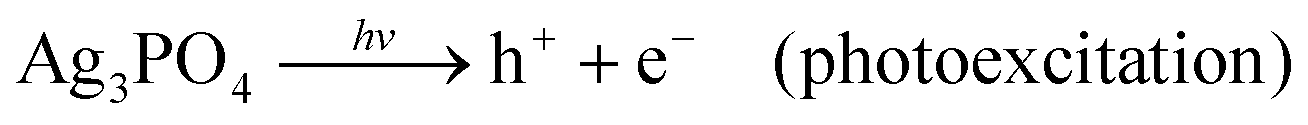DOI:
10.1039/D1MA01028J
(Paper)
Mater. Adv., 2022,
3, 2706-2715
A UV-visible-NIR active smart photocatalytic system based on NaYbF4:Tm3+ upconverting particles and Ag3PO4/H2O2 for photocatalytic processes under light on/light off conditions†
Received
4th November 2021
, Accepted 24th December 2021
First published on 24th December 2021
Abstract
The development of broad-spectrum photocatalytic materials that allow the use of a larger portion (UV to NIR) of the solar spectrum for photocatalytic processes has attracted great attention. Among visible light-active photocatalysts, silver phosphate (Ag3PO4) stands out prominent for its high photocatalytic activity towards the degradation of different pollutants. However, the full potential of this photocatalyst is limited by its inherent low photostability arising from self-photoreduction and its inability to efficiently use light beyond the UV-Vis range of the solar spectrum. To address these fundamental limitations of Ag3PO4, we have developed a smart photocatalytic system by combining the interesting upconversion property of NaYbF4:Tm3+ upconverting particles (UCPs) with the exceptionally high photocatalytic activity of Ag3PO4 and the oxidizing capacity of H2O2. In this UCPs/photocatalyst/oxidant (NaYbF4:Tm3+/Ag3PO4/H2O2) system, the NIR-to-UV/visible UCPs can convert low energy NIR photons into high energy UV-visible photons that can be absorbed by Ag3PO4, thus photo-exciting (activating) it indirectly under NIR illumination. Similarly, the oxidant (H2O2) prevents self-reduction and/or assists in regeneration of Ag3PO4 through a Fenton-like process, thus ensuring the photostability and recyclability of Ag3PO4. Interestingly, the Ag3PO4/H2O2 system remains active generating reactive oxygen species even after the photoexcitation process is turned off (dark conditions). The proposed broad spectrum photocatalytic system (UCPs/Ag3PO4/H2O2) was found to exhibit a high photocatalytic response (98% degradation of crystal violet dye in 90 min) under NIR illumination from a 980 nm laser and still a higher response (100% removal in less than 8 min) under direct visible light from low-cost blue emitting LEDs.
1. Introduction
Considering the growing environmental problems caused by anthropogenic activities as a global concern,1–4 the development of strategies and methods to counter these problems has become an active research area.5–12 Among environmental remediation strategies, heterogeneous photocatalysis based on semiconductor photocatalysts has so far been the most promising strategy with greater potential for combating environmental pollution on one hand and clean energy generation on the other.13–18 Unfortunately, most of the best known and commonly used semiconductor oxide photocatalysts are wide band semiconductors (TiO2 and ZnO, for example) and thus can only be photoactivated using ultraviolet (UV) radiation17,19–24 which is only around 5–6% of the solar spectrum. On the other hand, relatively narrow band photocatalysts (BiVO4, CdS, ZnSe, Ag3PO4 and Ag2O) exhibit photocatalytic activity under UV and visible (Vis) light,25–28 but often suffer from fast electron–hole recombination and limited photostability. For example, Ag3PO4 has been found to exhibit high photoactivity and thus high contaminant removal performance, but it shows low photostability and is photo(self)reduced (Ag+ + e− → Ag) during the photocatalytic process by the photoexcited conduction band (CB) electrons,29–32 thus limiting the recyclability of the photocatalysts.
One of the strategies to inhibit photocorrosion of Ag3PO4 is to prepare its composites with other electron-acceptor materials of adequate conductivity such as graphene oxide (GO) which can remove the photoexcited electrons from the CB of Ag3PO4. For instance, Q. Xiang and co-workers prepared GO-Ag3PO4 composites in which the photoactive component (Ag3PO4) exhibited improved photostability, a smaller particle size and reduced charge carrier recombination, thus leading to enhanced overall photocatalytic activity.33 The same results can be achieved and photo(self)reduction of Ag3PO4 can be prevented if the photocatalytic experiments are performed in an oxidizing environment, such as in the presence of H2O2 discussed in this article.
Furthermore, the presence of GO also provides additional active adsorption sites for effective adsorption of target molecules. Since adsorption plays an important role in photocatalytic reactions, enhanced photocatalytic degradation of pollutants has been reported for GO supported Ag3O4 and silica-supported TiO2 materials.33,34 Similarly, the enhanced CO2 photoreduction capability of nanosheet-assembled g-C3N4 has been associated with its high specific surface area and number of active sites and/or improved adsorption capacity towards CO2.35,36 Moreover, supported photocatalysts often exhibit increased active sites accessible for surface reactions. For instance, EPR studies of the mechanism of enhanced photocatalytic activity showed that the number of spins per gram (representing active sites) on the surface of silica-supported TiO2 (1.6 ± 0.3 × 1018 spin g−1 of TiO2) is higher as compared to pure or unsupported TiO2 (0.6 × 1018 spins g−1 of TiO2), which partly explains the higher photoactivity of the former.34
Like any photocatalyst, the photocatalytic activity of Ag3PO4 strongly depends on its particle size and the rate of charge carrier recombination, and decreasing these two parameters leads to improved photocatalytic activity.33 Although a narrow band gap is often useful for utilization of visible light, the position of the valence band (VB) and CB plays an important role in the formation of reactive oxygen species (ROS) and consequently the redox reactions of target molecules on the surface of the photocatalyst. For instance, the VB potential of Ag3PO4 is not positive enough to produce hydroxyl radicals (OH˙) from H2O/OH−, but Ag3PO4 can produce superoxide (O2˙−) radicals from the reduction of O2 present on or in the vicinity of the photocatalyst surface.33
Many attempts have been made to prepare new or modified and recyclable broad spectrum photocatalytic systems that can make use of a wider range of solar radiation (from UV to NIR), especially the NIR range which constitutes ∼50% of incident solar radiation.37–40 One promising strategy to extend the absorption/application range of UV-visible active photocatalysts is to couple them with lanthanide-based upconverting particles (UCPs) such as NaYbF4:Tm3+ and NaYF4:Yb3+/Tm3+.41–45 The UCPs can successively absorb two or more relatively low energy photons (NIR photons) via intermediate long-lived energy states and then emit higher energy photons (in the UV-Vis region) upon relaxation through a non-linear optical process known as upconversion.41–45 During this anti-Stokes (λemission < λexcitation) process, the UCPs are excited to a higher energy level through the population of intermediate energy levels and subsequently emit a higher energy photon.44–52 The photons emitted by the UCPs in the UV-Vis region can be exploited to photo-excite any photocatalysts with an adequate band gap (Eg) that matches the energy of the emitted photon (Eg = hv). Photoexcitation of the photocatalysts and interband transitions then result in the production of oxidative holes in VB and reductive electrons in the CB for the subsequent redox reaction on the photocatalyst surface responsible for the degradation of pollutants.45,53,54 Despite the theoretical feasibility of the NIR active UCPs/photocatalyst system, reported literature on this topic demonstrates the limited practical success and lower contaminant removal efficiency of the system, often requiring several hours of NIR illumination.54–58 The combination of different advanced oxidation processes such as photocatalysis and Fenton-like processes using H2O2 as an oxidizing agent is a possible strategy to address the problem of low photoactivity under NIR irradiation.59–61
In this work, we report a smart photocatalytic system based on NaYbF4:Tm3+ UCPs/Ag3PO4/H2O2 that can be activated under both direct visible light illumination from low-cost blue LEDs (460 ± 10 nm) as well as NIR illumination, thanks to the NIR-to-UV/visible upconversion by the UCPs. Moreover, not only the presence of H2O2 improves the photoactivity and photostability of Ag3PO4 (by oxidizing back/regenerating the photo-(self)reduced Ag back to Ag+), but also the process of regeneration/oxidation leads to the formation of reactive oxygen species through a Fenton-like process, even when the photoexcitation process is stopped (light off condition). The strategy proposed herein may be extended to other photocatalytic systems for improving their photocatalytic activity and recyclability under a broad range of solar radiation and illumination modes.
2. Experimental
2.1. Reagents
Yb2O3 (99.99%), Tm2O3 (99.99%), sodium fluoride (NaF) and bovine catalase enzyme (>10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 units per mg protein) were supplied by Sigma-Aldrich (USA). Ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA-Na2) was acquired from Merck. Nitric acid (HNO3) and crystal violet (CV) were purchased from Qhemis (Brazil). Hydrogen peroxide (H2O2, 50%), ethanol (C2H5OH) and silver acetate (CH3COOAg, 98%) were supplied by Synth (Brazil). All chemicals were used without further purification.
