
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrocatalytic water oxidation from a mixed linker MOF based on NU-1000 with an integrated ruthenium-based metallo-linker†
Andrew
Howe
ab,
Timofey
Liseev
a,
Marcos
Gil-Sepulcre
 b,
Carolina
Gimbert-Suriñach
b,
Jordi
Benet-Buchholz
b,
Antoni
Llobet
b and
Sascha
Ott
*a
b,
Carolina
Gimbert-Suriñach
b,
Jordi
Benet-Buchholz
b,
Antoni
Llobet
b and
Sascha
Ott
*a
aDepartment of Chemistry – Ångström Laboratory, Uppsala University, Box 523, 75120 Uppsala, Sweden. E-mail: sascha.ott@kemi.uu.se
bInstitute of Chemical Research of Catalonia (ICIQ), Barcelona Institute of Science and Technology (BIST), Av. Països Catalans 16, 43007, Tarragona, Spain
First published on 5th April 2022
Abstract
A novel tetratopic metallo-linker, [Ru(tda)(py(PhCOOH)2)2], 1, (tda = 2,2′:6′,2′′-terpyridine-6,6′′-dicarboxylate; py(PhCOOH)2 = (4,4′-(pyridine-3,5-diyl)dibenzoic acid), that is structurally based on one of the most active molecular water oxidation catalysts has been prepared and fully characterized, including single crystal X-ray diffraction. 1 bears geometric similarities to H4TBAPy (H4TBAPy = 4,4′,4′′,4′′′-(pyrene-1,3,6,8-tetrayl)tetrabenzoic acid), i.e. the native linker in NU-1000, which offers the possibility to synthesize NU-1000-Ru mixed linker MOFs solvothermally. Mixed linker MOF formation was demonstrated by powder X-ray diffraction (PXRD) and scanning electron microscopy (SEM), and Ru linker incorporation confirmed by FT-IR, energy-dispersive X-ray (EDX) spectroscopy and inductively coupled plasma optical emission spectroscopy (ICP-OES). It was found that the Ru contents in the final mixed linker MOFs correlate with the amount of Ru linker present during solvothermal synthesis, albeit not in a linear fashion. The cyclic voltammograms (CV) of the mixed linker MOFs are largely dominated by TBAPy-based oxidations with features attributed to 1. Interestingly, Ru linkers near the crystal surface are oxidized directly by interfacial hole transfer form the electrode, while those in the crystal interior can be oxidized indirectly from oxidized TBAPy linkers at more anodic potential. Upon repeated scanning, the CVs show the appearance of new waves that arise from irreversible TBAPy oxidation, as well as from the activation of the Ru-based water oxidation catalyst. Of the materials prepared, the one with the highest Ru content, NU-1000-Ruhigh, was shown to catalyze the electrochemical oxidation of water to dioxygen. The Faradaic efficiency (FE) of the construct is 37%, due to water oxidation being accompanied by oxidative transformations of the TBAPy linkers. Despite the low FE, NU-1000-Ruhigh is still among the best MOF-based water oxidation catalysts, operating by a unique co-linker mediated hole-transport mechanism to supply oxidizing equivalents also to catalysts in the crystal interior.
Introduction
Metal–organic frameworks (MOFs) are a class of crystalline materials composed of inorganic secondary building units (SBUs) that consist of metal cations or discrete clusters thereof, and that are interconnected in three dimensions by multidentate organic linkers. Due to their modular nature, the large number of unique combinations of SBUs and organic linkers gives rise to an almost endless variety of possible MOF materials. With the properties of MOFs being highly tuneable, they have been tailored, for example, for application in gas storage and separations,1–3 chemical sensing4 and catalysis.5–7For applications that require hydrolytic stability, MOFs with SBUs that consist of high-valent cations such as Zr4+ have moved into the centre of attention. Canonical members of this class of MOFs include UiO-66 (UiO = Universitet i Oslo),8 NU-1000 (NU = North Western University),9 or PCN-700 (PCN = Porous Coordination Network),10 all of which are known for possessing strong coordination bonds between Zr6O4(OH)4 cluster SBUs and carboxylate-containing linkers.
Benefitting from the high structural stability of Zr4+-based MOFs, their high internal surface areas, often with large pore diameters, and their synthetic tunability, these types of MOFs have become popular platforms for the incorporation of molecular electrocatalysts.11 Amongst other transformations, in particular catalysts of energy relevance, i.e. those that catalyse water oxidation, CO2 reduction or hydrogen evolution have attracted great attention in the field.12–15 The vast majority of catalyst motifs that have been investigated in this context are based on linkers that are either native to the MOF, or very similar in design to linkers in existing MOFs. Examples of the former strategy are MOFs that are composed of porphyrin-based linkers and that have been studied for oxygen reduction,16 CO2 reduction17 or hydrogen evolution.18 Examples of the latter strategy include exchanging 4,4′-biphenyldicarboxylates in UiO-67, or isoreticular version thereof, by geometrically identical 2,2′-bipyridine-5,5′-dicarboxylates. These metal binding sites have been used to host a variety of different metal fragments, including Ir and Ru species for water oxidation,19,20 or Mn and Re units for CO2 reduction.21
Examples of novel MOFs with metallo-linkers that are inspired by well-developed molecular electrocatalysts are still scarce.13 Such MOFs require substantial synthetic efforts to decorate the catalytic motif with suitable anchoring groups for SBU coordination. One such example is the cyclam-based VPI-100 (VPI = Virginia Polytechnic Institute), reported by Morris and co-workers (Fig. 1) for CO2 conversion chemistry.22 Another example is the cobaloxime-based UU-100 (UU = Uppsala University) for electrocatalytic hydrogen evolution23 and a report on a Ru-based water oxidation catalyst that has been incorporated into a UiO-type MOF by post-synthetic exchange.24
