DOI:
10.1039/D1MA01097B
(Paper)
Mater. Adv., 2022,
3, 2018-2025
Highly sensitive detection of phosphate using well-ordered crystalline cobalt oxide nanoparticles supported by multi-walled carbon nanotubes†
Received
20th November 2021
, Accepted 24th December 2021
First published on 28th December 2021
Abstract
Phosphates are well-known groundwater and surface water contaminants, with even modest increases in their concentration contributing to the eutrophication of lakes and coastal waterways and thus potentially harming the environment. Consequently, sensors capable of detecting phosphate ions at concentrations below the eutrophication threshold (0.1 μM) are highly sought after. Herein, cobalt oxide nanoparticles (CoONPs) supported by polybenzimidazole (PBI)-modified multi-walled carbon nanotubes (MWCNTs) were prepared and shown to feature uniform size (3.5–5.5 nm) and limited Co phase mainly containing hexagonal Co3O4 and a minor amount of CoO. The synthesized NPs exhibited better phosphate sensing performance than previously reported polycrystalline Co wires, i.e., the CoONPs/PBI/MWCNT-modified glassy carbon electrode could detect phosphate at pH 4 and 7 at levels of 0.1 to 100 nM, that is, below the eutrophication threshold of 0.1 μM.
Introduction
Phosphorus is involved in important physiological processes and is therefore an essential nutrient. In natural waters, phosphorus is typically present as phosphates, among which the inorganic orthophosphate form is more thermodynamically stable than other forms.1 However, phosphates are also well-known contaminants of groundwater and surface water2 and are mainly transported from terrestrial ecosystems to aquatic ones via wind erosion, surface runoff, and leaching. The eutrophication caused by this transport may be further accelerated by agricultural activities (supply of phosphorus to crops as a fertilizer), animal husbandry, and other human activities.3 Under the right conditions, even a modest increase in phosphate concentration due to influx from agricultural land or other sources can contribute to the eutrophication of lakes and coastal waterways. Eutrophication refers to the over-enrichment of natural waters in nutrients, such as phosphates, which leads to the excessive growth of algae and other aquatic plants and may cause the death of aquatic fauna due to the lack of oxygen.3,4 The eutrophication threshold concentrations of phosphates have been estimated to be 0.1–0.32 μM.5 Given that the increase in the phosphate levels in rivers over the past 50 years6 has negatively affected aquatic ecology and water quality, phosphate ion monitoring is crucial for ensuring environmental safety.
Phosphate is typically quantified using (i) high-performance liquid chromatography,7 (ii) the colorimetric method of Murphy and Riley,8–10 and (iii) spectroscopic methods relying on the molybdenum blue reaction or a series of complicated sample pretreatments.11,12 All the above-mentioned methods require complex processes and are therefore poorly suited for phosphate monitoring. Other classical techniques such as gravimetric methods,13 volumetric methods,14 and instrumental methods based on chromatographic measurements15,16 also require sample pre-treatment and can therefore be time-consuming, expensive, and/or produce toxic wastes. Such methods also had detection ranges higher than the eutrophication threshold of 0.1 μM.
Recent studies using colorimetric and ECL17 and fluorescence methods18 satisfy the requirement of lower detection range than the eutrophication threshold, however they are difficult to operate for on-site measurement of phosphate concentration. Hence, the development of inexpensive, easy to manufacture, and reliable sensors for monitoring phosphate concentration in agricultural wastewater and other aqueous systems is expected to benefit the mass production of food and agricultural products as well as help solve certain environmental problems.
In recent decades, considerable attention has been directed toward the development of electrochemical phosphate sensors in view of their simple operation, fast response, and high sensitivity.19 For example, a Co electrode has been successfully used as a base material for phosphate sensing,20 while the first Co-based phosphate sensor was reported in 1995 by Xiao et al.21 Co-based nanomaterials have attracted significant attention owing to their large specific surface area, which is reported to be >10 m2 g−1 (beneficial for increasing the number of active sites),22 excellent conductivity, catalytic activity, and ability to sense phosphate. A phosphate sensor fabricated by coating a glassy carbon electrode (GCE) with Co oxide nanoparticles (NPs) and reduced graphene oxide exhibited a good linear potentiometric response.23 However, its lowest detection limit was 1 μM, which is higher than the eutrophication threshold. Herein, we synthesized CoONPs supported by polybenzimidazole (PBI)-modified multi-walled carbon nanotubes (MWCNTs) to detect phosphate ions with high sensitivity. MWCNTs were selected owing to their large surface-to-volume ratio,24 good conductivity, which makes them suitable for catalytic and sensor applications,25–29 and relatively low cost.30 The CoONPs featured a narrow size, well-distribution and well-defined limited crystal planes that provided sites specifically interacting with phosphate ions and enabled their sensitive detection.
Experimental
Materials
N,N-Dimethylacetamide (DMAc), polyethylene glycol 400 (PEG400), Co(CH3COO)2·4H2O, Na2SO4, NaOH, and HClO4 were purchased from Fujifilm Wako Pure Chemical (Osaka, Japan). PBI fine powder was obtained from Sato Light Industrial Co., Ltd (Tokyo, Japan) and used as received. A polytetrafluoroethylene filter (pore size = 0.2 μm, Millipore) was used for filtration. The GCE (outer diameter = 6 mm, inner diameter = 3 mm) was purchased from BAS Inc. (Tokyo, Japan). MWCNTs (purity: ≥98%, length: 3–6 μm, outer diameter: 10 nm ± 1 nm, inner diameter: 4.5 nm ± 0.5 nm) were purchased from Sigma Aldrich (St. Louis, MO, USA). Co wire (purity: 99.99%, diameter: 0.50 mm) was sourced from Nilaco Co. (Tokyo, Japan). Phosphate solutions (phosphate-buffered saline, PBS) were prepared by mixing solutions of NaH2PO4 (Fujifilm Wako Pure Chemical) and Na2HPO4·12H2O (Fujifilm Wako Pure Chemical). The solution pH was adjusted to 4 and 7 with 0.1 M HClO4 and 0.1 M NaOH. All solutions were prepared with deionized water (resistivity >18.2 MΩ cm) obtained using a Milli-Q water purification system (Millipore, Billerica, MA).
