
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceArrangements of fluorophores in the salts of imidazole tethered anthracene derivatives with pyridinedicarboxylic acids influencing photoluminescence†
Abhay Pratap
Singh
and
Jubaraj B.
Baruah
 *
*
Department of Chemistry, Indian Institute of Technology Guwahati, Guwahati-781 039, Assam, India. E-mail: juba@iitg.ac.in; Fax: +91-361-2690762; Tel: +91-361-2582311
First published on 1st March 2022
Abstract
Self-assemblies of salts of 9-N-(3-imidazolylpropylamino)methylanthracene (Hanthraimida) with pyridinedicarboxylic acid and cocrystals with 1,3-dihydroxybenzenes were analysed to sort out their impacts on aggregation induced emissions in the solid state. Different structural features and proton transfers were observed in the salts and cocrystals. The salt of Hanthraimida with 2,6-pyridinedicarboxylic acid (H226pdc) had an unusual composition of 2[(H3anthraimida)]2+[(26pdc)2−·2(H26pdc)−]·(H226pdc)·CH3OH (1). This salt had hydrogen bonded anionic and neutral acid molecules that formed a chain-like arrangement, where the cations H3anthraimida2+ were accommodated in two distinct supramolecular environments. On the other hand, the corresponding salt of 3,5-pyridinedicarboxylic acid (H235pdc) with Hanthraimida had a conventional composition of (H3anthraimida)2+(35pdc)2−·4H2O. The self-assembly of this salt had octameric water clusters that were hydrogen bonded to 35pdc2− ions in its lattice. When such salts were prepared from solution in the presence of resorcinol, they yielded ionic cocrystals, namely [(H3anthraimida)]2+[(26pdc)]2−·resorc·H2O·CH3OH (3) and 2[(H3anthraimida)]+[(35pdc)]2−·2(resorc) (4). The self-assembly of the ionic cocrystal 3 had hydrogen bonded dimers of di-cations bridged by dicarboxylates. These dimers were bridged by water molecules to form chains, where resorcinol molecules were found as intervening molecules connecting such chains. Cocrystal 4 had two resorcinol guest molecules, one of which assembled with host ions to create space for the encapsulation of the other resorcinol molecule. DFT calculations were carried out on the neutral and ionic species to depict the possibilities of aggregation induced emissions in the solid state by 1–4. A change in the emission of the host caused by adding the two isomeric acids was indistinguishable in solution, but the corresponding solid samples of the salts had distinguishable emissions.
Introduction
Ionic cocrystals, in general, are supramolecular associations of neutral and ionic components where one or more components interact through charge-assisted hydrogen bonds or ionic interactions.1–4 They have emerged as an important sub-set of cocrystals with specific applications.2 Among them, the ionic cocrystals of organic salts crystallising together with one or more neutral molecules of acid or base or with one or more neutral components are known in the literature.5–10 However, beyond their structural features, their properties have not been explored in detail. In general, pKa differences among partner conjugate acid–base molecules play a decisive role in their crystallisation in a particular form.11 Hence, the proton transfer in such a process is a central point, and to a good extent, the pKa differences between the partner acid–base components are used to predict the proton transfer.12 It is attractive to utilise the intrinsic acidity or basicity of a poly-acid or a poly-base to design multicomponent cocrystals.13 A simple way to generate ionic cocrystals is to use the two H-bond donor or acceptor sites of a ditopic host such as 9-N-(3-imidazolylpropylamino)methylanthracene (abbreviated as Hanthraimida as shown in Fig. 1(i)) to utilise the proton transfer from or to the two distinguishable hydrogen bonding sites (NH or imidazole N). The binding of such hosts with and without a proton transfer in multiple ways leaves avenues to prepare different ionic cocrystals. This should be particularly true with Hanthraimida, as the ability of it to form cocrystals with phenolic and aromatic carboxylic acids has already been documented in the literature.14,15 Pyridinedicarboxylic acid is a nitrogen containing dicarboxylic acid that has provision to form mono-cationic salts as illustrated in A–D of Fig. 1(ii). Similarly, a di-anion will provide salts with different compositions as illustrated in E–G of Fig. 1(ii). Alternatively, neutral assembly may be formed by a zwitterion as illustrated in H–J of Fig. 1(ii). These aspects get further complicated by having mixed ionic species in the salts. For example, chain like hydrogen bonded carboxylate catemers of neutral, mono-protonated and di-deprotonated anions may bind cations in a distinguishable environment as shown in Fig. 2(i). Such possibilities will imbibe a symmetry independent relationship for the distinguishable ions to define their positions in a crystal lattice. This may translate to provide avenues to study them as symmetry non-equivalent16 cations in a unit cell. This may be conceived by the act of an interaction with or without an additional guest molecule as depicted in Fig. 2(i) and (ii). It is an established fact that different assemblies classified