
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApplication of ionic liquids for the functional materialization of chitin
Jun-ichi
Kadokawa

Graduate School of Science and Engineering, Kagoshima University, 1-21-40 Korimoto, Kagoshima 890-0065, Japan. E-mail: kadokawa@eng.kagoshima-u.ac.jp
First published on 14th March 2022
Abstract
Ionic liquids (ILs) have been shown to efficiently dissolve substrates with poor solubility. In particular, they act as powerful solvents for structural polysaccharides, which are difficult to dissolve using conventional solvents. Chitin is a representative structural polysaccharide and an important biomass resource because of its large annual production in nature. However, it is mostly unutilized owing its poor solubility and processability. Since 2008, some ILs, such as imidazolium carboxylates and 1-allyl-3-methylimidazolium bromide (AMIMBr), have been found to dissolve chitin. This perspective article overviews the applicability of these ILs for the functional materialization of chitin, for example, the fabrication of gels, films, and composites, through its dissolution in ILs. Bottom-up self-assembly processes from solutions and gels with ILs, that is, regeneration and electrospinning methods, for the fabrication of chitin nanofibers are also presented. Moreover, AMIMBr is shown to act as a reaction medium for chitin acylation. The resulting derivatives are subsequently used for graft polymerization to provide additional functions to chitin, such as thermoplasticity.
 Jun-ichi Kadokawa | Jun-ichi Kadokawa received his PhD at Tohoku University in 1992. He then joined Yamagata University as a Research Associate. From 1996 to 1997, he worked as a visiting scientist at the Max-Planck-Institute for Polymer Research in Germany. In 1999, he became an Associate Professor at Yamagata University and moved to Tohoku University in 2002. He was appointed as a Professor of Kagoshima University in 2004. He received the Award for Encouragement of Research in Polymer Science (1997), the Cellulose Society of Japan Award (2010), the Royalty Award from Institute of Materials, Malaysia (2016), and IAAM Medal (2016). |
Introduction
Ionic liquids (ILs) are low melting point molten salts that form liquids at room temperature or even at temperatures below the boiling point of water.1 This specific property is attributed to the fact that the liquid state is thermodynamically favorable because of the large size (mainly the cations) and conformational flexibility of the ions, which results in small lattice enthalpies and large entropy changes that favor the liquid state (Fig. 1). ILs have been identified as powerful solvents for various organic and inorganic materials with poor solubilities. For example, ILs have been used to dissolve natural polysaccharides, which are often insoluble in water and common organic solvents.2 | ||
| Fig. 1 Large ion sizes induce liquid state of ionic material. | ||
Natural polysaccharides are the most abundant organic substances and biomass resources on the earth and mainly serve two important roles: structural materials and suppliers of water and energy.3,4 Cellulose and chitin are two representative structural polysaccharides with huge annual productions, which are mainly present in the cell walls of plants and the exoskeletons of crustaceans, respectively.5–8 These polysaccharides are composed of repeating D-glucose and N-acetyl-D-glucosamine units, respectively, which are linked by β(1 → 4)-glycosidic linkages (Fig. 2). These specific structures allow for the formation of numerous regularly controlled intra- and intermolecular hydrogen bonds, resulting in high crystallinity and extended fibrous chain packing. Therefore, structural polysaccharides exhibit poor solubility in common solvents compared to that of energy storage polysaccharides, such as starch. Cellulose has traditionally been used in fibrous and structural applications, such as wood, paper, textiles, and furniture. Compared with cellulose, chitin is an unused biomass resource, mainly because of the presence of acetamido groups at the C-2 position in the N-acetyl-D-glucosamine units, which form strong intermolecular hydrogen bonds. Therefore, the solubility of chitin is inferior to that of cellulose, leading to poor processability and underutilization. However, chitin market was valued at $42.29 million in 2020 and is projected to reach $69.297 billion in 2028, growing at a CAGR (Compound Annual Growth Rate) of 5.07% from 2021 to 2028.9 The market demand is basically owing to the market growth of biopolymer processing, principally from its biological properties.10 Rise in the chitin demand of uses as nanomaterials is also the key market driver.
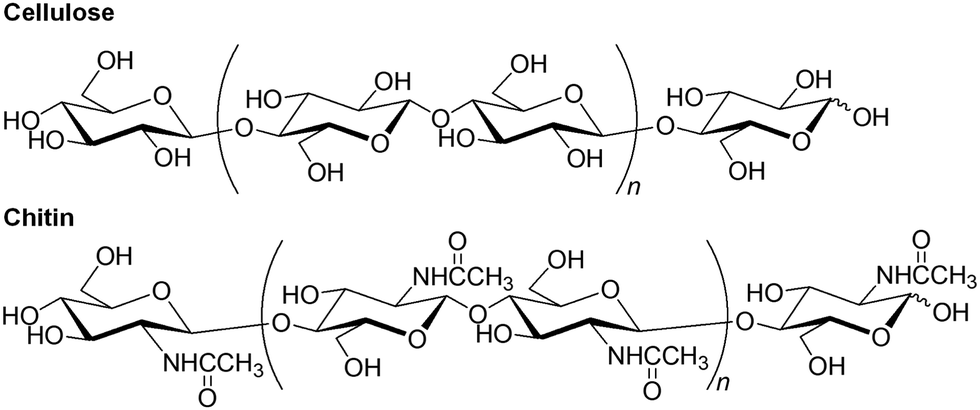 | ||
| Fig. 2 Structures of cellulose and chitin. | ||
Since an IL, 1-butyl-3-methylimidazolium chloride (BMIMCl), was reported to dissolve cellulose in 2002,11 numerous ILs have been used as solvents for cellulose and other polysaccharides.12–21 Furthermore, ILs have been used as media for the derivatization, modification, and functionalization of cellulose.14,17,18 Owing to its aforementioned poor solubility, ILs that can dissolve chitin were not found until 2008.22–24 However, some ILs have been reported to dissolve chitin.25–28 Moreover, these ILs are used to modify chitin, such as soft materialization, nanomaterialization, derivatization, and modification, to provide new functionalities on chitin.26,29–34 This perspective article overviews recent progress in the efficient application of ILs for the functional materialization of chitin. Processability of chitin in different ILs into different materials and their potential applications are concisely summarized in the previously reported review articles.26,32 Although deep eutectic solvents, analogous of ILs, have also been employed for the dissolution and subsequent materialization of chitin,35–39 ILs have some advantages based on the viewpoint of a structural variety with wide range of properties.