Fabrication of the CoONPs/PBI/MWCNT/GCE electrode
A schematic of the synthesis of the CoONPs/PBI/MWCNT composite is shown in Fig. 1. PBI (40 mg) was dissolved in DMAc (10 mL) with 20 min ultrasonication (probe-type BRANSON 5520 sonicator, Kanagawa, Japan), and the solution was supplemented with MWCNTs (10 mg) and sonicated for another 20 min to prepare the PBI/MWCNT dispersion (Fig. S1, ESI†). This dispersion was treated with 3 mL of the solution prepared by dissolving Co(CH3COO)2·4H2O (500 mg) in deionized water (10 mL) to obtain a ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Co(CH3COO)2·4H2O to PBI/MWCNT. The reaction mixture was supplemented with PEG400 (reducing agent), heated at 130 °C for 6 h in an oil bath upon stirring, filtered, and washed with DMAc to remove excess PBI and PEG400. The GCE was polished with a 0.05 μm alumina suspension on a polishing pad, rinsed with Milli-Q water, and sonicated in Milli-Q water for 5 min. Subsequently, a 10 μL (10 mg, see ESI† for surface area) aliquot of the CoONPs/PBI/MWCNT dispersion was cast on the polished GCE and dried under vacuum (∼0.06 MPa). A Co wire electrode (prepared by polishing with emery paper (#2000) and rinsed with Milli-Q water) was used as a reference for comparison with the modified electrode.
:1 Co(CH3COO)2·4H2O to PBI/MWCNT. The reaction mixture was supplemented with PEG400 (reducing agent), heated at 130 °C for 6 h in an oil bath upon stirring, filtered, and washed with DMAc to remove excess PBI and PEG400. The GCE was polished with a 0.05 μm alumina suspension on a polishing pad, rinsed with Milli-Q water, and sonicated in Milli-Q water for 5 min. Subsequently, a 10 μL (10 mg, see ESI† for surface area) aliquot of the CoONPs/PBI/MWCNT dispersion was cast on the polished GCE and dried under vacuum (∼0.06 MPa). A Co wire electrode (prepared by polishing with emery paper (#2000) and rinsed with Milli-Q water) was used as a reference for comparison with the modified electrode.
 |
| | Fig. 1 Schematic of the CoONPs/PBI/MWCNT composite. | |
Materials characterizations
Fourier transform infrared (FTIR) spectra were recorded on a VERTEX 70v spectrometer (Bruker Corp., Billerica, MA, USA) using the KBr method. Thermogravimetric analysis (TGA) was performed using an SII EXSTAR 6000 instrument (Seiko Instruments, Inc., Chiba, Japan) operated in conditioned air from 25 to 700 °C at a heating rate of 10 °C min−1. Transmission electron microscopy (TEM) measurements were conducted using an FEI Technai F20 microscope (Tokyo, Japan). X-Ray diffraction (XRD) patterns were recorded using a micro/thin film diffractometer (Rigaku SmartLab) with Cu Kα radiation (λ = 1.54059 Å) at 45 kV, 200 mA, a scan speed of 0.5° min−1, and an amorphous Si plate as a sample holder. X-Ray photoelectron spectroscopy (XPS) measurements were conducted using a Shimadzu Kratos AXIS-ULTRA instrument with Mg Kα radiation (hν = 1253.6 eV). Peak calibration was performed using the peaks of pure In (99.999%) at 665.2 eV and 443.7 eV (In 3d3/2 and 3d5/2, respectively).
Electrochemical measurements and ICP measurements
Electrochemical measurements were conducted using an electrochemical analyzer (model 700B, ALS Co., Ltd, Tokyo, Japan). pH was recorded using a pH meter (AUT-501, DKK-TOA Corp., Tokyo, Japan). Cyclic voltammetry (CV) measurements were performed using a three-electrode system with Ag|AgCl|sat. KCl and a Pt wire as reference and counter electrodes, respectively. Prior to measurements, high-purity O2 (99.99%) was bubbled through the solutions for at least 30 min. Open-circuit potentials (OCPs) were measured using a two-electrode system with the CoONP-modified electrode as the working electrode and Ag|AgCl|sat. KCl as the reference electrode. Inductively coupled plasma (ICP) measurements were performed using a Shimadzu ICPS-8100 instrument (Kyoto, Japan) to confirm the results of OCP measurements for real samples. Prior to real sample measurements, the pH of the solution was adjusted to pH 4 and 7.
Results and discussion
Characterization of CoONPs/PBI/MWCNTs
TEM imaging of the CoONPs/PBI/MWCNTs composite (Fig. 2) revealed black dots attached to the MWCNTs (as indicated by the red arrows), which indicated that the CoONPs were uniformly deposited on the MWCNTs without forming aggregates. The zoomed-in image of the CoONPs/PBI/MWCNTs reveals that the NPs were closely contacted the MWCNT surface, together with the Co exposure over the PBI layer.