as H and J aggregates contribute to shift emissions; for these reasons, the understanding of solid-state structures is useful.17 However, the photoluminescence properties of assemblies having arrays of symmetry non-equivalent molecules16 have not been dealt with. Besides these, the photoluminescence of cocrystals is influenced by the local environment,18,19 which also helps in ion recognitions.8 As a matter of fact, 2,6-pyridinedicarboxylic acid has the ability to modulate the photoluminescence of nanomaterials.20 Thus, self-assemblies of such an acid with a fluorescent host would modulate the emission properties based on crystal packing. The type of modulation of hydrogen bonds is a challenge when the differences in the pKa (ΔpKa) of the interacting molecules (acid–base counterparts) fall in the range of 0–3.21–23 Such an uncertainty in the proton transfer influencing the photoluminescence properties requires structural characterisation.24 The structural aspects and photoluminescence properties with DFT calculations of various species of the assemblies of the salts of Hanthraimida with two positional isomers 2,6- or 3,5-H2pdc and 1,3-dihydroxybenzene (resorc) are reported herein.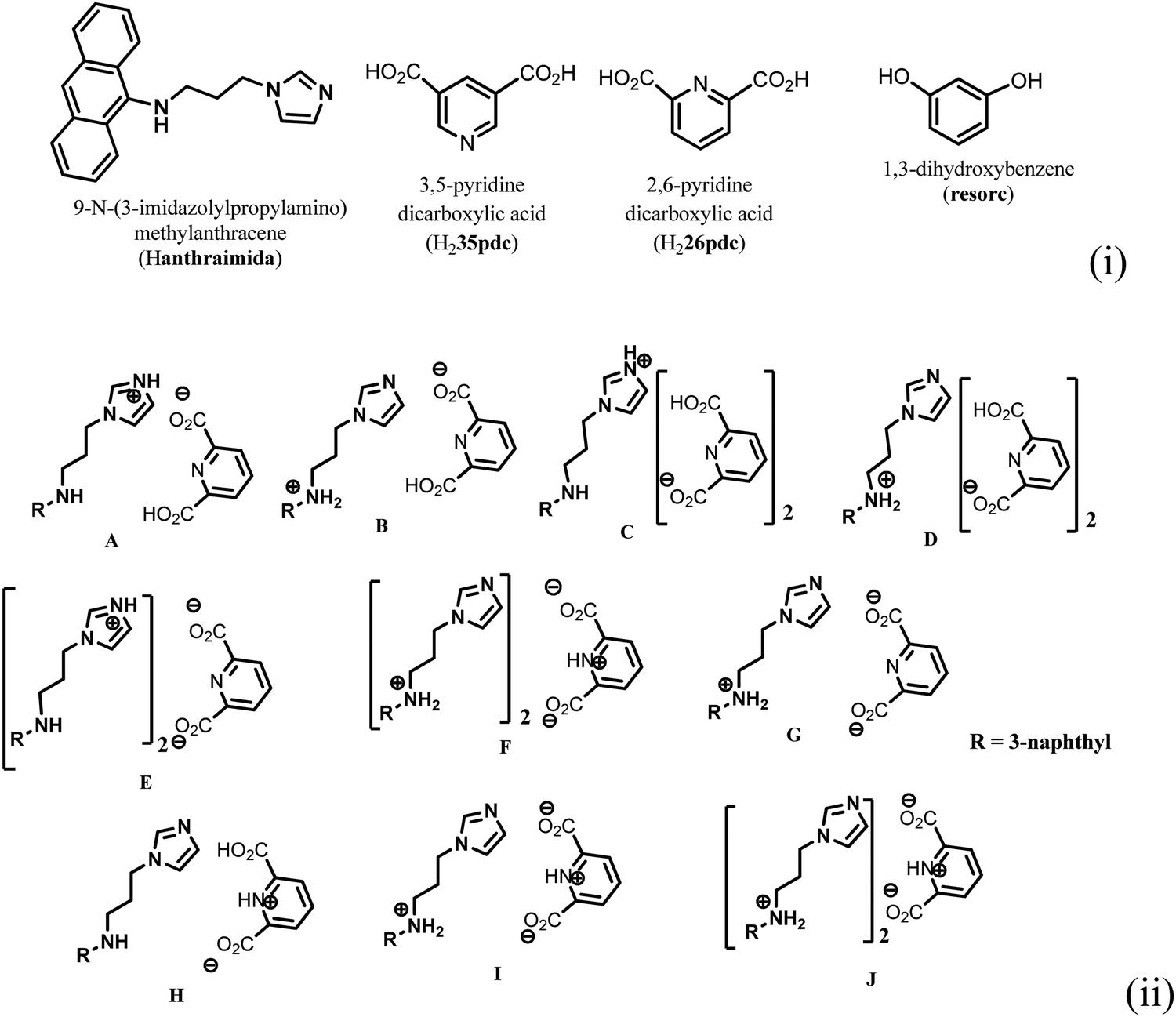 | ||
| Fig. 1 (i) Structures of Hanthraimida and the partner molecules. (ii) Some probable cocrystals and salts that may be expected from Hanthraimida reacting with pyridinedicarboxylic acid. | ||
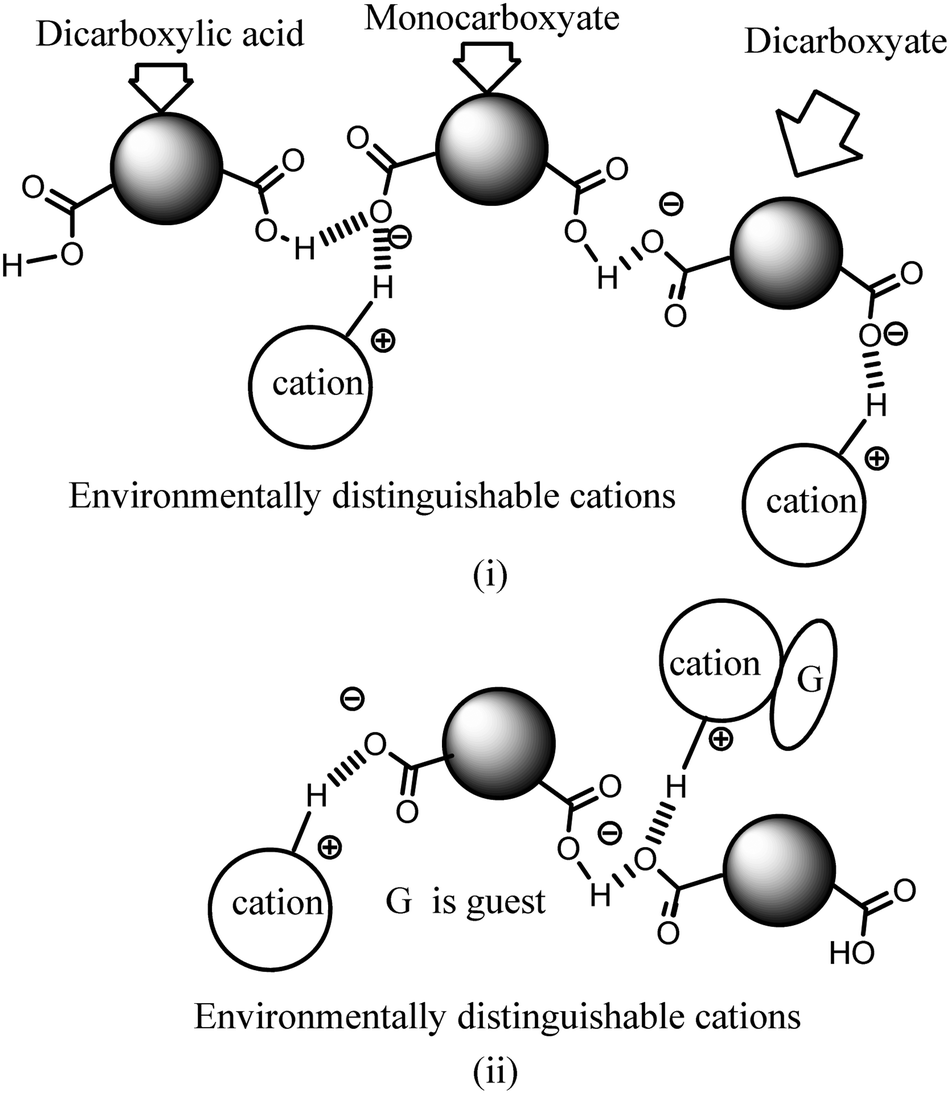 | ||
| Fig. 2 Certain possibilities in the non-equivalent environment of cations in the assemblies of dicarboxylates due to (i) assemblies of a parent neutral species with different anions, and (ii) uneven distributions of interacting guest molecules with respect to cations. | ||
Experimental
General
Infrared spectra of solid samples were recorded using a PerkinElmer Spectrum-Two FT-IR spectrophotometer in the region of 4000–400 cm−1 using the attenuated total reflectance method. Powder X-ray diffraction patterns were recorded using a Bruker powder X-ray diffractometer D2 phaser. 1H-NMR spectra of ligands were recorded using a BRUKER Ascend-600 MHz NMR spectrometer using TMS as the internal standard. A PerkinElmer Lamda-750 spectrometer was used to record the solid state UV-visible spectra by diffuse reflectance. Fluorescence emissions were measured using a Horiba Jobin Yvon Fluoromax-4C spectrofluorometer or a Horiba Jobin Yvon Fluoromax-4P spectrofluorometer by taking the specified amount of solutions as described in each figure caption, given in the text or the solid sample (20–30 mg) and exciting at a required wavelength. Lifetime decay profiles of the solid sample (finely ground) were measured using an Edinburgh Instrument, model: FSP920. The samples for scanning electron micrographs were prepared by the drop-cast method by placing a drop of the salt dissolved in DMSO and allowing it to evaporate slowly. FESEM images were recorded using a Gemini 300 FESEM. The melting points were recorded using Buchi melting point B-540 apparatus. Thermogravimetric analyses were performed using a PerkinElmer TGA 4000 at a heating rate of 10 °C per minute under a nitrogen gas flow.Preparation of Hanthraimida, salts and cocrystals
The compound Hanthraimida was prepared according to the reported procedure.14,15 The ionic cocrystals of the H226pdc and H235pdc salts were prepared by dissolving equimolar (1 mmol) amounts of Hanthraimida and either H226pdc or H235pdc in 25 mL of methanol at room temperature. The respective solution was kept undisturbed for two days, which yielded the crystals of the respective salts. The ionic cocrystals with resorcinol were prepared in methanol (25 mL) by dissolving equimolar (1 mmol) amounts of the three components, namely, Hanthraimida, H226pdc or H235pdc and resorcinol. Upon standing of the respective solution in open air, the crystals of the respective ionic salts 3 and 4 were crystallised. The respective supernatant solutions were decanted to collect the crystals and the crystals were dried over a filter paper. The spectroscopic details of the salts and ionic cocrystals are listed in the ESI.†Crystallographic study
The X-ray single crystal diffraction data for the salts were collected using an Oxford SuperNova diffractometer at room temperature. Data refinement and cell reductions were carried out using the CrysAlisPro.118 SMART software. Data reduction and cell refinements were performed using SAINT and XPREP software. Structures were solved by direct methods using SHELXS-14 and were refined by full-matrix least-squares on F2 using SHELXL-14. All non-hydrogen atoms were refined in the anisotropic approximation against F2 of all reflections. Hydrogen atoms were placed at their geometric positions by riding and refined in the isotropic approximation. The crystallographic parameters are listed in Table 1.| Parameters | 2[(H3anthraimida)]2+[(26pdc)2−·2(H26pdc)−] ·(H226pdc)·CH3OH | [(H3anthraimida)]2+[(26pdc)]2−·resorc·CH3OH·H2O | (H3anthraimida)2+(35pdc)2−·4H2O | 2[(H3anthraimida)]+[(35pdc)]2−·2resorc |
|---|---|---|---|---|
| Formula | C71H66N10O17 | C35H38N4O8 | C28H34N4O8 | C61H59N7O8 |
| CCDC | 2099505 | 2099504 | 2099935 | 2099936 |