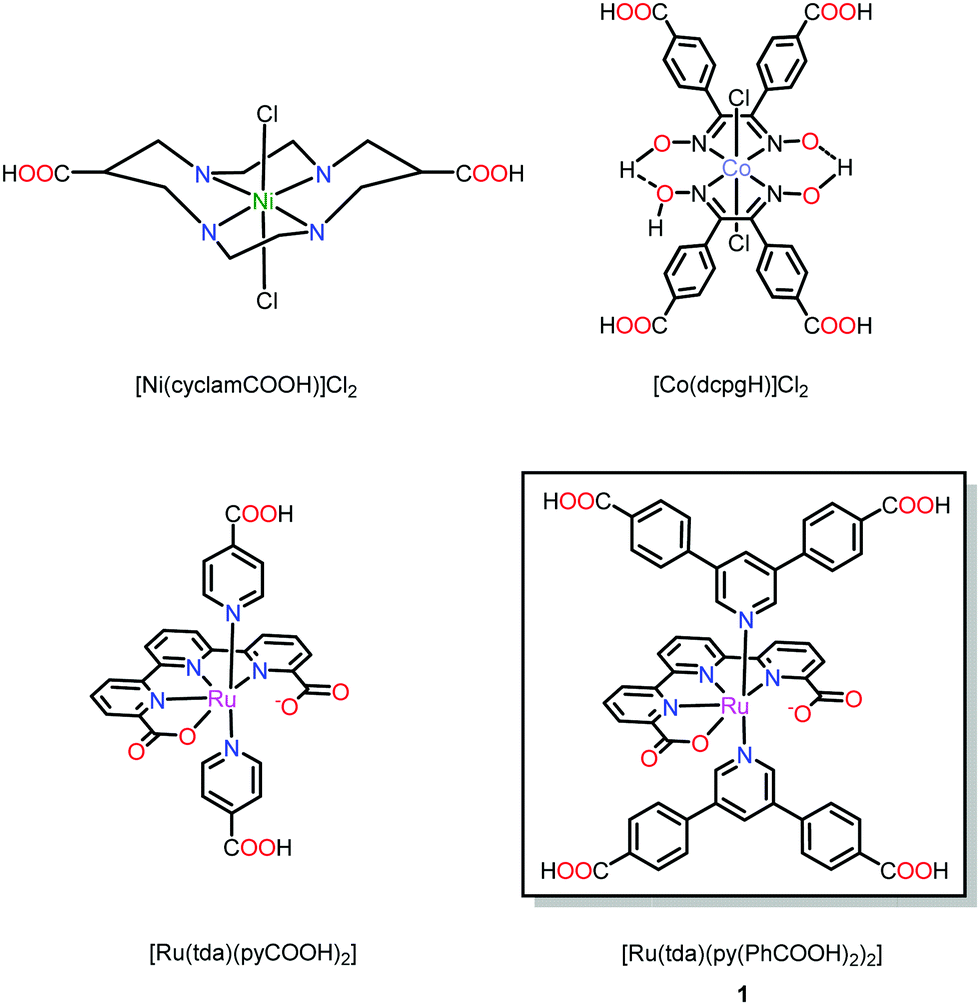 | ||
| Fig. 1 Metallo-linkers based on prominent homogenous catalyst motifs, specifically decorated with anchoring groups for MOF fabrication. | ||
Preparing new MOFs with custom-designed linkers is far from trivial, especially since the constituting linkers are often conformationally flexible and carry functional groups that are important for catalyst function, but may interfere with MOF fabrication. In the current work, we designed and synthesized a new Ru-based linker, 1, that is decorated with four carboxylic acid groups for SBU coordination, making it a tetratopic metallo-linker (Fig. 1). The new linker bears obvious geometric resemblance to a tetra-benzoic acid-decorated pyrene (H4TBAPy) linker that interconnects Zr6O4(OH)4 SBUs in NU-1000. Given the close geometric similarities between H4TBAPy and 1, we explored the possibility to prepare mixed linker MOFs that contain both of the linkers. Such mixed linker MOFs have been reported before, but are usually limited to systems with rather small variations in linker structure.25
From a thermodynamic viewpoint, the use of TBAPy as a co-linker to the catalytic complex 1 is promising as the formal potential for TBAPy oxidation in NU-1000 has been reported as ca. 1.1 V and 1.4 V vs. Ag/AgCl in CH2Cl226 and CH3CN,27 respectively. TBAPy-based oxidations in NU-1000 are thus sufficiently anodic to not interfere with the Ru(III/II) and Ru(IV/III) couples of the [Ru(tda)(py)2]-based linker which are expected to be cathodic of 1.0 V. At such potentials, the native NU-1000 framework is expected to behave as an inert matrix.27–29 The situation can however be expected to change at potentials at which water oxidation occurs, typically at onset potentials just beyond 1.0 V vs. NHE. At these potentials, TBAPy oxidation in NU-1000 can be expected. Thus, the TBAPy linkers may provide a hole transport pathway to efficiently deliver oxidizing equivalents to 1 for efficient electrochemical water oxidation.
Results and discussion
Metallo-linker synthesis and characterization
The synthetic sequence to 1 is outlined in Scheme 1. The axial ligand that provides the coordination bonds to the SBUs was prepared via a Pd-mediated Suzuki cross coupling reaction between 3,5-dibromopyridine and (4-(methoxycarbonyl)phenyl)boronic acid in refluxing dioxane/water (11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (Scheme 1). The obtained methyl ester was saponified by refluxing in an aqueous sodium hydroxide solution to afford the unprotected ligand in 71% yield over both steps. The target metallo-linker 1 was assembled by heating two equivalents of the ligand py(PhCOOH)2 with [RuII(tda-κ-N3O)(dmso)(OH2)] in water with a small quantity of NaOH to reflux for one day. Complex 1 was obtained in an isolated yield of 34% (see Section S1 in the ESI†). Single crystals of 1 could be obtained by slow diffusion of diethyl ether into a solution of the complex in a DMSO/DMF mixture. Structure elucidation by single crystal X-ray diffraction (Fig. 2) revealed an octahedral geometry and the expected coordination environment around the ruthenium centre, where tda2− coordinates the Ru centre in a κ-N3O1 manner, similar to those previously described in literature.30 The two carboxylates of the equatorial tda2− are weakly bonded with a long Ru–O distance of 2.19 Å. The N1–Ru, N2–Ru bond distances are 1.95 Å and 1.94 Å respectively, with the N3–Ru bond distance being larger at 2.13 Å. The angles between the carboxylate and ruthenium are 77.72°, whereas N1–Ru–N2 and N2–Ru–N3 are 81.79° and 78.99°, respectively. The phenyl rings that contain the carboxylic acid groups for SBU coordination are twisted relative to the axial pyridine with an angle of 118.75° and 121.81°, respectively.
:1) (Scheme 1). The obtained methyl ester was saponified by refluxing in an aqueous sodium hydroxide solution to afford the unprotected ligand in 71% yield over both steps. The target metallo-linker 1 was assembled by heating two equivalents of the ligand py(PhCOOH)2 with [RuII(tda-κ-N3O)(dmso)(OH2)] in water with a small quantity of NaOH to reflux for one day. Complex 1 was obtained in an isolated yield of 34% (see Section S1 in the ESI†). Single crystals of 1 could be obtained by slow diffusion of diethyl ether into a solution of the complex in a DMSO/DMF mixture. Structure elucidation by single crystal X-ray diffraction (Fig. 2) revealed an octahedral geometry and the expected coordination environment around the ruthenium centre, where tda2− coordinates the Ru centre in a κ-N3O1 manner, similar to those previously described in literature.30 The two carboxylates of the equatorial tda2− are weakly bonded with a long Ru–O distance of 2.19 Å. The N1–Ru, N2–Ru bond distances are 1.95 Å and 1.94 Å respectively, with the N3–Ru bond distance being larger at 2.13 Å. The angles between the carboxylate and ruthenium are 77.72°, whereas N1–Ru–N2 and N2–Ru–N3 are 81.79° and 78.99°, respectively. The phenyl rings that contain the carboxylic acid groups for SBU coordination are twisted relative to the axial pyridine with an angle of 118.75° and 121.81°, respectively.
