
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction of a drug-containing microenvironment for in situ bone regeneration†
Zhen
Wu
a,
Zhou
Zhong
b,
Wenchao
He
a,
Yanmei
Wu
a,
Yuyan
Cai
a,
Huilin
Yang
c and
Youliang
Hong
 *a
*a
aNational Engineering Research Centre for Biomaterials; Department of Biomedical Engineering, Sichuan University, Chengdu, 610064, P. R. China. E-mail: hyl@scu.edu.cn
bDepartment of Orthopaedic Surgery, West China Hospital, Sichuan University, Chengdu 610041, P. R. China
cDepartment of Orthopaedics, The first Hospital Affiliated to Suzhou University, Suzhou, 215006, P. R. China
First published on 31st March 2022
Abstract
The construction of bone scaffolds to treat bone defects caused by infection is a major challenge because such bone scaffolds need to both prevent infection from recurring and regenerate bone defects. To this end, we constructed bioactive glass (BG)-coated, hierarchical porous tricalcium phosphate (TCP) ceramics as both bone scaffolds and drug delivery devices to treat S. aureus-infected bone defects. In vitro simulation experiments confirmed that the as-prepared porous ceramics could construct a dynamic microenvironment, in which the concentration of the released antibiotics and calcium ions always changed and an apatite layer could grow on the ceramic surfaces. In vitro experiments demonstrated that antibiotic-loaded ceramics possessed good cytocompatibility and the microenvironment established by the antibiotic-loaded ceramics could mediate the osteogenic commitment of mesenchymal stem cells (MSCs), and at the same time, the antibiotic-loaded ceramics presented excellent and relatively-lasting antibacterial activity. In vivo experiments demonstrated that the antibiotic-loaded ceramics not only could prevent the S. aureus-infected bone defects from recurring, but also could boost the endogenous regeneration of bone.
1. Introduction
It is of clinical significance to tailor biomaterials into porous scaffolds that can create a microenvironment (also called a niche) to mediate in situ bone regeneration,1–6 and thus the exploration of the microenvironmental factors that can stimulate/activate an endogenous regenerative procedure of bones becomes an important topic in the bone tissue engineering field. To this end, some microenvironmental factors that could be administrated by biomaterials, e.g., pH,7–9 the concentration of calcium/phosphate ions,9–11 the formation of an apatite layer,12–15 surface topographies,16,17etc., have been demonstrated to be able to mediate the osteogenic commitment of MSCs and in situ bone regeneration. In particular, our recent work has demonstrated that a dynamic microenvironment established by a gypsum-coated TCP scaffold could promote native healing cascades of bone defects.18Still, current experiments mainly focused on exploring the scaffold-constructed microenvironment to regenerate bone defects arising from healthy bone. In actual clinical cases, however, plenty of bone defects arise from specific diseases, e.g., tumours, bone infection, etc. To treat disease-caused bone defects, two medical problems need to be tackled: (i) prevent the tumour/infection from recurring, and (ii) regenerate bone defects. Therefore, it is found that the scaffold-created microenvironment not only can mediate bone regeneration, but also can prevent disease recurrence.
Currently, the strategy of local drug administration,19–22 the construction of local delivery devices using biomaterials (e.g., PMMA, bioactive glass, calcium sulfates, and calcium phosphates),23 and the loading of antibacterial materials (e.g., graphene oxide nanosheets/polydopamine and polydopamine/silver nanoparticles) onto the scaffolds24–26 are well developed to treat the disease-caused bone defects. Still, no attention has been paid to the microenvironment formed by the drug-loaded devices. In particular, it is unclear and thus needs to be investigated whether the introduction of drugs into the microenvironment created by scaffolds will affect the endogenous bone regeneration.
Here, our aim is to study the endogenous bone regeneration mediated by a drug-containing microenvironment. To this end, we design triple hierarchical porous bioceramics as bone scaffolds and drug-loaded devices. In detail, we select bioactive materials, TCP and BG, as tailoring objectives, and combine multiple manufacturing techniques, including 3D printing, gelcasting, and nanoporous sol–gel coating techniques, to construct triple hierarchical porous bioceramics. The manufacturing process for the desired products is illustrated in Scheme 1. Bioceramics were designed into hierarchical porous structures because studies of Hollister27 and our group18 have demonstrated that the bone scaffolds with right porous structures play important roles in constructing a microenvironment of bone growth, especially, abundant nanopores (e.g., <50 nm) distributed within scaffolds can both improve the bioactivity of scaffolds and serve as delivery vehicles for drugs.10–14 TCP is selected because it is similar to the mineral components of hard tissues and has now become one of the optimal bone filling materials in clinical practice. Also, BG has been widely applied as a bone repair material.28 More importantly, BG can release metal ions (e.g., Ca2+) rapidly and possibly establishes a weak alkaline microenvironment to affect bone regeneration.29,30 In addition, BG can be processed into a thin membrane with abundant nanopores, which can load drugs.31–33 Finally, gentamicin sulphate (GS), a kind of broad-spectrum antibiotic, is used in this work to construct a drug-containing microenvironment.
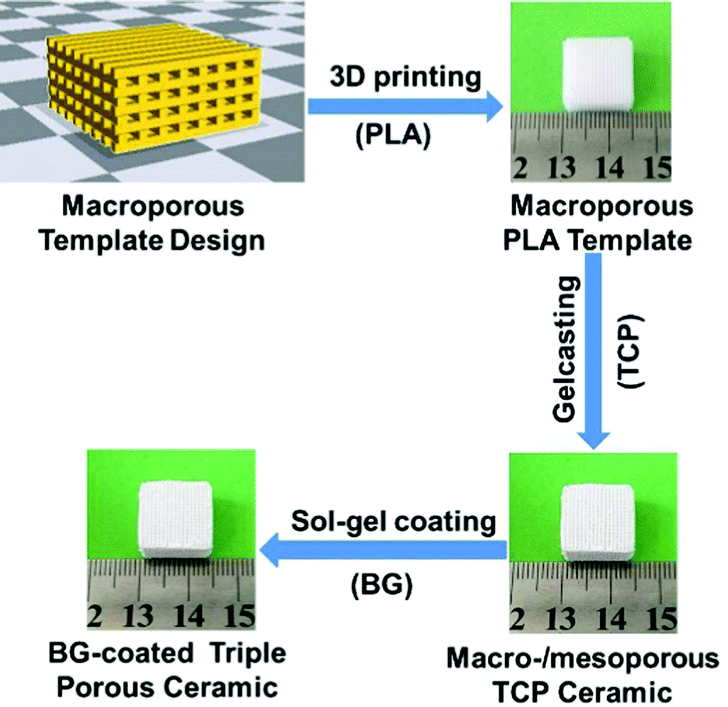 | ||
| Scheme 1 The illustrative procedure of the preparation of the BG-coated triple hierarchical porous bioceramics. | ||
2. Experimental
2.1. Materials
Absolute alcohol, dichloromethane (DCM), polymethacrylate (PMA), acrylamide (AM), hydrochloric acid (HCl), N,N,N′,N′-tetramethylethylenediamine (TMED), ammonium persulfate (APS), N,N′-methylene bisacrylamide (MBAM), tetraethyl orthosilicate (TEOS), Ca(NO3)2·4H2O, and non-ionic surfactant P123 (poly(ethylene oxide)20–poly(propylene oxide)70–poly(ethyl oxide)20 all are of analytical reagent (AR) grade and were purchased from Chengdu Kelong Reagent Co., China. Hydroxypropyl methylcellulose (HPMC) was purchased from Shangdong Weifang Lite Composite Materials Co., China. TCP powders were prepared using a co-precipitation method and were sintered at 700 °C for 2 h.34 GS is purchased from Sichuan Changzheng Pharmaceutical Co. Water used in all experiments has been purified using a reverse osmosis (RO) water system.2.2. Preparation of hierarchical porous bioceramics
At first, porous TCP bioceramics were prepared in light of our previous methods.18 In brief, a fused deposition modelling (FDM) 3D printer (JGAURORA, Shenzhen Aurora Technology Co.) was used to print porous PLA scaffolds according to the designed STL format files as the ceramic templates. According to different experimental requirements, scaffold prototypes (pore: 0.4 mm and rod: 0.4 mm) with various shapes (e.g., cylinders and cubes) were designed using the 3D design software Solidworks. Then, the TCP slurry was prepared according to the protocol (Table S1, ESI†) and was cast into porous PLA templates. After the TCP slurry solidified, porous PLA templates were removed and the remaining porous TCP green bodies were sintered at 1100 °C in air for 4 h to obtain bimodal porous TCP ceramics. Finally, the surface coating of BG to TCP ceramics was performed. In detail, BG precursors were prepared in light of the protocol (Table S2, ESI†) and then the as-prepared TCP ceramics were soaked under vacuum into BG precursors. 10 min later, the TCP ceramics were removed from the BG precursors and dried in air and then were sintered at 700 °C for 10 h to remove the organic components.2.3. Drug loading and release
The BG-coated TCP (BG-TCP) ceramics were soaked into GS aqueous solution (the GS concentration was 40 mg ml−1) in a vacuum. After 10 min, the ceramics were removed and dried in air. The GS loading amount in ceramics was analysed by measuring the GS concentration before and after porous ceramic soaking through fluorescence spectroscopy using an o-phthaldialdehyde (OPA) reagent. It reacts with the amino groups of GS to produce fluorescence that was determined (excitation wavelength = 340 nm and emission wavelength = 441 nm) by fluorescence spectroscopy and then by using the following formula, LG% = [(Cb/Vb − Ca/Va)/Wc] ×100, where Cb and Ca are the GS concentration before and after porous ceramic soaking, Vb and Va are the volume of the GS solution before and after porous ceramic soaking, and Wc is the weight of porous ceramics.The GS release dynamics were analysed by soaking six parallel GS-loaded ceramics (160 mg) into RO water (20 ml)/simulated body fluid (SBF) (20 ml). The soaking liquid was incubated at 37 °C in a thermostat shaker. At appropriate intervals part of the incubated liquid (10 ml) was removed for monitoring by fluorescence spectroscopy as described above, and at the same time, the soaking liquid was replenished with 10 ml fresh liquid. The quantity of GS released was calculated to determine the drug release kinetics of GS.