| Mol. wt | 1331.33 | 642.69 | 554.59 | 1018.15 |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
P |
| a (Å) | 8.9684(14) | 10.662(8) | 10.541(5) | 10.8880(9) |
| b (Å) | 14.022(2) | 12.684(9) | 10.714(5) | 12.0827(10) |
| c (Å) | 26.653(4) | 14.241(11) | 13.132(7) | 20.6944(19) |
| α (°) | 101.059(5) | 110.387(13) | 89.016(14) | 91.546(3) |
| β (°) | 94.792(5) | 111.87(3) | 70.669(13) | 92.109(3) |
| γ (°) | 95.101(5) | 92.60(2) | 81.670(14) | 107.956(3) |
| V (Å3) | 3259.2(9) | 1641(2) | 1384.0(12) | 2586.0(4) |
| Density, g cm−3 | 1.357 | 1.301 | 1.331 | 1.308 |
| Abs. coeff., mm−1 | 0.099 | 0.093 | 0.098 | 0.088 |
| F(000) | 1396 | 680 | 588 | 1076 |
| Total no. of reflections | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 550 550 |
5778 | 4899 | 9156 |
| Reflections, I > 2σ(I) | 7298 | 3886 | 2414 | 5940 |
| Max. θ/° | 25.048 | 25.048 | 25.049 | 25.050 |
| Ranges (h, k, l) | −10 ≤ h ≤ 10 | −12 ≤ h ≤ 12 | −12 ≤ h ≤ 12 | −12 ≤ h ≤ 12 |
| −16 ≤ k ≤ 16 | −15 ≤ k ≤ 15 | −12 ≤ k ≤ 12 | −14 ≤ k ≤ 14 | |
| −31 ≤ l ≤ 31 | −16 ≤ l ≤ 16 | −15 ≤ l ≤ 15 | −24 ≤ l ≤ 24 | |
| Complete to 2θ (%) | 100 | 99.6 | 99.8 | 100 |
| Data/restraints/parameters | 11550/4/905 |
5778/4/436 | 4899/0/361 | 9156/5/692 |
| GooF (F2) | 1.258 | 1.044 | 1.050 | 1.034 |
| R indices [I > 2σ(I)] | 0.0659 | 0.0742 | 0.1004 | 0.0637 |
| wR2 [I > 2σ(I)] | 0.1769 | 0.2108 | 0.2063 | 0.1606 |
| R indices (all data) | 0.1119 | 0.1051 | 0.1917 | 0.1030 |
| wR2 (all data) | 0.2174 | 0.2390 | 0.2439 | 0.1849 |
Results and discussion
The structure of the salt of Hanthraimida with H226pdc was discerned by X-ray crystallography and had the composition of 2[(H3anthraimida)]2+[(26pdc)2−·2(H26pdc)−]·(H226pdc)·CH3OH (1), whereas the salt of Hanthraimida with H235pdc was a conventional tetrahydrate having a composition of (H3anthraimida)2+(35pdc)2−·4H2O (2). On the other hand, three component reactions of Hanthraimida and resorc with the respective positional isomer of H2pdc in methanol yielded the ionic cocrystals [(H3anthraimida)]2+[(26pdc)]2−·resorc·H2O·CH3OH (3) and 2[(H3anthraimida)]+[(35pdc)]2−·2(resorc) (4) (Scheme 1), respectively. The conventional salts of Hanthraimida,14,15 reported so far in the literature, were mono-cationic salts. Accordingly, we also expected a similar mono-cation (H2anthraimida+) in each salt, but we found that only cocrystal 4 had such a cation and other salts 1–3 had a dication (H3anthraimida2+) as the respective cations.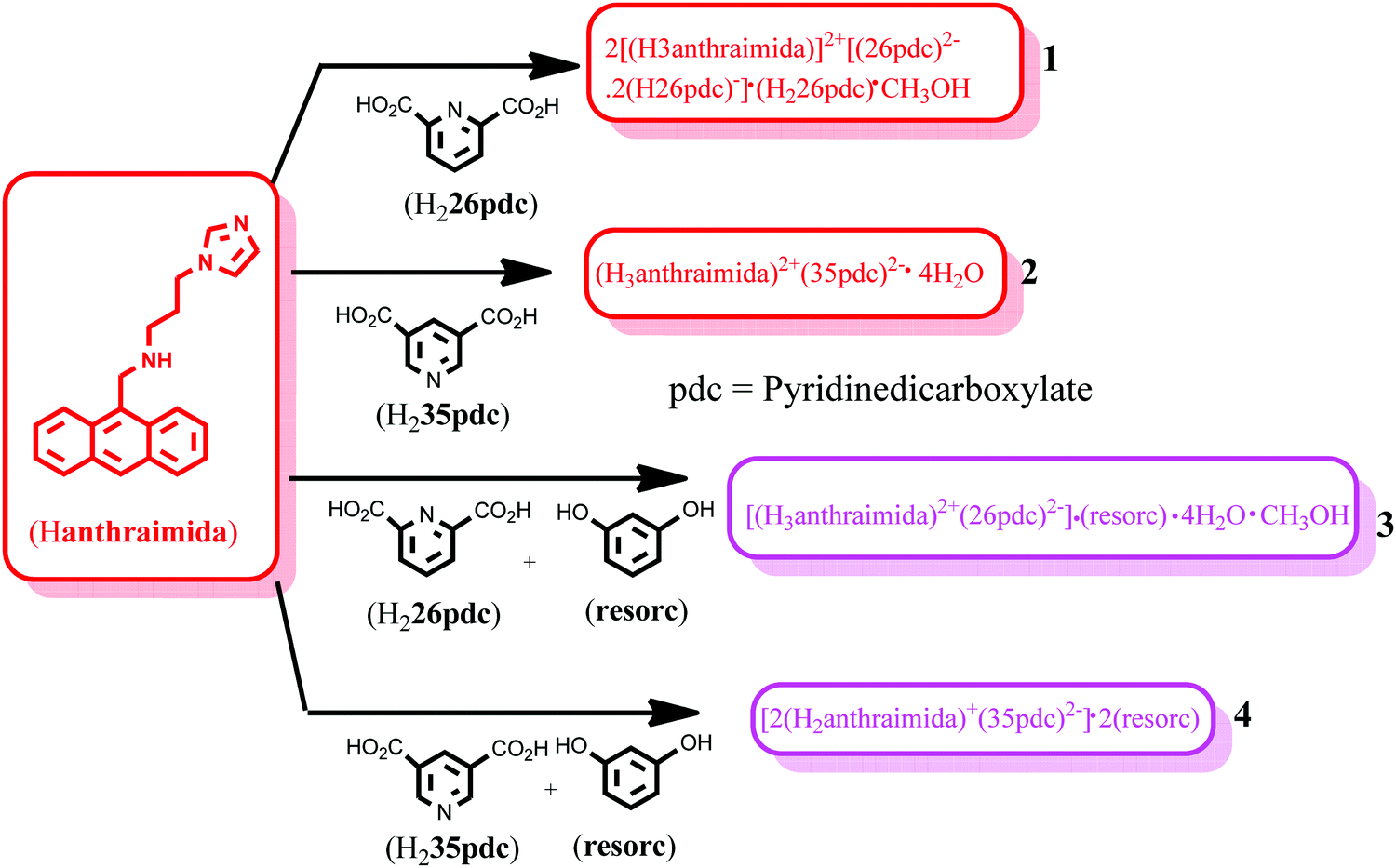 | ||
| Scheme 1 Reactants, products, salts and ionic cocrystals of Hanthraimida. | ||
The structure of 1 (Fig. 3a) has two symmetry independent cations in its unit cell, and these are illustrated in Fig. 3b. The self-assembly had chains of hydrogen bonded catemers (Fig. 3c) formed between the neutral species and anions of H226pdc. Each pyridine dicarboxylate of the chain was flanked by two pyridine monocarboxylates. Such units were hydrogen bonded to a neutral H226pdc forming a chain with ⋯H2pdc⋯HPdc⋯pdc⋯HPdc⋯ as repeat units. There are reports on sickle- or concave-shaped species having symmetry non-equivalent species in their unit cells.25,26H3anthraimida2+ had a sickle shaped geometry; possibly due to such a geometry it could manage to have two symmetry independent cations in the unit cell of salt 1. These cations were hydrogen bonded to the catemeric carboxylate–carboxylic acid chain in two distinguishable environments. The chains were hydrogen bonded between them and were located at translated positions with respect to each other. The spaces in between the chains accommodated the imidazolium portion of the di-cation. The imidazolium N+–H bond acted as the hydrogen bond donor from an oxygen atom to carboxylate. However, the –NH2–+ moiety formed N3+–H⋯O13 and N3+–H⋯O12 hydrogen bonds to link another chain (Fig. 3c).
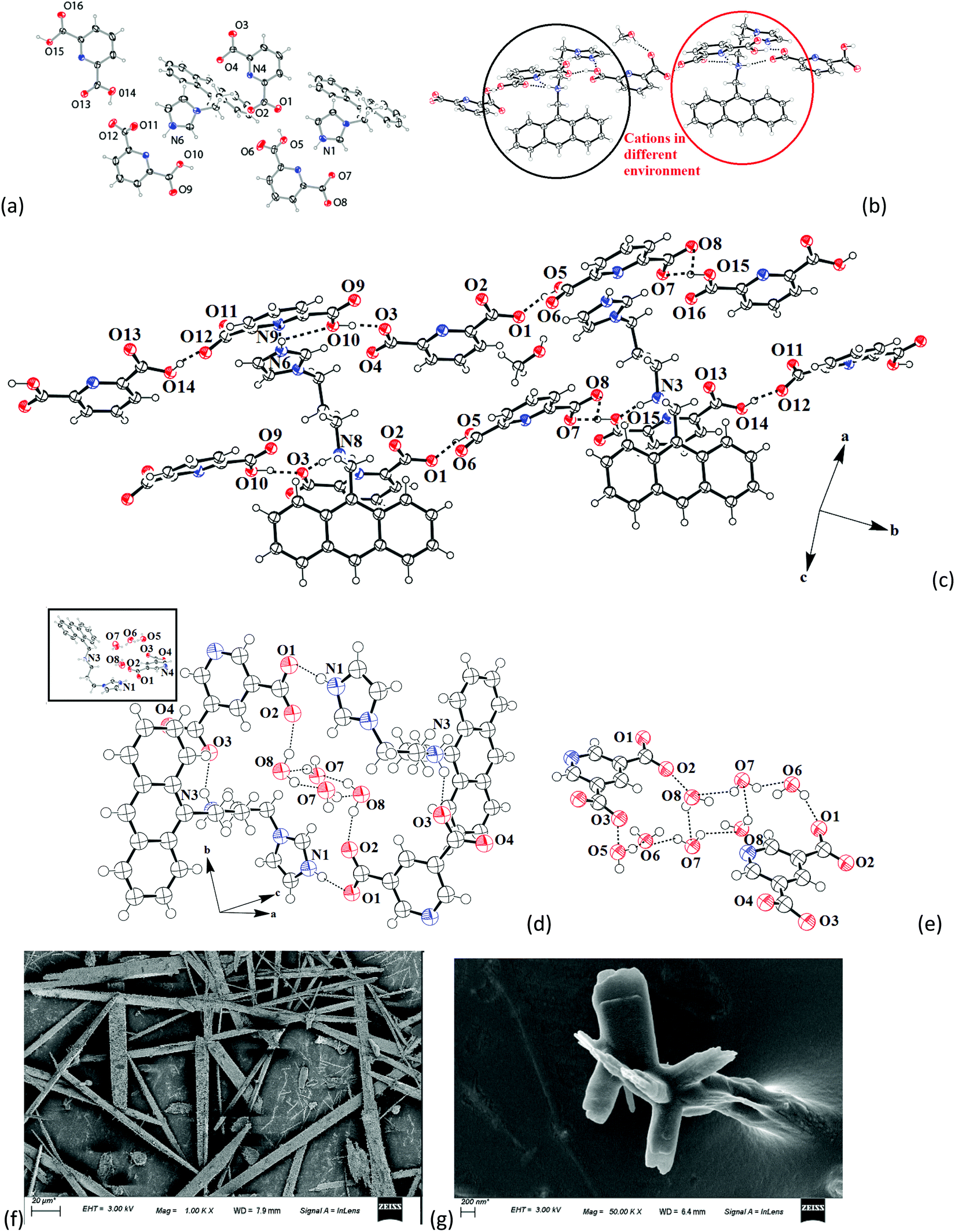 | ||
| Fig. 3 The crystal structure of (a) 2[(H3anthraimida)]2+[(26pdc)2−·2(H26pdc)−]·(H226pdc)·(CH3OH) (1). (The solvent molecules are omitted for clarity.) (b) Two distinct environments of the symmetry independent di-cations marked in red and black circles. (c) The prominent hydrogen bonds in the self-assembly. (d) The hydrogen bonded assembly of (H3anthraimida)2+(35pdc)2−·4H2O (2) (the inset is the crystal structure of 2). (e) Octameric water cluster in 2. (Thermal ellipsoids are with 50% probability.) (f) and (g) are the scanning electron micrographs of the crystals of 1 and 2, respectively. | ||
