
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A water-mediated approach for the preparation of conductive poly(3,4-ethylenedioxythiophene)-decorated poly(methyl methacrylate) microcomposites†
P. A.
Saeed
a,
R.
Shilpa
b and
A.
Sujith
 *a
*a
aMaterials Research Laboratory, Department of Chemistry, National Institute of Technology Calicut, Calicut-673601, Kerala, India. E-mail: athiyanathil.sujith@gmail.com; Fax: +91495 2287250; Tel: +91 9846475675
bSt. Thomas College, Ranni, Pathanamthitta, 689673, Kerala, India
First published on 24th March 2022
Abstract
The water-mediated synthesis of poly(3,4-ethylenedioxythiophene) (PEDOT) on the surface of poly(methyl methacrylate) (PMMA) microspheres leads to the formation of segregated electrically conductive composites. This work demonstrates the successful combination of environmentally benign acoustic emulsification, surfactant-free emulsion polymerization, and in situ oxidative polymerization. Acoustic emulsification creates 3,4-ethylenedioxythiophene (EDOT) monomers with an anionic surface and surfactant-free emulsion polymerization generates cationic spherical PMMA latex particles. Then, the in situ oxidative polymerization of the EDOT emulsion generates PEDOT-decorated PMMA microspheres. The composite shows synergic properties from PEDOT and PMMA, with good electrical, thermal, and morphological properties. The electrical conductivity of the composite was increased ∼109-fold compared with the PMMA matrix. The electrical conductivity of the PMMA–PEDOT composite reaches 0.30 S m−1 when the PEDOT loading is 10.20 wt%. Additionally, it provides a ∼105-fold increase in electrical conductivity compared to samples prepared without the acoustic emulsification of EDOT. This method provides new planning strategies for the integration of various conducting polymers into insulating polymers.
Introduction
Electrically conducting conjugated polymers, also called synthetic metals, have received widespread attention in recent academic and industrial research.1–3 They function as key components for display devices, electromagnetic shielding, sensors, and capacitors due to their inherent combination of useful chemical and physical properties.4–6 Poly(3,4-ethylenedioxythiophene) (PEDOT) is one such conjugated polymer with remarkably high conductivity and environmental stability.7–13 Generally, the applications of conducting polymers alone have been limited because of their high brittleness, owing to the rigid backbone structure of the molecules, and their poor processability. Therefore, the integration of conducting polymers into insulating polymers has shown great potential, exhibiting combined properties in the form of a hybrid material. This can meet the multitudinous demands presented by electronic devices, antistatic coatings, electromagnetic interference shielding, electrodes, and sensors.14–20 Among the various insulating polymers, poly(methyl methacrylate) (PMMA) has been widely investigated for various applications, including in the electronics industry, due to its excellent mechanical strength and optical transparency.21,22 It can form latex particles, which play an important role in colloid and materials science. Its structural backbone can support interfacial interactions between the PEDOT polymer and the polymer matrix via dipole–dipole interactions. The interfacial compatibility is crucial for obtaining superior electrical, thermal, and stability properties in composite materials. Various in situ and/or ex situ approaches have been used to disperse different fillers in PMMA matrices for the fabrication of nano- and microcomposites.19,22–28 However, there are some major challenges in this field. Firstly, there is the fact that synthetic processes need to use toxic and harmful solvents and surfactants, potentially introducing significant environmental issues.27,29–32 Secondly, it is a challenge to achieve the efficient incorporation of conducting polymers into a matrix polymer without compromising the desired properties.33 Moreover, if the ratio of conducting polymer is too low, then the hybrid material will show poor electrical properties, whereas when the ratio of conducting polymer is too high, the materials turn out to be rigid with low processability.34–38To overcome this challenge, several strategies have been developed in the past few decades for producing conducting polymeric nanocomposites.18,23,39,40 So far, the most exploited and frequently used process is latex-based colloidal blending, which provides interstitial spaces and forces the nanofiller particles into the spaces, finally forming a segregated conductive network.23,34,41 Unfortunately, the main drawback of this method is the essential need for surfactant to disperse the polymers in aqueous media, which decreases the conductivity. It also increases the cost and difficulty during the purification of the resulting hybrid composite material.22
On the other hand, acoustic emulsification produces a stable emulsion without the use of surfactants.42,43 This is considered a green emulsification method based on ultrasonic irradiation, utilizing the mechanical forces originating from acoustic cavitation at the liquid/liquid phase boundary.44–46 Emulsion droplets with a milky white to transparent appearance have been synthesised via ultrasonic irradiation at frequencies ranging from 20 kHz to 1.0 MHz.47–50 Recently, the effects of sequential ultrasonication or tandem acoustic emulsification were studied in terms of the stability and size of nanoemulsions upon the elongation of the sonication time. Unfortunately, the exact reason for the formation of stable acoustic emulsions without the need for surfactants has not been revealed completely at present. However, Fogler et al. proposed that the preferential adsorption of OH− ions from aqueous media stabilizes the acoustic emulsion.51–53
Water is a benign, low-cost, and safe medium for synthetic chemistry, but many organic monomers and polymers are water-insoluble or only sparingly soluble. Surfactant-free emulsion polymerization represents an aqueous process that has high potential in industrial research.23
Our aim is to demonstrate a green strategy for the preparation of an electrically conductive PEDOT-decorated PMMA composite without using harmful solvents or surfactants. For this goal, we ensure the successful integration of acoustic emulsification, surfactant-free emulsion polymerization, and in situ oxidative polymerization. Individually, these methods are greener, and harmful solvents and surfactants are not involved in the process. The successful integration of these methods is challenging, but it results in an environmentally benign conductive PEDOT-decorated PMMA composite material.
