the migration of electrons and holes to the more electropositive CB and electronegative VB, respectively, ascribed to the nature of charge transfer, as shown in Fig. 3b. In the type II heterojunction system, electrons and holes accumulate in the less negative CB and the more positive VB of the photocatalyst, respectively, resulting in weakened reduction and oxidation potentials.
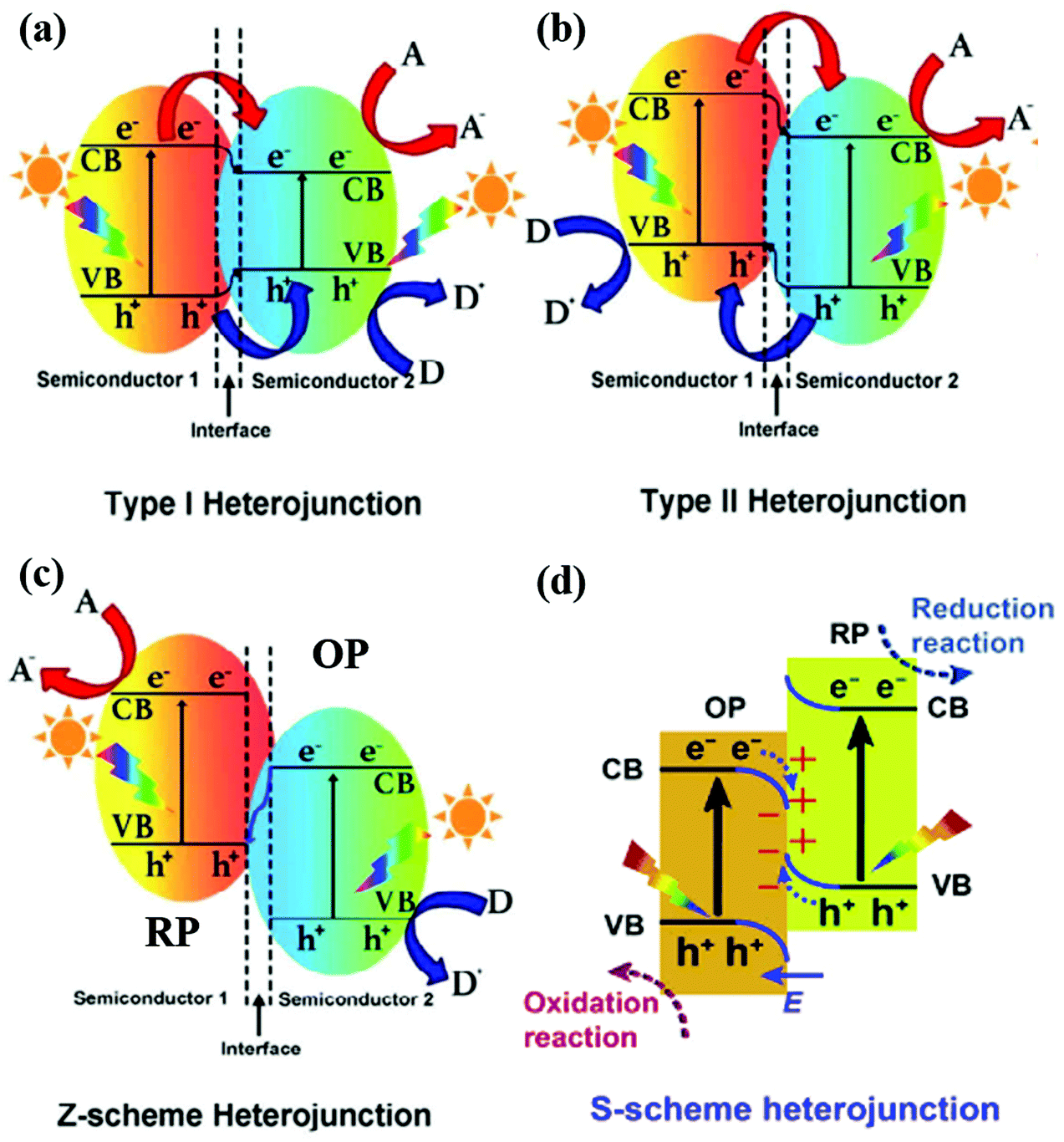 | ||
| Fig. 3 Band structures of various types of heterojunctions: (a) type I heterojunction, (b) type II heterojunction, and (c) Z-scheme heterojunction. Reproduced with permission.90 Copyright 2019 Elsevier. (d) S-scheme heterojunction. Reprinted with permission.88 Copyright 2019 Elsevier. A, D and E, F represent the electron acceptor, electron donor and Fermi level, respectively. | ||
Inspired by natural photosynthesis, Bard et al.78 proposed a Z-scheme system in 1979 to optimize the redox potentials of heterojunctions. As compared to a type-II heterojunction, a Z-scheme photocatalyst has the same band structure configuration and a distinctly different charge carrier transfer mode. However, the Z-scheme heterojunction can maintain the redox ability of photogenerated charge carriers after charge transfer. By adjusting electron mediators, the charge transfer path changes and the type-II heterojunction can be transformed to the Z-scheme system.79–81 The conventional Z-scheme photocatalytic system consists of an H2 production photocatalyst (HPP) or reduction photocatalyst (RP), A; an O2 production photocatalyst (OPP) or oxidation photocatalyst (OP), B; and redox mediators.82 The electrons in the CB of the OP combine with the holes in the VB of the RP through an electron mediator, facilitating the photogenerated electron–hole pairs on the OP and RP but maintaining their reduction and oxidation potentials. But the Z-scheme system still has some problems, such as redox mediator reverse (back) reactions, light-shielding effects, and low charge transfer by ion diffusion. Later, Tada et al. proposed all-solid-state Z-scheme photocatalytic systems using solid conductors as electron mediators in 2006,83 such as noble-metal particles84 and reduced graphene oxide (RGO).85 The selection of an appropriate electron conductor is crucial because it can not only efficiently transfer the photogenerated charge carriers but also effectively improve the stability of the photocatalysts.
The direct Z-scheme photocatalytic system includes only an HPP and an OPP without electron mediators as shown in Fig. 3c. The photogenerated electrons in the lower-level CB of semiconductor 2 can directly recombine with the photoinduced holes in the higher-level VB of semiconductor 1. The higher CB and VB of semiconductor 1 have lower work functions, but higher Fermi levels than semiconductor 2. When semiconductors 1 and 2 are in contact, free electrons keep moving from semiconductor 1 to semiconductor 2 until they reach an equilibrated Fermi level with negative and positive charges at interfaces. Furthermore, a built-in electric field is formed and band edge bending occurs. This Z-scheme system can effectively inhibit the backward reaction of H2 and O2 into H2O.86,87 It is worth noting that the induced electric field can also suppress the recombination between the photogenerated electrons of semiconductors in the Z-scheme and also maintained the oxidation and reduction reactions in a timely manner.
A step-scheme (S-scheme) heterojunction is similar to a type-II heterojunction with an OP and an RP but with a completely different charge-transfer route as shown in Fig. 3d.88,89 The RP has higher CB and VB positions and a higher Fermi level with respect to the OP. Thus, the electrons spontaneously transfer from the RP to OP until the Fermi level equilibrium is reached, resulting in an internal electric field (IEF). The charge-transfer route from the low CB of the OP to the high CB of the RP resembles a “step” in the S-scheme system. The alignment of the Fermi energies of the RP and OP to the same level leads to upward and downward shifts in the Fermi levels of the RP and OP, respectively. Therefore, the S-scheme system typically has the features of the built-in IEF and the band bending effect. The photoexcited electron–hole pairs are spatially separated in the S-scheme system due to the IEF, located in the CB of the RP and the VB of the OP, respectively. On the other side, the unused electrons in the CB of the OP and the holes of the VB of the RP recombine and disappear. Thus, the S-scheme heterostructure reserves the photogenerated electron–hole pairs from both the RP and OP, resulting in a powerful redox ability for photocatalytic water splitting.
Both Z-scheme and S-scheme photocatalysts are composed of two tandem structured photosystems that can meet the requirements of higher wavelength absorption and wideband energy gaps between the conduction and valence bands. However, the two-photon excitation mechanism reduces half of the efficiency of the system. Notably, the coupling of two semiconductors in which semiconductor 1 has a higher Fermi level will induce a negatively charged interface, which eventually leads to the formation of a direct Z-scheme system. Conversely, when semiconductor 1 has a lower Fermi level, a type-II heterojunction will be formed. The quasi-continuous energy state at the solid–solid interface of the photocatalyst system can bestow an internal electric field for vectorial electron flow with low resistance.
Other heterostructures such as p–n junctions combining p-type and n-type semiconductors and Schottky or ohmic junctions coupling semiconductors and metal-like materials can also promote charge transfer and separation as well as photocatalytic performance. The specific function and working mechanism of these heterojunctions based on various 2D materials for photocatalytic H2 generation are illustrated in Section 3.
3 Various 2D material based heterostructures
3.1 Black phosphorus-based heterostructures
Black phosphorus (BP) has received enormous research interest in recent years owing to its anisotropic 2D layered structure, tuneable gap energy (0.3–2.0 eV), and high charge carrier mobility.91–93 The crystal structure of monolayer BP consists of a repeated puckered honeycomb structure along the armchair direction as shown in Fig. 4a. The vdW forces hold interactions between layers along the crystallographic zigzag direction. Depending on the thickness or the number of layers as shown in Fig. 4b, the band gap between and the positions of the valence band maximum (VBM) and the conduction band minimum (CBM) can be adjusted. The unique structure and extraordinary electronic properties make BP a promising catalyst especially for solar light photocatalytic water splitting.30,94 By constructing heterojunctions of BP with other materials such as TiO2,95 WO3,96 CdS,97 and TMD materials98,99 as shown in Fig. 4c, the excitation energy of these heterostructures can be extended to the visible light and near-infrared regions.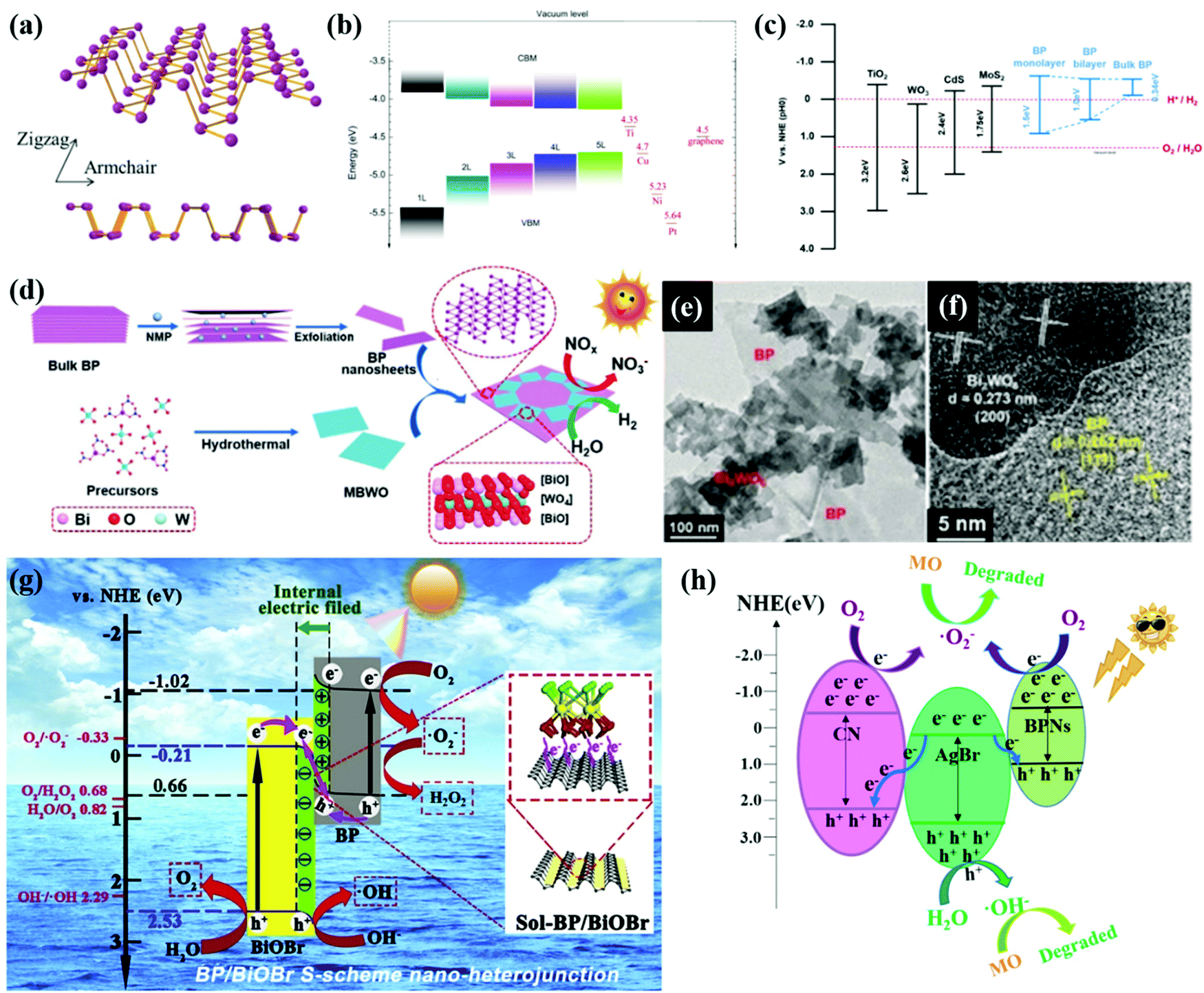 | ||
| Fig. 4 (a) The top view and cross-sectional view of the atomic structure of monolayer BP. Reproduced with permission.118 Copyright 2018, Wiley-VCH. (b) Variation of the VBM and CBM for few-layer phosphorene with the number of layers (from 1 L to 5 L) determined from HSE06 calculation. Reprinted with permission.119 Copyright 2014, Nature. (c) Reprinted with permission.32 Copyright 2016, MDPI. (d) Illustration of the fabrication, (e) morphology and (f) HR-TEM of the BP/Bi2WO6 heterojunction. Reproduced with permission.115 Copyright 2019, Wiley-VCH. (g) S-scheme photocatalytic charge transfer and reaction mechanism between BiOBr and BP. Reproduced with permission.116 Copyright 2020, Elsevier. (h) The proposed photocatalytic mechanism of CN/AgBr/BPN photocatalysts. Reproduced with permission.117 Copyright 2021, Elsevier. | ||
Zhu et al.104 reported a BP/CN binary nanohybrid as a metal-free photocatalyst for efficient H2 production (428.9 μmol h−1 g−1 and 102.2 μmol h−1 g−1) under both >420 and >780 nm light irradiation, respectively. The BP/CN nanocomposite also exhibited a promising photocatalytic H2 evolution of 571 μmol h−1 g−1 under visible light.105 Wen et al. synthesized a low-cost (0.235 Euro per gram) BP/CN heterostructure with abundant active sites leading to superior photocatalytic performance for H2 production, 786 μmol h−1 g−1.106 The BP can act as an electron acceptor and provide transfer channels of electrons from the CB of CN to the CB of BP. Moreover, the p–n defect sites existing at the interface of BP and CN help in trapping electrons in the CB of BP, inhibiting the recombination of electron–hole pairs. Therefore, the heterojunction of BP and CN largely promotes the kinetics of photocatalytic redox reactions. In addition to the preferable carrier migration, the strong and intimate interface interaction of BP and CN can ensure the long-term stability of photocatalytic activity. Besides, adding transition metal phosphides as co-catalysts can further improve the photocatalytic activity of BP and CN. For example, a novel heterostructure of 2D-Ni2P@BP and CN was found to exhibit a remarkable H2 production rate of 858.2 μmol h−1 g−1, which is much higher than that of Pt loaded CN owing to the rational design of the co-catalyst system and the synergetic effect among BP, Ni2P and CN.107 Obviously, 2D–2D heterostructures of BP and CN with type-I heterojunctions show impressive charge separation and migration as well as high photocatalytic performance.
Ran et al.111 for the first time achieved the application of type-II heterojunctions of phosphorene/CdS, Zn0.8Cd0.2S and ZnS guided by density functional calculations. The excellent photocatalytic activity for H2 production (11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 192 μmol h−1 g−1) and highly improved solar light utilization efficiency (quantum yield of 34.7% at 420 nm) were attributed to the strong electronic coupling between phosphorene and CdS and the high charge mobility of the heterostructure.
192 μmol h−1 g−1) and highly improved solar light utilization efficiency (quantum yield of 34.7% at 420 nm) were attributed to the strong electronic coupling between phosphorene and CdS and the high charge mobility of the heterostructure.
Elbanna et al. reported a type-II heterostructure of BP nanosheets (NS) and TiO2 meso-crystals, which exhibited visible-NIR-driven photocatalytic activity for H2 evolution (1.9 and 0.41 μmol h−1 under visible (420–1800 nm) and NIR (780–1800 nm) irradiation, respectively).109 The type-II charge transfer at the interface of BP and TiO2 facilitates the electron migration from BP to TiO2. Moreover, femtosecond time-resolved diffuse reflectance spectroscopy (fs-TRDRS) and photoelectrochemical measurements not only confirmed the importance of improved charge transfer and separation but also illustrated the enhanced electron injection from the BP NS to TiO2 by decreasing the thickness of the BP NS.
Bulk BP was normally exfoliated into a few layers, assisted by NMP-intercalation as shown in Fig. 4e. By a simple hydrothermal process of precursors of Bi2WO6 and ultrathin BP nanosheets, Hu et al.115 synthesized a Z-scheme heterojunction of BP monolayer Bi2WO6 (MBWO) with intimate face-to-face contact as shown in Fig. 4f and g. The photogenerated electrons in the CB of MBWO combine with the holes in the VB of BP, while electrons that remain in the CB of BP are used to produce H2 from water splitting. The highest H2 evolution rate of BP/MBWO can reach 21042 μmol g−1, which is more than 9 times that of pristine MBWO. In this work, the photoelectrochemical measurements and electrochemical impedance spectroscopy (EIS) spectra confirmed the excellent separation ability and efficient charge transfer of BP/MBWO.
Wang et al.117 reported a AgBr/CN/BP double S-scheme heterojunction with excellent photostability and photocatalytic activity. As shown in Fig. 4h, the Fermi level of the main photocatalyst AgBr is lower than those of CN and BP, leading to transfer of electrons from CN and BP to AgBr. IEFs exist at both the CN/AgBr and AgBr/BP contact surfaces. Driven by the IEF and band bending, the photogenerated electrons migrate from the CB of AgBr to the VB of CN and BP, forming a double S-scheme which facilitates the photocatalytic reactions. The research of S-scheme heterostructures especially based on BP materials for photocatalytic water splitting is still at an early stage. An in-depth understanding of charge transfer routes and rules in S-scheme systems and further application of S-scheme heterostructures is expected. Owing to the large contact area and superior charge mobility of 2D materials including BP, more efforts should be devoted to 2D material-based S-scheme heterojunctions for photocatalytic water splitting.