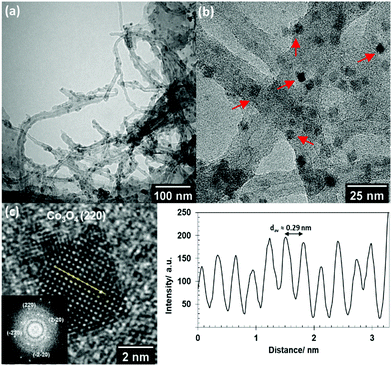 |
| | Fig. 2 (a) TEM plane views of CoONPs/PBI/MWCNTs, (b) zoomed-in TEM plane views of CoONPs/PBI/MWCNTs with red arrows pointing toward the CoONPs and (c) estimation of the interplanar distance in CoONPs where the intensity pattern shown is across the yellow line in the high-resolution lattice fringe image (HRLFI). The inset figure in (c) shows the corresponding fast Fourier transform (FFT) pattern. | |
FTIR measurements were conducted to verify the MWCNT functionalization with PBI (Fig. S4 and Table S1, ESI†). The amount of PBI was determined to be 7.4 wt% from the weight loss differences (TGA) between MWCNTs and PBI/MWCNTs (Fig. S5, ESI†). FTIR measurements and TGA demonstrated the successful incorporation of PBI onto the MWCNTs. The thin wrapping of PBI around the MWCNTs enabled the efficient and homogeneous loading of Co on the latter. Without the presence of PBI, well-defined crystalline CoO nanoparticles with no aggregation failed to form (Fig. S6, ESI†). Moreover, PBI is known to effectively solubilize CNTs by acting as an exfoliator and individually wrapping CNTs based on π–π interactions.31 PBI also serves as an active binding site, as it is strongly adsorbed at the surface of pristine graphitized carbon (e.g., MWCNTs), and can strongly bind metal ions32 such as Au,33 Pd34 and Pt35,36 by coordinating them via nitrogen.37
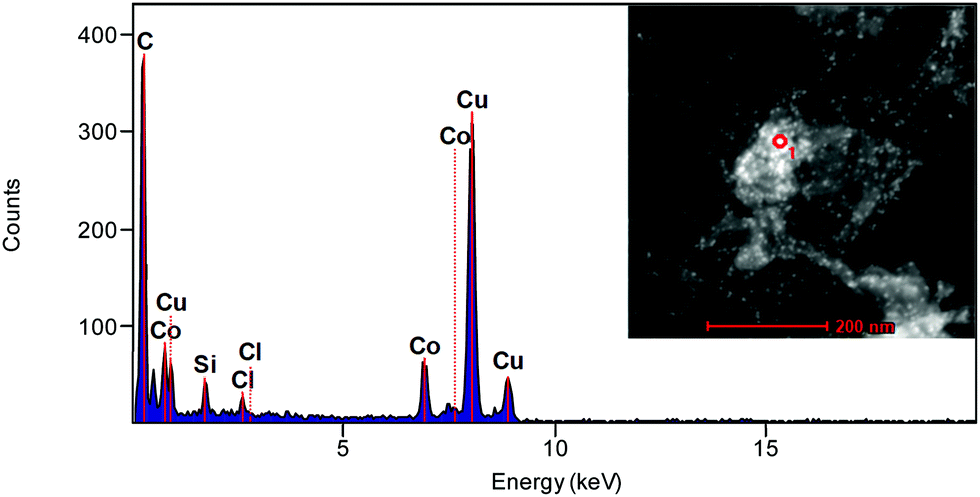
Through high-resolution TEM imaging (Fig. 2c), the average lattice spacing of CoONPs was estimated to be ∼0.29 nm, which corresponds to the (220) plane of hexagonal Co3O4. This result agreed with the fast Fourier transform imaging and diffraction simulation results (Fig. S7, ESI†), confirming the validity of our crystal phase assignment. The elemental compositions of the synthesized materials were probed by energy-dispersive X-ray spectroscopy (Fig. 3). The obtained composite was shown to contain C (from MWCNTs), Co (from CoONPs), and other elements such as Cu, Si, and Cl (from the grid support film) (Table S2, ESI†). The CoONPs appeared to have a uniform size with an average diameter of 3.5–5.5 nm (Fig. 4). The narrow size distribution and well-defined limited crystal surface of the CoONPs provide the advantages of a large specific surface area (with no aggregation) and high sensitivity to phosphate. Given that the properties of metal NPs are often strongly size-dependent, the narrow size distribution of the CoONPs was expected to limit the effect of nanoparticle size variation on phosphate ion detection. The crystal phases and Co valence states were further probed by XRD (Fig. 5) and XPS (Fig. 6).
 |
| | Fig. 3 Energy-dispersive X-ray spectrum of CoONPs/PBI/MWCNTs. Inset image shows the area measured (red circle) from the TEM microphotograph of CoONPs/PBI/MWCNTs. | |
 |
| | Fig. 4 Size distribution of CoONPs. | |
 |
| | Fig. 5 XRD patterns of (black line) pristine MWCNTs and (red line) CoONPs/PBI/MWCNTs. | |
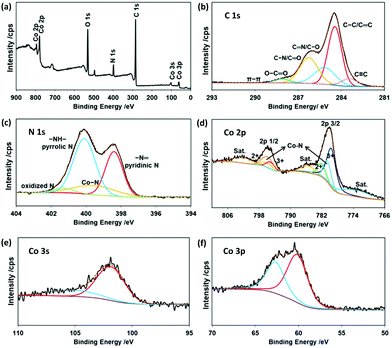 |
| | Fig. 6 (a) Survey, (b) C 1s, (c) N 1s, (d) Co 2p, (e) Co 3s, and (f) Co 3p X-ray photoelectron spectra of CoONPs/PBI/MWCNTs. | |
Diffraction peaks from Fig. 5 at 2θ = 25.58° and 42.85° characteristic of the (002) and (101) planes of the MWCNTs (JCPDS Card No. 75-1621) were observed for all samples, along with those of the CoONPs. Although the diffraction peaks for CoONPs appeared rather small and noisy, at least every peak could be distinguished and all of them agreed well with the crystal structures of Co3O4 (JCPDS Card No. 74-1657) and cubic CoO (JCPDS Card No. 78-0431). The peak at 36.21° was ascribed to the (111) plane of CoO, while those at 31.70, 36.55, 44.65, and 59.09° were attributed to the (220), (311), (400), and (511) planes of Co3O4, respectively. Thus, the CoONPs largely contained Co3O4 with a small amount of cubic CoO, i.e., they exhibited a rather simple biphasic composition.