Dissolution of chitin in ILs
The dissolution of chitin in an IL was first reported using 1-butyl-3-methylimidazolium acetate (BMIMOAc, Fig. 3) in 2008.40 Chitins of various origins and molecular weights were dissolved in BMIMOAc at 110 °C at satisfactory concentrations (maximum of 6 wt% chitin from crab shells). By cooling the chitin/BMIMOAc solutions to the ambient temperature, the corresponding gels were formed, which were further converted into chitin sponge and film materials by regeneration using a water or methanol coagulant. The dissolution behaviors of chitin in a series of alkylimidazolium chloride, dimethyl phosphate, and 1-allyl-3-methylimidazolium acetate (AMIMOAc, Fig. 3) ILs was explored. The former two series of ILs did not dissolve satisfactory amounts of chitin (less than 1.5 wt%), while the latter IL dissolved chitin to 5 wt%.22 Ethyl-3-methylimidazolium acetate (EMIMOAc, Fig. 3) was also reported to dissolve chitin to certain concentrations,41 and it was used to extract chitin from raw crustacean shells, such as shrimp shells. 1-Ethyl-3-methylimidazolium alkanoates with different chain lengths and 1-ethyl-3-methylimidazolium/tetrabutylphosphonium amino acid salts were also found to dissolve chitin.42 The dissolution of chitin using cyclic ammonium acetates and lactates with different substituents was explored, which formed solutions containing 1–9 wt% chitin.43 Chitin was also extracted from shrimp shells using several (hydroxy)ammonium acetate ILs.44,45 Overall, the above dissolution studies of chitin in ILs suggest that ILs with carboxylate anions generally act as good solvents for chitin. | ||
| Fig. 3 Representative ionic liquids (ILs), which dissolve chitin. | ||
Another type of IL, 1-allyl-3-methylimidazolium bromide (AMIMBr, Fig. 3), was used to dissolve chitin in concentrations of up to 4.8 wt% at 100 °C (Fig. 4a).46 This IL can be prepared by quaternarization of 1-methylimidazole with ally bromide in good yield, which is the more facile procedure than that for the abovementioned imidazolium carboxylates.47 The presence of a small quantity of bromide anions in 1-allyl-3-methylimidazolium chloride, which were generated by in situ anion exchange with 2-bromoethyl acetate as a bromide generator, enhanced the dissolution of chitin.48 The facile extraction of chitin from crab shells was achieved using AMIMBr, followed by demineralization using citric acid.49 Solubility of different chitin types in various ILs is comprehensively listed in the review article by Shamshina.27
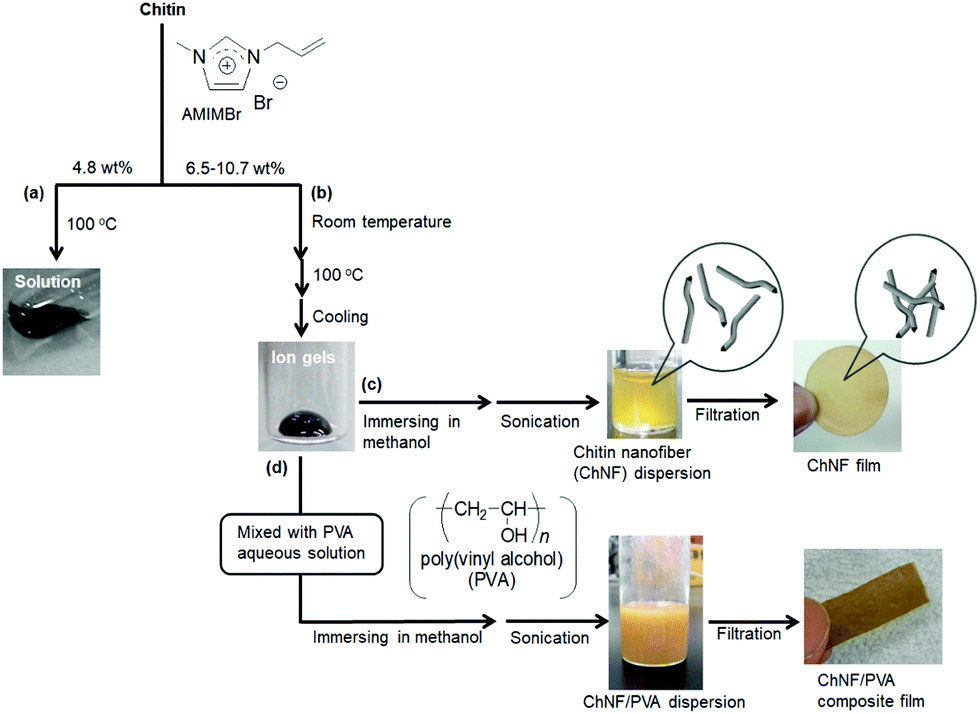 | ||
| Fig. 4 (a) Dissolution and (b) gelation of chitin with AMIMBr and preparation of (c) self-assembled ChNF dispersion/film and (d) ChNF/PVA composite film (reprinted with permission from ref. 33. Copyright 2020 Elsevier; DOI: 10.1016/B978-0-12-817968-0.00002-0). | ||
Molecular dynamics (MD) simulations were conducted to evaluate the dissolution behavior of chitin crystals in AMIMBr,48 and the results suggested that the chitin chains were peeled from the crystal surface, accompanied by cleavage of hydrogen bonds. The MD results also revealed the subsequent dissolution process, in which Br− contributed to the cleavage of intermolecular hydrogen bonds, mostly by acetamido groups, whereas AMIM+ prevented the return to the crystalline phase after peeling (Fig. 5). On the other hand, the MD results indicated that molecular chain peeling was not observed in BMIMCl. Although the weak hydrogen bonds by hydroxy groups were cleaved and division of a crystal into blocks occurred, the stacked chains were still maintained in BMIMCl, probably due to weaker capability of Cl− than Br− to cleave hydrogen bonds by acetamido groups.
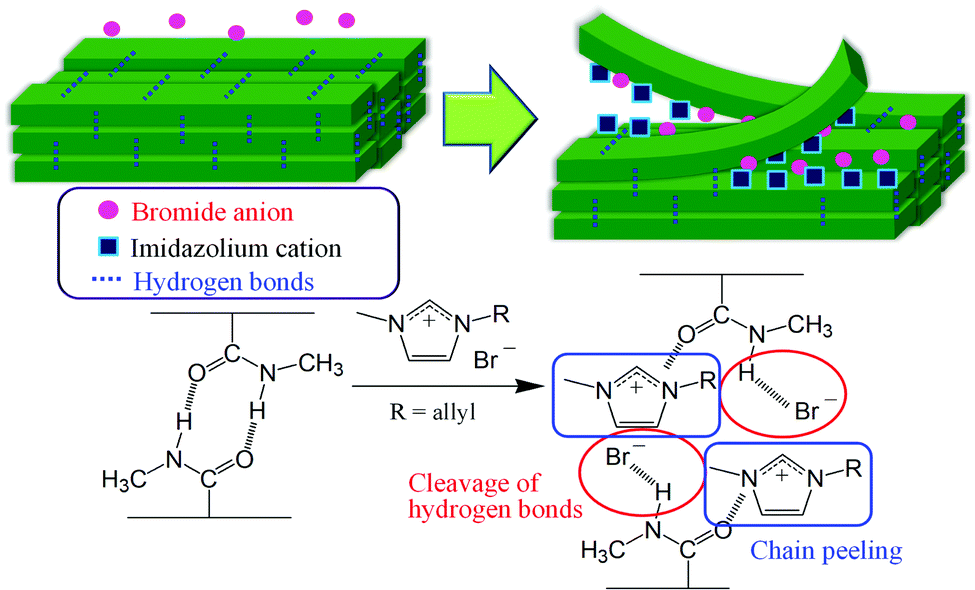 | ||
| Fig. 5 Plausible dissolution process of chitin in AMIMBr predicted by molecular dynamics (MD) simulations. | ||
When larger amounts of chitin (6.5–10.7 wt%) were successively immersed in AMIMBr at room temperature, heated at 100 °C, and cooled to room temperature, gel-like materials (ion gels) with high viscosities formed (Fig. 4b).46 The dynamic rheological measurements showed that the AMIMBr ILs containing 4.8 and 6.5 wt% chitin behaved as weak gels. ILs with hydroxide anions have been used to dissolve chitin by heating and subsequent deacetylation in solution.50,51
Soft materialization of chitin using ILs
Soft materials, such as gels and films, containing chitin have been fabricated through dissolution in ILs. As mentioned above, gels were obtained from chitin and BMIMOAc solutions (Fig. 6), which were further transformed into chitin sponges and films by regeneration using water and methanol coagulants, respectively.40 Furthermore, the rheological properties of chitin ion gels containing BMIMOAc were investigated.52 The chain rigidity of chitin and the hydrogen bonds between chitin and BMIMOAc greatly affected the rheological behavior of the ion gels. EMIMOAc has also been used to produce gels, films, membranes, and fibers from chitin.53–56 For example, chitin hydrogels were prepared through solution molding with EMIMOAc, followed by water coagulation and washing.54 ILs with alkanoate and amino acid anions induced the formation of chitin foils and coatings.42 | ||
| Fig. 6 Photograph of chitin gel prepared from solution in BMIMOAc (reprinted with permission from ref. 40. Copyright 2008 Elsevier; DOI: 10.1016/j.polymer.2008.03.027). | ||
Moreover, chitin-containing composite soft materials have been fabricated using ILs. For example, imidazolium acetates and alkanoates have been used to fabricate chitin/cellulose composite yarns, films, and hydrogels (Fig. 7).57–60 Chitin/cellulose composite hydrogels prepared using EMIMOAc were investigated as novel electrolytes for electrochemical capacitors.61 Chitin was also blended with poly(L-lactic acid) (PLLA) by co-dissolution in EMIMOAc, followed by extrusion into a water coagulation bath.62 Biocomposites containing chitin, PLLA, and hydroxyapatite were prepared by co-dissolution in EMIMOAc, and their biocompatibility was evaluated.63 The composites were appropriate for the growth and proliferation of osteocytes. Composite fibers composed of chitin and calcium alginate were fabricated by co-dissolution of chitin with alginic acid in EMIMOAc, followed by extrusion into a calcium chloride aqueous solution.64 The resulting fibers were used as wound healing patches and exhibited excellent biocompatibility, fast reepithelization, and accelerated healing ability. BMIMOAc-mediated processing was investigated to fabricate sucrose acetate isobutyrate (SAIB)–chitin scaffolds (Fig. 8).65 Human adipose stem cells were able to spread in the scaffolds, indicating the positive biological effect of SAIB. The result revealed that this type of scaffolds is practically applicable as a biomedical material.