The self-assembly of (H3anthraimida)2+(35pdc)2−·4H2O is shown in Fig. 3d, and it had self-assembled hydrogen bonded dimers of the salts. Each 35pdc2− was bridged by di-cations to form dimers. For this, one carboxylate group of 35pdc2− was hydrogen bonded to the imidazolium cation, whereas the second carboxylate group of the same pdc2− formed a hydrogen bond with NH2+ (Fig. 3d). The di-cation H3anthraimida2+ had a sickle shaped geometry. The self-assembly between the two di-cations took place by keeping the concave faces of the di-cations facing each other. The assembly was done with the help of hydrogen bonds with two dicarboxylate anions, which provided space to accommodate four water molecules in the void. The water molecules were observed as hydrogen bonded octameric water clusters as illustrated in Fig. 3e. Each water octamer was held in the lattice hydrogen bonds with two dicarboxylates. The cluster contributed to form the tight packed structure of the salt by filling the possible voids in the absence of them and also provided the additional hydrogen bonds to the self-assembly, contributing to the overall stabilization. The presence of water molecules in the salt was also confirmed by themogravimetry; salt 2 loses water molecules upon heating at 70–100 °C. Some imidazoles derived as ancillary ligands or as a part of carboxylate ligands in metal complexes stabilize different water clusters.27–29 Depending on the directional behaviors of ligands, central metal ion and substrates, different geometrical arrangements of the octamers of water clusters were observed.27,28 In the present case, four water molecules were assembled to form a cyclic synthon, which was connected to two pairs of hydrogen bonded water molecules each linked at the two terminals of the cyclic synthon. Certain concave shaped molecules having water clusters form a gel.30 However, we did not observe a stable gel in the present case but we found that salt 2 was difficult to crystallize, and the crude sample obtained by solvent evaporation was a sticky semi-solid; however, upon re-dissolving, it provided micro-crystals. The microcrystals grown in different directions with sizes less than 200 nm were observed in the scanning electron micrograph of 2, whereas the SEM image of salt 1 showed micrometer size needle shaped crystals (Fig. 3f and g).
The ionic cocrystal 3, namely [(H3anthraimida)]2+[(26pdc)]2−·resorc·H2O·CH3OH (Fig. 4a), had the 1,3-dihydroxybenzene partner molecule and the solvent of crystallization methanol and water molecules. One of the oxygen atoms (O4) and the nitrogen atom (N4) of one dicarboxylate were hydrogen bonded to two N–H bonds of NH2+. Another carboxylate of the same 26pdc formed hydrogen bonds with another N–H bond of a neighboring dication. Such assembling provided robust hydrogen bonded R44(12) synthons31 (Fig. 4b). The literature suggests that such synthons are useful in generating extended assemblies and contribute to molecular recognition.32 These synthons were connected to the N+–H bond of the imidazolium cationic part. One oxygen atom from each of the other carboxylate groups of two independent 35pdc ions acted as bridging atoms to two N+–H of the two independent di-cations. Therefore, R44(8) synthons composed of N+–H⋯O bonds were formed (Fig. 4b). These synthons had knitted the anions with the di-cations through charge-assisted hydrogen bonds. Such interactions have yielded layer-like arrangements in the self-assembly of the ionic cocrystal. The resorcinol molecules were hydrogen bonded to the O3 oxygen atom of dicarboxylate by O6–H⋯O3 hydrogen bonds. The structure may thus be explained in a simplified manner as a domain expanded carboxylate catemer by the two aqua bridges. Earlier we reported that depending on the directional hydrogen bonds, the partner provides avenues for robust synthons to have expansion of the domain.33,34 In the present case, the water molecules have played the role to provide robust R44(12) synthons, each of which holds 1,3-dihydroxybenzene guests at two sides. In the self-assembly of 3, the space between the self-assembled layers of cations and anions was occupied by the resorc molecules. The methanol molecules also occupied the interstitial positions and were weakly C–H⋯O hydrogen bonded.
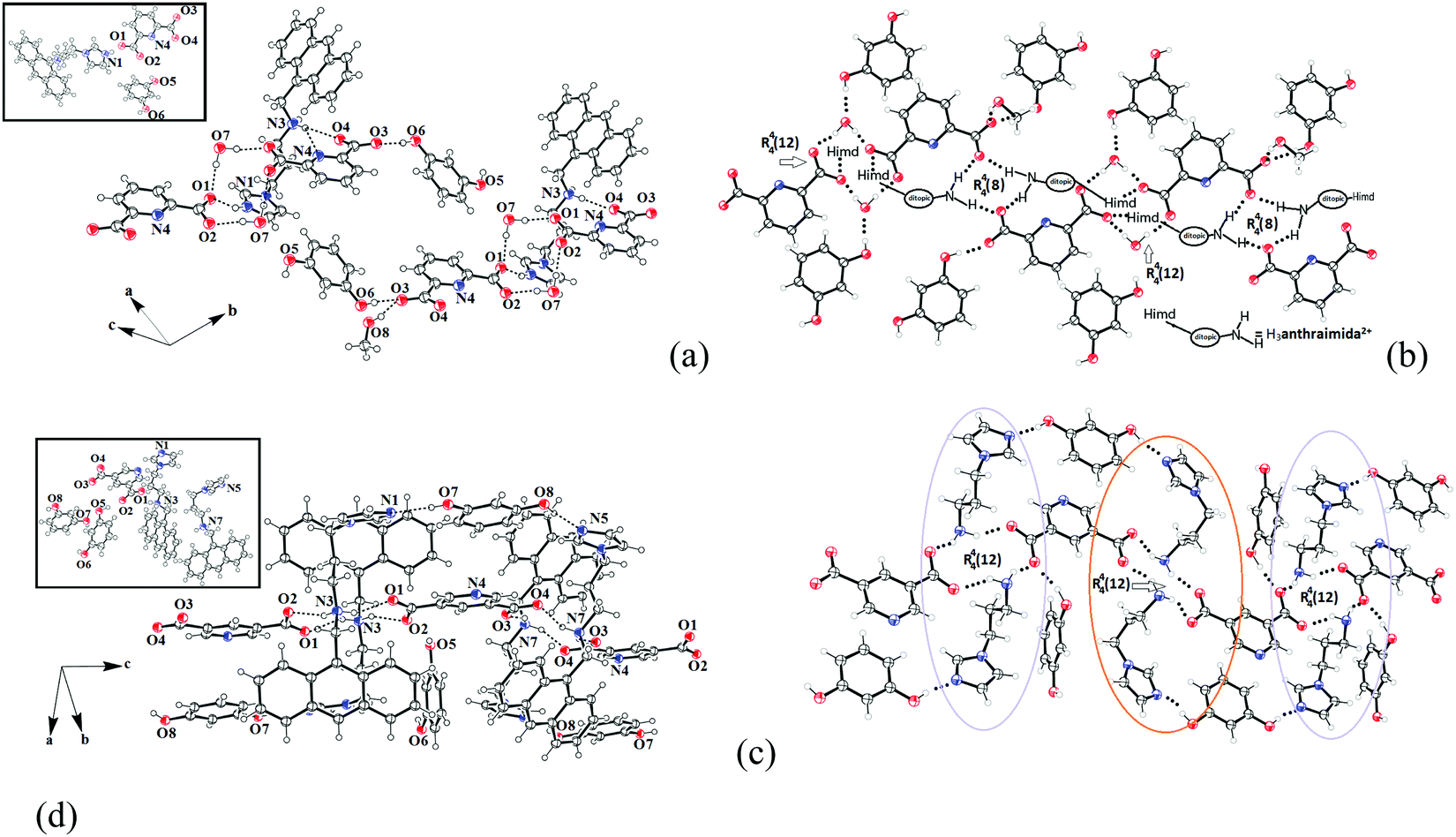 | ||
| Fig. 4 The self-assembly of (a) [(H3anthraimida)]2+[(26pdc)]2−·resorc·H2O·CH3OH (3). (b) The aqua-bridged hydrogen bonded carboxylate dimers held together by di-cations in the self-assembly of 3. (c) The self-assembly of 2[(H2anthraimida)]+[(35pdc)]2−·2(resorc) (4) and (d) two distinguishable environments of the cations of 4 drawn by omitting the anthracene unit. (The insets in (a and c) are the respective crystals structures and thermal ellipsoids are drawn with 50% probability.) | ||
The crystal structure of 2[(H2anthraimida)]+[(35pdc)]2−·2(resorc) (4) is shown in Fig. 4c and had hydrogen bonded mono-cations in pairs. These cations were arranged in head to tail orientations with respect to each other by hydrogen bonded bridges of two carboxylate anions. These provided robust hydrogen bonded R44(12) synthons (Fig. 4d). A set of resorc molecules connected the two such dimers to form cleft-like arrangements. Another set of resorc molecules were encapsulated in such a cleft. Thus, one of the resorc guests interacted with the dimers to form a secondary host to accommodate the guest, namely another resorc molecule. There are examples of assembling of a host with a guest to accommodate additional guest molecules.35,36 Cocrystal 4 is an example where the association of a guest molecule (resorc) with the host cations encapsulates an additional guest resorc molecule. While the guest resorcinol was present at the alternative positions between the dimeric units, there were two types of the surrounding supramolecular environment for the cations as shown in Fig. 4d.