3.2 CN-based heterostructures
Graphitic carbon nitride (CN) has a 2D structure of tri-s-triazine connected via tertiary amines (see Fig. 5a) with high thermal stability and chemical stability. It is identified to be one of the most promising photocatalysts for solar energy conversion owing to its appropriate electronic band structure, low cost, superior stability, facile preparation, and good compatibility with other materials.101,120–123 To improve the STH efficiency of CN, constructing heterostructures of CN is the main strategy to enhance the light utilization and carrier mobility and the resulting photocatalytic activity.124–126 CN-based heterostructures not only boost spatial charge separation but also allow for novel features originating from the synergistic effects between the components of the heterostructures.127 The rational design of CN-based heterostructures can be classified into conventional type II, Z-scheme, S-scheme and other heterostructures such as Schottky and PN junctions.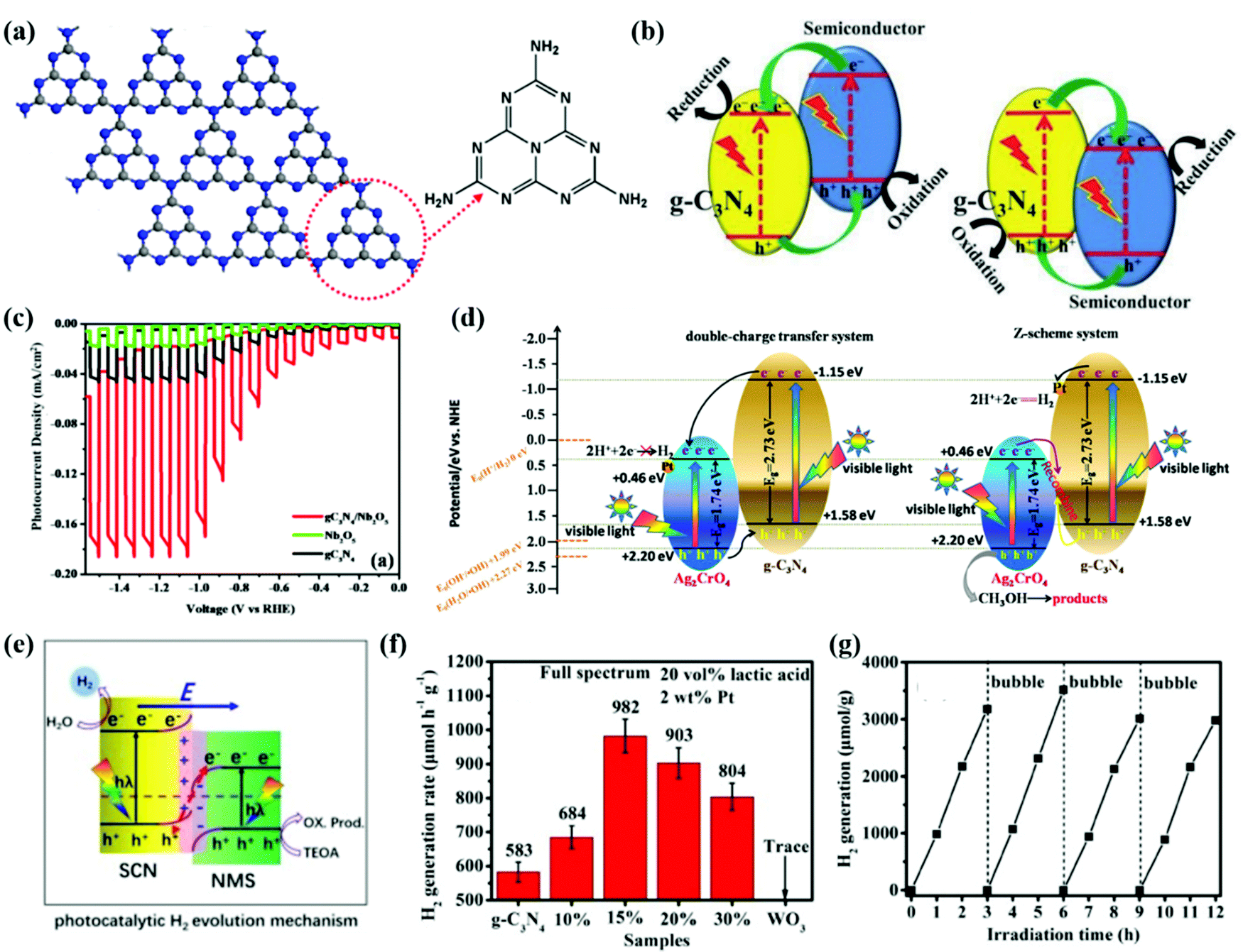 | ||
| Fig. 5 (a) Tri-s-triazine-based two-dimensional structure of CN. Color scheme: C, gray; N, blue. Reprinted with permission.155 Copyright 2014, American Chemical Society. (b) Charge transfer in conventional type II CN-based heterojunction systems. Reprinted with permission.124 Copyright 2017 Wiley-VCH. (c) PEC water splitting measurements of CN, Nb2O5 and CN/Nb2O5 photoanodes. Reprinted with permission.139 Copyright 2018 American Chemical Society. (d) Schematic representation of the charge migration and H2 production mechanism of the Z-scheme g-CN/Ag2CrO4 heterostructure. Reprinted with permission.146 Copyright 2018, Nature. (e) Proposed photocatalytic H2 evolution mechanism over the NMS/SCN S-scheme heterojunction. Reprinted with permission.156 Copyright 2021, Elsevier. (f) Comparison of the H2 generation activities of the as-prepared samples. (g) Time courses of photocatalytic H2 generation of 15%WO3/CN. Reprinted with permission.153 Copyright 2020 Elsevier. | ||
The engineered CN/Nb2O5 type-II heterojunction exhibited a significantly enhanced photocurrent response, which reached –0.17 mA cm−2, with good stability in the photoelectrochemical (PEC) water splitting measurements as presented using red lines in Fig. 5c.139 The Mott–Schottky (MS) analysis confirmed the negative shift of the CBM of the heterojunction between CN and Nb2O5 towards a lower potential of the hydrogen evolution reaction (HER). The EIS measurements further verified the efficient charge transfer derived from the suppressed recombination of photogenerated electrons and holes. Therefore, the CN/Nb2O5 heterojunction practically facilitates the migration of charge carriers, prolongs the lifetime of electron–hole pairs, and enhances the H2 production from PEC water splitting. Qin et al.140 reported a Ni2P-Cd0.9Zn0.1S/CN heterostructure with a high-quantum-yield of photocatalytic H2 production. The CN photocatalyst with a more negative CBM acts as an OP, while the Cd0.9Zn0.1S act as an RP, forming a type-II heterojunction. The Ni2P nanoparticles act as the active sites on Cd0.9Zn0.1S, accumulating the photoinduced electrons for reduction reaction. The Ni2P-Cd0.9Zn0.1S/CN ternary heterostructure resulted in a high H2 evolution rate of 2100 μmol h−1 g−1 and a corresponding apparent quantum yield (AQY) of 73.2% at 420 nm. Due to the superior photostability of CN, the photo-corrosion of Cd0.9Zn0.1S could be alleviated, leading to a stable PEC performance for 90 h.
A semiconductor with a narrow bandgap can not only achieve a type II band alignment when composited with CN for spatial charge separation but also act as a light sensitizer for visible light absorption. Yu et al.141 designed conjugated polymers, (N-annulated perylene/P3)/CN heterojunctions, with an increased light capture ability and charge separation for enhanced H2 production under visible light. Since the lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO) levels of the polymers are higher than the CBM and VBM of CN, P3/CN forms a type-II heterojunction with the function of spatial charge separation. The polymer P3 with a strong electron donor unit (N-annulated perylene) has a narrow optical gap of 1.85 eV, which extends the light response to the visible and even near-infrared regions of the P3/CN heterostructure. With a broader visible-light response range and more effective charge separation, the P3/CN heterostructure exhibits a significantly increased H2 production rate of 13.0 mmol h−1 g−1 with an AQY of 27.32% at 520 nm.
With efficient charge transfer and spatial separation of type II heterojunctions and improved light utilization, a highly enhanced photocatalytic performance for H2 production can be obtained over CN-based heterostructures.
The successful employment of heterostructures for photocatalysis depends on not only band engineering but also the construction of intimate contact between the components. You et al.148 constructed a Z-scheme heterostructure of CN and Bi4NbO8Cl with intimate interfaces realized an H2 production rate 6.9 and 67.2 times that of bare CN and Bi4NbO8Cl, respectively. The PEC measurement and time-resolved photoluminescence decay (TRPLD) results clearly indicate efficient charge transfer and separation in the Z-scheme CN/Bi4NbO8Cl heterostructure. The electron spin resonance (ESR) spectra of CN/Bi4NbO8Cl further detected signals from both superoxide radicals (˙O2˙−) and hydroxyl radicals (˙OH˙), directly verifying the Z-scheme charge transfer route. Ye et al.149 also reported a Z-scheme heterostructure of CoTiO3 and CN, which achieved an impressive H2-evolution rate of 858 μmol h−1 g−1 under solar light.
Apparently, the construction of CN-based Z-scheme heterostructures facilitates charge transfer for the subsequent photocatalytic reduction and oxidation reactions. In addition to the improved solar light utilization and optimal redox potential, the H2 production rate of these heterostructures is highly promoted under solar light.
By enlarging the contact area of CN with a counterpart photocatalyst, the interfacial charge transfer rate can be further enhanced. Fu et al.153 designed a heterostructure of ultrathin WO3 and CN nanosheets of 2.5–3.5 nm (equivalent to 5–8 atomic or molecular layer thickness). Fig. 5f presents the photocatalytic performance of the WO3/CN heterostructure and reference samples. Apparently, the 15% WO3/CN photocatalyst sample exhibits the highest H2-production rate of 982 μmol h−1 g−1, which is about 1.7 times higher than that of pristine CN nanosheets. The MS measurements determine the band structures of the CN and WO3 in the heterostructure. The CBM and VBM positions of CN are more negative than those of WO3. On the other hand, EPR analysis implies the existence of photogenerated electrons in the CB of CN and in the VB of WO3. Thus, the S-scheme charge transfer mechanism of the WO3/CN heterojunction is confirmed. Under light irradiation, the photogenerated electrons transfer from the CB of CN to the CB of WO3 resulting in an IEF and band bending at the interface of the WO3/CN heterojunction. Both the IEF and band bending accelerate the charge transfer and separation, promoting photocatalytic reduction for H2 production. Moreover, the interfacial contact can also be moderated to reduce interface contact resistance and facilitate charge migration and separation.154
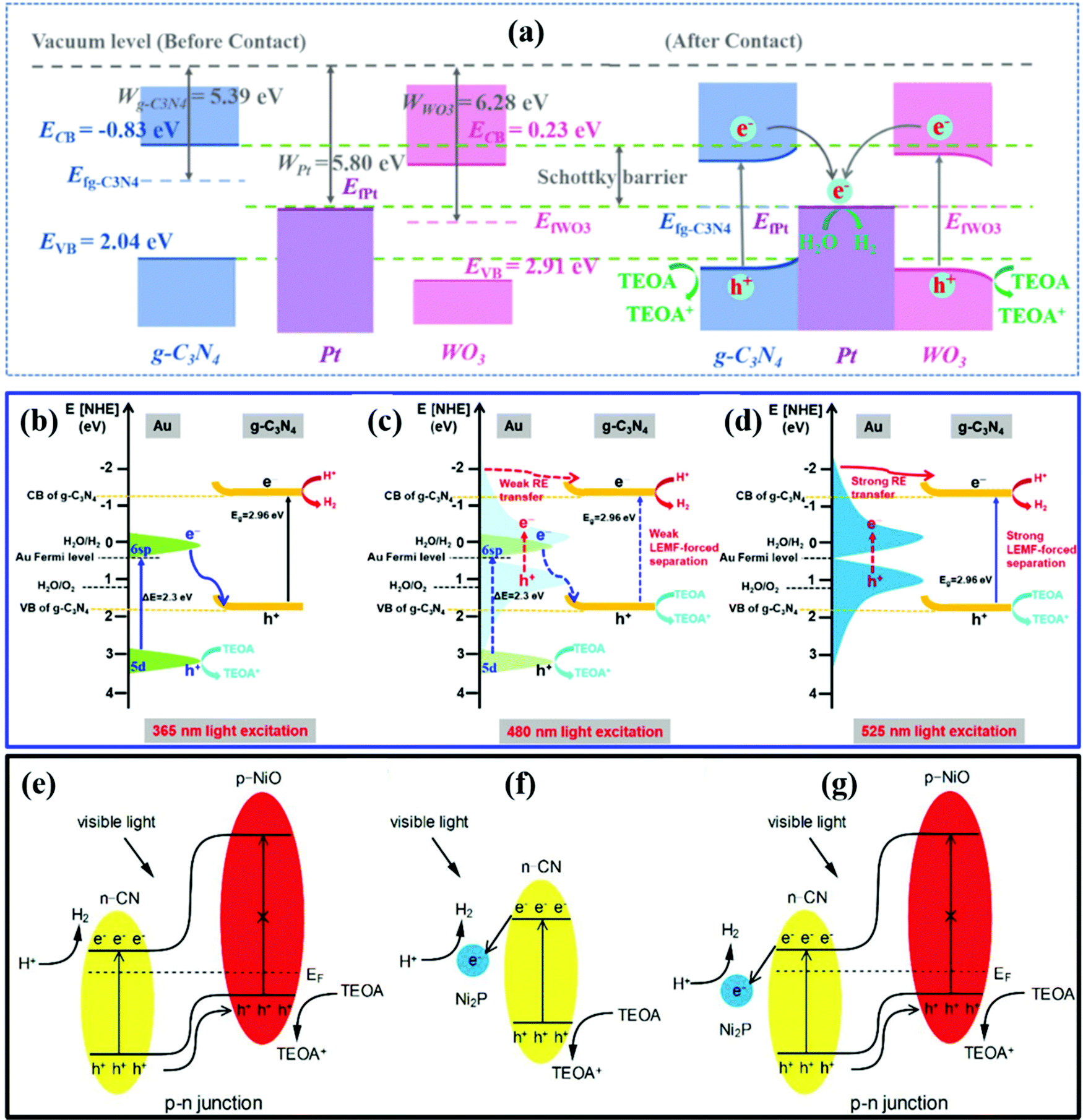 | ||
| Fig. 6 (a) The photocatalytic H2 evolution mechanism of the WPC system. Reprinted with permission.157 Copyright 2021, Elsevier. (b–d) Schematic illustrations of the different mechanisms of SCN/Au5 in producing H2. Reprinted with permission.160 Copyright 2020, Elsevier. The separation and transfer of electron–hole pairs in NiO/CN (e), Ni2P/CN (f), and NiO/Ni2P/CN (g). Reprinted with permission.162 Copyright 2019, Elsevier. | ||
In addition, the localized surface plasmon resonance (LSPR) effect and inter-band transitions of some metals (such as Ag, Au, and Bi) can largely enhance light absorption.159 Wang et al.160 reported a SiO2-CN/Au (SCN) hybrid photocatalyst generating hot carriers for enhanced photocatalytic H2 production. As presented in Fig. 6b–d, hot electrons that originated from inter-band transitions (d → sp) and LSPR inter-band transitions at the Au surface combined with holes in the VB of SCN. Due to the Schottky barrier, the electrons in the CB of SCN would not transfer back to the Au surface. Spatial charge separation was successfully achieved. The d → sp inter-band transitions occur at energies higher than 2.3 eV. Thus, the d → sp inter-band transitions can be ignored for 525 nm light excitation. The Au nanoparticles of small sizes (lower than ∼2 nm) have stronger inter-band transitions and show relatively weak LSPR absorption than the large Au NPs. By employing small sized Au particles, the AQE driven by the LSPR effect (0.28%) is much lower than that driven by the d → sp inter-band transitions (4.29%) under 525 nm light irradiation.
A p–n heterojunction also has an IEF promoting spatial charge separation.161 Shi et al.162 synthesized a Ni2P decorated NiO/CN p–n heterojunction for enhanced photocatalytic H2 production. As presented in Fig. 6e–g, the IEF helps the separation of electron–hole pairs by driving the holes in the VB of CN to the VB of NiO. The Ni2P acts as the cocatalyst to capture the electrons and enhance the light absorption. With a low overpotential, the water reduction reaction occurs on the surface of Ni2P. The NiO/Ni2P/CN p–n heterostructure exhibited an H2 evolution rate 126 times higher than that of pristine CN under visible-light irradiation (λ > 420 nm). The p–n heterojunctions accelerate the charge separation and improve the photocatalytic performance of CN.
3.3 TMD-based heterostructures
The family of TMD materials defined as MX2 (M = transition metals; X = S, Se or Te) ranges from insulators (group-IV TMDs HfS2, etc.), semiconductors (group-VI TMDs MoS2 and WS2, and group-VII TMDs ReS2, etc.), semi-metals (such as TiSe2), to metals (such as group-V TMDs NbS2 and VSe2).163,164 The properties of TMD materials highly depend on the number of d-electrons, chalcogen size and the number of layers. With their tuneable electronic and thermoelectric properties, TMD materials have attracted considerable attention in the application of energy conversion during the past few years.165–167 The physicochemical properties of TMD materials can be derived from the monolayered unit. The weak vdW force connection between layers results in different structures of TMDs as shown in Fig. 7a. Monolayer TMDs mainly include the trigonal prismatic and octahedral phases (the 1H and 1T phases with D3h and D3d symmetry, respectively). The 1T′ and 1T′′ phases are derived from the distorted structures of the 1T phase, while the 2H and 3R phases have resulted from different stacking arrangements of the 1H phase.168 These physicochemical properties, including the electronic structure, thermodynamic stability, and photocatalytic activity, of TMDs differ in various phases. For example, the trigonal prismatic phases of (Mo,W)S2 and (Mo,W)Se2 exhibit better stability than their octahedral phases.169 The 1T and 2H phases of MoS2 exhibit superconductivity, magnetism, ferroelectricity and memristive behavior, which could be applied in electronics, catalysis and energy storage.170 And the 3R-phases of MoS2 and WS2 are often considered to be better catalysts compared to the 2H-phases.171Fig. 7b summarizes the band structures of conventional semiconductor photocatalysts and TMD materials. Apparently, TMD materials have narrow bandgaps, and absorb a wide range of light energy. With effective light absorption, TMD photoelectrodes are expected to realize promising STH efficiencies higher than those of most traditional metal oxides as illustrated in Fig. 7c. By constructing heterostructures, the CBM and VBM positions of these TMDs can be further modified as shown in Fig. 7d. Therefore, the oxidation and reduction reactions of water splitting can be more feasible.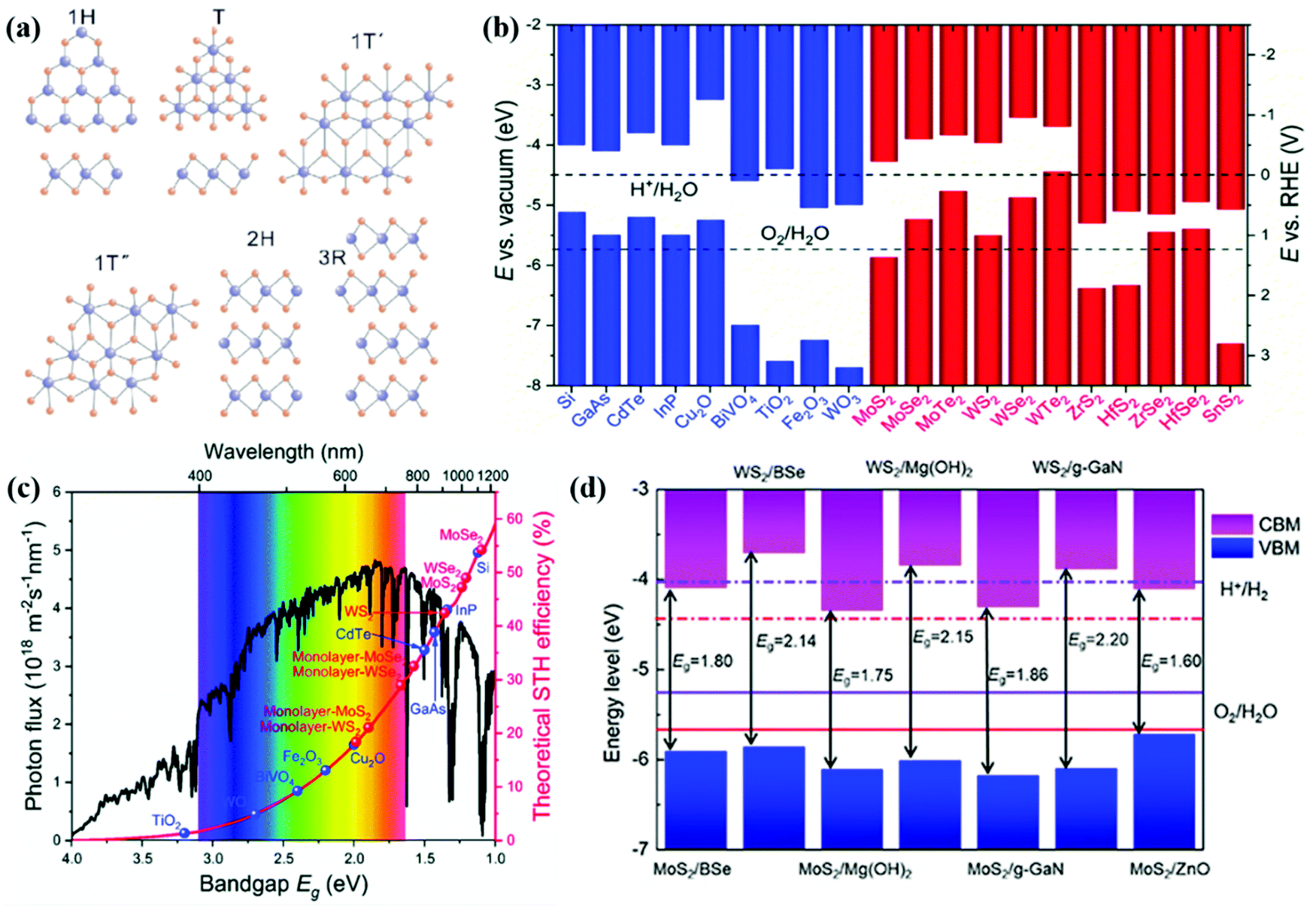 | ||
| Fig. 7 (a) Structures of 2D TMDs. Reproduced with permission.172 Copyright 2019, Science in China Press. (b) Band alignments for conventional semiconductors and 2D TMD monolayers. (c) Theoretical maximum solar-to-hydrogen (STH) conversion efficiency as a function of the bandgaps of the semiconductors based on the AM 1.5G solar spectrum, with the assumption that all the semiconductor photoelectrodes have an IPCE of 100% in the absorption range. Reproduced with permission.166 Copyright 2018, Royal Society of Chemistry. (d) The band structures of some TMD based vdW heterostructures. Reprinted with permission.38 Copyright 2020, Institute of Physics Publishing. | ||
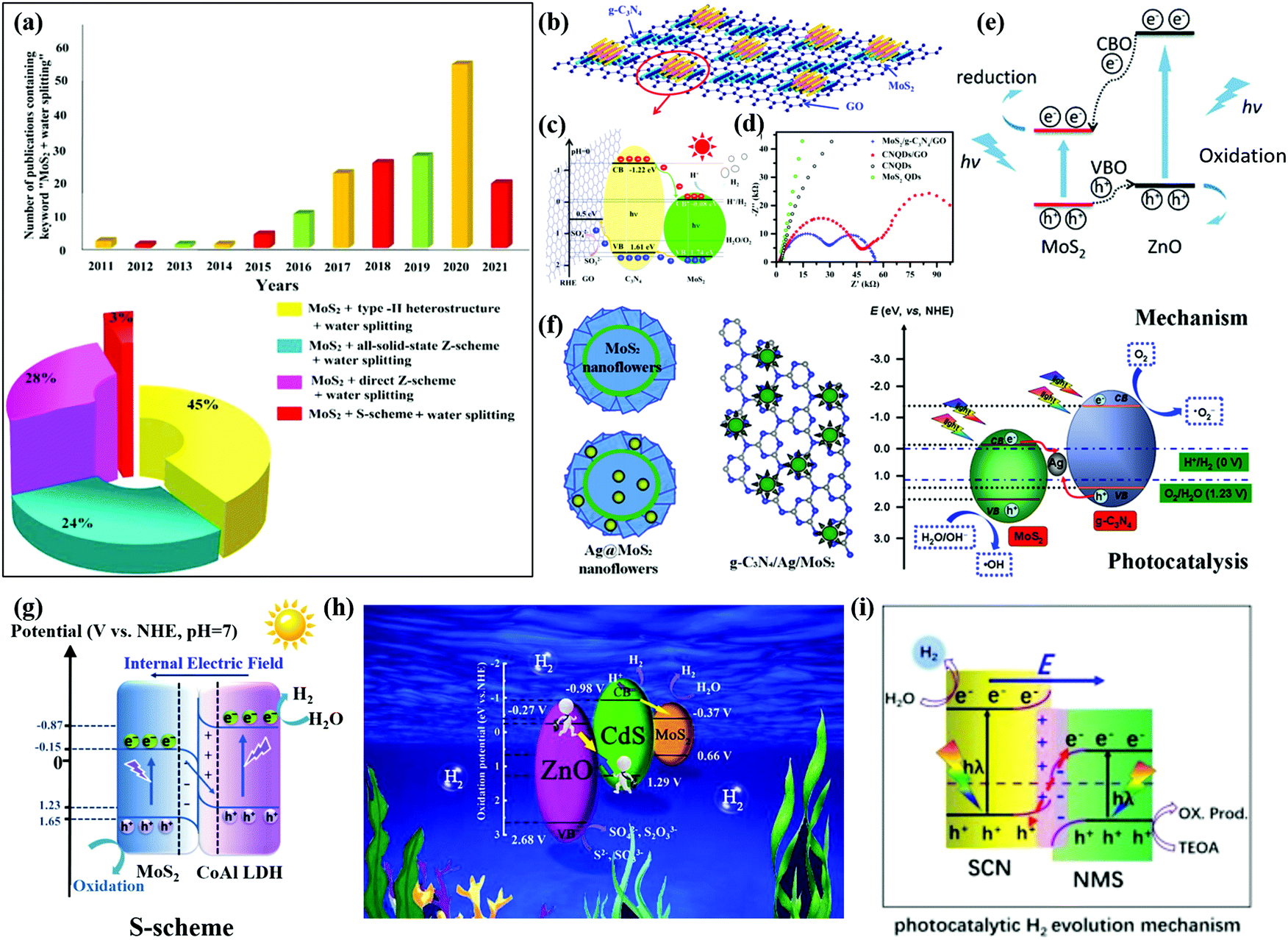 | ||
| Fig. 8 (a) Number of publications per year on “MoS2 + photocatalysts” from 2011 to 2021 and different MoS2-based heterostructure photocatalysts particularly for the application of water splitting photosystems. Reproduced with permission.183 Copyright 2021, Elsevier. (b) Schematic drawing of the MoS2/CN/GO composite, (c) schematic illustration of charge carrier separation in a MoS2/CN/GO junction, and (d) Nyquist plots of these samples. Reprinted with permission.175 Copyright 2017, American Chemical Society. (e) Schematic illustration of the migration of photogenerated electrons and holes at the MoS2/ZnO interface. Reprinted with permission.176 Copyright 2018, Royal Society of Chemistry. (f) Schematic illustration of CN and Ag co-modified MoS2 nanoflowers and the related photocatalytic mechanism. Reprinted with permission.178 Copyright 2017, American Chemical Society. (g) S-scheme charge transfer mechanism of the MoS2/CoAl LDH composite for photocatalytic H2 generation. Reprinted with permission.181 Copyright 2021, Elsevier. (h) S-scheme mechanism for the enhanced photocatalytic H2 evolution. Reprinted with permission.182 Copyright 2021, Elsevier. (i) S-scheme heterojunction. Reprinted with permission.156 Copyright 2021, Elsevier. | ||
Type II heterostructures. Wang et al.175 reported a MoS2/CN/graphene oxide (GO) ternary nanojunction for effective water splitting as shown in Fig. 8b. The type II MoS2/CN/GO heterostructure had a band alignment which facilitates collection of electrons in MoS2 and holes in CN as indicated in Fig. 8c. The effective charge transfer and separation allow for retardation of charge recombination. The MoS2/CN/GO photocatalyst exhibited a highly enhanced H2 production rate of 1.06 mmol h−1 g−1 (4.3 times higher than that of MoS2) using Na2SO3 as a sacrificial agent under simulated AM1.5G solar irradiation. The ESI spectra further confirm the efficiency of the trapping, transfer, and separation of charge carriers of the MoS2/CN/GO heterostructure as illustrated in Fig. 8d. The MoS2/ZnO vdW heterostructure also constructs a type-II band alignment with a large built-in electric field,176 which enables enhanced efficiency of optical absorption especially under visible light as shown in Fig. 8e. The type-II MoS2/GaN heterostructure moves up the band edges of MoS2 for ∼0.5 eV by the deployment of interface nitridation.177 The increased conduction band offset (CBO) could result in improved capability of electron accumulation on the GaN side. The nitridation interfacial layer in the MoS2/GaN 2D/3D heterostructure leads to a substantial optical absorption ability and subsequent photocatalytic activity for water splitting using solar energy.
