
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interface modification of NASICON-type Li-ion conducting ceramic electrolytes: a critical evaluation
Nurbol
Tolganbek
a,
Assel
Serikkazyyeva
a,
Sandugash
Kalybekkyzy
b,
Madina
Sarsembina
a,
Kiyoshi
Kanamura
c,
Zhumabay
Bakenov
 *ab and
Almagul
Mentbayeva
*a
*ab and
Almagul
Mentbayeva
*a
aDepartment of Chemical and Material Engineering, School of Engineering and Digital Sciences, Nazarbayev University, Nur-Sultan, Kazakhstan. E-mail: almagul.mentbayeva@nu.edu.kz
bNational Laboratory Astana, Nazarbayev University, Nur-Sultan, Kazakhstan
cGraduate School of Urban Environmental Sciences, Tokyo Metropolitan University, Tokyo, Japan
First published on 22nd February 2022
Abstract
This review article focuses on the methods to solve the critical issue of reduction in NASICON-type solid electrolytes such as Li1+xAlxTi2−x(PO4)3 and Li1+xAlxGe2−x(PO4)3 by Li metal. The formation of a reduced phase on the surface of solid electrolytes degrades ionic conductivity. Numerous research efforts were devoted to resolve this issue by depositing various types of artificial protecting layers on the materials’ surface. Inorganics such as lithium salts, oxides, and composites, solid polymer electrolytes, as well as hybrid composite membranes have been applied to protect the material from the side reaction by lithium metal. Besides prevention, these layers improved the interface between solid electrolyte and electrode. Particularly, a coating with a polymer layer prevented the lithium dendrite growth by evenly distributing lithium. The assessments of these layers and their effect on the performance of solid electrolytes are discussed to establish a pathway for further improvement of solid-state electrolytes and encourage their implementation in all-solid-state Li metal batteries. Through this critical evaluation, the present review provides a summary of the most recent works in this area, allowing more efficient and target-oriented research on improving solid-state ion conductors and their interfaces.
Introduction
The ever growing ecological concerns arising from exhaust emissions from fossil fuel based vehicles have imposed more strict regulations and requirements on the car industry enforcing the production of electricity powered and hybrid electric vehicles (EV, HV).1,2 EVs are mostly powered by lithium-ion batteries (LIB), the history of which started with the development and commercialization of this technology by Sony in 1990.3 The advantages of LIBs such as high energy density, long cycle life and no memory effect made them superior compared to other battery technologies.4–8 This enabled LIB application in smaller electronics, electric/hybrid vehicles and stationary energy storage systems. However, the further expansion of LIB's market is hindered by a significant number of safety concerns, which is mainly resulting from the use of organic solvent-based liquid electrolytes.9–11 In addition, liquid electrolytes may cause leakage problems. Taking into consideration these weaknesses of current commercial lithium ion batteries, the approaches for new battery technologies are under development. These so-called next generation Li-ion batteries rely on the design and advancement of solid electrolytes12–14 and cathode materials.15 Therefore, various solid electrolytes based on ion conducting polymers and ceramics have been developed. These solid electrolytes showed many advantages, for instance, a high lithium transference number (t+) value close to one superior to liquid organic electrolytes characterized with a t+ value in the range of 0.2–0.5.16–18 Among the other benefits are increased lifetime and improved safety of the battery towards high operating potential.19–23 Moreover, solid electrolytes have enhanced thermal stability and diminished flammability.24 By all means, rapid preparation and implementation of solid electrolytes to replace liquid electrolytes will be a breakthrough in the progress of Li-ion batteries. Currently, most of the developed and advanced solid polymer electrolytes are based on poly(ethylene oxide) (PEO),25 poly(acrylic acid) (PAA),26 poly(vinylidene fluoride) (PVdF),27 poly(methyl methacrylate) (PMMA),27etc.28 Promising representatives of ceramic-based solid electrolytes are LISICON-type,29,30 argyrodite,31,32 garnet,33–35 Li-nitride,36,37 perovskite,38–40 antiperovskite41,42 and NASICON-type.43–46 Despite a large number of solid electrolytes being investigated, extensive experimental studies still have been conducted to develop electrolytes with increased ionic conductivity due to structural and compositional tuning.47 Certainly, with all provided preferences, they are promising to advance the next generation batteries, and one of the most up-and-coming solid electrolytes is NASICON-type ceramic materials. They have the general formula of LiM2(PO4)3,48 where M can be a transition metal such as zirconium (Zr),49,50 hafnium (Hf),51 germanium (Ge),52,53 and titanium (Ti).54The framework of NASICON structured crystals has a rhombohedral unit cell with the space group of R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c. The crystal of LiM2(PO4)3 consists of MO6 octahedra joined in the corner with PO4 tetrahedra in an alternating sequence.55–57 The ionic conductivity of LiTi2(PO4)3 and LiGe2(PO4)3 materials is insufficient, and hence other transition metals are introduced into the structure in order to improve it. This was achieved by a partial substitution of titanium and germanium with d-elements that influenced the lithium ion migration and bottleneck parameters within the crystal structure.47 Because of the size of the dopants, the formula was revised to Li1+xM4+2−xM′3+x(PO4)3, where M is Zr, Hf, Sn, Ge and Ti and M’ is Cr, Ga, Sc, Y, In, La and Al.52,58,59 One of the most successful dopants that had an encouraging influence on ionic conductivity was aluminum.60 The solid electrolytes with the composition of Li1+xAlxTi2−x(PO4)3 (LATP) and Li1+xAlxGe2−x(PO4)3 (LAGP) have the highest room temperature ionic conductivity of ∼10−3 S cm−1.61–64 The ionic conductivity depends on the dopant level as well, and it should not exceed 15% to prevent the formation of a secondary phase.52,55,58–60 The addition of the aliovalent Al3+ dopant allowed the structure to accommodate more lithium. The increase of lithium content in the structure positively affected the overall ionic conductivity of the material. A summary of the designed LATP materials with various dopants and their ionic conductivity is shown in Table 1.
c. The crystal of LiM2(PO4)3 consists of MO6 octahedra joined in the corner with PO4 tetrahedra in an alternating sequence.55–57 The ionic conductivity of LiTi2(PO4)3 and LiGe2(PO4)3 materials is insufficient, and hence other transition metals are introduced into the structure in order to improve it. This was achieved by a partial substitution of titanium and germanium with d-elements that influenced the lithium ion migration and bottleneck parameters within the crystal structure.47 Because of the size of the dopants, the formula was revised to Li1+xM4+2−xM′3+x(PO4)3, where M is Zr, Hf, Sn, Ge and Ti and M’ is Cr, Ga, Sc, Y, In, La and Al.52,58,59 One of the most successful dopants that had an encouraging influence on ionic conductivity was aluminum.60 The solid electrolytes with the composition of Li1+xAlxTi2−x(PO4)3 (LATP) and Li1+xAlxGe2−x(PO4)3 (LAGP) have the highest room temperature ionic conductivity of ∼10−3 S cm−1.61–64 The ionic conductivity depends on the dopant level as well, and it should not exceed 15% to prevent the formation of a secondary phase.52,55,58–60 The addition of the aliovalent Al3+ dopant allowed the structure to accommodate more lithium. The increase of lithium content in the structure positively affected the overall ionic conductivity of the material. A summary of the designed LATP materials with various dopants and their ionic conductivity is shown in Table 1.
| Model | Composition | Ionic conductivity, S cm−1, at RT | Ref. |
|---|---|---|---|
| LiTi2(PO4)3 | LiTi2(PO4)3–0.2Li2BO3 | 3 × 10−4 | 65 |
| Li1.3Al0.3Ti1.7(PO4)3 | 7 × 10−4 | 48 | |
| Li1.3Sc0.3Ti1.7(PO4)3 | 7 × 10−4 | 66 | |
| 100[Li1.5Cr0.5Ti1.5(PO4)3]–5SiO2 | 2.14 × 10−2 | 43 | |
| 2[Li1.4Ti2Si0.4P2.6O12]–AlPO4 | 1.3 × 10−3 | 67 | |
| LiGe2(PO4)3 | Li1.5Al0.5Ge1.5(PO4)3 | 4 × 10−4 | 68 |
| Li1.5Al0.5Ge1.5(PO4)3–0.05Li2O | 7.25 × 10−4 | 69 | |
| Li1.5Al0.4Cr0.1Ge1.5(PO4)3 | 6.65 × 10−3 | 70 |
Although NASICON has encouraging dominance over other types of electrolytes regarding chemical and electrochemical stability and ionic conductivity, LATP and LAGP ceramics easily reduce (Me4+ to Me3+) while contacting with lithium metal forming a lithium hindering phase.44,71,72 Interestingly many other researchers working on solid electrolytes have also reported the electrochemical or chemical instability of inorganic solid electrolytes due to Li metal. Particularly, titanium, germanium, tantalum, and sulfide containing all-solid-state electrolytes demonstrated dramatic outcomes.73–75 For instance, LATP and LAGP electrolytes have high reactivity with Li metal, instantly forming a lithium ion blocking but high electron conductive layer between electrolyte and electrode (Fig. 1), which is described as a mixed (ionic/electronic) conducting interphase (MCI).75 The general reduction reactions can be represented as follows:76–78
| 2LiGe2(PO4)3 + 4Li ⇒ 3GeO2 + 6LiPO3 + Ge |
| LiTi2(PO4)3 + Li ⇒ Li2Ti2(PO4)3 |
 | ||
| Fig. 1 LATP pellets before and after the contact with the Li metal chip and SEM cross-section image of LATGP after contact with Li metal.75 | ||
In the case of mixed (both electronic and ionic) conduction, the interfacial phenomena are not straightforward, rather they are complex and complicated.74 While Hartman et al. stated that the formation of MCI was not continuous,75 Zhu et al. observed volumetric expansion between electrode and electrolyte that led to high resistance.79 The reactivity of LATP and LAGP was calculated based on free enthalpy of reaction with lithium metal at different temperatures. The result showed that Ge4+ was completely reduced to Ge0 while Ti4+ was reduced to Ti3+. The chemical transformation was identified at the electrolyte and electrode interface during the lithiation of LATP, and experimental and calculation studies proved that the MCI had enhanced electronic conductivity but the chemical diffusion coefficient of lithium was reduced.75,79 The formation of an additional phase that hinders lithium hopping within the structure can be resolved by designing a protection layer between the electrolyte and lithium metal. A protective artificial layer between ceramic electrolyte and Li metal can suppress chemical reaction and dendrite formation, improve interfacial conductivity and uniformity, and enhance cyclability of a cell. Introduction of these artificial buffer layers has been carried out using various types of lithium ion conducting materials. It should be taken into consideration that implementation of these artificial layers is also of current interest for other advanced types of solid electrolytes such as garnet and sulfide.80,81 The essence of addition of these layers is mainly to prevent dendrite growth and to improve the interface between the solid phases of electrolyte and electrode. Recently, a number of review papers have been published focusing on the challenges of advanced electrolytes and issues concerning the interfaces for all-solid-state batteries.82–85 Despite the existence of numerous reviews on the analysis of solid electrolytes, a particular review work on determining the problems of NASICON-type electrolyte reduction phenomena has not been reported, while resolving this dramatic issue of LATP and LAGP has gained a considerable amount of attention in recent years. Therefore, a comprehensive analytical summary of works on prevention of undesired reduction of LATP and LAGP and their improvements is necessary. This report has systematically compiled the findings on overcoming the above-mentioned issue by either depositing various types of novel materials on the solid electrolytes via different coating techniques or designing composite electrolytes by combining lithium ion conducting materials. Besides, the perspectives of NASICON-type electrolytes are discussed with a broad and extensive point of view.