The survey X-ray photoelectron spectrum of CoONPs/PBI/MWCNTs (Fig. 6a) showed that this nanocomposite mainly contained Co, O, N (due to PBI), and C. The C 1s spectrum (Fig. 6b) was deconvoluted into six peaks at 283.43, 284.45, 285.15, 286.26, 288.14, and 291.02 eV, which were attributed to C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C, main peak of sp2 hybridized carbon C–C/C
C, main peak of sp2 hybridized carbon C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C,38 CN/C–O which may arise from the nitrogenous carbon precursors and formation of interfacial C–O–Co bonds from combination between carbon materials with Co oxide,39,40 C–N/CO, O–CO bonds, and π–π interactions, respectively.
C,38 CN/C–O which may arise from the nitrogenous carbon precursors and formation of interfacial C–O–Co bonds from combination between carbon materials with Co oxide,39,40 C–N/CO, O–CO bonds, and π–π interactions, respectively.
The N 1s spectrum (Fig. 6c) was deconvoluted into four peaks at 398.4, 399.5, 400.1, and 401.3 eV, which were attributed to –N = pyridinic N, –NH– pyrrolic N, metal–nitrogen bonds of Co–N, and oxidized N for the PBI, respectively. Fig. 6d displays the related Co 2p spectrum, revealing two main spin orbit lines at 780.1 eV (Co 2p3/2) and 795.68 eV (Co 2p1/2), in line with the Co3+ and Co2+ states expected for Co3O4 and CoO. Each of these main peaks was deconvoluted into three characteristic peaks. Peaks at 780.6 and 795.9 eV were assigned for Co–N, originated from the interactions of Co with N of the PBI believed to generate high active sites for oxidation reaction.41 The above results agreed well with those of XRD and TEM. Satellite peaks near the Co 2p3/2 and Co 2p1/2 signals were observed at 773.5, 785.16, and 802.39 eV, corresponding to spin orbit components. Satellites are commonly observed for transition metals.42 Therefore, in the case of Co3O4, which contains both Co(II) and Co(III), we expected to see some satellite features due to these states.43 The Co 3s spectrum of the prepared composite (Fig. 6e) featured a peak at 102.08 eV corresponding to a low-spin Co3+ state. Finally, the Co 3p spectrum (Fig. 6f) featured a peak at 61.33 eV. The ratio of 3:1 was calculated as amount differences between Co3O4 and CoO obtained from each Co3+ and Co2+ peak area from Co 2p spectra. The presence of Co3O4 and CoO was expected to result in high sensitivity to phosphate. Transition metal (e.g., Co) oxides are widely used for sensing because of their surface redox properties.44,45 Specifically, the Co2+ and Co3+ states of Co found in our composite are readily accessible and thermodynamically stable.46
Electrochemical characterization
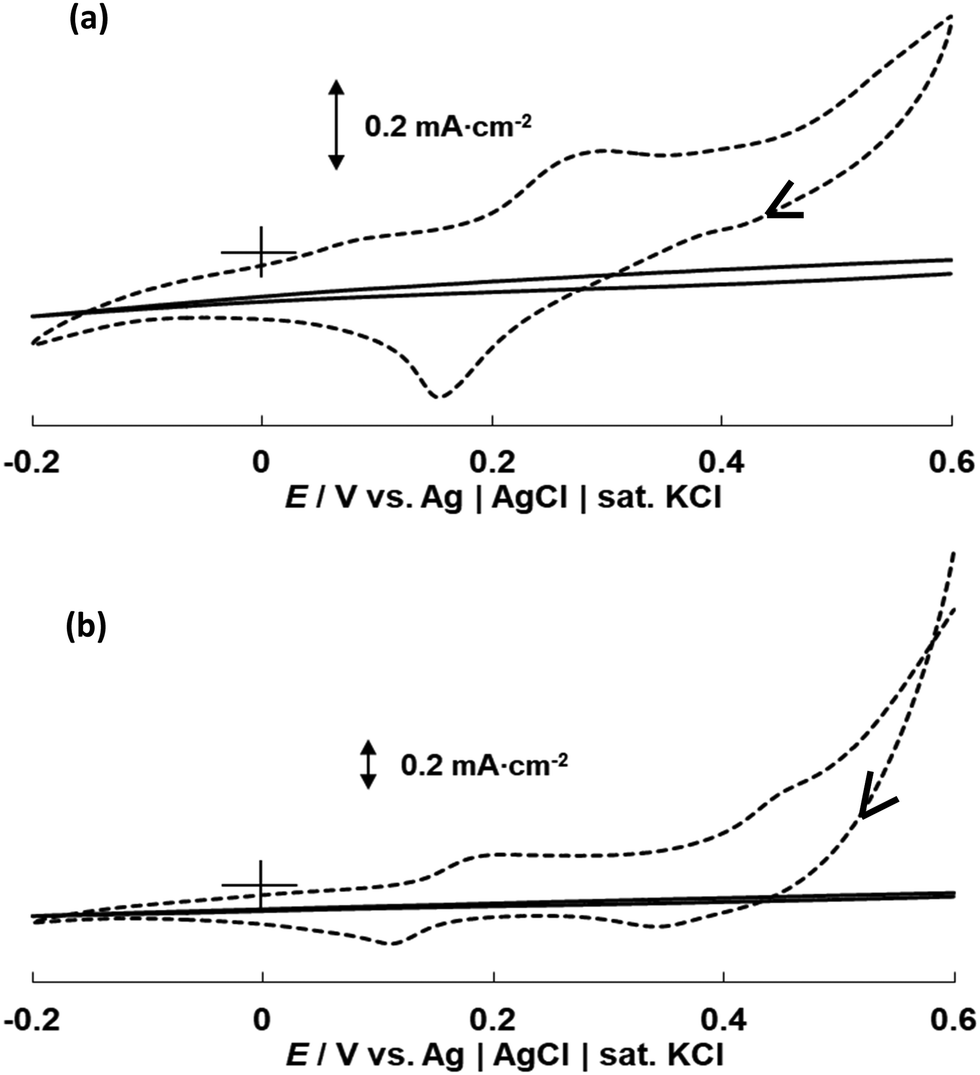
CV curves in phosphate solution were recorded for both the bulk Co wire electrode and CoONPs/PBI/MWCNTs/GCE (Fig. 7). The actual current vs. potential of the CV curves shown on Fig. S8 (ESI†). The curve of the Co wire electrode recorded in 0.1 M PBS at pH 7 featured one anodic peak around −0.35 V and two cathodic peaks around −0.8 V and −1.1 V (Fig. 7a). Given that this behavior was similar to that previously reported by Xu et al.,20 the first cathodic peak was identified as that observed in the absence of H2PO4− for Co, while the second cathodic peak was attributed to the reduction of Co(H2PO4)2 to Co. The following reactions are believed to occur on the surface of the Co electrode in the tested potential region.| | | Co(OH)2 + 2e− ⇌ Co + 2OH−, | (2) |