000 units per mg protein) were supplied by Sigma-Aldrich (USA). Ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA-Na2) was acquired from Merck. Nitric acid (HNO3) and crystal violet (CV) were purchased from Qhemis (Brazil). Hydrogen peroxide (H2O2, 50%), ethanol (C2H5OH) and silver acetate (CH3COOAg, 98%) were supplied by Synth (Brazil). All chemicals were used without further purification.
2.2. Synthesis of the Ag3PO4 photocatalyst
The Ag3PO4 photocatalyst was synthesized by a facile precipitation process at room temperature,62 followed by thermal treatment at 200 °C.63 Briefly, 125 mL (7.5 mmol) of silver acetate solution was added dropwise under continuous stirring to a 100 mL (2.5 mmol) Na2HPO4·12H2O solution. The resulting yellow suspension was stirred for 30 min and the Ag3PO4 particles were then recovered by centrifugation at 7500 rpm. The precipitated particles were washed several times with deionized water, dried in a vacuum oven at 80 °C and finally calcined at 200 °C for 1 h.
2.3. Synthesis of NaYbF4:Tm3+(0.5) UCPs
The NaYbF4:Tm3+(0.5) UCPs were synthesized by a microwave-assisted hydrothermal methodology developed by our group.44,45 A commercial microwave synthesizer (Discover SP model 909150, CEM Corp.) with a 35 mL pressure vessel (CEM Part no. 909036) covered with a special cap (CEM part no. 909235) provided by the manufacturer was used. Briefly, Ln(NO3)3.6H2O solutions (Ln = Yb3+, Tm3+) were prepared separately from the reaction of the respective oxides with concentrated nitric acid, followed by repeated addition/evaporation of water until the pH of the solution was around 3. The resulting solutions were standardized by titrating with a standard 0.01 mol L−1 EDTA solution. Then, appropriate amounts of Ln(NO3)3.6H2O solutions (total Ln3+ content = 1 mmol) were added to 10 mL (2 mmol) of EDTA solution. After stirring for 30 min, 15 mL (15 mmol) of NaF was added to the above Ln3+–EDTA solution and the resulting mixture was vigorously stirred for 30 min. The obtained colloidal suspension was subjected to microwave-assisted hydrothermal treatment for 1 h at 190 °C, 250 psi and 200 W of microwave power. The NaYbF4:Tm3+(0.5) UCPs were recovered by centrifugation at 7500 rpm and then vacuum-dried after being washed once with ethanol and twice with deionized water following centrifugation–resuspension cycles. To remove EDTA from the particle surface, the UCPs were calcined at 400 °C for 90 min.
2.4. Photocatalytic experiments
The photocatalytic experiments were carried out under two different illumination modes, namely blue LED illumination (direct photoexcitation of Ag3PO4) and 980 nm laser irradiation (indirect photoexcitation of Ag3PO4 by the unconverted UV-visible radiation from UCPs).
2.4.1. Visible light photocatalytic activity under 460 ± 10 nm LED irradiation.
The photocatalytic activity of the synthesized Ag3PO4 photocatalyst (25 mg) under blue light irradiation provided by LEDs (460 ± 10 nm) was evaluated through photodegradation of CV dye solution (7.5 mg L−1, 30 mL). The photocatalyst/CV mixture in a glass-cylindrical-reactor (10.4 cm in diameter, 5.9 cm in height) was allowed to stir in the dark for 8 min and then exposed to blue light for another 8 min. An aliquot of 1.2 mL was collected every 2 min and centrifuged to monitor changes in the electronic absorption spectrum of the supernatant part. The experiments in the presence of H2O2 were carried out using the same procedure but with the addition of 110 μL or 200 μL of 1 mol L−1 H2O2 (final H2O2 concentration of 3.6 or 6.6 mmol L−1) to the 30 mL CV–Ag3PO4 mixture just before starting irradiation. Sample aliquots were withdrawn at regular intervals and 30–40 μL of bovine catalase solution (0.4 g L−1) was added to each aliquot withdrawn to consume any residual H2O2 and interrupt the reaction. Regeneration of the Ag3PO4 photocatalysts once used in the dye degradation test (in the absence of H2O2 in the reaction mixture) was performed. For this purpose, 200 μL of H2O2 was added to the photocatalytic mixture at the end of degradation experiments and the photocatalyst/H2O2 mixture was kept under stirring for several hours. Finally, the regenerated photocatalyst was recovered by centrifugation and washed several times with ethanol and water.
2.4.2. NIR photocatalytic activity evaluation.
The photocatalytic activity of the UCPs/Ag3PO4 photocatalyst and UCPs/Ag3PO4/H2O2 systems under NIR excitation (980 nm laser) was evaluated using 3 mL of CV dye solution (4 mg L−1) and 10 mg of photocatalysts (mixture of UCPs + Ag3PO4 in different ratios) in a 4 mL glass vial. The CV/UCPs/Ag3PO4 mixture was left in the dark overnight and then irradiated with NIR radiation using a 980 nm diode laser (DMC, Brazil) coupled to a 1.5 mm diameter optical fiber at a 2 W laser power. Sample aliquots were collected for the measurement of UV-visible absorption spectra as described in Section 2.4.1, after which the sample was transferred back to the photoreactor. Different ratios of UCPs/Ag3PO4 were considered to find the optimal UCPs/photocatalyst ratio for effective dye degradation under 980 nm laser irradiation. Once the optimum ratio was found (6 mg UCPs/4 mg Ag3PO4), further experiments were carried out using this ratio in the presence of 6.6 mmol L−1 of H2O2 to analyze its influence on the degradation of CV dye under 980 nm laser irradiation and the stability/recyclability of Ag3PO4.
2.5. Characterization techniques
Scanning electron microscopy (SEM) and energy dispersive X-ray (EDX) spectroscopy measurements were performed using a field emission scanning electron microscope (FEG-SEM) (JEOL, model JSM-7500 F). Powder X-ray diffractograms (XRD) were acquired using a D8 Advance X-ray diffractometer (Bruker) operating at 40 mA and 40 kV and employing Ni-filtered Cu Kα X-ray radiation (λ = 1.540 Å). Fourier transform infrared (FTIR) spectra were obtained using a PerkinElmer Spectrum 1000 FTIR spectrometer with a resolution of 2 cm−1 and averaged over four scans. Raman spectra were measured in the 100–1000 cm−1 range with an acquisition time of 50 s and 2 cycles using a LabRAM HR 800 Raman spectrophotometer (Horiba Jobin Yvon) equipped with a CCD detector (model DU420A-OE-325) and a He–Ne laser (632.81 nm). The diffuse reflectance spectra (DRS) of the powder samples against a background of MgO were obtained using a Cary 5000 UV-Vis-NIR spectrophotometer (Varian). The upconversion luminescence (UCL) spectra of UCPs (8 mg of NaYbF4:Tm3+(0.5) in 10 mL of water) with an optical spectral resolution of 0.5 nm were acquired using a Horiba Jobin Yvon spectrofluorometer (Fluorolog-3 model FL3-122) equipped with a Hamamatsu R-928 photomultiplier tube. A 980 nm laser diode (DMC, Brazil) coupled with an optical fiber (1.5 mm diameter) was used as the NIR excitation source.
3. Results and discussion
3.1. Characterization of the Ag3PO4 photocatalyst
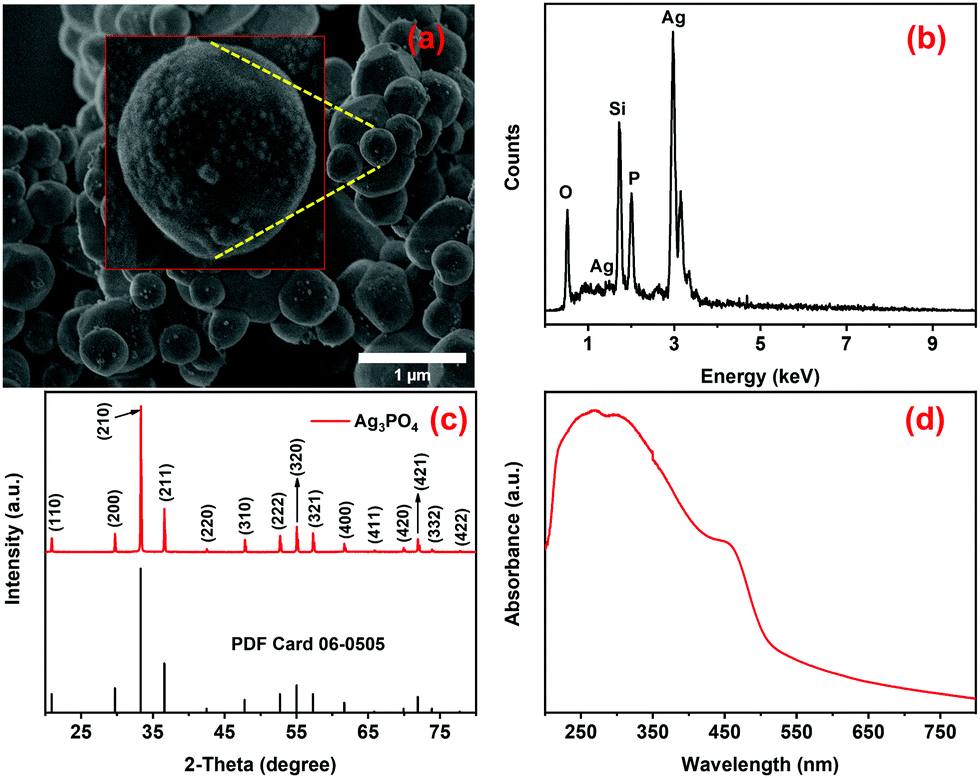
The morphological, compositional, crystallographic, and optical properties of the calcined Ag3PO4 particles, previously prepared by a facile room temperature method, were studied using SEM, EDX, XRD, and DRS, respectively. The Ag3PO4 particles are micrometric (mean: 475 ± 182 nm) with their surface decorated with small (approximately 16 ± 4 nm size) nanospheres (Fig. 1a), as also previously observed by P. Dong and collaborators.63 They reported the formation of small Ag nanoparticles, promoted by heat treatment, on the surface of calcinated Ag3PO4.63 The EDS spectrum (Fig. 1b) shows clear emission lines of Ag, P and O, confirming the presence of these elements in the samples.