Experimental
Materials
Methyl methacrylate (MMA), 3,4-ethylenedioxythiophene (EDOT), 2,2′-azobis(2-methylpropionamidine) dihydrochloride (AIBA), and ammonium persulfate (APS) were purchased from Sigma-Aldrich. Deionized water (DI) was used as a solvent.Emulsification treatment of EDOT
The acoustic emulsification of EDOT was conducted via modifying a reported procedure.50 10 mL of DI water was taken in a beaker and the EDOT monomer (100 μL) was dropped to it. Then, ultrasonication of the immiscible liquid was carried out with a probe sonicator (5 mm diameter, 20 kHz oscillator, titanium alloy) for 120 s. The probe was vertically placed 2.0 cm from the bottom of the beaker. Also, the experimental set-up was supported by a cooling bath (22 ± 2 °C) in order to dissipate the heat produced during sonication.Preparation of positively charged PMMA latex
Cationic PMMA latex was synthesised via surfactant-free emulsion polymerization.23 87.5 mL of DI water was taken in a three-neck RB flask and nitrogen gas was used to purge this for 15 min. Then, the temperature was raised to 75 °C under constant stirring at 350 rpm. Then, 10 g of MMA was added dropwise and, again, nitrogen gas was used for purging for 15 min. After that, 0.015 g of AIBA dissolved in 2.5 mL of DI water was added carefully, followed by stirring for 5 h.Preparation of the PMMA–PEDOT microcomposite
The desired volume of EDOT emulsion was gradually added to 10 g of PMMA latex under constant stirring for 15 min. The resultant PMMA–EDOT colloidal suspension was polymerised in situ with APS at 25 °C for 24 h. The colour of the reaction mixture slowly changed from white to light blue and finally to dark blue. The prepared bluish coloured PMMA–PEDOT composite was washed with DI water, unreacted EDOT monomer was washed out using methanol, and finally the composite particles were dried at 60 °C for 24 h.The PMMA–PEDOT composite was then crushed into a fine powder using a mortar and pestle. The composite was further subjected to compression molding in a hydraulic pellet press (KP 672) at 50 bar for 5 min to create a PMMA–PEDOT composite thin pellet. The thickness of the pellet was controlled based on the feed amount of powder.
Characterization
The morphologies of PMMA, PMMA–PEDOT, and the polymer composite films were analysed via SEM (FE-SEM, Hitachi 6600) and HRTEM (JOEL, JEM-2100F). EDX analysis was conducted with Jeol 6390LA/Oxford XMX N apparatus. X-ray photoelectron spectroscopy (XPS) spectra were obtained using a PHI 5000 versaprobe scanning ESCA microprobe electron spectrometer from Physical Electronics, USA, with monochromatic Al Kα radiation. XRD analysis was carried out using a Rigaku MiniFlex 600 diffractometer (Japan, using a Cu Ka radiation source). Fourier-transform infrared (FT-IR) spectroscopy analysis was performed using Nicolet 5700 apparatus. The thermal properties of samples were analyzed via TGA (Q50, TA Instruments, N2 atmosphere, heating rate of 10 °C min−1) and DSC (Q20, TA Instruments, N2 atmosphere, heating rate of 10 °C min−1). Zeta potentials of the PMMA latex and EDOT emulsion were analysed using Zetasizer apparatus (Nano ZS, Malvern Instruments). The electrical resistance of PMMA–PEDOT pellets was studied using the four-point probe method (Keithley 2450 instrument).Results and discussion
PEDOT-decorated PMMA microcomposite
PMMA–PEDOT composites were synthesized in a stepwise manner through a green protocol via acoustic emulsification, surfactant free emulsion polymerization, and in situ oxidative polymerization. A schematic diagram of the synthesis process is shown in Fig. 1 and an overview is given below.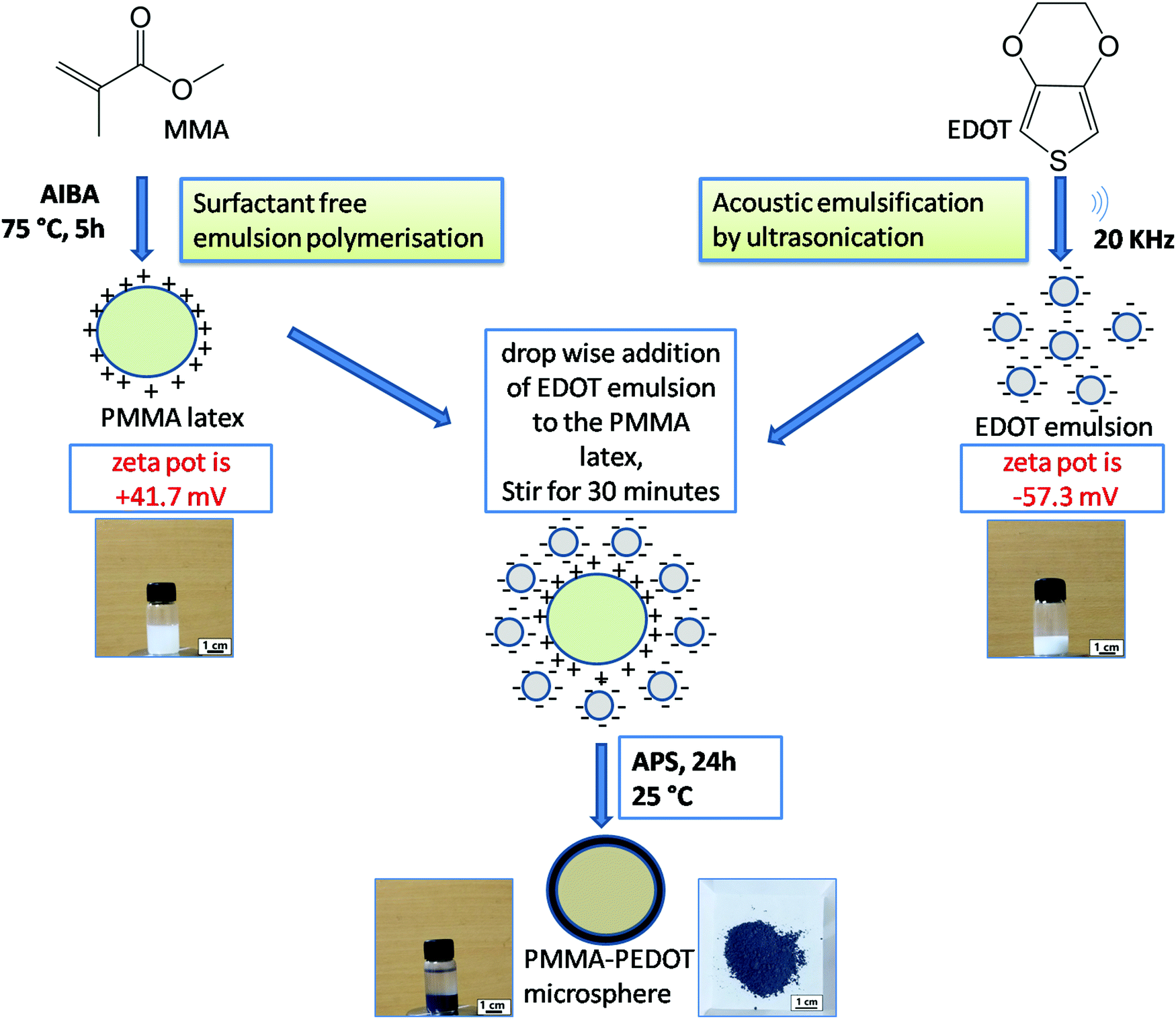 | ||
| Fig. 1 A schematic diagram of the self-assembly of PMMA latex and EDOT emulsion, followed by the oxidative polymerisation of EDOT by APS, with photographs of PMMA latex, EDOT emulsion, and the PMMA–PEDOT composite (suspension and powder). | ||