From the structural study, it is clear that the constituent cation of the salts of Hanthraimida depends on the partner molecules and carboxylic acid used. This suggests that the exception can be made over the type of salts of Hanthraimida with organic carboxylic acids where in general the mono-cationic salts dominated.17,18 There are limited examples of assemblies containing the conjugate acid or the base part as neutral components in organic salts37–39 and inorganic complexes.40 Any of the three forms of pyridinedicarboxylic acids, namely, the neutral, mono-carboxylate or dicarboxylate, could get incorporated in the salts either as partner molecules or anions. Having neutral pyridinedicarboxylic acid in the ionic cocrystal is a new observation and suggest that compositions of salts should have priority while dealing with the salts of polycarboxylic aids with the polytopic base. The pKa value of H226pdc (pKa = 2.16) and that of H235pdc (pKa = 2.80) are comparable, whereas the pKa values of Hanthraimida (in DMF/water) are 5.9 and 4.1, respectively. From such a small difference of pKa, one would expect a similar cocrystal from the two positional isomers of H2pdc,11,12 but it did not occur. So, it was not possible to make a prior prediction on the composition of the crystalline products. A series of independent pH titrations with the parent compound with aqueous hydrochloric acid and with the respective H2pdc were carried out. The variation of the pH of the parent compound upon the addition of H2pdc to the solution of Hanthraimida or it together with resorcinol was the same, indicating that the pH change profiles in each case were similar. This clearly showed that the formations of these salts and ionic cocrystals were not primarily guided by pKa. The transfers of protons were to confer stability by forming tight-packed structures. In these examples, we could not forecast the observed compositions, but had to depend on the structural and spectroscopic study to formulate each composition. In general, densely packed cocrystals with stronger hydrogen bonds are more stable,41 and the ordered π-stacks of aromatic rings in ionic cocrystals contribute to tightly packed structures.42,43 However, in the present examples, the stacking among anthracene rings was not observed. Thus, the interplay of hydrogen bonds with electrostatic interactions guided the tightly packed structures in the present case, resulted in the partial or complete proton transfer from H2pdc to realise the stable structures with varied compositions.
Photoluminescence properties
The detection of pyridinedicarboxylates by fluorescence spectroscopy has attracted interest from the biological point of view.44–46 Variations on the compositions of the salts discussed above had provided the scope to study the emission properties of them in solid and solution states. Hence, fluorescence titrations of Hanthraimida with the two positional isomers of H2pdc (λex, 365 nm) were carried out. Both titration profiles had showed a continuous increase in the emission of the conventional vibrational distinct π*–π transitions of Hanthraimida occurring at 418 nm, 441 nm and 471 nm (Fig. 5 (left)). The trends from both complemented the earlier observation caused by other aromatic carboxylic acids or phenolic compounds on the emission spectra of Hanthraimida.14,15 The observed fluorescence enhancement upon interactions was due to the protonation of the N–H group of Hanthraimida by organic carboxylic acids, in these cases by the two isomers of H2pdc. This was due to a photo-induced electron-transfer (PET) effect that was originally present in Hanthraimida, and PET was affected by protonation. Since the emission changes caused by two positional isomers were identical in the respective emission spectroscopic titration, no selectivity between them was observed.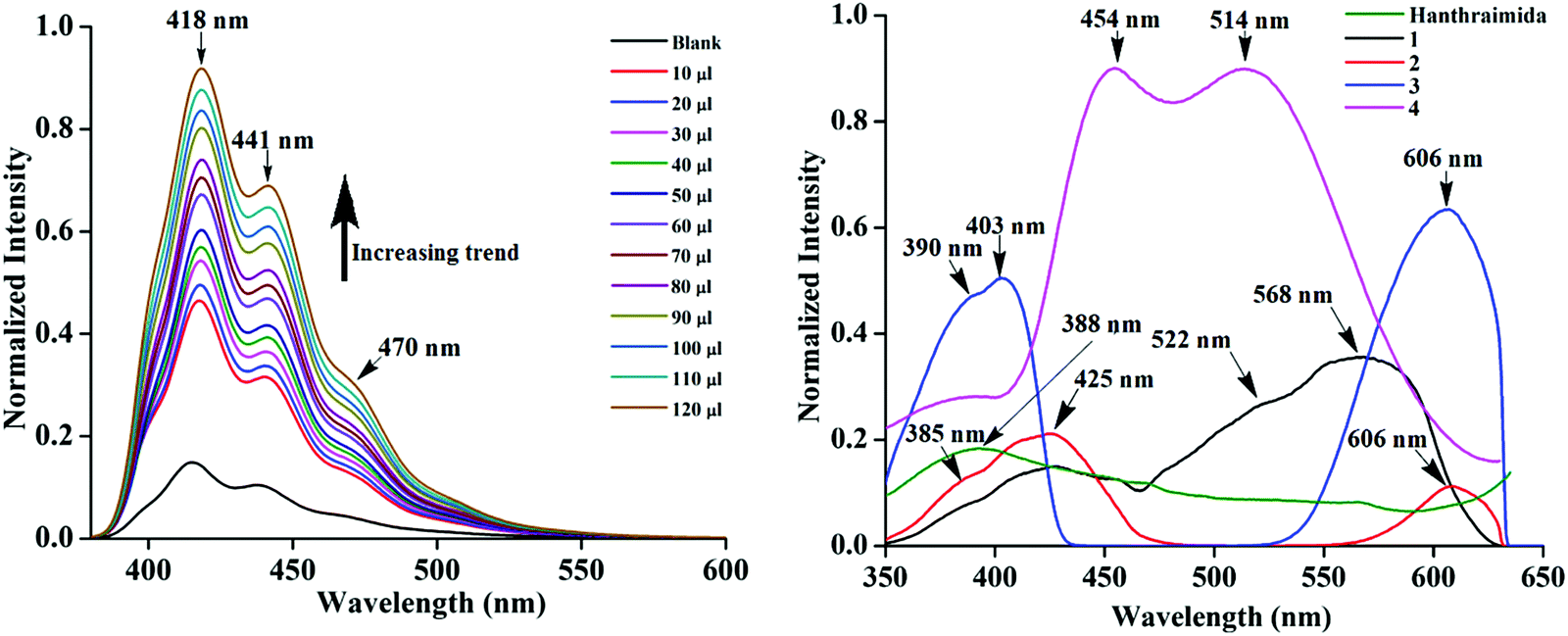 | ||
| Fig. 5 Fluorescence titration (λex, 365 nm) of Hanthraimida (10−5 M in ethanol, 2 mL) with (left) H226pdc (10 μl aliquot of 10−5 M in ethanol). (right) Photoluminescence spectra of the solid samples of Hanthraimida 1–4 (excitation at 325 nm). | ||
The solid sample of Hanthraimida had a broad emission at 388 nm (λex = 325 nm), and this broad emission was due to the π*–π transition, which is in accordance with the DFT calculated energy gap of HOMO–LUMO (357.41 nm), whereas the solid samples of 1–4 had shown completely different emission features, which showed multiple emission peaks as illustrated in Fig. 5 (right).