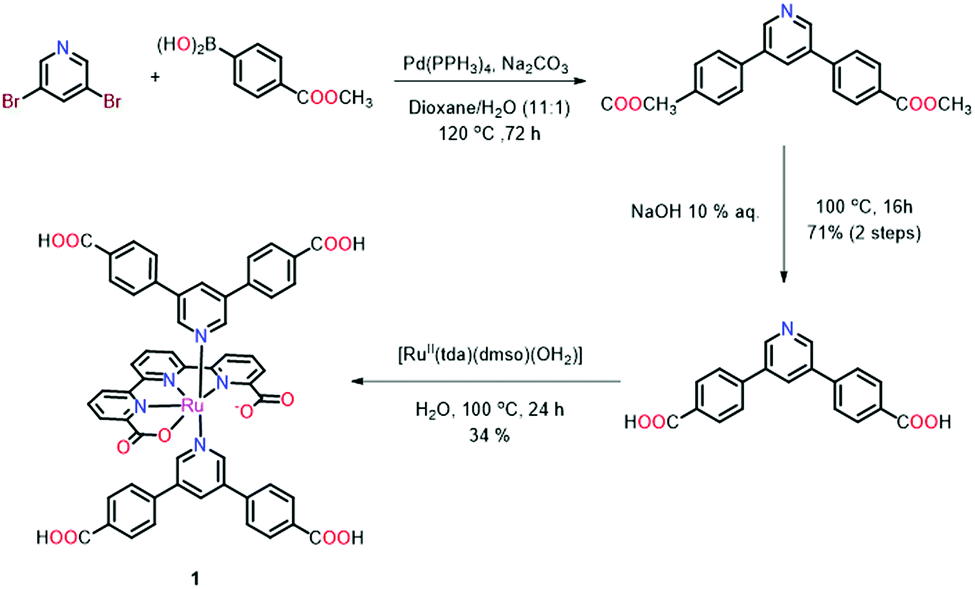 | ||
| Scheme 1 Synthetic scheme for the synthesis of complex 1. | ||
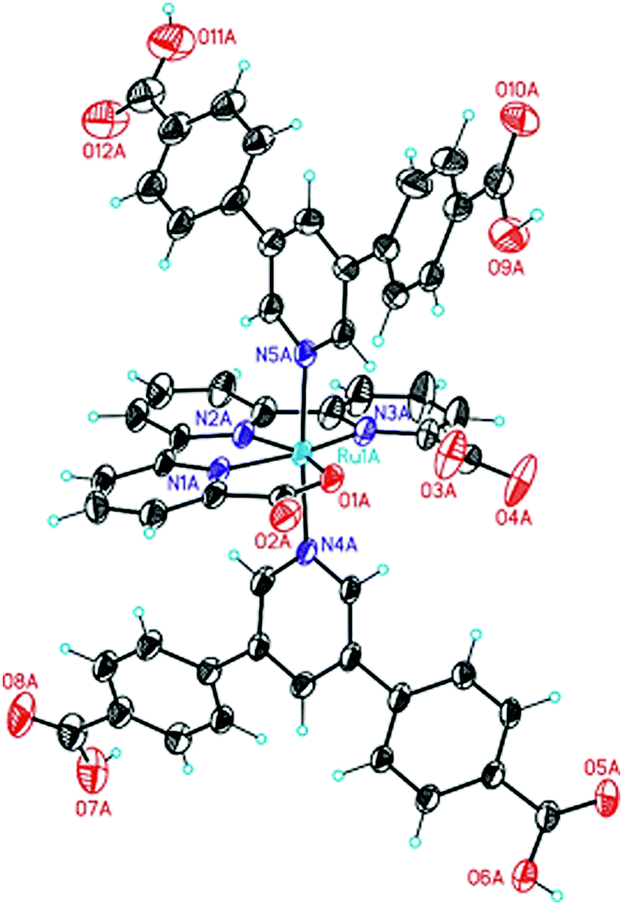 | ||
| Fig. 2 X-Ray crystal structure of 1. | ||
Being a new member of the Ru(tda) family of water oxidation catalysts, its electrochemical behaviour in water was investigated using a glassy carbon disk as working electrode, a platinum mesh counter electrode and Ag/AgCl (3 M NaCl) as a reference electrode. Unless otherwise stated, all CVs described in this work are measured under these conditions and the potentials are reported vs. NHE throughout the manuscript (by adding 0.2 V to measured potential).
Initial cyclic voltammograms of aqueous solutions of complex 1 in 0.1 M sodium phosphate buffer (pH 7) gave inconclusive results due to the hydrophobicity of the complex and its associated low solubility. Thus, complex 1 was suspended with multi-walled carbon nanotubes (MWCNTs) in THF, and subsequently drop-casted onto glassy carbon disk electrodes and used as working electrode. The CV of 1 at pH = 7 shows two reversible waves at E1/2 = 0.61 V and E1/2 = 1.09 V that are assigned to the Ru(III/II) and Ru(IV/III) couple, respectively (see Fig. 3).
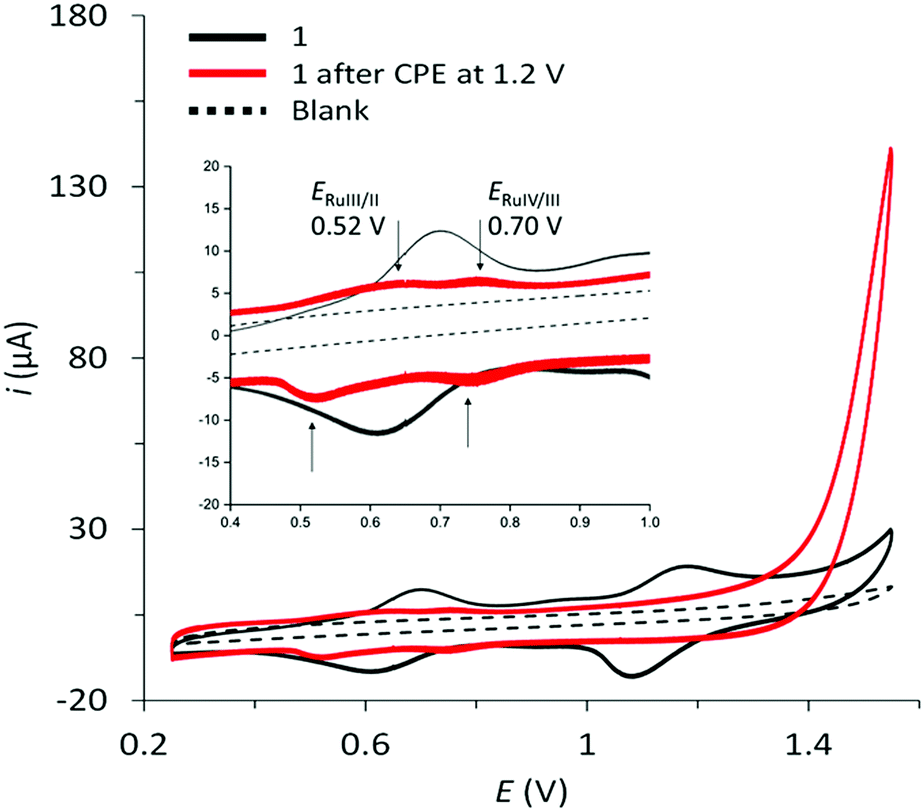 | ||
| Fig. 3 Cyclic voltammogram of 1 immobilized with MWCNTs on a glassy carbon electrode prior to (black trace) and after (red trace) the activation procedure (controlled potential electrolysis at 1.2 V). Background glassy carbon electrode (black dotted trace). 0.1 M phosphate buffer electrolyte; pH = 7; ν = 100 mV s−1. Inset shows a zoom into the oxidation waves observed after catalyst activation. | ||
As for all complexes of the Ru(tda) family, also complex 1 is a pre-catalyst that needs to be activated under oxidizing conditions to reveal its full catalytic potential. This oxidative activation converts the complex to a RuIV state and introduces the indispensable Ru–OH2 unit.31 Full activation of surface-immobilized 1 was achieved by controlled potential electrolysis (CPE) at 1.4 V in a pH 12 phosphate buffer solution (0.1 M) for a period of 40 minutes. As a result, a shift of the redox waves associated with the Ru(III/II) and Ru(IV/III) couples to E1/2 = 0.52 V and E1/2 = 0.70 V is observed, respectively, followed by a large catalytic current associated with electrocatalytic oxidation of water to dioxygen at an onset potential of around 1.19 V (Fig. 3, red trace). The lower currents of the Ru(III/II) and Ru(IV/III) waves after activation are presumably due to partial leaching of the complex from the electrode, which becomes more soluble in water after deprotonation of carboxylic acid functionalities during the activation step at pH 12.