| | | Co(H2PO4)2 + 2e− ⇌ Co + 2H2PO4−. | (3) |
 |
| | Fig. 7 CV curves recorded at 5 mV s−1 for (a) the bulk Co wire electrode (broken line) and CoONPs/PBI/MWCNTs/GCE (solid line) in 0.1 M PBS with pH 7 and (b) CoONPs/PBI/MWCNTs/GCE in 0.1 M Na2SO4 (broken line) and 0.1 M PBS with pH 7 (solid line). | |
The curve of CoONPs/PBI/MWCNTs/GCE featured one anodic peak around −0.35 V (the same as that observed for the Co wire electrode) and only one cathodic peak at around −0.9 V. Notably, this cathodic peak was not observed when the measurement was performed in 0.1 M Na2SO4 (i.e., in the absence of phosphate) (Fig. 7b), and therefore reflected the presence of phosphate ions. Specifically, the cathodic peak was ascribed to the occurrence of reactions (2) and (3), in which case the potential shifted to values more positive than that of the bulk Co wire electrode. This different behavior was also presumably attributed to differences in the exposed crystal planes in bulk Co wire and CoONPs/PBI/MWCNTs, i.e., the CoONPs in CoONPs/PBI/MWCNTs were thought to react with phosphate ions in a different way than polycrystalline bulk Co. TEM, XRD, and XPS results also suggested that the CoONPs contained Co3O4 and CoO phases, which ensured a considerably higher sensitivity and selectivity for phosphate ions than that attained for polycrystalline bulk Co. We assumed Co3O4 might play the main role in sensing phosphate ion as the main crystal phase. In particular, the detection of phosphate by the CoONPs–based electrode was not hindered by the presence of other anions such as SO42−, Cl−, CH3COO−, and NO3−, unlike that by the Co wire electrode (Fig. S9 and S10, ESI†). Fig. S11 and S12 (ESI†) depict the performances of both electrodes at various pH values.
Potentiometric response
The dynamic responses of the composite electrode to changes in phosphate concentration at pH 4 and 7 were measured using OCP in a two-electrode system (Fig. 8). These pH values were selected because the electrode worked best under acidic conditions (i.e., pH 4–7). The practical response time was recorded by varying the concentration of the phosphate solution from 1.0 × 10−10 to 1.0 × 10−7 M (equivalent to 0.1–100 nM) every 3 min. A solution (pH 7) obtained by mixing 0.1 M NaOH and 0.1 M HClO4 was used as the initial solution. Measurements were conducted in triplicate to confirm the response repeatability.
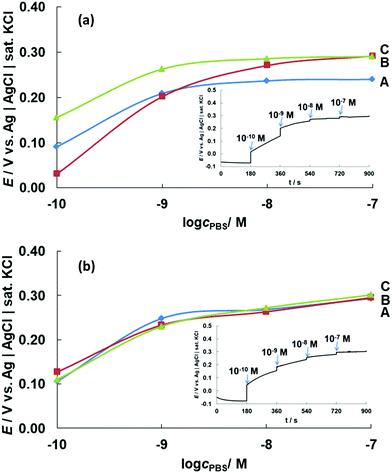 |
| | Fig. 8 OCPs (E) obtained for CoONPs/PBI/MWCNTs/GCE at different phosphate concentrations (cPBS = 1.0 × 10−10 to 1.0 × 10−7 M) at pH (a) 4 and (b) 7. The results of triplicate measurements are marked as A, B, and C and indicate high repeatability. | |
Fig. 8a shows that the potential commenced at 0.05–0.15 V for a 0.1 nM phosphate solution at pH 4, increasing to 0.20–0.25 V as the phosphate concentration increased to 1 nM. This significant potential change was observed until the phosphate level reached ∼10 nM. At higher concentrations, the response curve was almost flat, although the potential still shifted to a more positive value with increasing phosphate concentration. Notably, phosphate detection was impossible in the absence of Co, i.e., for MWCNTs/GCE (Fig. S13, ESI†) and the absence of PBI for CoONPs/MWCNT/GCE showed disproportionate relationship of potential value with the concentration change unlike the one with PBI (Fig. S14, ESI†).