The charge-assisted hydrogen bonds usually modulate π-stacking42 and influence photoluminescence properties in the solid state, but none of the assemblies of 1–4 had π-stacking among the anthryl fluorophores. The UV-visible spectra of 1–4 in the solution did not show the shift of the peaks from the parent Hanthraimida (Fig. S28, ESI†); however, the solid samples of 2 and 3 clearly showed a 32 nm shift of peaks (Fig. S32, ESI†) towards a higher side with respect to the absorption peak of Hanthraimida. This suggested a charge-transfer in these two examples. In general, the effects of different weak interactions and proton transfers are averaged out in solution and the interactions of solvent molecules at the excited and ground states play important roles in emission spectroscopy.47 In the solid state, the structures are rigid; hence, it provide scope to understand the properties from a fixed geometry. The HOMO–LUMO gaps calculated using DFT are used to elucidate different mechanisms such as the excited state proton transfer mechanism.48,49 So, we examined the molecular orbitals of the cations (Fig. 6a) and anions (Fig. 6b) using DFT calculations with the B3LYP functional using 6-311++G (d,p) as the basis set. There can be two forms of the protonated states of the monocation, namely the anthryl-ammonium form represented as H2anthraimida+ had a HOMO–LUMO gap of 360.22 nm (Fig. 7a), whereas the other form having the imidazolium cation [H2anthraimida]+(imidazole) had such an energy gap of 643.81 nm and a HOMO–LUMO+1 energy gap of 422.28 nm (Fig. 7a). Accordingly, the two forms of mono-cations had a large difference with their respective π* to π transition. Hanthraimida and H2anthraimida+ had orbitals in the HOMO and LUMO and were localized at the anthryl group. Hence, in these cases, the excitation of HOMO to LUMO was for π–π* transition. Moreover, in the form [H2anthraimida]+(imidazole) and in H3anthraimida2+, the respective HOMO was localized at the anthryl group, whereas the LUMO was localized at the imidazole unit (Fig. 6a). Thus, the lowest π–π* emission in these species had to be from LUMO+1 to HOMO. The dication H3anthraimida2+ had the calculated HOMO–LUMO energy gap of 499.98 nm, and the calculated HOMO and LUMO+1 was 365.4 nm. The calculated energy differences between the HOMO and LUMO of H226pdc, H26pdc−, 26pdc2−, were 231 nm, 403 nm and 373 nm, respectively (Fig. 6b). The same gap for 35pdc2− was 281.20 nm. Thus, the calculated π* to π emission for H3anthraimida2+ at 365.4 nm was comparable to that of 26pdc2− (373 nm). The similar energy gaps of these two species of the salts had suggested the possibility of the Förster resonance energy transfer.50 The excitation spectra were shown in each case and the excitation took place at 325 nm. Hence, by comparing the theoretical π* to π emissions, there was a possibility to observe the emission from either H3anthraimida2+ or H2anthraimida+ or 26pdc2−, but the observed emission in each case was at longer wavelengths having a large Stokes shift. Salt 1 had two H3anthraimida2+ cations with an independent crystallographic symmetry in the unit cell; in the solid state, it showed emissions at 522 nm and 568 nm. The degenerated HOMO and LUMO of the two symmetry independent di-cations split by mixing of orbitals, leading to emissions at 522 nm and 568 nm, whereas the peaks at 385 nm and 425 nm were from π* to π transitions of H3anthraimida2+ and H26pdc−, respectively. The broad unresolved emissions in 390–403 nm were observed for 2, which is assigned to H3anthraimida2+. Salt 3 had emissions in the region of 385–425 nm from the emissions of H3anthraimida2+ and 26pdc2−. The salt and cocrystals 2 and 3 had a common emission at 606 nm. This emission was attributed to the formation of the mono-cation by the excited state proton transfer from the di-cation to the pdc anion. The path E shown in Fig. 7a is one of the possible path. This suggestion is based on the fact that different cations have small energy barriers and they could have easily transformed to one another by the excited state proton transfer. On the other hand, the ionic cocrystal 4 showed a weak emission at 385 nm and two relatively sharper emissions at 454 nm and 515 nm. The 385 nm emission was from π* to π transition of H2anthraimida+ and the latter two emissions were due to the mixing of the orbitals of the two symmetry independent H2anthraimida+, as shown in Fig. 7b. The emission spectra of 1 and 2 were recorded by changing the irradiations to 330, 340, 360 and 370 nm in independent experiments. It was found that as the irradiation wavelength was increased, the emissions at the higher wavelengths were shifted to further higher wavelengths and the intensities of the emissions at shorter wavelengths were reduced drastically (Fig. S19 and S20, ESI†). In aggregates, the vibrational levels play a major role to shift the spectral positions.16 The excitation to higher vibrational levels was reduced by increasing the irradiated wavelength, and the emission occurred from the lower vibrational level of the excited state in such a situation which caused the shifting of the emissions towards a higher wavelength side to show a left-skewed shape of the spectral peak at longer wavelengths. The possibility of stray light interference in photoluminescence was checked by recording the data in two sets of independent experiments performed on cocrystal 2 and cocrystal 3 by using two different models of the spectro-fluorimeter (Horiba Jobin Yvon Fluoromax-4P and Fluoromax-4C). Consistencies of the emission peaks in the respective spectra obtained from both the equipments under analogous conditions were observed. We also had carried out experiments by changing the slit widths, as illustrated in Fig. S26 and S27 (ESI†). As expected, the independent samples of cocrystal 2 and that of cocrystal 3 showed the two anticipated emission peaks with a change in slit widths having a band-pass from 5 nm to 4 nm. However, the spectra were with slight differences in the intensities of emissions. However, when the band-pass was 2 nm, there was a drastic change in the emission peaks. A single peak was observed in both the cases by using this slit (2 nm). It is understandable from the manual of the equipment as well as from the conventional understanding that lowering the slit width is useful to obtain better peaks but at the cost of other peaks. In the present examples, we have dealt with multi-component cocrystals to observe photoluminescence as a collective effect from the aggregates. Hence, the consistent data on the dual emissions with larger slits are attributed to covering the domain (repeat units in the lattice as discussed in structural analysis) of the aggregate while performing irradiations. The dual emissions in different materials may occur through different paths, and some of these are reported to violate the Kasha rule.51 Our systems do violate the Kasha rule in showing the higher wavelength emissions.