2.4. Ion release analysis and pH measurements
The as-prepared ceramics (20 mg) were soaked in 37 °C SBF (20 ml). At appropriate intervals part of the incubated SBF (10 ml) was removed for pH and ion concentration analysis and the same volume of fresh SBF was replenished. The calcium/phosphate ion concentration in the soaked SBF was measured by using a calcium colorimetric assay kit (Beyotime S10635) and a tissue inorganic phosphorus content assay kit (Solarbio BC2845). The silica concentration was measured using inductively coupled plasma atomic emission spectrometry (ICP-AES) (PerkinElmer Optima, 7000DV), and before analysis, the organic components in the soaked liquid were removed by the heating and redissolving method (i.e., the soaked SBF was heated at 80 °C and sintered at 800 °C for 4 h, and the formed sediment was dissolved in 5 ml of 1 M HCl).2.5. The growth of the apatite layer on the ceramic surfaces
Porous TCP, BG-TCP, and GS-loaded BG-TCP (GS/BG-TCP) ceramics were soaked in a simulated body fluid (SBF) over time to grow a bone-like nanoapatite layer. The process of nanoapatite layer growth was observed using a Scanning Electron Microscope (SEM) (Hitachi S-4800).2.6. Physicochemical characterization
2.7 In vitro biological functions of porous ceramics
2.8. Antibacterial activity assessment of porous ceramics
To assess the antibacterial activity, Gram-positive bacteria Staphylococcus aureus (S. aureus, ATCC 29213) and Gram-negative bacteria Escherichia coli (E. coli, ATCC 25922) were used, and the experiments were conducted in an ultraclean table. To determine the antibacterial activity of the ceramics, the methods of both bacterial suspension and disk diffusion (i.e., inhibition zone) were used, and at the same time, a method of sample transfer was created here, i.e., a sample was incubated in turn for 24 h in the fresh bacterial suspensions/disks.In the bacterial suspension culture, different porous ceramics with three parallel samples were placed into 24-well culture plates, and an LB liquid medium was used as a control. The bacterial liquid (106 CFU mL−1), which had been grown in a tryptic soy broth (TSB, BD) overnight in an incubator at 37 °C, was dropped into the wells (200 μL per well). After 24 h of incubation, the porous ceramics were transferred repeatedly into fresh bacterial suspensions to incubate for another 24 h, and the OD value of the cultured bacterial liquid was detected at 600 nm using a microplate reader. The antibacterial rate of the samples was calculated using the formula A = (ODC − ODS)/(ODC − ODB) × 100%, where A denotes the antibacterial rate, ODS is the absorption value of the bacterial liquid with samples, ODB is the absorption value of the LB medium, and ODC is the absorption value of the antibacterial liquid without samples.
In the disk diffusion method, the bacterial suspension (100 μL, 106 CFU mL−1) was evenly coated on a solid medium, and then ceramics were placed on the solid medium. After being incubated for 24 h at 37 °C, the culture dish with ceramics was observed, and the porous ceramics were transferred repeatedly into fresh bacterium-containing solid medium to incubate for another 24 h.
2.9. In vivo biological functions of hierarchical porous ceramics
9 adult male New Zealand rabbits were anesthetized by intravenous injection of 1.5 wt% pentobarbital sodium (2 mg kg−1) and the operations were performed under sterile conditions. Then the femoral bone defects (5 mm) were established using an electric drill. The rabbits were evenly divided into 3 groups. Group 1: two porous TCP ceramics were implanted into the marrow cavity and bone defects respectively; Group 2: two BG-TCP porous ceramics were implanted into the marrow cavity and bone defects respectively (Fig. S1, ESI†); and Group 3: the GS/BG-TCP porous ceramics were implanted into bone defects. The sizes of all porous ceramics were the same (∅: 5 mm, high: 5 mm). After the ceramics were implanted, the muscle and skin were sealed. GS was injected daily after operation (800![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 U kg−1) for 3 consecutive days. The rabbits with implanted ceramics were sacrificed at 6 weeks. The harvested specimens were fixed with 10% formalin for further histological analysis.
000 U kg−1) for 3 consecutive days. The rabbits with implanted ceramics were sacrificed at 6 weeks. The harvested specimens were fixed with 10% formalin for further histological analysis.
2.10. Statistical analysis
All data are reported in this work as mean ± standard deviations (s.d.). All data were analyzed using an unpaired Student t-test. Confidence levels of >95% (*p < 0.05) were considered to be statistically significant.3. Results and discussion
3.1. Preparation of hierarchical porous BG-TCP bioceramics