Z-scheme heterostructures. Lu et al.178 synthesized a CN/Ag/MoS2 ternary plasmonic photocatalyst in a flowerlike architecture forming a Z-scheme heterojunction as shown in Fig. 8f. The Z-scheme-assisted rapid charge separation and suppressed recombination of photoexcited electron–hole pairs significantly boost the photocatalytic H2 production rate of CN/Ag/MoS2. The CN/Ag/MoS2 photocatalyst has an H2 production rate of 10.40 μmol h−1, which is approximately 8.78 times, 3.51 times and 2.08 times that of Ag/MoS2, pure CN, and CN/MoS2, respectively. A solid-state Ag NP-decorated MoS2/reduced graphene oxide (RGO)/NiWO4 Z-scheme photosystem was constructed.179 In addition to the Z-scheme function, the RGO mediator and the plasmonic effect of Ag NPs facilitate the charge transfer to enhance photocatalytic efficiency. Nagajyothi et al.180 reported a ZnFe2O4/MoS2 Z-scheme heterostructure achieved an H2 production rate of 142.1 μmol h−1 g−1, which is 2.3 times higher than that of pristine MoS2. The MoS2/CaTiO3 Z-scheme heterostructure was developed for efficient H2 production from water splitting. The high transient current density of ZnFe2O4/MoS2 verifies the efficient separation of electron–hole pairs. And the low radian impedance of ZnFe2O4/MoS2 from EIS analysis further confirmed the inhibited recombination of electron–hole pairs.
S-scheme heterostructures. A MoS2/CoAl LDH heterostructure was reported with the advantages of the S-scheme heterojunction driving photocatalytic H2 generation under visible-light irradiation.181 The IEF across the interface of MoS2 and CoAl LDH accounts for the rapid detachment of the electron–hole pairs and a strong redox ability. The XPS and DFT results indicate an upward shifted energy band edge of CoAl LDH and a downward shifted energy band edge of MoS2, as illustrated in Fig. 8g. Due to the IEF-induced S-scheme system, the H2 generation rate of the MoS2/CoAl LDH could reach 17.1 μmol g−1 h−1, which is 8 times that of the pure CoAl LDH. The S-scheme heterojunction not only boosts the charge separation but also allows for the preservation of strong reducibility. Jia et al.182 reported a new S-scheme heterojunction, ZnO/CdS/MoS2 (ZCM), with suppressed charge recombination and greatly enhanced photocatalytic H2 evolution performance. As presented in Fig. 8h, the CdS plays as the bridge between ZnO and MoS2, which promotes charge separation through an intimate interface. The UV-vis diffuse reflectance spectra (DRS) and surface photo-voltage (SPV) spectra prove the strong visible light absorption of the ZCM sample. The transient photocurrent (TPC) spectra and EIS spectra further confirmed the efficient charge separation ability of ZCM. Therefore, the obtained ZCM exhibited superior photocatalytic activity for water splitting over the individual or binary components. An S-scheme heterojunction of N-doped MoS2 (NMS) and S-doped CN (SCN) was also found to show a high H2 generation rate of 658.5 μmol g−1 h−1 (23 and 38 times that of pure SCN and NMS, respectively) using TEOA as a sacrificial reagent. As presented in Fig. 8i, the electrons transfer from SCN with a high Fermi level to NMS with a lower Fermi level, forming an IEF until the Fermi level equilibrium is reached. Thus, the photogenerated electrons in the CB of SCN and the photogenerated holes in the VB of NMS with a maximized redox ability are preserved, while photogenerated holes in the VB of SCN combine with photogenerated electrons in the CB of NMS. Therefore, the driving force for charge transfer and separation provided by the S-scheme leads to highly enhanced photocatalytic activity of the NMS/SCN heterostructure.
Type II heterostructures. The photoactivity of wide band gap oxides, such as WO3 and TiO2, can be extended to the visible light region by forming type-II heterojunctions with WS2.186 Xiao et al.187 reported TiO2/WS2 (MoS2) heterostructures integrating the merits of both components for photocatalytic water splitting under solar light. Notably, the TiO2/WS2 (or MoS2) heterostructures have a type-II band alignment but with a built-in IEF. As shown in Fig. 9a and b, the work function of WS2 is smaller than that of TiO2, which results in an electron flow from WS2 to TiO2. The formation of the built-in IEF facilitates the migration of carriers. Under solar light irradiation, the photoexcited electrons in the CB of WS2 transfer to the CB of TiO2 for the reduction reaction of water splitting. On the other side, the photoexcited holes migrate from the VB of TiO2 to the VB of WS2 for the oxidation reaction of water splitting. Therefore, the TiO2/WS2 heterostructure achieves the spatial separation of photogenerated electron–hole pairs. The maximum conversion efficiencies for WS2/TiO2 and MoS2/TiO2 could reach 11.5% and 13.5%, respectively, according to the DFT calculation.
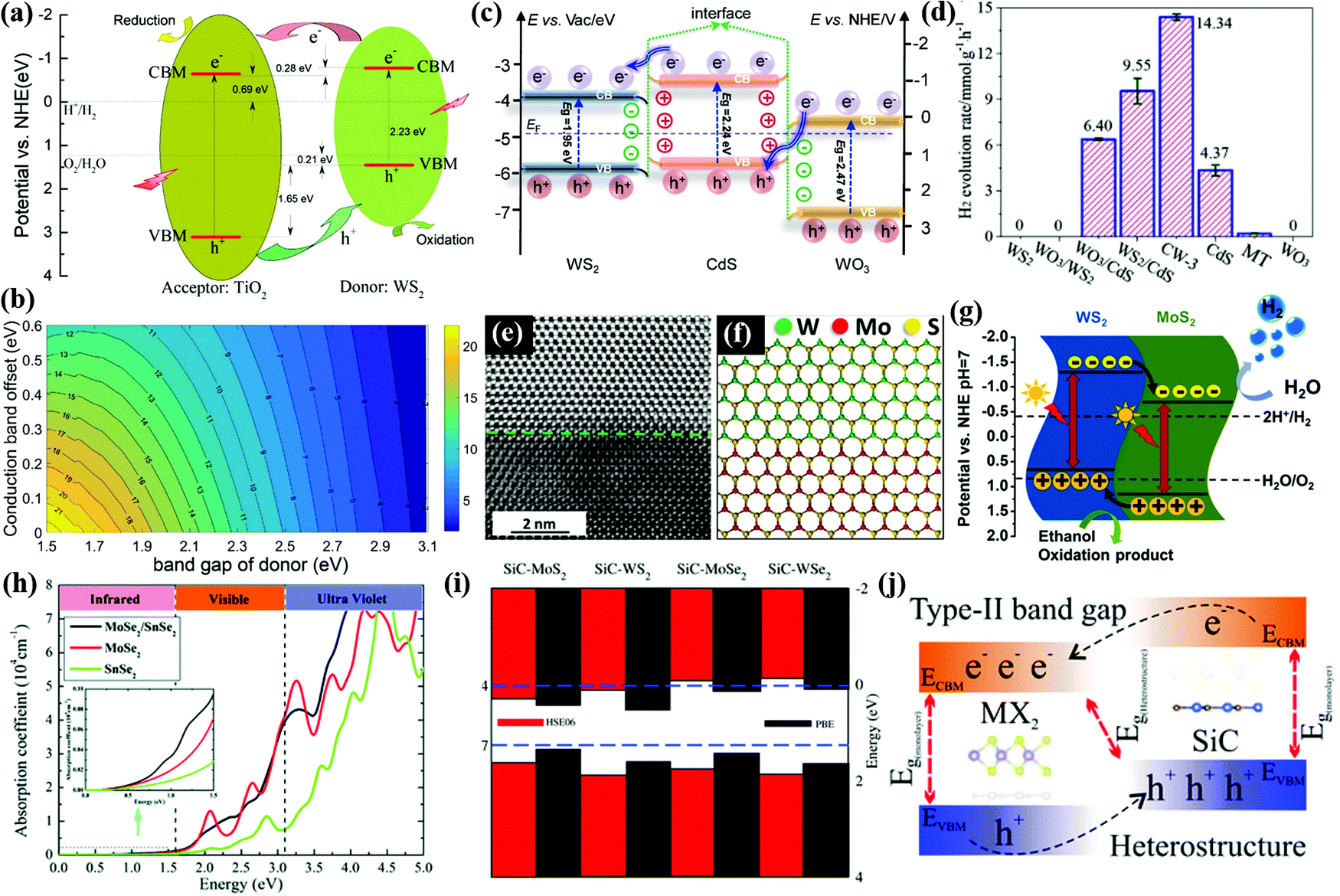 | ||
| Fig. 9 (a) Schematic illustration of type-II band-alignments in the WS2/TiO2 heterojunction. (b) Contour plots showing the energy-conversion efficiency versus the donor bandgap and the conduction band offset. All the energy levels are referenced to the reduction (H+/H2) potential. Reprinted with permission.187 Copyright 2020, Elsevier. (c) Charge transfer behavior of the CW heterostructure, (d) H2 evolution rates over different samples under visible light irradiation (λ > 420 nm). Reprinted with permission.189 Copyright 2020, Elsevier. (e) The top part is the WS2, while the bottom is the MoS2. (f) Atomic model of the WS2–MoS2 interface. (g) Schematic representation of the proposed mechanism for photocatalytic H2 evolution over a WS2–MoS2 heterostructure. Reproduced with permission.190 Copyright 2021, Elsevier. (h) Calculated absorption coefficients of the MoSe2 (WSe2), SnSe2 monolayer, and MoSe2/SnSe2 (WSe2/SnSe2) heterostructure. Reprinted with permission.191 Copyright 2019, Royal Society of Chemistry. (i) VBM and CBM for the heterostructures. (j) Type-II band alignment in heterostructures. Reprinted with permission.192 Copyright 2018, Royal Society of Chemistry. | ||
Z-scheme heterostructures. WS2 based Z-scheme heterojunctions offered excellent H2 production from photocatalytic water splitting by optimization of pristine WS2, including the recombination of charge pairs, active centres and photostability.188 Xue et al.189 constructed a direct Z-scheme WO3/WS2/CdS tandem heterojunction with a high photocatalytic H2 evolution rate of 14.34 mmol h−1 g−1 and an outstanding apparent quantum efficiency (AQE) of 22.96% at 435 nm as shown in Fig. 9c and d. The electrons transfer from the CB of CdS to the CB of adjacent WO3 and WS2, forming a dual built-in IEF at the heterointerfaces of the WS2/CdS and WO3/CdS. The in situ irradiated XPS (ISI-XPS) measurement and ultraviolet photoelectron spectral (UPS) analysis results confirmed the route of interfacial charge transfer in the hybrid system. Apparently, the energy-level alignment of the direct Z-scheme system facilitates the spatial separation for enhanced H2 production performance. A WS2/MoS2 in-plane few-layer heterostructure was reported with rapid charge separation and a promising H2 generation rate of 9.83 mmol g−1 h−1.190 As presented in Fig. 9e and f, the WS2 and MoS2 are located in the upper and lower parts of the images, respectively. Due to the difference of atomic numbers in WS2 and MoS2, the interface of the two materials is distinguishable. The WS2/MoS2 heterostructure forms a type II heterojunction as shown in Fig. 9g, acting as an electron sink to prevent the recombination of electron–hole pairs. Thus, the WS2/MoS2 heterostructure exhibits much higher photocatalytic activity than the individual components.
3.4 LDH-based heterostructures
LDH materials consist of brucite like metal hydroxide layers and water molecules balanced by interlayer anions196,197 as shown in Fig. 10a. The general formula of an LDH can be expressed as [M2+(1−x)M3+x(OH)2]x+(An−)x/n·mH2O, where M2+ and M3+ represent bivalent (Mg2+, Cu2+, Zn2+, Mn2+) and trivalent (Al3+, Fe3+, Ti3+, Cr3+) cations, respectively. An− is the interlayer anion of valence n (CO32−, SO42−, NO3−, Cl−, F−, etc.) and x (0.2–0.4) represents the molar ratio of trivalent cations to the total cation content. LDH materials have high adsorption capacity, tunable bandgaps, and abundant reaction sites for water splitting reactions.198 Moreover, the rich components and various derivatives make LDHs flexible to construct heterostructures with different semiconductors and cocatalysts such as CN, TiO2, CdS, and some noble metals.199–201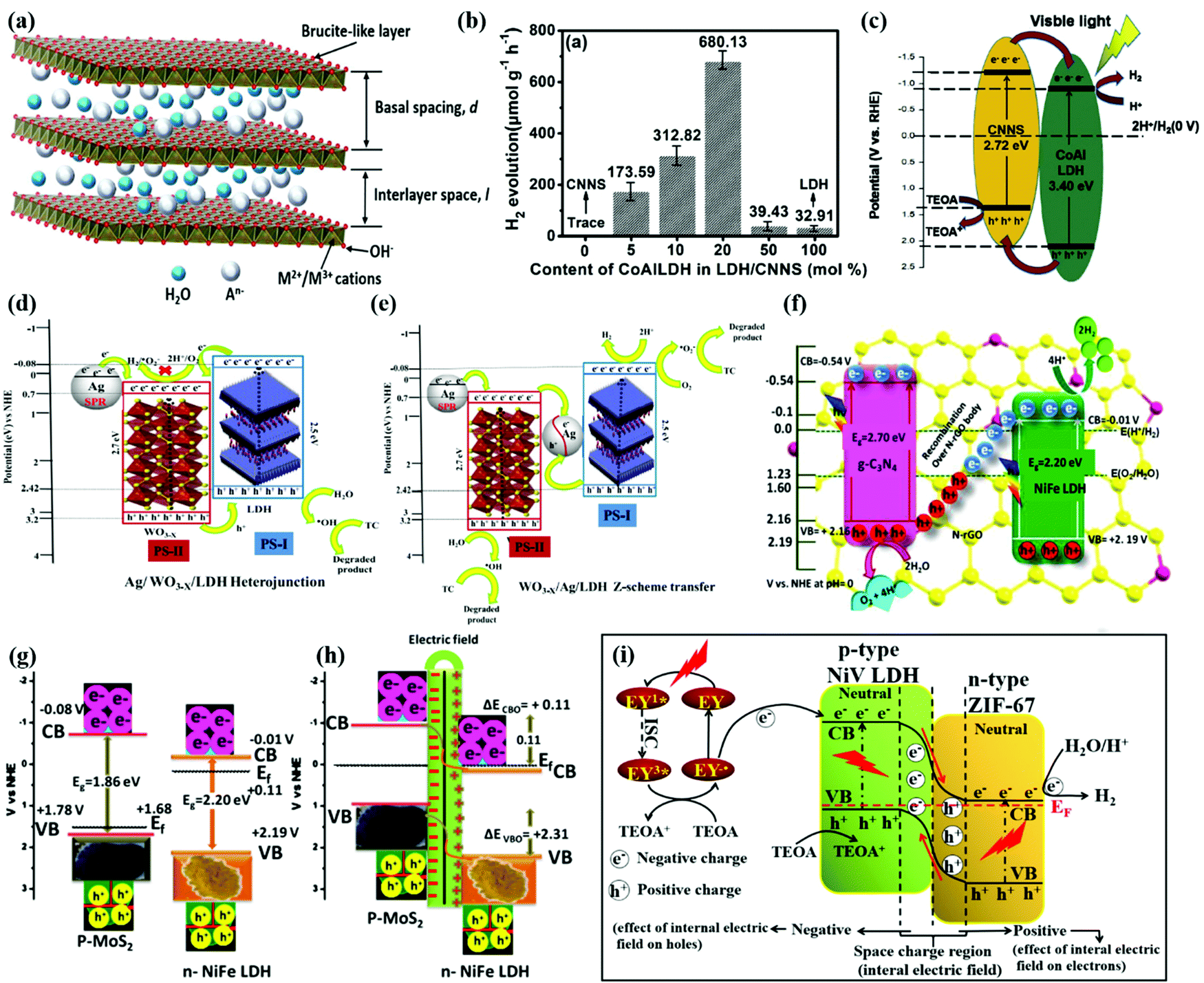 | ||
| Fig. 10 (a) Representative structure of LDH materials. Reprinted with permission.198 Copyright 2022, Elsevier. (b) The H2 evolution efficiency with different ratios of CoAl-LDH loading on the CNNS. (c) Mechanism for hydrogen evolution over CoAl-LDH/CNNS under visible light irradiation. Reprinted with permission.202 Copyright 2020, Elsevier. Schematic illustration of the proposed reaction mechanism in the (d) Ag/WO3−x/LDH heterostructure and (e) WO3−x/Ag/LDH-based reaction systems toward TC degradation and H2 evolution under visible light irradiation. Reprinted with permission.209 Copyright 2019, American Chemical Society. (f) Z-scheme mechanism for mineralization of dyes with H2 and O2 evolution over the CNNG3LDH heterostructure. Reprinted with permission.210 Copyright 2019, Nature. (g and h) Potential edge alignment for n-type NiFe LDH and p-type MoS2. Reprinted with permission.218 Copyright 2019, American Chemical Society. (i) Internal migration mechanism of electrons in the p–n junction. Reprinted with permission.221 Copyright 2020, Elsevier. | ||
CdS also forms type-II heterojunctions with LDH materials.204 Li et al.205 reported a novel type-II heterojunction based on NiCo-LDH/P doped CdS (P-CdS) with an H2 production rate of 8.665 mmol h−1 g−1 under visible light, which was 45 times higher than that of pure CdS. The DFT results indicated that the work function value of P-CdS is higher than that of NiCo-LDH, leading to an electron flow from NiCo-LDH to CdS. The space charge layer and IEF at the interface between NiCo-LDH and CdS help the charge separation. In addition, the P doping induces a mid-gap between the CB and VB of CdS, trapping photogenerated electrons. Due to the synergy of the NiCo-LDH/P-CdS heterostructure, the efficiency of photocatalytic H2 production was highly enhanced. Zhou et al.199in situ synthesized CdS on the surface of NiFe LDH, forming a nanoscale heterojunction. The TPR results of the CdS/NiFe LDH clearly showed a reduced onset potential and increased photocurrent density. The efficient separation of photogenerated electron–hole pairs was confirmed in the CdS/NiFe LDH due to the type II band alignment between the CdS and NiFe LDH. The NiFe LDH acted as an electron sink absorbing protons and accumulating electrons for the HER. Thus, the CdS/NiFe LDH exhibited notable H2 evolution kinetics (469 μmol h−1 g−1), which was much higher than those of individual CdS and NiFe LDH.