Mixed linker MOF synthesis and characterization
Initial attempts to produce a crystalline material by direct solvothermal synthesis using 1 as the sole linker were not successful, despite the testing of a broad range of different conditions. Considering the geometric similarities between 1 and the pyrene-based H4TBAPy linker in NU-1000 (Scheme 2), the possibility to dope NU-1000 with complex 1 during the solvothermal synthesis was explored.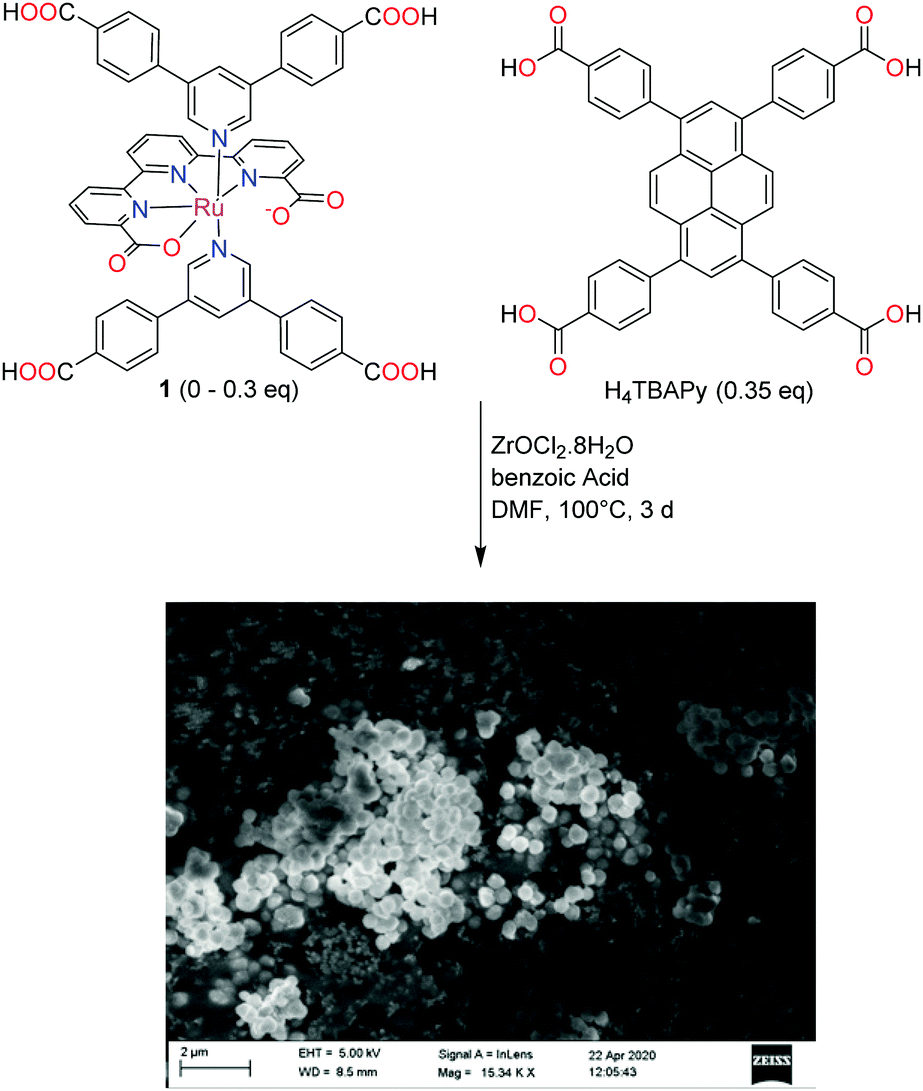 | ||
| Scheme 2 Synthesis of the mixed-linker MOF and an SEM micrograph of one of the obtained mixed linker MOFs (5.7 mol% of 1). | ||
Using the optimized conditions for phase-pure NU-1000 preparation,32 the H4TBAPy linker was complemented by 1 in the solvothermal synthesis. In total, seven materials were prepared by increasing the proportions of the metallo-linker in increments of 5% from 0 to 30% relative to the ZrOCl2 precursor (Scheme 2). NU-1000 synthesis in the absence of any Ru dopant confirmed the reproducibility of the synthetic procedure in our hands, and provided a material to which all Ru-doped materials were compared to.
All mixed-linker solvothermal syntheses produced crystalline materials, as visible by their powder X-ray diffraction patterns (Fig. 4). The reflections of the PXRD are largely located in similar positions as those found in NU-1000,9 with each sample displaying three clearly visible reflections at 2θ = 5.2, 7.5 and 10.4°. Upon increasing proportions of the Ru linker in the solvothermal syntheses, a gradual decrease in the intensities of the PXRD reflections can be observed, which can be assigned to decreased crystallinity of the samples. In fact, gradually decreasing crystallinity is consistent with further experiments in which the Ru linker content was as high as 75 to 100% relative to ZrOCl2, and that do not produce any crystalline material.
 | ||
| Fig. 4 Powder X-ray Diffraction (PXRD) patterns of the mixed linker MOFs. NU-1000 (black trace) and mixed-linker NU-1000-Ru with increasing proportions (from 5 to 30%) of 1 as indicated by the arrow. | ||
A similar picture emerges from an inspection of the scanning electron micrographs of the mixed linker MOFs. While the preparations with low Ru content yielded rod-shaped crystals similar in morphology and size to NU-1000, increasing Ru contents lead to a significant smoothening of the crystal contours, and a decrease in crystal size (Fig. 5). In the most extreme case, the particles deviate significantly from hexagonal rods as in case of NU-1000, and are better described as almost spherical.
 | ||
| Fig. 5 Scanning electron micrographs of the obtained mixed linker MOF materials: from top left to bottom right are shown the MOFs obtained from solvothermal synthesis where the content of Ru(tda)(py(PhCOOH)2)2 was systematically increased from 5 to 30 mol% (in steps of 5%) relative to the used ZrOCl2. | ||
The presence of complex 1 in the mixed linker MOFs was further evidenced by FTIR spectroscopy which showed peaks that are assigned to the metallo-linker. Transitions at 1697 cm−1, 1097 cm−1 (C–O stretch), 865 cm−1, 784 cm−1 and 717 cm−1 correspond to those found in the FTIR spectrum of the homogenous linker (see Fig. S8, ESI†). Additionally, Ru doping was also examined by energy-dispersive X-ray spectroscopy (EDX), which further confirmed the presence of ruthenium throughout the materials (see Fig. S12 and S13, ESI†).