Inorganic protective layers
Improvements without compromising the safety and ionic conductivity of solid electrolytes are the main tasks in material design in order to obtain compelling stable performance and increased lifespan of a cell. Yet, physical contact between electrode and solid electrolyte requires further advancement for constant lithium ion transport from electrode to electrolyte and vice versa through intermediate phase. The progress in this field also aids in preventing the side reaction of LATP and LAGP with Li metal. In recent years, prohibition of the side reactions between Li-ion conducting NASICON-type solid electrolytes and Li metal has been carried out by applying a wide range of lithium ion conducting materials as an interlayer. These interlayers are designed using either inorganics, polymers or their combinations. The choice of inorganics such as lithium salts, oxides, and other types of solid electrolytes as the buffer layer is justified by their robustness and high chemical and mechanical stability towards the active lithium metal. Most importantly, these interlayers act as a physical barrier and should be able to form conducting phases of Li2S, Li2O, LiF, Li3N, and LiAl5O8 during the initial cycles with lithium that can prevent the mixed conductive interphase (reductive decomposition) formation. These interlayer compounds can shield solid electrolytes from reductive degradation in case they are sufficiently electron insulating and reduce mechanical stress amplification at rough and faulty surface enabling lithium plating to be more stable.74,86,87 Actually, the widely used inorganic salt as the interlayer was lithium phosphate (Li3PO4). Initially, the salt was utilized as an artificial SEI layer in Li/LiFePO4 cells to suppress lithium dendrite formation and improve cyclability of the battery with Li metal anode (Fig. 2a).88 Young's modulus of the artificial SEI was generally assumed to be higher than 6 GPa which was satisfactory to prevent lithium dendrite growth. The authors stated that lithium ion conductivity of the salt was sufficient and it positively influenced the kinetics of a full cell.88 Taking all these advantages into consideration, the salt showed great potential as a thin film interlayer between LATP and Li metal to improve the interface stability along with the prevention of dendrite growth.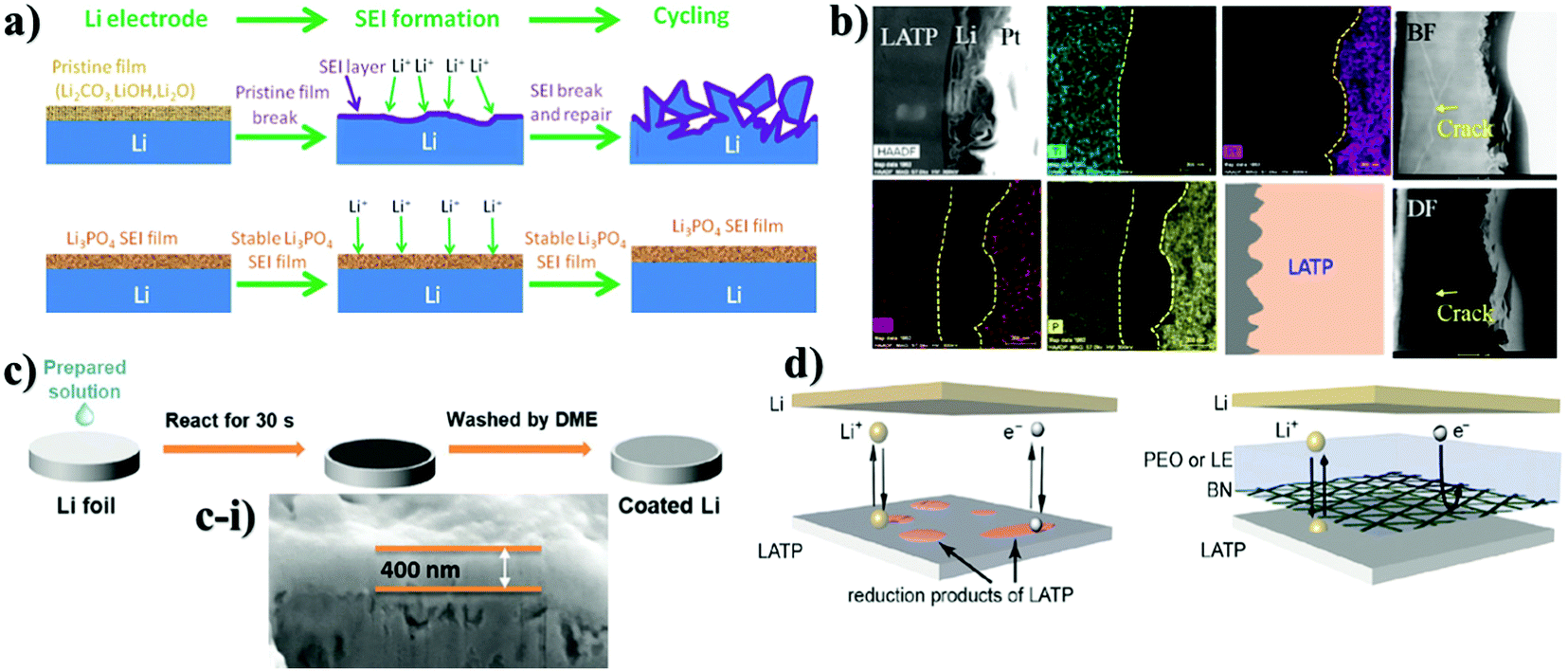 | ||
| Fig. 2 Illustration of the deposition of various inorganic protective layers on LATP and their TEM and SEM images: (a) explanation of the role of the SEI layer;88 (b) TEM-EDS of Li3PO4 deposited LATP after 100 cycles;89 (c) nanocomposite layer formation through the dropping of solution; (c-i) cross-sectional view of the nanocomposite layer on LATP;90 (d) scheme of BN and polymer combined thin layer on LATP.91 | ||
There are a few works on lithium salts deposited as a buffer layer for NASICON-structured electrolytes as well. Basically, the distinctness of these reports was the implementation of protective substances by two diverse methods resulting in different thicknesses. Fig. 2 illustrates the cell assembly and cross sectional images of the interlayers. J. Liu et al. designed an interlayer through a one-step reaction process where polished Li metal was placed in a phosphoric acid solution to form a 200 nm artificial SEI layer.92 On the other hand, other authors (Y. Liu et al.) deposited the salt with a thickness of 15 nm on LATP by an atomic layer deposition (ALD) technique.89 The effect of these SEI layers was determined via electrochemical stripping and plating tests of symmetric cells and their performing time period. The outcome of these reports showed that the cells behaved totally different. The cell where the layer was coated through a chemical reaction performed stably for 200 hours.88 It is worth noting that during the cell preparation, a liquid electrolyte was also added. In the case of another cell, symmetric cells performed for 600 hours; however, starting from 50 hours, the sample began to show instability when the overpotential started to increase, showing definitely the same performance as bare LATP.89 According to the authors, the poor performance of the salt was explained by lower ionic conductivity of Li3PO4 compared to LATP and degradation of the thin layer during repeated Li stripping and plating at high current density (above 0.5 mA cm−2) (Fig. 2b). Nevertheless, the different influence of Li3PO4 on the performance of the solid electrolyte can be explained either by its thickness or deposition technique. However, addition of a small amount of a commercial liquid electrolyte has to be taken into consideration in the work presented by J. Liu, where steady performance can be attributed to the effect of the liquid electrolyte.92 Another possible explanation for the constant behavior of the cell could be the thicker layer which was durable enough for longer cyclability. An interesting approach where the solution containing LiBF4 and Mg(ClO4)2 was cast on lithium to form metal fluorides as a protective layer was reported by Yang et al. (Fig. 2c).90 The reaction between lithium metal and applied solutions enabled the production of three substances of LiF, MgF2 and B2O3 which was confirmed via X-ray photoelectron spectroscopy (XPS). Unfortunately, the chemistry behind the reaction is not clear yet. Generally, this nanocomposite layer was formed within 30 seconds and had a thickness of approximately 400 nm, which was estimated by SEM (Fig. 2c-i). The benefits of the process were explained by facile and instant layer formation, and the great contribution of metal fluorides in the formation of a stable SEI layer in conventional lithium-ion batteries. The effect of coating on electrochemical performance was tested via EIS with a non-blocking lithium electrode, a Li stripping and plating test in a symmetric cell, and galvanostatic charge and discharge of a full cell with LiFePO4 cathode. During the cell preparation, the ether-based liquid electrolyte of lithium bis(fluorosulfonyl)imide (LiFSI) in dimethoxy ethane (DME) was applied to the surface of LATP. Consequently, the spectra of electrochemical impedance showed a dramatic decrease of resistivity and the symmetric cell exhibited 300 hours of continuous lithium stripping and plating.90 Application of nanocomposites was stated in the research of Cheng et al. as well.91 In this work, mechanically robust and chemically inert film of boron nitride (BN) was deposited for interfacial protection against reduction of Li1.3Al0.3Ti1.7(PO4)3 (LATP by low pressure chemical vapor deposition (LPCVD) technique) (Fig. 2d). Moreover, BN was combined with polyethylene oxide (PEO) and coated with a thickness of ∼1–2 μm to create a hybrid protective layer. This layer served as a barrier, which prevented LATP reduction and demonstrated steady cycling behavior of around 500 hours in a symmetric cell of Li/LATP/Li. Contrastingly, the sample with bare LATP was degraded after 81 h.91 Intensive research on stabilizing the interface of the Li1.3Al0.3Ti1.7(PO4)3 solid electrolyte towards lithium metal was undertaken through oxides of d-elements and their doping derivatives as well. The choice of oxides depended on their robustness and high mechanical and chemical stability towards lithium metal. The deposition process and cross-sectional TEM and SEM images with electrochemical performance are shown in Fig. 3. Y. Lui et al. used Al2O3 as an artificial layer, and also utilized ALD to deposit on LATP with a thickness of 15 nm (Fig. 3a).89 A work by Bai et al. introduced a 60 nm alumina doped ZnO (AZO) (Fig. 3b) interlayer via the magnetron sputtering technique (MST).93 Hao et al. utilized 200 nm ZnO (Fig. 3c-i) as a protective layer for LATP by MST as well (Fig. 3c).94 Significant differences in the performance between Li3PO4 and oxide coatings on LATP can be observed. A large overpotential is monitored in the initial cycles due to the insulating nature of the oxide materials. Overcoming this issue was carried out by electrochemical precycling and heating the cells to form a lithium ion conducting phase. Lui et al. stated that the Li–Al–O conducting layer formed only after 50 hours, and further performance up to 600 hours was balanced.89 Bai et al. believed that this activation period of time could be solved by combining two oxides where interfacial resistivity between LATP and an artificial layer can be suppressed. The symmetric cell of AZO coated LATP showed 1000 hours of permanent performance (Fig. 3b).93 Although Bai et al. claimed insufficiency of bare ZnO as the buffer layer, a study conducted by Hao and co-workers with the same sputtering technique and material proved otherwise by achieving an outstanding performance of 2000 hours for symmetric cells (Fig. 3c-i).94 According to the study, the lithiophilicity of ZnO allowed it to react with lithium and then form an SEI layer (Fig. 3c). The activation of the SEI layer on the cell required heating at 170 °C where the reaction between lithium metal and oxide material occurred. Oxide layers proved to increase the lifetime of LATP and protect it from reacting with lithium, but there were some issues to achieve that. For instance, it took some time to form the lithium ion conducting phase. Some researchers tried to combine LATP with additional lithium containing chemicals that also might help to achieve an improved cyclability.