To prepare hierarchical porous BG-TCP bioceramics, two processing steps were carried out: porous β-TCP bioceramics were prepared at first, and then the as-prepared TCP bioceramics were coated by BG. In our previous work, we have demonstrated that porous TCP bioceramics with regular macropores could be well prepared by combining the 3D printing and gelcasting techniques.21 Herein, such a strategy was continuously used to prepare porous TCP bioceramics. Fig. 1a(I) shows a prepared cylindrical porous β-TCP ceramic. SEM images and MIP measurements (Fig. 1a(II–V) and Table 1) show that the as-prepared TCP ceramic was composed of regular macropores (266 μm) and abundant micropores (average diameter: 0.7 μm), and the macroporous and microporous volumes of ceramics were 1.5 and 1.6 cm3 g−1, respectively. The compressive strength of the TCP bioceramics measured was 2.78 MPa (Table 1).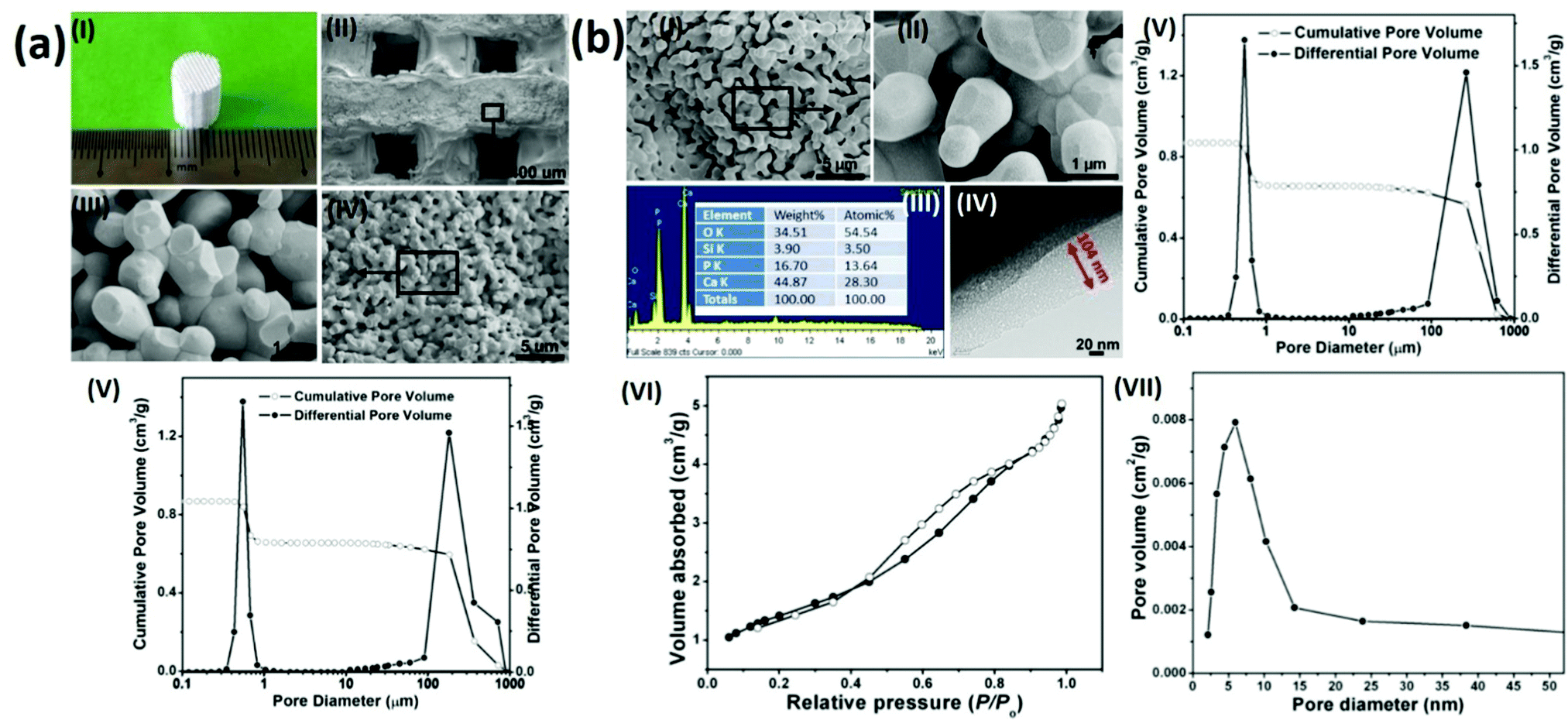 | ||
| Fig. 1 Preparation of hierarchical porous BG-TCP bioceramics. (a) Preparation of porous TCP bioceramics. (I) Photography, (II–IV) low-, middle- and highly-magnified SEM images, and (V) pore size distribution measured from MIP. (b) The surface coating of BG on porous BG-TCP bioceramics. (I and II) Middle- and highly-magnified SEM images, (III) EDX, (IV) TEM image, (V) MIP, (VI) N2 adsorption–desorption, and (VII) BJH pore size distribution. | ||
| TCP ceramics | BG-TCP ceramics | |
|---|---|---|
| Macropore diameter (μm)/volume (cm3 g−1) | 266/1.5 | 265/1.5 |
| Micropore diameter (μm)/volume (cm3 g−1) | 0.55/1.6 | 0.45/1.2 |
| Nanopore diameter (nm) | — | 6.5 |
| Compressive strength (MPa) | 2.78 | 3.26 |
Subsequently, a sol–gel BG coating onto porous TCP bioceramics was performed to achieve BG-coated TCP ceramics, as shown in Fig. 1(b). SEM images (Fig. 1b(I and II)) show that a thin amorphous layer covered the TCP grain surfaces, and no cracks were displayed in the coating layer. The EDX result (Fig. 1b(III)) shows that besides elements Ca, P, and O, Si was also expressed, demonstrating that the coated amorphous layer was BG. In addition, no element C was present in the EDX curve, demonstrating that no residual organic component remained in the BG matrix. The TEM image (Fig. 1b(IV)) shows that the thickness of the coated BG was ∼104 nm with abundant nanopores (∼6 nm), and the formed nanopores were worm-like and were distributed disorderly in the BG matrix. The MIP result (Fig. 1b(V) and Table 1) shows that the diameter (265 μm) and pore volume (1.5 cm3 g−1) of the macropores of the ceramic did not change in essence, but both the diameter (0.45 μm) and pore volume (1.2 cm3 g−1) of the micropores of the ceramic decreased significantly in comparison with those of the TCP bioceramics without the BG coating (macroporous diameter/volume: 266 μm/1.5 cm3 g−1; microporous diameter/volume: 0.55 μm/1.6 cm3 g−1, see cm3 g−1 that the porosity of the BG-TCP ceramics also dropped to 69.5%). Still, Table 1 shows that the compressive strength of the BG-TCP bioceramics (3.26 MPa) increased significantly.
The N2 adsorption–desorption isotherm (Fig. 1(VI)) indicates that the isotherm of the as-prepared samples was type IV, and its corresponding Barrett–Joyner–Halenda (BJH) pore size distribution (Fig. 1b(VII)) shows that the average diameter of the nanopores was 6.5 nm, almost in agreement with the TEM observation. The above results well demonstrated that a layer of BG with abundant nanopores (6.5 nm) was coated on the TCP grain surfaces, and the used sol concentration here was suitable for the BG coating.
3.2. In vitro microenvironments established by the drug-loaded bioceramics
To understand the osteogenic activity of the GS-loaded bioceramics, especially, to understand the ability of the GS-loaded bioceramics in mediating endogenous bone regeneration, it is essential to evaluate the microenvironment established by the GS-loaded bioceramics. To this end, the experiments of the GS loading and the ceramic soaking (in 37 °C SBF) were carried out at first. The experiments demonstrated that 7.25 wt% GS (the GS/ceramic weight ratio) could be loaded into porous BG-TCP bioceramics. Then, the release dynamics of GS, calcium ions, phosphate ions and silica, pH of the soaked SBF, and the growth of the apatite layer on the ceramic surfaces were investigated in detail.Fig. 2(a) shows the in vitro release profile of GS from the BG-TCP ceramics in SBF. At the initial stage (within 40 h), the average GS release curve shows that the burst release of GS occurred at first, and then (40 h later) the GS release rate decreased. After ∼132 h, the GS release almost ceased from the ceramics. At the releasing peak value (∼ at 40 h), the GS amount in the SBF became 50%. The cumulative release of GS shows that the amount of GS released was ca 83.5% when the release of GS from the ceramics ceased (at 160 h), suggesting that 16.5% GS remained in the ceramics. For comparison, Fig. 2(a) shows that the release of GS was faster in water than in SBF. In particular, at the initial stage, the burst release of GS in water was sharper than that in SBF, and after 12 h, the amount of GS released became ∼60%. 100 h later, all GS almost released from the ceramics. The above results suggest that the mineralization reaction of ceramics occurred in SBF, thus impeding the release of GS.