First, PMMA latex particles were prepared using the cationic free radical initiator 2,2′-azobis(2-methylpropionamidine) dihydrochloride, which produced positive charges on the surface of the PMMA particles. The generation of a positive surface is due to the accumulation of cationic fragments from the decomposition of the AIBA initiator.54,55 The EDOT emulsion was prepared via acoustic emulsification through probe ultrasonication (20 kHz oscillator) of the EDOT/water mixture for 120 s, creating negative charges on the surface of the EDOT molecules. This is due to the preferential adsorption of OH− ions from the aqueous medium, which stabilizes the acoustic emulsion.51–53 The zeta potential of the PMMA latex and EDOT emulsion were measured to be +41.7 mV and −57.3 mV (Fig. S1 and S2, ESI†), respectively. Then, the oxidative in situ polymerisation of EDOT on positively charged PMMA microspheres was carried out via the slow addition of an aqueous solution of APS and further stirring for 24 h. The PMMA–PEDOT composite turned light blue immediately, indicating the formation of PEDOT. Finally, a dark-blue-coloured PMMA–PEDOT composite was formed. During this process, the PMMA microparticles serve as a template for the in situ polymerisation of PEDOT. Conducting polymers such as PEDOT generally undergo strong interchain interactions. PEDOT was also synthesized under similar conditions using APS as the initiator. The use of water as a medium for the overall procedure makes the method low-cost and simple.
A comparison between the water-mediated strategy for PMMA–PEDOT composite formation and other methods for polymer–PEDOT composites is given in Table 1. To date, several methods have been demonstrated for preparing polymer–PEDOT composites. The main disadvantages of these techniques include the use of toxic surfactants and organic solvents, which is undesirable from an environmental aspect. Interestingly, our newly designed strategy for the preparation of a PMMA–PEDOT composite without using surfactants and organic solvents can satisfy the requirements of green scientific research.
| No. | Composite | Preparation of matrix polymer | Integration of conducting polymer | Solvent or surfactants used for preparation |
|---|---|---|---|---|
| 1 | PMMA–PEDOT nanofiber mat19 | Electrospinning | Oxidative polymerization | DMF |
| 2 | PS–PEDOT nanospheres32 | Dispersion polymerization | Oxidative polymerization | PVP, propanol, methanol |
| 3 | PMMA–PEDOT polymer sheet33 | PMMA film | Oxidative polymerization | Pyridine, butanol |
| 4 | PAN–PEDOT nanofiber mat29 | Electrospinning | Vapour-phase deposition | DMF, pyridine |
| 5 | PMMA–PEDOT polymer film27 | PMMA granules | Mini-emulsion polymerization | Surfactants (DBSA), CH2Cl2 |
| 6 | PS–PEDOT nanofiber mat31 | Electrospinning | Vapour deposition polymerization | THF, acetone, butanol |
| 7 | PMMA–PEDOT thin fiber mat30 | Electrospinning | Solid-state polymerization of DBEDOT | CHCl3, acetic acid, hexane, CH2Cl2, DMF, THF |
| 8 | PMMA–PEDOT polymer film27 | PMMA granules | Electrochemical polymerization | LiClO4, CH2Cl2 |
| 9 | PMMA–PEDOT microspheres (our strategy) | Surfactant-free polymerisation | Acoustic emulsification | H 2 O |
Morphology of PMMA–PEDOT microcomposites
TEM analysis was conducted to examine the decoration of PEDOT on the surface of PMMA microspheres. Fig. 2 shows TEM images of PMMA (Fig. 2a) and the PMMA–PEDOT composites formed via the polymerization of anionic EDOT emulsion on the surface of cationic PMMA latex particles with 7.08 wt% PEDOT loading (Fig. 2b) and 10.20 wt% PEDOT loading (Fig. 2c). The PMMA particles are clean, smooth, and spherical, with an average size of 200 nm, as shown in Fig. 2a. | ||
| Fig. 2 TEM images of (a) PMMA, and PMMA–PEDOT composites with (b) 7.08 wt% and (c) 10.20 wt% PEDOT loading. SEM images of (d) PMMA, (e) the PMMA–PEDOT composite with 7.08 wt% PEDOT loading, and (f) the PMMA–PEDOT composite formed from the direct addition of EDOT with 10.48 wt% PEDOT loading. | ||
Surprisingly, the PMMA–PEDOT composites also exhibit spherical morphology but with a totally different surface; a continuous rugged morphology was observed, as shown in Fig. 2b and c. Indeed, this morphology suggests the deposition of segregated PEDOT, preferably at the surface, whereas the majority of the volume consists of PMMA matrix. As seen in Fig. 2c, for 10.20 wt% PEDOT loading, the tightly coated PEDOT particles are interstitially spaced between PMMA particles forming a conductive network. This type of network is very crucial for electrical conduction throughout the composite, consequently promising a low percolation threshold of around 7.08 wt% PEDOT loading.
We believe that the in situ polymerisation of EDOT occurs on the surface of the spherical PMMA matrix. Initially, oligomeric chains of EDOT were generated at the surface of PMMA particles. Later, the spontaneous assembly of EDOT oligomers was carried out and finally the formation of PEDOT occurred on the surface of the PMMA particles.