The amount of Ru doping was quantified by ICP-OES analysis of samples that had been digested in concentrated H2O2/HNO3. While the amount of Ru present in the mixed linker MOFs increased with increasing proportions of the metallo-linker in the solvothermal syntheses, a linear correlation could not be established.
The mixed-linker MOF materials can be grouped into two categories, those that have a low Ru content, ≤1.5%, and those with a higher content of ≥4% relative to Zr. A transition between these two groups is observed when the proportion of 1 exceeds 15 mol% relative to ZrOCl2 in the solvothermal syntheses (Table 1). The fact that the ratio between the different linkers in the solvothermal synthesis is not directly mirrored in their abundance in the mixed linker MOF has ample precedence in the literature.
| 1 used in solvothermal synthesis in mol% relative to ZrOCl2 | 1 in final mixed linker MOF in mol% relative to Zr, as determined by ICP-OES |
|---|---|
| 5 | <0.1 |
| 10 | 0.6 |
| 15 | 1.6 |
| 20 | 4.8 |
| 25 | 4.1 |
| 30 | 5.7 |
The phenomenon is usually ascribed to a situation where different linkers have different propensities to form MOFs, for example, as a result of different pKa values of coordinating carboxylates.33 However, in such cases, a linear correlation between linker proportion in the synthesis and their presence in the MOF is still observed. In the present case, the co-incorporation of the 1 and H4TBAPy linkers seems to be disturbed by additional factors. One such factor may be the poor solubility of 1 under the solvothermal conditions, or its higher degree of rotational freedom around the axial N–Ru–N vector.
With NU-1000 and mixed linker MOF materials in hand, focus was directed towards their electrocatalytic water oxidation activity. Similar to the measurements of the homogenous 1, the MOFs were suspended and sonicated in THF, mixed with a separately prepared suspension of MWCNTs, and drop-casted onto glassy carbon working electrodes (see Fig. S1, ESI†).
Fig. 6 A shows multiple CV scans of NU-1000 carried out at pH 7 in a phosphate buffer (0.1 M). The first CV scan (in blue) shows an anodic feature in the range of 1.0–1.4 V that is assigned to the oxidation of the TBAPy linker. In the aqueous phosphate buffer used, this oxidation is electrochemically irreversible, and the corresponding cathodic feature in the reverse scan of the CV is largely absent. Consequently, consecutive CV cycles show a continuous decrease of the TBAPy oxidation and new waves appear in the range of −0.2 to 0.3 V. The appearance of these features is assigned to TBAPy-derived products that are formed in the electrochemically irreversible oxidation of the linkers.
The CV of the mixed linker MOF with the highest content of 1 (5.7%), NU-1000-Ruhigh, is shown in Fig. 6B together with that of NU-1000. For NU-1000-Ruhigh, an additional anodic wave can be observed at Ep,a = 0.8 V that is absent in the parent MOF. Consequently, this wave is assigned to the incorporated Ru linker. Finally, Fig. 6C shows the 20th CV scan for both NU-1000 and NU-1000-Ruhigh. While the CVs of both materials show the TBAPy-derived product linkers at potentials more negative than 0.3 V, the 20th CV scan of NU-1000-Ruhigh shows the evolution of additional waves at E1/2 = 0.55 and 0.71 V. At the same time, the anodic feature at Ep,a = 0.8 V is absent. The new waves are typical for activated forms of Ru(tda) complexes,31 and are a further demonstration for the successful incorporation of the Ru-linker into NU-1000 material.
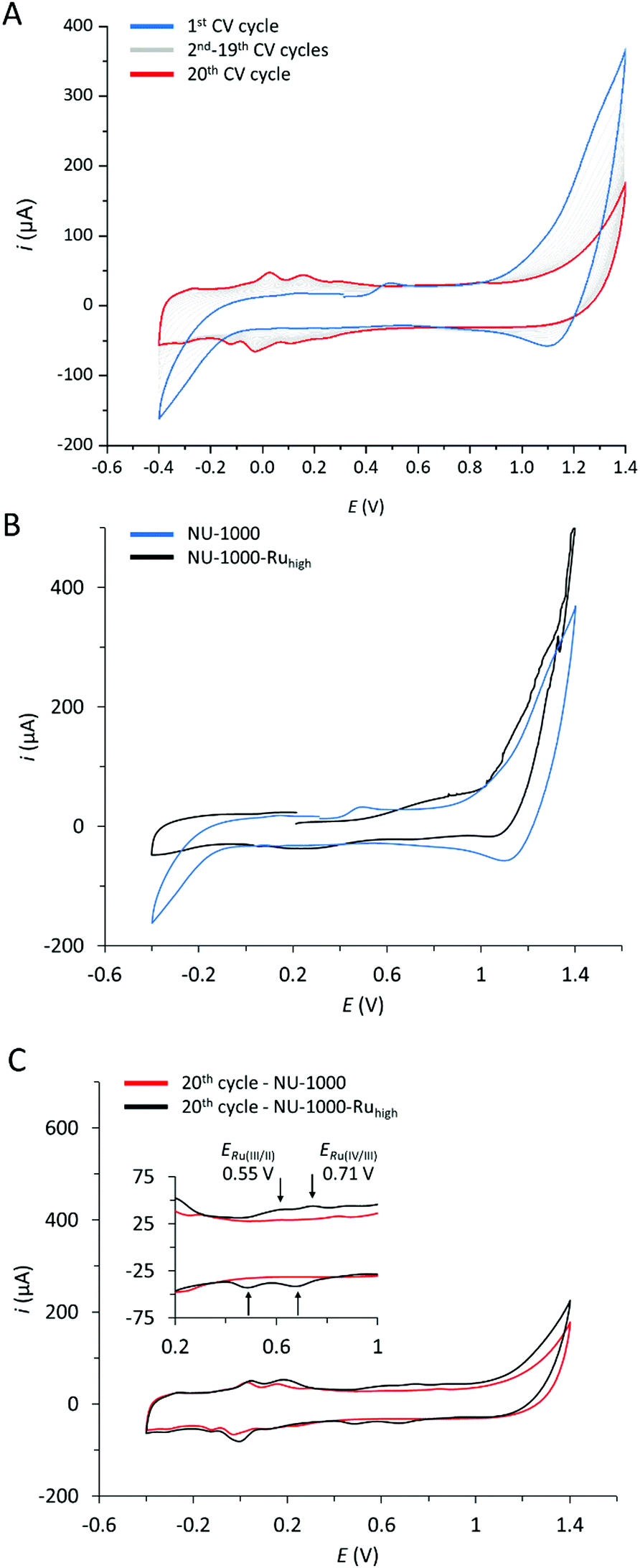 | ||