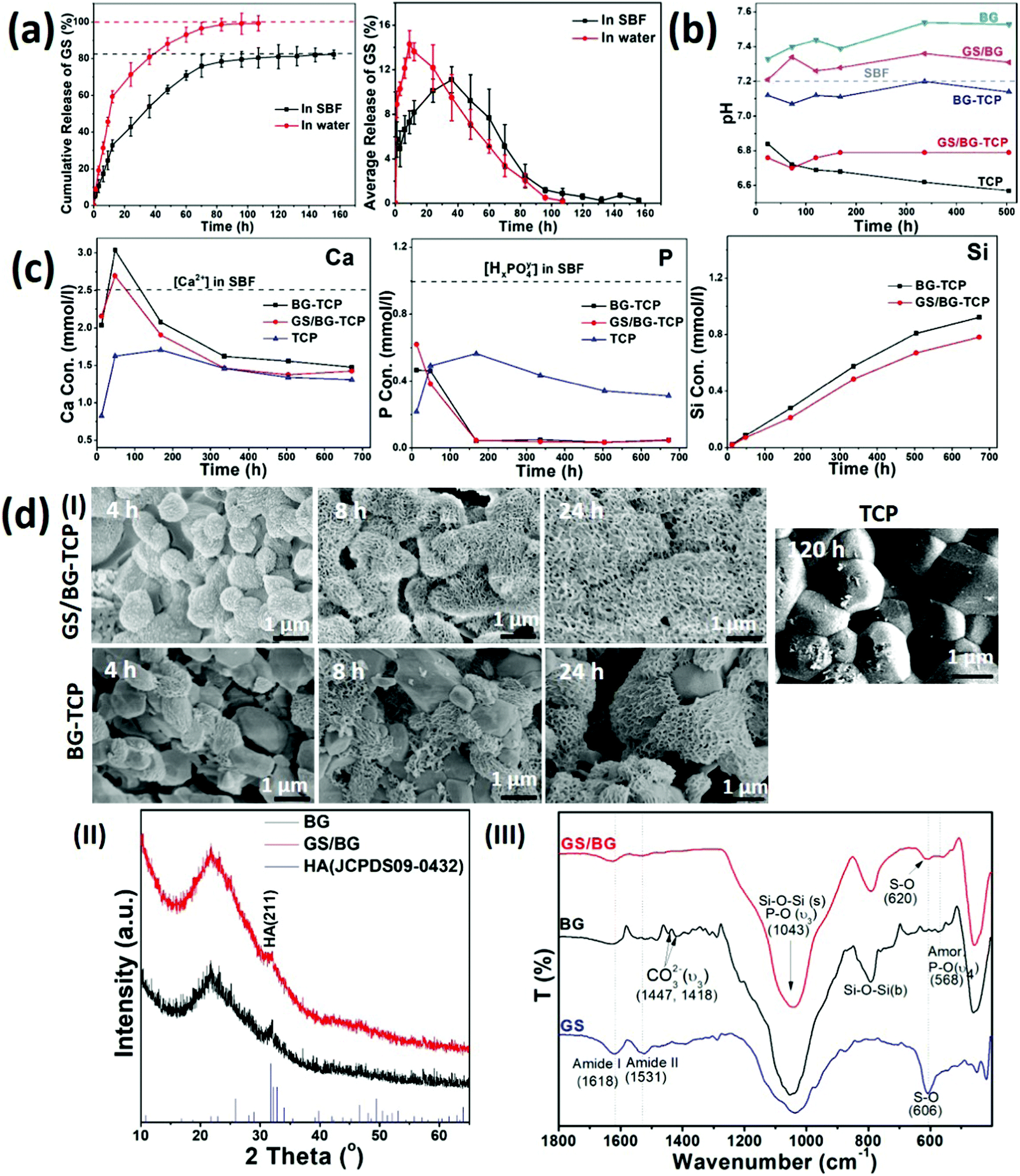 | ||
| Fig. 2 In vitro analysis of the microenvironments constructed by the GS-loaded BG-TCP ceramics in SBF. (a) Cumulative and average release of GS from the BG-TCP ceramics in SBF. Also, the release of GS in water was analysed as a control. (b) The pH value of SBF soaking different samples. (c) The Ca/P/Si release profile from the BG-TCP ceramics with/without GS loading. (d) Surface biomineralization: (I) SEM images of the BG-TCP ceramics with and without GS loading soaked in SBF for 4, 8, and 24 h. For comparison, pure TCP ceramics were soaked in SBF for 120 h. (II) XRD pattern of the BG-TCP ceramics with and without GS loading soaked in SBF for 72 h. (III) FTIR of GS, the SBF soaked BG (soaked in SBF for 72 h), and the SBF soaked GS/BG (72 h). | ||
Then, the pH of the soaked SBF was tested. Fig. 2(b) shows that the pH value of the SBF containing the GS/BG-TCP was almost that of weak acid (pH: 6.8). To understand the formation of a weak acid environment, the BG, GS/BG, TCP and BG-TCP ceramics were soaked in SBF respectively. Fig. 2(b) shows that the BG and GS/BG ceramic soaking caused the pH of SBF to increase, but the TCP and BG-TCP ceramic soaking led to a pH decrease of SBF. The above results showed that BG is a kind of alkaline ceramic but TCP is an acidic one,18,26,27 and in the BG-TCP ceramics, although a layer of BG was coated on the TCP surfaces, TCP predominated in controlling the pH of the ceramics. In the GS-loaded ceramics, because GS was an acid antibiotic (pH: 6), it deceased the pH of the ceramics. Then, the pH of the soaked SBF was tested. Fig. 2(b) shows that the pH value of the SBF containing the GS/BG-TCP was almost that of weak acid (pH: 6.8). To understand the formation of a weak acid environment, the BG, GS/BG, TCP and BG-TCP ceramics were soaked in SBF respectively. Fig. 2(b) shows that the BG and GS/BG ceramic soaking caused the pH of SBF to increase, but the TCP and BG-TCP ceramic soaking led to the pH decrease of SBF. The above results showed that BG is a kind of alkaline ceramic but TCP is an acidic one,18,26,27 and in the BG-TCP ceramics, although a layer of BG was coated on the TCP surfaces, TCP predominated in controlling the pH of the ceramics. In the GS-loaded ceramics, because GS is an acid antibiotic (pH: 6), it deceased the pH of the ceramics.
Thirdly, the release dynamics of calcium/phosphate ions and silica were analysed. Fig. 2(c) shows that the concentration of Ca2+ increased at first, and then decreased, and at the initial stage, the Ca2+ concentration was higher than 2.5 mM (the Ca2+ concentration in SBF was 2.5 mM). The Ca2+ concentration higher than that in SBF at the initial stage was significant because Olszak et al. have reported that high [Ca2+]o can recruit immunocytes and MSCs.35 In contrast, the concentration of phosphate ions was always far lower than 1.0 mM. In comparison with the calcium/phosphate ions, the concentration of silica increased continuously during the entire release process. The decrease of [Ca2+] and [HxPO4y−] in SBF could be attributed to the formation of a nano-apatite layer as will be demonstrated afterwards. For comparison, the BG-TCP ceramics without drug loading had a slightly higher [Ca2+] and silica release. The above results suggested that the existence of GS would affect the release of inorganic components.
Finally, the surface mineralization of the GS/BG-TCP ceramics was analysed. Fig. 2d(I) shows that after 4 h SBF immersion, some small nanocrystals homogeneously grew on all grain surfaces. After 8 h soaking, a large amount of nanoplates instead of small nanocrystals covered on all grain surfaces, and 24 h later, more nanoplates formed and the grain profiles could not be observed. In the BG-TCP ceramics without GS loading, however, some nanoplates randomly grew on a few BG-TCP grain surfaces after 4 h SBF immersion, and with the increase of soaking time, more and more apatite nanoplates formed on the surfaces of more BG-TCP grains. Still, the amount of grown nanocrystals was obviously lower than that in the GS-loaded ceramics. For comparison, the pure TCP ceramics were also measured. Fig. 2d(I) shows that only a small number of needle-like nanocrystals formed and deposited on the TCP grain surfaces after 120 h SBF soaking. Such results further confirmed the bioactivity of BG, i.e., the formation of the apatite nanoplates on the ceramic surface was induced by BG. The above results suggested that GS could obviously promote the growth of apatite nanoplates.
To understand the growth of the apatite nanoplates, BG and GS loaded BG (GS/BG) ceramics were prepared and were soaked in SBF for 72 h. The XRD patterns ((Fig. 2d(II)) show that the grown apatite nanoplates in both samples had a low crystal degree. Still, both apatite plates were the hydroxyapatite (HA) phase (JCPDS 09-0432). The FTIR results (Fig. 2d(III)) show that in the GS/BG ceramics, the peaks of amide I/II groups (at 1629/1531 cm−1) and P–O groups (the υ4 modes of amorphous phosphate adsorption at 568 cm−1 and the υ3 modes of P–O at 1043 cm−1)36 were expressed. Furthermore, the peak at 606 cm−1, assigned to the S–O groups of GS, shifted to 620 cm−1, suggesting that sulfate groups bonded with Ca. The above results demonstrated that the formed nanoplates were the low crystal GS-containing HA, and the existence of GS would take part in the mineralization reaction, thus suggesting that GS could promote the growth of nanoplates and affect the release of GS (Fig. 2a). In the BG ceramics, besides the peaks of the P–O groups, the peaks of the carbonate groups (at 1447 and 1418 cm−1) were present, indicating that the grown nanoplates were low crystal carbonated HA. Such a result suggested that the existence of GS would inhibit the carbonate groups to take part in the mineralization reaction.