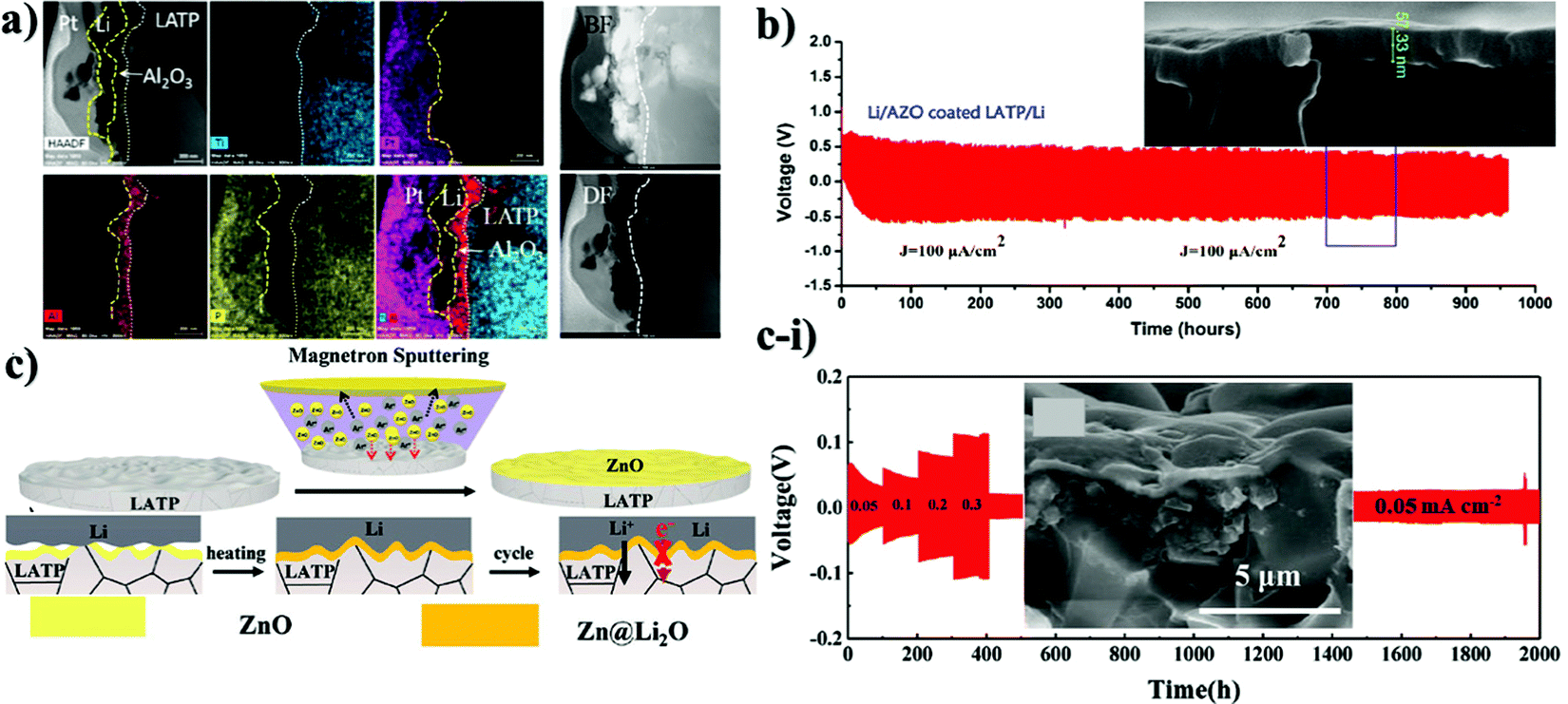 | ||
| Fig. 3 Illustration of the deposition of various inorganic protective layers on LATP and their TEM and SEM images as well as Li stripping and plating results: (a) TEM-EDS of Al2O3 deposited LATP after 100 cycles;89 (b) SEM image of AZO deposited LATP and its electrochemical performance;93 (c) illustration of magnetron sputtering of ZnO on LATP; (c-i) SEM image of ZnO deposited LATP and the electrochemical behavior of the ZnO deposited LATP symmetric cell with Li.94 | ||
Advancing NASICON-structured materials and solving their problems were also accomplished by using other types of solid electrolytes. For instance, Guhl and co-workers investigated the LIPON interlayer which was grown on the substrates of LATP and LATGP by the magnetron sputtering technique (Fig. 4a).95 The thickness of the LiPON film was 200 nm. A comprehensive stepwise coating technique was accompanied via XPS to confirm interface formation. As it is known that Ge4+ is also unstable towards lithium, the authors stated that due to electrochemical potential differences this metal is more challenging to be reduced by the negative electrode. Thus, Li and colleagues suggested an unusual way to improve the stability between electrode and electrolyte through amorphous and nanocrystalline Li1.5Al0.5Ge1.5(PO4)3 (a-LAGP) as a protective layer on the surface of the LATP solid electrolyte.96 A similar deposition technique to that in the previous report was used to create a thin layer less than 100 nm (Fig. 4b). Yet, these two reports have obtained valuable findings regarding the durability of NASICON-type electrolytes. Despite electrochemical potential differences of Ge4+ and Ti4+, during the continuous lithium ion insertion and extraction the materials went through reduction. As an illustration, nanocrystalline LATGP coated LATP symmetric cells performed for a modest 250 hours, where a linear increase in the overpotential was observed, and Fourier transform infrared (FTIR) spectra showed the presence of a small amount of GeO2, which is the product of reduction.96 The same phenomenon was observed in the case of deposited LiPON on LATP and LATGP. XPS depicted the chemical structure change on the surface of these substrates, possibly indicating that reduction began at the early stages of the deposition process.95 The authors stated that the reduction occurred because of the lithium containing environment during the deposition procedure. The process and electrochemical performance with SEM images are demonstrated in Fig. 4. Furthermore, from the electrochemical analysis result (Fig. 4a-i), it can be clearly seen that increased impedance is related to reduced phase of lithium titanate, and LiPON coating. By all means, the lower ionic conductivity of LiPON corrupted the overall impedance of the system. Yet, interesting information regarding the early reduction phenomena of titanium was obtained, and it could be taken into consideration for further advancements in the field of interface between Li metal and NASICON-type solid electrolytes. Regardless of significant research on LATP, there were a few reports on solving the problem of LAGP too. Exceptionally, the reduction issue of the material was solved by using pure metals. Liu et al. studied an amorphous Ge thin film as a protective layer for Li1.5Al0.5Ge0.5P3O12.78 A thin film of 60 nm was sputtered on the surface of the solid electrolyte with a precision etching coating system (Gatan 682) (Fig. 4c). Cortes and colleagues tested the influence of bare chromium (Cr) as a metallic protective layer for LAGP with a thickness of 30 nm via a Unifilm Sputtering system.97 Compared to Ge metal with 200 hours of performance, Cr deposition demonstrated prolonged cyclability where the symmetric cells reached up to 850 hours of operation. In both cases, a slight increase in the overpotential in the initial cycles could be observed due to the lithium ion insulating properties of metals. In further cycles, it started to decrease due to the formation of a lithium ion conducting phase/alloy. The observation from XPS confirmed that after the Li stripping and plating process, there was a secondary phase formation. Generally, the results from electrochemical analysis indicated that the impedance of Ge coated LAGP with non-blocking electrodes was dramatically lower due to its wettability with lithium metal.78 Therefore, the artificial interface layer provided intimate contact between LAGP electrolyte and Li metal anode. In the case of the Cr protection layer, the role of the metal was only as a physical barrier, which hindered Li plating and stripping.97 Thus, symmetric cells were conditioned to cycle at 0.1 mA cm−2 for 5 min in each direction for 16 hours before electrochemical measurements. Clearly, this pre-electrochemical activation aided in achieving further stable performance without having large overpotentials. Despite its stability the Cr metal suffered from degradation at 0.3 mA cm−2 and higher current densities, which limited the performance of the cells. A outstanding performing time of 1500 hours for the LAGP solid electrolyte was obtained by Matic's98 and Hu's99 (Fig. 4d-ii) research groups. Such performance was delivered by designing a quasi-solid state paste from LAGP and an ionic liquid (IL) (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50) and an interlayer of the Li bis(trifluoromethylsulfonyl)imide/poly(vinylene carbonate) (LiBTFMSI/PVC) polymer electrolyte with SiO2 microspheres (Fig. 4d).98 0.1LiFSI/0.9BIB-FSI was used as the IL.
:50) and an interlayer of the Li bis(trifluoromethylsulfonyl)imide/poly(vinylene carbonate) (LiBTFMSI/PVC) polymer electrolyte with SiO2 microspheres (Fig. 4d).98 0.1LiFSI/0.9BIB-FSI was used as the IL.
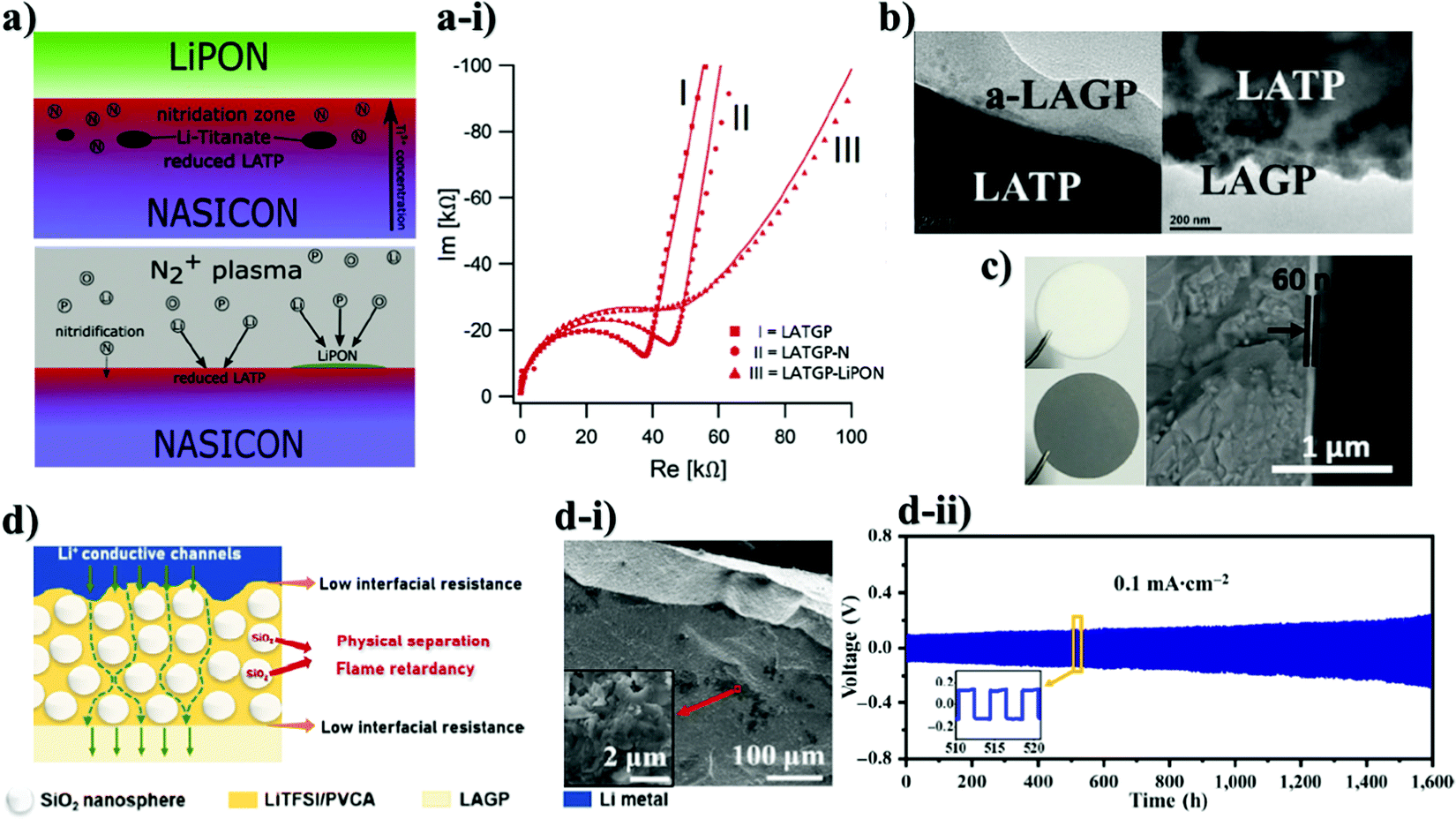 | ||
| Fig. 4 The illustration of the deposition and SEM images of various samples with electrochemical performance: (a) the illustration of the NASICON-LiPON interface;95 (a-i) EIS of LiPON coated LAGTP;95 (b) SEM images of LAGP coated LATP;96 (c) images of pristine and Ge coated LAGP and SEM image of Ge coated LAGP;78 (d) the ionic conducting mechanism in the interface;99 (d-i) SEM images of the hybrid layer with SiO2 nanopraticles;99 (d-ii) result of Li stripping and plating performance.99 | ||
As a result, the combined LAGP-IL created a chemically stable interface with low resistance. Deep analysis revealed the formation of an additional SEI layer because of the reaction between IL and lithium. Yet, the authors believed that the evolution of this layer stabilized the interface between electrolyte and electrode enhancing the ionic conductivity. The double layer allowed the cell to perform at higher current density up to 1 mA cm−2, while the outstanding performance was obtained with 0.3 mA cm−2. The efficient split of LAGP and Li prevented the side reaction and lithium dendrite nucleation. Still, the use of IL is not beneficial in terms of high cost. The hybrid interlayer also resulted in a decrease in the interfacial resistance between lithium metal electrode and LAGP electrolyte where SiO2 physically separated and acted as a filler to reduce the degree of crystallinity of the polymer (Fig. 4d and d-i).99Table 2 provides a summary of inorganic buffer layers and their electrochemical performance with LATP and LAGP.
| Solid electrolyte | Artificial layer | Thickness of the artificial layer | Symmetric cell performance, hours | Full cell performance | Condition | Ref. |
|---|---|---|---|---|---|---|
| LATP | Li3PO4 | 200 nm | 200 | 50 cycles | 0.1C, 95.2% capacity retention, LFP cathode | 92 |
| LATP | Li3PO4 | 15 nm | 100 | 89 | ||
| LATP | Mixture of LiF, MgF2 and B2O3 nanocomposite | 400 nm | 300 | 500 cycles | 0.13 mA cm−2 1.5C, LFP | 90 |
| LATP | BN and PEO hybrid layer | ∼1–2 μm | 500 | 500 cycles | 0.3 mA cm−2, 96.6% capacity retention | 91 |
| LATP | Al2O3 | 15 nm | 600 | 89 | ||
| LATP | AZO | 60 nm | 960 | 50 cycles | 50 cycles at 60 °C at 0.1 mA cm−2 | 93 |
| LATP | ZnO | 200 nm | 2000 | 200 cycles | 0.05 mA cm−2, 200 cycles at 0.02C | 94 |
| LATP | LiPON | 200 nm | 95 | |||
| LATP | a-LAGP | Less than 100 nm | 250 | 0.01 mA cm−2 | 96 | |
| LAGP | Cr | 30 nm | 850 | 0.2 mA cm−2 | 97 | |
| LAGP | Ge | 60 nm | 200 | 0.1 mA cm−2 | 78 | |
| LAGP | IL-LAGP | 1500 | 0.3 mA cm−2 | 98 | ||
| LAGP | (LiBTFMSI/PVC) and SiO2 | 1500 | 100 cycles, LFP, 0.075 mA cm−2 | 0.1 mA cm−2 | 99 |
A considerable number of inorganic materials were introduced as artificial buffer layers to solve the critical problem of LATP and LAGP solid electrolytes’ reduction by Li metal. The role of these layers varies from one to several. Regardless of the protection this layer provides intimate contact between electrode and solid electrolyte that influences the cells' longer cyclability and kinetics. According to many reported research studies, the artificial layers can be designed via various types of materials such as metal oxides, lithium salts, nanocomposites, bare metals and even other solid electrolytes. Each material had unique chemistry and mostly some of them formed alloys and others remained inert with electrodes. To form lithium ion conducting phases, the vast majority of them required pre-cycling or heating beforehand. So far the most promising inorganic compound as a protective layer has been ZnO. The most prolonged cyclability of around 2000 hours was obtained with a lithium symmetric cell. Even though it showed outstanding long performance, ZnO was not stable at high currents. Low current operation applied not only for ZnO, but also to other artificial buffer layers. An increase in current led to decomposition of these layers, leading to poor performance and undesired contact between two solid materials. Another promising interlayer was LAGP-IL quasi-solid material, which exhibited excellent performance under high current density. Due to the formation of the SEI layer on top of the protective layer, it enhanced the cell behavior allowing continuous stripping and plating and evenly distributing lithium flux preventing dendrite growth.