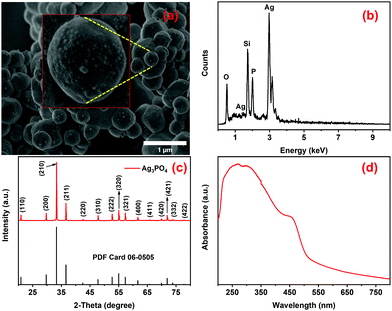 |
| | Fig. 1 (a) SEM image, (b) EDS spectrum, (c) XRD pattern and (d) UV-Vis-NIR absorption spectrum of the Ag3PO4 photocatalyst. The Si signal in the EDX spectrum comes from the sample holder. The inset in a shows a magnified view of the Ag3PO4 photocatalyst. | |
As shown in Fig. 1c, all the diffraction peaks in the diffractogram of the sample perfectly match with those of the body-centered cubic phase of Ag3PO4 with the space group P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3n (PDF no. 06-0505). The optical absorption property is an important factor that can determine the photocatalytic efficiency of materials under irradiation in a certain part of the electromagnetic spectrum and, therefore, the absorption spectrum of Ag3PO4 powder was measured by diffuse reflectance spectroscopy (Fig. 1d). The absorption transformed DRS spectrum of the Ag3PO4 particles shows strong absorption in the UV-Vis region (Fig. 1d), attributed to the intrinsic charge transfer of Ag3PO4 from the VB to the CB,29,63–65 with an indirect band-gap of 2.30 eV, as estimated from the Kubelka–Munk plot shown in Fig. S1a (ESI†). The strong absorption in the visible region (540–400 nm) overlaps well with the emission from blue LEDs (460 ± 10 nm), allowing effective photoexcitation of Ag3PO3 and photodegradation of pollutants using low-cost commercial LEDs as the illumination source (vide infra). The Ag3PO4 samples were also characterized by vibrational spectroscopy. The FTIR spectrum of the sample exhibits four typical FTIR bands of Ag3PO4 (Fig. S1b, ESI†), two of which lying around 560 cm−1 and 1010 cm−1 are of medium/high intensity and can be attributed to the O
3n (PDF no. 06-0505). The optical absorption property is an important factor that can determine the photocatalytic efficiency of materials under irradiation in a certain part of the electromagnetic spectrum and, therefore, the absorption spectrum of Ag3PO4 powder was measured by diffuse reflectance spectroscopy (Fig. 1d). The absorption transformed DRS spectrum of the Ag3PO4 particles shows strong absorption in the UV-Vis region (Fig. 1d), attributed to the intrinsic charge transfer of Ag3PO4 from the VB to the CB,29,63–65 with an indirect band-gap of 2.30 eV, as estimated from the Kubelka–Munk plot shown in Fig. S1a (ESI†). The strong absorption in the visible region (540–400 nm) overlaps well with the emission from blue LEDs (460 ± 10 nm), allowing effective photoexcitation of Ag3PO3 and photodegradation of pollutants using low-cost commercial LEDs as the illumination source (vide infra). The Ag3PO4 samples were also characterized by vibrational spectroscopy. The FTIR spectrum of the sample exhibits four typical FTIR bands of Ag3PO4 (Fig. S1b, ESI†), two of which lying around 560 cm−1 and 1010 cm−1 are of medium/high intensity and can be attributed to the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P–O bending vibration in the plane and the asymmetric stretching of P–O–P groups, respectively.66–68 The other two bands of low intensity, situated at 850 and 1385 cm−1, are related to the symmetric stretching vibration of P–O–P bonds and the stretching vibration of the PO bonds in the Ag3PO4 structure.67,69 Additionally, the vibrational bands around 3390 cm−1 and 1650 cm−1 are attributed to the stretching vibrations of O–H groups and H–O–H bending vibrations of the adsorbed water molecules, respectively.67,69 Similarly, the Raman spectrum of Ag3PO4 samples exhibits the characteristic vibrational modes of the PO43− groups in a Td symmetry around 405, 550, 907, 950 and 1000 cm−1,64,70 confirming the formation of Ag3PO4 (Fig. S1c, ESI†).
P–O bending vibration in the plane and the asymmetric stretching of P–O–P groups, respectively.66–68 The other two bands of low intensity, situated at 850 and 1385 cm−1, are related to the symmetric stretching vibration of P–O–P bonds and the stretching vibration of the PO bonds in the Ag3PO4 structure.67,69 Additionally, the vibrational bands around 3390 cm−1 and 1650 cm−1 are attributed to the stretching vibrations of O–H groups and H–O–H bending vibrations of the adsorbed water molecules, respectively.67,69 Similarly, the Raman spectrum of Ag3PO4 samples exhibits the characteristic vibrational modes of the PO43− groups in a Td symmetry around 405, 550, 907, 950 and 1000 cm−1,64,70 confirming the formation of Ag3PO4 (Fig. S1c, ESI†).
3.2. Morphological/structural characterization and optical properties of NaYbF4:Tm3+(0.5) UCPs
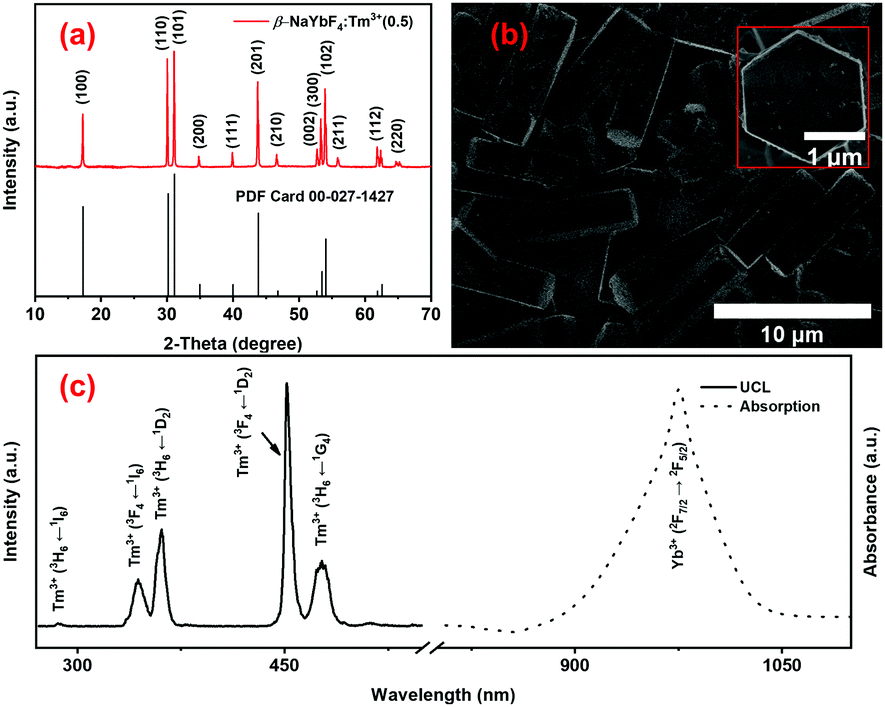
The structural, morphological and optical properties of the NaYbF4:Tm3+(0.5) UCPs, synthesized by a facile microwave-assisted hydrothermal method,44,45 were studied using XRD, SEM and photoluminescence (PL) spectroscopy, respectively (Fig. 2). The X-ray diffraction patterns of UCPs perfectly match with the standard diffraction pattern of the hexagonal β-NaYbF4 phase (PDF no. 27-1427) (Fig. 2a) which is usually associated with stronger UCL than its cubic (α-NaYbF4) counterpart. This UCP sample consists of microrods of around 7.9 ± 1.4 μm length and 2.5 ± 0.5 μm diameter (Fig. 2b). A magnified view of a vertically oriented microrod reveals its hexagonal shape (inset in Fig. 2b), in accordance with the results of the XRD analysis (Fig. 2a). The presence of Yb3+ ions and/or NIR absorption properties of the UCPs are evident from the characteristic absorption peak of Yb3+ at around 975 nm in the electronic absorption spectrum, obtained by measuring the diffuse reflectance from the powder sample (Fig. 2c, dotted line).71 The presence of Yb3+ thus allows the UCPs to absorb the NIR photons from a 980 nm laser while doping with Tm3+ ions as the emitters assists in obtaining upconverted light in the UV and visible region of the spectrum (Fig. 2c, solid line). The UCL spectrum of an aqueous suspension of UCPs, measured under 980 nm laser excitation, thus shows emission peaks centered around 286, 344, 361, 452, and 477 nm which are, respectively, attributed to the 1I6 → 3H6, 1I6 → 3F4, 1D2 →3H6, 1D2 → 3F4 and 1G4 → 3H6 transitions of Tm3+ ions (Fig. 2c, solid line),44–48 as explained later.