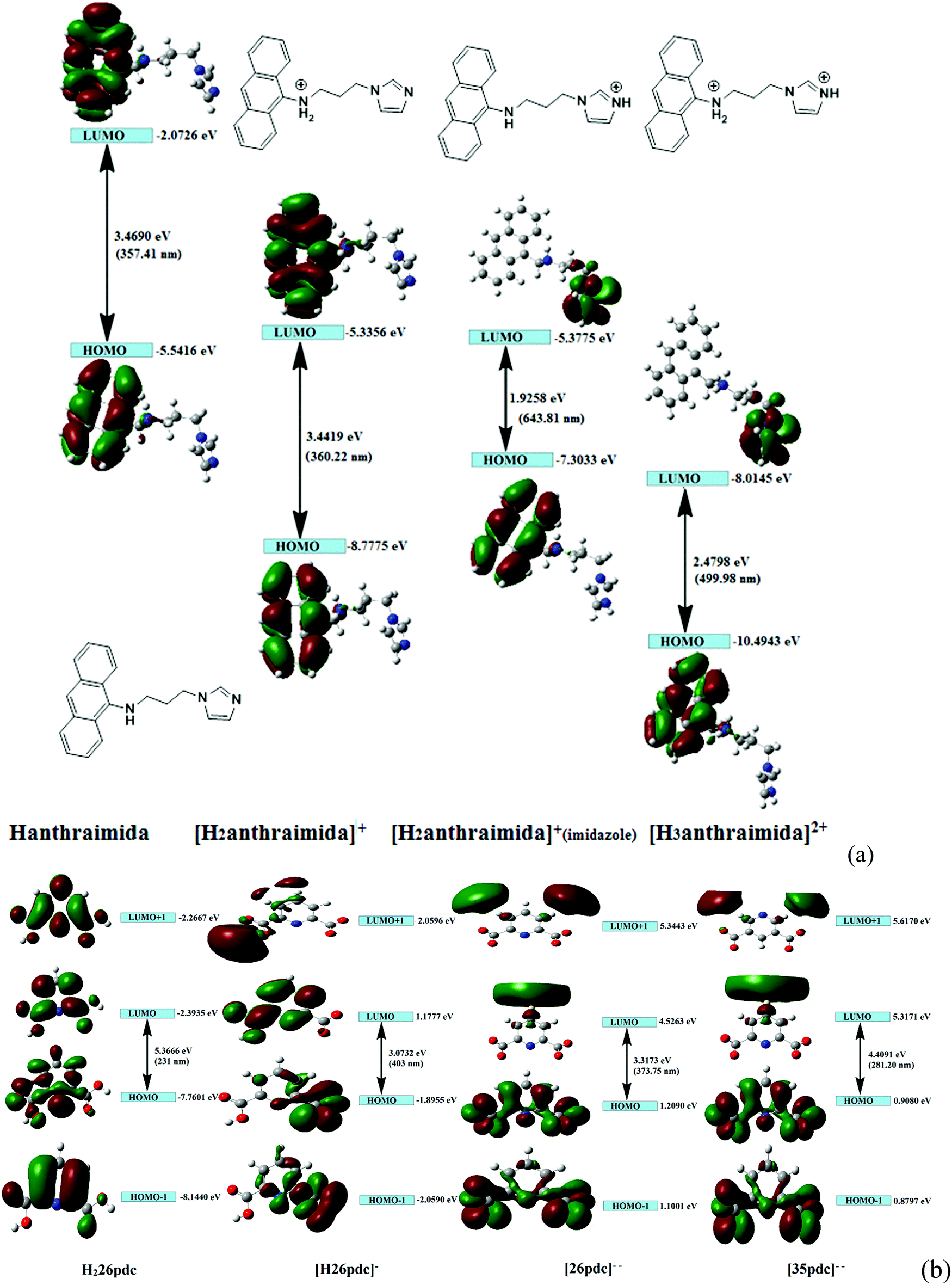 | ||
| Fig. 6 Energy details of the HOMO and LUMO from DFT calculations by the B3LYP functional using 6-311++G(d,p) as the basis set of the different species of (a) Hanthraimida, [H2anthraimida]+, [H2anthraimida]+(imidazole), and [H3anthraimida]2+ cations. (b) HOMO−1, HOMO, LUMO and LUMO+1 energy states of H226pdc, H26pdc−, 26pdc2−, and 35pdc2−. | ||
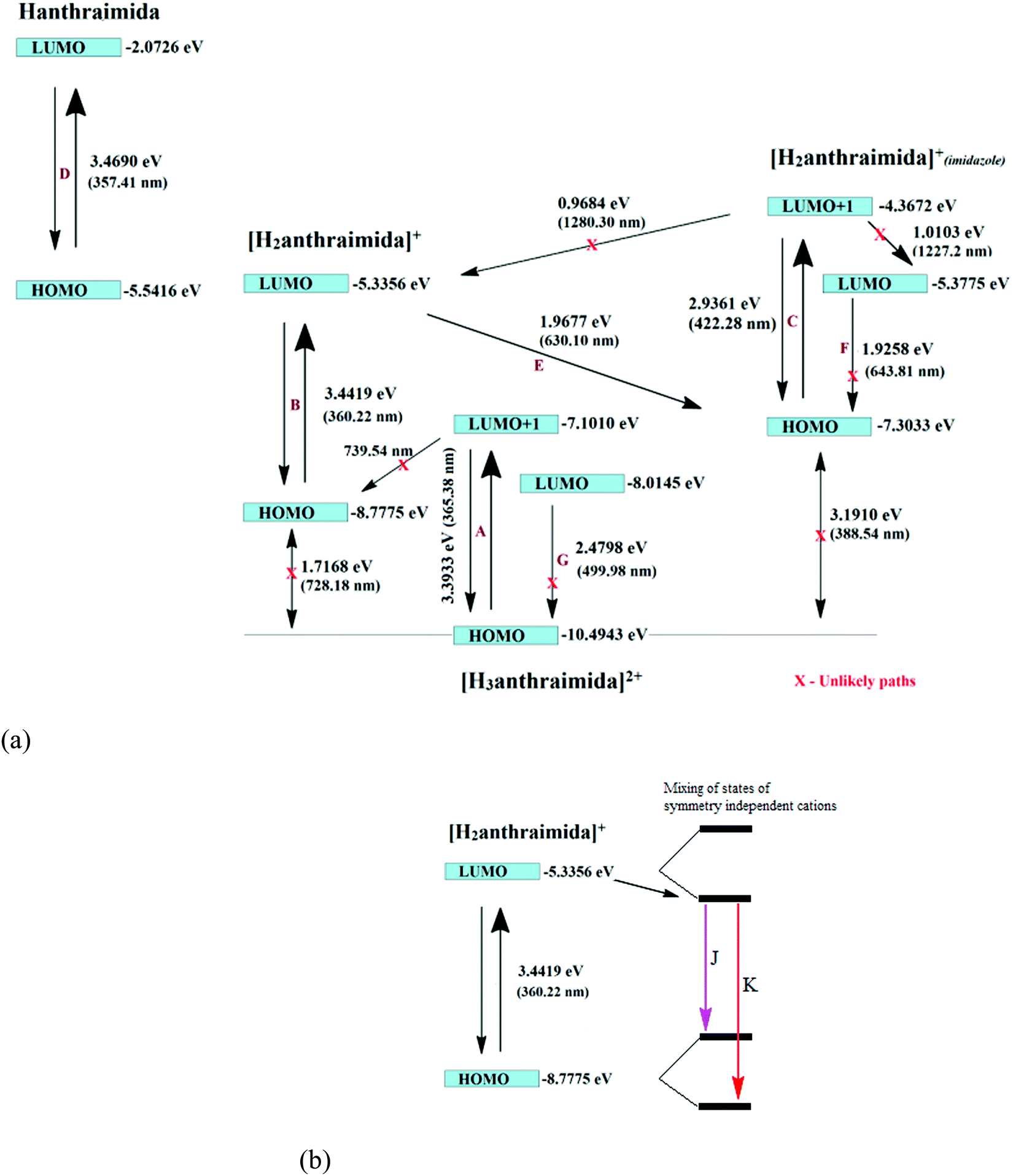 | ||
| Fig. 7 (a) Different emission paths in salts having the H3anthraimida2+ cation yielding the mono-cation through the excited state intermolecular proton transfer to anions (assuming the invariant role of the anions to accept protons at the excited state). (b) The emission paths of symmetry non-equivalent H2anthraimida+ cations due to splitting caused by mixing of orbitals. | ||
The emission decay profile of 1 was tri-exponential and had lifetimes of 0.65 ns, 5.25 ns and 13.70 ns, where the second and third paths were followed by about 75% species. However, in the other three cases 2–4, each had a short lifetime path having a lifetime in the range of 2.06–3.47 ns, two relatively longer lifetime paths with 20.95–27.48 ns for one set and 45.25–120.90 ns for another. The relatively longer lifetimes were followed by a major proportion of species (>80%) attributed to the intra-molecular excited state proton transfers. This was due to the transfer of protons from the dication to the anion. As a result of such a proton transfer, the mono-cation transforms from one form to another as in the path E in 1–3 and in the case of 4 the splitting of the HOMO and LUMO took place. This caused the emissions in the visible region.
In conclusion, a combined structural and theoretical study on the observed photoluminescence of salts and ionic cocrystals having different compositions and also having different cationic forms of the same fluorophore has shown emissions at longer wavelengths. Salt 1 had provided an unusual example on the combinations of neutral and two different anionic forms. Here, the catemeric carboxylic acid–carboxylate had provided the template for the cations at symmetry independent positions. In these selected examples, symmetric non-equivalent cations in the lattice were observed due to the different local non-covalent environments. There was a larger difference in the emission of Hanthraimida in the presence of pdc in solution than that of the corresponding salt in the solid state. The DFT calculated energy gaps of different forms of ions could explain the emissions observed at shorter wavelengths. The multiple fluorescence emissions in the visible-region of the salts in the solid state were attributed to the mixing of degenerate energy-levels of the symmetry independent ions and intermolecular proton transfers at the excited state.
Conflicts of interest
The authors declare that there is no coflict of interest.Acknowledgements
The authors thank the Ministry of human resource and development (India) (Grant No. F. No. 5-1/2014-TS.VII), the North-east centre for biological sciences and healthcare engineering, IIT-Guwahati, and Indian Institute of Technology, Guwahati, for providing the facilities.References
- D. J. Berry and J. W. Steed, Pharmaceutical cocrystals, salts and multicomponent systems; intermolecular interactions and property based design, Adv. Drug Delivery Rev., 2017, 117, 3–24 CrossRef CAS PubMed.
- O. Shemchuk, D. Braga, F. Grepioni and R. Turner, Co-crystallization of antibacterials with inorganic salts: paving the way to activity enhancement, RSC Adv., 2020, 10, 2146 RSC.
- O. Shemchuk, F. Grepioni and D. Braga, Co-crystallization of racemic amino acids with ZnCl2: An investigation of chiral selectivity upon coordination to the metal centre, CrystEngComm, 2020, 22, 5613–5619 RSC.
- S. Mohamed, A. A. Alwan, T. Friscic, A. J. Morris and M. Arhangelskis, Towards the systematic crystallisation of molecular ionic cocrystals: insights from computed crystal form landscapes, Faraday Discuss., 2018, 211, 401–424 RSC.
- Z. X. Ng, D. Tan, W. L. Teo, F. Leon, X. Shi, Y. Sim, Y. Li, R. Ganguly, Y. Zhao, S. Mohamed and F. García, Mechanosynthesis of higher-order cocrystals: tuning order, functionality and size in cocrystal design, Angew. Chem., Int. Ed., 2021, 133, 17622–17631 CrossRef.
- A. F. Shunnar, B. Dhokale, D. P. Karothu, D. H. Bowskill, I. J. Sugden, H. H. Hernandez, P. Naumov and S. Mohame, Efficient screening for ternary molecular ionic cocrystals using a complementary mechanosynthesis and computational structure prediction approach, Chem. – Eur. J., 2020, 26, 4752–4765 CrossRef CAS PubMed.
- C. B. Aakeröy, N. C. Schultheiss, A. Rajbanshi, J. Desper and C. Moore, Supramolecular synthesis based on a combination of hydrogen and halogen bonds, Cryst. Growth Des., 2009, 9(1), 432–441 CrossRef PubMed.
- M. Karimi-Jafari, L. Padrela, G. M. Walker and D. M. Croker, Creating cocrystals: a review of pharmaceutical cocrystal preparation routes and applications, Cryst. Growth Des., 2018, 18(10), 6370–6387 CrossRef CAS.
- G. R. Desiraju, in The supramolecular synthon in crystal engineering in stimulating concepts in chemistry, ed. F. Vögtle, J. F. Stoddart and M. Shibasaki, Wiley-VCH, Weinheim, Germany, 2000, pp. 293–306 Search PubMed.
- P. Kavuru, D. Aboarayes, K. K. Arora, H. D. Clarke, A. Kennedy, L. Marshall, T. T. Ong, J. Perman, T. Pujari, L. Wojtas and M. J. Zaworotko, Hierarchy of supramolecular synthons: persistent hydrogen bonds between carboxylates and weakly acidic hydroxyl moieties in cocrystals of zwitterions, Cryst. Growth Des., 2010, 10, 3568–3584 CrossRef CAS.
- S. L. Childs, G. P. Stahly and A. Park, The salt-cocrystal continuum: the influence of crystal structure on ionization state, Mol. Phramaceutics, 2007, 4, 323–338 CrossRef CAS PubMed.
- A. J. Cruz-Cabeza, Acid–base crystalline complexes and the pKa rule, CrystEngComm, 2012, 14, 6362–6365 RSC.
- N. A. Mir, R. Dubey and G. R. Desiraju, Strategy and methodology in the synthesis of multicomponent molecular solids: the quest for higher cocrystals, Acc. Chem. Res., 2019, 52, 2210–2220 CrossRef CAS PubMed.
- M. P. Singh, A. Tarai and J. B. Baruah, Photo-physical properties of salts of a di-topic imidazole-tethered anthracene derivative in solid and solution, CrystEngComm, 2019, 21, 4898–4909 RSC.
- A. Pandith, A. Kumar and H.-S. Kim, 9-N-Alkylaminomethylanthracene probes for selective fluorescence sensing of pentafluorophenol, RSC Adv., 2015, 5, 81808–81816 RSC.
- N. J. Hestan and F. C. Spano, Expanded theory of H- and J-molecular aggregates: the effects of vibronic coupling and intermolecular charge transfer, Chem. Rev., 2018, 118, 7069–7163 CrossRef PubMed.
- K. M. Steed and J. W. Steed, Packing problems: high Z′ crystal structures and their relationship to cocrystals, inclusion compounds, and polymorphism, Chem. Rev., 2015, 115, 2895–2933 CrossRef CAS PubMed.