| Fig. 6 Cyclic voltammetry of MOFs immobilized together with MWCNTs on glassy carbon working electrodes (0.1 M phosphate buffer electrolyte; pH = 7; ν = 100 mV s−1.). (A) Multiple scans of NU-1000. Initial cycle of NU-1000 (black trace), cycles 2–19 (grey trace) and final cycle of NU-1000 (red trace). (B) Initial cycles of NU-1000 (black trace), and NU-1000-Ruhigh (blue trace). (C) 20th CV scans of NU-1000 and NU-1000-Ruhigh; inset shows a zoom into the Ru-based oxidations of NU-1000-Ruhigh. | ||
The currents for the Ru-based oxidations at the glassy carbon electrode are relatively low, as the number of Ru-linkers that are in close contact to the MWCNT is small, and Ru–to–Ru electron hopping charge transport can be expected to be very slow due to the low incorporation yield of 1 and resulting large spatial separation.34 The TBAPy linkers are not conducting at potentials below 1.0 V either,35 and do thus not provide hole transport pathways at potentials of, for example, the Ru(III/II) couple.36 This situation however changes at more positive potentials at which TBAPy oxidation occurs and hole hopping transport throughout the whole crystalline material is enabled. Every oxidized TBAPy linker is a sufficiently strong oxidant to drive the oxidation of the Ru linkers through a mediated process. In other words, while the direct oxidation of 1 in CV experiments may be limited to near-surface site, potentially all Ru-linkers can be oxidized via the TBAPy moieties at higher applied potentials. Consequently, the Ru(III/II) and Ru(IV/III) oxidations may only feature as very small waves in the CVs of NU-1000-Ruhigh, but will nevertheless occur in a TBAPy-mediated process at more positive applied potentials.
With the possibility to engage a large proportion of the metallo-linkers in catalysis, CPE experiments were conducted with NU-1000-Ruhigh to evaluate its capacity to catalyse electrochemical water oxidation. NU-1000-Ruhigh was used as the working electrode in a gas-tight two-compartment electrochemical cell that was coupled to a Clark-type electrode to measure oxygen evolution. Fig. 7 shows the oxygen evolution as a function of time at an applied potential of 1.30 V. After 80 minutes of CPE, NU-1000-Ruhigh (black trace) shows the generation of 1.94 μmols of O2. With a total charge of 2.006 C having passed through the cell, a faradaic efficiency (FE) of 37% can be calculated. The low FE is most likely due to TBAPy oxidations that occur at the applied potential as described above and clearly observed in the repetitive CVs in Fig. 6A. A control experiment with parent NU-1000 generates a negligible amount of oxygen (Fig. 7, red trace), manifesting the role of the Ru metallo-linker as water oxidation catalyst.
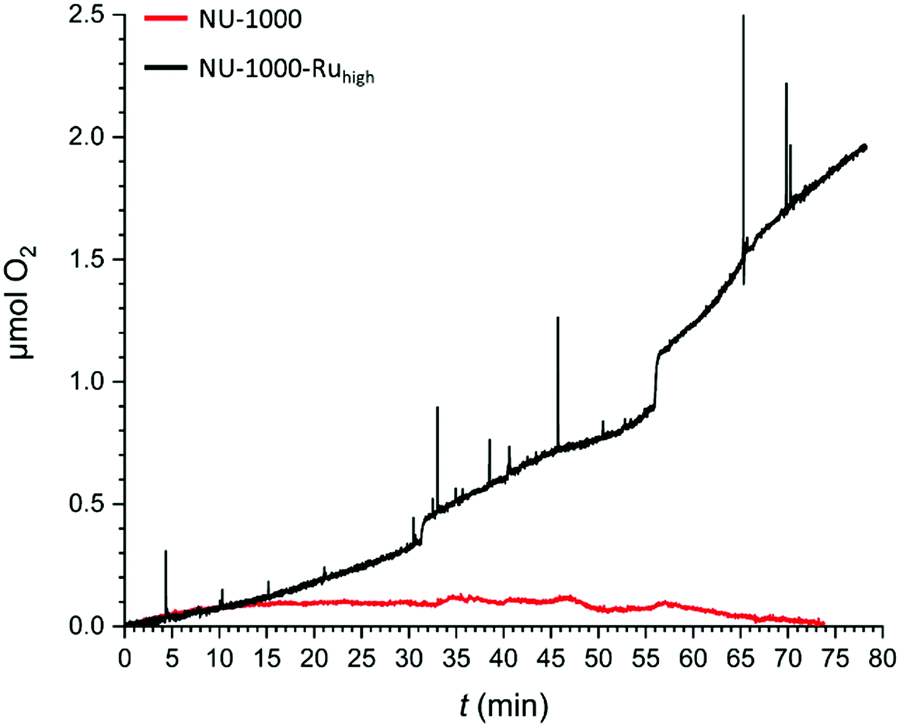 | ||
| Fig. 7 Controlled potential electrolysis (CPE) coupled to oxygen evolution measurement of NU-1000 (red) and NU-1000-Ruhigh (black). Oxygen evolution vs. time traces were recorded at an applied potential of 1.3 V vs. NHE in a 0.1 M phosphate buffer (pH 7), monitored with a gas phase Clark electrode. A faradaic efficiency of 37% was calculated at 80 minutes for NU-1000-Ruhigh. | ||
As Ru linkers in the interior of the MOF crystals can only be oxidized in a TBAPy-mediated mechanism, an independent determination of electroactive Ru sites in NU-1000-Ruhigh is not possible. Assuming that catalysis occurs only at the near-surface Ru sites that also give rise to the CV response in Fig. 6C, a TON of 44600 can be calculated. It is however important to note that this number should only be seen as an upper limit, as most likely also Ru linkers in the interior of the MOF crystals contribute to catalysis, which would give rise to significantly lower TONs.