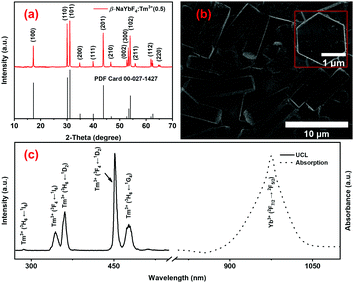 |
| | Fig. 2 (a) XRD pattern, (b) SEM image and (c) UV-Vis-NIR absorption spectrum (dotted line) and UCL spectrum under 980 nm laser irradiation (black solid line) of NaYbF4:Tm3+ UCPs. The inset in b shows a magnified view of a single NaYbF4:Tm3+ UCP. | |
3.3. Photocatalytic activity evaluation
Two main objectives of the present study were to improve the photostability of Ag3PO4 (by employing H2O2) and extend the application of this photocatalyst beyond the UV-visible region and into the NIR region (by coupling UCPs with Ag3PO4). Thus, detailed photocatalytic assays were performed employing Ag3PO4 and Ag3PO4-UCP samples in the presence and absence of H2O2 using either visible light from low-cost blue (460 ± 10 nm) commercial LEDs or NIR light from a laser. Evolution of the photocatalytic degradation process by the Ag3PO4–H2O2 system under light ON/light OFF modes was also studied.
3.3.1. Photocatalytic activity of Ag3PO4/H2O2 under visible light from blue LEDs.
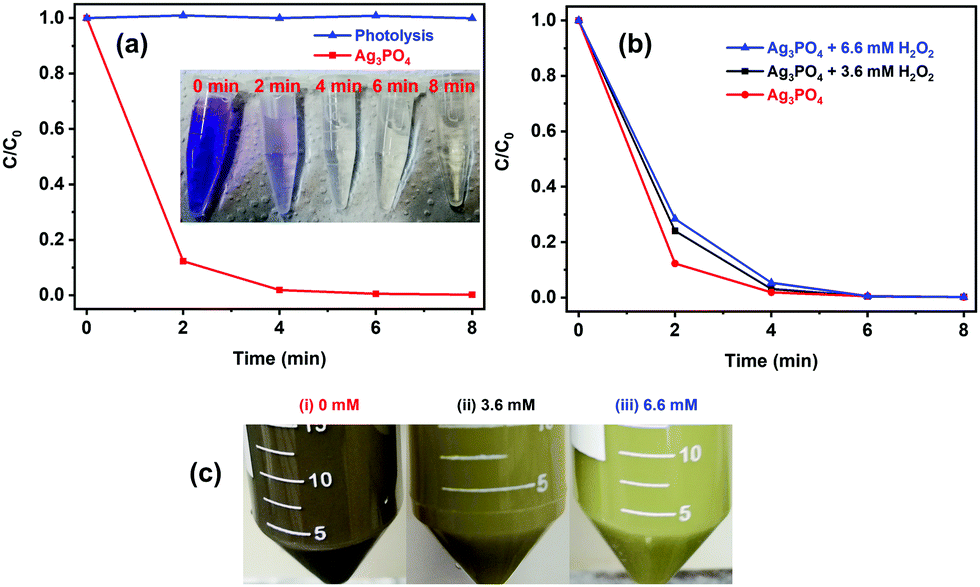
The visible photocatalytic activity of Ag3PO4 particles was examined under direct excitation from blue (460 ± 10 nm) LEDs (Fig. 3). No significant decrease in the absorbance of CV dye was observed in the absence of Ag3PO4, suggesting that direct photolysis does not degrade the dye. On the other hand, in the presence of Ag3PO4, the normalized absorbance of the dye at around 582 nm exhibits a drastic decrease within just 2 min of LED illumination (Fig. 3a, red line) and almost 100% dye discoloration is achieved in less than 8 min (see the inset in Fig. 3a), indicating the high visible light photocatalytic activity of the prepared Ag3PO4 particles.
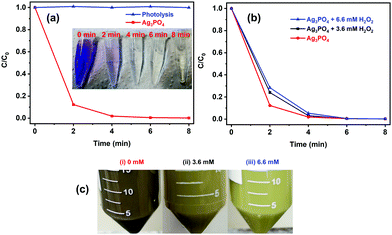 |
| | Fig. 3 Temporal change in the concentration of CV dye as a function of irradiation time with blue LEDs in the (a) absence (blue line) and presence (red line) of the Ag3PO4 photocatalyst, and in the (b) absence and presence of H2O2 in the dye–Ag3PO4 mixture. The inset in (a) shows the digital images of CV dye solution at different irradiation times (marked on the images) in the presence of the Ag3PO4 photocatalyst. (c) Digital images of aqueous dispersions of the Ag3PO4 photocatalyst recovered and washed after four photocatalytic cycles corresponding to the experiments carried out in the absence of H2O2 (i) and with the addition of 3.6 mmol L−1 (ii) and 6.6 mmol L−1 (iii) of H2O2. | |
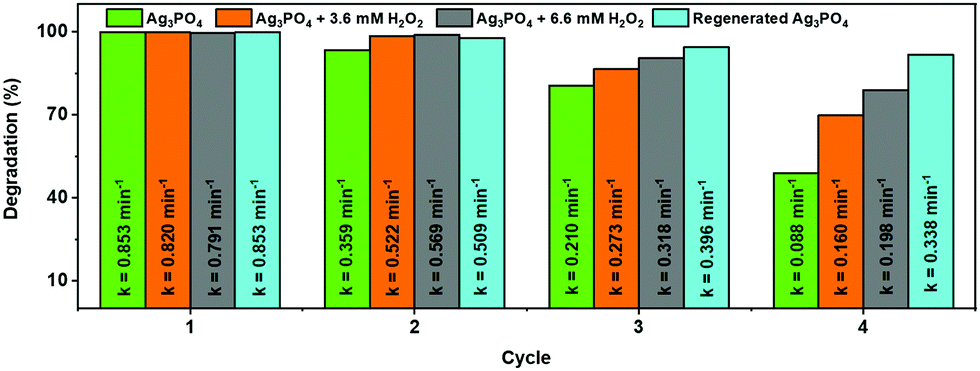
Unfortunately, the high photocatalytic activity was accompanied by a change in both the morphology (Fig. S2a and b, ESI†) and color (from yellow to black) of the used photocatalyst (Fig. 3c(i)), suggesting the decomposition of the photocatalysts and/or self-reduction of Ag+ to Ag by the photoexcited CB electron.29 The presence of H2O2 in the photocatalytic reaction media, however, prevented any change in the morphology of the particles (Fig. S2c and d, ESI†) as well as the darkening of the photocatalyst suspension during the photocatalytic experiments (Fig. 3c(ii/iii)), without significantly affecting the original photocatalytic activity of the sample (Fig. 3b). The change in the color and morphology of the Ag3PO4 photocatalyst during the photocatalytic experiments in the absence of H2O2 is also accompanied by a loss in photocatalytic activity (green bar in Fig. 4), thus negatively affecting the recyclability of the photocatalyst. In fact, the observed photodegradation rate constant (k) decreases around 10 times, going from 0.853 min−1 (first cycle) to 0.088 min−1 (4th cycle). The loss in photocatalytic activity in the absence of H2O2 after repeated photodegradation cycles is attributed to the photo-assisted self-reduction of Ag+ to Ag, mediated by the photoexcited electrons in the CB of Ag3PO4 (eqn (1)–(5)).29–32
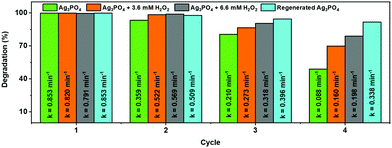 |
| | Fig. 4 Evaluation of the effect of H2O2 as an auxiliary oxidizing agent on the photostability and recyclability of Ag3PO4 during repeated photocatalytic degradation cycles under blue LED irradiation. | |
The photoassisted self-reduction of Ag3PO4 occurs according to the following reactions:
| | 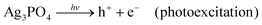 | (1) |
| | | Ag3PO4 + e− → Ag + Ag2PO4− | (2) |
| | | Ag2PO4− + e− → Ag + AgPO42− | (3) |
| | | AgPO42− + e− → Ag + PO43− | (4) |
| | | Ag3PO4 + 3e− → 3Ag + PO43− (self-reduction/photocorrosion) | (5) |
In fact, the XRD patterns of the used Ag
3PO
4 show the presence of a diffraction peak of Ag
0 at around 38.1° (PDF no. 65-2871) when no H
2O
2 is used during the photocatalytic tests (Fig. S4, ESI
†). However, the intensity of this peak is much lower when H
2O
2 is added to the Ag
3PO
4/dye mixture during photocatalytic tests (Fig. S4, ESI
†), indicating the importance of H
2O
2 for the photostability and recyclability of Ag
3PO
4.