- M. P. Singh, A. Tarai and J. B. Baruah, Neutral, zwitterion, ionic forms of 5-aminoisophthalic acid in cocrystals, salts and their optical properties, ChemistrySelect, 2019, 19, 5427–5436 CrossRef.
- D. Yan, A. Delori, G. O. Lloyd, T. Friscic, G. M. Day, W. Jones, J. Lu, M. Wei, D. G. Evans and X. Duan, A cocrystal strategy to tune the luminescent properties of stilbene-type organic solid-State materials, Angew. Chem., Int. Ed., 2011, 50, 12483–12486 CrossRef CAS PubMed.
- R. Gui, H. Jin, X. Bu, Y. Fu and Z. Wang, Recent advances in dual-emission ratiometric fluorescence probes for chemo/biosensing and bioimaging of biomarkers, Coord. Chem. Rev., 2019, 383, 82–103 CrossRef CAS.
- M. L. Cheney, N. Shan, E. R. Healey, M. Hanna, L. Wojtas, M. J. Zaworotko, V. Sava, S. J. Song and J. R. Sanchez-Ramos, Effects of crystal form on solubility and pharmacokinetics: a crystal engineering case study of lamotrigine, Cryst. Growth Des., 2010, 10, 394–405 CrossRef CAS.
- M. L. Cheney, D. R. Weyna, N. Shan, M. Hanna, L. Wojtas and M. J. Zaworotko, Supramolecular architectures of meloxicam carboxylic acid cocrystals, a crystal engineering case study, Cryst. Growth Des., 2010, 10, 4401–4413 CrossRef CAS.
- A. Delori and W. Jones, A hydrogen bonded cocrystal with an unusual interweaving between the adjacent triple-helices, CrystEngComm, 2011, 13, 6315–6318 RSC.
- Y. Yano, T. Ono, S. Hatanaka, D. T. Grykoc and Y. Hisaeda, Salt–cocrystal continuum for photo function modulation: stimuli-responsive fluorescence color-tuning of pyridine-modified intramolecular charge-transfer dyes and acid complexes, J. Mater. Chem. C, 2019, 7, 8847–8854 RSC.
- D. Kalita, R. Sarma and J. B. Baruah, Formation of symmetry non-equivalent molecules in urea and carbamate derivatives: role of anion, CrystEngComm, 2009, 11, 803–810 RSC.
- N. Barooah, A. Karmakar and J. B. Baruah, Solvent induced symmetry non-equivalence in crystal lattice of 7-carboxymethyl-1,3,6,8-tetraoxo-3,6,7,8-tetrahydro-1H-benzo[lmn][3,8]phenathrolin-2-yl) acetic acid, CrystEngComm, 2008, 10, 151–154 RSC.
- H.-J. Hao, D. Sun, F.-J. Liu, R. B. Huang and L. S. Zheng, Discrete octamer water cluster and 1D T5(2) water tape trapped in two luminescent Zn(II)/1,2-bis(imidazol-1′-yl)ethane/dicarboxylate hosts: from 2D (4,4) net to 3D 5-fold interpenetrated diamond network, Cryst. Growth Des., 2011, 11, 5475–5482 CrossRef CAS.
- N. Kumar, S. Khullar and S. K. Mandal, Encapsulation of a water octamer chain in a chiral 2D sheet like supramolecular coordination network composed of dinickel–dicarboxylate subunits, ACS Omega, 2018, 3, 11062–11070 CrossRef CAS PubMed.
- N. Saraei, O. Hietsoi, C. S. Mullins, A. J. Gupta, B. C. Frye, M. S. Mashuta, R. M. Buchanan and C. A. Grapperhaus, Streams, cascades, and pools: various water cluster motifs in structurally similar Ni (II) complexes, CrystEngComm, 2018, 20, 7071–7081 RSC.
- A. Karmakar, R. J. Sarma and J. B. Baruah, Structural aspects and properties of salt and inclusion compounds of 8-hydroxyquinoline based amides, CrystEngComm, 2007, 9, 379–389 RSC.
- M. C. Etter, J. C. MacDonald and J. Bernstein, Graph-set analysis of hydrogen-bond patterns in organic crystals, Acta Crystallogr., Sect. B: Struct. Sci., 1990, 46, 256–262 CrossRef PubMed.
- A. P. Singh, M. P. Singh and J. B. Baruah, Changes in the proportions of an active pharmaceutical through cocrystals, Drug Dev. Res., 2021, 1–10 Search PubMed.
- B. Lou, S. R. Perumalla and C. C. Sun, Significant expansion of the solid state landscape of salicylic acid based on charge-assisted hydrogen bonding interactions, Cryst. Growth Des., 2015, 15, 24–28 CrossRef CAS.
- A. Tarai and J. B. Baruah, Quaternary and senary sub-assemblies in cocrystals and salts of quinoline-4-carbaldoxime with aromatic carboxylic acids, CrystEngComm, 2016, 18, 9095–9102 RSC.
- V. Chandrasekhar, P. Thilagar and A. Steiner, Guest-assisted self-assembly of organostannoxane nanotubules on a mica surface, Cryst. Growth Des., 2011, 11, 1446–1449 CrossRef CAS.
- D. Singh and J. B. Baruah, Solid state assemblies of cyclic imides tethered hydroxy benzoic acids with pyridine and quinoline: toward the formation of channels and cavities, Cryst. Growth Des., 2012, 12, 3169–3180 CrossRef CAS.
- L. Sun, K. Hu, S. Jin, Y. Lu, C. Xu, B. Liu, D. Wang and G. Xia, Single-crystal and molecular structures of five hydrogen-bonding supramolecular salts based on 4-aminobenzoic acid, 2-aminobenzoic acid and acidic components, J. Mol. Struct., 2019, 1178, 229–241 CrossRef CAS.
- G. Smith, D. E. Lynch, K. A. Byriel and C. H. L. Kennard, Molecular cocrystals of carboxylic acids. XX. the crystal structures of 3,5-dinitrosalicylic acid and its adducts with the isomeric monoaminobenzoic acids, Aust. J. Chem., 1995, 48, 1133–1149 CrossRef CAS.
- A. O. L. E. Vora, C. E. S. Bernardes, M. F. M. Piedade, A. C. L. Conceic and M. E. M. da Piedade, Energetics of glycine cocrystal or salt formation with two regioisomers: fumaric acid and maleic acid, Cryst. Growth Des., 2019, 19, 5054–5064 CrossRef.
- J. B. Baruah, Inorganic molecular-complexes: potential for growth of a new subject-area in self-assembly, Top. Curr. Chem., 2020, 378, 30 CrossRef CAS PubMed.
- C. R. Taylor and G. M. Day, Evaluating the energetic driving force for cocrystal formation, Cryst. Growth Des., 2018, 18, 892–904 CrossRef CAS PubMed.
- S. P. Yelgaonkar, D. Kiani, J. Baltrusaitis and L. R. MacGillivray, Superstructural diversity in salt-cocrystals: higher-order hydrogen-bonded assemblies formed using U-shaped di-cations and with assistance of π–π stacking, Chem. Commun., 2020, 56, 6708–6710 RSC.
- J. Sendh, M. P. Singh and J. B. Baruah, 5-[(Pyren-9-ylmethyl)amino]isophthalic acid with nitrogen containing heterocylces: stacking, N–H⋯π interactions and photoluminescence, CrystEngComm, 2021, 23, 6952–6966 RSC.
- Y. Cui, R. Liu, F. Ye and S. Zhao, Single-excitation, dual-emission biomass quantum dots: preparation and application for ratiometric fluorescence imaging of coenzyme A in living cells, Nanoscale, 2019, 11, 9270–9275 RSC.
- M. L. Liu, B. B. Chen, J. H. He, C. M. Li, Y. F. Li and C. Z. Huang, Anthrax biomarker: An ultrasensitive fluorescent ratiometry of dipicolinic acid by using terbium(III)-modified carbon dots, Talanta, 2019, 191, 443–448 CrossRef CAS PubMed.
- J. Yuan, X. Dong, B. Zhang, Q. Zhou, S. Lu, Q. Wang, Y. Liao, Y. Yang and H. Wang, Tunable dual emission of fluorescence-phosphorescence at room temperature based on pure organic supramolecular gels, Dyes Pig., 2020, 181, 108506 CrossRef CAS.
- A. Tarai and J. B. Baruah, A solution and solid state study on the recognition of hydroxyaromatic aldoximes by nitrogen containing compounds, Cryst. Growth Des., 2016, 16, 126–135 CrossRef CAS.
- H. Dong, J. Zhao, H. Yang and Y. Zheng, The mechanism of ratiometric fluoride sensing and the ESIPT process for 2,6-dibenzothiazolylphenol and its derivative, Org. Chem. Front., 2018, 5, 1241–1247 RSC.
- Y. Chen, Y. Yang, Y. Zhao, S. Liu and Y. Li, The effect of different environments on excited-state intramolecular proton transfer in 4′-methoxy-3-hydroxyflavone, Org. Chem. Front., 2019, 6, 218–225 RSC.
- A. Kaur, P. Kaur and S. Ahuja, Förster resonance energy transfer (FRET) and applications thereof, Anal. Methods, 2020, 12, 5532–5550 RSC.
- S. K. Behera, S. Y. Park and J. Gierschner, Dual emission: classes, mechanisms, and conditions, Angew. Chem., Int. Ed., 2021, 60, 22624–22638 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The spectroscopic details of the salts, PXRD, pH titrations, fluorescence titrations, and the table of hydrogen-bond parameters. CCDC 2099504, 2099505, 2099935 and 2099936. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2ma00075j |
| This journal is © The Royal Society of Chemistry 2022 |