Taken together, the above results showed that the GS-loaded BG-TCP ceramics established a dynamic microenvironment, in which the pH and the concentrations of calcium ions, phosphate ions, GS, and silica in SBF always changed. These changes mainly resulted from the release of calcium ions, phosphate ions, GS, and silica and the growth of apatite nanoplates on the ceramic surfaces. At the same time, some chemical reactions possibly occurred during the release of ions and the growth of nanoplates.
| CaO (in BG) + H2O → [Ca2+] + 2[OH−] | (1) |
| 8.8–10[Ca2+] + 6[HxPO4y−] → Ca8.8–10(PO4)6(OH)2 + 6x[H+] | (2) |
| [Ca2+] + G-H2SO4 (GS) → CaSO4-G + 2[H+] | (3) |
| (2) + (3) → Ca8.8–10(PO4)6(OH)2·CaSO4-G | (4) |
3.3. In vitro biological properties of the drug-loaded bioceramics
Although the biological properties of TCP and BG have been fully studied,5,6,37 it has not been well understood how the microenvironment established by the drug-loaded bioceramics mediates the biological behaviours of MSCs. Herein, BM-MSCs were employed to assess the in vitro biological properties of the GS-loaded BG-TCP ceramics. At the same time, the TCP/BG-TCP ceramics were investigated as a control. BM-MSCs were used because they were osteogenic cell lines and the in vitro biological behaviours of BM-MSCs mediated by bioceramics can reflect in vivo bone regeneration events. To better understand the biological functions of ceramics in vivo, a surface mineralization experiment was performed by soaking bioceramics into SBF for 36 h before cell culture (when implanted into the body, bioceramics surely contact body liquid at first and likely react with the body liquid to grow the apatite nanoplate layers on the ceramic surfaces, and thus the cells in vivo in fact interact with the apatite nanoplate layers). SEM images show that the apatite nanoplate layer formed on the BG-TCP and GS/BG-TCP ceramic surfaces, but not on the TCP ceramic surface (Fig. S4, ESI†).At first, the survival and proliferation of MSCs on different ceramic surfaces were tested. The dead–live staining (Fig. 3a(I and II)) shows that only a few dead cells (red cells) were shown on day 1 on all ceramic surfaces and then there were hardly dead cells on all ceramic surfaces during the subsequent cell culture. However, the number of cells on the TCP ceramic surface appears to be significantly higher than that on the BG-TCP and GS/BG-TCP ceramic surfaces. A quantitative CCK8 analysis further confirmed such a result, as can be seen from Fig. 3a(III). The cells cultured on the TCP ceramics indicated a significantly higher O.D. value than those on the BG-TCP/GS/BG-TCP ceramics. Still, the proliferation tendency of cells on all ceramic surfaces was similar. The above results showed that the introduction of GS into ceramics did not cause cytotoxicity. The low survival of cells on the mineralized surfaces at the initial stage presumably resulted from a relatively high Ca2+ concentration released from the BG-containing ceramics (see Fig. 2c), which weakened the activity of integrins (a kind of adhesive protein),38 and thus affected the adhesion of some cells on the mineralized surfaces during cell seeding.
 | ||
| Fig. 3 In vitro biological behaviours of the BM-MSCs mediated by the porous ceramics. (a) Survival and proliferation of MSCs: (I and II) a dead–live staining (n = 3), (III) a quantitative CCK8 analysis (n = 3). (b) Cell adhesion and spreading: (I) SEM and phalloidin staining of BM-MSCs, and (II) the spreading area of cells on different ceramic surfaces (n = 7). (c) Osteogenic commitment of BM-MSCs. (I) ALP (day 4), OCN (day 14) and mineral nodule expression (day 14), and (II) RT-PCR analysis of the bone-relative gene expression (n = 3). | ||
Secondly, the adhesion behaviours of the MSCs cultured on the ceramic surfaces were analysed. SEM images (Fig. 3b(I)) show that the cells could well adhere and spread on the surfaces of all samples. Still, the spreading degree of the cells on different surfaces was different from each other (see Fig. 3b(I and II)). On the TCP surface, the cells expressed the strongest adhesion tension, and abundant filopodia and stress fibres were expressed. In contrast, the cells on the apatite nanoplate layer surfaces (BG-TCP and GS/BG-TCP) assembled few filopodia and stress fibres, and thus resulted in a small spreading area in comparison with those on the TCP surface. Furthermore, Fig. 3b(II) shows that the existence of GS further affected the spread of cells.
Good adhesion of the cells on the TCP surface arose from the Ca-/P-sites on the TCP surface, which could interact non-specifically with the cell membrane proteins.39,40 The high adhesion tension further triggered the cells to assemble the filopodia and stress fibers.41 The decrease of the cytoskeleton tension in the cells mediated by the BG-TCP and GS/BG-TCP surfaces presumably could be attributed to the aligned nanoplates, which reduced the contact area of the cells (see Fig. 3b). In addition, the release of Ca2+ from the BG-containing ceramics (see Fig. 2c) would also possibly affect the activity of the integrins distributed on the cell membranes (it has well been demonstrated that integrins were Ca2+-sensitive and high [Ca2+]o could inhibit the activity of integrins),38,42 and thus weaken the assembly of the cytoskeleton proteins.38,42,43 As regards a further decrease of the cytoskeleton tension in the GS-mediated cells, a possible explanation is that the released GS would interact with membrane proteins (GS has abundant amide groups), and therefore interfering with the adhesion of the cells on the mineralized surfaces.44
Finally, the osteogenic commitment of MSCs mediated by the ceramics was assessed, and the corresponding results are shown in Fig. 3c(I and II). ALP (an early bone-relative protein),45 OCN (the mineral-relative proteins and can only be expressed by osteoblasts),46 and mineral nodes all were secreted by the cells cultured on the mineralized BG-TCP and GS/BG-TCP surfaces, but not by those adhered on the TCP surface. Also, the quantitative (q-RT) PCR results show that in the cells cultured with the surface-mineralized ceramics, some important osteoblast-related genes, including RUNX-2, OPN and OCN, were activated in the correct order to permit differentiation, but the expression of these genes was weak in the cells cultured with the TCP ceramics. Furthermore, Fig. 3c(I and II) shows that the existence of GS did not affect the osteogenic commitment of MSCs.
Now, different signalling cues have been demonstrated to be able to mediate the osteogenic commitment of MSCs, including physical (e.g., surface stiffness47 and special topographies48), chemical (e.g., [Ca2+]o49,50 and [HxPO4y−]o51) and biological (a variety of growth factors, e.g., BMP-2/752,53) factors. In our samples, the cues that could mediate the osteogenic commitment of MSCs mainly came from the physical (the surface stiffness and the grain-/nanoplate-constructed topography) and chemical (the Ca2+ release) factors. The physical cues can mediate the osteogenic differentiation of MSCs because the physical cues can mediate MSCs to generate a strong traction force to trigger the contraction-driving Rho A kinase (ROCK), which is central to the derivation of the high tensional state required for the osteogenic differentiation through activation of Rho A-initiating actin/myosin cytoskeletal contraction.54 In our example, no osteogenic differentiation of MSCs that adhered on the TCP surfaces suggested that the traction force mediated by the TCP surface was not strong enough to activate ROCK. Also, ROCK in the cells adhered on the mineralized surfaces could not be activated because the mineralized surfaces mediated MSCs to form a smaller traction force. Therefore, the osteogenic differentiation of MSCs on the mineralized surfaces surely arose from the release of Ca2+ (see Fig. 2c). Maeno and our work well demonstrated that high concentration [Ca2+]o could regulate the osteogenic differentiation of MSCs.18,55 In addition, our results showed that the weak acidic microenvironment did not mediate the osteogenic commitment of MSCs (the TCP ceramics could cause a weak acidic environment, see Fig. 2b).
3.4. Antibacterial activity of the drug-loaded ceramics
To understand the antibacterial activity of the GS-loaded porous ceramics, Gram-positive bacteria S. aureus and Gram-negative bacteria E. coli were used here because most bone infections are caused by these bacteria.56 At the same time, the TCP and BG-TCP porous ceramics were also assessed as a control. To determine the antibacterial activity of the ceramics, a sample transferring method was created in this work, i.e., a porous ceramic was incubated for 24 h in turn in fresh bacterial suspensions/disks.At first, a quantitative analysis of the antibacterial activity was performed using the bacterial suspension assay.57,58Fig. 4(a) shows that the GS-loaded ceramics had very strong antibacterial activity. After the ceramics had been incubated in 4 fresh bacterial suspensions, their antibacterial rate was still kept at 100% against two kinds of bacteria. Only after the fifth incubation, the antibacterial ability of the GS-loaded ceramics began to decrease. Such a result suggested that GS could be released under control and was well in accordance with the release profiles of GS (Fig. 2a). In contrast, the TCP and BG-TCP ceramics hardly presented the antibacterial activity in the S. aureus/E. coli suspensions.
 | ||
| Fig. 4 In vitro antibacterial activity of the as-prepared porous ceramics. (a) Quantitative bacterial suspension assay (the individual ceramics were incubated in turn for 24 h in the 6 fresh bacterial suspensions. I–VI indicate the number of times the ceramics were incubated in the fresh bacterial suspension) (n = 3); (b) disk diffusion method to assess three kinds of ceramics (incubated for 24 h) (top disks: in order to observe the inhibition zone length, the incubated ceramics remained in the bacterial disks. Bottom disks: in order to observe the inhibition in ceramics, the incubated ceramics were removed); and (c) the disk diffusion method to assess the GS-loaded BG-TCP ceramics (the individual ceramics were incubated in turn for 24 h in the 6 fresh bacterial disks. I–VI indicate the number of times the ceramics were incubated in the fresh bacterial disk) (top disks: to observe the inhibition zone length, the incubated ceramics remained in the bacterial disks. Bottom disks: to observe the inhibition in ceramics, the incubated ceramics were removed). | ||
A disk diffusion method was further performed to assess the antibacterial activity of the as-prepared ceramics (i.e., the inhibitory effect was tested on the basis of the clear zone surrounding the circular-shaped sample).57,58Fig. 4(b) shows that after 24 h incubation in the S. aureus/E. coli colony, a huge inhibition zone surrounding the GS-loaded ceramic (3.6 mm in the S. aureus colony, 3.7 mm in the E. coli colony) was formed. In contrast, in the TCP/BG-TCP ceramics, no inhibition zone was formed in the S. aureus/E. coli colony. Furthermore, the GS-loaded ceramic was transferred continuously to the disks with a fresh S. aureus/E. coli colony, and Fig. 4(c) shows that the inhibition zone reduced gradually, and during the sixth transfer of the GS-loaded ceramic, no inhibition zone was present around the ceramic. The disk diffusion method further confirmed that the release of the GS loaded in ceramics could be controlled.