During the photocatalyst regeneration process, H2O2 can oxidize the Ag back to Ag+ and thus reform Ag3PO4 through a Fenton-like process (eqn (6)–(9)),72–76 as shown below.
| | | Ag + H2O2 → Ag–O(H)–OH | (6) |
| | | Ag–O(H)–OH + H+ → Ag+ + HO˙ + H2O | (7) |
| | | Ag–O(H)–OH + H2O2 → Ag+ + O2˙− + 2H2O | (8) |
| | | 3Ag+ + PO43− → Ag3PO4 (regeneration step) | (9) |
Control experiments in the absence of Ag
3PO
4 in the reaction mixture or in the absence of LED illumination showed no significant decreases in the absorbance of the CV dye in 8 min (Fig. S3, ESI
†). Although the presence of H
2O
2 slightly decreased the dye degradation rate in the first photocatalytic cycle (
Fig. 3b), it significantly improved the structural stability (Fig. S2 and S4, ESI
†) and recyclability of Ag
3PO
4 as mentioned above (
Fig. 3c, 4 and Fig. S2, S4, ESI
†). These results demonstrate that the presence of H
2O
2 had a positive influence on the photocatalytic performance during repeated cycles of use (orange and gray bars in
Fig. 4). The slight decrease in the photoactivity of the Ag
3PO
4/H
2O
2 system after repeated photocatalytic cycles may be related to the incomplete recovery of the photocatalyst or partial photoreduction of Ag
3PO
4 to Ag
0. Previous studies have shown that the oxygen vacancies present on the surface of Ag
3PO
4 play an important role in its photocatalytic activity and the slight decrease in photoactivity upon repeated use may also be related to the loss of these vacancies.
63 Furthermore, the Ag
3PO
4 used in the degradation experiments, without the addition of H
2O
2, was recovered and then regenerated with H
2O
2 in an
ex situ manner (Fig. S2e and f, ESI
†). This regenerated photocatalyst was as efficient as the freshly prepared new photocatalysts (cyan bar in
Fig. 4), thus demonstrating a remarkable recovery of its photocatalytic activity.
3.3.2. Photocatalytic activity of the Ag3PO4/H2O2 system after turning off the excitation light (dark).
Fig. 5 shows that, in the presence of both Ag3PO4 and H2O2, the absorbance or concentration of CV dye continues to decrease even when the LED illumination is turned off at the end of the 4th photodegradation cycle. On the other hand, no significant post-irradiation change in concentration was observed in the absence of H2O2 in the reaction mixture. Since Ag3PO4 can be charged with photoinduced reductive energy by the excitation light from LEDs (eqn (1)–(5)), this energy can then be used to produce reactive oxygen species (ROS) in the presence of H2O2, even in the dark, as can be observed from Fig. 5. The results of Fig. 5 can thus be explained based on eqn (6)–(8)72–76 where the peroxide-induced oxidation of Ag0 to Ag+ or regeneration of Ag3PO4 is accompanied by the generation of ROS (HO˙ and O2˙−) that can take part in the photodegradation of the dye even after the excitation light is turned off. Although previous studies suggest that Ag3PO4 cannot produce HO˙ radicals from H2O/OH− as its VB edge potential is not enough, HO˙ radicals can be produced in the presence of H2O2, even in the dark (eqn (7)). Under adequate light illumination, Ag3PO4 has been found to produce superoxide radicals (O2˙−) and oxidative holes that degrade the organic dye molecules.33 The presence of H2O2 thus not only ensures the improved photostability and recyclability of Ag3PO4, but also allows photoinduced electron storage and harnessing of the stored chemical energy for redox processes even after the photoexcitation light is turned off.
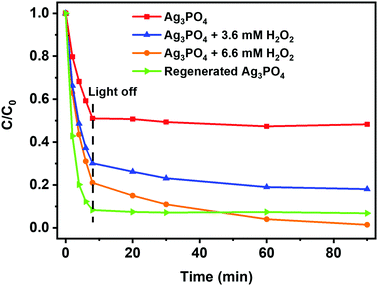 |
| | Fig. 5 Changes in the concentration of CV dye during the fourth photocatalytic cycle under irradiation (the first 8 min) and in the dark (after 8 min) using the photocatalyst recovered in the experiments performed in the absence of H2O2 (red line) and in the presence of 3.6 mmol L−1 (blue line) and 6.6 mmol L−1 of H2O2 (orange line) and the regenerated material (green line). | |
3.3.3. Photocatalytic activity of UCPs/Ag3PO4/H2O2 under NIR illumination.
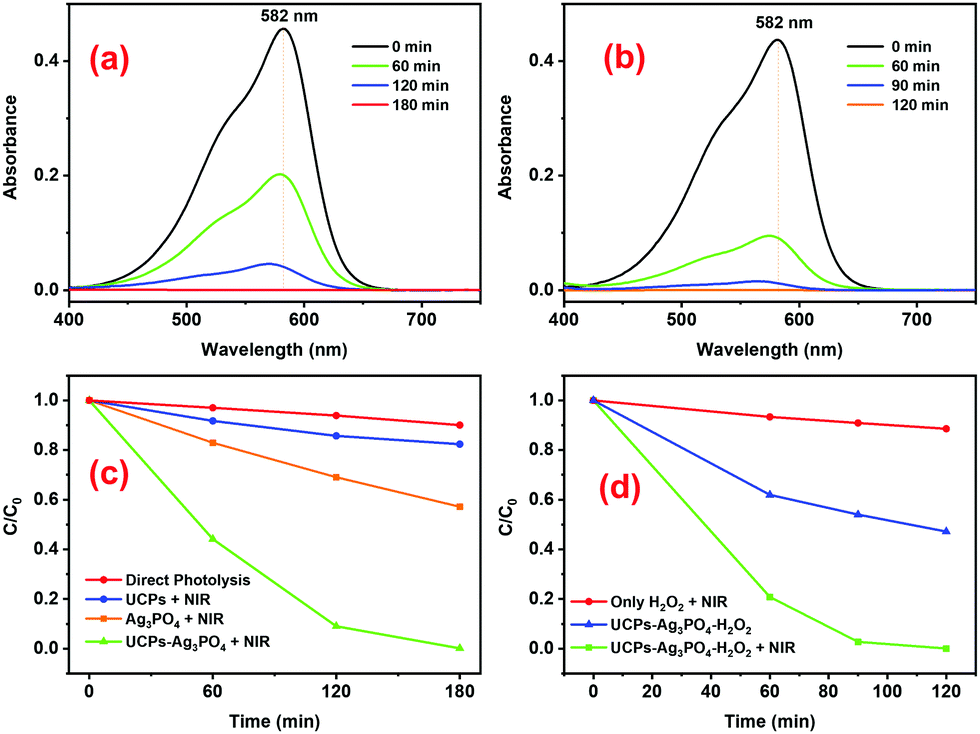
After confirming the excellent inherent photocatalytic activity of the prepared Ag3PO4 under visible light, we evaluated the NIR photoactivity of the UCPs–Ag3PO4 system under 980 nm illumination from a laser (Fig. 6). A clear decrease in the absorbance of CV dyes as a function of NIR irradiation time in the presence of UCPs–Ag3PO4 (Fig. 6a) and/or UCPs–Ag3PO4–H2O2 (Fig. 6b) confirms the NIR-based photoactivity of the systems.