 | ||
| Fig. 7 Preparation procedure for chitin/cellulose composite film (reprinted from ref. 60 (Elsevier). This is an open access article distributed under the terms of the Creative Commons CC-BY license; DOI: 10.1016/j.eurpolymj.2021.110681). | ||
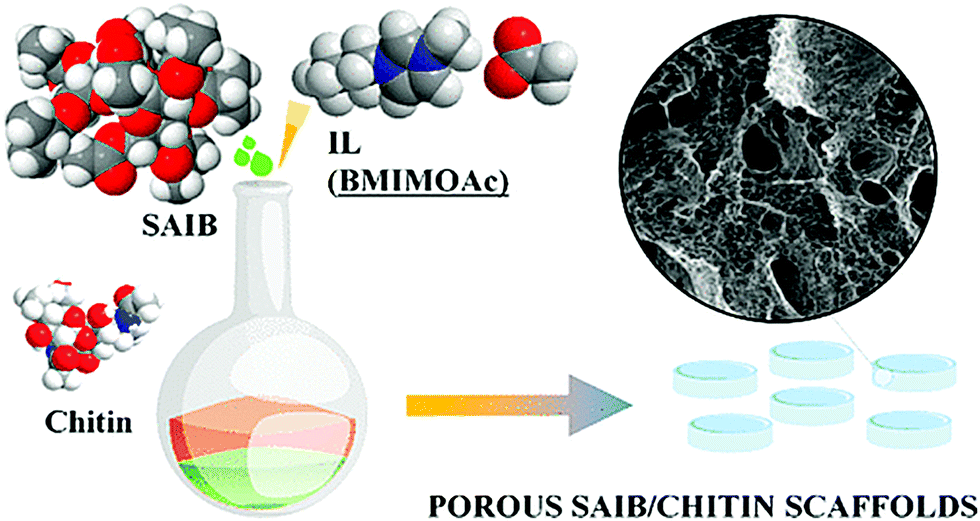 | ||
| Fig. 8 Schematic representation for preparation of sucrose acetate isobutyrate (SAIB)-chitin scaffolds (reprinted with permission from ref. 65. Copyright 2020 American Chemical Society; DOI: 10.1021/acssuschemeng.0c00385). | ||
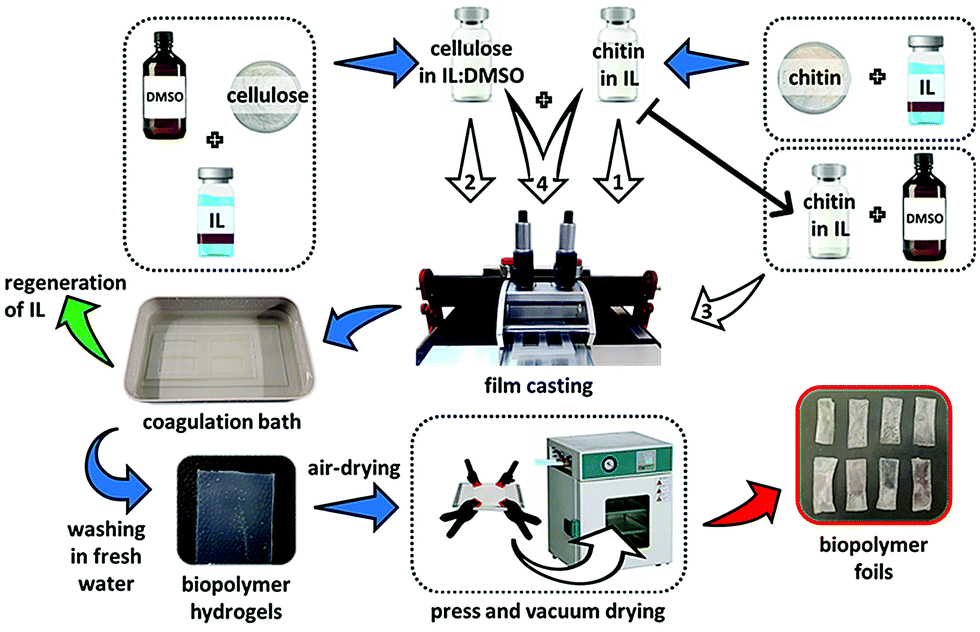
A chitin/cellulose composite ion gel was fabricated from a solution containing two ILs, AMIMBr and BMIMCl (Fig. 9a).66 An AMIMBr solution containing 4.8 wt% chitin and a BMIMCl solution containing 9.1 wt% cellulose were first mixed at 100 °C to form a homogeneous solution. The chitin/cellulose composite ion gel was obtained by eliminating the excess ILs by incubating the solution at room temperature, followed by washing with ethanol. The composite ion gel was employed as a novel electrolyte for an electric double-layer capacitor after conversion into the corresponding acidic gel by treatment with a 2.0 mol L−1 H2SO4 aqueous solution.67–69 The acidic composite gel electrolyte exhibited an excellent high-rate discharge capability over a wide range of current densities as well as in a H2SO4 aqueous solution. In addition, the discharge capacitance of the test cell retained over 80% of its initial value after 105 cycles, even at a high current density of 5000 mA g−1. Chitin materials combined with ILs, which have excellent ionic conductivity, are thus suitable for electrochemical application. Casting the aforementioned homogeneous solution containing chitin/cellulose and AMIMBr/BMIMCl on a glass plate, followed by coagulation with ethanol and water, yielded a chitin/cellulose composite film (Fig. 9b).66,70
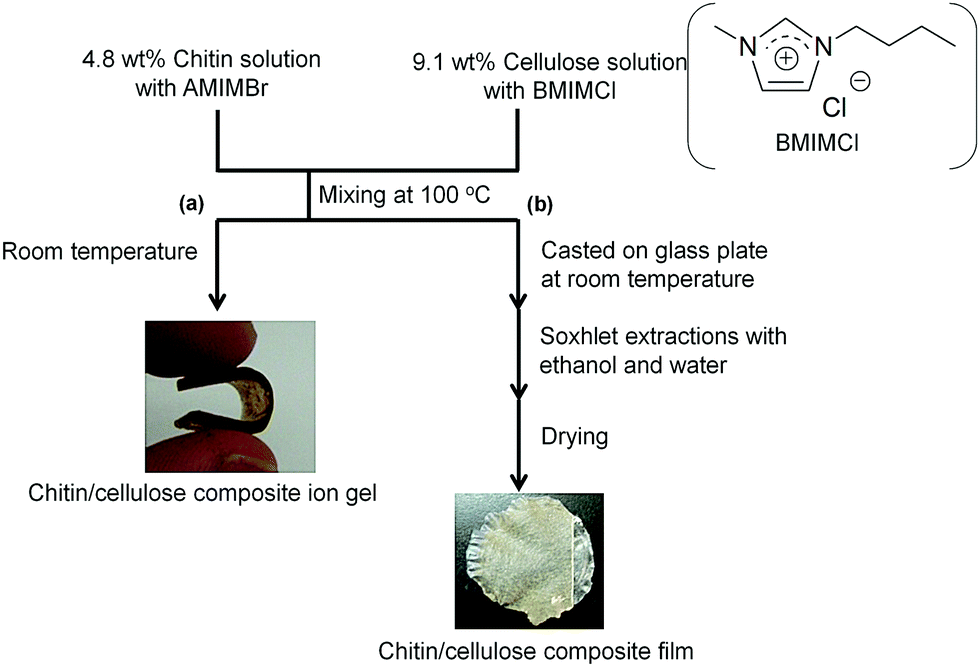 | ||
| Fig. 9 Preparation of (a) chitin/cellulose composite ion gel and (b) chitin/cellulose composite film using ionic liquids (reprinted with permission from ref. 33. Copyright 2020 Elsevier; DOI: 10.1016/B978-0-12-817968-0.00002-0). | ||
Chitin nanomaterials from IL solutions and gels
The preparation of nanoscale chitin assemblies, such as nanofibers and nanocrystals, is one of the most useful methods to practically transform chitin sources into functional materials because of the exceptional properties of bio-based nanomaterials, such as their lightweight, high tensile strength, low thermal expansion coefficient, biocompatibility, and nanosheet formability for sensing and electronic devices.71–76 In addition to the conventional top-down approach,77–83 regenerative bottom-up approaches from solutions/gels with ILs have also been used to fabricate chitin nanofibers (ChNFs).24,33,84–86ChNF dispersions were obtained by immersing the aforementioned 9.1–10.7 wt% chitin ion gels with AMIMBr in methanol at room temperature for 24 h to slowly regenerate chitin, followed by sonication (Fig. 4c).87 Nanofibers approximately 20–60 nm in width and several hundred nanometers in length were observed by scanning electron microscopy of the methanol dispersion, supporting the controlled self-assembly of ChNFs through regeneration from the ion gels. High entanglement of ChNFs occurred through the filtration of the dispersion to form a ChNF film. Self-assembled ChNFs with higher aspect ratios were obtained by regeneration using a CaBr2·2H2O/methanol solution at an adjusted concentration.88 Transmission electron microscopy observations of the self-assembled ChNFs identified bundle-like nanofiber morphologies, indicating that they were hierarchically constructed through the assembly of thinner fibrils.89 When the self-assembled ChNF film was treated with aqueous NaOH for partial deacetylation of acetamido groups and subsequently treated in a 1.0 mol L−1 aqueous acetic acid with ultrasonication, the bundles disintegrated through cationization and electrostatic repulsion of the generated amino groups, yielding a dispersion of thinner nanofibers, named “scaled-down (SD)-ChNFs.”90 The SD-ChNFs were isolated through filtration of the dispersion to fabricate a highly flexible film that bent and twisted easily.