Polymer based protective layers
Along with the inorganic protection layer coatings, there have been numerous works that endeavored to use polymer films that have high ionic conductivity and electron insulating nature as a physical barrier for this purpose as well, which offer additional benefits because of their unique properties such as flexibility, high degree of adhesion and dendrite growth prevention.100,101 The polymers containing Li salts are promising candidates for interface advancements in all-solid-state batteries owing to their high stability towards Li metal and space-charge suppression at the interface.102–104 Salt containing/liquid containing polymer materials wet the surface of Li metal and minimize interfacial resistance and create very intimate contact and fill the gap at the electrolyte and electrode surface.74 Li ions of dissolved Li salts coordinate with the electron-donor groups of the polymer host in a polar polymer matrix. Ion conduction in polymer coating occurs by the electric field which forces ions to hop from one coordinating site to another basically in the amorphous region.105 Various strategies have been applied to address these issues of ion conductive polymers and nowadays there are a wide range of systems with different strategies proposed for application in LIBs.106 The combination of polymers with ceramic electrolytes is also one of the promising ways of solid state electrolyte advancement. Early in order, one of the pioneering groups led by Goodenough presented a polymer/ceramic/polymer sandwich electrolyte.107 A cross-linked poly(ethylene glycol)methyl ether acrylate 3D framework formed a flexible and low-cost membrane which was pressed on either side of LATP pellets. Integrated polymer-ceramic design allowed solving the problem of each electrolyte, where the ceramic blocked anion transport, while the polymer suppressed dendrite nucleation. However, the combination of polymer and ceramic increased the overall resistivity of the cell. Despite the increase in impedance, the prevention of side reactions and dendrite formation led to its feasibility for solid-state batteries. The effect of the polymer was characterized by assembling a full cell with LFP cathode and the cell performed steadily for 640 cycles.107 The sandwich system created many other opportunities in terms of use of other polymer materials. Currently, the most widely investigated polymer material in energy storage application is polyethylene oxide because of its high ionic conductivity, high liquid absorption, flexibility and compatibility with lithium metal.102,104,108 Yet, polymer materials used in lithium batteries met requirements such as high mechanical strength, excellent fire resistance and excellent thermal stability. Thus, there have been works that put stress on solving the problem of the NASICON-type electrolyte towards Li by coating it with PEO.The role of polymer layers and their cross-sectional SEM views with electrochemical behavior are shown in Fig. 5. Cai et al. architected a flexible modified PEO coating on LATP (Fig. 5a and a-i) for ambient-temperature solid state Li batteries.109 Jin et al. engineered the interface based on the multifunctional layer of PEO with LiTFSI salt (Fig. 5b).110 Koizumi et al. designed a comprehensive composite layer out of PEO, an ionic liquid and oxide filler of BaTiO3 (or SiO2) for LAGTP.100 All three reports concentrated on decreasing the interfacial impedance of Li and solid electrolyte along with addressing the side reaction issue. Cai et al. achieved a decrease of interfacial impedance from 30000 to 280 ohm cm−1 by drop casting the polymer on the ceramic pellet. However, the performing period of the symmetric cell was only 200 hours at 0.1 mA cm−2. The cell assembled by Jin and colleagues with the polymer layer showed significant improvements where the reduction of interfacial resistance was monitored and the symmetric cell performed for 400 hours at low current density (0.05 mA cm−2).110 The symmetric cell with the composite interlayer exhibited cycling only for 100 hours with a higher current density of 0.5 mA cm−2, and this was reached at 60 °C.100 There have been reports on other types of complex polymer materials. For instance, Chen et al. tried to resolve the problem of LATP by an ultrathin poly[2,3-bis(2,2,6,6-tetramethylpiperidine-N-oxycarbonyl)-norbornene] (PTNB) polymer coating with a high Li concentration ionic liquid electrolyte (ILE) (0.4LiFSI–0.6Pyr14FSI) on Li metal.114 LATP with PTNB coating performed for nearly 800 hours at 0.1 mA cm−2 current density. On the other hand, the full cell with LiNi0.8Co0.1Mn0.1O2 (NCM811) cathode performed for 200 cycles with a slight decrease in capacity. Yu et al. worked on the coating of LATP ceramic material with a protective polymer layer, the formulation of which was as follows: a mixture of 65.3 wt% poly[bis(2-(2-methoxyethoxy)ethoxy)phosphazene] (MEEP) polymer, 21.7 wt% poly(vinylidene fluoride-co-hexafluoropropylene) (PVDF-HFP) and 13.0 wt% lithium bis(oxalato)borate (LiBOB) (Fig. 5c).111 This composite had a thickness of around 120 μm in dry state. The final assembled symmetric cell showed a stable performance of 1500 hours at 0.13 mA cm−2 current density. The full cell was assembled with Li3V2(PO4)3 and carbon nanotube (CNT) composite cathode. As a result, 500 cycles with 100% coulombic efficiency performance were achieved. In fact, the prolonged time and good electrochemical outcome were achieved at an elevated temperature of 50 °C. Both works demonstrated that polymer coatings on LATP can prevent the side reaction and provide longer cycle life for LATP towards lithium metal. Another work applied one component system of PVDF-HFP which was simply coated on the LATP pellet (Fig. 5d).112 The research's goal was to stabilize the electrolyte by keeping the impedance at one point. By all means, up to 10 days, the sandwiched LATP exhibited the same impedance value. A full cell with LiCoO2 cathode material and modified solid electrolyte performed for over 140 cycles with a discharge capacity of around 145 mA h g−1 at 0.1C. The further development in this field also was researched by Lu and co-workers.113 A LAGP solid electrolyte was covered with poly(acrylic acid-co-maleic acid) (P(AA-co-MA)) via spraying (Fig. 5e). As a lithium source LiCl was utilized. The concentration of the salt also played a crucial role in decreasing the interfacial impedance. Although the interlayer slightly decreased the impedance between the electrolyte and the Li metal, the performance of the symmetric cell was moderate at only 165 hours at 0.7 mA cm−2. This poor performance was due to the negligible changes in the LAGP solid electrolyte during the continuous stripping and plating. The authors claimed that this reaction led to the reduction of Ge4+ to Ge0, and some of them formed alloys with Li metal. Despite obtaining lower interfacial resistivity, the performance of these interlayers still requires further improvements in order to increase the stability towards lithium metal. The use of PEO coated LATP has gained significant attention in the field of lithium–sulfur batteries as well. The coating of the protection layer not only protects LATP from reduction, but also addresses the problem of the polysulfide shuttle effect in sulfur-containing batteries. The illustrations of battery components and SEM images of interlayers with electrochemical results are depicted in Fig. 6. Wang et al. did research on combination of solid electrolytes and sulfur-based electrodes where NASICON-type ceramic and sulfurized polyacrylonitrile cathodes were used (Fig. 6a).101 The direct contact between LATP and anode was solved via PEO that contained LiTFSI (Fig. 6a-i). The solid polymer electrolyte (SPE) precursor solution was simply drop coated onto LATP and dried. This kind of cell with lithium metal on both sides performed for more than 250 hours, and according to the authors it prevented the dendrite growth. Further, a full solid cell with sulfur/polyacrylonitrile (SPAN) cathode had an initial discharge capacity of 1793 mA h g−1, which dropped to 784 mA h g−1 after 120 cycles. However, the electrochemical characterization of solid cells was conducted at 75 °C, where PEO operates beneficially well. Manthiram's group tried to solve the problem of sulfur batteries by introducing LATP as the electrolyte which was modified by a layer of polymer with intrinsic nanoporosity (PIN) (Fig. 6b-i and b-ii).115 PIN with precisely designed nanoporosity had primarily roles such as an electrical insulator and provider of a lithium ion transport path, and at the same time LATP aided in suppressing the polysulfide shuttle effect.
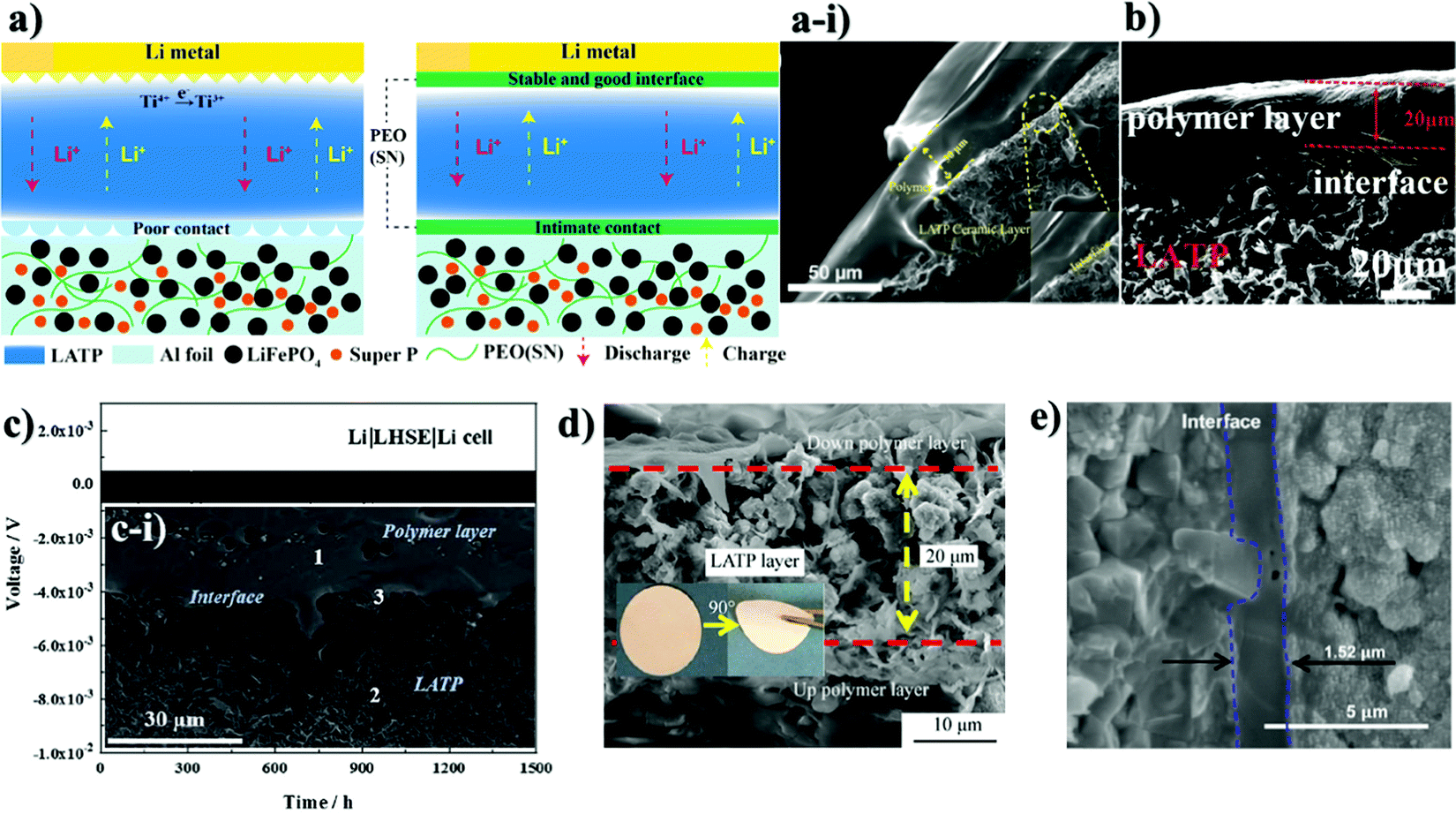 | ||
| Fig. 5 The cross section illustration and SEM images of various samples with electrochemical performance: (a) the illustration of lithium ion conduction on PEO coated LATP; (a-i) PEO coated LATP cross section;109 (b) SEM image of the PEO interlayer;110 (c) illustration of polymer coating and galvanostatic profile of the full cell;111 (d) SEM image of MEEP-PVDF-HFP polymer coating on LATP;112 (e) (P(AA-co-MA)) interlayer.113 | ||
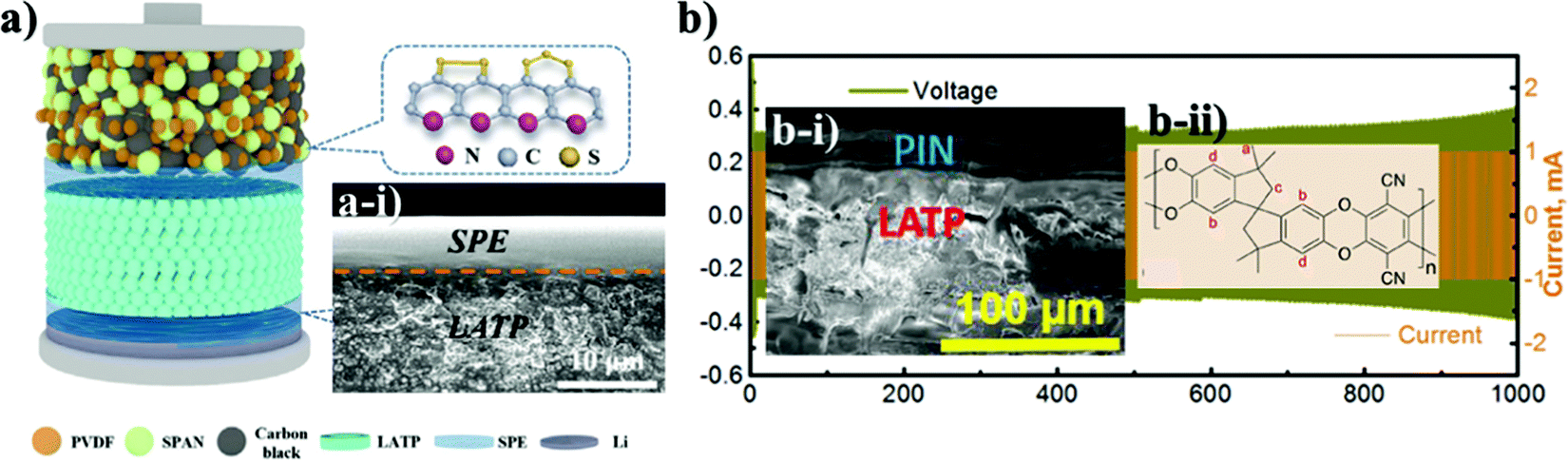 | ||
| Fig. 6 SEM images and illustrations of various cells with their electrochemical performance: (a) solid polymer layer on LATP; (a-i) illustration of cell characterization; (a-ii) electrochemical behavior of the cell;101 (b) the formula of PIN; (b-i) PIN coated LATP; (b-ii) the symmetric cell performance of PIN coated LATP.115 | ||
PIN was deep coated on LATP (Ohara Inc.), where the bond between them was mechanical anchoring and interhooking (Fig. 6b-i). Yet, a small amount of liquid electrolyte was added to the 20–25 μm thick layer of PIN and a cyclability of around 1000 hours for a Li/PIN/LATP/PIN/Li symmetric cell was achieved with stable potential (Fig. 6b). Electrochemical results are depicted in Table 3.