Conclusions
In summary, we have designed and synthesized a new tetratopic linker 1, which is based on one of the most active water oxidation catalysts reported to date.30 Complex 1 has been characterized by spectroscopic techniques and single crystal X-ray diffraction. The linker was further characterized by cyclic voltammetry, and was shown to behave like all other members of this family of water oxidation catalysts. Given the geometric similarities between 1 and H4TBAPy, i.e. the native linker in NU-1000, the preparation of mixed-linker MOFs was explored. Successful MOF formation was confirmed by PXRD and SEM, while Ru linker incorporation was specifically shown by FTIR, EDX and ICP-OES analyses. It was found that the Ru contents in the final mixed linker MOFs correlate with the amount of Ru linker present during solvothermal syntheses, albeit not in a linear fashion. The lack of linear correlation may be due to vastly different solubility of the two linkers during solvothermal synthesis, slight differences in size, and a higher rotational flexibility of 1.The Ru centres in NU-1000-Ruhigh can be activated like any other Ru(tda)-based water oxidation catalyst, as evidenced by the emergence of new voltammetric waves from the activated catalyst. These CV features arise from Ru linkers that are positioned near the crystal surface, in close contact to the MWCNT electrode. The majority of the Ru linkers in the crystal are not directly accessible electrochemically, but can be oxidized indirectly through prior oxidation of the TBAPy linkers.
Such mediated electron transport is a phenomenon that has precedence on the cathodic side,37,38 but, to the best of our knowledge, is unprecedented for anodic processes. The process bears some resemblance to DNA mediated hole transport that can operate over multiple nanometers.39
Once activated, the Ru metallo-linkers catalyse electrochemical water oxidation at an applied potential of 1.30 V vs. NHE. Unfortunately, in the aqueous electrolyte, the TBAPy co-linkers are oxidatively transformed at the same potential, giving rise to a low Faradaic efficiency for water oxidation catalysed by 1 of 37%. The strategy to supply oxidizing equivalents to catalytic centres through redox-active co-linkers is an interesting strategy to “dilute” catalytically active sites while not compromising the charge transport properties of the material. By doing so, the two functions can be optimized separately, and the MOF-borne catalysis may become more effective as larger proportions of the material actually engage in catalysis.40 Mediated electron/hole transport pathways thus offers exciting new avenues in MOF research, and we foresee increasing interest on the topic in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the European Research Council through a Consolidator Grant (ERC-CoG2015-681895_MOFcat) and the EU-funded ITN eSCALED (Grant agreement ID: 765376) are gratefully acknowledged. AL gratefully acknowledges the support from Ministerio de Ciencia e Innovacion and FEDER (PID2019-111617RB-I00 and SO-CEX2019-000925-S).References
- J. R. Li, R. J. Kuppler and H. C. Zhou, Selective gas adsorption and separation in metal-organic frameworks, Chem. Soc. Rev., 2009, 38(5), 1477–1504 RSC.
- L. J. Murray, M. Dinca and J. R. Long, Hydrogen storage in metal-organic frameworks, Chem. Soc. Rev., 2009, 38(5), 1294–1314 RSC.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O’Keeffe and O. M. Yaghi, Systematic design of pore size and functionality in isoreticular MOFs and their application in methane storage, Science, 2002, 295(5554), 469–472 CrossRef CAS PubMed.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Metal–organic framework materials as chemical sensors, Chem. Rev., 2012, 112(2), 1105–1125 CrossRef CAS PubMed.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C. Y. Su, Applications of metal-organic frameworks in heterogeneous supramolecular catalysis, Chem. Soc. Rev., 2014, 43(16), 6011–6061 RSC.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Metal–organic framework materials as catalysts, Chem. Soc. Rev., 2009, 38(5), 1450–1459 RSC.
- L. Ma, C. Abney and W. Lin, Enantioselective catalysis with homochiral metal-organic frameworks, Chem. Soc. Rev., 2009, 38(5), 1248–1256 RSC.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, A new zirconium inorganic building brick forming metal organic frameworks with exceptional stability, J. Am. Chem. Soc., 2008, 130(42), 13850–13851 CrossRef PubMed.
- J. E. Mondloch, W. Bury, D. Fairen-Jimenez, S. Kwon, E. J. DeMarco, M. H. Weston, A. A. Sarjeant, S. T. Nguyen, P. C. Stair, R. Q. Snurr, O. K. Farha and J. T. Hupp, Vapor-phase metalation by atomic layer deposition in a metal-organic framework, J. Am. Chem. Soc., 2013, 135(28), 10294–10297 CrossRef CAS PubMed.
- S. Yuan, W. Lu, Y. P. Chen, Q. Zhang, T. F. Liu, D. Feng, X. Wang, J. Qin and H. C. Zhou, Sequential linker installation: precise placement of functional groups in multivariate metal-organic frameworks, J. Am. Chem. Soc., 2015, 137(9), 3177–3180 CrossRef CAS PubMed.
- B. D. McCarthy, A. M. Beiler, B. A. Johnson, T. Liseev, A. T. Castner and S. Ott, Analysis of Electrocatalytic Metal–Organic Frameworks, Coord. Chem. Rev., 2020, 406 Search PubMed.
- T. Zhang and W. Lin, Metal–organic frameworks for artificial photosynthesis and photocatalysis, Chem. Soc. Rev., 2014, 43(16), 5982–5993 RSC.
- S. Banerjee, R. I. Anayah, C. S. Gerke and V. S. Thoi, From Molecules to Porous Materials: Integrating Discrete Electrocatalytic Active Sites into Extended Frameworks, ACS Cent. Sci., 2020, 6(10), 1671–1684 CrossRef CAS PubMed.
- Y. H. Budnikova, Recent advances in metal-organic frameworks for electrocatalytic hydrogen evolution and overall water splitting reactions, Dalton Trans., 2020, 49(36), 12483–12502 RSC.
- W. Wang, X. M. Xu, W. Zhou and Z. P. Shao, Recent Progress in Metal–Organic Frameworks for Applications in Electrocatalytic and Photocatalytic Water Splitting, Adv. Sci., 2017, 4, 1600371 CrossRef PubMed.
- I. Liberman, R. Shimoni, R. Ifraemov, I. Rozenberg, C. Singh and I. Hod, Active-Site Modulation in an Fe-Porphyrin-Based Metal–Organic Framework through Ligand Axial Coordination: Accelerating Electrocatalysis and Charge-Transport Kinetics, J. Am. Chem. Soc., 2020, 142(4), 1933–1940 CrossRef CAS PubMed.
- N. Kornienko, Y. Zhao, C. S. Kley, C. Zhu, D. Kim, S. Lin, C. J. Chang, O. M. Yaghi and P. Yang, Metal–Organic Frameworks for Electrocatalytic Reduction of Carbon Dioxide, J. Am. Chem. Soc., 2015, 137(44), 14129–14135 CrossRef CAS PubMed.
- D. Micheroni, G. Lan and W. Lin, Efficient Electrocatalytic Proton Reduction with Carbon Nanotube-Supported Metal–Organic Frameworks, J. Am. Chem. Soc., 2018, 140(46), 15591–15595 CrossRef CAS PubMed.
- S. Lin, Y. Pineda-Galvan, W. A. Maza, C. C. Epley, J. Zhu, M. C. Kessinger, Y. Pushkar and A. J. Morris, Electrochemical Water Oxidation by a Catalyst-Modified Metal–Organic Framework Thin Film, ChemSusChem, 2017, 10(3), 514–522 CrossRef CAS PubMed.
- B. A. Johnson, A. Bhunia and S. Ott, Electrocatalytic water oxidation by a molecular catalyst incorporated into a metal-organic framework thin film, Dalton Trans., 2017, 46(5), 1382–1388 RSC.