Nano-scaled materials, such as ChNFs, are good candidates to be used in composites both physically and chemically combined with other polymeric components. For example, the self-assembled ChNFs were composited with poly(vinyl alcohol) (PVA) to fabricate composite films (Fig. 4d).87 Solutions comprising the desired amounts of PVA in hot water were first added to the chitin ion gel with AMIMBr. The same regeneration and filtration procedures as described above were used to produce ChNF/PVA composite films. Tensile testing of the composite films indicated the mechanical properties improved with increasing ratio of PVA to chitin.
Self-assembled ChNFs have been employed as stabilizers for the Pickering emulsion polymerization of styrene,91 in which Pickering emulsions of any type (oil-in-water, water-in-oil, or even multicomponent emulsions) are stabilized by solid particles or other types of solid materials in place of the surfactants in general emulsions. Prior to emulsion polymerization, anionic carboxylate groups (maleyloyl groups) were introduced on ChNFs by reaction with maleic anhydride in the presence of perchloric acid to enhance dispersibility in aqueous ammonia. Radical polymerization using potassium persulfate as an initiator was then performed at 70 °C in the emulsion, in which styrene droplets were stably surrounded by ChNFs in aqueous ammonia, to fabricate composite particles. When the Pickering emulsion polymerization of styrene was conducted using the aforementioned SD-ChNFs as stabilizers, the composite particles were smaller than those produced using the self-assembled ChNFs.92 The composite particles were easily converted into hollow ChNF-based particles by solubilizing the styrene core with toluene. In addition to the introduction of anionic maleyloyl groups on the ChNFs, polymerizable methacryloyl groups were substituted on the ChNFs as a secondary functionalization, which could be copolymerized with styrene to stabilize the hollow structure. Hollow particles were fabricated by using toluene to solubilize the polystyrene inside the composite particles fabricated using the bifunctional ChNF stabilizers, which stably redispersed in water.93
Self-assembled ChNFs have been employed as reinforcements through combination with other polymers.86 Because chitin is regarded as a cationic polymer owing to the presence of several percent of free amino groups in the total repeating units due to deacetylation of acetamido groups, self-assembled ChNFs were composited with an anionic polysaccharide, carboxymethyl cellulose (CMC), through electrostatic interactions.94 Cast CMC films were immersed in self-assembled ChNF/methanol dispersions with different contents, followed by centrifugation and drying, to fabricate the ChNF-reinforced CMC films. The quantity of ChNFs in the composite film strongly affected the enhancement of the mechanical properties under tension, supporting their reinforcement effect. The reinforcement effect of the self-assembled ChNFs was also observed by adding them to hydrogels of another anionic polysaccharide, xanthan gum.95 A composite film of the aforementioned SD-ChNFs with ι-carrageenan, a sulfated anionic polysaccharide, was also produced via electrostatic interactions.90
Self-assembled ChNF-reinforced cellulose and natural rubber (NR) films/sheets have also been fabricated.96,97 When cellulose/BMIMCl ion gels, which easily formed from solution,98 were immersed in self-assembled ChNF/methanol dispersions, the two polysaccharides were composited through the regeneration of cellulose to fabricate ChNF-reinforced cellulose films. Self-assembled ChNF dispersions with aqueous ammonia were mixed with NR latex stabilized with aqueous ammonia, followed by drying under reduced pressure, to obtain ChNF-reinforced NR sheets. Tensile testing of the ChNF/cellulose films and ChNF/NR sheets demonstrated the reinforcement effect of ChNFs. The above ChNF/NR dispersions in aqueous ammonia were heated to evaporate the ammonia, followed by lyophilization, resulting in a porous material.97 By evaporating the ammonia stabilizer, ChNFs aggregated with the NR and then agglomerated to form spaces between them, resulting in the porous morphology.
Self-assembled ChNF-graft-synthetic polymer composite films were fabricated by surface-initiated graft polymerization (Fig. 10).84 ChNF-graft-biodegradable polyester and polypeptide composite films were prepared by surface-initiated ring-opening graft polymerization of the corresponding cyclic monomers, that is, L-lactide (LA)/ε-caprolactone (CL) and γ-benzyl L-glutamate-NCA (BLG-NCA), initiated from hydroxy and amino groups, respectively, present on the self-assembled ChNF films.99,100 Surface-initiated graft atom transfer radical polymerization (ATRP) from ChNF macroinitiators has also been conducted. ATRP is a versatile living radical polymerization technique, which has been employed to synthesize a wide range of well-defined polymeric materials.101,102 Because ATRP is initiated by α-haloalkylacyl groups, the ChNF macroinitiator film with the initiating groups was prepared by the reaction of hydroxy groups on the self-assembled ChNFs with α-bromoisobutyryl bromide. The surface-initiated ATRP of 2-hydroxyethyl acrylate (HEA) from the resulting macroinitiator film was then demonstrated in the presence of CuBr (catalyst)/2,2’-bipyridine (ligand) in 3 wt% LiCl/N,N-dimethylacetamide at 60 °C to fabricate ChNF-graft-polyHEA films.103 Tensile testing of the fabricated films indicated larger elongation values at break compared to that of the original ChNF film, which increased with increasing graft chain length. Surface-initiated graft ATRP of methyl methacrylate (MMA) on the ChNF macroinitiator under dispersion conditions was also attempted, followed by entanglement of the products by filtration, to fabricate ChNF-graft-polyMMA films.104
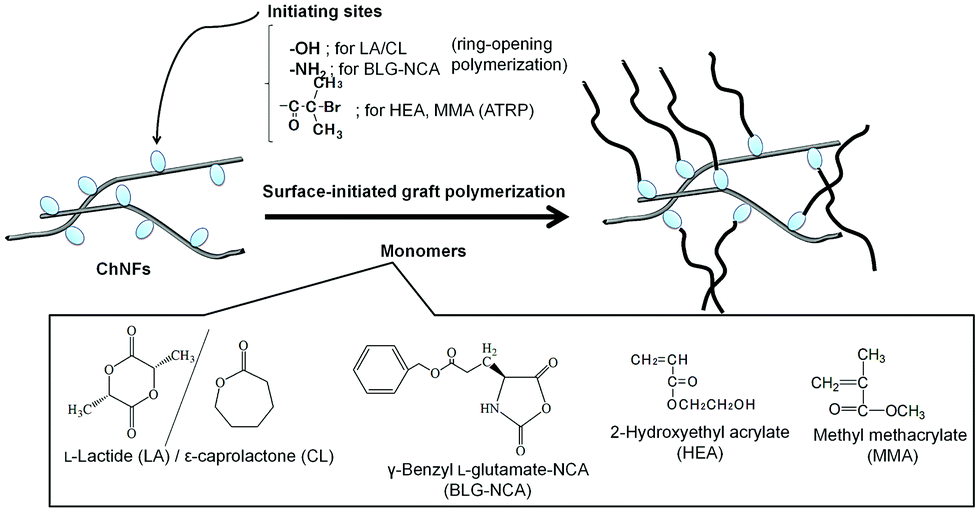 | ||
| Fig. 10 Surface-initiated graft ring opening (co)polymerization and atom transfer radical (ATRP) polymerization from appropriate initiating sites on self-assembled chitin nanofibers (ChNF) (reprinted with permission from ref. 33. Copyright 2020 Elsevier; DOI: 10.1016/B978-0-12-817968-0.00002-0). | ||
Chitin solutions in EMIMOAc have been electrospun to fabricate ChNFs via a bottom-up process.105–107 Because EMIMOAc does not evaporate, a coagulation bath is required to remove EMIMOAc and precipitate ChNFs. The high viscosities of the solutions allow electrospinning of solutions with low chitin concentrations (less than 1 wt%) to produce thin fibers without adjusting the electrospinning parameters. This process has been extended to the electrospinning of chitin composites with other polymers, such as lignin, cellulose, and PLLA (Fig. 11).62,108 These results strongly suggest that besides regeneration as above mentioned, electrospinning is also a powerful technique for controlled nano-assembly from chitin according to bottom-up approach.