3.5. In situ bone regeneration mediated by the drug-loaded ceramics
To assess the endogenous regenerative procedure of the bones mediated by the GS-loaded ceramics, circular bone defects (∅: 5 mm) were created in the rabbit femurs (see Fig. S1, ESI†), and at the same time, the femoral marrow cavities of the rabbits were used as the ceramic implanting sites. Correspondingly, cylindrical ceramics were prepared (∅: 5 mm, high: 5 mm). The femoral marrow cavity was selected to assess the osteogenic activity of implants because the femoral marrow cavity has abundant MSCs, which are the same in the cell lineage as bone cells and are important osteogenic cells for bone growth/remodelling, and at the same time, the femoral marrow cavity shares the microenvironment with bone tissue.18Fig. 5(a) shows the histological sections of three kinds of ceramics that have implanted into the femoral marrow cavity for 6 weeks. The magnified images show that in the porosity of all ceramics a little new bone tissue formed, and some newly grown bone embedded into the micropores distributed on the ceramic substrates. The above results demonstrated that the BM-MSCs could migrate into ceramics and differentiate into bone tissue (it should be noted that in the TCP ceramics, new bone tissue could form in vivo but not in vitro (see Fig. 3c). Such a distinct result suggested that the in vitro MSC culture time was not long enough, or unknown cues existed in vivo, which could mediate the osteogenic differentiation of MSCs, e.g., myeloid cells or OsteoMac could induce the differentiation of BM-MSCs).59 Still, the quantified bone mass (Fig. 5a) shows that the newly formed bone mass in the GS-loaded ceramics was significantly higher than that in the TCP ceramics, but was significantly lower than that in the GS-TCP ceramics. A possible explanation is that the difference of the released [Ca2+] among three kinds of ceramics (see Fig. 2c) resulted in the ceramics with different ability to recruit MSCs into the ceramic pores.35,60 Furthermore, our results, i.e., the grown bone embedded into the substrate micropores, also suggested that the existence of micropores could promote bone regeneration.
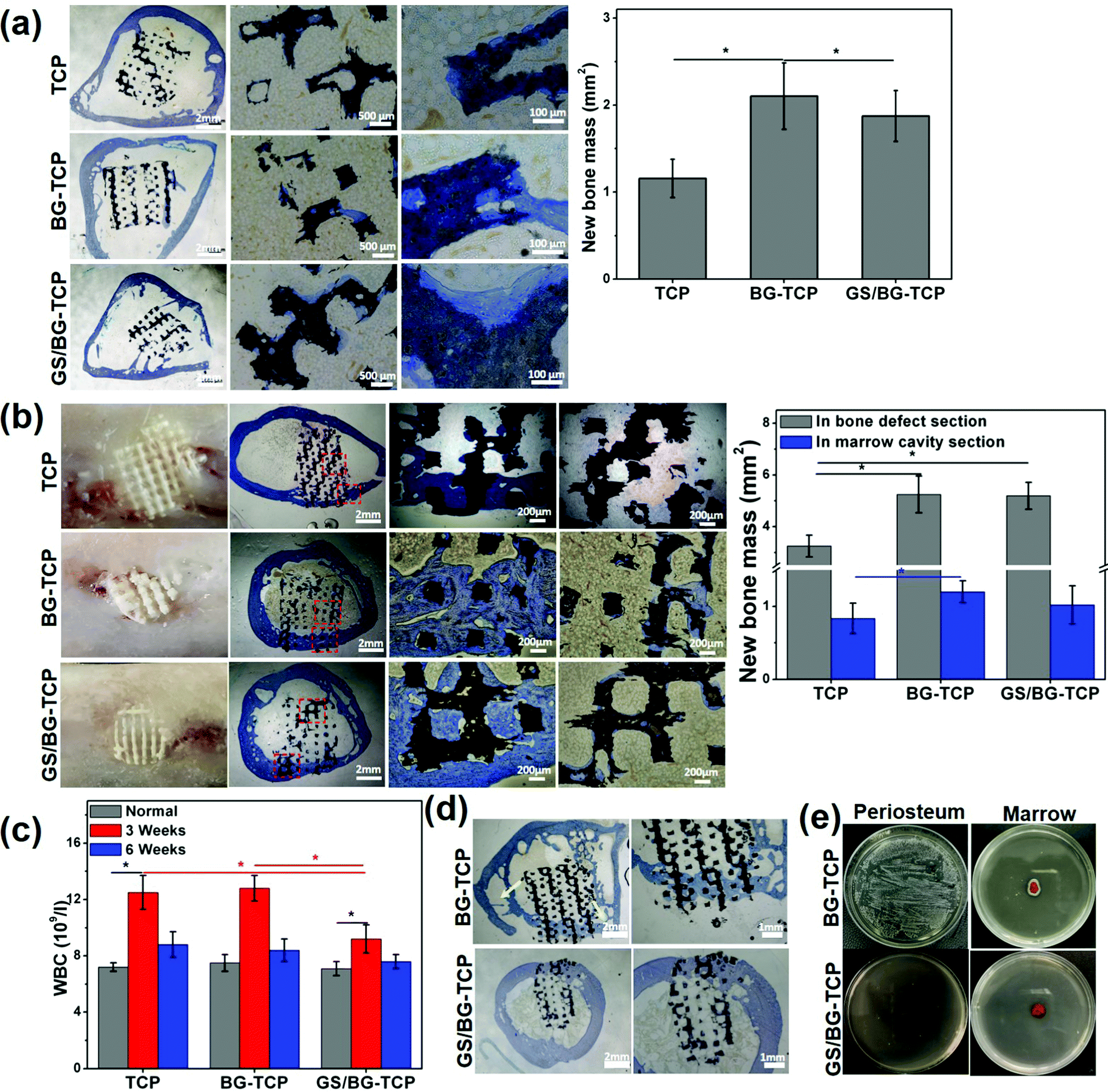 | ||
| Fig. 5 The porous ceramic-mediated in situ regeneration of bone. (a) The TB-stained histological sections of the porous ceramics implanted into the femoral marrow cavities for 6 weeks (n = 3). (b) The TB-stained histological sections of the ceramics implanted into the femoral defects (5 mm) for 6 weeks (n = 3). (c) WBC of the rabbits in the normal and ceramic-implanted (3/6 weeks) state (n =3). (d) The ceramic-mediated in situ regeneration of bone in the S. aureus-infected bone defects (6 weeks) (the yellow arrows indicate the formation of cavitary defects). (e) The marrows harvested from the 6 week-ceramic implanted femurs were incubated for 24 h in the solid medium. | ||
In the sites of bone defects, Fig. 5(b) shows that new bone formed in all defects implanted by different ceramics (6 weeks). The photographs show that the ceramics were embedded well in the newly grown bone tissue, and new bone did not grow in the section of the ceramics that exceeded the bone surfaces. The histological sections show that in all samples new bone tissue grew well in the section of the ceramic embedded in the bone defect, but in the section of ceramics embedded in the marrow only a little bit of newly grown bone tissue adhered on the porous surfaces of the ceramics, and a clear bone-marrow interface was present in all ceramics. The quantitative bone mass shows that in the section of the ceramics distributed in bone defects, the bone mass grown in the GS-loaded ceramics was similar to that in the BG-TCP ceramics, but was significantly higher than that in the TCP ceramics. In the section of the ceramics in the marrow, the bone mass grown in three kinds of ceramics was similar in ratio to that grown in the whole ceramics implanted in marrow cavities (Fig. 5a). The above results further demonstrated that it was the released Ca2+ that promoted the in situ regeneration of bone.