| Solid electrolyte | Artificial layer | Thickness of the layer | Striping and plating time | Full cell performance | Ref. |
|---|---|---|---|---|---|
| CR, capacity retention. CE, Coulombic efficiency. | |||||
| LATP_8 wt% of Nb2O5 | PEO | 30 μm | 200 h at 0.5 mA cm−2 | 160 mA g h−1 at 0.1C, LFP, CR = 82% | 109 |
| LATP | PEO | 20 μm | 400 h at 0.05 mA cm−2 | 139.5 mA g h−1 at 0.05C | 110 |
| LAGTP | PEO_BaTiO3 (or SiO2) | 50 μm | 100 h at 0.5 mA cm−2 | 100 | |
| LATP | PTNB | 792 h at 0.1 mA cm−2 | 200 mA g h−1 at 0.05C (Fig. 5d), CR is 99% | 114 | |
| LATP | MEEP_PVDF-HFP and LiBOB salt | ∼120 μm | 1500 h at 0.13 mA cm−2 and 50 °C | 125 mA g h−1 at 0.2C, CE is 100% | 111 |
| LATP | PVDF-HFP | ∼2 μm | 145 mA g h−1 at 0.1C | 112 | |
| LAGP | P(AA-co-MA) | ∼1.5 μm | 55 h at 0.7 mA cm−2 | 113 | |
| LATP | PEO | ∼7 μm | 220 h at 0.25 mA cm−2 at 75 °C | 1793 mA h g−1 at 75 °C | 101 |
| LATP | PIN | 20–25 μm | 1000 h at 1 mA cm−2 | 1110 mA h g−1 at 0.1C | 115 |
Polymer coating is considered to be a moderate choice towards preventing the reduction of solid electrolytes in contact with the Li anode. The reasons for such candidacy are wettability, ability of polymers to adhere well on the surface of ceramics and presence of lithium ions in their matrices for high lithium ion conductivity. Comparably, except in a few cases, polymer coatings performed weaker than inorganic protection layers. However, it should be taken into consideration that currently available polymer electrolytes are limited by low ionic conductivity and high operating temperature and judging their perspectives as interlayers is still questionable. Still, utilization of NASICON type electrolytes with polymer coating greatly attributed to the progress of Li–S batteries, where ceramics only conducted lithium ions and blocked the migration of polysulfides, and polymer coating was applied to advance the interface between the ion conducting electrolyte and the lithium metal. The robust nature of inorganics steadily protects while the polymer coating deteriorates at certain current densities. Yet, the same issue was reported for inorganic buffer layers too. Compared to inorganics, in most studies, it was revealed that polymer coatings have a superior advantage over inorganics where they evenly distribute lithium deposition by preventing lithium dendrite nucleation.
Composite electrolytes
The application of a NASICON-type electrolyte in an all-solid-state energy storage system could be also realized through blending with lithium ion conducting polymer materials. By doing so, the direct contact of ceramic and lithium metal is compensated by polymers. To overcome the drawbacks of single-phase inorganic and polymer solid-state electrolytes (SSEs) while maximizing their benefits, several recent studies have shown that combining inorganics and polymers to form composite solid-state electrolytes (CSSEs) resulted in considerable performance improvement.116 Interestingly, there is a debate regarding the role of each component in ionic conductivity. Some say that enhancement of ionic conductivity in composite electrolytes is due to the reduced crystallinity of the polymer matrix by fillers (ceramics) while others believe that the role of these active fillers is more complicated. Thus, among researchers several hypotheses have been proposed. The first one is based on the formation of a highly conductive interface between polymer matrix and inorganic fillers which acts as a primary conduction pathway for Li-ions. The second hypothesis suggests that ion conduction occurs through a continues network of ceramic particles that are well linked to each other where the Li ion can hop without obstacles.117 A study conducted by Lim and colleagues claimed that an increase in ionic conductivity and an improvement in the thermal stability of solid electrolytes were achieved by adding ceramics (NASICON) in the polymer matrix or by copolymerization with other materials.115 These two modifications aid in decreasing the glass transition temperature and slowing polymer crystallization. In this regard, a number of studies on composite electrolytes with NASICON-type structure were carried out in recent years. As polymers polyethylene oxide (PEO), polyvinylidene fluoride (PVDF), poly methacrylic acid (PMAA), polyvinyl chloride acetate (PVCA) and hexafluoropropylene (HFP) were used. The majority of works demonstrated the advancements through electrochemical impedance spectroscopy and performance of full cells. A group of authors designed composites with PVDF binder. The choice of the polymer was due to the good impact strength, high thermal and chemical resistance, as well as uniform dispersion of LATP particles in its matrix. Composite membranes and their fabrication with electrochemical performance are shown in Fig. 7. Shi et al.118 and Liang et al.119 investigated the electrochemical properties of the LATP/PVDF membrane for rechargeable lithium ion batteries. Shi and coworkers synthesized LATP by a solution method and dispersed it in poly(vinylidene fluoride) (PVDF) to form a composite material by casting.118 The authors investigated the compatibility of the LATP ceramic and the binder, according to which the lithium-ion conductivity of CEM (9.6 × 10−4 S cm−1) increased two times compared to bare LATP and an electrochemical stability window up to 5.67 V versus Li+/Li was achieved due to the plasticization effect of LATP particles in PVDF. This was achieved by adding highly crystalline LATP in large proportions (2:1), but exceeding this ratio led to agglomeration and negatively affected the surface effect. Liang's group investigated the ratio of PVDF and LATP to advance electrolyte performance.119 In contrast to the previously reported work, it was revealed that by increasing the mass of LATP by almost 90 wt% in a hybrid solid electrolyte, the ionic conductivity and electrochemical stability were also improved.
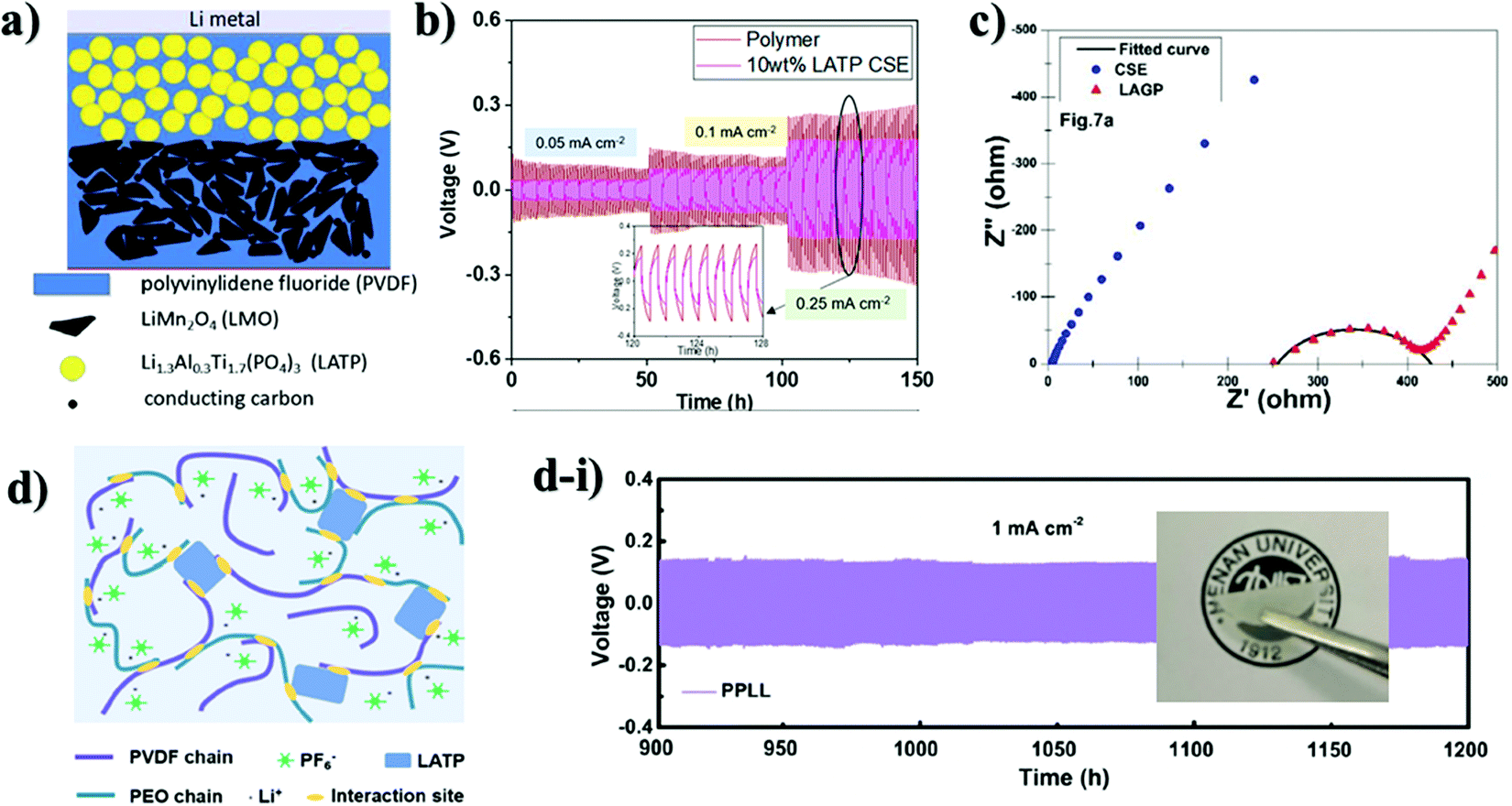 | ||
| Fig. 7 Images of composite electrolytes and their preparation scheme and performances: (a) schematic presentation of the solid-state Li/LMO cell assembled with the composite polymer electrolyte;119 (b) Li stripping and plating performance of LATP/PVdF-HFP at various current densities;120 (c) LAGP/HFP-PVdF membranes’ electrochemical impedance spectra;121 (d) illustration of the LATP/PVdF–PEO hybrid electrolyte and the membrane; (d-i) with Li stripping and plating performance.122 | ||
Improved results were observed with LATP ceramic which acted as a gel center, which increased the amorphous phase of the polymer and the concentration of the carrier, which favorably affected the synergism of Li-ion migration and increased ionic conductivity. The authors achieved a high lithium transference number value of 0.86 by using a high ratio of LATP ceramic. Further, the full cell with LiMn2O4 cathode, composite electrolyte and lithium metal was assembled (Fig. 7a), and it provided a high discharge capacity of 128 mA h g−1 with 98.3% capacity retention after 200 cycles. Research led by Li and Wang120 focused on the same combination but with an additional polymer of HFP into PVDF. The uniqueness of the work was the performance of hybrid electrolytes without absorption of the liquid electrolyte. The combination of PVDF with HFP has a number of preferable characteristics such as high dielectric constant, low degree of crystallinity due to the HFP unit, as well as excellent thermal stability and compatibility with electrodes. Consequently, LATP (10 wt%) ceramic embedded into the PVDF-HFP matrix exhibited quite a high ionic conductivity of 2.3 × 10−4 S cm−1. The symmetric cell at the lowest current density performed steadily, while the increase in current density led to voltage instability (Fig. 7b). During the Li plating/stripping process, it was found that the interface between the Li anode and the CSE membrane remained stable at various current densities, and the PVDF-HFP polymer protected LATP from reaction with the Li anode. The work by Das and colleagues121 introduced another NASICON type solid electrolyte, 20 wt% LAGP, as a filler for a similar polymer matrix as in the previous report, HFP-PVDF, and lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) and 1-ethyl-3-methylimidazolium bis-(trifluoromethylsulfonyl)-imide (EMITFSI) were also used. Addition of an ionic liquid and LAGP increased the amorphous regions and improved the channels for the migration of Li ions. As a result, the composite solid electrolytes have shown the highest ionic conductivity (4.49 ± 0.31) × 10−3 S cm−1 at room temperature (Fig. 7c). Yi et al. used the cross-linked structure of PVDF and PEO polymers and combined with LATP to improve the ability to transfer lithium ions and enhance the mechanical properties of solid-state batteries.122 In the research they stated that a combination of these two polymer materials and addition of ceramic resulted in a lower crystal dual-matrix system (Fig. 7d). The improvement of the mechanical strength of the matrix and the formation of amorphous areas that contribute to the transport of Li ions upon cycling were caused by abundant and irregular bonds in the composite electrolytes. In addition, a liquid electrolyte LiPF6 was added into the composite, which improved the performance of the system. As a result, the symmetric cell performed for prolonged 1000 hours and a relatively high ionic conductivity of 5.24 × 10−4 S cm−1 was obtained at RT (Fig. 7d-i). The full cell with LFP performed steadily up to 500 cycles at 1C. Furthermore, an approach of the application of composite polymer electrolytes was described by Singh et al., where they synthesized 5LiCF3SO3–95[PEO1−xLATPx] in a wide range of combinations.123 Equally dispersed LATP crystallites with the composition of 30 PEO/70 LATP led to a slight increase in Li-ion conductivity (10−4 S cm−1) and an increase in the Li ion transfer number from ∼0.12 for bare PEO to ∼0.46 for 70 LATP and 30 PEO. The full information regarding the electrochemical performance is provided in Table 4.