 | ||
| Fig. 11 Image for fabrication of ChNF-poly(lactic acid) (PLA) nanocomposite fibers and stress–strain curves under tensile mode (reprinted with permission from ref. 62. Copyright 2018 American Chemical Society; DOI: 10.1021/acssuschemeng.8b01554). | ||
Derivatization and modification of chitin in ILs
Derivatization of polysaccharides, such as acylation (ester derivatization), has been identified as a useful functional materialization, which provides high-performance properties and is used in practical applications such as bio-based thermoplastics, as represented by cellulose acylates.109–111 Chitin acylates have not been employed in practical applications because efficient and practical acylation methods for chitin from native sources have not been sufficiently developed.6,112–115 AMIMBr has been found to act as a good reaction medium for the acylation of chitin.116 For example, the acetylation of chitin in AMIMBr was investigated to obtain chitin acetate.117 Reactions using acetic anhydride (20 equiv. with a repeating unit) in a 2 wt% AMIMBr solution at 60–100 °C for 24 h yielded degrees of derivatization (DSs) of 1.82 to 1.90 (Fig. 12a, m = 0), indicating the occurrence of almost quantitative acetylation (the highest DS value for chitin is 2). In contrast, ILs with carboxylate anions, such as acetate and alkanoates, are not considered as suitable media for acylation because of their higher basicity compared to that of bromide in AMIMBr, resulting in potential reactivity with acylation reagents. Accordingly, acetylation of chitin using 20 equiv. of acetic anhydride in BMIMOAc at 100 °C for 24 h yielded a lower DS value (0.81) than that obtained with AMIMBr (1.90) under the same conditions.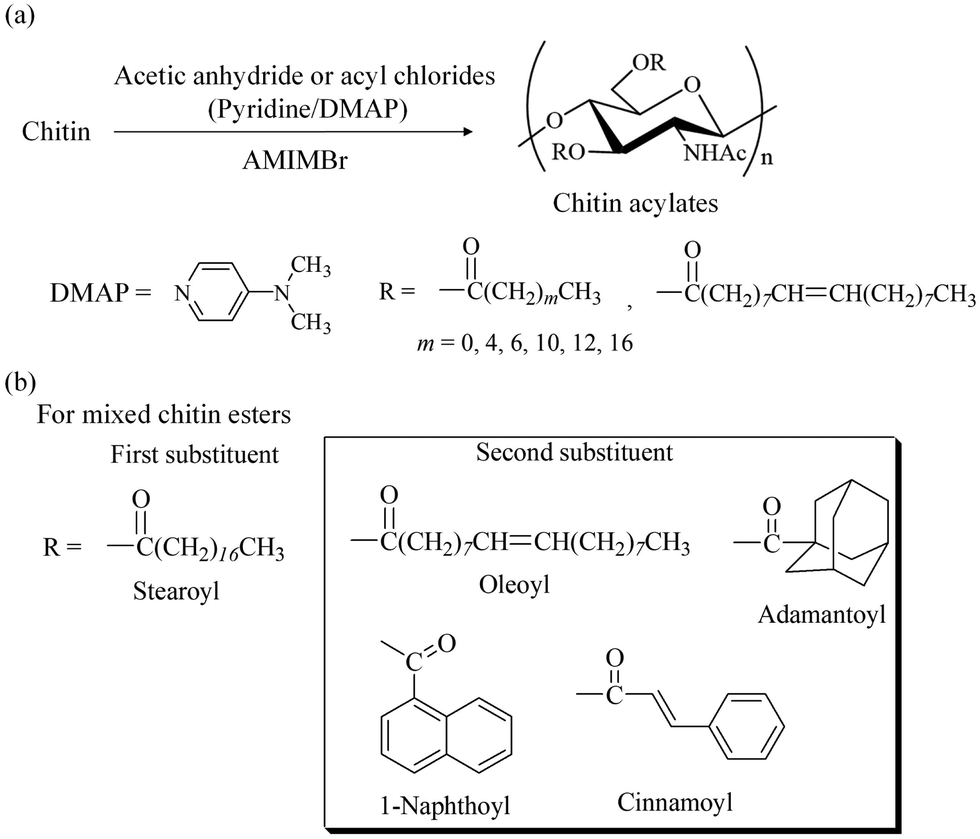 | ||
| Fig. 12 (a) Acylation of chitin in AMIMBr and (b) substituents for mixed chitin esters. | ||
When the synthesis of various chitin acylates with different substituents was attempted in AMIMBr, the following optimal conditions were found. Using acyl chlorides in the presence of pyridine as a base and N,N-dimethyl-4-aminopyridine (DMAP) as a catalyst at 100 °C for 24 h produced derivatives with quantitative DS values (Fig. 12a).118 To confirm the efficient acylation of chitin using acyl chlorides in AMIMBr, a mixture of hexanoyl chloride and AMIMBr was heated at 100 °C in the absence of chitin.1191H NMR analysis supported the occurrence of halogen exchange between hexanoyl chloride and AMIMBr in the mixture to produce hexanoyl bromide in situ (Fig. 13). Accordingly, acyl bromides efficiently react with hydroxy groups in chitin owing to their higher reactivity compared to that of acyl chlorides to obtain high-DS products.
 | ||
| Fig. 13 Halogen exchange reaction between hexanoyl chloride and AMIMBr to produce hexanoyl bromide (reprinted with permission from ref. 119. Copyright 2021 Elsevier; DOI: 10.1016/j.ijbiomac.2021.09.044). | ||
The above chitin acylates (single chitin esters) did not exhibit specific processability, such as film formability. The synthesis of mixed chitin esters with two different substituents was then conducted using the same acylation manner in AMIMBr to provide derivatives with specific processability.119 This attempt was based on the fact that mixed cellulose esters exhibit a lower viscous flow temperature compared to that of single cellulose esters, leading to superior melt processability and thermoplasticity.120 A stearoyl group was selected as the first substituent, which was combined with different long fatty and bulky acyl groups as the second substituent (Fig. 12b). The almost quantitative DS values achieved by stearoyl with oleoyl or some bulky acyl groups resulted from acylation using the corresponding acyl chlorides in the presence of pyridine/DMAP in AMIMBr. All products exhibited film formability by casting from solutions in chloroform or chloroform/trifluoroacetic acid solvents, but they did not exhibit thermoplasticity. There is a possibility for exhibiting thermostasticity from mixed chitin esters with different substituents and their DSs by additional investigations in the future.
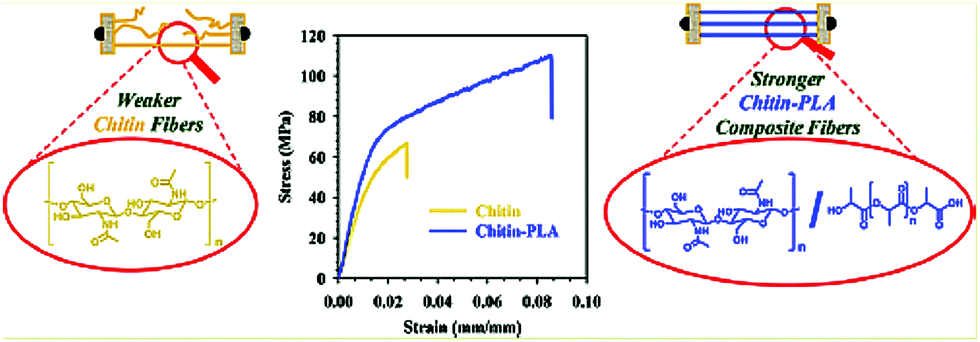
To achieve thermoplasticity, surface-initiated ring-opening graft polymerizations of CL from the hydroxy groups of chitin hexanoates with DS values in the 1.4–1.8 range, which were prepared by the acylation of chitin with hexanoyl chloride in AMIMBr, were performed (Fig. 14a).121 Surface-initiated ring-opening graft polymerization of CL was conducted in the presence of tin(II) 2-ethylhexanoate as a catalyst at 100 °C to produce chitin hexanoate-graft-poly(ε-caprolactone) (ChHex-g-PCL). The reaction conditions strongly affected the molar substitution and degree of polymerization of the PCL graft chains. Longer PCL graft chains formed crystalline structures, and the ChHex-g-PCL materials largely contained uncrystallized chitin chains. Accordingly, these ChHex-g-PCL materials exhibited melting points associated with the PCL graft chain length, allowing the formation of films by melt pressing at 80 °C and 1 MPa for 30 min (Fig. 14b). This study provides the first example of thermoplastic chitin derivatives.