Furthermore, the WBC number of the ceramic-implanted rabbits was measured. The purpose of measuring the WBC number is that it has been well acknowledged that the normal inflammatory response played important roles in mediating the endogenous regenerative procedure of tissues (normal tissue inflammation can recruit and activate a variety of cells, e.g., immunocytes, endothelial cells, stem cells, etc., to orchestrate tissue regeneration).61 The change in the WBC number can reflect the inflammatory response of rabbits to the implanted ceramics. Fig. 5c shows the WBC value of normal and postoperative (3/6 weeks) rabbits. In comparison with the normal WBC value of rabbits, the WBC number increased significantly in all rabbits implanted with porous ceramics for 3 weeks. Still, the WBC value in the rabbits implanted with the GS-loaded ceramics expressed the lowest WBC number. Such a result showed that the implanting sites with the high concentration of GS could suppress the inflammatory response. After 6 weeks, Fig. 5c shows that the WBC number of all rabbits almost returned to the normal value. The above results demonstrated that more bone mass grown from the BG-containing ceramics did not arise from the tissue inflammation.
Taken together, our in vivo implanting experiments showed that all porous ceramics could mediate the endogenous regeneration of bone. In particular, in the bone defect sites, a clear bone-marrow interface within ceramics could form due to the as-prepared ceramics with regular porosity (our previous work has demonstrated that the ceramics with irregular porosity inhibited the formation of the bone-marrow interface).18 Still, our experiments showed that the BG-containing porous ceramics promoted the body to regenerate more new bone. Such a difference was attributable to the dynamic microenvironment constructed by the BG-containing ceramics, in which the apatite nanoplate layer could grow on the ceramic surfaces. Previously, Bohner et al. and our group demonstrated the growth of an apatite nanoplate layer acting as an inorganic mineral ion adjuster in the microenvironment, and the change in the Ca2+ concentration has made a significant contribution to boosting the endogenous regeneration of bone.6 In addition, our experiments showed that the weak acidic microenvironment did not boost bone growth. A possible explanation is that the acidic degree did not arrive at the critical value to trigger the extra inflammation of bone (a high WBC number expressed in all rabbits within 3 weeks should be attributed to the surgical operation and the implanted ceramics).62 Also, our experiments also showed that the relatively high concentration of GS in the microenvironment (the highest release concentration of GS was <0.15 mg ml−1) did not affect the endogenous regeneration of bone. Such a result is attributable to two factors: (i) GS was cytocompatibility (Fig. 3a), and (ii) GS in the microenvironment did not affect in essence the growth of the apatite nanoplate layer (in fact GS promoted the growth of the apatite nanoplate layer, see Fig. 2d).
Finally, the in vivo antibacterial activity of the as-prepared ceramics was checked by creating the S. aureus-infected bone defects as the implanting sites (see Fig. S3, ESI†). After the infected bone defects were washed, the BG-TCP ceramics and the GS/BG-TCP ceramics were implanted respectively in the bone defects for 6 weeks. The histological sections (Fig. 5d) show that new bone could grow in both ceramics. Nevertheless, in the BG-TCP ceramic treated bone defect, osteolysis seems to have occurred in the femur (the arrows indicated the formation of cavitary defects). This suggested that some S. aureus still resided in the bone marrow because the existence of bacteria in the body would cause the inflammation response and thus macrophages/osteoclasts were recruited or stimulated to absorb bone due to the strong stimulatory effect by bacterial toxins.58,63 To demonstrate this speculation, the periostea and marrows were harvested from the ceramic implanted femurs. Fig. 5(e) shows that some S. aureus indeed resided in the periosteum and marrow of the BG-TCP ceramic implanted femur. In contrast, in the GS-loaded ceramic-treated specimens, Fig. 5(e) shows that no bacteria were indicated. The above results demonstrated that our GS-loaded ceramics could prevent the bacterial infection from recurring.
Taken together, our in vivo experiments well demonstrated that the GS-loaded BG-TCP ceramics not only could mediate the in situ regeneration of bone, but also possessed high antibacterial activity against S. aureus. In particular, because the used biomaterials (BG and TCP) and drugs (GS) all have been widely applied in clinical practice, our samples have the potential to be exploited in commercial products.
4. Conclusions
BG-coated hierarchical porous TCP bioceramics were successfully prepared by combining 3D printing, gelcasting, and sol–gel coating methods. Abundant pores distributed hierarchically in ceramics endowed the ceramics with multiple biological functions. The macropores with regular porous structures allowed the growth of new bone only in the bone defect section and the formation of a clear bone-marrow interface. The nanopores endowed the ceramics with the drug delivery ability. More importantly, the coating of BG on the TCP ceramic surfaces constructed a dynamic microenvironment which could boost the endogenous regeneration of bone. The existence of a relatively high concentration of antibacterial drug GS (<0.15 mg ml−1) in the microenvironment did not affect the endogenous regeneration of bone because the released GS in the microenvironment did not affect in essence the growth of an apatite nanoplate layer. Our experiments further demonstrated that the GS-loaded ceramics could prevent the bacterium-ever infected bone defects from recurring. Because the used ceramic materials, TCP and BG, have used widely in clinical practice, our porous ceramics have the potential for treating infected bone defects.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We deeply acknowledge the financial support provided by the Key Research and Development Project (Social Development) of Jiangsu Province (Grant No. SBE2019750209) and the Innovation Huohua Project of Sichuan University (2019SCUH0018).Notes and references
- G. L. Koons, M. Diba and A. G. Mikos, Nat. Rev. Mater., 2020, 5, 584–603 CrossRef CAS.
- A. K. Gaharwar, I. Singh and A. Khademhosseini, Nat. Rev. Mater., 2020, 5, 686–705 CrossRef CAS.
- K. L. Christman, Science, 2019, 363, 340–341 CrossRef CAS PubMed.
- Y. Zhou, C. Wu and J. Chang, Mater. Today, 2019, 24, 41–56 CrossRef CAS.
- Y. Hong, H. Fan, B. Li, B. Guo, M. Liu and X. Zhang, Mater. Sci. Eng. R, 2010, 70, 225–242 CrossRef.
- M. Bohner and R. J. Miron, Mater. Today, 2019, 22, 132–141 CrossRef CAS.
- D. Zhang, M. Hupa and L. Hupa, Acta Biomater., 2008, 4, 1498–1505 CrossRef CAS PubMed.
- Y. Shen, W. Liu, C. Wen, H. Pan, T. Wang, B. W. Darvell, W. W. Lu and W. Huang, J. Mater. Chem., 2012, 22, 8662–8670 RSC.
- Y. Maazouz, I. Rentsch, B. Lu, B. L. G. Santoni, N. Doebelin and M. Bohner, Acta Biomater., 2020, 102, 440–457 CrossRef CAS PubMed.
- U. Ripamonti, R. M. Klar, L. F. Renton and C. Ferretti, Biomaterials, 2010, 31, 6400–6410 CrossRef CAS PubMed.
- P. Habibovic, H. Yuan, C. M. van der Valk, G. Meijer, C. A. van Blitterswijk and K. de Groot, Biomaterials, 2005, 26, 3565–3575 CrossRef CAS PubMed.
- S. Fujibayashi, M. Neo, H. M. Kim, T. Kokubo and T. Nakamura, Biomaterials, 2004, 25, 443–450 CrossRef CAS PubMed.
- D. Barbieri, A. J. S. Renard, J. D. de Bruijn and H. Yuan, Eur. Cells Mater., 2010, 19, 252–261 CrossRef CAS PubMed.
- Y. Chen, Z. Sun, Y. Li and Y. Hong, J. Mater. Chem. B, 2017, 5, 807–816 RSC.
- C. Zhou, Y. Jiang, Z. Sun, Y. Li, B. Guo and Y. Hong, J. Mater. Chem. B, 2018, 6, 5621–5632 RSC.
- Y. Chen, Z. Sun, Y. Li and Y. Hong, J. Mater. Chem. B, 2017, 5, 807–816 RSC.
- M. J. Dalby, N. Gadegaard and R. O. C. Oreffo, Nat. Mater., 2014, 13, 558–569 CrossRef CAS PubMed.
- W. He, Z. Wu, Y. Wu, Z. Zhong and Y. Hong, ACS Appl. Mater. Interfaces, 2021, 13, 31527–31541 CrossRef CAS PubMed.