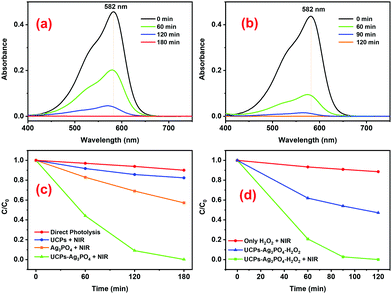 |
| | Fig. 6 Temporal evolution of the absorbance of CV under 980 nm laser irradiation in the presence of (a) UCPs–Ag3PO4 and (b) and UCPs–Ag3PO4–H2O2 systems. Concentration profiles showing a decrease in the concentration of CV dye as a function of NIR irradiation time in the presence of (c) UCPs–Ag3PO4 and (d) UCPs–Ag3PO4–H2O2 systems. Different controls are also shown in (c and d). | |
An important factor determining the degradation efficiency of a certain pollutant using the UCPs–Ag3PO4 photocatalytic system is the ratio between the UCPs and the Ag3PO4 photocatalyst. A suitable UCP/Ag3PO4 ratio may allow efficient absorption of the excitation (NIR) light by the UCPs, leading to their strong UCL emission which, in turn, may efficiently photoactivate the photocatalyst particles for effective photodegradation of the pollutants in the vicinity of UCP–Ag3PO4 particles. Therefore, we evaluated the efficiency of different UCP/Ag3PO4 ratios keeping the total amount of the photocatalyst system (UCPs + Ag3PO4) fixed at 10 mg (Fig. S5, ESI†). It turns out that this ratio really plays an important role in the degradation of CV dye under 980 nm laser irradiation (Fig. S5, ESI†) and an increase in the degradation rate was clearly observed until reaching a maximum for the 6/4 UCPs/Ag3PO4 ratio (Fig. S5e, ESI†). Thus, further photocatalytic experiments presented in Fig. 6 were performed using the 6/4 ratio of UCPs/Ag3PO4 and an almost 100% degradation of the dye was achieved in 180 min (Fig. 6c). Additionally, the presence of H2O2 led to a considerable improvement in the degradation of the dye under 980 nm laser irradiation, reaching almost 100% degradation of the dye in just 90 min of NIR illumination (Fig. 6b and d).
Control experiments were performed to better understand the photocatalytic process and evaluate the contribution of different possible factors to it. For example, it may be noted that direct photolysis (DP) of the dye by the NIR laser, irrespective of the absence or presence of H2O2 (red lines in Fig. 6c and d, respectively), is insignificant. Similarly, we observed an around 40% decrease in the concentration of CV dye in the presence of only Ag3PO4 (orange line in Fig. 6c), possibly due to direct excitation of the highly photoactive Ag3PO4 by the NIR laser. Likewise, a significant reduction in the concentration of CV dye by the UCPs–Ag3PO4–H2O2 system in the absence of NIR illumination was observed (blue line in Fig. 6d), which might be attributed to the production of ROS by a Fenton-like reaction between H2O272–76 and the Ag particles on the surface of Ag3PO4 formed during the calcination process.63,77 In either case, the decrease in the concentration of CV dye in the presence of UCPs–Ag3PO4 and/or UCPs–Ag3PO4–H2O2 systems under NIR illumination is much higher (100%), confirming the NIR photocatalytic activity of these two systems.
3.3.4. Mechanism of the NIR based photocatalytic activity.
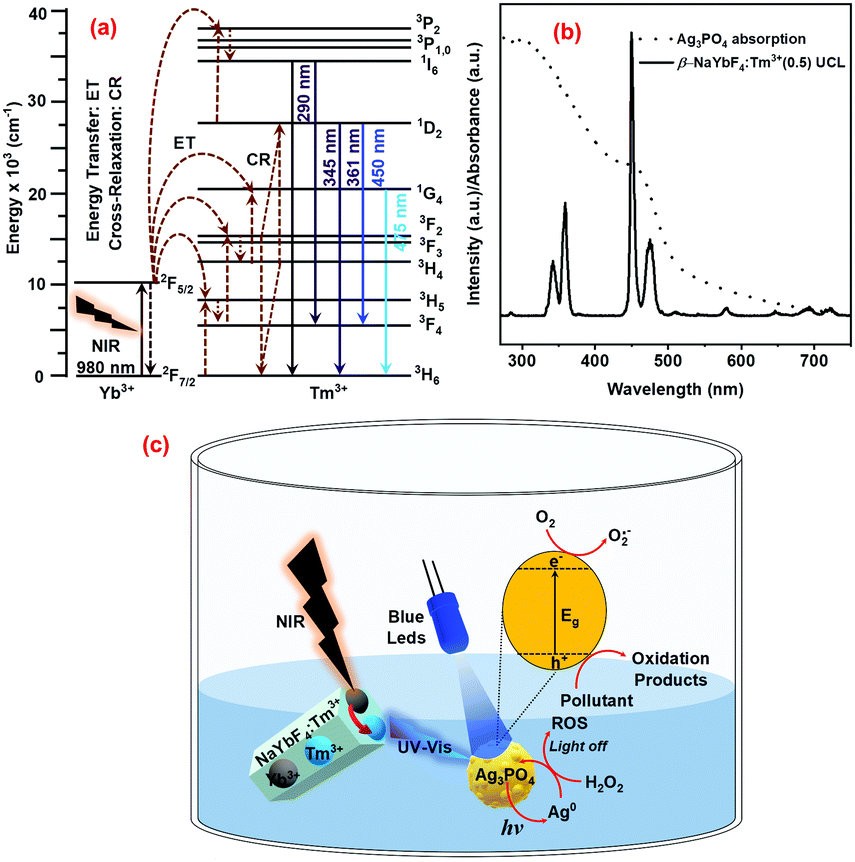
As shown earlier in Fig. 2c, the NaYbF4:Tm3+(0.5) UCPs could efficiently convert the 980 nm NIR light into UV-visible light at 286, 344, 361, 452, and 477 nm, thanks to the presence of Yb3+ ions (sensitizer/NIR absorber) and Tm3+ ions (UV-visible emitters). As schematically presented in Fig. 7a, the Yb3+ ion absorbs the NIR photons (980 nm) from the laser leading to population of the higher energy state (2F5/2) as a result of the 2F7/2 → 2F5/2 transition. The excited Yb3+ transfers its energy to the emitter Tm3+ ion through successive energy transfer (ET) steps, resulting in the population of its higher energy levels. The excited Tm3+ ion then relaxes by emitting upconverted photons of shorter wavelengths (than the excitation wavelength of 980 nm) at around 286, 344, 361, 452, and 477 nm which are, respectively, attributed to the 1I6 → 3H6, 1I6 → 3F4, 1D2 → 3H6, 1D2 → 3F4 and 1G4 → 3H6 transitions of Tm3+ ions (Fig. 7a).44–48
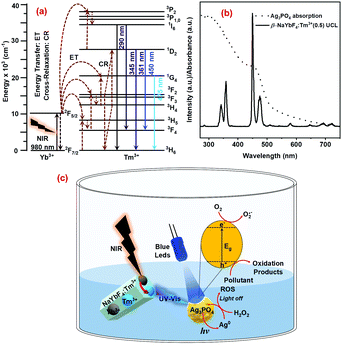 |
| | Fig. 7 (a) Upconversion mechanism of Tm3+ doped NaYbF4 UCPs under 980nm laser excitation, (b) comparison between the absorption spectrum of the Ag3PO4 photocatalyst and the UCL spectrum of NaYbF4:Tm3+(0.5) UCPs, and (c) photoactivation of the Ag3PO4 photocatalyst under UV-Vis/NIR irradiation and its regeneration with H2O2. | |
These upconverted photons with a wavelength shorter than 500 nm can be absorbed by Ag3PO4 (Eg = 2.3 eV), as suggested by the excellent overlap between the absorption spectrum of Ag3PO4 and the UCL spectrum of UCPs (Fig. 7b). Absorption of the upconverted photons of suitable energy by Ag3PO4 is then followed by its photoexcitation, generating oxidative holes (h+) and reductive electrons (e−) as a result of typical interband transitions. The oxidative holes in the VB and reductive electrons in the CB then take part in a series of redox processes on the surface of the semiconductor photocatalyst particles which ultimately leads to photodegradation of pollutants. It may be noted that the reductive electrons take part not only in redox processes (such as the formation of superoxide radicals, O2 + e− → O2˙−) but also in the self-reduction of Ag3PO4 (see eqn (5)), leading to the formation of metallic Ag and hence the photocorrosion of the photocatalysts (Fig. 7c). Then the addition of H2O2 not only prevents photocorrosion and/or assists in photocatalyst regeneration, but also produces ROS through a Fenton-like process, even in the dark. Fig. 7 schematically summarizes the mechanism of the photocatalytic activity of the UCPs–Ag3PO4 system under direct UV-vis light (direct excitation from LEDs) and/or upconverted UV-vis light obtained from UCPs under NIR illumination (indirect excitation), as well the process of photocatalyst regeneration using H2O2.