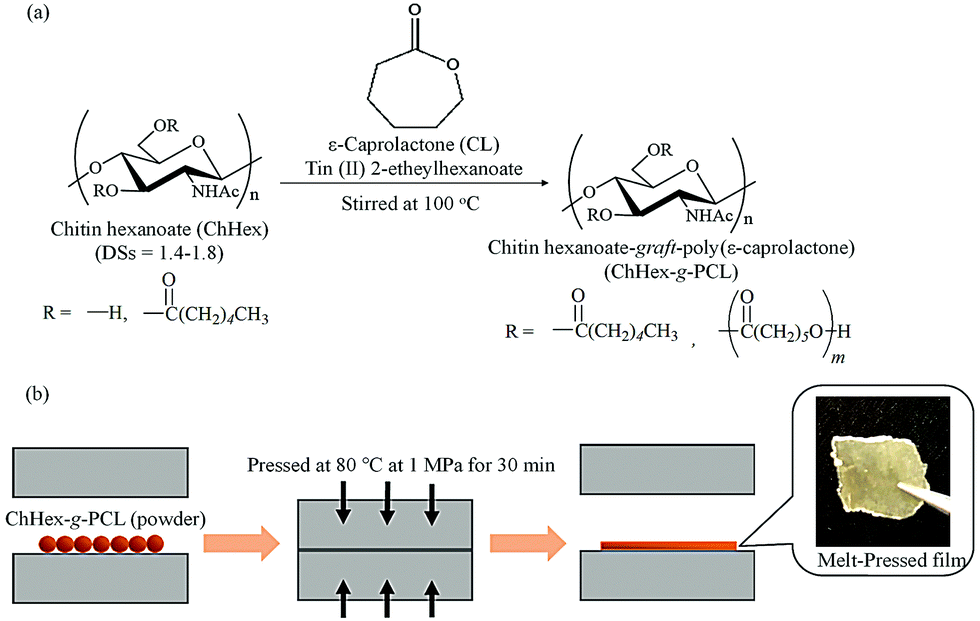 | ||
| Fig. 14 (a) Surface-initiated ring-opening graft polymerization of ε-caprolactone (CL) from chitin hexanoates (ChHexs) to obtain chitin hexanoate-graft-poly(ε-caprolactone) (ChHex-g-PCL), and (b) melt-pressing experiment of ChHex-g-PCL (reprinted with permission from ref. 121. Copyright 2022 Elsevier; DOI: 10.1016/j.carbpol.2021.119024). | ||
The above acylation method for chitin in AMIMBr was extended to synthesize a chitin macroinitiator for subsequent graft ATRP.122 The chitin macroinitiator modified with initiating groups was synthesized by the acylation of hydroxy groups in chitin with 2-bromopropyl bromide in AMIMBr according to the above reaction mechanism (Fig. 15a). The DS value of the product, which was synthesized by a reaction using 30 equiv. of 2-bromopropyl bromide with a repeating unit at 100 °C for 24 h, was estimated to be 1.86 by 1H NMR analysis. The graft polymerization of styrene was then conducted by ATRP from the resulting chitin macroinitiator using CuBr and N,N,N′N′,N′′,N′′-pentamethyldiethylene triamine (PMDETA) as the catalyst system to obtain chitin-graft-polystyrene (Fig. 15b). The grafted polystyrene chains were separated from the products by alkaline hydrolysis of ester linkages at the root of the chitin chain to estimate their Mn values using size exclusion chromatography (SEC). The SEC results suggested the Mn value increased with increasing monomer/initiating site feed ratio, indicating graft ATRP progressed in a living manner.
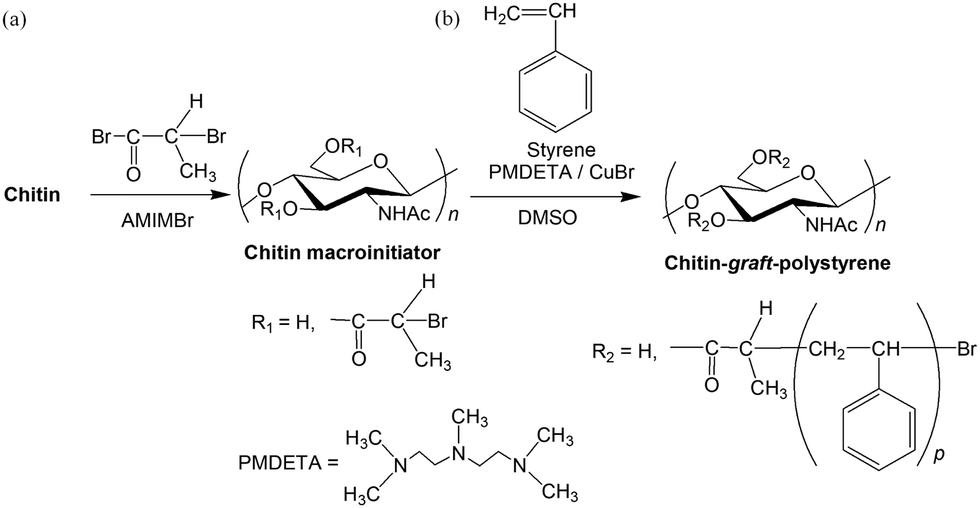 | ||
| Fig. 15 (a) Synthesis of chitin macroinitiator in AMIMBr and (b) following graft atom transfer radical polymerization (ATRP) of styrene (reprinted with permission from ref. 116. Copyright 2019 Elsevier; DOI: 10.1016/j.ijbiomac.2018.11.165). | ||
Conclusions
This perspective article presents the efficient functional materialization of chitin through dissolution in ILs. Because of the poor solubility and processability of chitin, ILs have attracted increasing attention for the development of new uses and applications of chitin. Unique forms, such as gels and films, can be fabricated from chitin solutions in ILs. The regeneration and electrospinning of solutions/gels provide a bottom-up self-assembly approach to easily fabricate ChNFs. The resulting chitin materials have been used to prepare composite materials. Acylation of chitin efficiently occurs in AMIMBr to produce various chitin acylates. This article illustrates that the possible applications of chitin as a material have been widely extended through the use of ILs, which are now identified as an important research field regarding chitin. Indeed, the dissolution of chitin in ILs has provided a certain number of materials with tailored and improved properties, enhancing the potential use of chitin in different applications, such as in biomedical, tissue engineering, and green/sustainable fields as some previous review articles have stated.26,28,32–34,86 Based on environmentally aspect, harmful chemicals, currently used in industrial processes, should be replaced by solvents that show suitability for the green chemistry approach, such as ILs, and that the use of greener ILs in applications of chitin in functional material field will gradually increase. Overall, further research will be conducted, including the development of new greener ILs for chitin solvents and additional approaches to add new functions, which can be employed in practical materials and can lead to their commercialization in the future.Author contributions
The author designed and wrote the manuscript.Conflicts of interest
The author declares no conflict of interest.Acknowledgements
The author is indebted to the co-workers, whose names are found in references from his papers, for their enthusiastic collaborations. The author would like to thank Editage (www.editage.com) for English language editing.References
- J. G. Yao, P. S. Fennell and J. P. Hallett, RSC Energy and Environment Series, 2020, vol. 2020, pp. 69–105 Search PubMed.
- X. Yang, C. Qiao, Y. Li and T. Li, React. Funct. Polym., 2016, 100, 181–190 CrossRef CAS.
- C. Schuerch, in Encyclopedia of Polymer Science and Engineering, ed. H. F. Mark, N. Bilkales and C. G. Overberger, John Wiley & Sons, New York, 2nd edn, 1986, vol. 13, pp. 87–162 Search PubMed.
- E. H. Song, J. Shang and D. M. Ratner, in Polymer Science: A Comprehensive Reference, ed. K. Matyjaszewski and M. Möller, Elsevier, Amsterdam, 2012, pp. 137–155 Search PubMed.
- D. Klemm, B. Heublein, H. P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- K. Kurita, Mar. Biotechnol., 2006, 8, 203–226 CrossRef CAS PubMed.
- M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603–632 CrossRef CAS.
- C. K. S. Pillai, W. Paul and C. P. Sharma, Prog. Polym. Sci., 2009, 34, 641–678 CrossRef CAS.
- Global Chitin Market Size By Derivative Type, By End-User, By Geographic Scope And Forecast, Verified Market Research, Lewws, DE, USA, 2021.
- R. A. A. Muzzarelli, M. El Mehtedi and M. Mattioli-Belmonte, Mar. Drugs, 2014, 12, 5468–5502 CrossRef CAS PubMed.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- O. A. El Seoud, A. Koschella, L. C. Fidale, S. Dorn and T. Heinze, Biomacromolecules, 2007, 8, 2629–2647 CrossRef CAS PubMed.
- T. Liebert and T. Heinze, BioResources, 2008, 3, 576–601 Search PubMed.
- L. Feng and Z. I. Chen, J. Mol. Liq., 2008, 142, 1–5 CrossRef.
- A. Pinkert, K. N. Marsh, S. S. Pang and M. P. Staiger, Chem. Rev., 2009, 109, 6712–6728 CrossRef CAS PubMed.
- M. E. Zakrzewska, E. Bogel-Łukasik and R. Bogel-Łukasik, Energy Fuels, 2010, 24, 737–745 CrossRef CAS.
- M. Gericke, P. Fardim and T. Heinze, Molecules, 2012, 17, 7458–7502 CrossRef PubMed.
- M. Isik, H. Sardon and D. Mecerreyes, Int. J. Mol. Sci., 2014, 15, 11922–11940 CrossRef CAS PubMed.