| Composite electrolyte | Cathode | Performance mA h g−1 | Condition | Ref. |
|---|---|---|---|---|
| CR, capacity retention. CE, Coulombic efficiency. AE, activation energy. | ||||
| LATP/PVDF | LFP | 163.2 | 0.1C | 118 |
| First discharge capacity | 155.3 | 0.2C | ||
| 150.0 | 0.5C | |||
| 141.2 | 1C | |||
| After 50 cycles | 154.7 | 0.1C | ||
| CR is 94.8% | ||||
| 146.8 | 0.2C | |||
| CR is 94.5% | ||||
| 127.1 | 0.5C | |||
| CR is 84.7% | ||||
| 118.8 | 1C | |||
| CR is 84.1% | ||||
| 200 cycles | 147.1 | 0.1C | ||
| LFP | CR is 90.1% | |||
| LATP/PVDF | LMO | 127.8 | 0.1C | 119 |
| In the beginning | ||||
| 119.9 | 0.2C | |||
| 113.8 | 0.5C | |||
| 104.5 | 1C | |||
| 92.1 | 2C | |||
| 118 | 0.2C | |||
| CR is 98.3% | ||||
| 107.4 | 0.2C | |||
| CE is 100% | ||||
| LATP/PVdF-HFP | LFP | 155 | 120 | |
| 5th cycle | 130 | |||
| After 50 cycles | ||||
| LAGP/HFP-PVdF | LFP | 151 | 0.05C | 121 |
| After 50 cycles | CR is 100% | |||
| LATP/PVdF-PEO | LFP | CR is 93.95% | 0.1C | 122 |
| After 500 cycles | CR is 84.77% | 1C | ||
| After 550 cycles | ||||
| 5LiCF3SO3–95[PEO1−xLATPx] | AE is 0.49 eV | 123 | ||
Composite electrolytes have gained a significant amount of attention due to their versatile characteristics. Fabrication of this hybrid system reduced cons of each electrolyte type. As already mentioned above, the combination of ceramics and polymer matrix significantly improves the performance, where LATP decreases the crystallinity of the polymer and aids in maintaining the amorphous phase while the polymer helps in preventing direct contact with lithium. In addition, the ion-conducting mechanism in composite solid electrolytes is highly dependent on the composition. Ion conductive pathways were in interfacial regions, where inorganic active filler concentration was less than 5%, while an increase in the filler content led to ionic conductivity through Li-conducting ceramics or on the interface as well. However, it should be taken into consideration that a large amount of active fillers negatively influenced, as fillers started to agglomerate and damaged the percolated network within the electrolyte. Regarding the transference number value, it was leveled up with LATP, as ideally ceramics have the transference number of one. Consequently, a combination of these electrolytes allowed one to obtain high lithium ion conducting composite solid membranes. The highly crystalline LATP with high stability under various conditions turned out to be the outstanding candidate for composite electrolytes by solving several dramatic issues.
Summary and future perspectives
The progress of next generation energy storage systems enabled scientists to expand their vision regarding the fabrication of battery components and solving their current problems. The implementation of solid electrolytes into battery packages is under progress, and the exact duration of this process is a matter of time and other factors. As the object of this review, NASICON structured LATP and LAGP ceramics are gaining attention due to their stability towards air and moisture under ambient conditions; these materials are very promising electrolytes for the next generation batteries including all-solid-state systems. By all means, the synthesis routes enable control of the morphology and grain size of these materials that significantly influence their ionic conductivity.45 Further improvements of these solid electrolytes include applying interlayers that reduce the interfacial impedance, prevent dendrite nucleation and, most importantly, protect the electrolyte from the side reactions. Summarizing the above paragraphs, it was clear that various types of coating materials on LATP influence differently, and the performing status of these layers and composite electrolytes is depicted in Fig. 8.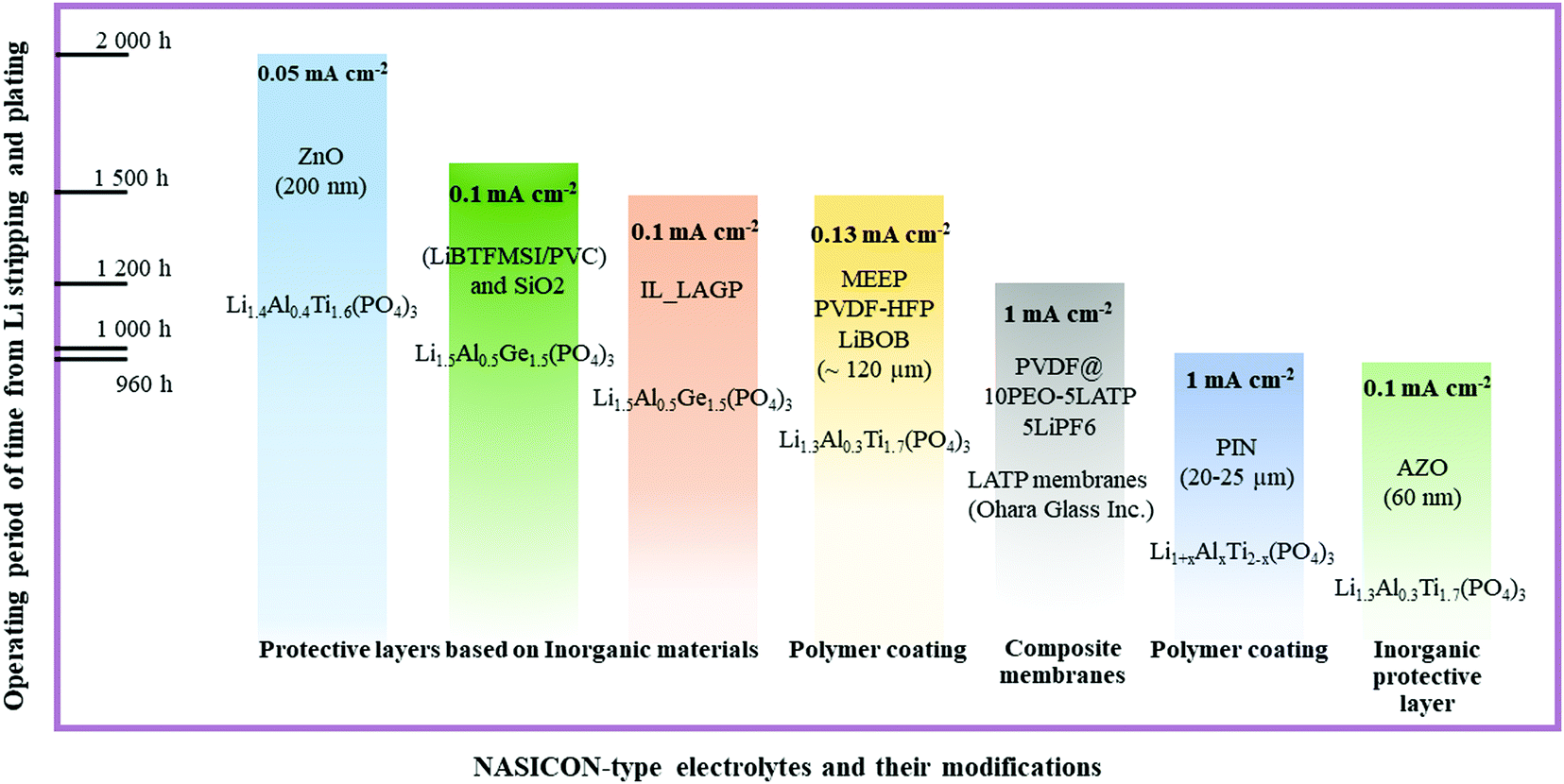 | ||
| Fig. 8 Advancement of NASICON-type electrolyte's interface through coating with various types of materials and composite electrolytes and their current status. | ||
As inorganic buffer layers, lithium ion conducting and non-conducting materials such as lithium salts, nanocomposites, oxides and hybrid layers with SiO2 nanoparticles, bare metals, and other types of solid electrolytes were utilized. In the class of inorganic protective layers, an excellent performing time period exceeding 2000 hours was attained with the ZnO2 protective layer with a thickness of 200 nm (Fig. 8). Generally, non-lithium conducting oxide materials and their doping derivatives exhibited a long functioning time at low current density, because of their robustness and high stability. Still, the activation of these layers requires precycling or heat treatment to activate the lithium ion conducting phase. On the other hand, the excellent performance in polymers was achieved by MEEP in PVDF-HFP with LiBOB film. It should be noted that polymer coatings are tested at higher current densities than inorganics and they could achieve a performance of 1500 hours at 0.13 mA cm−2 (Fig. 8). The major advantage of the polymer coating is prevention of dendrite nucleation by evenly distributing the lithium deposition. In contrast to inorganic protective films where comprehensive operating deposition tools are used, the polymer films do not require such complicated procedures. Simple casting of films is highly beneficial in terms of cost. Some other option to prevent the side reactions was a combination of inorganic and polymers to fabricate hybrid composite lithium ion conducting materials. Particularly, the high ion conducting phase was obtained by adding LATP or LAGP into the polymer matrix which increased the transference number significantly. One of the excellent performing periods of 1200 hours at 1 mA cm−2 was obtained by the PVDF@PEO_LATP composite membrane with LiPF6 liquid electrolyte (Fig. 8). The ionic conductivity was escalated, because ceramics reduced the crystallinity of the polymer phase and the direct contact between NASICON and lithium metal was prevented by the presence of polymer. The overall evaluation of each approach is given in Fig. 9, and depending on their uniqueness they displayed benefits and drawbacks.