4. Conclusions
Taking advantage of the high photocatalytic activity of Ag3PO4, the interesting NIR-to-UV/vis upconversion ability of NaYbF4:Tm3+ UCPs and the oxidizing power of H2O2, we successfully developed a smart photocatalytic system that shows distinct features including (i) high photoactivity under both NIR and UV-vis light, (ii) better photostability of the otherwise photounstable Ag3PO4 and its regeneration for effective recycling and (iii) the possibility of achieving photocatalytic processes after the excitation light is turned off (light off mode). Highly photoactive Ag3PO4 particles were prepared by a simple precipitation method at room temperature. The UCPs were prepared by a facile MW-assisted hydrothermal method which allowed their crystallization in the hexagonal β-phase with a high NIR-to-UV/vis upconversion luminescence intensity under 980 nm laser irradiation. The upconverted UV-vis light obtained from UCPs under NIR excitation was successfully used to photoactivate the Ag3PO4 photocatalyst, achieving a high degradation rate of crystal violet dye. It was demonstrated that the presence of H2O2 has a positive influence on the stability of Ag3PO4, preventing its photocorrosion and regenerating the used photocatalysts. Importantly, the photocatalyst regeneration process also simultaneously produces reactive oxygen species in a Fenton-like manner which help in the degradation of the pollutants even when the excitation light is turned off. Moreover, the presence of H2O2 also increased the degradation rate of the crystal violet (CV) dye under NIR irradiation, reaching an almost complete degradation of the dye in 90 min. These results indicate that the coupling between the upconverting particles and suitable photocatalysts, in combination with other advanced oxidation processes, may allow the application of photocatalytic processes in an extended region of the electromagnetic spectrum.
Author contributions
Y. E. Serge Correales: conceptualization, methodology, investigation, visualization, writing – original draft, writing – review & editing. S. Ullah: conceptualization, methodology, visualization, writing – review & editing, supervision. E. P. Ferreira-Neto: methodology, writing – review & editing. H. D. Rojas-Mantilla: design and construction of the blue emitting LED reactor. C. Hazra: conceptualization, methodology. S. J. Lima Ribeiro: writing – review & editing, supervision, acquisition of funds, project management.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial assistance from Brazilian agencies FAPESP (grant no. 2015/22828-6), CNPq and CAPES is acknowledged. York E. Serge-Correales and Elias P. Ferreira-Neto acknowledge the financial support from FAPESP under fellowship grant numbers 2018/15791-7 and 2018/01934-0, respectively, and CNPq (166682/2020-9). S. Ullah acknowledges the financial support from Higher Education Commission (HEC) Pakistan (Project no. 9286). We also acknowledge Prof. Regina C. G. Frem from IQ-UNESP for the access to the microwave synthesizer.
Notes and references
- M. Patel, R. Kumar, K. Kishor, T. Mlsna, C. U. Pittman and D. Mohan, Chem. Rev., 2019, 119, 3510–3673 CrossRef CAS PubMed.
- R. S. Prosser and P. K. Sibley, Environ. Int., 2015, 75 Search PubMed.
- S. H. A. Koop and C. J. van Leeuwen, Environ. Dev. Sustainable, 2017, 19, 385–418 CrossRef.
- A. N. Rahmasary, S. Robert, I.-S. Chang, W. Jing, J. Park, B. Bluemling, S. Koop and K. van Leeuwen, Environ. Manage., 2019, 63, 520–535 CrossRef PubMed.
- E. P. Ferreira-Neto, S. Ullah, T. C. A. da Silva, R. R. Domeneguetti, A. P. Perissinotto, F. S. de Vicente, U. P. Rodrigues-Filho and S. J. L. Ribeiro, ACS Appl. Mater. Interfaces, 2020, 12, 41627–41643 CrossRef CAS PubMed.
- J. Kim, T. Zhang, W. Liu, P. Du, J. T. Dobson and C.-H. Huang, Environ. Sci. Technol., 2019, 53, 13312–13322 CrossRef PubMed.
- R. Kumar, M. Patel, P. Singh, J. Bundschuh, C. U. Pittman, L. Trakal and D. Mohan, Sci. Total Environ., 2019, 694, 133427 CrossRef CAS PubMed.
- T. Do Minh, J. Song, A. Deb, L. Cha, V. Srivastava and M. Sillanpää, Chem. Eng. J., 2020, 394, 124856 CrossRef CAS.
- J. M. Galindo-Miranda, C. Guízar-González, E. J. Becerril-Bravo, G. Moeller-Chávez, E. León-Becerril and R. Vallejo-Rodríguez, Water Supply, 2019, 19, 1871–1884 CrossRef.
- A. M. Gorito, J. F. J. R. Pesqueira, N. F. F. Moreira, A. R. Ribeiro, M. F. R. Pereira, O. C. Nunes, C. M. R. Almeida and A. M. T. Silva, J. Environ. Chem. Eng., 2021, 9, 105315 CrossRef CAS.
- S. F. Ahmed, M. Mofijur, S. Nuzhat, A. T. Chowdhury, N. Rafa, M. A. Uddin, A. Inayat, T. M. I. Mahlia, H. C. Ong, W. Y. Chia and P. L. Show, J. Hazard. Mater., 2021, 416, 125912 CrossRef CAS PubMed.
- S. F. Ahmed, M. Mofijur, T. A. Parisa, N. Islam, F. Kusumo, A. Inayat, V. G. Le, I. A. Badruddin, T. M. Y. Khan and H. C. Ong, Chemosphere, 2022, 286, 131656 CrossRef CAS PubMed.
- R. Ahmad, Z. Ahmad, A. U. Khan, N. R. Mastoi, M. Aslam and J. Kim, J. Environ. Chem. Eng., 2016, 4, 4143–4164 CrossRef CAS.
- S. Hu and X. Wang, Chem. Soc. Rev., 2013, 42, 5577 RSC.
- I. Y. Habib, J. Burhan, F. Jaladi, C. M. Lim, A. Usman, N. T. R. N. Kumara, S. C. E. Tsang and A. H. Mahadi, Catal. Today, 2021, 375, 506–513 CrossRef CAS.
- M. R. Al-Mamun, S. Kader, M. S. Islam and M. Z. H. Khan, J. Environ. Chem. Eng., 2019, 7, 103248 CrossRef CAS.
- D. Chen, Y. Cheng, N. Zhou, P. Chen, Y. Wang, K. Li, S. Huo, P. Cheng, P. Peng, R. Zhang, L. Wang, H. Liu, Y. Liu and R. Ruan, J. Clean. Prod., 2020, 268, 121725 CrossRef CAS.
- S. Younis and K.-H. Kim, Catalysts, 2020, 10, 1109 CrossRef CAS.
- K. Wetchakun, N. Wetchakun and S. Sakulsermsuk, J. Ind. Eng. Chem., 2019, 71, 19–49 CrossRef CAS.
- C. P. Sajan, A. Naik and H. N. Girish, Int. J. Environ. Sci. Technol., 2017, 14, 1513–1524 CrossRef CAS.
- F. Shi, J.-X. Liu, X. Huang, L. Yu, S.-H. Liu, X. Feng, X.-K. Wang, G.-L. Shao, S.-C. Hu, B. Yang and C.-Y. Fan, Adv. Powder Technol., 2015, 26, 1435–1441 CrossRef CAS.
- V. G. Parale, T. Kim, V. D. Phadtare, W. Han, K. Y. Lee, H. N. R. Jung, H. Choi, Y. Kim, H. M. Yadav and H. H. Park, J. Mol. Liq., 2019, 287, 110990 CrossRef CAS.
- L. Zheng, C. Chen, Y. Zheng, Y. Zhan, Y. Cao, X. Lin, Q. Zheng, K. Wei and J. Zhu, Appl. Catal., B, 2014, 148–149, 44–50 CrossRef CAS.
- X. Chen, X. Wang and X. Fu, Energy Environ. Sci., 2009, 2, 872 RSC.
- G. Xie, H. Wang, Y. Zhou, Y. Du, C. Liang, L. Long, K. Lai, W. Li, X. Tan, Q. Jin, G. Qiu, D. Zhou, H. Huo, X. Hu and X. Xu, J. Taiwan Inst. Chem. Eng., 2020, 112, 357–365 CrossRef CAS.
- D. Kandi, A. Behera, S. Sahoo and K. Parida, Sep. Purif. Technol., 2020, 253, 117523 CrossRef CAS.
- X. Yan, Z. Wu, C. Huang, K. Liu and W. Shi, Ceram. Int., 2017, 43, 5388–5395 CrossRef CAS.
- S. Ullah, Fayeza, A. A. Khan, A. Jan, S. Q. Aain, E. P. F. Neto, Y. E. Serge-Correales, R. Parveen, H. Wender, U. P. Rodrigues-Filho and S. J. L. Ribeiro, Colloids Surf., A, 2020, 600, 124946 CrossRef CAS.
- W. Wang, B. Cheng, J. Yu, G. Liu and W. Fan, Chem. – Asian J., 2012, 7, 1902–1908 CrossRef CAS PubMed.
- L. Luo, Y. Li, J. Hou and Y. Yang, Appl. Surf. Sci., 2014, 319, 332–338 CrossRef CAS.
- J. Cao, B. Luo, H. Lin, B. Xu and S. Chen, J. Hazard. Mater., 2012, 217–218, 107–115 CrossRef CAS PubMed.