Fig. 2d shows the SEM image of spherical PMMA particles. Fig. 2e shows the PMMA–PEDOT composite formed via the polymerization of an anionic EDOT emulsion on the surface of cationic PMMA latex particles with 7.08 wt% PEDOT loading, exhibiting the uniform coating of PEDOT onto the surfaces of PMMA microspheres, as seen in the TEM images. Specifically, it highlights the core–shell morphology, in which in situ polymerised PEDOT particles were accumulated on the insulating PMMA matrix. More specifically, this strategy ensures strong improvements in the electrical and thermal properties of the polymer composites. Fig. 2f shows the PMMA–PEDOT composite formed from the direct addition of the EDOT, with 10.48 wt% PEDOT loading; it exhibited an aggregated structure with a non-uniform PEDOT coating on the surface of the PMMA microspheres, which limits the overall conductivity of the composite due to the absence of segregated morphology, and this is verified via electrical conductivity studies.
Structural characteristics
The measured IR spectra of PMMA, PEDOT, and PMMA–PEDOT are presented in Fig. 3a. For PEDOT, the exhibited peaks could be assigned to symmetric and asymmetric C–H (2852 and 2920 cm−1), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (1527 cm−1), C–C (1345 cm−1), C–O (1090 cm−1), and C–S (981 cm−1 and 836 cm−1).19,56,57 PMMA exhibited characteristic peaks at 1719 cm−1 (CO stretching) and 1145 cm−1 (CO stretching).22 However, the FT-IR spectrum of the PMMA–PEDOT composite exhibited the characteristic peaks of both PEDOT and PMMA.
C (1527 cm−1), C–C (1345 cm−1), C–O (1090 cm−1), and C–S (981 cm−1 and 836 cm−1).19,56,57 PMMA exhibited characteristic peaks at 1719 cm−1 (CO stretching) and 1145 cm−1 (CO stretching).22 However, the FT-IR spectrum of the PMMA–PEDOT composite exhibited the characteristic peaks of both PEDOT and PMMA.
 | ||
| Fig. 3 Characterization of the PMMA–PEDOT composite: (a) FTIR spectra, (b) XRD patterns, and (c) XPS survey spectra. | ||
Distinguishable peaks were exhibited at 1731 cm−1 (CO stretching), 1527 cm−1 (CC stretching), and 981 cm−1 (C–S stretching). These assigned peaks were correlated to the characteristic bonds of PMMA and PEDOT. The shifting of the characteristic peaks was connected to electrostatic and interfacial interactions between PMMA and PEDOT. In addition, PEDOT particles were shown to be physically entrapped with the matrix PMMA microspheres rather than chemically bonded.
Fig. 3b shows the XRD patterns of PMMA, PEDOT, and PMMA–PEDOT. For PEDOT, the diffraction peak observed at 2θ = 6.6° seems to be the (100) reflection of the polymer backbone and the other two peaks at 2θ = 11.8° and 2θ = 25.5° correspond to (200) and (020) reflections, respectively.56–58 PMMA exhibited two broad peaks at 2θ = 14.5° and 30.1°, indicating an amorphous structure.59 The XRD pattern of the PMMA–PEDOT composite, showing diffraction peaks at 2θ scattering angles of 14.2° and 25.1°, confirmed the incorporation of PEDOT onto the PMMA particles. The successful incorporation of PEDOT onto the PMMA particles was also demonstrated via XPS analysis. Fig. 3c shows the XPS survey spectra of PMMA, PEDOT, and the PMMA–PEDOT composite. In PMMA–PEDOT, apart from the signals of carbon and oxygen elements, signals due to sulphur confirm the presence of PEDOT in the polymer matrix.
Composite characteristics
A group of PEDOT-decorated PMMA composites was prepared in which the PEDOT loading was varied from 4.38 wt% to 16.66 wt%. Fig. 4 shows the C 1s XPS spectra of PMMA (Fig. 4a), PEDOT (Fig. 4b), and PEDOT-decorated PMMA composites with 4.38 wt%, 7.08 wt%, and 10.20 wt% PEDOT loading (Fig. 4c–e). For PMMA, the dominant peaks are centred at 283.59 eV (C–C/C–H), 285.29 eV (C–C–O), 286.79 eV (C–O), and 288.41 eV (O–CO).60,61 For PEDOT, the main peaks are seen at 283.54 eV (C–C/C–H) and 284.88 eV (C–S), and a smaller peak is located at 286.04 eV (C–O).19,62–64 Additionally, a weak π → π* shake-up signal is seen at 290.6 eV from the heteroaromatic thiophene ring.62 For the composites, some of the subpeaks overlap based on the reference spectra obtained from PMMA and PEDOT particles. However, the composite with higher PEDOT loading exhibits comparatively similar signals to the PEDOT particles, clearly revealing that PEDOT is heavily concentrated at the surface of the PMMA microspheres, which was also supported by TEM images.