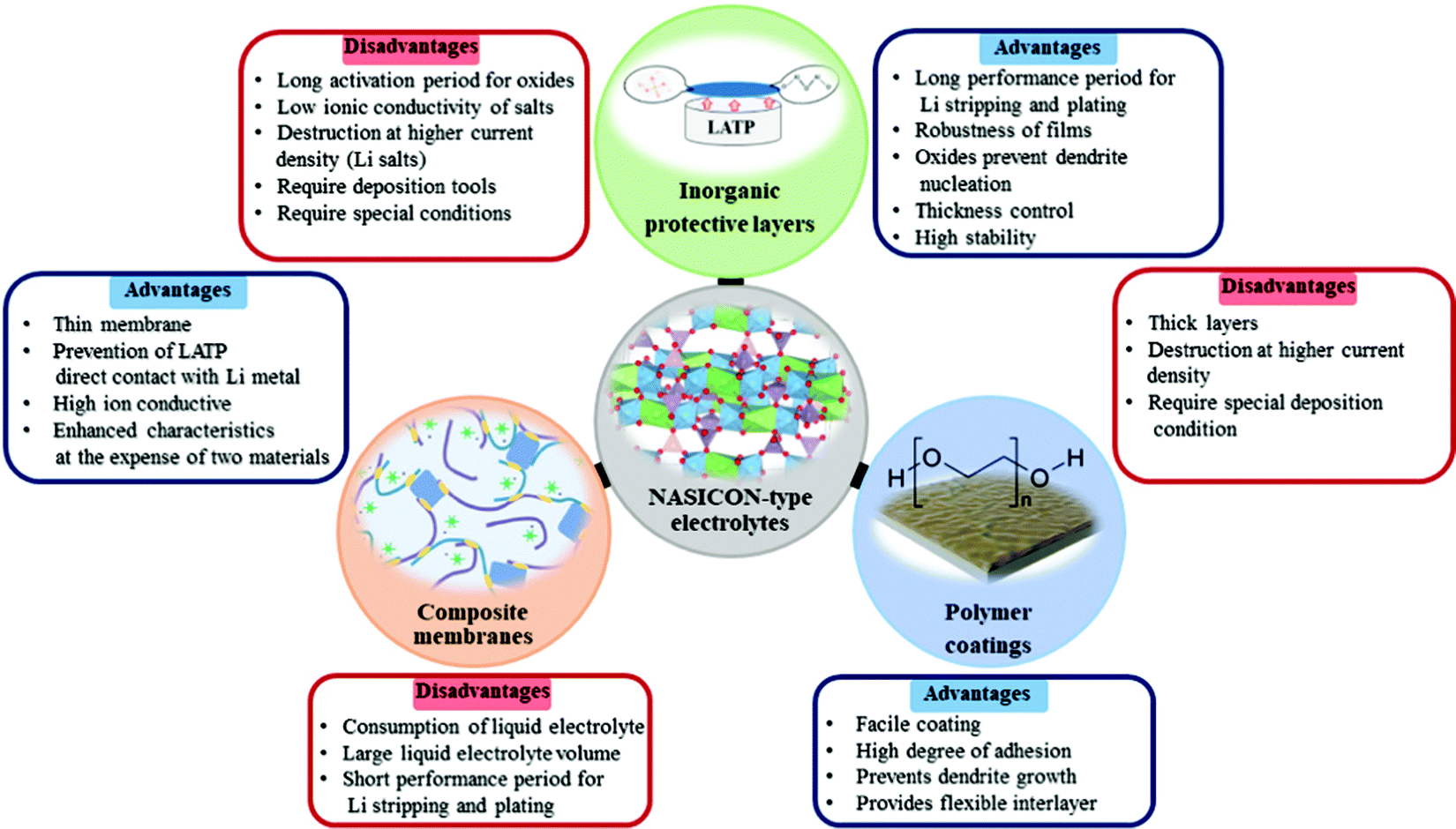 | ||
| Fig. 9 Pros and cons of inorganic and polymer coatings as well as composite electrolytes. | ||
It is very important to have a variety of alternatives to solve the emerged problem of solid electrolytes. Despite robustness, the inorganic protective layers tended to have a low or non-ionic conductive nature that requires a longer period to turn them into a conducting phase. Plus, the deposition tools usually operate under certain conditions which is time consuming. Polymer coating as an interlayer and composite electrolytes were the most optimal option to improve the interphase. The lower ionic conductivity is easily solved by using ion conductive copolymers or addition of fillers. Their coating and fabrication were straightforward without complex operating tool involvement. The wettability of polymers played a crucial role providing an intimate contact between electrode and electrolyte, and preventing the most dramatic issue of lithium metal batteries, the dendrite growth. Yet, the mechanical strength of polymers needs to be further engineered for longer cyclability at higher current density.
Many research studies endeavored to promote the ionic conductivity of solid electrolytes without putting effort on designing solid state full cells. There have been a number of research studies regarding solving the interphase problem of solid electrolytes. Most importantly, the majority of works described deposition techniques and materials by observing the electrochemical behavior of cells. Instead, the fundamental research on the lithium ion transport mechanism between interlayers and solid electrolytes could be the subject for further investigations. This kind of work could be more beneficial with in situ or operando characterization techniques to fully understand the process in detail. Deterioration of protective layers can be monitored by analyzing structural and chemical stability and for this purpose modelling techniques could be applied. Simulations may provide which kind of approach would be the most beneficial for advancing NASICON type solid electrolytes regarding the stability towards various conditions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the research grant #51763/IIIIϕ-MIIPOAII PK-19 “New materials and devices for defense and aerospace applications” from MDDIAI Republic of Kazakhstan.References
- W. Jing, Y. Yan, I. Kim and M. Sarvi, Adv. Mech. Eng., 2016, 8, 1–8 CrossRef.
- M. Wada, J. Environ. Sci., 2009, 21, 745–749 CrossRef CAS.
- ed. Y. Kato, Z. Ogumi and J. M. P. Martín, Lithium-Ion Batteries, Jenny Stanford Publishing, 2019 Search PubMed.
- N. Tolganbek, A. Mentbayeva, N. Serik, N. Batyrgali, M. Naizakarayev, K. Kanamura and Z. Bakenov, J. Power Sources, 2021, 493, 229686 CrossRef CAS.
- F. M. Hassan, Z. Chen, A. Yu, Z. Chen and X. Xiao, Electrochim. Acta, 2013, 87, 844–852 CrossRef CAS.
- J. Y. Liao, D. Higgins, G. Lui, V. Chabot, X. Xiao and Z. Chen, Nano Lett., 2013, 13, 5467–5473 CrossRef CAS PubMed.
- N. Liu, H. Wu, M. T. McDowell, Y. Yao, C. Wang and Y. Cui, Nano Lett., 2012, 12, 3315–3321 CrossRef CAS PubMed.
- D. Ahn, X. Xiao, Y. Li, A. K. Sachdev, H. W. Park, A. Yu and Z. Chen, J. Power Sources, 2012, 212, 66–72 CrossRef CAS.
- A. Luntz, J. Phys. Chem. Lett., 2015, 6, 300–301 CrossRef CAS PubMed.
- K. Liu, Y. Liu, D. Lin, A. Pei and Y. Cui, Sci. Adv., 2018, 4, eaas9820 CrossRef PubMed.
- A. Abouimrane, J. Ding and I. J. Davidson, J. Power Sources, 2009, 189, 693–696 CrossRef CAS.
- H. Dai, W. Xu, Z. Hu, Y. Chen, X. Wei, B. Yang, Z. Chen, J. Gu, D. Yang, F. Xie, W. Zhang, R. Guo, G. Zhang and W. Wei, Front. Energy Res., 2020, 8, 1–7 CrossRef.
- Z. A. Grady, C. J. Wilkinson, C. A. Randall and J. C. Mauro, Front. Energy Res., 2020, 8, 1–23 CrossRef.
- F. Bai, K. Kakimoto, X. Shang, D. Mori, S. Taminato, M. Matsumoto, Y. Takeda, O. Yamamoto, H. Izumi, H. Minami and N. Imanishi, Front. Energy Res., 2020, 8, 2–10 CrossRef.
- N. Tolganbek, Y. Yerkinbekova, S. Kalybekkyzy, Z. Bakenov and A. Mentbayeva, J. Alloys Compd., 2021, 882, 160774 CrossRef CAS.
- S. Zugmann, M. Fleischmann, M. Amereller, R. M. Gschwind, H. D. Wiemhöfer and H. J. Gores, Electrochim. Acta, 2011, 56, 3926–3933 CrossRef CAS.
- N. Kamaya, K. Homma, Y. Yamakawa, M. Hirayama, R. Kanno, M. Yonemura, T. Kamiyama, Y. Kato, S. Hama, K. Kawamoto and A. Mitsui, Nat. Mater., 2011, 10, 682–686 CrossRef CAS PubMed.
- K. Xu, Chem. Rev., 2004, 104, 4303–4417 CrossRef CAS PubMed.
- J. Hassoun, R. Verrelli, P. Reale, S. Panero, G. Mariotto, S. Greenbaum and B. Scrosati, J. Power Sources, 2013, 229, 117–122 CrossRef CAS.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- Y. C. Jung, S. K. Kim, M. S. Kim, J. H. Lee, M. S. Han, D. H. Kim, W. C. Shin, M. Ue and D. W. Kim, J. Power Sources, 2015, 293, 675–683 CrossRef CAS.
- M. Park, X. Zhang, M. Chung, G. B. Less and A. M. Sastry, J. Power Sources, 2010, 195, 7904–7929 CrossRef CAS.
- A. Patil, V. Patil, D. Wook Shin, J. W. Choi, D. S. Paik and S. J. Yoon, Mater. Res. Bull., 2008, 43, 1913–1942 CrossRef CAS.
- E. P. Roth and C. J. Orendorff, Electrochem. Soc. Interface, 2012, 21, 45–49 CrossRef CAS.
- C. C. Yang and S. J. Lin, J. Power Sources, 2002, 112, 497–503 CrossRef CAS.
- W. Kam, C. W. Liew, J. Y. Lim and S. Ramesh, Ionics, 2014, 20, 665–674 CrossRef CAS.
- M. Sethupathy, V. Sethuraman and P. Manisankar, Soft Nanosci. Lett., 2013, 03, 37–43 CrossRef.
- S. Kalybekkyzy, A. F. Kopzhassar, M. V. Kahraman, A. Mentbayeva and Z. Bakenov, Polymers, 2021, 23, 1–12 Search PubMed.
- Y.-W. Hu, I. D. Raistrick and R. A. Huggins, J. Electrochem. Soc., 1977, 124, 1240–1242 CrossRef CAS.
- J. Kuwano and A. R. West, MRS Bull., 1980, 15, 1661–1667 CrossRef CAS.
- S. Boulineau, M. Courty, J. M. Tarascon and V. Viallet, Solid State Ionics, 2012, 221, 1–5 CrossRef CAS.
- C. Yu, L. van Eijck, S. Ganapathy and M. Wagemaker, Electrochim. Acta, 2016, 215, 93–99 CrossRef CAS.
- R. Murugan, S. Ramakumar and N. Janani, Electrochem. Commun., 2011, 13, 1373–1375 CrossRef CAS.
- S. Ohta, T. Kobayashi and T. Asaoka, J. Power Sources, 2011, 196, 3342–3345 CrossRef CAS.
- R. Murugan, V. Thangadurai and W. Weppner, Angew. Chem., Int. Ed., 2007, 46, 7778–7781 CrossRef CAS PubMed.
- R. A. Huggins, Electrochim. Acta, 1977, 22, 773–781 CrossRef CAS.
- B. A. Boukamp and R. A. Huggins, Phys. Lett. A, 1976, 58, 231–233 CrossRef.
- V. Thangadurai, A. K. Shukla and J. Gopalakrishnan, Chem. Mater., 1999, 11, 835–839 CrossRef CAS.
- C. H. Chen and K. Amine, Solid State Ionics, 2001, 144, 51–57 CrossRef CAS.
- M. Itoh, Y. Inaguma, W. Jung, L. Chen and T. Nakamura, Solid State Ionics, 1994, 70–71, 203–207 CrossRef CAS.
- X. Lü, J. W. Howard, A. Chen, J. Zhu, S. Li, G. Wu, P. Dowden, H. Xu, Y. Zhao and Q. Jia, Adv. Sci., 2016, 1500359 CrossRef PubMed.
- Y. Zhao and L. L. Daemen, J. Am. Chem. Soc., 2012, 134, 15042–15047 CrossRef CAS PubMed.
- P. Goharian, B. Eftekhari Yekta, A. R. Aghaei and S. Banijamali, J. Non. Cryst. Solids, 2015, 409, 120–125 CrossRef CAS.
- A. Aatiq, M. Ménétrier, L. Croguennec, E. Suard and C. Delmas, J. Mater. Chem., 2002, 12, 2971–2978 RSC.
- N. Tolganbek, Y. Yerkinbekova, A. Khairullin, Z. Bakenov, K. Kanamura and A. Mentbayeva, Ceram. Int., 2021, 47, 18188–18195 CrossRef CAS.
- N. Tolganbek, A. Mentbayeva, B. Uzakbaiuly, K. Kanamura and Z. Bakenov, Mater. Today Proc., 2019, 25, 97–100 CrossRef.
- J. C. Bachman, S. Muy, A. Grimaud, H. H. Chang, N. Pour, S. F. Lux, O. Paschos, F. Maglia, S. Lupart, P. Lamp, L. Giordano and Y. Shao-Horn, Chem. Rev., 2016, 116, 140–162 CrossRef CAS PubMed.
- H. Aono, N. Imanaka and G. Ya Adachi, Acc. Chem. Res., 1994, 27, 265–270 CrossRef CAS.
- D. Petit, P. Colomban, G. Collin and J. P. Boilot, Mater. Res. Bull., 1986, 21, 365–371 CrossRef CAS.
- B. E. Taylor, A. D. English and T. Berzins, Mater. Res. Bull., 1977, 12, 171–181 CrossRef CAS.
- H. Aono, E. Sugimoto, Y. Sadaoka, N. Imanaka and G. Ya Adachi, Solid State Ionics, 1993, 62, 309–316 CrossRef CAS.