The CoONPs-modified electrode could detect phosphate ions at very low concentrations, with the detection range at pH 7 determined as 0.1–100 nM (Fig. 8b). The wide detection range at both pH 4 and 7 enabled the sensing of phosphate at concentrations lower than the eutrophication threshold of ∼0.1 μM,5 and our electrode was therefore suitable for phosphate monitoring in environmental samples. While previous studies using similar electrochemical methods reported a response time of 15–60 s, our composite electrode featured a fast response time of ∼5 s. The potential response for 10 nM phosphate solution pH 7 remained almost unchanged d for 1 month using same electrode stored inside an initial mixed solution of 0.1 M NaOH and 0.1 M HClO4 when not in use under the condition of room temperature (Fig. S15, ESI†). The proposed sensor was compared with other types of detection for phosphate ions with respect to the detection range, type of electrode/reagent, and response time (Table 1).
Table 1 Comparison of the analytical performances obtained with different detection types for phosphate ions
| Detection range |
Type of detection |
Electrode/reagent |
Response time |
Real sample |
Ref. |
|
Co nanoparticles and reduced graphene oxide/glassy carbon electrode.
|
| 60 nM–3 μM |
Spectrophotometric (880 nm) |
Molybdenum blue |
Up to 60 s |
— |
47
|
| 50 nM–5 μM |
Fluorescence (590 nm) |
Enzymatic system + amplex red |
15 min |
— |
48
|
| Up to 0.434 nM |
Colorimetric and ECL |
CoOOH Nanosheet |
10 min |
Tap and river water |
17
|
| 0.15–5 μM |
Fluorescence (520 nm) |
CoFe Nanoparticles |
60–120 min |
SBF and HBS |
18
|
| 1 μM–10 mM |
Spectrophotometric |
Co3O4 Film electrode (transmitted at 620 nm) |
50 s |
— |
49
|
| 10 μM–0.1 M |
Electrochemical |
Co-Based phosphate microelectrode |
<60 s |
Microbial flocs |
50
|
| 10 μM–0.1 M |
Electrochemical |
Cobalt phosphate coated Co |
30 s |
— |
20
|
| 1 μM–10 mM |
Electrochemical |
Co NPs/rGO/GCEa |
15 s |
Tap and well water |
23
|
| 10 μM–10 mM |
Electrochemical |
Co Microelectrode |
30–60 s |
Standard ATP and ADT |
51
|
| 10 μM–0.1 M |
Electrochemical |
Co Electrode |
<40 s |
Waste water |
52
|
| 0.1 nM–0.1 μM |
Electrochemical |
CoONPs/PBI/MWCNT/GCE |
∼5 s |
Creek water |
This paper |
To confirm the fast response of the modified electrode, we performed CV measurements in the presence and absence of phosphate (Fig. 9). The actual current vs. potential of the CV curves shown on Fig. S16 (ESI†). Redox peaks at 0.15 and 0.27 V were initially observed for the CoONPs-modified electrode in a mixed solution (pH 4) of 0.1 M NaOH and 0.1 M HClO4 but disappeared after the addition of 0.1 nM phosphate ions (Fig. 9a). The same behavior was also observed for pH 7 (Fig. 9b), in which case the initial peaks were observed at 0.1 and 0.19 V. The addition of phosphate seemed to strongly influence the double-layer capacitance. Thus, phosphate binding occurred on the CoONPs surface even at low phosphate concentrations, which suggested that our composite electrode is capable of highly sensitive phosphate detection.
 |
| | Fig. 9 CV curves of CoONPs/PBI/MWCNTs/GCE recorded in a mixed solution of 0.1 M NaOH and 0.1 M HClO4 (broken line) and in 0.1 nM PBS (solid line) at (a) pH 4 and (b) pH 7 using a scan rate of 5 mV s−1. | |
As mentioned before, cobalt oxides react with phosphate ions in solution to form cobalt phosphate precipitates at the electrode surface, with the precipitate nature depending on pH. Soluble phosphorus in natural water is largely present as four orthophosphates (H3PO4, H2PO4−, HPO42−, and PO43−), with the proportions of these species depending on pH (Fig. S17, ESI†).53 Hem investigated the distribution of orthophosphate species at 25 °C, revealing that no more than two types are present at any pH.54 The phosphate species formed herein at pH 4 is thought to be H2PO4− (eqn (4)), while H2PO4− and/or HPO42− might form at pH 7 (eqn (4) and (5)).
| | | 3CoO + 2H2PO4− + 2H+ ⇌ Co3(PO4)2 + 3H2O, | (4) |
| | | 3CoO + 2HPO42− + H2O ⇌ Co3(PO4)2 + 4OH−. | (5) |
Phosphate ion monitoring in real water samples
The CoONPs/PBI/MWCNT electrode was used to monitor phosphate ions in creek water samples collected around Saga City, Japan. Before measurements, the sample pH was adjusted to pH 4 or 7 to ensure that the electrode worked under optimal conditions, and phosphate levels were determined by matching the potential values to the OCPs obtained at these pH values. The final concentration of phosphate was determined by adding phosphate to the analyzed samples to obtain a total concentration of 10 μM, and the validity of the obtained concentration was confirmed by ICP measurements. The phosphate additions were performed because the exceptionally low concentration of phosphate in the analyzed samples complicated its analysis by ICP with limited range detection for comparison and validation.