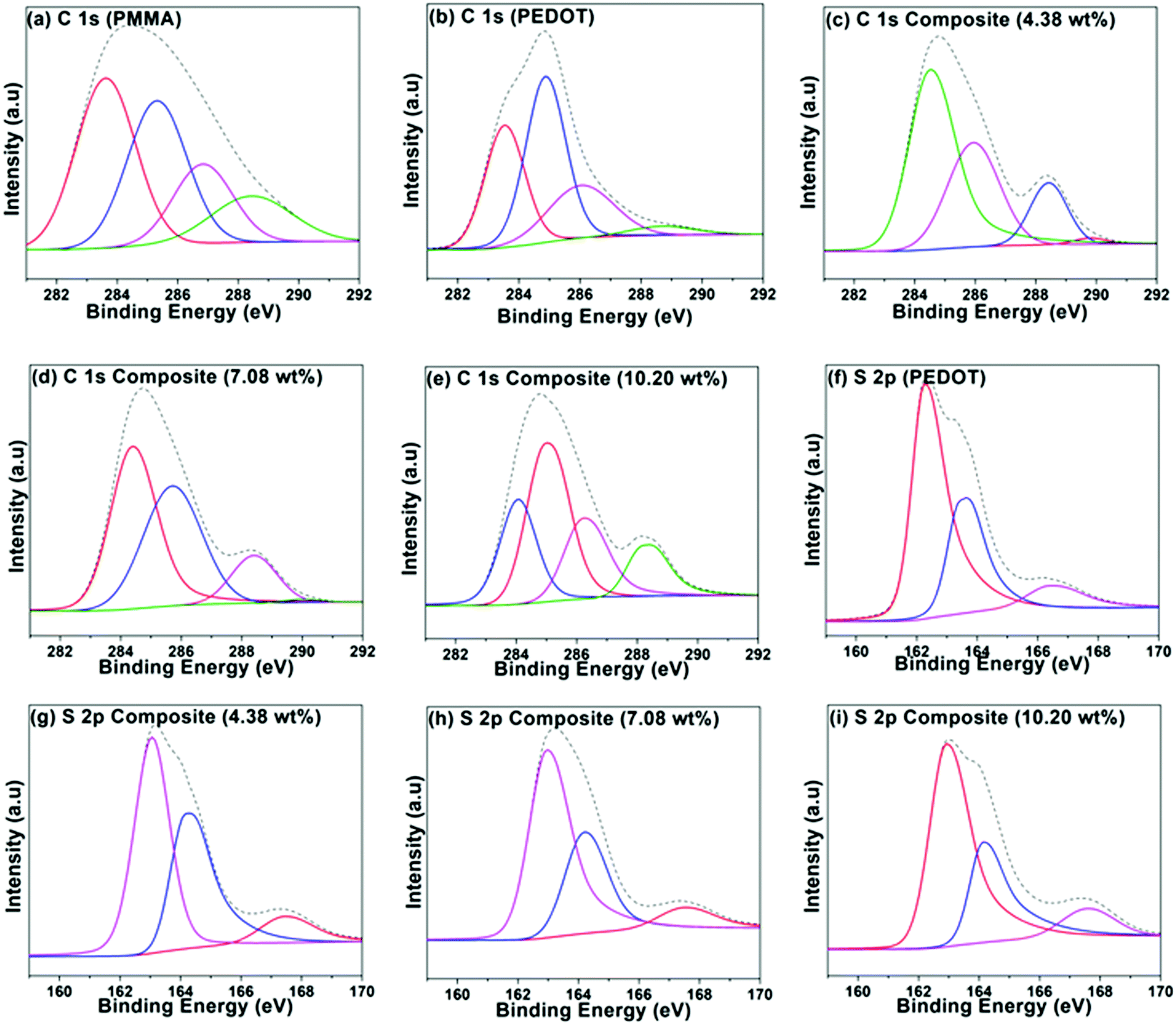 | ||
| Fig. 4 XPS spectra: C 1s spectra of (a) PMMA, (b) PEDOT, and PMMA–PEDOT composites with (c) 4.38 wt%, (d) 7.08 wt%, and (e) 10.20 wt% PEDOT loading; S 2p spectra of (f) PEDOT, and PMMA–PEDOT composites with (g) 4.38 wt%, (h) 7.08 wt%, and (i) 10.20 wt% PEDOT loading. | ||
Fig. 4f–i shows the S 2p spectra for PEDOT particles and PEDOT-decorated PMMA composites with PEDOT loading levels of 4.38, 7.08, and 10.20 wt%. The dominant signals can be fitted to neutral S (162.6–163.01 eV), cationic S+ (163.44–164.28 eV) assigned to the PEDOT backbone, and sulphate dopant (166.41–167.77 eV).62–68 It is possible to determine the doping levels via analysing the peak area of sulphate dopant compared to those of the other two signals, and it was indicated that PEDOT was doped with roughly 11.7 ± 1.6% sulphate. Approximately similar low doping levels of the PEDOT in the composite were recorded based on XPS analysis.
Compositional analysis of the PMMA–PEDOT composites was also carried out based on energy dispersive X-ray (EDX) analysis (see Fig. S3, ESI†). The intensity of the S signal increases as the PEDOT composition in the composite increases. The signal consists of carbon, oxygen, and sulphur. Examining the elemental composition from EDX studies and the percentage of sulphate dopant in PEDOT, five different samples were analysed. The PEDOT loading levels in the different composites were calculated to be 4.38 wt%, 7.08 wt%, 9.77 wt%, 10.20 wt%, and 16.66 wt%.
Table 2 summarizes the elemental analysis results from XPS and EDX analysis for five PEDOT–decorated PMMA composite samples, including the doping level percentage, the relative proportion of PEDOT at the surface, and its loading in the composites. The relative proportion of PEDOT at the surface of the composites was calculated based on the atomic ratio between the S 2p signals from the coated composites and PEDOT bulk powder, assuming that the composition is homogeneous in the volume analysed via XPS. The PEDOT loading in the composite was quantified via taking the weight ratio between sulphur in the composite and the PEDOT bulk, resulting from EDX analysis. In both calculations, sulphur content due to sulphate dopant was excluded.
| Composite | S (at%) from XPS | % of sulphate dopant | Doping level (%) | PEDOT coverage at the surface (at%) | S (wt%) from EDX | PEDOT (wt%) |
|---|---|---|---|---|---|---|
| Sample I | 2.58 | 10.25 | 11.42 | 20.88 | 0.98 | 4.38 |
| Sample II | 2.61 | 9.40 | 10.37 | 21.37 | 1.59 | 7.08 |
| Sample III | 3.33 | 11.85 | 13.40 | 26.49 | 2.20 | 9.77 |
| Sample IV | 4.36 | 9.10 | 10.01 | 35.80 | 2.30 | 10.20 |
| Sample V | 4.65 | 9.87 | 10.95 | 37.89 | 3.74 | 16.66 |
The relative proportion of PEDOT at the surface increases as PEDOT loading increases, suggesting that the PMMA surface is supportive of EDOT polymerisation. However, the relatively low proportion of PEDOT at the surface of the composite particles shows that the PEDOT coating is non-uniform, and this is supported by the morphological analysis. Ultimately, the PEDOT-rich surface relative to the composite bulk ensures a segregated morphology, providing higher composite conductivity.