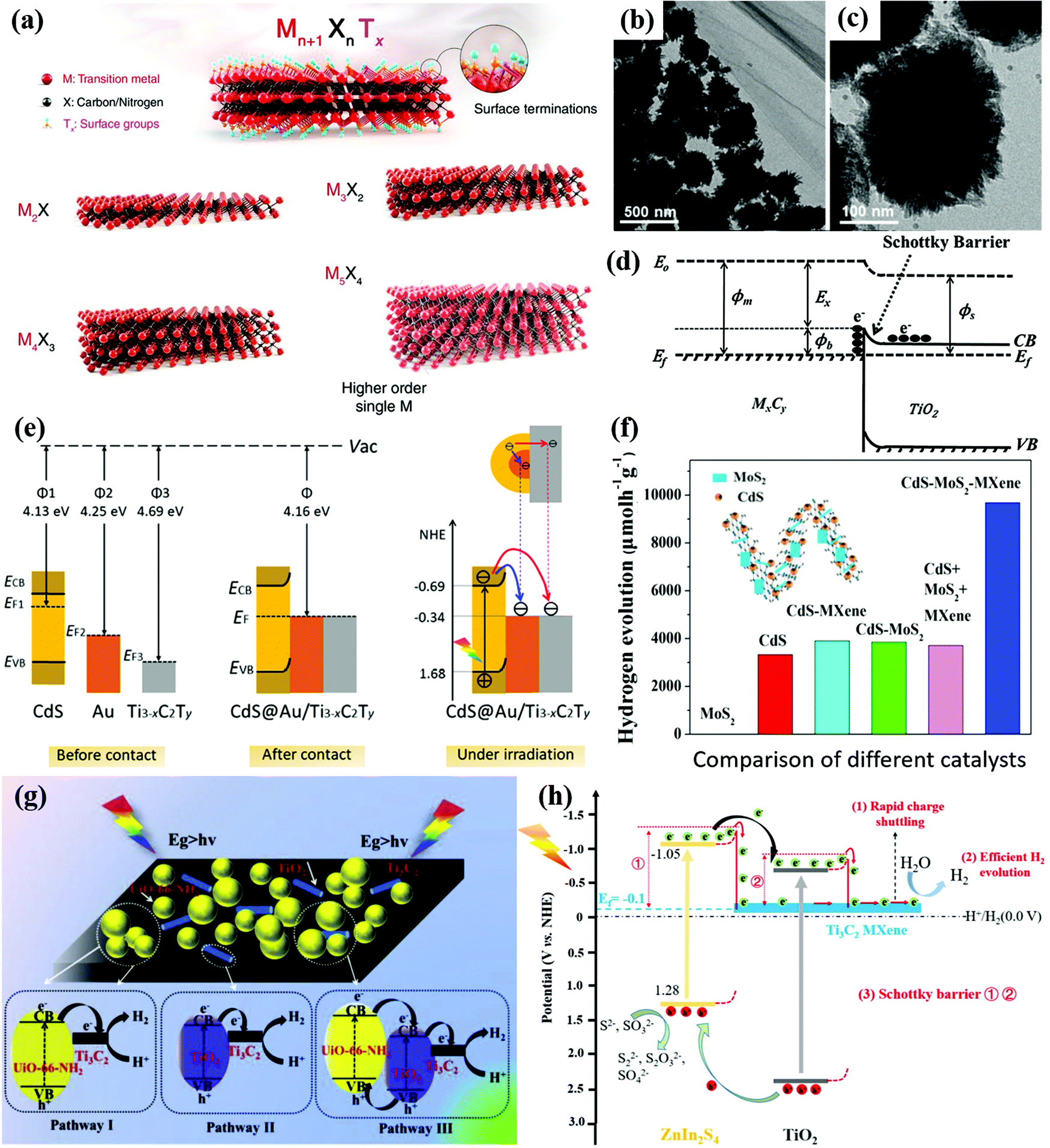 | ||
| Fig. 11 (a) Schematic illustration of the MXene structures. Reproduced with permission.238 Copyright 2021, Science. (b and c) TEM images of TiO2/Ti3C2Tx (5 wt%). (d) Formation of a Schottky barrier at the MXene/TiO2 interface. Reprinted with permission.231 Copyright 2016, Wiley-VCH. (e) Energy level diagram and the photogenerated charge carrier transfer process of CdS, Au, and Ti3−xC2Ty. Reprinted with permission.234 Copyright 2021, American Chemical Society. (f) Photocatalytic H2 evolution over different catalysts. Reprinted with permission.235 Copyright 2019, Elsevier. (g) Schematic illustration of the charge-transfer pathways for Ti3C2/TiO2/UiO-66-NH2. Reprinted with permission.236 Copyright 2019, Elsevier. (h) Proposed process of photoinduced charge transfer process on the M@T/ZIS-50 ternary hybrid. Reprinted with permission.237 Copyright 2020, Elsevier. | ||
Ternary heterostructures with two Z-scheme channels for enhanced photocatalytic H2 production are often reported.211,212 Megala et al. synthesised NiAl-LDH/CN/Ag3PO4 (AP) ternary composites, where CN has the lowest CB potential and AP has the highest VB potential, which result in a strong redox ability.213 In the dual Z-scheme, the holes from the lower potential VB combine with electrons from the higher potential CB, which separates photogenerated electron–hole pairs. The electrons from the lowest potential CB reduce protons to H2 and those from the highest CB potential VB oxidise water into O2. By band alignment between NiAl-LDH, CN, and AP, the most efficient charge transfer route can be achieved, leading to high photocatalytic performance.
The interface charge transfer between the two components in the Z-scheme system is a crucible for the separation and subsequent migration of photogenerated electron–hole pairs.214 A donor–bridge–acceptor structure bonding two components at atomic and electronic levels in a heterostructure can optimize the charge migration at the heterojunction interface.215 Wang et al. constructed a covalent-bonding-bridge structure at the Cu2O@ZnCr-LDH heterointerface, with a thin-layer Cu2O core and a S2O32− anion intercalated ZnCr-LDH shell.208 The extended X-ray absorption fine structure (EXAFS) fitting and coincidence Doppler broadened positron annihilation spectroscopy (CDB-PAS) confirmed the formation of a Cu−(S2O32−)/LDH bridge structure and its function at the heterointerface. The bridge-type bonding helps the passage of carriers and promotes the charge transfer, which thus highly enhances the utilization of photogenerated carriers. The transient current–potential measurement and PL spectra further revealed improved charge transfer efficiency of photoexcited carriers in the Cu2O@ZnCr-LDH heterostructure. Thus, the Cu2O@ZnCr-LDH heterostructure displayed an acceptable H2 production rate from pure water without using any co-catalysts under visible light.
Cobalt-based semiconductors such as Co3O4, CoO and Co(OH)2 are typical p-type semiconductors with attractive electronic properties and narrow band gaps (about 1.2–2.1 eV). By constructing heterojunctions with n-type LDH materials, the disadvantages of Co-based catalysts, such as low light harvesting efficiency and charge transfer capability, can be compensated for. Sahoo et al.219 coupled Co(OH)2 (CH) platelets with an n-type ZnCr LDH, achieving remarkable photocatalytic performance. The MS plots determined the p–n heterojunction in the CH/ZnCr LDH heterostructure. The EIS spectra and photocurrent response proved the efficient charge transfer and separation in the heterostructure. High electron/hole densities are found in the CB of the LDH and the VB of CH, respectively. These concentrated electrons/holes take part in water reduction for H2 production and water oxidation for O2 evolution, respectively.
A Co-based zeolitic imidazolate framework (ZIF-67) is another p-type catalyst with large surface area and rich active sites for the HER under visible light irradiation.220 Wang et al.221 constructed a p–n heterojunction of ZIF-67 and NiV LDH, showing excellent photocatalytic performances for the HER. The ZIF-67/NiV LDH heterostructure had large specific surface area and enhanced light absorption. The photocurrent density of ZIF-67/NiV LDH indicated the high efficiency of separation and migration of photogenerated charges. Owing to the space charge region and IEF at the interface of the p–n junction as shown in Fig. 11i, the charge transfer is accelerated and the recombination of electron–hole pairs is suppressed. The high-density electrons in the CB of ZIF-67 react with protons to produce H2, while the sacrificial reagent TEOA consumes high-density holes in the VB of NiV LDH. With multiple advantages, the ZIF-67/NiV LDH heterostructure shows a superior photocatalytic H2 evolution rate, which is 9.5 and 5.9 times higher than those of ZIF-67 and NiV LDH, respectively.
3.5 MXene-based heterostructures
The large MXene family has been proved to be the one of the most attractive photocatalysts with multiple virtues of large interlayer spacing, nontoxicity, large surface area, superior chemical stability, outstanding oxidation resistance, etc.222–224 The general formula of MXenes is Mn+1XnTx (n = 1–4), where M represents a transition metal (such as Ti, V, Cr, Nb, and Mo), X stands for carbon and/or nitrogen elements, and Tx represents terminal groups (such as O, OH, and F) depending on the synthesis methods as shown in Fig. 11a. For example, the complete oxygen- or chlorine-terminated titanium carbide MXene with two layers of transition metal (n = 1) can be written as Ti2CO2 or Ti2CCl2, respectively.225 The strong interaction through the bonding group successfully avoids cleavage fractures, resulting in a unique 2D structure with novel properties.226 Even though most MXene materials are semiconductors with indirect bandgaps, MXene based heterostructures with other photoactive materials such as TiO2, CdS, and CN exhibit efficient charge transfer, long-term chemical stability and photocatalytic activity.227–229Ti3C2Tx materials were the first and the most investigated MXene materials applied in photocatalysis with a good hydrophilic ability, electrical conductivity, tunable bandgaps in the visible region, and ultrahigh catalytic activity towards the HER.230 Wang et al.231 reported Ti3C2Tx materials encapsulated within TiO2 nanoparticles as shown in Fig. 11b and c. Due to the rapid charge transfer from TiO2 to Ti3C2Tx, the Ti3C2Tx/TiO2 heterostructure showed a remarkably H2 yield. The conductive Ti3C2Tx can serve as an electron sink attracting electrons from the CB of TiO2. The Schottky barrier would form at the interface of Ti3C2Tx and TiO2, which improves the charge separation as illustrated in Fig. 11d. Ultra-thin Ti3C2 nanosheets can provide versatile platforms for photocatalytic reactions with large surface areas and abundant surface-active sites. The CdS–Ti3C2Tx heterostructure was also reported for enhanced photocatalytic H2 production under visible light.232 The matched energy level alignment and intimate interfacial contact between conductive Ti3C2Tx and CdS allow rapid charge transfer and enhanced charge separation.233
Ultrathin Ti3-xC2Ty (where x refers to the Ti defects and Ty refers to the surface functional groups) with reductive Ti vacancies was composited with CdS@Au core–shell nanojunctions.234 In the Ti3-xC2Ty/CdS/Au ternary heterostructure, the work function values follow the order: CdS (4.69 eV) > Au (4.25 eV) > Ti3−xC2Ty (4.13 eV). Thus, the photogenerated electrons in the CB of CdS transfer to Au and Ti3−xC2Ty, resulting in dual Schottky barriers at the CdS/Au and CdS/MXene interfaces as illustrated in Fig. 11e. The Au and Ti3−xC2Ty successfully trap the electrons and inhibit the recombination of electron–hole pairs. The metallic Au nanoparticles transmit electrons to the ultrathin layered MXene for the HER. The transient photocurrent response (TPR) and EIS measurements further confirmed the rapid charge transfer and efficient charge separation in the Ti3−xC2Ty/CdS/Au system. With 1.0 wt% Ti3−xC2Ty and 0.1 wt% Au in CdS, the ternary composites achieved an H2 production rate of 5371 μmol g−1 h−1 under visible-light irradiation in lactic acid aqueous solution, which is approximately 26.6 times higher than that of pristine CdS. Another ternary composite, CdS–MoS2–MXene (Ti3C2Tx),235 also exhibited a prominent H2 production rate of 9679 μmol g−1 h−1 under visible light (λ ≥ 420 nm) irradiation, which is much higher than those of the bare components and a physical mixture of the three components as shown in Fig. 11f. Due to the high carrier mobility of MXenes, the effective transmission of electrons to MoS2 and CdS for HER reactions can be realized. MXene materials significantly facilitate the separation of photoexcited electron–hole pairs and the subsequent HER reactions.
Ti3C2 MXene materials can maintain their layered structure after a facile annealing process but also provide Ti to form TiO2 as a new platform for photocatalytic reactions. TiO2 originated from Ti3C2 MXene in close contact with metallic Ti3C2, which benefits the rapid transfer of photogenerated charges. By introducing annealed Ti3C2Tx MXenes over water-stable Zr-MOFs (UiO-66-NH2), the Ti3C2/TiO2/UiO-66-NH2 ternary heterostructure was obtained.236Fig. 11g presents the possible contact interfaces including Ti3C2/UiO-66-NH2 and Ti3C2/TiO2 Schottky junctions in Ti3C2/TiO2/UiO-66-NH2. The photogenerated electrons in the CB of UiO-66-NH2 and TiO2 can separately transfer to Ti3C2 following pathways I and II, respectively. Since the UiO-66-NH2 has a higher CB position than TiO2, photogenerated electrons in the CB of UiO-66-NH2 can migrate to the CB of TiO2 first and then to Ti3C2 following pathway III. Owing to the synergistic effects of Schottky junctions of Ti3C2/UiO-66-NH2, Ti3C2/TiO2 in Ti3C2/TiO2/UiO-66-NH2, the photocatalytic H2 performance of Ti3C2/TiO2/UiO-66-NH2 reached 1980 μmol h−1 g−1, which is 2.1 times higher than that of pristine UiO-66-NH2 under simulated sunlight irradiation. Huang et al.237 also in situ synthesized a Ti3C2 MXene embedded with TiO2 nanosheets (M@TiO2). By compositing M@TiO2 with ZnIn2S4 (ZIS), a type-II heterojunction of ZIS and TiO2 and a Schottky junction of ZIS and metallic Ti3C2 were formed as shown in Fig. 11h. The obtained Ti3C2@TiO2/ZIS ternary heterostructure exhibited superior visible light absorption, and charge separation and transfer. Therefore, Ti3C2@TiO2/ZIS achieved a high photocatalytic H2 production rate of 1185.8 μmol g−1 h−1, which is 9.1 and 4.6 times higher than those of M@TiO2 and pure ZIS, respectively.
Ti3C2Tx as a typical MXene material shows remarkable photocatalytic properties for H2 production when composited with other semiconductors. The MXene based heterostructures often exhibit rapid charge transfer and separation due to the function of the Schottky junction and synergy with the component cocatalysts.
3.6 Other 2D layered materials
Bismuth oxyhalides (BiOX, X = Cl, Br, and I) are also considered as superior photocatalysts owing to their layered structure and excellent physicochemical properties.239,240 However, most BiOX photocatalysts are applied in photocatalytic degradation of pollutants or water treatment. The positive CB positions of BiOX highly limit their applications for photocatalytic H2 production. By constructing heterostructures with semiconductors that have strong reduction power, effective photocatalytic H2 production over BiOX can be realized. Liu et al.241 synthesized novel SnO/BiOX bilayer heterojunctions with narrow band gaps and type-II band alignment. The HSE06 hybrid DFT results confirm that the band structures of SnO/BiOCl and SnO/BiOBr bilayers are desirable for the redox water-splitting reactions as shown in Fig. 12a. The type-II heterojunctions with an IEF facilitate charge separation as illustrated in Fig. 12b. SnO/BiOCl and SnO/BiOBr heterostructures with strong optical absorption of solar light, engineered band gaps, and improved charge transfer exhibit remarkably enhanced photocatalytic activity for water splitting.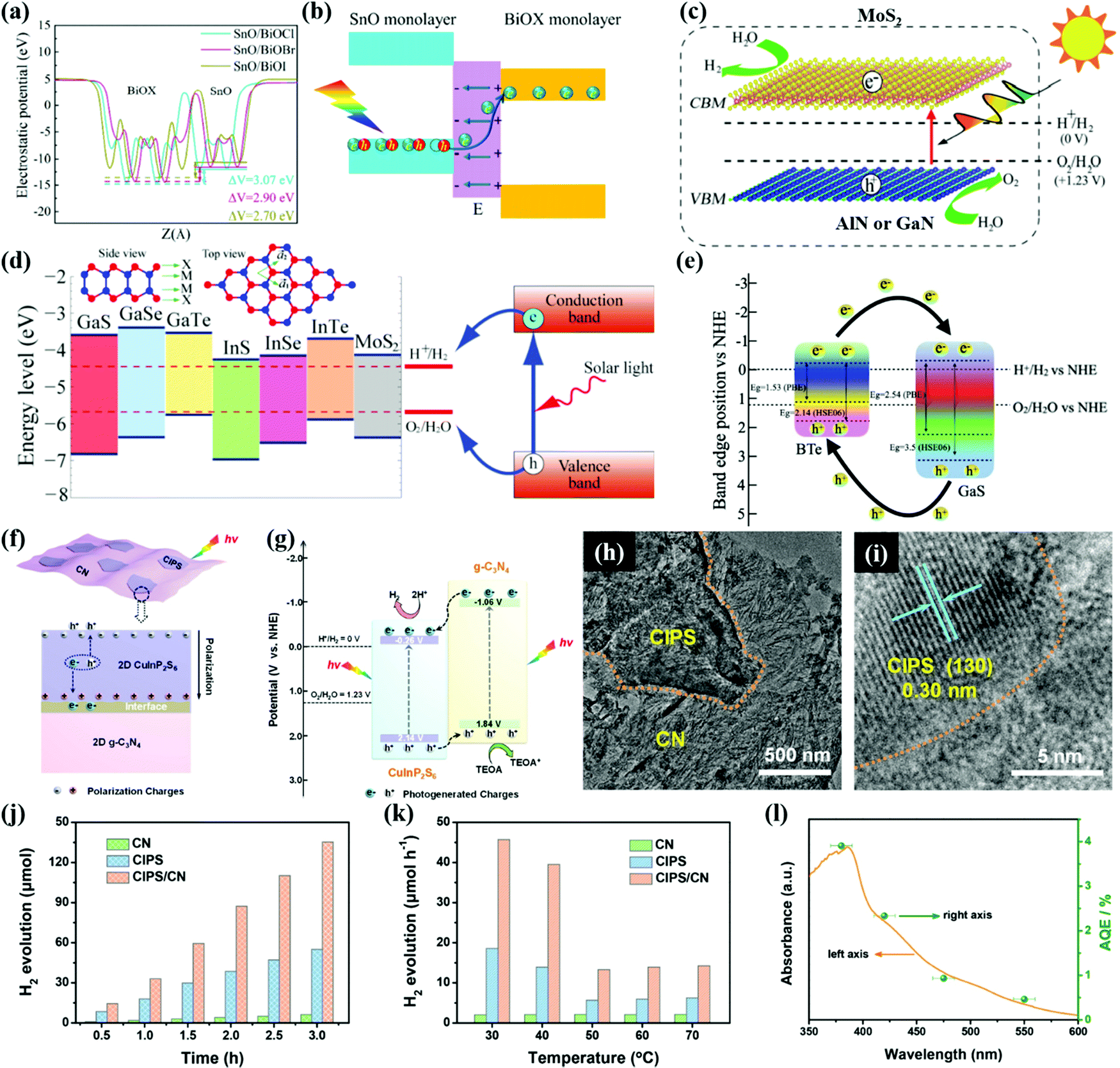 | ||
| Fig. 12 (a) Calculated electrostatic potential and (b) the diagrammatic view illustrating the photoexcitation process under a built-in electric field of the SnO/BiOX bilayer. Reprinted with permission.241 Copyright 2020, American Chemical Society. (c) Schematic illustration for MoS2/AlN(GaN) vdW heterostructures. Reproduced with permission.245 Copyright 2014, American Chemical Society. (d) Band edge positions of single-layer MX relative to the vacuum level at zero strain calculated with the HSE06 functional. The band edge positions of single-layer MoS2 and the standard redox potentials for water splitting at pH 0 are shown for comparison. Reprinted with permission.49 Copyright 2013, American Chemical Society. (e) Heterojunction charge transfer mechanism of the GaS/BTe vdW heterostructure. Reprinted with permission.246 Copyright 2021, Elsevier. Schematic illustration of the transfer of photogenerated charges in (f) the interior of 2D ferroelectric CuInP2S6 and (g) the 2D/2D CuInP2S6/CN heterojunction system for photocatalytic H2 evolution, (h and i) TEM images of CIPS/CN, (j) time-dependent photocatalytic H2 evolution and (k) temperature-dependent photocatalytic H2 evolution rates of different samples under visible-light irradiation (λ > 420 nm). (l) Wavelength-dependent AQE for photocatalytic H2 evolution over CIPS/CN. Reprinted with permission.248 Copyright 2020, Elsevier. | ||
Many new classes of 2D materials have emerged, such as group III nitrides (AlN and GaN), group-III metal monochalcogenides, MX (M = Ga, In, X = S, Se), and metal phosphorus trichalcogenides (MPTs) with large potential application in solar energy conversion.242–244 For instance, AlN and GaN have hexagonal crystal structures and only 2% lattice mismatch with MoS2. AlN or GaN and MoS2 could form intimate heterostructures as reported by Liao et al.245 The band structure characterization determines that MoS2/AlN and MoS2/GaN vdW heterostructures have narrow band gaps of 1.62 and 1.52 eV, respectively. And the CBM originates from the Mo dz2 states in the MoS2 monolayer, which is more negative than the water reduction potential as shown in Fig. 12c. The VBM results from the N pz states in the AlN(GaN) monolayer are more positive than the water oxidation potential. Therefore, both the optical absorption and water splitting reactions can be facilitated in the AlN(GaN)/MoS2 heterostructure.
As illustrated in Fig. 12d, single-layer group-III monochalcogenides all have low formation energies like MoS2, which are suitable for photocatalytic water splitting reactions.49 Singh et al.246 reported a GaS/BTe type-II vdW heterostructure with a large electric field at the heterointerface. The existence of the transverse electric field from 1.0 to 1.0 eV Å−1 effectively achieves the band alignment of the heterostructure and improves optical absorption in the visible light region. Fig. 12e presents the charge transfer process in the heterostructure. The photogenerated electrons in the CB of BTe migrate to the CB of the GaS, while the holes in the VB of GaS transfer to the VB of BTe. The efficient charge transfer route in the GaS/BTe heterojunction is beneficial to the HER. Thus, the GaS/BTe heterostructure could exhibit better photocatalytic performance for H2 production than the individual components.