An OCP of 0.28 V was obtained at pH 4, indicating that the phosphate concentration was between 1.0 × 10−2 and 10 μM. Furthermore, the OCP obtained at pH 7 (0.31 V) indicated that the phosphate concentration was between 1 and 10 μM. The combined results suggested that the level of phosphate in the analyzed sample was approximately 10 μM. This result was in good agreement with that of the ICP measurements, indicating the suitability of our electrode for purely electrochemical phosphate quantitation (Table 2).
Table 2 Concentrations of phosphate ions in real water samples
| Sample no. |
pH |
Intended conc./μM |
ICP result/ppm |
ICP conc./μM |
E
OCP/V |
OCP conc./μM |
| 1 |
4 |
5–55 |
0.45 |
9.4 |
0.28 |
∼10 |
| 2 |
7 |
5–55 |
0.36 |
7.5 |
0.31 |
∼10 |
Conclusions
A new phosphate ion sensor based on CoONPs hybridized with PBI and MWCNTs was developed. The synthesized CoONPs were shown to be well dispersed on the MWCNTs and exhibit a uniform size of 3.5–5.5 nm, mainly comprising hexagonal Co3O4 with a fraction of CoO. The effects of morphology and microstructure on electrochemical performance were investigated to reveal that the CoONPs–containing electrode exhibited a considerably higher sensitivity than that of the bulk Co wire electrode. Specifically, the former electrode could sense phosphate at pH 4 and 7 and at concentrations (0.1–100 nM) lower than the eutrophication threshold. The CoONPs/PBI/MWCNT electrode was successfully used to detect phosphate in actual creek water samples.
Author contributions
Shaimah Rinda Sari: data curation, formal analysis, investigation, methodology, writing – original draft; Masayuki Tsushida: resources, formal analysis, investigation, validation; Tetsuya Sato: resources, formal analysis, investigation, validation; Masato Tominaga: conceptualization, investigation, funding acquisition, writing – review & editing, supervision.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful to Professor Keisuke Ohto (Saga University) for help with ICP measurements. This work was supported by the Nanotechnology Platform Program (Molecule and Material Synthesis) of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan (Grant no. JPMXP09 S20KU0040, JPMXP09 S21KU0041). We also acknowledge support in the form of research equipment sharing by the MEXT Project for Promoting the Public Utilization of Advanced Research Infrastructure (Program for Supporting the Introduction of the New Sharing System; Grant no. JPMXS0422400020).
Notes and references
-
R. G. Wetzel, Limnology: Lake and River Ecosystems, Academic Press, San Diego, 3rd edn, 2001, pp. 239–288 Search PubMed.
-
J. L. Domagalski and H. Johnson, Phosphorus and Groundwater: Establishing Links Between Agricultural Use and Transport to Streams, U.S. Geological Survey Fact Sheet 2012–3004, 2012, p. 4.
- S. O. Engblom, Biosens. Bioelectron., 1998, 13, 981–994 CrossRef CAS PubMed.
- N. C. Hanson, T. C. Daniel, A. N. Sharpley and J. L. Lemunyon, J. Soil Water Conserv., 2002, 57, 408–417 Search PubMed.
- C. Forano, H. Farhat and C. Mousty, Curr. Opin. Electrochem., 2018, 11, 55–61 CrossRef CAS.
-
R. Tirado and M. Allsopp, Greenpeace Research Laboratories Technical Report (Review) 02-2012, https://www.greenpeace.to/greenpeace/wp-content/uploads/2012/06/tirado-and-allsopp-2012-phosphorus-in-agriculture-technical-report-02-2012.pdf (accessed May 2021).
- J. L. Haberer and J. A. Brandes, Mar. Chem., 2003, 82, 185–196 CrossRef CAS.
- J. Murphy and J. P. Riley, Anal. Chim. Acta, 1962, 27, 31–36 CrossRef CAS.
- S. Lee, K. K. Y. Yuen, K. A. Jolliffe and J. Yoon, Chem. Soc. Rev., 2015, 44, 1749–1762 RSC.
- V. E. Zwicker, G. E. Sergeant, E. J. New and K. A. Jolliffe, Org. Biomol. Chem., 2021, 19, 1017–1021 RSC.
- J. Cleary, C. Slater, C. McGraw and D. Diamond, IEEE Sens. J., 2008, 8, 508–515 CAS.
- E. Climent, R. Casasús, M. D. Marcos, R. Martínez-Máñez, F. Sancenón and J. Soto, Dalton Trans., 2009, 4806–4814 RSC.
- L. A. Shaver, J. Chem. Educ., 2008, 85, 1097–1098 CrossRef CAS.
- L. Szekers, E. Kardos and G. L. Szekeres, Microchem. J., 1966, 11, 1–12 CrossRef.
- P. J. Antony, S. Karthikeyan and C. S. P. Iyer, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2002, 767, 363–368 CrossRef CAS.
- M. T. Galceran, M. Diez and L. Paniagua, J. Chromatogr. A, 1993, 657, 77–85 CrossRef CAS.
- H. Cheng, P. Hui, J. Peng, W. Li, W. Ma, H. Wang, J. Huang, X. He and K. Wang, Anal. Chem., 2021, 93(17), 6770–6778 CrossRef CAS PubMed.
- G. P. Ratkovski, K. T. O. do Nascimento, G. C. Pedro, D. R. Ratkovski, F. D. S. Gorza, R. J. da Silva, B. G. Maciel, L. C. Mojica-Sánchez and C. P. de Melo, Langmuir, 2020, 36(11), 2920–2929 CrossRef CAS PubMed.
- A. T. Lawal and S. B. Adeloju, Talanta, 2013, 114, 191–203 CrossRef CAS PubMed.
- K. Xu, Y. Kitazumi, K. Kano and O. Shirai, Electrochim. Acta, 2018, 282, 242–246 CrossRef CAS.