Thermal and electrical properties
Thermogravimetric analysis (TGA) was conducted to test the thermal properties of the composites. Fig. 5a shows the TGA curves of PEDOT, PMMA, and PEDOT-decorated PMMA composites with different PEDOT loading levels. Fig. 5b shows the derivative thermal gravimetric (DTG) curves of samples. | ||
| Fig. 5 (a) TGA, (b) DTG, (c) DSC, and (d) electrical conductivity curves of PMMA, PEDOT, and PMMA–PEDOT composites with different PEDOT loading levels. | ||
Table 3 summarises the initial decomposition temperature (Ti) and temperature of the maximum decomposition rate (Tmax) values of PMMA and the composites with different PEDOT loadings. Initially, the weight loss between 25 and 110 °C (about 2–3 wt%) corresponds to the vaporisation of adsorbed water. PEDOT powder is stable up to 190 °C and then continuous degradation occurs up to 400 °C due to the degradation of the ethylenedioxy groups in the PEDOT molecules. From 400 °C to 600 °C, a smaller degradation step is seen due to the decomposition of the polymer backbone. There was around 57.4 wt% left after heating to 600 °C.58 For PMMA, the two stages of weight loss appeared in the temperature ranges of 110–220 °C and 254–410 °C, and these may be attributed to the degradation of unsaturated end groups and main-chain pyrolysis.23 In the case of the PMMA–PEDOT composites, the onset decomposition temperature and pyrolysis shifted toward a higher temperature compared to the PMMA matrix. Specifically, the onset of decomposition was increased by about 28.2 °C and 37.7 °C for the composites with PEDOT loading levels of 7.08 wt% and 10.20 wt%, respectively. The final weight fractions of PMMA and PEDOT remaining at 410 °C were 9.6% and 65.2%, respectively. Interestingly, the final weight fractions of the PMMA–PEDOT composites increased dramatically compared to the matrix polymer. They increased to 43.5% and 48.6% for the composites with PEDOT loading levels of 9.77 wt% and 10.20 wt%, respectively. The improved thermal stability when compared to the PMMA matrix could be attributed to the formation of a segregated structure with PEDOT on the surface of PMMA. Fig. 5c exhibits the DSC thermograms of the PMMA and PMMA–PEDOT composites with different PEDOT loading levels, providing evidence for the formation of segregated conductive-filler networks. According to the results, the PEDOT morphology in the PMMA–PEDOT composites affects the glass transition temperature value of the PMMA matrix. A Tg value of 111.3 °C was observed for pure PMMA particles.23,69
| Sample | T i (°C) | T max (°C) | T g (°C) |
|---|---|---|---|
| PMMA | 254.4 | 351.6 | 111.3 |
| 4.38 wt% | 259.5 | 360.2 | 123.0 |
| 7.08 wt% | 283.2 | 359.1 | 125.7 |
| 9.77 wt% | 284.3 | 367.1 | 126.6 |
| 10.20 wt% | 292.1 | 357.7 | 126.8 |
| 16.66 wt% | 258.6 | 355.9 | 126.9 |
The addition of PEDOT to PMMA caused an increase in Tg (see Table 3), revealing that the decoration of PEDOT onto the surface of PMMA limits the segmental mobility of the matrix for the glass transition. Also, this supports the concept of interfacial interactions between PEDOT and the matrix polymer and the compatibility between the two polymers. Moreover, SEM and TEM images of composites with the thick decoration of protruding PEDOT on the PMMA surface validate the interfacial interactions between components which can limit the relaxation behaviour of PMMA.
Electrical conductivity is one of the most useful characteristics of PEDOT. About a 109-fold increase in the electrical conductivity of the PMMA–PEDOT composite was obtained compared to the polymer matrix.22,23Fig. 5d shows the electrical conductivity of the composites as a function of PEDOT loading (wt%) at 25 °C, demonstrating the percolation behaviour. The electrical conductivity data for PMMA and composites with different PEDOT loading levels are shown in Table 4. Interestingly, a sudden increase in electrical conductivity, also termed the percolation threshold, is reached when the PEDOT loading was about 7.08 wt%, specifying the generation of a conductive network in PEDOT-decorated PMMA. The electrical conductivity of the PMMA–PEDOT composite reaches 0.30 S m−1 when the PEDOT loading is 10.20 wt%. It is worth noting that the conductivity of the composite consisting of directly added EDOT with 10.48 wt% PEDOT loading is only 3.21 × 10−6 S m−1. In contrast, our new strategy ensures an almost similar level of conductivity of 3.16 × 10−6 S m−1 when the PEDOT loading was only 4.38 wt%. The very low conductivity of the former is most likely due to the absence of a conductive network, which was limited by the uneven agglomeration of PEDOT over PMMA particles, as seen in SEM imaging. The PMMA–PEDOT composite formed via the polymerization of the anionic EDOT emulsion on the surface of cationic PMMA latex particles has a conductive network with the interstitial spacing of PEDOT between the PMMA particles. Moreover, the surfactant-free water-mediated process incorporating acoustic emulsification and latex technology ensured that the good electrical properties of PEDOT were maintained in the composite material.
| Sample | Electrical conductivity (S m−1) |
|---|---|
| PMMA | ∼10−10 |
| 4.38 wt% | 3.16 × 10−6 |
| 7.08 wt% | 3.37 × 10−5 |
| 9.77 wt% | 3.37 × 10−2 |
| 10.20 wt% | 0.30 |
| 16.66 wt% | 0.37 |
Conclusions
In this work, we report a simple and green strategy for the synthesis of conductive-PEDOT-decorated PMMA composites involving acoustic emulsification, surfactant-free emulsion polymerization, and in situ polymerization. The EDOT emulsion was prepared via green acoustic emulsification, which could create negative charge on the surface of the EDOT monomer. In addition, cationic PMMA latex particles were synthesised via surfactant-free emulsion polymerization. Finally, the in situ oxidative polymerization of EDOT generated PEDOT-decorated PMMA microspheres. Morphological analysis shows that PEDOT particles were efficiently decorated onto the surface of PMMA particles, forming a segregated network. The prepared PMMA–PEDOT composite exhibits better electrical conductivity and thermal stability than related materials. The electrical conductivity of the composite when the PEDOT loading was ∼10.20 wt% is increased ∼109-fold over that of the matrix polymer and ∼105-fold compared with a composite that was prepared conventionally. With 10.20 wt% PEDOT loading, the electrical conductivity of the composite exceeded 0.30 S m−1, which is a sufficient level of conductivity for many electrical applications, including electromagnetic interference shielding. Also, the onset decomposition temperature and pyrolysis temperature of the composite were shifted toward higher temperatures compared to the PMMA matrix. Specifically, the onset decomposition temperature increased by about 28.2 °C and 37.7 °C for composites with PEDOT loading levels of 7.08 wt% and 10.20 wt%, respectively. The improved thermal stability when compared to the PMMA matrix could be attributed to the formation of the segregated PEDOT structure on the surface of PMMA particles. The significant improvement of Tg at low PEDOT loading levels indicates the strong interfacial interactions between PEDOT particles and the PMMA matrix, and this also validates the good compatibility between the two polymers. This method opens up new paths in sustainable materials research for the synthesis of conductive polymeric composites. We strongly recommend that our green process and the related composite be used for the preparation and fabrication of electronic devices, antistatic coatings, gas sensors, and energy-storage systems. In addition, the composite can be utilized as an effective conductive filler for the preparation of highly transparent conductive thin films.Conflicts of interest
There are no conflicts to declare.References
- A. J. Heeger, Angew. Chem., Int. Ed., 2001, 40, 2591–2611 CrossRef CAS.