MPTs include MIIPX3 such as FePS3, NiPS3, and CoPSe3 and MIMIIIP2X6 (X represents S and Se) such as CuInP2S6, AgAlP2Se6, and AgInP2S6,242,247 which are known as ferroelectric materials with hexagonal lamellar structures and spontaneous electric polarization. Huang et al.244 demonstrated that ferroelectric vdW heterostructures can result in type-II band alignment by switching the direction of polarization via an electric field. Both visible light absorption and charge separation can be improved in CuInP2S6 (CIPS)/Mn2P2S6 and CuInP2S6/Zn2P2Se6 ferroelectric heterostructures. The inherent polarization electric field of MPTs can promote fast transfer and spatial separation of charge carriers, which are promising virtues for photocatalytic water splitting. A CIPS/CN type-II heterojunction was constructed as a photocatalyst for H2 production (see Fig. 12f and g). The unique 2D/2D CIPS/CN heterojunction with CuInP2S6 nanosheets loaded onto the surfaces of the CN ultrathin flakes as shown in Fig. 12h and i is beneficial to the surface mass transport and charge transfer of the composite system. Owing to the synergy of the IEF and type-II band alignment, the H2 evolution rate of CIPS/CN (451.0 μmol h−1 g−1) was 2.5 and 21.5 times higher than those of individual CIPS and CN, respectively, as exhibited in Fig. 12j. As the temperature increases, the difference of H2 evolution rate between CIPS/CN and its components decreases (see Fig. 12k) due to the loss of ferroelectricity. Furthermore, the wavelength-dependent apparent quantum efficiency (AQE) for H2 evolution over CIPS/CN as shown in Fig. 12l can reach 2.35% at 420 nm, surpassing those of many CN heterojunctions. Apparently, the ferroelectric effect of CIPS plays an important role in the photocatalytic H2 evolution.
Based on the above, 2D materials can form various heterostructures, which greatly facilitate the photocatalytic reactions for water splitting. Thanks to the tunable electronic structures of 2D materials, different types of heterojunctions can be constructed based on the same materials. For instance, layered BP and CN often form a nested type I structure owing to their band structures.249 However, BP quantum dots (BPQDs) exhibit a CBM higher than that of CN due to the quantum confinement effect.250 Thus, the BPQDs form a type-II heterostructure with CN. In the heterostructure of BP and Bi2WO6, the electrons in the CB of Bi2WO6 combine with the holes in the VB of BP, forming a Z-scheme heterojunction. However, the n-type Bi2WO6 shows a lower Fermi level near its CB, while BP shows a higher Fermi level near its VB. Once these two EF levels reach the equilibrium level, an IEF is formed and the Z-scheme is converted to the S-scheme.251 The photocatalytic H2 production activity of various 2D material based heterostructures is summarized in Table 1.
| Photocatalysts | Morphology | Heterojunction type | Light source and intensity | Electrolyte or scavenger | Published year | H2 evolution rate | Ref. |
|---|---|---|---|---|---|---|---|
| BP/CN | 2D/2D | Type-I | Xe lamp (λ ≥ 420 nm)/300 mW cm−2 | Methanol | 2017 | 1.3 mmol h−1 g−1 | 100 |
| BP/CdS | 2D/0D | Type-II | Xe lamp (λ ≥ 420 nm)/80 mW cm−2 | Lactic acid | 2017 | 11.2 mmol h−1 g−1 | 111 |
| BPQDs/CN | 0D/2D | Type-II | λ ≥ 420 nm | Methanol | 2018 | 271.0 μmol h−1 g−1 | 250 |
| BP/BiVO4 | 2D/1.5D | Z-scheme | Xe lamp (λ ≥ 420 nm)/400 mW cm−2 | Ethylenediaminetetraacetic acid (EDTA) | 2018 | 160.0 μmol h−1 g−1 | 29 |
| BP/Au/CdS | 2D/3D | Schottky junction | Xe lamp (full solar spectrum)/300 mW cm−2 | Na2SO3/Na2S | 2018 | 8.6 mmol h−1 g−1 | 97 |
| BP/CoP | 0D/2D | - | Xe lamp (λ ≥ 420 nm) | Oxalic acid | 2018 | 694 μmol h−1 g−1 | 252 |
| BP/TiO2 | 2D/3D | Z-scheme | Sunlight lamp/100 mW cm−2 | Chloroplatinic acid | 2018 | 2.0 mmol h−1 g−1 | 87 |
| BP-MoS2/CdS | 3D/2D/2D | — | Solar simulator/100 mW cm−2 | Lactic acid | 2019 | 183.2 mmol h−1 g−1 | 98 |
| MoS2-BP/GO | 2D/2D/3D | — | Xe lamp (λ ≥ 420 nm) | Methanol | 2019 | 8.7 mmol h−1 g−1 | 253 |
| BP/Bi2WO6 | 2D/2D | Z-scheme | Xe lamp (λ ≥ 420 nm) | TEOA | 2019 | 4.2 mmol h−1 g−1 | 115 |
| BP/CN | 2D/2D | Type-I | Xe lamp (λ ≥ 420 nm) | TEOA | 2019 | 384.2 μmol h−1 g−1 | 102 |
| BP/Co2P | 2D/3D | - | Xe lamp (λ ≥ 420 nm) | Na2SO3/Na2S | 2019 | 1.2 mmol h−1 g−1 | 254 |
| BP/RP | 2D/2D | Z-scheme | LED light source (λ ≥ 420 nm) | Pure water | 2019 | 3.0 mmol h−1 g−1 | 255 |
| BP/MoS2 | 2D/2D | Type-II | Xe lamp (λ ≥ 420 nm)/150 mW cm−2 | Na2SO3/Na2S | 2021 | 575.4 μmol h−1 g−1 | 256 |
| CN/CoTiO3 | 2D/3D | Z-scheme | Xe lamp (simulated solar light)/190 mW cm−2 | Ethanol solution | 2016 | 858.0 μmol h−1 g−1 | 149 |
| Ni(OH)2-CdS/CN | Core–shell | — | Xe lamp (λ ≥ 420 nm) | Na2SO3/Na2S | 2016 | 115.2 mmol h−1 g−1 | 257 |
| CN/MoS2/TiO2 | 2D/3D | — | 300W Xe arc lamp (λ ≥ 400 nm) | Methanol | 2016 | 1.3 mmol h−1 g−1 | 258 |
| CN/CuS/CdS | 2D/3D | p–n junctions | Xe arc lamp (λ ≥ 400 nm)/70 mW cm−2 | Na2SO3/Na2S | 2017 | 1.2 mmol h−1 g−1 | 259 |
| CN/Ni2P-Cd0.9Zn0.1S | 2D/1D | Type-II | Xe lamp (λ ≥ 420 nm) | TEOA | 2017 | 2.1 mmol h−1 g−1 | 140 |
| CN/PSi | 2D/3D | Z-scheme | Xe lamp (λ ≥ 400 nm) 100 mW cm−2 | TEOA | 2017 | 870.4 μmol h−1 g−1 | 260 |
| CN/rGO | 2D/2D | - | Xe lamp (λ > 420 nm) | TEOA | 2018 | 715.0 μmol h−1 g−1 | 261 |
| CN/Ni2P/ZrO2 | 2D/3D | Type-II | Xe lamp (λ ≥ 420 nm) | TEOA | 2018 | 10.0 mmol h−1 g−1 | 262 |
| CN/phosphorene | 2D/2D | Type-I | Xe lamp (λ ≥ 400 nm) | Lactic acid | 2018 | 571.0 μmol h−1 g−1 | 101 |
| CN/TiO2/NixP | 2D/3D | Type-II | Xe lamp (λ ≥ 420 nm)/57.1 mW cm−2 | TEOA | 2018 | 5.4 mmol h−1 g−1 | 263 |
| CN/WO3 | 2D/1.5D | S-scheme | Xe lamp (full solar spectrum) | Lactic acid | 2019 | 982.0 μmol h−1 g−1 | 153 |
| CN/MoS2 | 2D/2D | Type-I | Xe lamp (λ ≥ 420 nm) | TEOA | 2019 | 1.2 mmol h−1 g−1 | 120 |
| CN/S-PAN-2 | 2D/3D | — | Xe lamp (λ ≥ 420 nm) | TEOA | 2019 | 764.2 μmol h−1 g−1 | 264 |
| CN/CdS | 2D/3D | Type-II | Xe arc lamp (λ ≥ 400 nm) | TEOA | 2019 | 5.4 mmol h−1 g−1 | 265 |
| CN/ ZnxCd1−xIn2S4 | 2D/2D | Type-II | Xe lamp (λ ≥ 420 nm) | TEOA | 2019 | 170.3 μmol h−1 g−1 | 266 |
| RP/CN | 2D/2D | Type-II | Xe lamp (full arc irradiation) | Pure water | 2020 | 367.0 μmol h−1 g−1 | 267 |
| NiCoP/CN | 3D/2D | — | Xe lamp (λ ≥ 420 nm) | TEOA | 2020 | 1.4 mmol h−1 g−1 | 268 |
| CN/carbon dots/CoO | 2D/0D | Type-II | Xe lamp (λ ≥ 420 nm) | TEOA | 2020 | 987.4 μmol h−1 g−1 | 269 |
| CN/SiOC | 2D/3D | Type-II | Xe lamp (simulated sunlight) | TEOA | 2020 | 1.0 mmol h−1 g−1 | 270 |
| CN/CoFe2O4 | 2D/3D | Type-II | Direct sunlight | Methanol | 2020 | 2.7 mmol h−1 g−1 | 271 |
| CN/Mn0.5Cd0.5Se | 2D/3D | S-scheme | Xe lamp (full solar spectrum) | Na2SO3/Na2S | 2021 | 5.9 mmol h−1 g−1 | 272 |
| CN/SnO2 | 2D/0D | Type-II | Xe lamp (λ ≥ 400 nm) | TEOA | 2021 | 2.5 mmol h−1 g−1 | 273 |
| CN/ Cd4−xZnxIn16S35 | 2D/0D | Z-scheme | Xe lamp (full solar spectrum) | TEOA | 2021 | 288.0 μmol h−1 g−1 | 274 |
| CN/LaFeO3 | 2D/0D | Z-scheme | Xe lamp (λ ≥ 420 nm)/100 mW cm−2 | TEOA | 2021 | 1.3 mmol h−1 g−1 | 275 |
| MoS2/TiO2 | 2D/1D | — | Xe lamp (full solar spectrum) | Ethanol | 2016 | 4.3 mmol h−1 g−1 | 276 |
| MoS2/CdS | 2D/1D | — | Xe lamp (λ ≥ 420 nm)/100 mW cm−2 | Lactic acid | 2016 | 49.8 mmol h−1 g−1 | 277 |
| MoS2/CuInS2 | 2D/3D | — | Xe lamp (λ > 420 nm) | Na2SO3/Na2S | 2016 | 316.0 μmol h−1 g−1 | 278 |
| WS2/CdS | 2D/1D | — | Xe arc lamp (λ > 420 nm) | Na2SO3/Na2S | 2016 | 1.8 mmol h−1 g−1 | 279 |
| WS2/CdS | 2D/1D | — | Arc lamp (λ ≥ 400 nm) | Lactic acid | 2016 | 1.2 mmol h−1 g−1 | 280 |
| MoS2/CdS | 2D/1D | — | Xe lamp (λ > 400 nm) | Lactic acid | 2016 | 17.5 mmol h−1 g−1 | 281 |
| MoS2/Mo/CdS | 2D/3D | — | Xe lamp (λ ≥ 420 nm)/52.2 mW cm−2 | Na2SO3/Na2S | 2017 | 4.5 mmol h−1 g−1 | 282 |
| MoS2/rGO/CdS | 2D/3D | — | Xe lamp (λ ≥ 400 nm)/100 mW cm−2 | Lactic acid | 2017 | 5.3 mmol h−1 g−1 | 283 |
| WS2-MoS2/CdS | 2D/1D | — | AM 1.5G solar simulator/100 mW cm−2 | Lactic acid | 2017 | 209.8 mmol h−1 g−1 | 284 |
| MoS2/CdS | 2D/1D | — | Xe lamp (λ > 420 nm)/160 mW cm−2 | Na2SO3/Na2S | 2017 | 9.7 mmol h−1 g−1 | 285 |
| BiOI-MoS2/CdS | 2D/3D | — | Xe lamp (λ > 420 nm) | Lactic acid | 2017 | 9.7 mmol h−1 g−1 | 286 |
| MoS2/AgInZnS | 2D/3D | — | Xe lamp (λ ≥ 420 nm) | Lactic acid | 2017 | 19.9 mmol h−1 g−1 | 287 |
| MoSe2/MoS2 | 2D/2D | — | Xe lamp (λ > 420 nm) | TEOA | 2017 | 4.6 mmol h−1 g−1 | 288 |
| MoS2-CdS/RGO | 2D/3D | — | Xe arc lamp (λ ≥ 420 nm)/100 mW cm−2 | Lactic acid | 2018 | 36.7 mmol h−1 g−1 | 289 |
| MoS2/CdS | Core–shell | — | Xe lamp (λ > 420 nm) | Lactic acid | 2018 | 1.4 mmol h−1 g−1 | 290 |
| MoS2/ZnIn2S4 | 2D/3D | Type-I | Xe lamp (λ > 420 nm) | Na2SO3/Na2S | 2018 | 3.9 mmol h−1 g−1 | 291 |
| WS2/CN/CdS | 2D/3D | Type-II | Xe lamp (λ > 420 nm) | TEOA | 2018 | 1.2 mmol h−1 g−1 | 292 |
| WS2/CdS | 2D/3D | — | Xe lamp (λ > 400 nm) | Lactic acid | 2018 | 14.1 mmol h−1 g−1 | 293 |
| WS2/ZnIn2S4 | 2D/3D | — | Xe lamp (λ ≥ 420 nm)/15 mW cm−2 | Na2SO3/Na2S | 2018 | 199.1 μmol h−1 g−1 | 294 |
| MoS2/Mn0.2Cd0.8S/MnS | 2D/3D | Type-II | Xe lamp (λ ≥ 420 nm) | Na2SO3/Na2S | 2019 | 19.9 mmol h−1 g−1 | 295 |
| MoS2/PyP | 2D/3D | Type-II | Xe lamp (λ ≥ 420 nm) | Methanol | 2019 | 540 μmol h−1 g−1 | 296 |
| MoS2/CdS | 2D/1D | — | Xe lamp (λ ≥ 420 nm) | Lactic acid | 2019 | 3.1 mmol h−1 g−1 | 297 |
| Au-MoS2/ZnIn2S4 | 2D/3D | P−N heterojunction | Xe arc lamp (λ > 400 nm) | Na2SO3/Na2S | 2019 | 2.8 mmol h−1 g−1 | 298 |
| MoS2/ZnO | 2D/1D | — | Natural solar light | Na2SO4/Na2S | 2019 | 17.3 mmol h−1 g−1 | 299 |
| In2S3-MoS2/MnS | 2D/1D | — | Xe lamp (λ > 400 nm) | Na2SO3/Na2S | 2019 | 49.6 mmol h−1 g−1 | 300 |
| MoS2/SnNb2O6 | 2D/2D | — | Xe lamp (λ ≥ 420 nm)/180 mW cm−2 | Methanol | 2019 | 322.5 μmol h−1 g−1 | 301 |
| MoS2/Mn0.2Cd0.8S/Co3O4 | 2D/3D | Type-II | Xe lamp (λ ≥ 420 nm) | Na2SO3/Na2S | 2020 | 16.5 mmol h−1 g−1 | 302 |
| MoS2/Mn0.2Cd0.8S | 2D/3D | — | Xe lamp (λ ≥ 420 nm) | Na2SO3/Na2S | 2020 | 2 mmol h−1 g−1 | 303 |
| MoS2/LiNb3O8 | 2D/1D | — | Hg lamp (185–500 nm) | Na2SO3/Na2S | 2020 | 900.0 μmol h−1 g−1 | 304 |
| MoS2/Ti3C2 | 2D/2D | — | Xe lamp (λ > 400 nm) | Methanol | 2020 | 6.1 mmol h−1 g−1 | 228 |
| WS2/WO3/CdS/ | 2D/0D | Z-scheme | Xe lamp (λ > 420 nm) | Lactic acid | 2020 | 14.3 mmol h−1 g−1 | 189 |
| WS2/ZnIn2S4 | 2D/2D | — | Xe lamp (λ ≥ 420 nm) | Lactic acid | 2020 | 2.6 mmol h−1 g−1 | 305 |
| WS2/WO3 | 2D/1.5D | Z-scheme | Xe lamp (full solar spectrum) | Lactic acid | 2020 | 680.0 μmol h−1 g−1 | 306 |
| WS2/CdS | 2D/1D | Type-I | Xe arc lamp (λ > 395 nm) | Lactic acid | 2020 | 19.2 mmol h−1 g−1 | 307 |
| MoS2/CdS | 2D/1D | Type-I | Xe arc lamp (λ > 395 nm) | Lactic acid | 2020 | 9.7 mmol h−1 g−1 | 307 |
| MoS2/Ag–AgVO3 | 2D/3D | Z-scheme | 250W Xe-arc lamp (λ > 420 nm) | 25 vol% methanol | 2020 | 19.3 mmol h−1 g−1 | 308 |
| N-MoS2/MoO3 | 2D/3D | Z-scheme | 400 W Xenon lamp (λ > 420 nm) | — | 2021 | 118 μ mol h−1g−1 | 309 |
| P-MoS2/WO3 | 2D/3D | Z-scheme | 300 W simulated solar light source | Na2SO3/Na2S | 2021 | 73.8 μ mol h−1g−1 | 310 |
| MoS2/MAPbI3 | 2D/3D | Type-II | Xe lamp (λ ≥ 420 nm) | Ethyl acetate | 2021 | 30.0 mmol h−1 g−1 | 311 |
| MoS2/CdS | 2D/3D | — | Xe lamp (λ > 420 nm) | Na2SO3/Na2S | 2021 | 34.1 mmol h−1 g−1 | 312 |
| WS2-WO3·H2O/CN | 2D/3D | Z-scheme | Xe lamp (λ ≥ 420 nm) | Lactic acid | 2021 | 1.3 mmol h−1 g−1 | 313 |
| MoS2/CdS | 2D/3D | — | Xe lamp (λ ≥ 420 nm) | Lactic acid | 2021 | 54.1 mmol h−1 g−1 | 314 |
| WS2/MoS2 | 2D/2D | Type-II | Xe lamp (full solar spectrum) | Ethanol | 2021 | 9.8 mmol h−1 g−1 | 190 |
| MgAl-LDH/CeO2 | 2D/3D | — | Hg lamp (λ ≥ 420 nm) | Methanol | 2016 | 16.5 mmol h−1 g−1 | 315 |
| ZnCr-LDH/CN | 2D/2D | Type-II | Xe lamp (λ ≥ 420 nm) | TEOA | 2017 | 680.0 μmol h−1 g−1 | 316 |
| NiFe-LDH/rGO/La2Ti2O7 | 2D/2D | — | AM 1.5/100 mW cm−2 | TEOA | 2018 | 532.2 μmol h−1 g−1 | 317 |
| NiFe-LDH/CdS | 2D/3D | — | Simulated solar light | Methanol | 2018 | 469.0 μmol h−1 g−1 | 199 |
| ZnCr-LDH/Co(OH)2 | 2D/2D | P–N heterojunction | Hg lamp (λ > 400 nm) | Methanol | 2018 | 1.1 mmol h−1 g−1 | 219 |
| MgCr-LDH/MgO/MgCr2O4 | 2D/3D | — | Hg lamp (λ > 400 nm)/100 mW cm−2 | Methanol | 2018 | 420.0 μmol h−1 g−1 | 318 |
| NiFe-LDH/MoS2 | 2D/2D | Z-scheme | Hg lamp (λ > 400 nm)/100 mW cm−2 | Methanol | 2019 | 18.4 mmol h−1 g−1 | 218 |
| ZnTi-LDH/CN | 2D/2D | — | AM1.5G | Methanol | 2019 | 161.9 μmol h−1 g−1 | 319 |
| CoAl-LDH/CN | 2D/2D | Type-II | Xe lamp (λ ≥ 420 nm) | TEOA | 2020 | 680.1 μmol h−1 g−1 | 202 |
| CoAl-LDH/ZnxCd1-xS | 2D/2D | — | LED white light (full solar spectrum) | Lactic acid | 2020 | 30.3 mmol h−1 g−1 | 320 |
| NiAl-LDH/CN | 2D/2D | — | Quartz tungsten halogen lamp (simulated solar light)/300 mW cm−2 | TEOA | 2020 | 3.2 mmol h−1 g−1 | 321 |
| NiFe-LDH/CN | 2D/2D | Type-II | AM 1.5 | TEOA | 2021 | 3.1 mmol h−1 g−1 | 322 |
| NiFe-LDH/Zn0.5Cd0.5S | 2D/3D | — | Xe lamp (λ ≥ 420 nm) | Lactic acid | 2021 | 1.0 mmol h−1 g−1 | 323 |
| NiCo-LDH/NiCoP/Cu3P | 2D/3D | P-N heterojunction | λ > 420 nm | TEOA | 2021 | 8.9 mmol h−1 g−1 | 216 |
| NiCo-LDH/CoO | 2D/1D | Type-II | AM 1.5/100 mW cm−2 | Na2SO3/Na2S | 2021 | 1.5 mmol h−1 g−1 | 203 |
| NFe-LDH/ZnIn2S4 | 2D/1D | Type-II | Xe lamp (λ ≥ 420 nm) | TEOA | 2021 | 2.0 mmol h−1 g−1 | 324 |
| NiCo-LDH/RP | 2D/3D | S-scheme | Visible light | TEOA | 2021 | 7.4 mmol h−1 g−1 | 325 |
| NiAl-LDH/NiP2/Cu3P | 2D/3D | — | λ ≥ 420 nm | TEOA | 2021 | 6.8 mmol h−1 g−1 | 326 |
| Ti3C2Tx/TiO2 | 2D/3D | Schottky junction | Hg lamp (λ > 400 nm) | Methanol | 2016 | 17.8 μmol h−1 g−1 | 231 |
| Ti3C2 MXene/CN | 2D/2D | — | Xe lamp (simulated solar light) | TEOA | 2018 | 5.1 mmol h−1 g−1 | 327 |
| Ti3C2Tx/TiO2/Cu2O(001) | 2D/3D | — | Xe lamp (simulated solar light) | Methanol | 2019 | 1.5 mmol h−1 g−1 | 328 |
| Ti3C2Tx MXenes/UiO-66-NH2 | 2D/3D | Schottky junction | Xe lamp (350 < λ < 780 nm) | Na2SO3/Na2S | 2019 | 2.0 mmol h−1 g−1 | 236 |
| Ti3C2 MXene/MoS2/TiO2 | 2D/2D/1.5D | — | Xe lamp/AM1.5 | Acetone | 2019 | 5.1 mmol h−1 g−1 | 329 |
| Ti3C2 MXene/CdS | 2D/1D | Schottky junction | Xe lamp (λ ≥ 420 nm) | Lactic acid | 2020 | 2.4 mmol h−1 g−1 | 330 |
| Ti3C2 MXene/TiO2/ZnIn2S4 | 2D/2D | — | Xe lamp (full solar spectrum) | Na2SO3/Na2S | 2020 | 1.2 mmol h−1 g−1 | 237 |
| Ti3C2Tx MXene/CdS | 2D/1D | — | Xe lamp (λ ≥ 420 nm) | Methanol | 2020 | 15.4 mmol h−1 g−1 | 232 |
| Ti3-xC2Ty/CdS/Au | 2D/3D | Schottky junction | LED lamp (λ ≥ 420 nm) | Lactic acid | 2021 | 5.4 mmol h−1 g−1 | 234 |
| Ti3C2Tx MXene/CdS | 2D/1.5D | Schottky junction | Xe lamp (λ ≥ 420 nm) | Lactic acid | 2021 | 3.2 mmol h−1 g−1 | 230 |
| BiOCl-PbS | 2D/3D | Type-II | Xe lamp (full solar spectrum) | Na2SO3/Na2S | 2019 | 16.0 μmol h−1 g−1 | 331 |
| CuInP2S6/CN | 2D/2D | Type-II | Xe lamp (λ ≥ 420 nm) | TEOA | 2020 | 451.0 μmol h−1 g−1 | 248 |
4 Summary and prospects
In summary, the very recent efforts on the design and construction of various 2D material-based heterostructures, their photocatalytic performances for H2 production under solar light and the action mechanisms of different heterojunctions are reviewed. The band alignments in different types of heterojunctions, especially type-II, Z-scheme and S-scheme, successfully promote visible light absorption and charges separation in the water splitting reaction. As illustrated in a large number of studies, 2D material-based heterostructures have excellent photocatalytic H2 production rates owing to the heterojunction configuration and synergy of the components in the heterostructures.However, challenges remain which hinder the practical application of 2D material-based photocatalytic systems for H2 production from water splitting. (1) The structure and morphology of 2D hetero-materials usually have a significant effect on the photocatalytic performance.26,332 Nevertheless, controllable, large-scale, economical and efficient synthesis approaches are still waiting to be developed. The band structure and surface states of 2D semiconductors can be modulated by metal or non-metal doping, optimizing the charge transfer route at the interface of the heterojunction.333 The doping in 2D material based heterostructures with controllable dopant distribution and composition should be more explored for efficient photocatalyst design. (2) The aggregation of 2D photocatalyst particles in a liquid photocatalytic system will decrease the surface area and active sites of photocatalysts, resulting in weakened photocatalytic activity and stability.39,40 Except for the self-assembly or in situ integration of 2D thin materials with substrates, other functional and effective optimization approaches should be investigated. (3) It is still difficult to track the migration behaviors of the electrons or holes and intermediates during photocatalytic reactions. The investigation of the main bottleneck in photocatalytic reactions by catching the intrinsic features and charge transport pathway will offer superior guidance for designing highly efficient 2D material-based heterojunctions.334,335 Thus, more advanced in situ or time-resolved characterization techniques should be developed and employed in the research of photocatalytic reactions over 2D material based photocatalysts. (4) The theoretical calculation can help in deconstructing the electronic structure of 2D materials in more depth and predict the electronic properties of 2D material-based heterojunctions.336–339 Thus, theoretical simulations should be more employed to understand the mechanism behind the chemical reactions and predict novel functional photocatalysts.