- H. Fei, M. D. Sampson, Y. Lee, C. P. Kubiak and S. M. Cohen, Photocatalytic CO2 Reduction to Formate Using a Mn(I) Molecular Catalyst in a Robust Metal–Organic Framework, Inorg. Chem., 2015, 54(14), 6821–6828 CrossRef CAS PubMed.
- J. Zhu, P. M. Usov, W. Xu, P. J. Celis-Salazar, S. Lin, M. C. Kessinger, C. Landaverde-Alvarado, M. Cai, A. M. May, C. Slebodnick, D. Zhu, S. D. Senanayake and A. J. Morris, A New Class of Metal-Cyclam-Based Zirconium Metal–Organic Frameworks for CO2 Adsorption and Chemical Fixation, J. Am. Chem. Soc., 2018, 140(3), 993–1003 CrossRef CAS PubMed.
- S. Roy, Z. Huang, A. Bhunia, A. Castner, A. K. Gupta, X. Zou and S. Ott, Electrocatalytic Hydrogen Evolution from a Cobaloxime-Based Metal–Organic Framework Thin Film, J. Am. Chem. Soc., 2019, 141(40), 15942–15950 CrossRef CAS PubMed.
- T. Liseev, A. Howe, M. A. Hoque, C. Gimbert-Surinach, A. Llobet and S. Ott, Synthetic strategies to incorporate Ru-terpyridyl water oxidation catalysts into MOFs: direct synthesis vs. post-synthetic approach, Dalton Trans., 2020, 49(39), 13753–13759 RSC.
- H. Deng, C. J. Doonan, H. Furukawa, R. B. Ferreira, J. Towne, C. B. Knobler, B. Wang and O. M. Yaghi, Multiple functional groups of varying ratios in metal-organic frameworks, Science, 2010, 327(5967), 846–850 CrossRef CAS PubMed.
- I. Hod, W. Bury, D. M. Karlin, P. Deria, C. W. Kung, M. J. Katz, M. So, B. Klahr, D. Jin, Y. W. Chung, T. W. Odom, O. K. Farha and J. T. Hupp, Directed growth of electroactive metal-organic framework thin films using electrophoretic deposition, Adv. Mater., 2014, 26(36), 6295–6300 CrossRef CAS PubMed.
- I. Hod, W. Bury, D. M. Gardner, P. Deria, V. Roznyatovskiy, M. R. Wasielewski, O. K. Farha and J. T. Hupp, Bias-Switchable Permselectivity and Redox Catalytic Activity of a Ferrocene-Functionalized, Thin-Film Metal–Organic Framework Compound, J. Phys. Chem. Lett., 2015, 6(4), 586–591 CrossRef CAS PubMed.
- Q. Chen, J. Sun, P. Li, I. Hod, P. Z. Moghadam, Z. S. Kean, R. Q. Snurr, J. T. Hupp, O. K. Farha and J. F. Stoddart, A Redox-Active Bistable Molecular Switch Mounted inside a Metal–Organic Framework, J. Am. Chem. Soc., 2016, 138(43), 14242–14245 CrossRef CAS PubMed.
- S. Goswami, I. Hod, J. D. Duan, C.-W. Kung, M. Rimoldi, C. D. Malliakas, R. H. Palmer, O. K. Farha and J. T. Hupp, Anisotropic Redox Conductivity within a Metal–Organic Framework Material, J. Am. Chem. Soc., 2019, 141(44), 17696–17702 CrossRef CAS PubMed.
- R. Matheu, M. Z. Ertem, C. Gimbert-Surinach, X. Sala and A. Llobet, Seven Coordinated Molecular Ruthenium-Water Oxidation Catalysts: A Coordination Chemistry Journey, Chem. Rev., 2019, 119(6), 3453–3471 CrossRef CAS PubMed.
- R. Matheu, M. Z. Ertem, J. Benet-Buchholz, E. Coronado, V. S. Batista, X. Sala and A. Llobet, Intramolecular Proton Transfer Boosts Water Oxidation Catalyzed by a Ru Complex, J. Am. Chem. Soc., 2015, 137(33), 10786–10795 CrossRef CAS PubMed.
- T. Islamoglu, K.-i. Otake, P. Li, C. T. Buru, A. W. Peters, I. Akpinar, S. J. Garibay and O. K. Farha, Revisiting the structural homogeneity of NU-1000, a Zr-based metal–organic framework, CrystEngComm, 2018, 20(39), 5913–5918 RSC.
- A. D. Burrows, L. C. Fisher, C. Richardson and S. P. Rigby, Selective incorporation of functional dicarboxylates into zinc metal-organic frameworks, Chem. Commun., 2011, 47(12), 3380–3382 RSC.
- S. Lin, P. M. Usov and A. J. Morris, The role of redox hopping in metal-organic framework electrocatalysis, Chem. Commun., 2018, 54(51), 6965–6974 RSC.
- E. Mijangos, S. Roy, S. Pullen, R. Lomoth and S. Ott, Evaluation of two- and three-dimensional electrode platforms for the electrochemical characterization of organometallic catalysts incorporated in non-conducting metal-organic frameworks, Dalton Trans., 2017, 46(15), 4907–4911 RSC.
- G. S. Mohammad-Pour, K. O. Hatfield, D. C. Fairchild, K. Hernandez-Burgos, J. Rodriguez-Lopez and F. J. Uribe-Romo, A Solid-Solution Approach for Redox Active Metal–Organic Frameworks with Tunable Redox Conductivity, J. Am. Chem. Soc., 2019, 141(51), 19978–19982 CrossRef CAS PubMed.
- A. T. Castner, B. A. Johnson, S. M. Cohen and S. Ott, Mimicking the Electron Transport Chain and Active Site of [FeFe] Hydrogenases in One Metal–Organic Framework: Factors That Influence Charge Transport, J. Am. Chem. Soc., 2021, 143(21), 7991–7999 CrossRef CAS PubMed.
- H. Noh, C.-W. Kung, K.-I. Otake, A. W. Peters, Z. Li, Y. Liao, X. Gong, O. K. Farha and J. T. Hupp, Redox-Mediator-Assisted Electrocatalytic Hydrogen Evolution from Water by a Molybdenum Sulfide-Functionalized Metal–Organic Framework. ACS, Catalysis, 2018, 8(10), 9848–9858 CAS.
- J. D. Slinker, N. B. Muren, S. E. Renfrew and J. K. Barton, DNA charge transport over 34 nm, Nat. Chem., 2011, 3(3), 228–233 CrossRef CAS PubMed.
- B. A. Johnson, A. M. Beiler, B. D. McCarthy and S. Ott, Transport Phenomena: Challenges and Opportunities for Molecular Catalysis in Metal–Organic Frameworks, J. Am. Chem. Soc., 2020, 142(28), 11941–11956 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details: synthesis, characterization, spectroscopic and electrochemical analyses. CCDC 2128791. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ma00128d |
| This journal is © The Royal Society of Chemistry 2022 |