- D. Xiao, H.-Y. Yuan, J. Li and R.-Q. Yu, Anal. Chem., 1995, 67, 288–291 CrossRef CAS.
- S. G. Hosseini, S. J. H. Toloti and K. Babaei,
et al.
, J. Therm. Anal. Calorim., 2016, 124, 1243–1254 CrossRef CAS.
- K. Kargosha, P. Hemmatkhah and S. H. Ahmadi, Anal. Bioanal. Electrochem., 2017, 9, 521–534 CAS.
- E. T. Thostenson, C. Li and T. W. Chou, Compos. Sci. Technol., 2005, 65, 491–516 CrossRef CAS.
- L. Li and Y. Xing, J. Power Sources, 2008, 178, 75–79 CrossRef CAS.
- W. Li, C. Liang, J. Qiu, W. Zhou, H. Han, Z. Wei, G. Sun and Q. Xin, Carbon, 2002, 40, 787–790 CrossRef.
- Y. Lin, F. Lu, Y. Tu and Z. Ren, Nano Lett., 2004, 4, 191–195 CrossRef CAS.
- Y. Pan, Y. Chen, Y. Lin, P. Cui, K. Sun, Y. Liu and C. Liu, J. Mater. Chem. A, 2016, 4, 14675–14686 RSC.
- J. Tian, Q. Liu, A. M. Asiri and X. Sun, J. Am. Chem. Soc., 2014, 136, 7587–7590 CrossRef CAS PubMed.
- C. Gao, Z. Guo, J.-H. Liu and X.-J. Huang, Nanoscale, 2012, 4, 1948–1963 RSC.
- T. Fujigaya and N. Nakashima, Adv. Mater., 2013, 25, 1666–1681 CrossRef CAS PubMed.
- A. A. D'Archivio, L. Galantini, A. Biffis, K. Jeřábek and B. Corain, Chem. – Eur. J., 2000, 6, 794–799 CrossRef.
- M.-Y. Hua, H.-C. Chen, R.-Y. Tsai and C.-S. Lai, Talanta, 2011, 85, 631–637 CrossRef CAS PubMed.
- L. F. Villalobos, R. Hilke, F. H. Akhtar and K.-V. Peinemann, Adv. Energy Mater., 2018, 8, 1701567 CrossRef.
- M. R. Berber, H. Elkhenany, I. H. Hafez, A. El-Badawy, M. Essawy and N. El-Badri, Nanomedicine, 2020, 15, 793–808 CrossRef CAS PubMed.
- E. O. Eren, N. Özkan and Y. Devrim, Int. J. Hydrogen Energy, 2021, 46, 29556–29567 CrossRef CAS.
- M. Okamoto, T. Fujigaya and N. Nakashima, Adv. Funct. Mater., 2008, 18, 1776–1782 CrossRef CAS.
- R. Li, Z. Wei, X. Gou and W. Xu, RSC Adv., 2013, 3, 9978–9984 RSC.
- M. M. Zhang, R. Li, X. X. Chang, C. Xue and X. L. Gou, J. Power Sources, 2015, 290, 25–34 CrossRef CAS.
- Y. Meng, G. Wang, M. Xiao, C. Duan, C. Wang, F. Zhu and Y. Zhang, J. Mater. Sci., 2017, 52, 13192–13202 CrossRef CAS.
- Y. Chen, S. Jie, C. Yang and Z. Liu, Appl. Surf. Sci., 2017, 419, 98–106 CrossRef CAS.
- S. Chenakin and N. Kruse, Appl. Surf. Sci., 2020, 515, 146041 CrossRef CAS.
- S. Farid, W. Qiu, J. Zhao, X. Song, Q. Mao, S. Ren and C. Hao, J. Electroanal. Chem., 2020, 858, 113768 CrossRef CAS.
- A. Zecchina, D. Scarano, S. Bordiga, G. Spoto and C. Lamberti, Adv. Catal., 2001, 46, 265–397 CAS.
- N. Barsan, D. Koziej and U. Weimar, Sens. Actuators, B, 2007, 121, 18–35 CrossRef CAS.
- S. C. Petitto, E. M. Marsh, G. A. Carson and M. A. Langell, J. Mol. Catal. A: Chem., 2008, 281, 49–58 CrossRef CAS.
- L. Drummond and W. Maher, Anal. Chim. Acta, 1995, 302, 69–74 CrossRef CAS.
- M. J. Vazquez, B. Rodriguez, C. Zapatero and D. G. Tew, Anal. Biochem., 2003, 320, 292–298 CrossRef CAS PubMed.
- Y. Shimizu and Y. Furuta, Solid State Ionics, 1998, 241, 113–115 Search PubMed.
- W. H. Lee, Y. W. Seo and P. L. Bishop, Sens. Actuators, B, 2009, 137, 121–128 CrossRef PubMed.
- Z. Zou, J. Han, A. Jang, P. L. Bishop and C. H. Ahn, Biosens. Bioelectron., 2007, 22, 1902–1907 CrossRef CAS PubMed.
- L. Zhu, X. Zhou and H. Shi, Front. Environ. Sci. Eng., 2014, 8, 945–951 CrossRef CAS.
- S. Engblom, Plant Soil, 1999, 206, 173–179 CrossRef.
-
J. Hem, Study and Interpretation of the Chemical Characteristics of Natural Water, U.S. Geological Survey Water Supply Paper, 1992, p. 2254.
Footnote |
| † Electronic supplementary information (ESI) available: Additional CoONP characterization data. See DOI: 10.1039/d1ma01097b |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Masayuki
Tsushida
b,
Tetsuya
Sato
b and
Masato
Tominaga
a,
Masayuki
Tsushida
b,
Tetsuya
Sato
b and
Masato
Tominaga