Overall, 2D material-based photocatalysts are highly promising in H2 production from water splitting under solar light to solve the energy crisis and environmental issues. The architectures of 2D material-based heterostructures with fine-tuned compositions, structures and interfacial reactions sites greatly promote light harvesting, charge separation and transfer, and the resulting photocatalytic performances. To realize optimized 2D photocatalysts, the electronic structure, surface states, energy band alignments with other materials, etc., should be comprehensively considered based on both in-depth experimental and theoretical research studies, in which more signs of progress are yet to come. Ideal 2D based photocatalysts consist of two semiconductors with sufficient oxidative and reductive band edges and couple with intercalated atoms and adjustors for directional charge flow. Among various 2D material based heterostructures, CN and TMD based heterostructures are the most promising photocatalysts owing to their advantages, including large-scale synthesis and high efficiency for photocatalytic water splitting. On the other hand, the application of BP for H2 production from water splitting is also promising thanks to its thickness dependent band structure. However, the investigation on BP as a photocatalyst is still in its infancy with challenges to be overcome, such as the stability issue.
We believe that the present report will encourage the photocatalytic community to gain a depth understanding of the field and excel in the knowledge to develop resilient and sustainable 2D-material photocatalysts for future applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 12074265), the Shenzhen Science and Technology Project (Grant No. JCYJ20180507182246321, JCYJ20210324095611032), and the Natural Science Foundation of Guangdong Province.References
- T. H. Oh, M. Hasanuzzaman, J. Selvaraj, S. C. Teo and S. C. J. R. Chua, Renewable Sustainable Energy Rev., 2018, 81, 3021–3031 CrossRef.
- N. A. Ludin, N. I. Mustafa, M. M. Hanafiah, M. A. Ibrahim, M. A. M. Teridi, S. Sepeai, A. Zaharim and K. J. R. Sopian, Renewable Sustainable Energy Rev., 2018, 96, 11–28 CrossRef.
- E. Kabir, P. Kumar, S. Kumar, A. A. Adelodun and K.-H. J. R. Kim, Renewable Sustainable Energy Rev., 2018, 82, 894–900 CrossRef.
- K. Khanafer and K. J. R. E. Vafai, Renewable Energy, 2018, 123, 398–406 CrossRef CAS.
- A. Fujishima and K. J. n. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- A. Kudo and Y. J. C. S. R. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- X. Chen, C. Li, M. Grätzel, R. Kostecki and S. S. J. C. S. R. Mao, Chem. Soc. Rev., 2012, 41, 7909–7937 RSC.
- T. Hisatomi, J. Kubota and K. J. C. S. R. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- J. Ran, J. Zhang, J. Yu, M. Jaroniec and S. Z. J. C. S. R. Qiao, Chem. Soc. Rev., 2014, 43, 7787–7812 RSC.
- K. J. J. o. P. Maeda, J. Photochem. Photobiol. C: Photochem. Rev., 2011, 12, 237–268 CrossRef CAS.
- S. Chen, T. Takata and K. J. N. R. M. Domen, Nat. Rev. Mater., 2017, 2, 1–17 Search PubMed.
- H. Wang, L. Zhang, Z. Chen, J. Hu, S. Li, Z. Wang, J. Liu and X. Wang, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC.
- X. Li, R. Shen, S. Ma, X. Chen and J. Xie, Appl. Surf. Sci., 2018, 430, 53–107 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. J. S. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- J. C. Garcia, D. B. De Lima, L. V. Assali and J. F. Justo, J. Phys. Chem. C, 2011, 115, 13242–13246 CrossRef CAS.
- B. Luo, G. Liu and L. J. N. Wang, Nanoscale, 2016, 8, 6904–6920 RSC.
- A. Zhao, H. Li, X. Hu, C. Wang, H. Zhang, J. Lu, S. Ruan and Y.-J. Zeng, J. Phys. D: Appl. Phys., 2020, 53, 293002 CrossRef CAS.
- A. Zhao, L. Zhang, Y. Guo, H. Li, S. Ruan and Y.-J. Zeng, 2D Mater., 2021, 8, 012004 CrossRef CAS.
- C. Tan, X. Cao, X.-J. Wu, Q. He, J. Yang, X. Zhang, J. Chen, W. Zhao, S. Han and G.-H. J. C. R. Nam, Chem. Rev., 2017, 117, 6225–6331 CrossRef CAS.
- D. Deng, K. Novoselov, Q. Fu, N. Zheng, Z. Tian and X. J. N. N. Bao, Nat. Nanotechnol., 2016, 11, 218 CrossRef CAS.
- M.-Y. Li, C.-H. Chen, Y. Shi and L.-J. J. M. T. Li, Mater. Today, 2016, 19, 322–335 CrossRef CAS.
- X. Li, Y. Dai, M. Li, W. Wei and B. Huang, J. Mater. Chem. A, 2015, 3, 24055–24063 RSC.
- X. Lv, W. Wei, Q. Sun, F. Li, B. Huang and Y. J. A. C. B. E. Dai, Appl. Catal., B, 2017, 217, 275–284 CrossRef CAS.
- Z. Guo, J. Zhou, L. Zhu and Z. J. J. o. M. C. A. Sun, J. Mater. Chem. A, 2016, 4, 11446–11452 RSC.
- J. Low, S. Cao, J. Yu and S. Wageh, Chem. Commun., 2014, 50, 10768–10777 RSC.
- T. A. Shifa, F. Wang, Y. Liu and J. He, Adv. Mater., 2019, 31, 1804828 CrossRef CAS.
- B. Song and S. Jin, Joule, 2017, 1, 220–221 CrossRef.
- D. Voiry, J. Yang and M. Chhowalla, Adv. Mater., 2016, 28, 6197–6206 CrossRef CAS.
- M. Zhu, Z. Sun, M. Fujitsuka and T. Majima, Angew. Chem., Int. Ed., 2018, 57, 2160–2164 CrossRef CAS.
- J. Yan, P. Verma, Y. Kuwahara, K. Mori and H. Yamashita, Small, 2018, 2, 1800212 CrossRef.
- Z.-K. Shen, Y.-J. Yuan, L. Pei, Z.-T. Yu and Z. Zou, Chem. Eng. J., 2020, 386, 123997 CrossRef CAS.
- T. H. Lee, S. Y. Kim and H. W. Jang, Nanomaterials, 2016, 6, 194 CrossRef PubMed.
- A. Mishra, A. Mehta, S. Basu, N. P. Shetti, K. R. Reddy and T. M. Aminabhavi, Carbon, 2019, 149, 693–721 CrossRef CAS.
- M. Ismael, J. Alloys Compd., 2020, 846, 156446 CrossRef CAS.
- M. Tekalgne, A. Hasani, Q. Van Le and S. Y. Kim, Funct. Compos. Struct., 2019, 1, 012001 CrossRef CAS.
- Q. Lu, Y. Yu, Q. Ma, B. Chen and H. Zhang, Adv. Mater., 2016, 28, 1917–1933 CrossRef CAS PubMed.
- V.-H. Nguyen, T. P. Nguyen, T.-H. Le, D.-V. N. Vo, D. L. Nguyen, Q. T. Trinh, I. T. Kim and Q. V. Le, J. Chem. Technol. Biotechnol., 2020, 95, 2597–2607 CAS.
- K. Ren, K. Wang, Y. Cheng, W. Tang and G. Zhang, Nano Futures, 2020, 4, 032006 CrossRef CAS.
- B. Luo, G. Liu and L. Wang, Nanoscale, 2016, 8, 6904–6920 RSC.
- P. Ganguly, M. Harb, Z. Cao, L. Cavallo, A. Breen, S. Dervin, D. D. Dionysiou and S. C. Pillai, ACS Energy Lett., 2019, 4, 1687–1709 CrossRef CAS.
- Q. Weng, X. Wang, X. Wang, Y. Bando and D. Golberg, Chem. Soc. Rev., 2016, 45, 3989–4012 RSC.
- L. H. Li and Y. Chen, Adv. Funct. Mater., 2016, 26, 2594–2608 CrossRef CAS.
- J. Zhang, Y. Chen and X. Wang, Energ. Environ. Sci., 2015, 8, 3092–3108 RSC.
- C. Zhi, Y. Bando, C. Tang, H. Kuwahara and D. Golberg, Adv. Mater., 2009, 21, 2889–2893 CrossRef CAS.
- Z. Jin, X. Jiang, Q. Zhang, S. Huang, L. Zhang, L. Huang, T. He, H. Zhang, T. Ohno, S. Ruan and Y.-J. Zeng, Commun. Mater., 2020, 1, 90 CrossRef.
- D. Voiry, A. Mohite and M. Chhowalla, Chem. Soc. Rev., 2015, 44, 2702–2712 RSC.
- W. Choi, N. Choudhary, G. H. Han, J. Park, D. Akinwande and Y. H. Lee, Mater. Today, 2017, 20, 116–130 CrossRef CAS.
- S. Demirci, N. Avazlı, E. Durgun and S. Cahangirov, Phys. Rev. B, 2017, 95, 115409 CrossRef.
- H. L. Zhuang and R. G. Hennig, Chem. Mater., 2013, 25, 3232–3238 CrossRef CAS.
- P. Li and I. Appelbaum, Phys. Rev. B, 2015, 92, 195129 CrossRef.
- X. Long, Z. Wang, S. Xiao, Y. An and S. Yang, Mater. Today, 2016, 19, 213–226 CrossRef CAS.
- L. Mohapatra and K. Parida, J. Mater. Chem. A, 2016, 4, 10744–10766 RSC.
- Y. Lee, J. H. Choi, H. J. Jeon, K. M. Choi, J. W. Lee and J. K. Kang, Energ. Environ. Sci., 2011, 4, 914–920 RSC.
- Y. Zhang, L. Zhang, L. Hu, S. Huang, Z. Jin, M. Zhang, X. Huang, J. Lu, S. Ruan and Y.-J. Zeng, Dalton Trans., 2019, 48, 426–434 RSC.
- L. Li, Y. Yu, G. J. Ye, Q. Ge, X. Ou, H. Wu, D. Feng, X. H. Chen and Y. Zhang, Nat. Nanotechnol., 2014, 9, 372 CrossRef CAS PubMed.
- H. Liu, Y. Du, Y. Deng and D. Y. Peide, Chem. Soc. Rev., 2015, 44, 2732–2743 RSC.
- L. Hu, J. Yuan, Y. Ren, Y. Wang, J.-Q. Yang, Y. Zhou, Y.-J. Zeng, S.-T. Han and S. Ruan, Adv. Mater., 2018, 30, 1801232 CrossRef PubMed.
- J. Ran, G. Gao, F.-T. Li, T.-Y. Ma, A. Du and S.-Z. Qiao, Nat. Commun., 2017, 8, 1–10 CrossRef PubMed.
- Y. Li, X. Deng, J. Tian, Z. Liang and H. Cui, Appl. Mater. Today, 2018, 13, 217–227 CrossRef.
- P. Vogt, P. De Padova, C. Quaresima, J. Avila, E. Frantzeskakis, M. C. Asensio, A. Resta, B. Ealet and G. Le Lay, Phys. Rev. Lett., 2012, 108, 155501 CrossRef PubMed.
- L. Tao, E. Cinquanta, D. Chiappe, C. Grazianetti, M. Fanciulli, M. Dubey, A. Molle and D. Akinwande, Nat. Nanotechnol., 2015, 10, 227–231 CrossRef CAS PubMed.
- Y. Peng, Y. Li, Y. Ban, H. Jin, W. Jiao, X. Liu and W. Yang, Science, 2014, 346, 1356–1359 CrossRef CAS PubMed.
- M. G. Campbell, S. F. Liu, T. M. Swager and M. Dincă, J. Am. Chem. Soc., 2015, 137, 13780–13783 Search PubMed.
- K. Zhao, S. Liu, G. Ye, Q. Gan, Z. Zhou and Z. He, J. Mater. Chem. A, 2018, 6, 2166–2175 RSC.
- J. W. Colson and W. R. Dichtel, Nat. Chem., 2013, 5, 453 CrossRef CAS PubMed.
- L. Wang, Y. Zhang, L. Chen, H. Xu and Y. Xiong, Adv. Mater., 2018, 30, 1801955 CrossRef PubMed.
- M. Zhang and X. Wang, Energ. Environ. Sci., 2014, 7, 1902–1906 RSC.
- S. J. A. Moniz, S. A. Shevlin, D. J. Martin, Z.-X. Guo and J. Tang, Energ. Environ. Sci., 2015, 8, 731–759 RSC.
- H. Uk Lee, S. C. Lee, J. Won, B.-C. Son, S. Choi, Y. Kim, S. Y. Park, H.-S. Kim, Y.-C. Lee and J. Lee, Sci. Rep., 2015, 5, 8691 CrossRef PubMed.
- Y. Liu, R. Cheng, L. Liao, H. Zhou, J. Bai, G. Liu, L. Liu, Y. Huang and X. Duan, Nat. Commun., 2011, 2, 579 CrossRef PubMed.
- C. R. Dean, A. F. Young, I. Meric, C. Lee, L. Wang, S. Sorgenfrei, K. Watanabe, T. Taniguchi, P. Kim, K. L. Shepard and J. Hone, Nat. Nanotechnol., 2010, 5, 722–726 CrossRef CAS PubMed.
- J. Zhang, H. Hong, J. Zhang, H. Fu, P. You, J. Lischner, K. Liu, E. Kaxiras and S. Meng, Nano Lett., 2018, 18, 6057–6063 CrossRef CAS PubMed.
- Z. Ji, H. Hong, J. Zhang, Q. Zhang, W. Huang, T. Cao, R. Qiao, C. Liu, J. Liang, C. Jin, L. Jiao, K. Shi, S. Meng and K. Liu, ACS Nano, 2017, 11, 12020–12026 CrossRef CAS PubMed.
- X. Niu, Y. Li, Y. Zhang, Q. Zheng, J. Zhao and J. Wang, J. Mater. Chem. C, 2019, 7, 1864–1870 RSC.
- K. Ren, J. Yu and W. Tang, J. Appl. Phys., 2019, 126, 065701 CrossRef.
- K. Ren, Y. Luo, J. Yu and W. Tang, Chem. Phys., 2020, 528, 110539 CrossRef CAS.
- Y. Liu, N. O. Weiss, X. Duan, H.-C. Cheng, Y. Huang and X. Duan, Nat. Rev. Mater., 2016, 1, 16042 CrossRef CAS.
- A. J. Bard, J. Photochem., 1979, 10, 59–75 CrossRef CAS.
- Y. Zou, J.-W. Shi, D. Ma, Z. Fan, L. Lu and C. Niu, Chem. Eng. J., 2017, 322, 435–444 CrossRef CAS.
- J.-W. Shi, Y. Zou, D. Ma, Z. Fan, L. Cheng, D. Sun, Z. Wang, C. Niu and L. Wang, Nanoscale, 2018, 10, 9292–9303 RSC.
- Y. Zou, J.-W. Shi, D. Ma, Z. Fan, C. Niu and L. Wang, ChemCatChem, 2017, 9, 3752–3761 CrossRef CAS.
- H. Kato, Y. Sasaki, N. Shirakura and A. Kudo, J. Mater. Chem. A, 2013, 1, 12327–12333 RSC.
- H. Tada, T. Mitsui, T. Kiyonaga, T. Akita and K. Tanaka, Nat. Mater., 2006, 5, 782–786 CrossRef CAS PubMed.
- X.-L. Yin, J. Liu, W.-J. Jiang, X. Zhang, J.-S. Hu and L.-J. Wan, Chem. Commun., 2015, 51, 13842–13845 RSC.
- K. Iwashina, A. Iwase, Y. H. Ng, R. Amal and A. Kudo, J. Am. Chem. Soc., 2015, 137, 604–607 CrossRef CAS PubMed.
- J. Yu, S. Wang, J. Low and W. Xiao, Phys. Chem. Chem. Phys., 2013, 15, 16883–16890 RSC.
- J. Wu, S. Huang, Z. Jin, J. Chen, L. Hu, Y. Long, J. Lu, S. Ruan and Y.-J. Zeng, J. Mater. Sci., 2018, 53, 16557–16566 CrossRef CAS.
- Q. Xu, L. Zhang, B. Cheng, J. Fan and J. Yu, Chem, 2020, 6, 1543–1559 CAS.
- J. Mu, F. Teng, H. Miao, Y. Wang and X. Hu, Appl. Surf. Sci., 2020, 501, 143974 CrossRef CAS.
- Y. Ren, D. Zeng and W.-J. Ong, Chinese J. Catal., 2019, 40, 289–319 CrossRef CAS.
- F. Xia, H. Wang and Y. Jia, Nat. Commun., 2014, 5, 4458 CrossRef CAS PubMed.
- J. Kim, S. S. Baik, S. H. Ryu, Y. Sohn, S. Park, B.-G. Park, J. Denlinger, Y. Yi, H. J. Choi and K. S. J. S. Kim, Science, 2015, 349, 723–726 CrossRef CAS PubMed.
- J. Qiao, X. Kong, Z.-X. Hu, F. Yang and W. Ji, Nat. Commun., 2014, 5, 4475 CrossRef CAS PubMed.
- P. Li, J. Lu, H. Cui, S. Ruan and Y.-J. Zeng, Mater. Adv., 2021, 2, 2483–2509 RSC.
- Y. Xu, X. Wang, M. Jin, K. Kempa and L. Shui, ChemElectroChem, 2020, 7, 96–104 CrossRef CAS.
- W. Gao, X. Bai, Y. Gao, J. Liu, H. He, Y. Yang, Q. Han, X. Wang, X. Wu, J. Wang, F. Fan, Y. Zhou, C. Li and Z. Zou, Chem. Commun., 2020, 56, 7777–7780 RSC.
- X. Cai, L. Mao, S. Yang, K. Han and J. Zhang, ACS Energy Lett., 2018, 3, 932–939 CrossRef CAS.
- D. A. Reddy, E. H. Kim, M. Gopannagari, Y. Kim, D. P. Kumar and T. K. Kim, Appl. Catal., B, 2019, 241, 491–498 CrossRef CAS.
- G. Wang, W. Zhao, M. Zhong, Y. Li, S. Xiao, S. Dang, C. Li, X. Long and W. Zhang, J. Phys.: Condens. Matter, 2019, 31, 465002 CrossRef CAS PubMed.
- M. Zhu, S. Kim, L. Mao, M. Fujitsuka, J. Zhang, X. Wang and T. Majima, J. Am. Chem. Soc., 2017, 139, 13234–13242 CrossRef CAS PubMed.
- J. Ran, W. Guo, H. Wang, B. Zhu, J. Yu and S.-Z. Qiao, Adv. Mater., 2018, 30, 1800128 CrossRef PubMed.
- Q. Zhang, S. Huang, J. Deng, D. T. Gangadharan, F. Yang, Z. Xu, G. Giorgi, M. Palummo, M. Chaker and D. Ma, Adv. Funct. Mater., 2019, 29, 1902486 CrossRef.
- Y. Zheng, Y. Chen, B. Gao, B. Lin and X. J. A. F. M. Wang, Adv. Funct. Mater., 2020, 30, 2002021 CrossRef CAS.