- S. K. Nandi, S. Bandyopadhyay, P. Das, I. Samanta, P. Mukherjee, S. Roy and B. Kundu, Biotechnol. Adv., 2016, 34, 1305–1317 CrossRef CAS PubMed.
- N. H. Besheli, F. Mottaghitalab, M. Eslami, M. Gholami, S. C. Kundu, D. L. Kaplan and M. Farokhi, ACS Appl. Mater. Interfaces, 2017, 9, 5128–5138 CrossRef PubMed.
- P. Wei, W. Jing, Z. Yuan, Y. Huang, B. Guan, W. Zhang, X. Zhang, J. Mao, Q. Cai, D. Chen and X. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 30596–30609 CrossRef CAS PubMed.
- I. Qayoom, A. K. Teotia, A. Panjla, S. Verma and A. Kumar, ACS Infect. Dis., 2020, 6, 2938–2949 CrossRef CAS PubMed.
- J. Geurts, J. J. Chris Arts and G. H. Walenkamp, Injury, 2011, 42, S82–S86 CrossRef PubMed.
- Y. Deng, X. Shi, Y. Chen, W. Yang, Y. Ma, X.-L. Shi, P. Song, M. S. Dargusch and Z.-G. Chen, Ind. Eng. Chem. Res., 2020, 59, 12123–12135 CrossRef CAS.
- Y. Deng, X. Gao, X.-L. Shi, S. Lu, W. Yang, C. Duan and Z.-G. Chen, Chem. Mater., 2020, 32, 2180–2193 CrossRef CAS.
- S. Wang, C. Duan, W. Yang, X. Gao, J. Shi, J. Kang, Y. Deng, X.-L. Shi and Z.-G. Chen, Nanoscale, 2020, 12, 11936–11946 RSC.
- S. J. Hollister, Nat. Mater., 2005, 4, 518–524 CrossRef CAS PubMed.
- L. L. Hench, J. Am. Ceram. Soc., 1998, 81, 1705–1728 CrossRef CAS.
- W. Liu, T. Wang, X. Zhao, X. Dan, W. W. Lu and H. Pan, Bioact. Mater., 2016, 1, 151–159 CrossRef PubMed.
- Y. Shen, W. Liu, K. Lin, H. Pan, B. W. Darvell, S. Peng, C. Wen, L. Deng, W. W. Lu and J. Chang, Langmuir, 2011, 27, 2701–2708 CrossRef CAS PubMed.
- Y. Hong, H. Fan and X. Zhang, J. Phys. Chem. B, 2009, 113, 5837–5842 CrossRef CAS PubMed.
- Y. Hong, X. Chen, X. Jing, H. Fan, B. Guo, Z. Gu and X. Zhang, Adv. Mater., 2010, 22, 754–758 CrossRef CAS PubMed.
- X. X. Yan, H. X. Deng, X. H. H. Huang, M. Lu, S. Qiao, D. Y. Zhao and C. Z. Yu, J. Non-Crystalline Solids, 2005, 351, 3209–3217 CrossRef CAS.
- Y. Chen, Z. Sun, Y. Li and Y. Hong, J. Mater. Chem. B, 2017, 5, 807–816 RSC.
- I. T. Olszak, M. C. Poznansky, R. H. Evans, D. Olson, C. Kos, M. R. Pollak, E. M. Brown and D. T. Scadden, J. Clin. Invest., 2000, 105, 1299–1305 CrossRef CAS PubMed.
- C. Tan, Z. Sun, Y. Hong, Y. Li, X. Chen and X. Zhang, J. Mater. Chem. B, 2013, 1, 3694–3704 RSC.
- L. L. Hench, J. Mater. Sci.: Mater. Med., 2015, 26, 86 CrossRef PubMed.
- K. Zhang and J. Chen, Cell Adhes. Migr., 2012, 6, 20–29 CrossRef PubMed.
- K. Kandori, T. Kuroda, S. Togashi and E. Katayama, J. Phys. Chem. B, 2011, 115, 653–659 CrossRef CAS PubMed.
- K. Kandori, K. Murata and T. Ishikawa, Langmuir, 2007, 23, 2064–2070 CrossRef CAS PubMed.
- A. L. Berrier and K. M. Yamada, J. Cell. Physiol., 2007, 213, 565–573 CrossRef CAS PubMed.
- D. D. Hu, J. R. Hoyer and J. W. Smith, J. Bio. Chem., 1995, 270, 9917–9925 CAS.
- M. Shimaoka, J. Takagi and T. A. Springer, Annu. Rev. Biophys. Biomol. Struct., 2002, 31, 485–516 CrossRef CAS PubMed.
- C. K. Miranti and J. S. Brugge, Nat. Cell Biol., 2002, 4, E83–E90 CrossRef CAS PubMed.
- N. D. Escudero and P. M. Mandalunis, Bone, 2007, 40, S7 CrossRef.
- P. Ducy, C. Desbois, B. Boyce, G. Pinero, B. Story, C. Dunstan, E. Smith, J. Bonadio, S. Goldstein, C. Gundberg, A. Bradley and G. Karsenty, Nature, 1996, 382, 448–452 CrossRef CAS PubMed.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS PubMed.
- M. J. Dalby, N. Gadegaard, R. Tare, A. Andar, M. O. Riehle, P. Herzyk, C. D. W. Wilkinson and R. O. C. Oreffo, Nat. Mater., 2007, 6, 997–1003 CrossRef CAS PubMed.
- T. Sugimoto, M. Kanatani, J. Kano, H. Kaji, T. Tsukamoto, T. Yamaguchi, M. Fukase and K. Chihara, J. Bone Miner. Res., 1993, 8, 1445–1452 CrossRef CAS PubMed.
- E. M. Brown and R. J. Macleod, Physiol. Rev., 2001, 81, 239–297 CrossRef CAS PubMed.
- Y.-R. V. Shih, Y. Hwang, A. Phadke, H. Kang, N. S. Hwang, E. J. Caro, S. Nguyen, M. Siu, E. A. Theodorakis, N. C. Gianneschi, K. S. Vecchio, S. Chien, O. K. Lee and S. Varghese, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 990–995 CrossRef CAS PubMed.
- U. Ripamonti and J.-C. Petit, Cytokine Growth Factor Rev., 2009, 20, 489–499 CrossRef CAS PubMed.
- U. Ripamonti, Biomaterials, 2006, 27, 807–822 CrossRef CAS PubMed.
- R. McBeath, D. M. Pirone, C. M. Nelson, K. Bhadriraju and C. S. Chen, Dev. Cell, 2004, 6, 483–495 CrossRef CAS PubMed.
- S. Maeno, Y. Niki, H. Matsumoto, H. Morioka, T. Yatabe, A. Funayama, Y. Toyama, T. Taguchi and J. Tanaka, Biomaterials, 2005, 26, 4847–4855 CrossRef CAS PubMed.
- G. Walter, M. Kemmerer, C. Kappler and R. Hoffmann, Dtsch. Arztebl. Int., 2012, 109, 257–264 Search PubMed.
- Z. Wu and Y. Hong, ACS Appl. Mater. Interfaces, 2019, 11, 33734–33747 CrossRef CAS PubMed.
- W. He, Z. Wu, Y. Wu, Y. Cai, Z. Cui, B. Yu and Y. Hong, ACS Appl. Bio Mater., 2021, 4, 6280–6293 CrossRef CAS PubMed.
- L. Batoon, S. M. Millard, L. J. Raggatt and A. R. Pettit, Curr. Osteoporos. Rep., 2017, 15, 385–395 CrossRef PubMed.
- T. Yamaguchi, I. Olozak, N. Chattopadhyay, R. R. Butters, O. Kifor, D. T. Scadden and E. M. Brown, Biochem. Biophys. Res. Commun., 1998, 246, 501–506 CrossRef CAS PubMed.
- T. A. Wynn and K. M. Vannella, Immunity, 2016, 44, 450–462 CrossRef CAS PubMed.
- J. M. Anderson, A. Rodriguez and D. T. Chang, Semin. Immunol., 2008, 20, 86–100 CrossRef CAS PubMed.
- J. Lorenzo, M. C. Horowitz, Y. Choi, H. Takayanagi and G. Schett, Osteoimmunology: Interactions of the Immune and Skeletal Systems, Academic Press, 2nd edn, 2015 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The chemical components of the TCP slurry and the BG precursors; detailed information about the primer sequences used for qRT-PCR; the created bone defects and the ceramic implanting experiments; bone infection model and the outcomes of the established bone infection; and the SEM images of the ceramics when soaked in SBF for 36 h. See DOI: https://doi.org/10.1039/d2ma00057a |
| This journal is © The Royal Society of Chemistry 2022 |