- Z. Yi, J. Ye, N. Kikugawa, T. Kako, S. Ouyang, H. Stuart-Williams, H. Yang, J. Cao, W. Luo, Z. Li, Y. Liu and R. L. Withers, Nat. Mater., 2010, 9, 559–564 CrossRef CAS PubMed.
- Q. Xiang, D. Lang, T. Shen and F. Liu, Appl. Catal., B, 2015, 162, 196–203 CrossRef CAS.
- S. Ullah, E. P. Ferreira-Neto, A. A. Pasa, C. C. J. Alcântara, J. J. S. Acuña, S. A. Bilmes, M. L. Martínez Ricci, R. Landers, T. Z. Fermino and U. P. Rodrigues-Filho, Appl. Catal., B, 2015, 179, 333–343 CrossRef CAS.
- F. Li, D. Zhang and Q. Xiang, Chem. Commun., 2020, 56, 2443–2446 RSC.
- Y. Li, X. Li, H. Zhang and Q. Xiang, Nanoscale Horiz., 2020, 5, 765–786 RSC.
- H. Guo, C.-G. Niu, D.-W. Huang, N. Tang, C. Liang, L. Zhang, X.-J. Wen, Y. Yang, W.-J. Wang and G.-M. Zeng, Chem. Eng. J., 2019, 360, 349–363 CrossRef CAS.
- Y. Li, L. Yao, Z. Yin, Z. Cheng, S. Yang and Y. Zhang, Inorg. Chem. Front., 2020, 7, 519–528 RSC.
- X. Zhang, M. Liu, Z. Kang, B. Wang, B. Wang, F. Jiang, X. Wang, D.-P. Yang and R. Luque, Chem. Eng. J., 2020, 388, 124304 CrossRef CAS.
- T. Ni, Q. Li, Y. Yan, Z. Yang, K. Chang and G. Liu, Front. Mater., 2021, 8, 649411 CrossRef.
- W. Fan, H. Bai and W. Shi, CrystEngComm, 2014, 16 Search PubMed.
- A. Jarosz-Duda, P. O’Callaghan, J. Kuncewicz, P. Łabuz and W. Macyk, Catalysts, 2020, 10, 232 CrossRef CAS.
- P. Acosta-Mora, K. Domen, T. Hisatomi, H. Lyu, J. Méndez-Ramos, J. C. Ruiz-Morales and N. M. Khaidukov, Opt. Mater., 2018, 83, 315–320 CrossRef CAS.
- S. Ullah, C. Hazra, E. P. Ferreira-Neto, T. C. Silva, U. P. Rodrigues-Filho and S. J. L. Ribeiro, CrystEngComm, 2017, 19, 3465–3475 RSC.
- S. Ullah, E. P. Ferreira-Neto, C. Hazra, R. Parveen, H. D. D. Rojas-Mantilla, M. L. L. Calegaro, Y. E. E. Serge-Correales, U. P. P. Rodrigues-Filho and S. J. L. J. L. Ribeiro, Appl. Catal., B, 2019, 243, 121–135 CrossRef CAS.
- G. Wang, W. Qin, J. Zhang, L. Wang, G. Wei, P. Zhu and R. Kim, J. Alloys Compd., 2009, 475, 452–455 CrossRef CAS.
- G. De, W. Qin, J. Zhang, J. Zhang, Y. Wang, C. Cao and Y. Cui, J. Lumin., 2007, 122–123, 128–130 CrossRef CAS.
- D. Chen, Y. Wang, Y. Yu and P. Huang, Appl. Phys. Lett., 2007, 91, 30–33 Search PubMed.
- S. Wen, J. Zhou, K. Zheng, A. Bednarkiewicz, X. Liu and D. Jin, Nat. Commun., 2018, 9, 2415 CrossRef PubMed.
- Y. E. Serge Correales, C. Hazra, S. Ullah, L. R. Lima and S. J. L. Ribeiro, Nanoscale Adv., 2019, 1, 1936–1947 RSC.
- Z. Zhang, Q. Han, J. W. Lau and B. Xing, ACS Mater. Lett., 2020, 2, 1516–1531 CrossRef CAS.
- X. Cheng, D. Tu, W. Zheng and X. Chen, Chem. Commun., 2020, 56, 15118–15132 RSC.
- L. Bai, W. Jiang, C. Gao, S. Zhong, L. Zhao, Z. Li and S. Bai, Nanoscale, 2016, 8, 19014–19024 RSC.
- H. Kwon, F. Marques Mota, K. Chung, Y. J. Jang, J. K. Hyun, J. Lee and D. H. Kim, ACS Sustainable Chem. Eng., 2018, 6, 1310–1317 CrossRef CAS.
- S. Wu, J. Lv, F. Wang, N. Duan, Q. Li and Z. Wang, Sci. Rep., 2017, 7, 14435 CrossRef PubMed.
- Y. Yang, W. Que, X. Zhang, X. Yin, Y. Xing, M. Que, H. Zhao and Y. Du, Appl. Catal., B, 2017, 200, 402–411 CrossRef CAS.
- J. Wang, H. Wang, S. Zuo, X. Jin, B. Zheng, R. Deng, W. Liu and J. Wang, Environ. Sci.: Nano, 2020, 7, 3333–3342 RSC.
- P. Mazierski, J. K. Roy, A. Mikolajczyk, E. Wyrzykowska, T. Grzyb, P. N. A. Caicedo, Z. Wei, E. Kowalska, A. Zaleska-Medynska and J. Nadolna, Appl. Surf. Sci., 2021, 536, 147805 CrossRef CAS.
- C. Dong, M. Xing and J. Zhang, Front. Environ. Chem., 2020, 1, 8 CrossRef.
- Y. Mokhbi, M. Korichi and Z. Akchiche, Appl. Water Sci., 2019, 9, 35 CrossRef.
- V. Augugliaro, M. Litter, L. Palmisano and J. Soria, J. Photochem. Photobiol., C, 2006, 7, 127–144 CrossRef CAS.
- Y. Bi, S. Ouyang, N. Umezawa, J. Cao and J. Ye, J. Am. Chem. Soc., 2011, 133, 6490–6492 CrossRef CAS PubMed.
- P. Dong, G. Hou, C. Liu, X. Zhang, H. Tian, F. Xu, X. Xi and R. Shao, Materials, 2016, 9, 968 CrossRef PubMed.
- G. Botelho, J. C. Sczancoski, J. Andres, L. Gracia and E. Longo, J. Phys. Chem. C, 2015, 119, 6293–6306 CrossRef CAS.
- X. Ma, B. Lu, D. Li, R. Shi, C. Pan and Y. Zhu, J. Phys. Chem. C, 2011, 115, 4680–4687 CrossRef CAS.
- Y. M. Moustafa and K. El-Egili, J. Non Cryst. Solids, 1998, 240, 144–153 CrossRef CAS.
- Q. Liang, Y. Shi, W. Ma, Z. Li and X. Yang, Phys. Chem. Chem. Phys., 2012, 14, 15657 RSC.
- L. Zhou, O. G. Alvarez, C. S. Mazon, L. Chen, H. Deng and M. Sui, Catal. Sci. Technol., 2016, 6, 5972–5981 RSC.
- J. Ma, Q. Liu, L. Zhu, J. Zou, K. Wang, M. Yang and S. Komarneni, Appl. Catal., B, 2016, 182, 26–32 CrossRef CAS.
- M. Mroczkowska, J. L. Nowinski, G. Z. Zukowska, A. Mroczkowska, J. E. Garbarczyk, M. Wasiucionek and S. Gierlotka, J. Power Sources, 2007, 173, 729–733 CrossRef CAS.
- A. S. Pinheiro, A. M. Freitas, G. H. Silva, M. J. V. Bell, V. Anjos, A. P. Carmo and N. O. Dantas, Chem. Phys. Lett., 2014, 592, 164–169 CrossRef CAS.
- D. He, A. M. Jones, S. Garg, A. N. Pham and T. D. Waite, J. Phys. Chem. C, 2011, 115, 5461–5468 CrossRef CAS.
- Y. Ono, T. Matsumura, N. Kitajima and S. I. Fukuzumi, J. Phys. Chem., 1977, 81, 1307–1311 CrossRef CAS.
- A. M. Jones, S. Garg, D. He, A. N. Pham and T. D. Waite, Environ. Sci. Technol., 2011, 45, 1428–1434 CrossRef CAS PubMed.
- D. He, C. J. Miller and T. D. Waite, J. Catal., 2014, 317, 198–205 CrossRef CAS.
- W. He, Y.-T. Zhou, W. G. Wamer, M. D. Boudreau and J.-J. Yin, Biomaterials, 2012, 33, 7547–7555 CrossRef CAS PubMed.
- T. Yan, W. Guan, J. Tian, P. Wang, W. Li, J. You and B. Huang, J. Alloys Compd., 2016, 680, 436–445 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma01028j |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Sajjad
Ullah
a,
Sajjad
Ullah