- M. Zhu, S. Kim, L. Mao, M. Fujitsuka, J. Zhang, X. Wang and T. J. J. o. t. A. C. S. Majima, J. Am. Chem. Soc., 2017, 139, 13234–13242 CrossRef CAS PubMed.
- J. Ran, W. Guo, H. Wang, B. Zhu, J. Yu and S. Z. J. A. m. Qiao, Adv. Mater., 2018, 30, 1800128 CrossRef PubMed.
- M. Wen, J. Wang, R. Tong, D. Liu, H. Huang, Y. Yu, Z.-K. Zhou, P. K. Chu and X.-F. Yu, Adv. Sci., 2019, 6, 1970007 CrossRef.
- R. Boppella, W. Yang, J. Tan, H.-C. Kwon, J. Park and J. Moon, Appl. Catal., B, 2019, 242, 422–430 CrossRef CAS.
- Y. Qu and X. J. C. S. R. Duan, Chem. Soc. Rev., 2013, 42, 2568–2580 RSC.
- O. Elbanna, M. Zhu, M. Fujitsuka and T. Majima, ACS Catal., 2019, 9, 3618–3626 CrossRef CAS.
- Y. Chen, T. Shi, P. Liu, X. Ma, L. Shui, C. Shang, Z. Chen, X. Wang, K. Kempa and G. Zhou, J. Mater. Chem. A, 2018, 6, 19167–19175 RSC.
- J. Ran, B. Zhu and S.-Z. Qiao, Angew. Chem., Int. Ed., 2017, 56, 10373–10377 CrossRef CAS.
- S. Zhang, G. Cheng, L. Guo, N. Wang, B. Tan and S. J. A. C. Jin, Angew. Chem., 2020, 132, 6063–6070 CrossRef.
- F. Liu, R. Shi, Z. Wang, Y. Weng, C. M. Che and Y. J. A. C. Chen, Angew. Chem., 2019, 131, 11917–11921 CrossRef.
- X. Chen, J. S. Ponraj, D. Fan and H. Zhang, Nanoscale, 2020, 12, 3513–3534 RSC.
- J. Hu, D. Chen, Z. Mo, N. Li, Q. Xu, H. Li, J. He, H. Xu and J. Lu, Angew. Chem., Int. Ed., 2019, 58, 2073–2077 CrossRef CAS PubMed.
- X. Li, J. Xiong, X. Gao, J. Ma, Z. Chen, B. Kang, J. Liu, H. Li, Z. Feng and J. Huang, J. Hazard. Mater., 2020, 387, 121690 CrossRef CAS PubMed.
- P. Wang, Y. Liu, N. Jiang, R. Jing, S. Li, Q. Zhang, H. Liu, J. Xiu, Z. Li and Y. Liu, J. Mol. Liq., 2021, 329, 115540 CrossRef CAS.
- Z. Yang and J. Hao, Small Methods, 2018, 2, 1700296 CrossRef.
- Y. Cai, G. Zhang and Y.-W. Zhang, Sci. Rep., 2014, 4, 6677 CrossRef CAS PubMed.
- Y.-J. Yuan, Z. Shen, S. Wu, Y. Su, L. Pei, Z. Ji, M. Ding, W. Bai, Y. Chen, Z.-T. Yu and Z. Zou, Appl. Catal., B, 2019, 246, 120–128 CrossRef CAS.
- H. Yu, H. Ma, X. Wu, X. Wang, J. Fan and J. Yu, Sol. RRL, 2021, 5, 2000372 CrossRef CAS.
- S. Huang, H. Yi, L. Zhang, Z. Jin, Y. Long, Y. Zhang, Q. Liao, J. Na, H. Cui, S. Ruan, Y. Yamauchi, T. Wakihara, Y. V. Kaneti and Y.-J. Zeng, J. Hazard. Mater., 2020, 393, 122324 CrossRef CAS PubMed.
- X. Zhou, Y. Liu, Z. Jin, M. Huang, F. Zhou, J. Song, J. Qu, Y.-J. Zeng, P.-C. Qian and W.-Y. Wong, Adv. Sci., 2021, 8, 2170020 CrossRef.
- J. Fu, J. Yu, C. Jiang and B. Cheng, Adv. Energy Mater., 2018, 8, 1701503 CrossRef.
- J. Wen, J. Xie, X. Chen and X. Li, Appl. Surf. Sci., 2017, 391, 72–123 CrossRef CAS.
- Z. Jin, Q. Zhang, J. Chen, S. Huang, L. Hu, Y.-J. Zeng, H. Zhang, S. Ruan and T. Ohno, Appl. Catal., B, 2018, 234, 198–205 CrossRef CAS.
- L. Zhang, Z. Jin, J. Huang, Y. Zhang, S. Huang, Z. Wang, Y.-J. Zeng, V. Malgras, S. Ruan and Y. Yamauchi, ACS Appl. Energy Mater., 2021, 4, 5677–5686 CrossRef CAS.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed.
- L. Zhang, Z. Jin, H. Lu, T. Lin, S. Ruan, X. S. Zhao and Y.-J. Zeng, ACS Omega, 2018, 3, 15009–15017 CrossRef CAS PubMed.
- T. Giannakopoulou, I. Papailias, N. Todorova, N. Boukos, Y. Liu, J. Yu and C. Trapalis, Chem. Eng. J., 2017, 310, 571–580 CrossRef CAS.
- B. Tao and Z. Yan, J. Colloid Interface Sci., 2016, 480, 118–125 CrossRef CAS PubMed.
- Q. Fan, Y. Huang, C. Zhang, J. Liu, L. Piao, Y. Yu, S. Zuo and B. Li, Catal. Today, 2016, 264, 250–256 CrossRef CAS.
- B. Xue, H.-Y. Jiang, T. Sun and F. Mao, Catal. Lett., 2016, 146, 2185–2192 CrossRef CAS.
- P. Qiu, J. Yao, H. Chen, F. Jiang and X. Xie, J. Hazard. Mater., 2016, 317, 158–168 CrossRef CAS PubMed.
- J. Di, J. Xia, S. Yin, H. Xu, L. Xu, Y. Xu, M. He and H. Li, J. Mater. Chem. A, 2014, 2, 5340–5351 RSC.
- H. Xu, J. Yan, Y. Xu, Y. Song, H. Li, J. Xia, C. Huang and H. Wan, Appl. Catal., B, 2013, 129, 182–193 CrossRef CAS.
- Y. Jiang, P. Liu, Y. Chen, Z. Zhou, H. Yang, Y. Hong, F. Li, L. Ni, Y. Yan and D. H. Gregory, Appl. Surf. Sci., 2017, 391, 392–403 CrossRef CAS.
- B. Wang, J. Zhang and F. Huang, Appl. Surf. Sci., 2017, 391, 449–456 CrossRef CAS.
- I. Khan, N. Baig and A. Qurashi, ACS Appl. Energy Mater., 2019, 2, 607–615 CrossRef CAS.
- Z. Qin, F. Xue, Y. Chen, S. Shen and L. Guo, Appl. Catal., B, 2017, 217, 551–559 CrossRef CAS.
- F. Yu, Z. Wang, S. Zhang, H. Ye, K. Kong, X. Gong, J. Hua and H. Tian, Adv. Funct. Mater., 2018, 28, 1804512 CrossRef.
- Y. Gong, X. Quan, H. Yu, S. Chen and H. Zhao, Appl. Catal., B, 2018, 237, 947–956 CrossRef CAS.
- C. Yang, Z. Xue, J. Qin, M. Sawangphruk, S. Rajendran, X. Zhang and R. Liu, J. Phys. Chem. C, 2019, 123, 4795–4804 CrossRef CAS.
- K. He, J. Xie, X. Luo, J. Wen, S. Ma, X. Li, Y. Fang and X. Zhang, Chinese J. Catal., 2017, 38, 240–252 CrossRef CAS.
- S. Huang, Y. Long, S. Ruan and Y.-J. Zeng, ACS Omega, 2019, 4, 15593–15599 CrossRef CAS PubMed.
- Y. Che, B. Lu, Q. Qi, H. Chang, J. Zhai, K. Wang and Z. Liu, Sci. Rep., 2018, 8, 16504 CrossRef PubMed.
- J. Wang, G. Wang, X. Wang, Y. Wu, Y. Su and H. Tang, Carbon, 2019, 149, 618–626 CrossRef CAS.
- Y. You, S. Wang, K. Xiao, T. Ma, Y. Zhang and H. Huang, ACS Sustainable Chem. Eng., 2018, 6, 16219–16227 CrossRef CAS.
- R. Ye, H. Fang, Y.-Z. Zheng, N. Li, Y. Wang and X. Tao, ACS Appl. Mater. Interfaces, 2016, 8, 13879–13889 CrossRef CAS PubMed.
- X. Chen, T. Hu, J. Zhang, C. Yang, K. Dai and C. Pan, J. Alloys Compd., 2021, 863, 158068 CrossRef CAS.
- T. Yan, H. Liu and Z. Jin, Energy Fuels, 2021, 35, 856–867 CrossRef CAS.
- B. Zhang, H. Shi, Y. Yan, C. Liu, X. Hu, E. Liu and J. Fan, Colloids Surf., A, 2021, 608, 125598 CrossRef CAS.
- J. Fu, Q. Xu, J. Low, C. Jiang and J. Yu, Appl. Catal., B, 2019, 243, 556–565 CrossRef CAS.
- X. Xu, J. Wang and Y. Shen, Langmuir, 2021, 37, 7254–7263 CrossRef CAS PubMed.
- S. Cao and J. Yu, J. Phys. Chem. Lett., 2014, 5, 2101–2107 CrossRef CAS PubMed.
- Y. Chen, F. Su, H. Xie, R. Wang, C. Ding, J. Huang, Y. Xu and L. Ye, Chem. Eng. J., 2021, 404, 126498 CrossRef CAS.
- Y. Qin, J. Lu, F. Meng, X. Lin, Y. Feng, Y. Yan and M. Meng, J. Colloid Interface Sci., 2021, 586, 576–587 CrossRef CAS PubMed.
- R. Shen, K. He, A. Zhang, N. Li, Y. H. Ng, P. Zhang, J. Hu and X. Li, Appl. Catal., B, 2021, 291, 120104 CrossRef CAS.
- X. Li, W. Bi, L. Zhang, S. Tao, W. Chu, Q. Zhang, Y. Luo, C. Wu and Y. Xie, Adv. Mater., 2016, 28, 2427–2431 CrossRef CAS PubMed.
- W. Wang, J. Fang and X. Huang, Appl. Surf. Sci., 2020, 513, 145830 CrossRef CAS.
- S. Wang, F. Wang, Z. Su, X. Wang, Y. Han, L. Zhang, J. Xiang, W. Du and N. Tang, Catalysts, 2019, 9, 439 CrossRef CAS.
- J.-W. Shi, Y. Zou, L. Cheng, D. Ma, D. Sun, S. Mao, L. Sun, C. He and Z. Wang, Chem. Eng. J., 2019, 378, 122161 CrossRef CAS.
- Q. H. Wang, K. Kalantar-Zadeh, A. Kis, J. N. Coleman and M. S. Strano, Nat. Nanotechnol., 2012, 7, 699–712 CrossRef CAS PubMed.
- M. Chhowalla, H. S. Shin, G. Eda, L. J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef PubMed.
- Y. Zhao, Y. Cai, L. Zhang, B. Li, G. Zhang and J. T. L. Thong, Adv. Funct. Mater., 2020, 30, 1903929 CrossRef CAS.
- C. Li, Q. Cao, F. Wang, Y. Xiao, Y. Li, J.-J. Delaunay and H. Zhu, Chem. Soc. Rev., 2018, 47, 4981–5037 RSC.
- M.-H. Chiu, C. Zhang, H.-W. Shiu, C.-P. Chuu, C.-H. Chen, C.-Y. S. Chang, C.-H. Chen, M.-Y. Chou, C.-K. Shih and L.-J. Li, Nat. Commun., 2015, 6, 7666 CrossRef CAS PubMed.
- J. Zhou, J. Lin, X. Huang, Y. Zhou, Y. Chen, J. Xia, H. Wang, Y. Xie, H. Yu, J. Lei, D. Wu, F. Liu, Q. Fu, Q. Zeng, C.-H. Hsu, C. Yang, L. Lu, T. Yu, Z. Shen, H. Lin, B. I. Yakobson, Q. Liu, K. Suenaga, G. Liu and Z. Liu, Nature, 2018, 556, 355–359 CrossRef CAS PubMed.
- K.-A. N. Duerloo, Y. Li and E. J. Reed, Nat. Commun., 2014, 5, 4214 CrossRef CAS PubMed.
- P. Cheng, K. Sun and Y. H. Hu, Nano Lett., 2016, 16, 572–576 CrossRef CAS PubMed.
- R. J. Toh, Z. Sofer, J. Luxa, D. Sedmidubský and M. J. C. C. Pumera, Chem. Commun., 2017, 53, 3054–3057 RSC.
- Y. Xiao, M. Zhou, J. Liu, J. Xu and L. Fu, Sci. China Mater., 2019, 62, 759–775 CrossRef CAS.
- X. Xu, G. Zhou, X. Dong and J. Hu, ACS Sustainable Chem. Eng., 2017, 5, 3829–3836 CrossRef CAS.
- A. Di Bartolomeo, Nanomaterials, 2020, 10, 579 CrossRef CAS PubMed.
- M. Wang, P. Ju, J. Li, Y. Zhao, X. Han and Z. Hao, ACS Sustainable Chem. Eng., 2017, 5, 7878–7886 CrossRef CAS.
- S. Wang, C. Ren, H. Tian, J. Yu and M. Sun, Phys. Chem. Chem. Phys., 2018, 20, 13394–13399 RSC.
- Z. Zhang, Q. Qian, B. Li and K. J. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 17419–17426 CrossRef CAS PubMed.
- D. Lu, H. Wang, X. Zhao, K. K. Kondamareddy, J. Ding, C. Li and P. Fang, ACS Sustainable Chem. Eng., 2017, 5, 1436–1445 CrossRef CAS.
- A. H. Hendi, A. M. Osman, I. Khan, T. A. Saleh, T. A. Kandiel, T. F. Qahtan and M. K. Hossain, ACS Omega, 2020, 5, 31644–31656 CrossRef CAS PubMed.
- P. C. Nagajyothi, K. C. Devarayapalli, J. Shim and S. V. Prabhakar Vattikuti, Int. J. Hydrogen Energy, 2020, 45, 32756–32769 CrossRef CAS.
- J. Tao, X. Yu, Q. Liu, G. Liu and H. Tang, J. Colloid Interface Sci., 2021, 585, 470–479 CrossRef CAS PubMed.
- Y. Jia, Z. Wang, X.-Q. Qiao, L. Huang, S. Gan, D. Hou, J. Zhao, C. Sun and D.-S. Li, Chem. Eng. J., 2021, 424, 130368 CrossRef CAS.
- P. Raizada, T. H. C. Nguyen, S. Patial, P. Singh, A. Bajpai, V.-H. Nguyen, D. L. T. Nguyen, X. Cuong Nguyen, A. Aslam Parwaz Khan, S. Rangabhashiyam, S. Young Kim and Q. V. Le, Fuel, 2021, 303, 121302 CrossRef CAS.
- Q. Luan, C.-L. Yang, M.-S. Wang and X.-G. Ma, Chin. J. Phys., 2017, 55, 1930–1937 CrossRef CAS.
- M. Thripuranthaka, R. V. Kashid, C. S. Rout and D. J. Late, Appl. Phys. Lett., 2014, 104, 081911 CrossRef.
- S. Ma, L. Zeng, L. Tao, C. Y. Tang, H. Yuan, H. Long, P. K. Cheng, Y. Chai, C. Chen, K. H. Fung, X. Zhang, S. P. Lau and Y. H. Tsang, Sci. Rep., 2017, 7, 3125 CrossRef PubMed.
- W.-Z. Xiao, L. Xu, Q.-Y. Rong, X.-Y. Dai, C.-P. Cheng and L.-L. Wang, Appl. Surf. Sci., 2020, 504, 144425 CrossRef CAS.
- Z. Thiehmed, A. Shakoor and T. Altahtamouni, Catalysts, 2021, 11, 1283 CrossRef CAS.
- C. Xue, P. Zhang, G. Shao and G. Yang, Chem. Eng. J., 2020, 398, 125602 CrossRef CAS.
- G.-J. Lai, L.-M. Lyu, Y.-S. Huang, G.-C. Lee, M.-P. Lu, T.-P. Perng, M.-Y. Lu and L.-J. Chen, Nano Energy, 2021, 81, 105608 CrossRef CAS.
- Y. Fan, J. Wang and M. Zhao, Nanoscale, 2019, 11, 14836–14843 RSC.
- H. U. Din, M. Idrees, G. Rehman, C. V. Nguyen, L.-Y. Gan, I. Ahmad, M. Maqbool and B. Amin, Phys. Chem. Chem. Phys., 2018, 20, 24168–24175 RSC.
- F. Wang, T. A. Shifa, X. Zhan, Y. Huang, K. Liu, Z. Cheng, C. Jiang and J. He, Nanoscale, 2015, 7, 19764–19788 RSC.
- S. Altaf, A. Haider, S. Naz, A. Ul-Hamid, J. Haider, M. Imran, A. Shahzadi, M. Naz, H. Ajaz and M. Ikram, Nanoscale Res. Lett., 2020, 15, 144 CrossRef CAS PubMed.
- X. Peng, Y. Yan, X. Jin, C. Huang, W. Jin, B. Gao and P. K. Chu, Nano Energy, 2020, 78, 105234 CrossRef CAS.
- P. J. Sideris, U. G. Nielsen, Z. Gan and C. P. Grey, Science, 2008, 321, 113 CrossRef CAS PubMed.
- A. Razzaq, S. Ali, M. Asif and S.-I. In, Catalysts, 2020, 10, 1185 CrossRef.
- H. Boumeriame, E. S. Da Silva, A. S. Cherevan, T. Chafik, J. L. Faria and D. Eder, J. Energy Chem., 2022, 64, 406–431 CrossRef.
- H. Zhou, Y. Song, Y. Liu, H. Li, W. Li and Z. Chang, Int. J. Hydrogen Energy, 2018, 43, 14328–14336 CrossRef CAS.
- G. Zheng, C. Wu, J. Wang, S. Mo, Y. Wang, Z. Zou, B. Zhou and F. Long, RSC Adv., 2019, 9, 24280–24290 RSC.
- S. Das, S. Patnaik and K. M. Parida, Inorg. Chem. Front., 2019, 6, 94–109 RSC.
- J. Zhang, Q. Zhu, L. Wang, M. Nasir, S.-H. Cho and J. Zhang, Int. J. Hydrogen Energy, 2020, 45, 21331–21340 CrossRef CAS.
- Y. Wang, S. Guo, X. Xin, Y. Zhang, B. Wang, S. Tang and X. Li, Appl. Surf. Sci., 2021, 549, 149108 CrossRef CAS.
- S. Mancipe, F. Tzompantzi and R. Gómez, Appl. Clay Sci., 2017, 136, 67–74 CrossRef CAS.
- S. Li, L. Wang, Y. Li, L. Zhang, A. Wang, N. Xiao, Y. Gao, N. Li, W. Song, L. Ge and J. Liu, Appl. Catal., B, 2019, 254, 145–155 CrossRef CAS.
- D. Kandi, D. P. Sahoo, S. Martha and K. Parida, Adv. Mater. Interfaces, 2019, 6, 1900370 CrossRef.
- C. Wang, B. Ma, S. Xu, D. Li, S. He, Y. Zhao, J. Han, M. Wei, D. G. Evans and X. Duan, Nano Energy, 2017, 32, 463–469 CrossRef CAS.
- C. Wang, B. Ma, X. Cao, S. He, J. Han, M. Wei, D. G. Evans and X. Duan, J. Mater. Chem. A, 2018, 6, 7871–7876 RSC.
- D. P. Sahoo, S. Patnaik and K. Parida, ACS Omega, 2019, 4, 14721–14741 CrossRef CAS PubMed.
- S. Nayak and K. M. Parida, Sci. Rep., 2019, 9, 2458 CrossRef PubMed.
- H. Gao, R. Cao, X. Xu, S. Zhang, H. Yongshun, H. Yang, X. Deng and J. Li, Appl. Catal., B, 2019, 245, 399–409 CrossRef CAS.
- A. Kumar, G. Sharma, A. Kumari, C. Guo, M. Naushad, D.-V. N. Vo, J. Iqbal and F. J. Stadler, Appl. Catal., B, 2021, 284, 119808 CrossRef CAS.
- S. Megala, P. Ravi, P. Maadeswaran, M. Navaneethan, M. Sathish and R. Ramesh, Nanoscale Adv., 2021, 3, 2075–2088 RSC.
- K. Iqbal, A. Iqbal, A. M. Kirillov, C. Shan, W. Liu and Y. Tang, J. Mater. Chem. A, 2018, 6, 4515–4524 RSC.
- K. Hu, A. D. Blair, E. J. Piechota, P. A. Schauer, R. N. Sampaio, F. G. L. Parlane, G. J. Meyer and C. P. Berlinguette, Nat. Chem., 2016, 8, 853–859 CrossRef CAS PubMed.
- M. Yang, Y. Li, T. Yan and Z. Jin, Nanoscale, 2021, 13, 13858–13872 RSC.
- L. Sun, Y. Zhuang, Y. Yuan, W. Zhan, X.-J. Wang, X. Han and Y. Zhao, Adv. Energy Mater., 2019, 9, 1902839 CrossRef CAS.
- S. Nayak, G. Swain and K. Parida, ACS Appl. Mater. Interfaces, 2019, 11, 20923–20942 CrossRef CAS PubMed.
- D. P. Sahoo, S. Nayak, K. H. Reddy, S. Martha and K. Parida, Inorg. Chem., 2018, 57, 3840–3854 CrossRef CAS PubMed.
- S. Wang, B. Y. Guan, X. Wang and X. W. D. Lou, J. Am. Chem. Soc., 2018, 140, 15145–15148 CrossRef CAS PubMed.
- G. Wang, Y. Li, L. Xu, Z. Jin and Y. Wang, Renewable Energy, 2020, 162, 535–549 CrossRef CAS.
- J. K. Im, E. J. Sohn, S. Kim, M. Jang, A. Son, K.-D. Zoh and Y. Yoon, Chemosphere, 2021, 270, 129478 CrossRef CAS PubMed.
- Q. Zhong, Y. Li and G. Zhang, Chem. Eng. J., 2021, 409, 128099 CrossRef CAS.