- L. V. Kayser and D. J. Lipomi, Adv. Mater., 2019, 31, 1–13 CrossRef PubMed.
- T. V. Vernitskaya and O. N. Efimov, Usp. Khim., 1997, 66, 502–505 Search PubMed.
- H. W. Park, T. Kim, J. Huh, M. Kang, J. E. Lee and H. Yoon, ACS Nano, 2012, 6, 7624–7633 CrossRef CAS PubMed.
- B. Winther-Jensen and K. West, Macromolecules, 2004, 37, 4538–4543 CrossRef CAS.
- K. Namsheer and C. S. Rout, RSC Adv., 2021, 11, 5659–5697 RSC.
- X. Bai, X. Hu, S. Zhou, J. Yan, C. Sun, P. Chen and L. Li, J. Mater. Chem., 2011, 21, 7123–7129 RSC.
- M. G. Han and S. H. Foulger, Chem. Commun., 2005, 3092–3094 RSC.
- H. Ling, R. Chen, Q. Huang, F. Shen, Y. Wang and X. Wang, Green Chem., 2020, 22, 3208–3215 RSC.
- B. L. Groenendaal, F. Jonas, D. Freitag, H. Pielartzik and J. R. Reynolds, Adv. Mater., 2000, 12, 481–494 CrossRef.
- B. G. Heywang and F. Jonas, Adv. Mater., 1992, 4, 116–118 CrossRef.
- P. A. Levermore, R. Jin, X. Wang, L. Chen, D. D. C. Bradley and J. C. De Mello, J. Mater. Chem., 2008, 18, 4414–4420 RSC.
- J. Kim, J. Lee, J. You, M. S. Park, M. S. Al Hossain, Y. Yamauchi and J. H. Kim, Mater. Horiz., 2016, 3, 517–535 RSC.
- S. Nair, E. Hsiao and S. H. Kim, J. Mater. Chem., 2008, 18, 5155–5161 RSC.
- H. Yoon and J. Jang, Adv. Funct. Mater., 2009, 19, 1567–1576 CrossRef CAS.
- J. Yang, R. Zhu, Z. Hong, Y. He, A. Kumar, Y. Li and Y. Yang, Adv. Mater., 2011, 23, 3465–3470 CrossRef CAS PubMed.
- M. Chang, D. Choi, G. Wang, N. Kleinhenz, N. Persson, B. Park and E. Reichmanis, ACS Appl. Mater. Interfaces, 2015, 7, 14095–14103 CrossRef CAS PubMed.
- H. Bai, L. Zhao, C. Lu, C. Li and G. Shi, Polymer, 2009, 50, 3292–3301 CrossRef CAS.
- H. Park, S. Jong, S. Kim, H. Woog, S. Hwan, H. Hee, I. Woo and J. Kim, Polymer, 2013, 54, 4155–4160 CrossRef CAS.
- M. Panapoy, N. Saengsil and B. Ksapabutr, Adv. Mater. Res., 2008, 55–57, 257–260 CAS.
- U. Ali, K. J. B. A. Karim and N. A. Buang, Polym. Rev., 2015, 55, 678–705 CrossRef CAS.
- B. J. Jang and J. H. Oh, Adv. Funct. Mater., 2005, 15, 494–502 CrossRef.
- V. H. Pham, T. T. Dang, S. H. Hur, E. J. Kim and J. S. Chung, ACS Appl. Mater. Interfaces, 2012, 4, 2630–2636 CrossRef CAS PubMed.
- O. S. Kwon, S. J. Park, H. W. Park, T. Kim, M. Kang, J. Jang and H. Yoon, Chem. Mater., 2012, 4, 4088–4092 CrossRef.
- S. H. Ryu, S. Kim, H. Kim, S. O. Kang and Y. H. Choa, RSC Adv., 2015, 5, 36456–36460 RSC.
- X. Zhao, J. P. Cao, J. Zhao, G. H. Hu and Z. M. Dang, J. Mater. Chem. A, 2014, 2, 10614–10622 RSC.
- M. J. Sanchis, B. Redondo-Foj, M. Carsí, P. Ortiz-Serna, M. Culebras, C. M. Gómez, A. Cantarero and R. Muñoz-Espí, RSC Adv., 2016, 6, 62024–62030 RSC.
- H. Abu Hassan Shaari, M. M. Ramli, M. N. Mohtar, N. Abdul Rahman and A. Ahmad, Polymers, 2021, 13, 1939–1966 CrossRef CAS PubMed.
- A. Laforgue and L. Robitaille, Chem. Mater., 2010, 22, 2474–2480 CrossRef CAS.
- T. Pisuchpen, N. Keaw-on, K. Kitikulvarakorn, S. Kusonsong, Y. Sritana-anant, P. Supaphol and V. P. Hoven, Eur. Polym. J., 2017, 96, 452–462 CrossRef CAS.
- S. Nair, E. Hsiao and S. H. Kim, Chem. Mater., 2009, 21, 115–121 CrossRef CAS.
- M. A. Khan and S. P. Armes, Langmuir, 1999, 15, 3469–3475 CrossRef CAS.