- J. Zhang, J. Wu, J. Yu, X. Zhang, J. He and J. Zhang, Mater. Chem. Front., 2017, 1, 1273–1290 RSC.
- F. Hermanutz, M. P. Vocht, N. Panzier and M. R. Buchmeiser, Macromol. Mater. Eng., 2019, 304, 1800450 CrossRef.
- C. Verma, A. Mishra, S. Chauhan, P. Verma, V. Srivastava, M. A. Quraishi and E. E. Ebenso, Sustainable Chem. Pharm., 2019, 13, 100162 CrossRef.
- W. T. Wang, J. Zhu, X. L. Wang, Y. Huang and Y. Z. Wang, J. Macromol. Sci. Phys., 2010, 49, 528–541 CrossRef CAS.
- J. Kadokawa, Green Sustainable Chem., 2013, 03, 19–25 CrossRef.
- J. Kadokawa, RSC Adv., 2015, 5, 12736–12746 RSC.
- M. M. Jaworska, T. Kozlecki and A. Gorak, J. Polym. Eng., 2012, 32, 67–69 CAS.
- S. S. Silva, J. F. Mano and R. L. Reis, Green Chem., 2017, 19, 1208–1220 RSC.
- J. L. Shamshina, Green Chem., 2019, 21, 3974–3993 RSC.
- S. K. Singh, Int. J. Biol. Macromol., 2019, 132, 265–277 CrossRef CAS PubMed.
- R. A. A. Muzzarelli, Mar. Drugs, 2011, 9, 1510–1533 CrossRef CAS PubMed.
- J. Kadokawa, in RSC Smart Materials, ed. A. Eftekhari, Royal Society of Chemistry, London, 2018, pp. 319–341 Search PubMed.
- J. L. Shamshina, O. Zavgorodnya and R. D. Rogers, Advances in. Biochemical Engineering/Biotechnology, 2019, vol. 168, pp. 177–198 Search PubMed.
- J. L. Shamshina, P. Berton and R. D. Rogers, ACS Sustainable Chem. Eng., 2019, 7, 6444–6457 CrossRef CAS.
- J. Kadokawa, in Handbook of Chitin and Chitosan, ed. S. Gopi, S. Thomas and A. Pius, Elsevier, 2020, pp. 47–60 Search PubMed.
- J. L. Shamshina, A. Kelly, T. Oldham and R. D. Rogers, Ebviron. Chem. Lett., 2020, 18, 53–60 CrossRef CAS.
- M. Sharma, C. Mukesh, D. Mondal and K. Prasad, RSC Adv., 2013, 3, 18149–18155 RSC.
- C. Mukesh, D. Mondal, M. Sharma and K. Prasad, Carbohydr. Polym., 2014, 103, 466–471 CrossRef CAS PubMed.
- S. Idenoue, K. Yamamoto and J. Kadokawa, ChemEngineering, 2019, 3, 90, DOI:10.3390/chemengineering3040090.
- J. Kadokawa, S. Idenoue and K. Yamamoto, ACS Sustainable Chem. Eng., 2020, 8, 8402–8408 CrossRef CAS.
- N. Özel and M. Elibol, Carbohydr. Polym., 2021, 262, 117942 CrossRef PubMed.
- Y. Wu, T. Sasaki, S. Irie and K. Sakurai, Polymer, 2008, 49, 2321–2327 CrossRef CAS.
- Y. Qin, X. M. Lu, N. Sun and R. D. Rogers, Green Chem., 2010, 12, 968–971 RSC.
- P. Walther, A. Ota, A. Müller, F. Hermanutz, F. Gähr and M. R. Buchmeiser, Macromol. Mater. Eng., 2016, 301, 1337–1344 CrossRef CAS.
- M. M. Jaworska, I. Stępniak, M. Galiński, D. Kasprzak, D. Biniaś and A. Górak, Carbohydr. Polym., 2018, 202, 397–403 CrossRef CAS PubMed.
- L. D. Tolesa, B. S. Gupta and M.-J. Lee, Int. J. Biol. Macromol., 2019, 130, 818–826 CrossRef CAS PubMed.
- J. L. Shamshina, P. S. Barber, G. Gurau, C. S. Griggs and R. D. Rogers, ACS Sustainable Chem. Eng., 2016, 4, 6072–6081 CrossRef CAS.
- K. Prasad, M. Murakami, Y. Kaneko, A. Takada, Y. Nakamura and J. Kadokawa, Int. J. Biol. Macromol., 2009, 45, 221–225 CrossRef CAS PubMed.
- H. Zhang, Y. Xu, Y. Li, Z. Lu, S. Cao, M. Fan, L. Huang and L. Chen, Polymers, 2017, 9, 526 CrossRef PubMed.
- T. Uto, S. Idenoue, K. Yamamoto and J. Kadokawa, Phys. Chem. Chem. Phys., 2018, 20, 20669–20677 RSC.
- T. Setoguchi, T. Kato, K. Yamamoto and J. I. Kadokawa, Int. J. Biol. Macromol., 2012, 50, 861–864 CrossRef CAS PubMed.
- M. Shimo, M. Abe and H. Ohno, ACS Sustainable Chem. Eng., 2016, 4, 3722–3727 CrossRef CAS.
- Q. Ma, X. Gao, X. Bi, Q. Han, L. Tu, Y. Yang, Y. Shen and M. Wang, Carbohydr. Polym., 2020, 230, 115605 CrossRef CAS PubMed.
- L. Deng and L.-M. Zhang, Colloids Surf., A, 2020, 586, 124220 CrossRef CAS.
- D. G. Ramírez-Wong, M. Ramírez-Cardona, R. J. Sánchez-Leija, A. Rugerio, R. A. Mauricio-Sánchez, M. A. Hernández-Landaverde, A. Carranza, J. A. Pojman, A. M. Garay-Tapia, E. Prokhorov, J. D. Mota-Morales and G. Luna-Bárcenas, Green Chem., 2016, 18, 4303–4311 RSC.
- X. Shen, J. L. Shamshina, P. Berton, J. Bandomir, H. Wang, G. Gurau and R. D. Rogers, ACS Sustainable Chem. Eng., 2016, 4, 471–480 CrossRef CAS.
- C. King, J. L. Shamshina, G. Gurau, P. Berton, N. F. A. F. Khan and R. D. Rogers, Green Chem., 2017, 19, 117–126 RSC.
- J. Chakravarty, M. F. Rabbi, N. Bach, V. Chalivendra, C.-L. Yang and C. J. Brigham, Carbohydr. Polym., 2018, 198, 443–451 CrossRef CAS PubMed.
- K. Mundsinger, A. Müller, R. Beyer, F. Hermanutz and M. R. Buchmeiser, Carbohydr. Polym., 2015, 131, 34–40 CrossRef CAS PubMed.
- Y. Duan, A. Freyburger, W. Kunz and C. Zollfrank, Carbohydr. Polym., 2018, 192, 159–165 CrossRef CAS PubMed.
- P. Berton, X. Shen, R. D. Rogers and J. L. Shamshina, Ind. Eng. Chem. Res., 2019, 58, 19862–19876 CrossRef CAS.
- D. Kasprzak and M. Galiński, Eur. Polym. J., 2021, 158, 110681 CrossRef CAS.
- D. Kasprzak and M. Galiński, J. Solid State Electrochem., 2021, 25, 2549–2563 CrossRef CAS.
- J. L. Shamshina, O. Zavgorodnya, P. Berton, P. K. Chhotaray, H. Choudhary and R. D. Rogers, ACS Sustainable Chem. Eng., 2018, 6, 10241–10251 CrossRef CAS.
- J. Chakravarty, M. F. Rabbi, V. Chalivendra, T. Ferreira and C. J. Brigham, Int. J. Biol. Macromol., 2020, 151, 1213–1223 CrossRef CAS PubMed.
- J. L. Shamshina, G. Gurau, L. E. Block, L. K. Hansen, C. Dingee, A. Walters and R. D. Rogers, J. Mater. Chem. B, 2014, 2, 3924–3936 RSC.
- C. Gonçalves, S. S. Silva, J. M. Gomes, I. M. Oliveira, R. F. Canadas, F. R. Maia, H. Radhouani, R. L. Reis and J. M. Oliveira, ACS Sustainable Chem. Eng., 2020, 8, 3986–3994 CrossRef.
- A. Takegawa, M. Murakami, Y. Kaneko and J. Kadokawa, Carbohydr. Polym., 2010, 79, 85–90 CrossRef CAS.
- S. Yamazaki, A. Takegawa, Y. Kaneko, J. Kadokawa, M. Yamagata and M. Ishikawa, Electrochem. Commun., 2009, 11, 68–70 CrossRef CAS.