- V. Sharma, A. Kumar and V. Krishnan, Handbook of Smart Photocatalytic Materials, Elsevier, 2020, pp. 247–267 Search PubMed.
- W. Huang, L. Hu, Y. Tang, Z. Xie and H. Zhang, Adv. Funct. Mater., 2020, 30, 2005223 CrossRef CAS.
- N. M. Abbasi, Y. Xiao, L. Zhang, L. Peng, Y. Duo, L. Wang, P. Yin, Y. Ge, H. Zhu, B. Zhang, N. Xie, Y. Duan, B. Wang and H. Zhang, J. Mater. Chem. C, 2021, 9, 8395–8465 RSC.
- Y. Cao, Y. Fang, X. Lei, B. Tan, X. Hu, B. Liu and Q. Chen, J. Hazard. Mater., 2020, 387, 122021 CrossRef CAS.
- J. Zhang, C. Xing and F. Shi, Int. J. Hydrogen Energy, 2020, 45, 6291–6301 CrossRef CAS.
- J. Zhou, D. Li, W. Zhao, B. Jing, Z. Ao and T. An, ACS Appl. Mater. Interfaces, 2021, 13, 23843–23852 CrossRef CAS PubMed.
- M. Ding, R. Xiao, C. Zhao, D. Bukhvalov, Z. Chen, H. Xu, H. Tang, J. Xu and X. Yang, Sol. RRL, 2021, 5, 2000414 CrossRef CAS.
- H. Wang, R. Peng, Z. D. Hood, M. Naguib, S. P. Adhikari and Z. Wu, ChemSusChem, 2016, 9, 1490–1497 CrossRef CAS PubMed.
- J.-Y. Li, Y.-H. Li, F. Zhang, Z.-R. Tang and Y.-J. Xu, Appl. Catal., B, 2020, 269, 118783 CrossRef CAS.
- M. Ding, R. Xiao, C. Zhao, D. Bukhvalov, Z. Chen, H. Xu, H. Tang, J. Xu and X. Yang, Sol. RRL, 2021, 5, 2000414 CrossRef CAS.
- Z. Li, W. Huang, J. Liu, K. Lv and Q. Li, ACS Catal., 2021, 11, 8510–8520 CrossRef CAS.
- R. Chen, P. Wang, J. Chen, C. Wang and Y. Ao, Appl. Surf. Sci., 2019, 473, 11–19 CrossRef CAS.
- P. Tian, X. He, L. Zhao, W. Li, W. Fang, H. Chen, F. Zhang, Z. Huang and H. Wang, Int. J. Hydrogen Energy, 2019, 44, 788–800 CrossRef CAS.
- K. Huang, C. Li and X. Meng, J. Colloid Interface Sci., 2020, 580, 669–680 CrossRef CAS PubMed.
- A. VahidMohammadi, J. Rosen and Y. Gogotsi, Science, 2021, 372, eabf1581 CrossRef CAS PubMed.
- G.-J. Lee, Y.-C. Zheng and J. J. Wu, Catal. Today, 2018, 307, 197–204 CrossRef CAS.
- J. Xiong, P. Song, J. Di and H. Li, J. Mater. Chem. A, 2020, 8, 21434–21454 RSC.
- Y. Liu, P. Lv, W. Zhou and J. Hong, J. Phys. Chem. C, 2020, 124, 9696–9702 CrossRef CAS.
- T. Rao, H. Wang, Y.-J. Zeng, Z. Guo, H. Zhang and W. Liao, Adv. Sci., 2021, 8, 2002284 CrossRef CAS PubMed.
- Y. Cui, L. Peng, L. Sun, Q. Qian and Y. Huang, J. Mater. Chem. A, 2018, 6, 22768–22777 RSC.
- S. Huang, Z. Shuai and D. Wang, J. Mater. Chem. A, 2021, 9, 2734–2741 RSC.
- J. Liao, B. Sa, J. Zhou, R. Ahuja and Z. Sun, J. Phys. Chem. C, 2014, 118, 17594–17599 CrossRef CAS.
- D. Singh, N. Khossossi, A. Ainane and R. Ahuja, Catal. Today, 2021, 370, 14–25 CrossRef CAS.
- X. Wang, K. Du, Y. Y. Fredrik Liu, P. Hu, J. Zhang, Q. Zhang, M. H. S. Owen, X. Lu, C. K. Gan, P. Sengupta, C. Kloc and Q. Xiong, 2D Mater., 2016, 3, 031009 CrossRef.
- B. Lin, A. Chaturvedi, J. Di, L. You, C. Lai, R. Duan, J. Zhou, B. Xu, Z. Chen, P. Song, J. Peng, B. Ma, H. Liu, P. Meng, G. Yang, H. Zhang, Z. Liu and F. Liu, Nano Energy, 2020, 76, 104972 CrossRef CAS.
- S. Li, Y. Zhang and H. Huang, J. Energy Chem., 2022, 67, 745–779 CrossRef.
- W. Lei, Y. Mi, R. Feng, P. Liu, S. Hu, J. Yu, X. Liu, J. A. Rodriguez, J.-o. Wang, L. Zheng, K. Tang, S. Zhu, G. Liu and M. Liu, Nano Energy, 2018, 50, 552–561 CrossRef CAS.
- C. Chen, J. Hu, X. Yang, T. Yang, J. Qu, C. Guo and C. M. Li, ACS Appl. Mater. Interfaces, 2021, 13, 20162–20173 CrossRef CAS PubMed.
- Q. Liang, F. Shi, X. Xiao, X. Wu, K. Huang and S. Feng, ChemCatChem, 2018, 10, 2179–2183 CrossRef CAS.
- M. Zhu, M. Fujitsuka, L. Zeng, M. Liu and T. Majima, Appl. Catal., B, 2019, 256, 117864 CrossRef CAS.
- Y.-J. Yuan, Z.-K. Shen, S. Song, J. Guan, L. Bao, L. Pei, Y. Su, S. Wu, W. Bai, Z.-T. Yu, Z. Ji and Z. Zou, ACS Catal., 2019, 9, 7801–7807 CrossRef CAS.
- F. Liu, R. Shi, Z. Wang, Y. Weng, C.-M. Che and Y. Chen, Angew. Chem., 2019, 58, 11791–11795 CrossRef CAS PubMed.
- Y. Huang, H. Lu, B. Wang, W. He, H. Dong, L. Sui, Z. Gan, S. Ma, B. Pang, L. Dong and L. Yu, Int. J. Hydrogen Energy, 2021, 46, 3530–3538 CrossRef CAS.
- Z. Yan, Z. Sun, X. Liu, H. Jia and P. Du, Nanoscale, 2016, 8, 4748–4756 RSC.
- X. Yang, H. Huang, M. Kubota, Z. He, N. Kobayashi, X. Zhou, B. Jin and J. Luo, Mater. Res. Bull., 2016, 76, 79–84 CrossRef CAS.
- F. Cheng, H. Yin and Q. Xiang, Appl. Surf. Sci., 2017, 391, 432–439 CrossRef CAS.
- Y. Shi, J. Chen, Z. Mao, B. D. Fahlman and D. Wang, J. Catal., 2017, 356, 22–31 CrossRef CAS.
- J. Wan, C. Pu, R. Wang, E. Liu, X. Du, X. Bai, J. Fan and X. Hu, Int. J. Hydrogen Energy, 2018, 43, 7007–7019 CrossRef CAS.
- J. Xu, J. Gao, Y. Qi, C. Wang and L. Wang, ChemCatChem, 2018, 10, 3327–3335 CrossRef CAS.
- M. Wu, J. Zhang, C. Liu, Y. Gong, R. Wang, B. He and H. Wang, ChemCatChem, 2018, 10, 3069–3077 CrossRef CAS.
- H. Che, C. Liu, H. Dong, C. Li, X. Liu and G. Che, Int. J. Hydrogen Energy, 2019, 44, 20029–20041 CrossRef CAS.
- Y. Wang, X. Zhang, Y. Liu, Y. Zhao, C. Xie, Y. Song and P. Yang, Int. J. Hydrogen Energy, 2019, 44, 30151–30159 CrossRef CAS.
- Y. Zou, J.-W. Shi, L. Sun, D. Ma, S. Mao, Y. Lv and Y. Cheng, Chem. Eng. J., 2019, 378, 122192 CrossRef CAS.
- M. Wang, Z. Qin, Z. Diao, R. Li, J. Zhong, D. Ma and Y. Chen, ACS Sustain. Chem. Eng., 2020, 8, 13459–13466 CrossRef CAS.
- C. Li, H. Wu, Y. Du, S. Xi, H. Dong, S. Wang and Y. Wang, ACS Sustain. Chem. Eng., 2020, 8, 12934–12943 CrossRef CAS.
- W. Shi, J. Wang, S. Yang, X. Lin, F. Guo and J. Shi, J. Chem. Technol. Biotechnol., 2020, 95, 2129–2138 CrossRef CAS.
- J. Pan, W. Shen, Y. Zhang, H. Tang, H. Sun, W. Zhong and X. Yan, Appl. Surf. Sci., 2020, 520, 146335 CrossRef CAS.
- M. Lallimathi, P. Kalisamy, M. Suryamathi, T. Alshahrani, M. Shkir, M. Venkatachalam and B. Palanivel, ChemistrySelect, 2020, 5, 10607–10617 CrossRef CAS.
- Y. Chen, Q. Wang, H. Huang, J. Kou, C. Lu and Z. Xu, Int. J. Hydrogen Energy, 2021, 46, 32514–32522 CrossRef CAS.
- J. Yan, Z. Song, H. Li, H. Xu and L. Y. S. Lee, Chem. Eng. J., 2021, 425, 131512 CrossRef CAS.
- Z. Wu, X.-L. Wang, X. Wang, X. Xu, D.-S. Li and T. Wu, Chem. Eng. J., 2021, 426, 131216 CrossRef CAS.
- Z. Tian, X. Yang, Y. Chen, X. Wang, T. Jiao, W. Zhao, H. Huang and J. Hu, J. Alloys Compd., 2021, 161850 Search PubMed.
- H. He, J. Lin, W. Fu, X. Wang, H. Wang, Q. Zeng, Q. Gu, Y. Li, C. Yan, B. K. Tay, C. Xue, X. Hu, S. T. Pantelides, W. Zhou and Z. Liu, Adv. Energy Mater., 2016, 6, 1600464 CrossRef.
- X.-L. Yin, L.-L. Li, W.-J. Jiang, Y. Zhang, X. Zhang, L.-J. Wan and J.-S. Hu, ACS Appl. Mater. Interfaces, 2016, 8, 15258–15266 CrossRef CAS PubMed.
- Y.-J. Yuan, D.-Q. Chen, Y.-W. Huang, Z.-T. Yu, J.-S. Zhong, T.-T. Chen, W.-G. Tu, Z.-J. Guan, D.-P. Cao and Z.-G. Zou, ChemSusChem, 2016, 9, 1003–1009 CrossRef CAS PubMed.
- Q. Xiang, F. Cheng and D. Lang, ChemSusChem, 2016, 9, 996–1002 CrossRef CAS PubMed.
- J. He, L. Chen, Z.-Q. Yi, C.-T. Au and S.-F. Yin, Ind. Eng. Chem. Res., 2016, 55, 8327–8333 CrossRef CAS.
- Q. Liu, Q. Shang, A. Khalil, Q. Fang, S. Chen, Q. He, T. Xiang, D. Liu, Q. Zhang, Y. Luo and L. Song, ChemCatChem, 2016, 8, 2614–2619 CrossRef CAS.
- L. Zhao, J. Jia, Z. Yang, J. Yu, A. Wang, Y. Sang, W. Zhou and H. Liu, Appl. Catal., B, 2017, 210, 290–296 CrossRef CAS.
- M. Ben Ali, W.-K. Jo, H. Elhouichet and R. Boukherroub, Int. J. Hydrogen Energy, 2017, 42, 16449–16458 CrossRef CAS.
- D. A. Reddy, H. Park, R. Ma, D. P. Kumar, M. Lim and T. K. Kim, ChemSusChem, 2017, 10, 1563–1570 CrossRef CAS PubMed.
- Y. Li, L. Wang, T. Cai, S. Zhang, Y. Liu, Y. Song, X. Dong and L. Hu, Chem. Eng. J., 2017, 321, 366–374 CrossRef CAS.
- C. Zhou, H. Yang, G. Wang, J. Chen, R. Wang and C. Jiang, Int. J. Hydrogen Energy, 2017, 42, 28337–28348 CrossRef CAS.
- T. Huang, W. Chen, T.-Y. Liu, Q.-L. Hao and X.-H. Liu, Int. J. Hydrogen Energy, 2017, 42, 12254–12261 CrossRef CAS.
- J. Chen, X.-J. Wu, Y. Gong, Y. Zhu, Z. Yang, B. Li, Q. Lu, Y. Yu, S. Han, Z. Zhang, Y. Zong, Y. Han, L. Gu and H. Zhang, J. Am. Chem. Soc., 2017, 139, 8653–8660 CrossRef CAS PubMed.
- X.-L. Yin, L.-L. Li, D.-C. Li and J.-M. Dou, Int. J. Hydrogen Energy, 2018, 43, 20382–20391 CrossRef CAS.
- Y. Liu, H. Niu, W. Gu, X. Cai, B. Mao, D. Li and W. Shi, Chem. Eng. J., 2018, 339, 117–124 CrossRef CAS.
- Z. Zhang, L. Huang, J. Zhang, F. Wang, Y. Xie, X. Shang, Y. Gu, H. Zhao and X. Wang, Appl. Catal., B, 2018, 233, 112–119 CrossRef CAS.
- Y. Zou, J.-W. Shi, D. Ma, Z. Fan, L. Cheng, D. Sun, Z. Wang and C. Niu, ChemSusChem, 2018, 11, 1187–1197 CrossRef CAS PubMed.
- K. Zhang, M. Fujitsuka, Y. Du and T. Majima, ACS Appl. Mater. Interfaces, 2018, 10, 20458–20466 CrossRef CAS PubMed.
- J. Zhou, D. Chen, L. Bai, L. Qin, X. Sun and Y. Huang, Int. J. Hydrogen Energy, 2018, 43, 18261–18269 CrossRef CAS.
- J. Wang, J. Luo, D. Liu, S. Chen and T. Peng, Appl. Catal., B, 2019, 241, 130–140 CrossRef CAS.
- S. Zang, G. Zhang, Z.-A. Lan, D. Zheng and X. Wang, Appl. Catal., B, 2019, 251, 102–111 CrossRef CAS.
- Z.-W. Zhang, Q.-H. Li, X.-Q. Qiao, D. Hou and D.-S. Li, Chinese J. Catal., 2019, 40, 371–379 CrossRef CAS.
- G. Swain, S. Sultana and K. Parida, Inorg. Chem., 2019, 58, 9941–9955 CrossRef CAS PubMed.
- S. Kumar, A. Kumar, V. Navakoteswara Rao, A. Kumar, M. V. Shankar and V. Krishnan, ACS Appl. Energy Mater., 2019, 2, 5622–5634 CrossRef CAS.
- M. Dan, J. Xiang, F. Wu, S. Yu, Q. Cai, L. Ye, Y. Ye and Y. Zhou, Appl. Catal., B, 2019, 256, 117870 CrossRef CAS.
- D. Jiang, B. Wen, Y. Zhang, Y. Jin, D. Li and M. Chen, J. Colloid Interface Sci., 2019, 536, 1–8 CrossRef CAS PubMed.
- H.-Q. Feng, Y. Xi, H.-Q. Xie, Y.-K. Li and Q.-Z. Huang, Int. J. Hydrogen Energy, 2020, 45, 10764–10774 CrossRef CAS.
- X. Liu, X. Chen, L. Xu, B. Wu, X. Tu, X. Luo, F. Yang and J. Lin, Int. J. Hydrogen Energy, 2020, 45, 26770–26784 CrossRef CAS.
- H. Zhai, J. Qi, Y. Tan, L. Yang, H. Li, Y. Kang, H. Liu, J. Shang and H. S. Park, Appl. Mater. Today, 2020, 18, 100536 CrossRef.
- M. Xiong, B. Chai, J. Yan, G. Fan and G. Song, Appl. Surf. Sci., 2020, 514, 145965 CrossRef CAS.
- S. Zhang, S. Chen, D. Liu, J. Zhang and T. Peng, Appl. Surf. Sci., 2020, 529, 147013 CrossRef CAS.
- B. Archana, N. Kottam, S. Nayak, K. B. Chandrasekhar and M. B. Sreedhara, J. Phys. Chem. C, 2020, 124, 14485–14495 CrossRef CAS.
- S. Bera, S. Ghosh and R. N. Basu, J. Alloys Compd., 2020, 830, 154527 CrossRef CAS.
- N. R. Khalid, M. Rizwan Kamal, M. B. Tahir, M. Rafique, N. A. Niaz, Y. Ali, M. Alzaid, H. Alrobei and S. Muhammad, Int. J. Hydrogen Energy, 2021, 46, 39822–39829 CrossRef CAS.
- X. Wei, X. Zhang, S. Ali, W. Han, D. He, J. Wang, S. Kong, Z. Feng, G. Zhang and J. Qi, J. Alloys Compd., 2021, 872, 159637 CrossRef CAS.
- W. Guan, Y. Li, Q. Zhong, H. Liu, J. Chen, H. Hu, K. Lv, J. Gong, Y. Xu, Z. Kang, M. Cao and Q. Zhang, Nano Lett., 2021, 21, 597–604 CrossRef CAS PubMed.
- J. Chen, X.-J. Wu, Q. Lu, M. Zhao, P.-F. Yin, Q. Ma, G.-H. Nam, B. Li, B. Chen and H. Zhang, Small, 2021, 17, 2006135 CrossRef CAS PubMed.
- X. Wang, G. Hai, B. Li, Q. Luan, W. Dong and G. Wang, Chem. Eng. J., 2021, 426, 130822 CrossRef CAS.
- L. Su, L. Luo, J. Wang, T. Song, W. Tu and Z.-J. Wang, Catal. Sci. Technol., 2021, 11, 1292–1297 RSC.
- S. Nayak and K. M. Parida, Int. J. Hydrogen Energy, 2016, 41, 21166–21180 CrossRef CAS.
- B. Luo, R. Song and D. Jing, Int. J. Hydrogen Energy, 2017, 42, 23427–23436 CrossRef CAS.
- R. Boppella, C. H. Choi, J. Moon and D. Ha Kim, Appl. Catal., B, 2018, 239, 178–186 CrossRef CAS.
- S. Nayak, A. C. Pradhan and K. M. Parida, Inorg. Chem., 2018, 57, 8646–8661 CrossRef CAS PubMed.
- D. Sun, D. Chi, Z. Yang, Z. Xing, J. Yin, Z. Li, Q. Zhu and W. Zhou, Int. J. Hydrogen Energy, 2019, 44, 16348–16358 CrossRef CAS.
- H. Li, X. Hao, Y. Liu, Y. Li and Z. Jin, J. Colloid Interface Sci., 2020, 572, 62–73 CrossRef CAS PubMed.
- S. Megala, M. Sathish, S. Harish, M. Navaneethan, S. Sohila, B. Liang and R. Ramesh, Appl. Surf. Sci., 2020, 509, 144656 CrossRef CAS.
- J. Yan, X. Zhang, W. Zheng and L. Y. S. Lee, ACS Appl. Mater. Interfaces, 2021, 13, 24723–24733 CrossRef CAS PubMed.
- Y. Sun, X. Wang, Q. Fu and C. Pan, ACS Appl. Mater. Interfaces, 2021, 13, 39331–39340 CrossRef CAS PubMed.
- S. Zhao, Q. Liang, W. Gao, M. Zhou, C. Yao, S. Xu and Z. Li, Inorg. Chem., 2021, 60, 9762–9772 CrossRef CAS PubMed.
- L. Hu, J. Xu, Y. Liu and S. Zhao, New J. Chem., 2021, 45, 17984–17995 RSC.
- X. Yan and Z. Jin, Chem. Eng. J., 2021, 420, 127682 CrossRef CAS.
- X. An, W. Wang, J. Wang, H. Duan, J. Shi and X. Yu, Phys. Chem. Chem. Phys., 2018, 20, 11405–11411 RSC.
- C. Peng, W. Xu, P. Wei, M. Liu, L. Guo, P. Wu, K. Zhang, Y. Cao, H. Wang, H. Yu, F. Peng and X. Yan, Int. J. Hydrogen Energy, 2019, 44, 29975–29985 CrossRef CAS.
- Y. Li, Z. Yin, G. Ji, Z. Liang, Y. Xue, Y. Guo, J. Tian, X. Wang and H. Cui, Appl. Catal., B, 2019, 246, 12–20 CrossRef CAS.
- R. Xiao, C. Zhao, Z. Zou, Z. Chen, L. Tian, H. Xu, H. Tang, Q. Liu, Z. Lin and X. Yang, Appl. Catal., B, 2020, 268, 118382 CrossRef CAS.
- J. Lu, Y. Chen, L. Li, X. Cai, S. Zhong, L. Wu, J. Chen and S. Bai, Chem. Eng. J., 2019, 362, 1–11 CrossRef CAS.
- R. Hu, G. Liao, Z. Huang, H. Qiao, H. Liu, Y. Shu, B. Wang and X. Qi, J. Hazard. Mater., 2021, 405, 124179 CrossRef CAS PubMed.
- Y.-Y. Wang, Y.-X. Chen, T. Barakat, Y.-J. Zeng, J. Liu, S. Siffert and B.-L. Su, J. Energy Chem., 2022, 66, 529–559 CrossRef.
- X. Liu, Q. Zhang and D. Ma, Sol. RRL, 2021, 5, 2000397 CrossRef CAS.
- C.-F. Fu, J. Sun, Q. Luo, X. Li, W. Hu and J. Yang, Nano Lett., 2018, 18, 6312–6317 CrossRef CAS PubMed.
- C.-F. Fu, X. Wu and J. Yang, Adv. Mater., 2018, 30, 1802106 CrossRef PubMed.
- A. K. Singh, K. Mathew, H. L. Zhuang and R. G. Hennig, J. Phys. Chem. Lett., 2015, 6, 1087–1098 CrossRef CAS PubMed.
- Y. Li, Y.-L. Li, B. Sa and R. Ahuja, Catal. Sci. Technol., 2017, 7, 545–559 RSC.
- X. Zhang, Z. Zhang, D. Wu, X. Zhang, X. Zhao and Z. Zhou, Small, 2018, 2, 1700359 CrossRef.
| This journal is © The Royal Society of Chemistry 2022 |