- T. Steen, K. West, O. Hassager and N. B. Larsen, Synth. Met., 2006, 156, 1203–1207 CrossRef.
- H. Pang, L. Xu, D. X. Yan and Z. M. Li, Prog. Polym. Sci., 2014, 39, 1908–1933 CrossRef CAS.
- M. Wang, P. Baek, A. Akbarinejad, D. Barker and J. Travas-Sejdic, J. Mater. Chem. C, 2019, 7, 5534–5552 RSC.
- D. W. Hatchett and M. Josowicz, Chem. Rev., 2008, 108, 746–769 CrossRef CAS PubMed.
- X. Lu, W. Zhang, C. Wang, T. Wen and Y. Wei, Prog. Polym. Sci., 2011, 36, 671–712 CrossRef CAS.
- H. Khatoon and S. Ahmad, J. Ind. Eng. Chem., 2017, 53, 1–22 CrossRef CAS.
- K. Deshmukh and G. M. Joshi, J. Mater. Sci.: Mater. Electron., 2015, 26, 5896–5909 CrossRef CAS.
- M. Yildirim, G. E. Demir, A. Çaǧlar, U. Cengiz and I. Kaya, Prog. Org. Coat., 2016, 97, 254–260 CrossRef CAS.
- F. M. Huijsa, F. F. Vercauterenb, B. De Ruiterb and D. Kalicharan, Synth. Met., 1999, 102, 1151–1152 CrossRef.
- T. S. H. Leong, G. J. O. Martin and M. Ashokkumar, Ultrason. Sonochem., 2017, 35, 605–614 CrossRef CAS PubMed.
- M. D. Tzirakis, R. Zambail, Y. Z. Tan, J. W. Chew, C. Adlhart and A. Honciuc, RSC Adv., 2015, 5, 103218–103228 RSC.
- M. Ashokkumar, SpringerBriefs in Green Chemistry for Sustainability, 2016, 17–40 Search PubMed.
- K. Nakabayashi, H. Yanagi and M. Atobe, RSC Adv., 2014, 4, 57608–57610 RSC.
- R. Asami, M. Atobe and T. Fuchigami, J. Am. Chem. Soc., 2005, 127, 13160–13161 CrossRef CAS PubMed.
- Y. Hirai, M. Koshino, Y. Matsumura and M. Atobe, Chem. Lett., 2015, 44, 1584–1585 CrossRef CAS.
- K. Nakabayashi, F. Amemiya, T. Fuchigami, K. Machida, S. Takeda, K. Tamamitsu and M. Atobe, Chem. Commun., 2011, 47, 5765–5767 RSC.
- K. Nakabayashi, M. Kojima, S. Inagi, Y. Hirai and M. Atobe, ACS Macro Lett., 2013, 2, 482–484 CrossRef CAS.
- R. Asami, T. Fuchigami and M. Atobe, Langmuir, 2006, 22, 10258–10263 CrossRef CAS PubMed.
- M. K. Li and H. S. Fogler, J. Fluid Mech., 1978, 88, 499–511 CrossRef CAS.
- M. K. Li and H. S. Fogler, J. Fluid Mech., 1978, 88, 513–528 CrossRef CAS.
- S. R. Reddy and H. S. Fogler, J. Phys. Chem., 1980, 84, 1570–1575 CrossRef CAS.
- A. M. Master, M. E. Rodriguez, M. E. Kenney, N. L. Oleinick and A. Sen Gupta, J. Pharm. Sci., 2010, 99, 2386–2398 CrossRef CAS PubMed.
- A. B. D. Nandiyanto, A. Suhendi, T. Ogi, T. Iwaki and K. Okuyama, Colloids Surf., A, 2012, 396, 96–105 CrossRef CAS.
- Q. Zhao, R. Jamal, L. Zhang, M. Wang and T. Abdiryim, Nanoscale Res. Lett., 2014, 9, 1–9 CrossRef CAS PubMed.
- N. Paradee and A. Sirivat, Polym. Int., 2014, 63, 106–113 CrossRef CAS.
- J. W. Choi, M. G. Han, S. Y. Kim, S. G. Oh and S. S. Im, Synth. Met., 2004, 141, 293–299 CrossRef CAS.
- V. K. Thakur, D. Vennerberg, S. A. Madbouly and M. R. Kessler, RSC Adv., 2014, 4, 6677–6684 RSC.
- R. Nathawat, A. Kumar, N. K. Acharya and Y. K. Vijay, Surf. Coat. Technol., 2009, 203, 2600–2604 CrossRef CAS.
- E. Yilmaz, H. Sezen and S. Suzer, Angew. Chem., 2012, 124, 5584–5588 CrossRef.
- C. P. M. Khan and S. Armes, Langmuir, 2000, 16, 4171–4179 CrossRef.
- J. S. Sohn, U. M. Patil, S. Kang, S. Kang and S. C. Jun, RSC Adv., 2015, 5, 107864–107871 RSC.
- E. Mitraka, M. J. Jafari, M. Vagin, X. Liu, M. Fahlman, T. Ederth, M. Berggren, M. P. Jonsson and X. Crispin, J. Mater. Chem. A, 2017, 5, 4404–4412 RSC.
- Y. Yan, P. Zhang, Z. Qu, M. Tong, S. Zhao, Z. Li, M. Liu and Z. Lin, Nano Lett., 2020, 20, 7662–7669 CrossRef CAS PubMed.
- M. Liu, P. Zhang, Z. Qu, Y. Yan, C. Lai, T. Liu and S. Zhang, Nat. Commun., 2019, 10, 3917–3928 CrossRef PubMed.
- Y. Yan, S. Liang, X. Wang, M. Zhang, S. M. Hao, X. Cui, Z. Li and Z. Lin, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, 1–7 Search PubMed.
- X. Feng, J. Yao, H. Li, Y. Fang, Y. Yoneyama, G. Yang and N. Tsubaki, Chem. Commun., 2019, 55, 1048–1051 RSC.
- K. H. Liao, S. Aoyama, A. A. Abdala and C. Macosko, Macromolecules, 2014, 47, 8311–8319 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Zeta potential distributions of PMMA latex and EDOT emulsion, and EDX spectra of PMMA-PEDOT composites. See DOI: 10.1039/d2ma00027j |
| This journal is © The Royal Society of Chemistry 2022 |