- H. Aono, E. Sugimoto, Y. Sadaoka, N. Imanaka and G. Adachi, Bull. Chem. Soc. Jpn., 1992, 65, 2200–2204 CrossRef CAS.
- S. Chun Li, J. Yi Cai and Z. Xiang Lin, Solid State Ionics, 1988, 28–30, 1265–1270 Search PubMed.
- M. Giarola, A. Sanson, F. Tietz, S. Pristat, E. Dashjav, D. Rettenwander, G. J. Redhammer and G. Mariotto, J. Phys. Chem. C, 2017, 121, 3697–3706 CrossRef CAS.
- L.-O. Hagman, P. Kierkegaard, P. Karvonen, A. I. Virtanen and J. Paasivirta, Acta Chem. Scand., 1968, 22, 1822–1832 CrossRef CAS.
- J. B. Goodenough, H.-P. Hong and J. A. Kafalas, Mater. Res. Bull., 1976, 11, 203–220 CrossRef CAS.
- H. Y. P. Hong, Mater. Res. Bull., 1976, 11, 173–182 CrossRef CAS.
- A. Martínez-Juárez, C. Pecharromán, J. E. Iglesias and J. M. Rojo, J. Phys. Chem. B, 1998, 102, 372–375 CrossRef.
- M. A. Subramanian, R. Subramanian and A. Clearfield, Solid State Ionics, 1986, 18–19, 562–569 CrossRef CAS.
- H. Aono, E. Sugimoto, Y. Sadaoka, N. Imanaka and G. Adachi, J. Electrochem. Soc., 1989, 136, 590–591 CrossRef CAS.
- T. Šalkus, A. Dindune, Z. Kanepe, J. Ronis, A. Určinskas, A. Kežionis and A. F. Orliukas, Solid State Ionics, 2007, 178, 1282–1287 CrossRef.
- M. Cretin and P. Fabry, J. Eur. Ceram. Soc., 1999, 19, 2931–2940 CrossRef CAS.
- C. Cao, Z. Bin Li, X. L. Wang, X. B. Zhao and W. Q. Han, Front. Energy Res., 2014, 2, 1–10 Search PubMed.
- C. J. Leo, G. V. Subba Rao and B. V. R. Chowdari, J. Mater. Chem., 2002, 12, 1848–1853 RSC.
- H. Aono, E. Sugimoto, Y. Sadaoka, N. Imanaka and G. Ya Adachi, Solid State Ionics, 1991, 47, 257–264 CrossRef CAS.
- O. G. Gromov, G. B. Kunshina, A. P. Kuz’min and V. T. Kalinnikov, Russ. J. Appl. Chem., 1996, 69, 385–388 Search PubMed.
- J. Fu, J. Am. Ceram. Soc., 1997, 80, 1901–1903 CrossRef CAS.
- J. Fu, Solid State Ionics, 1997, 104, 191–194 CrossRef CAS.
- X. Xu, Z. Wen, X. Wu, X. Yang and Z. Gu, J. Am. Ceram. Soc., 2007, 90, 2802–2806 CrossRef CAS.
- M. Illbeigi, A. Fazlali, M. Kazazi and A. H. Mohammadi, Solid State Ionics, 2016, 289, 180–187 CrossRef CAS.
- Y. Ren, K. Chen, R. Chen, T. Liu, Y. Zhang and C. W. Nan, J. Am. Ceram. Soc., 2015, 98, 3603–3623 CrossRef CAS.
- C. Delmas, A. Nadiri and J. L. Soubeyroux, Solid State Ionics, 1988, 28–30, 419–423 CrossRef.
- S. Stramare, V. Thangadurai and W. Weppner, Chem. Mater., 2003, 15, 3974–3990 CrossRef CAS.
- A. Jena, Y. Meesala, S. F. Hu, H. Chang and R. S. Liu, ACS Energy Lett., 2018, 3, 2775–2795 CrossRef CAS.
- P. Hartmann, T. Leichtweiss, M. R. Busche, M. Schneider, M. Reich, J. Sann, P. Adelhelm and J. Janek, J. Phys. Chem. C, 2013, 117, 21064–21074 CrossRef CAS.
- Y. Kee, S. S. Lee and H. Yun, Acta Crystallogr. Sect. E Struct. Reports Online, 2011, E67, i49 CrossRef PubMed.
- H. Chung and B. Kang, Chem. Mater., 2017, 29, 8611–8619 CrossRef CAS.
- Y. Liu, C. Li, B. Li, H. Song, Z. Cheng, M. Chen, P. He and H. Zhou, Adv. Energy Mater., 2018, 8, 1–7 Search PubMed.
- J. Zhu, J. Zhao, Y. Xiang, M. Lin, H. Wang, B. Zheng, H. He, Q. Wu, J. Y. Huang and Y. Yang, Chem. Mater., 2020, 32, 4998–5008 CrossRef CAS.
- J. Li and R. Wang, Ceram. Int., 2021, 47, 13280–13290 CrossRef CAS.
- A. Sakuda, A. Hayashi and M. Tatsumisago, Curr. Opin. Electrochem., 2017, 6, 108–114 CrossRef CAS.
- H. D. Lim, J. H. Park, H. J. Shin, J. Jeong, J. T. Kim, K. W. Nam, H. G. Jung and K. Y. Chung, Energy Storage Mater., 2020, 25, 224–250 CrossRef.
- Y. Chen, K. Wen, T. Chen, X. Zhang, M. Armand and S. Chen, Energy Storage Mater., 2020, 31, 401–433 CrossRef.
- X. Shen, Q. Zhang, T. Ning, T. Liu, Y. Luo, X. He, Z. Luo and A. Lu, Mater. Today Chem., 2020, 18, 100368 CrossRef CAS.
- L. Xu, S. Tang, Y. Cheng, K. Wang, J. Liang, C. Liu, Y. C. Cao, F. Wei and L. Mai, Joule, 2018, 2, 1991–2015 CrossRef CAS.
- T. Krauskopf, F. H. Richter, W. G. Zeier and J. Janek, Chem. Rev., 2020, 120, 7745–7794 CrossRef CAS PubMed.
- X. B. Cheng, C. Z. Zhao, Y. X. Yao, H. Liu and Q. Zhang, Chem, 2019, 5, 74–96 CAS.
- N. W. Li, Y. X. Yin, C. P. Yang and Y. G. Guo, Adv. Mater., 2016, 28, 1853–1858 CrossRef CAS PubMed.
- Y. Liu, Q. Sun, Y. Zhao, B. Wang, P. Kaghazchi, K. R. Adair, R. Li, C. Zhang, J. Liu, L. Y. Kuo, Y. Hu, T. K. Sham, L. Zhang, R. Yang, S. Lu, X. Song and X. Sun, ACS Appl. Mater. Interfaces, 2018, 10, 31240–31248 CrossRef CAS PubMed.
- L. Yang, Y. Song, H. Liu, Z. Wang, K. Yang, Q. Zhao, Y. Cui, J. Wen, W. Luo and F. Pan, Small Methods, 2020, 4, 1–7 Search PubMed.
- Q. Cheng, A. Li, N. Li, S. Li, A. Zangiabadi, T. De Li, W. Huang, A. C. Li, T. Jin, Q. Song, W. Xu, N. Ni, H. Zhai, M. Dontigny, K. Zaghib, X. Chuan, D. Su, K. Yan and Y. Yang, Joule, 2019, 3, 1510–1522 CrossRef CAS.
- J. Liu, T. Liu, Y. Pu, M. Guan, Z. Tang, F. Ding, Z. Xu and Y. Li, RSC Adv., 2017, 7, 46545–46552 RSC.
- H. Bai, J. Hu, Y. Duan, T. Kozawa, M. Naito, J. Zhang and S. Dong, Ceram. Int., 2019, 45, 14663–14668 CrossRef CAS.
- X. Hao, Q. Zhao, S. Su, S. Zhang, J. Ma, L. Shen, Q. Yu, L. Zhao, Y. Liu, F. Kang and Y. B. He, Adv. Energy Mater., 2019, 9, 1–8 Search PubMed.
- C. Guhl, M. Fingerle and R. Hausbrand, J. Power Sources, 2017, 362, 299–307 CrossRef CAS.
- L. Li, Z. Zhang, L. Luo, R. You, J. Jiao, W. Huang, J. Wang, C. Li, X. Han and S. Chen, Ionics, 2020, 26, 3815–3821 CrossRef CAS.
- F. J. Q. Cortes, J. A. Lewis, J. Tippens, T. S. Marchese and M. T. McDowell, J. Electrochem. Soc., 2020, 167, 050502 CrossRef CAS.
- S. Xiong, Y. Liu, P. Jankowski, Q. Liu, F. Nitze, K. Xie, J. Song and A. Matic, Adv. Funct. Mater., 2020, 30, 2001444 CrossRef CAS.
- Y. Hu, Y. Zhong, L. Qi and H. Wang, Nano Res., 2020, 13, 3230–3234 CrossRef CAS.
- Y. Koizumi, D. Mori, S. Taminato, O. Yamamoto, Y. Takeda and N. Imanishi, Solid State Ionics, 2019, 337, 101–106 CrossRef CAS.
- Y. Wang, G. Wang, P. He, J. Hu, J. Jiang and L. Z. Fan, Chem. Eng. J., 2020, 393, 124705 CrossRef CAS.
- H. V. Venkatasetty, Proc. Annu. Batter. Conf. Appl. Adv., 2000, 109–114 CAS.
- Y. Kumar, S. A. Hashmi and G. P. Pandey, Solid State Ionics, 2011, 201, 73–80 CrossRef CAS.
- B. Liang, S. Tang, Q. Jiang, C. Chen, X. Chen, S. Li and X. Yan, Electrochim. Acta, 2015, 169, 334–341 CrossRef CAS.
- M. Armand, Solid State Ionics, 1994, 69, 309–319 CrossRef CAS.
- G. S. MacGlashan and Y. G. Andreev, Nature, 1999, 398, 792–794 CrossRef CAS.
- W. Zhou, S. Wang, Y. Li, S. Xin, A. Manthiram and J. B. Goodenough, J. Am. Chem. Soc., 2016, 138, 9385–9388 CrossRef CAS PubMed.
- S. Ramesh, T. S. Yin and C. W. Liew, Ionics, 2011, 17, 705–713 CrossRef CAS.
- Z. Cai, R. Xiao and B. Jiang, Surf. Coatings Technol., 2021, 421, 127389 CrossRef CAS.
- Y. Jin, C. Liu, X. Zong, D. Li, M. Fu, S. Tan, Y. Xiong and J. Wei, J. Power Sources, 2020, 460, 228125 CrossRef CAS.
- S. Yu, S. Schmohl, Z. Liu, M. Hoffmeyer, N. Schön, F. Hausen, H. Tempel, H. Kungl, H. D. Wiemhöfer and R. A. Eichel, J. Mater. Chem. A, 2019, 7, 3882–3894 RSC.
- Z. Y. Kou, Y. Lu, C. Miao, J. Q. Li, C. J. Liu and W. Xiao, Rare Met., 2021, 40, 3175–3184 CrossRef CAS.
- L. He, Q. Sun, C. Chen, J. A. S. Oh, J. Sun, M. Li, W. Tu, H. Zhou, K. Zeng and L. Lu, ACS Appl. Mater. Interfaces, 2019, 11, 20895–20904 CrossRef CAS PubMed.
- Z. Chen, G. T. Kim, J. K. Kim, M. Zarrabeitia, M. Kuenzel, H. P. Liang, D. Geiger, U. Kaiser and S. Passerini, Adv. Energy Mater., 2021, 11, 2101339 CrossRef CAS.
- X. Yu and A. Manthiram, Adv. Funct. Mater., 2019, 29, 1–10 Search PubMed.
- M. Dirican, C. Yan, P. Zhu and X. Zhang, Mater. Sci. Eng., R, 2019, 136, 27–46 CrossRef.
- M. Dirican, C. Yan, P. Zhu and X. Zhang, Mater. Sci. Eng., R, 2019, 136, 27–46 CrossRef.
- X. Shi, N. Ma, Y. Wu, Y. Lu, Q. Xiao, Z. Li and G. Lei, Solid State Ionics, 2018, 325, 112–119 CrossRef CAS.
- X. Liang, D. Han, Y. Wang, L. Lan and J. Mao, RSC Adv., 2018, 8, 40498–40504 RSC.
- Y. Li and H. Wang, Ind. Eng. Chem. Res., 2021, 60, 1494–1500 CrossRef CAS.
- A. Das, M. Goswami, K. Illath, T. G. Ajithkumar, A. Arya and M. Krishnan, J. Non. Cryst. Solids, 2021, 558, 120654 CrossRef CAS.
- S. Yi, T. Xu, L. Li, M. Gao, K. Du, H. Zhao and Y. Bai, Solid State Ionics, 2020, 355, 115419 CrossRef CAS.
- M. D. Singh, A. Dalvi, D. M. Phase and Y. Kumar, J. Mater. Sci., 2020, 55, 3951–3963 CrossRef.
| This journal is © The Royal Society of Chemistry 2022 |