- S. Yamazaki, A. Takegawa, Y. Kaneko, J. Kadokawa, M. Yamagata and M. Ishikawa, J. Power Sources, 2010, 195, 6245–6249 CrossRef CAS.
- S. Yamazaki, A. Takegawa, Y. Kaneko, J. Kadokawa, M. Yamagata and M. Ishikawa, J. Electrochem. Soc., 2010, 157, A203–A208 CrossRef CAS.
- J. Kadokawa, K. Hirohama, S. Mine, T. Kato and K. Yamamoto, J. Polym. Environ., 2012, 20, 37–42 CrossRef CAS.
- J. You, M. Li, B. Ding, X. Wu and C. Li, Adv. Mater., 2017, 29, 1606895 CrossRef PubMed.
- M. Anraku, R. Tabuchi, S. Ifuku, T. Nagae, D. Iohara, H. Tomida, K. Uekama, T. Maruyama, S. Miyamura, F. Hirayama and M. Otagiri, Carbohydr. Polym., 2017, 161, 21–25 CrossRef CAS PubMed.
- R. Koizumi, K. Azuma, H. Izawa, M. Morimoto, K. Ochi, T. Tsuka, T. Imagawa, T. Osaki, N. Ito, Y. Okamoto, H. Saimoto and S. Ifuku, Int. J. Mol. Sci., 2017, 18, 11 Search PubMed.
- C. C. Satam, C. W. Irvin, A. W. Lang, J. C. R. Jallorina, M. L. Shofner, J. R. Reynolds and J. C. Meredith, ACS Sustainable Chem. Eng., 2018, 6, 10637–10644 CrossRef CAS.
- N. E. Mushi, T. Nishino, L. A. Berglund and Q. Zhou, ACS Sustainable Chem. Eng., 2019, 7, 1692–1697 CrossRef CAS.
- T. Naghdi, H. Golmohammadi, H. Yousefi, M. Hosseinifard, U. Kostiv, D. Horák and A. Merkoçi, ACS Appl. Mater. Interfaces, 2020, 12, 15538–15552 CrossRef CAS PubMed.
- T. Yang, H. Qi, P. Liu and K. Zhang, ChemPlusChem, 2020, 85, 1081–1088 CrossRef CAS PubMed.
- S. Ifuku and H. Saimoto, Nanoscale, 2012, 4, 3308–3318 RSC.
- S. Ifuku, Kobunshi Ronbunshu, 2012, 69, 460–467 CrossRef.
- S. Ifuku, Molecules, 2014, 19, 18367–18380 CrossRef PubMed.
- Y. Fan, T. Saito and A. Isogai, Biomacromolecules, 2008, 9, 192–198 CrossRef CAS PubMed.
- Y. M. Fan, T. Saito and A. Isogai, Carbohydr. Polym., 2009, 77, 832–838 CrossRef CAS.
- Y. M. Fan, H. Fukuzumi, T. Saito and A. Isogai, Int. J. Biol. Macromol., 2012, 50, 69–76 CrossRef CAS PubMed.
- J. Kadokawa, Coatings, 2016, 6, 27 CrossRef.
- J. Kadokawa, Pure Appl. Chem., 2016, 88, 621–629 CAS.
- J. Kadokawa, Polymers, 2021, 13, 3548 CrossRef CAS PubMed.
- J. Kadokawa, A. Takegawa, S. Mine and K. Prasad, Carbohydr. Polym., 2011, 84, 1408–1412 CrossRef CAS.
- R. Tajiri, T. Setoguchi, S. Wakizono, K. Yamamoto and J. Kadokawa, J. Biobased Mater. Bioenergy, 2013, 7, 655–659 CrossRef CAS.
- J. Kadokawa, A. Kawano and K. Yamamoto, ChemistrySelect, 2019, 4, 797–801 CrossRef CAS.
- T. Hashiguchi, K. Yamamoto and J. Kadokawa, Carbohydr. Polym., 2021, 270, 118369 CrossRef CAS PubMed.
- S. Noguchi, K. Sato, K. Yamamoto and J. Kadokawa, Int. J. Biol. Macromol., 2019, 126, 187–192 CrossRef CAS PubMed.
- R. Watanabe, K. Izaki, K. Yamamoto and J. Kadokawa, Coatings, 2021, 11, 672 CrossRef CAS.
- S. Noguchi, K. Yamamoto and J. Kadokawa, Int. J. Biol. Macromol., 2019, 157, 680–686 CrossRef PubMed.
- D. Hatanaka, K. Yamamoto and J. Kadokawa, Int. J. Biol. Macromol., 2014, 69, 35–38 CrossRef CAS PubMed.
- J. Kadokawa, S. Noguchi, T. Gotanda, A. Kawano and K. Yamamoto, Polym. Bull., 2020, 77, 4095–4103 CrossRef CAS.
- J. Kadokawa, R. Endo, D. Hatanaka and K. Yamamoto, J. Polym. Environ., 2015, 23, 348–355 CrossRef CAS.
- A. Kawano, K. Yamamoto and J. Kadokawa, Biomolecules, 2017, 7, 47 CrossRef PubMed.
- J. Kadokawa, M. Murakami and Y. Kaneko, Carbohydr. Res., 2008, 343, 769–772 CrossRef CAS PubMed.
- T. Setoguchi, K. Yamamoto and J. Kadokawa, Polymer, 2012, 53, 4977–4982 CrossRef CAS.
- J. Kadokawa, T. Setoguchi and K. Yamamoto, Polym. Bull., 2013, 70, 3279–3289 CrossRef CAS.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689–3745 CrossRef CAS PubMed.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- K. Yamamoto, S. Yoshida and J. Kadokawa, Carbohydr. Polym., 2014, 112, 119–124 CrossRef CAS PubMed.
- R. Endo, K. Yamamoto and J. Kadokawa, Fibers, 2015, 3, 338–347 CrossRef CAS.
- P. S. Barber, C. S. Griggs, J. R. Bonner and R. D. Rogers, Green Chem., 2013, 15, 601–607 RSC.
- J. L. Shamshina, O. Zavgorodnya, J. R. Bonner, G. Gurau, T. Di Nardo and R. D. Rogers, ChemSusChem, 2017, 10, 106–111 CrossRef CAS PubMed.
- O. Zavgorodnya, J. L. Shamshina, J. R. Bonner and R. D. Rogers, ACS Sustainable Chem. Eng., 2017, 5, 5512–5519 CrossRef CAS.
- J. L. Shamshina, O. Zavgorodnya, H. Choudhary, B. Frye, N. Newbury and R. D. Rogers, ACS Sustainable Chem. Eng., 2018, 6, 14713–14722 CrossRef CAS.
- Q. Tang and G. Huang, Mini-Rev. Med. Chem., 2016, 16, 1244–1257 CrossRef CAS PubMed.
- K. J. Edgar, C. M. Buchanan, J. S. Debenham, P. A. Rundquist, B. D. Seiler, M. C. Shelton and D. Tindall, Prog. Polym. Sci., 2001, 26, 1605–1688 CrossRef CAS.
- M. Kostag, M. Gericke, T. Heinze and O. A. El Seoud, Cellulose, 2019, 26, 139–184 CrossRef CAS.
- M. N. V. Ravi Kumar, React. Funct. Polym., 2000, 46, 1–27 CrossRef.
- H. Sashiwa and S.-I. Aiba, Prog. Polym. Sci., 2004, 29, 887–908 CrossRef CAS.
- K. Kurita, Prog. Polym. Sci., 2001, 26, 1921–1971 CrossRef CAS.
- M. Morimoto, H. Saimoto and Y. Shigemasa, Trends Glycosci. Glycotechnol., 2002, 14, 205–222 CrossRef CAS.
- J. Kadokawa, Int. J. Biol. Macromol., 2019, 123, 732–737 CrossRef CAS PubMed.
- S. Mine, H. Izawa, Y. Kaneko and J. Kadokawa, Carbohydr. Res., 2009, 344, 2263–2265 CrossRef CAS PubMed.
- H. Hirayama, J. Yoshida, K. Yamamoto and J. Kadokawa, Carbohydr. Polym., 2018, 200, 567–571 CrossRef CAS PubMed.
- K. Kohori, H. Hirayama, K. Yamamoto and J. Kadokawa, Int. J. Biol. Macromol., 2021, 190, 763–768 CrossRef CAS PubMed.
- X. Wang, Y. Wang, Y. Xia, S. Huang, Y. Wang and Y. Qiu, Text. Res. J., 2018, 88, 1491–1504 CrossRef CAS.
- A. Nakashima, K. Kohori, K. Yamamoto and J. Kadokawa, Carbohydr. Polym., 2022, 280, 119024 CrossRef CAS PubMed.
- K. Yamamoto, S. Yoshida, S. Mine and J. Kadokawa, Polym. Chem., 2013, 4, 3384–3389 RSC.
| This journal is © The Royal Society of Chemistry 2022 |